
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in metal-catalysed asymmetric sigmatropic rearrangements
Yangbin
Liu
a,
Xiaohua
Liu
 *b and
Xiaoming
Feng
*ab
*b and
Xiaoming
Feng
*ab
aInstitute of Chemical Biology, Shenzhen Bay Laboratory, Shenzhen 518132, China
bKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 23rd September 2022
Abstract
Asymmetric sigmatropic rearrangement is a powerful organic transformation via substrate-reorganization to efficiently increase molecular complexity from readily accessible starting materials. In particular, a high level of diastereo- and enantioselectivity can be readily accessed through well-defined and predictable transition states in [3,3], [2,3]-sigmatropic rearrangements, which have been widely applied in the synthesis of various chiral building blocks, natural products, and pharmaceuticals. In recent years, catalytic asymmetric sigmatropic rearrangements involving chiral metal complexes to induce stereocontrol have been intensively studied. This review presents an overview of metal-catalysed enantioselective versions of sigmatropic rearrangements in the past two decades, mainly focusing on [3,3], [2,3], and [1,3]-rearrangements, to show the development of substrate design, new catalyst exploitation, and novel cascade processes. In addition, their application in the asymmetric synthesis of complex natural products is also exemplified.
1. Introduction
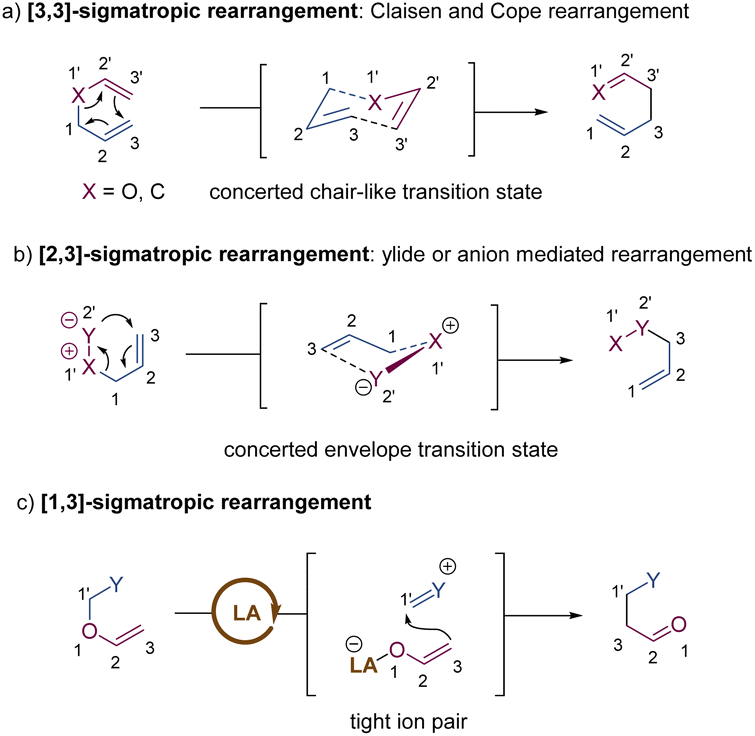
Sigmatropic rearrangements are defined by the migration of a σ-bond that is flanked by at least one π-system to a new position within the same molecule, without changing the total number of σ-bonds and π-bonds (Scheme 1).1 As one of the most important and fundamental transformations in organic chemistry, sigmatropic rearrangements can directly increase the complexity of the molecular structure from readily accessible starting materials.2 In particular, a high level of diastereo- and enantioselectivity can be well accessed through well-defined and predictable transition states in [3,3], [2,3]-sigmatropic rearrangements, which have been widely applied in the synthesis of various chiral building blocks, complex natural products, and pharmaceuticals.3 Although there are many well-established stereoselective sigmatropic rearrangements based on chiral auxiliaries or enantiomerically enriched starting materials, catalytic asymmetric sigmatropic rearrangements promoted by chiral catalysts are considered as the most attractive method, and significant advances have been made during the past decade. In general, the chiral catalyst-rearranged substrate adduct may undergo two diastereomeric pathways with different activation energies, in which the steric interactions between rearranged substrates and chiral catalysts primarily control the stereoselectivity, mostly following a less sterically hindered pathway. The Re or Si face of a prochiral π-bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) can be precisely recognized and a new σ-bond will be stereoselectively formed after migration, leading to one particular (R or S) enantiomer as the major product.4 Since the chiral catalysts are not consumed in the rearrangement process, substoichiometric chiral catalysts can be used to greatly improve the conversion efficiency and avoid waste.
C) can be precisely recognized and a new σ-bond will be stereoselectively formed after migration, leading to one particular (R or S) enantiomer as the major product.4 Since the chiral catalysts are not consumed in the rearrangement process, substoichiometric chiral catalysts can be used to greatly improve the conversion efficiency and avoid waste.
 | ||
| Scheme 1 [3,3], [2,3], and [1,3]-sigmatropic rearrangement. | ||
The catalytic asymmetric systems of sigmatropic rearrangement reactions can be broadly categorized into three main types: (a) enzyme catalysis, for instance, the chorismate mutase catalyses the [3,3]-rearrangement of the shikimate branch pathway specifically to furnish phenylalanine and tyrosine biosynthesis;5 (b) organocatalysis6 generally exerts asymmetric induction on sigmatropic rearrangements through multiple interactions between catalysts and substrates including but not limited to hydrogen bonds (Brønsted acid, thiourea, and guanidinium), or covalent bonds (organoamine, N-heterocyclic-carbene, and Lewis base); (c) metal-catalysis is the most intensively explored process in the field of catalytic stereoselective sigmatropic rearrangements.7 Based on the variation of the rearrangement type, the substrate structure or the innate properties of the metal catalyst, diverse models of metal-activation have been developed. In theory, the precursors of sigmatropic rearrangements containing heteroatom-substituents (O, N, S, etc.) or π-bonds (alkene, alkyne) at various positions with different electronic properties can significantly influence the reactivity of sigmatropic rearrangements. Therefore, a unique advantage of chiral metal catalysts is that they can serve as an electron acceptor to regulate the electronic properties of the heteroatom-substituents or π-bonds, thereby increasing the rate of rearrangement reactions via the charge-acceleration effect and providing effective stereochemical induction, simultaneously. Moreover, combination of various metals and chiral ligands can conveniently provide numerous modular chiral metal complex catalysts, which are more conducive to achieving high levels of reactivity and stereoselectivity in sigmatropic rearrangements.
This review article is to summarize recent progress made since 2010 in metal-catalysed asymmetric [3,3], [2,3], and [1,3]-rearrangement and discuss the related opportunities and challenges. We hope it will inspire worldwide researchers to work together in this area. To be noticed, it is not our original intention to give a complete list of all available literature. The purpose is to bring readers a concise review to have an overview of notable features in metal-catalysed sigmatropic rearrangements. Therefore, selected and representative literature that demonstrated pioneering breakthroughs or significant improvement will be covered in this article.
2. [3,3]-Sigmatropic rearrangements
Since the first discovery of the classical [3,3]-sigmatropic rearrangement–Claisen rearrangement reaction in 1912,8 a variety of its modified variants (including Cope rearrangement) have been one of the most useful tools for the formation of carbon–carbon bonds, enjoying unparalleled value in the synthesis of complex organic structures.9 The thermal [3,3]-sigmatropic rearrangement is proposed to undergo a concerted pericyclic mechanism with a chair-like transition state (Scheme 2a).10 As a result, the stereochemical information of substrates bearing stereocenters or chiral auxiliary groups can be effectively transferred into the products by minimizing unfavorable 1,3-diaxial interactions.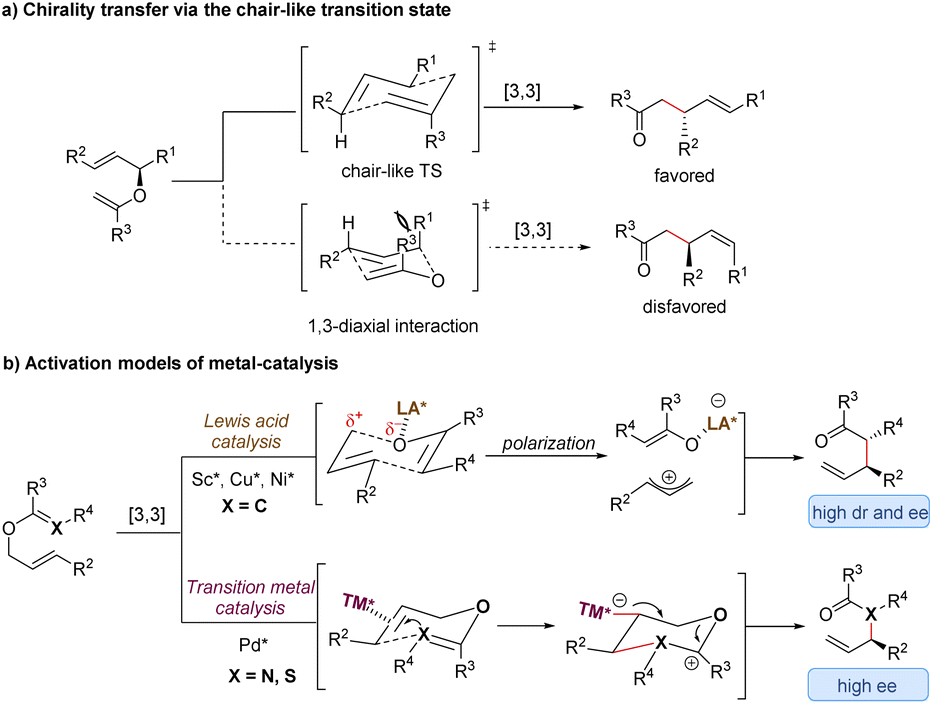 | ||
| Scheme 2 Stereochemical mechanism of [3,3]-sigmatropic rearrangement and activation models of metal-catalysis. TS = transition state; LA = Lewis acid; TM = transition metal. | ||
Generally, two activation models: Lewis acid catalysis and transition metal catalysis, can be classified for metal-catalysis in asymmetric [3,3]-sigmatropic rearrangements (Scheme 2b). Since ammonium chloride was first used to promote Claisen rearrangement, numerous other Brønsted acids and Lewis acids have demonstrated apparent catalytic activity. Theoretical and experimental studies on the mechanism of [3,3]-sigmatropic rearrangements have revealed a powerful charge-accelerated effect.11 The metal ion as a Lewis acid can coordinate with the ether oxygen atom of the substrate, and polarize the electrically neutral substrate to form a Lewis acid-stabilized enolate and an allyl cation. For the perspective of frontier molecular orbital theory, the LUMO energy level of the allyl moiety is lowered and the HOMO energy level of the enol moiety is raised. Consequently, the Lewis acid-catalysed [3,3]-rearrangement is remarkably accelerated through a charge-delocalized transition state, and the catalytic asymmetric version also becomes possible by the coordination of chiral ligands with Lewis acids (Scheme 2b, up).12
In addition, some late transition metals as π-acids can exert influence upon the mechanism of [3,3]-sigmatropic rearrangements, changing it from a concerted process into a stepwise cyclization process (Scheme 2b, down). One classical example is the well-known Overman rearrangement. Mechanistically, the double bond of the alkene unit coordinates with a chiral transition metal–palladium complex, followed by an intramolecular enantioselective anti-attack to generate a six-membered intermediate. Subsequently, the β-elimination releases the transition metal catalyst and yields the corresponding optically active rearrangement product. Due to space limitations of this review, the stepwise [3,3]-rearrangements catalysed by chiral transition-metal complexes will not be covered in this article. The reader can follow several pertinent reviews for details.13
2.1. Direct asymmetric [3,3]-rearrangements
Allyl vinyl ether derivatives are typical substrates for [3,3]-rearrangements, and well-designed functional groups (ester, amide) in the precursors could be introduced to improve the reactivity and enantioselectivity via a bidentate chelation with a Lewis-acid metal, which renders the catalytic system simple and controllable.Previously, stoichiometric chiral Lewis acid catalysts were generally required to overcome the high activation barrier and suppress the racemic thermal background reaction in [3,3]-sigmatropic rearrangements. In 2001, Hiersemann and co-workers reported the first asymmetric Claisen rearrangement of 2-ester substituted allyl vinyl ethers 2 in the presence of a catalytic amount of the chiral Lewis acid, bis(oxazoline)–copper(II) complex (Scheme 3).14 In this case, an extra ester functional group was essential for the bidentate coordination with the chiral Lewis acid complex to increase rearrangement reactivity and provide better stereocontrol. On the basis of the stereochemical analysis proposed by Hiersemann, a stereocontrol model was speculated (not yet confirmed): allyl vinyl ether 2 coordinated with the Cu(II) ion in a bidentate fashion, forming a relatively rigid chair-like conformation. To minimize steric repulsion, the allyl unit preferred to approach the vinyl-ether double bond on the side opposite to the phenyl group of the BOX ligand, leading to Re-face attack on the Z-configured vinyl ether to deliver a (3R)-configuration product. Diastereoselectivity was determined by the relative locations of Me and nPr in the chair-like transition state. This approach not only represents the first example of a truly catalytic asymmetric Claisen rearrangement but it also opens up new avenues in the development of chiral Lewis-acid-metal catalysis in asymmetric [3,3]-rearrangements.
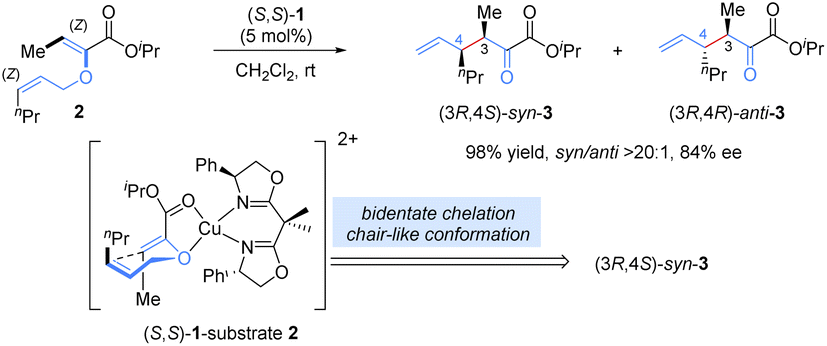 | ||
| Scheme 3 Copper-catalysed asymmetric Claisen rearrangement of enolesters. | ||
To demonstrate the synthetic potential of chiral Lewis-acid-mediated enantioselective Claisen rearrangements, the same group disclosed an asymmetric total synthesis of (−)-ecklonialactone B and its analogs (Scheme 4).15 The enantiomerically enriched α-ketoester anti-6 was readily prepared on a gram scale through the asymmetric Claisen rearrangement of the enolester (E,Z)-4 catalysed by a bis(oxazoline)–copper(II) complex (S,S)-5. Subsequently, a highly diastereoselective reduction was subjected by the bulky K-selectride reducing agent to afford the α-hydroxyl ester 7 as a single diastereomer (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.). After reductive homology and ring-closing metathesis, the desired chiral cyclopentene-like building block 8 was generated, which was further transformed into 14-membered (−)-ecklonialactone B.
:1 d.r.). After reductive homology and ring-closing metathesis, the desired chiral cyclopentene-like building block 8 was generated, which was further transformed into 14-membered (−)-ecklonialactone B.
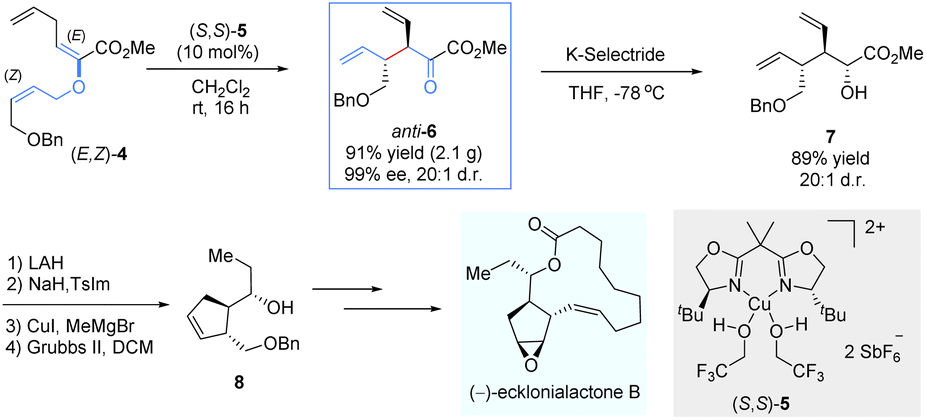 | ||
| Scheme 4 Enantioselective total synthesis of (−)-ecklonialactone B. | ||
Yamamoto and co-workers developed a similar approach for catalytic enantioselective rearrangement of allyl vinyl ethers containing an extra phosphonate group (Scheme 5).16 As shown in a plausible transition-state model, using (R,R)-1 as the catalyst, the phosphonate group can provide an additional binding site to tightly coordinate with the copper(II) ion, resulting in a good enantioface differentiation. A wide range of α-keto-phosphonate derivatives 10 with contiguous tertiary and quaternary carbon centers were afforded in excellent yields and enantioselectivities. Previous [3,3]-rearrangements of allyl vinyl ethers14,15 relied heavily on α-ketocarboxylic acid derivatives as substrates, thus affording products with limited generality and obvious difficulty in further functionalization. By contrast, the present products, α-ketophosphonates, are more synthetically useful and can be transformed into other functional groups with ease and efficiency.17
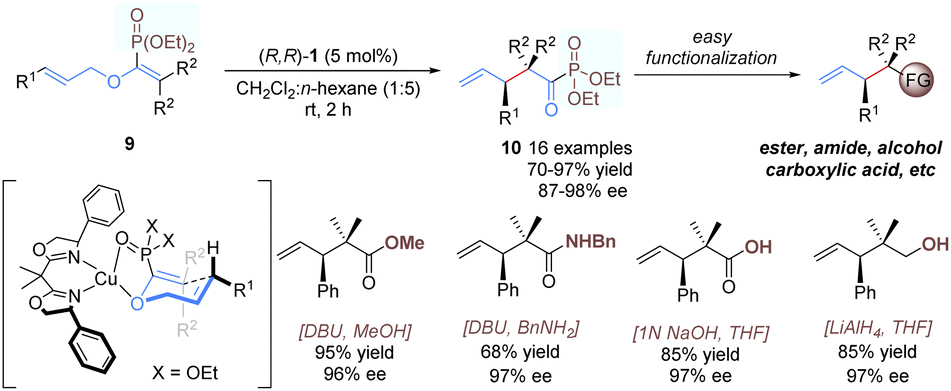 | ||
| Scheme 5 Copper-catalysed asymmetric Claisen rearrangement of enol-phosphonates. | ||
Oxindoles bearing C3-all-carbon quaternary stereocenters are central building blocks for the construction of indole alkaloids, and have attracted widespread attention in organic synthesis.18 The Kozlowski group reported an elegant design for preparing such chiral oxindoles derivatives 12, based on an enantioselective Meerwein–Eschenmoser Claisen rearrangement of indole-containing substrates 11. This reaction was remarkably accelerated by the chiral palladium(II) complex, occurring within 5–30 min at 0 °C, and almost no background reaction was detected in the absence of catalysts (Scheme 6a).19 In view of the control experiment that no deuterium scrambling occurred from labeled substrate d-11, both the Lewis acidic catalysis and the stepwise pathway via π-acid activation were considered to be possible, excluding the π-allyl cation palladium(II) (Scheme 6b). Later, the author carried out detailed computational (DFT) studies,20 revealing that the concerted Lewis acid catalysis was more favorable. Therefore, a stereochemical model based on a two-point chelating coordination of the substrate-amidoester moiety to the chiral palladium(II) complex was proposed.
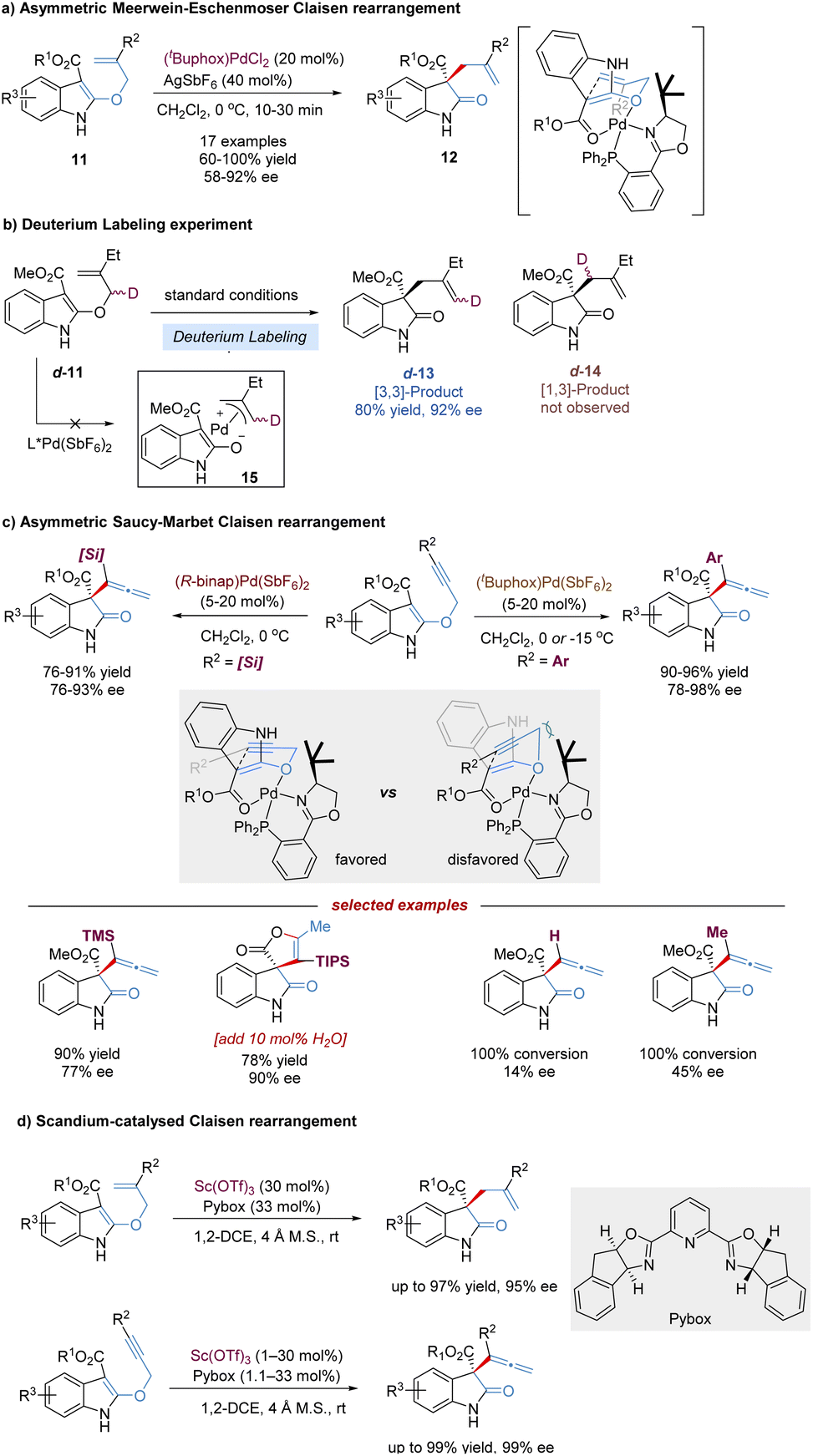 | ||
| Scheme 6 Palladium and scandium-catalysed asymmetric Claisen rearrangement. | ||
Propargyl Claisen rearrangements are attractive platforms for the generation of synthetically valuable functionalized allenes. But enantioselective catalytic versions of the propargyl vinyl rearrangement have been a challenging task and remain relatively rare, probably due to the cylindrically symmetric alkyne resulting in difficulties in facial discrimination. In 2012, Kozlowski disclosed the first Pd-catalysed asymmetric propargyl [3,3]-rearrangement of 3-ester substituted indoles, named Saucy–Marbet Claisen rearrangement, for the synthesis of allenyl oxindoles and spirolactones (Scheme 6c).21 As expected, the alkyne terminal substituents significantly affect the enantioselectivity of the propargyl Claisen rearrangement, thereby leading to the tuning of various chiral ligands to achieve better stereodifferentiation. Alkynes with ortho-substituted aryl groups at R2 afforded high levels of enantioselection (96–98% ee), while smaller groups, such as H- and Me, gave relatively poor selectivities (14–45% ee). With the use of a [Pd(II)binap] complex, larger silyl groups such as TBS, TES and TIPS (excluding TMS) allowed the enantioselectivity to be retained (89–90% ee). Interestingly, for a much larger TIPS substrate, the addition of a small amount of water provided a spirolactone product in one step with high efficiency, which might involve Pd(II)-catalysed allene hydration to form a ketone followed by an intermolecular cyclization. However, in 2018, the Kozlowski group examined the detailed mechanism of this Pd(II)-catalysed sigmatropic shift of 3-propargyloxyindoles using density functional theory.20 These calculations revealed a surprising reaction pathway, where π-acid activation predominated for alkyne [3,3]-sigmatropic rearrangement, not the Lewis acid coordination.
In 2017, You and co-workers developed a Sc(OTf)3–Pybox catalysis system instead of the precious Pd(II) complex to improve the reactivity and enantioselectivity of asymmetric Claisen rearrangement reactions of 2-allyloxyindoles and 2-propargyloxyindoles.22 Diverse 3-allyloxyindoles and 3-allenyloxindoles were provided in excellent yields (up to 99%) and enantioselectivities (up to 99% ee) under mild reaction conditions (Scheme 6d).
Chiral N,N′-dioxide ligands23 developed by the Feng group are confirmed to serve as unique tetradentate oxygen ligands and can coordinate with various metal ions to form well-defined chiral Lewis acid catalysts, which are found to be extremely efficient for diverse asymmetric transformations including sigmatropic rearrangement reactions (Scheme 7a). As such, the Feng group disclosed that the readily cheap nickel (instead of previous noble metals, Pd or Au) with a chiral N,N′-dioxide ligand (L3-PiMe2) also smoothly enabled the enantioselective propargyl Claisen rearrangements of cyclic β-ketoesters (Scheme 7b).24 This approach demonstrated an excellent scope of the alkyne moiety and exhibited exceptional functional group compatibility at R1 (aryl, hetero-aryl, alkyl, H, and TMS). A variety of allenes with a quaternary all-carbon stereocenter were obtained in excellent yields and enantiomer excesses. Control experiments indicated that the presence of a 3-ester group was crucial for both reactivity and stereoselectivity. Meanwhile, a bidentate coordination model was also proposed through the ether oxygen atom and the carbonyl group. The propargyl unit preferentially approached the enolate ether from the Re face to avoid steric hindrance with the amide group in the ligand.
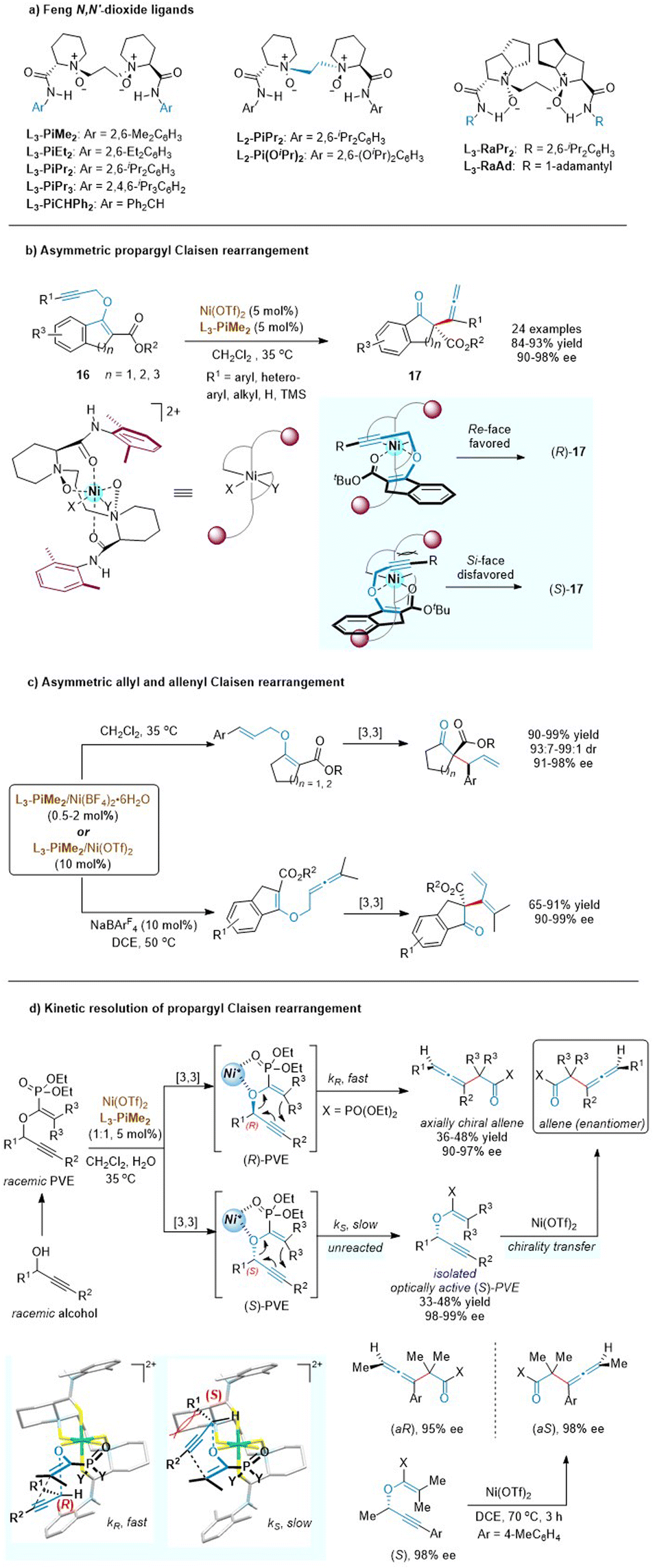 | ||
| Scheme 7 Structures of Feng N,N′-dioxide ligands, asymmetric Claisen rearrangement and kinetic resolution. | ||
The availability and generality of the chiral N,N′-dioxide/nickel(II) complex were further investigated in asymmetric allyl and allenyl Claisen rearrangements based on similar cyclic β-ketoester variants (Scheme 7c).24,25 Although the substrate scope of O-allyl β-ketoesters was mostly limited to aryl substituents, the catalyst loading was lowered to 0.5 mol% without deterioration of the outcomes. This exciting result changes the previously inherent view that high loadings of chiral Lewis acids are required in asymmetric Claisen rearrangements due to the tight complexation between the Lewis acid and the carbonyl group of the product. Furthermore, the less developed Claisen rearrangement of allenyl vinyl ethers was first realized in a catalytic asymmetric version, to access 1,3-butadienyl substituted quaternary stereocenters. Comparatively, the allenyl Claisen rearrangement required a higher temperature to overcome the activation energy, rendering the starting material to decompose under such harsh conditions. The addition of a noncoordinating tetraarylborate counterion (NaBArF4) could significantly improve the conversion to 90% while maintaining an ee value of 99%. This study fills a gap in the field of asymmetric allenyl Claisen rearrangement.
Generally, the central chirality of enantioenriched propargyl vinyl ethers (PVEs) can be completely transferred into the axial chirality of the rearranged products via a concerted mechanism in alkyne Claisen rearrangement.26 Based on this stereochemical feature, the Feng group developed a novel kinetic resolution process of propargyl Claisen rearrangement (Scheme 7d).27 In the presence of a chiral L3-PiMe2/nickel(II) complex, the R-enantiomer of the racemic PVE would undergo a [3,3]-rearrangement reaction faster than the S-enantiomer to generate an enantiomerically enriched axially chiral allene. Meanwhile, the unreacted S-enantiomer of PVE could be isolated by column chromatography in an optically active form, which was easily converted into the opposite enantiomer of the chiral allene product obtained in the kinetic-resolution system. As depicted in stereochemical models, enolphosphonates are tightly coordinated to Ni(II) via a bidentate chelation. The rearrangement rate of the (S)-PVE-catalyst complex is relatively slow because there is an obvious steric interaction between the substituents (R1) at the stereocenter of the ether and the upper piperidine backbone of L3-PiMe2.
The Claisen rearrangement is an efficient approach for the assembly of sterically congested structures by a single step. In 2008, Feng and co-workers reported a catalytic enantioselective dearomatization Claisen rearrangement of allyl furyl ethers to achieve adjacent quaternary–quaternary stereocenters with high steric hindrance (Scheme 8).28 It was found that the diastereoselectivity could be significantly improved by using a bulky silyl protecting group (TIPS) and reducing the temperature to 35 °C, but leading to prolonging of the reaction time to 4 days. By adjusting the (E)-/(Z)-geometry of the double bond in the allyl unit of substrates 18 with matched enantiomers of the ligands, all four stereoisomers of the rearranged products 19 were achieved in good yields with excellent diastereo- and enantioselectivities. Of note, the diastereomeric products (S,S)-19 and (S,R)-19 could undergo one-pot deprotection/lactonization to deliver hyperlactone C and epi-hyperlactone C in almost quantitative yields, which are a family of spirolactone natural products isolated from Hypericum chinese L., exhibiting biological activity against HIV replication.29
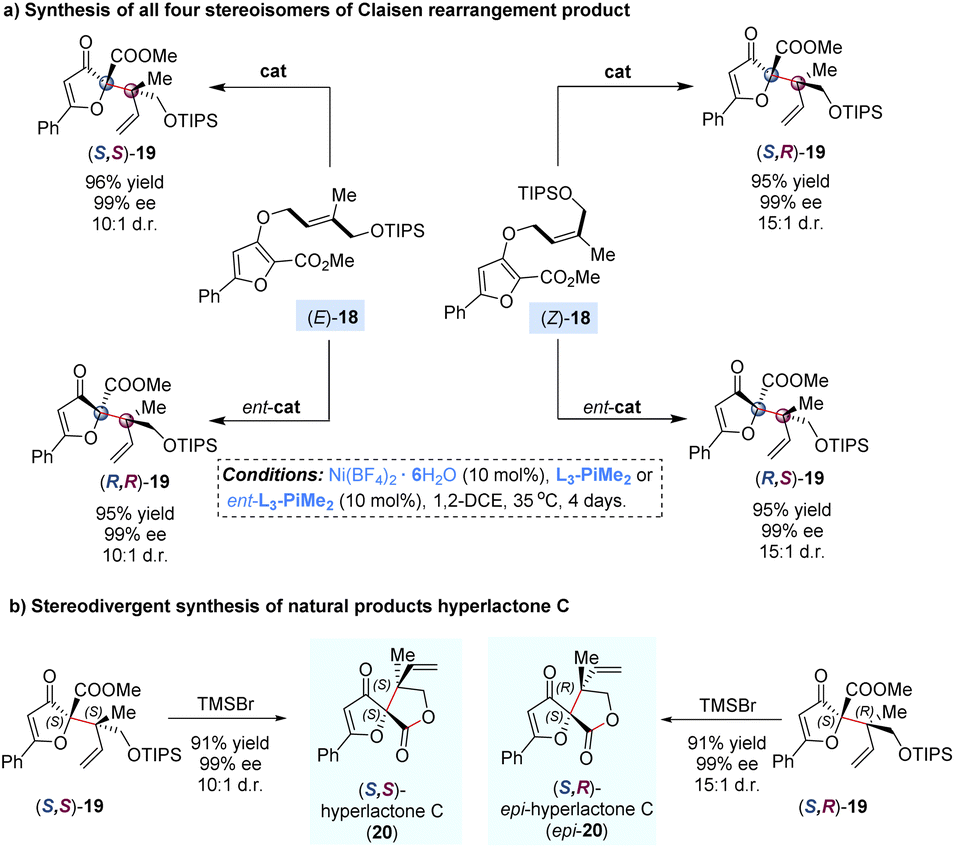 | ||
| Scheme 8 Nickel-catalysed Claisen rearrangement and stereodivergent synthesis of hyperlactone C and its diastereoisomer. | ||
2.2. Tandem asymmetric [3,3]-rearrangements
Tandem reactions are highly attractive in organic transformations because diverse complicated substances could be easily accessed with exquisite control and efficiency from relatively simple starting materials.30 As such, tandem [3,3]-rearrangement reactions have also achieved remarkable advances over the past few years. The incorporation of suitable transformation and [3,3]-rearrangement not only realizes the in situ preparation of rearrangement precursors avoiding the sometimes tedious synthesis and handling of substrates, but also makes the diversified transformations possible. Generally, there are mainly two types of asymmetric tandem [3,3]-rearrangements. First, the starting materials undergo various transformations to generate achiral precursors in situ, subsequently followed by the catalytic enantioselective [3,3]-rearrangement in the presence of chiral metal catalysts (Scheme 9a). Second, optically active chiral precursors are prepared via initial transformations, which would retain the stereochemical information of the chiral precursors in the corresponding rearranged products (Scheme 9b). Considering the brevity and relevance of the article, the second category of studies is not discussed here.31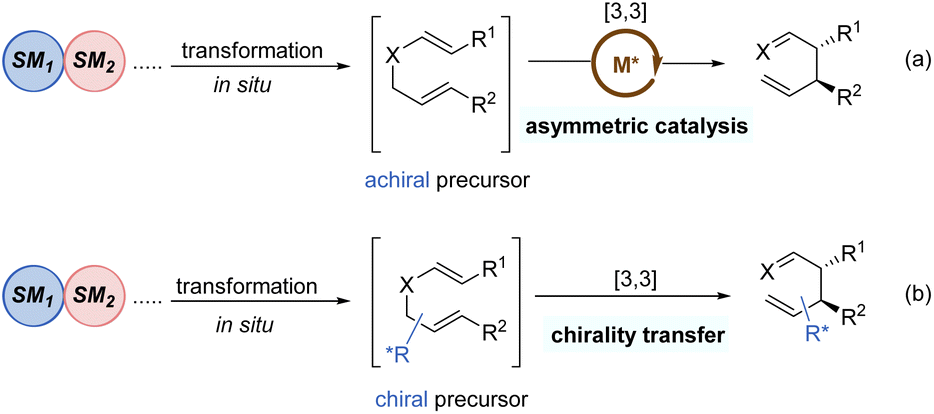 | ||
| Scheme 9 Tandem asymmetric [3,3]-rearrangement. | ||
Gold(I)-catalysed nucleophilic alkoxylation reactions of alkynes have been the subject of numerous investigations. For example, addition of allylic alcohols or ethers to highly electrophilic alkynes promoted by a Au(I) catalyst would generate in situ reactive allyl vinyl ethers, followed by Claisen rearrangement to engender both C–O and C–C bonds in a single step. In 2015, Toste and co-workers implemented an elegantly gold(I)-catalysed desymmetrization of 1,4-dienes through the asymmetric tandem alkoxylation/Claisen rearrangement (Scheme 10).32 This reaction system was highly selective and efficient for the synthesis of chiral multi-substituted cycloheptenes in good yields and enantioselectivities, which are common skeletons in natural products and bioactive molecules. It is worth mentioning that the enantiocontrol of this process does not rely on the nucleophilic addition of alkyne activated by the gold(I)-complex because this step only forms an achiral intermediate 23. Instead, the chiral gold(I) catalyst differentiates these two symmetric alkenyl substituents in transition state 24 following a chirality transfer process. This study inspires future organic chemists to develop more chiral gold(I) complexes for enantioselective [3,3]-rearrangements.
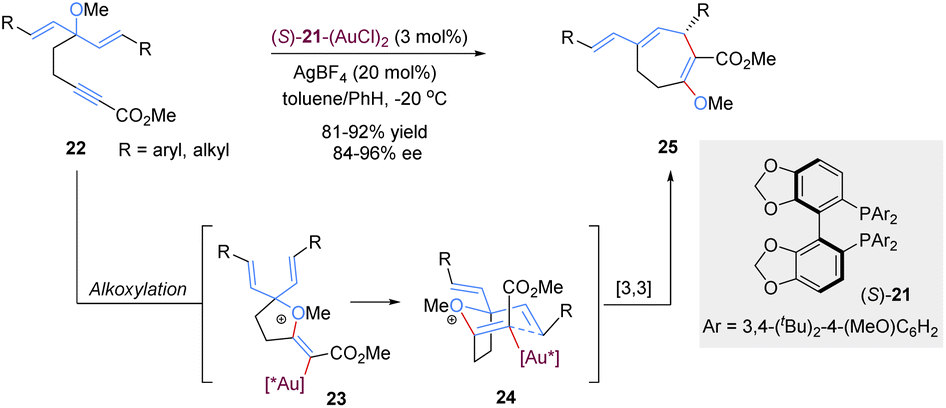 | ||
| Scheme 10 Gold-catalysed tandem alkoxylation/Claisen rearrangement. | ||
Recently, the Shin group described the first gold-catalysed enantioselective thioallylation propiolates with allyl sulfides via a charge-induced thio-Claisen rearrangement (Scheme 11).33 Comparing with allylic ethers which are less reactive in the intermolecular alkoxylation, the sulfur atom of allylic thioethers shows stronger nucleophilicity in gold-activated alkynes, and can facilitate the challenging intermolecular addition process, furthermore, due to the less polarized C–S bond, the undesired allyl dissociation from the sulfonium intermediate can be mitigated. This protocol featured the remarkable scope of allyl thioethers, allowing the synthesis of diverse enantioenriched 1,4-dienes embedded with β-thioacrylates bearing a tertiary (27a), or quaternary stereocenter (27b), even a chiral lactone scaffold (27c) via a spontaneous auro-lactonization in one-pot. The vinyl sulfide moiety in the product was transformed into various useful groups. Likewise, the vinyl sulfide was easily oxidized by mCPBA to give a sulfone, which could serve as an excellent functional handle for desulfonation, Suzuki-cross coupling, substitution with alcohols, and Cu-catalysed coupling with Grignard reagent.
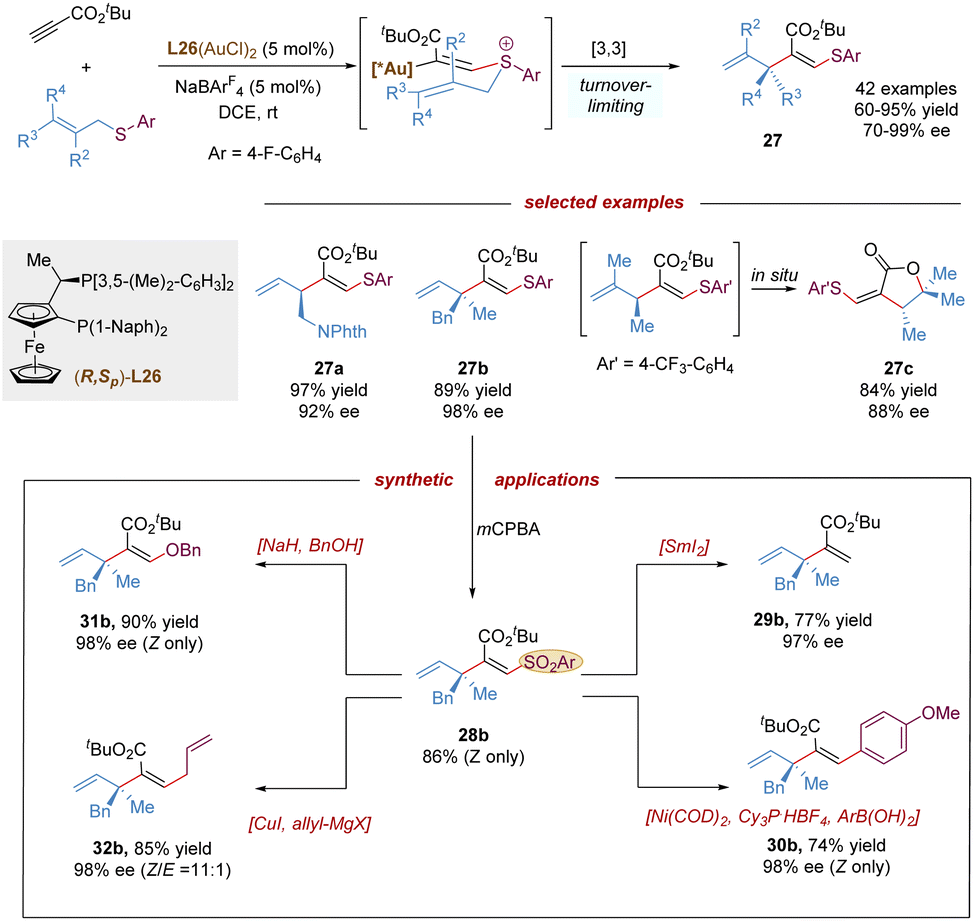 | ||
| Scheme 11 Gold-catalysed asymmetric thio-Claisen rearrangement. | ||
In addition to single chiral gold catalysts that have been used to promote [3,3]-sigmatropic rearrangements, the cooperative combination of two metal catalysts also remarkedly enriches the diversity of asymmetric tandem Claisen rearrangements. In this field, the Feng group demonstrated an intermolecular tandem reaction of alkynyl esters with allylic alcohols, by merging achiral gold(I)-catalysed nucleophilic hydroalkoxylation with a chiral Lewis acid–nickel(II)-catalysed Claisen rearrangement (Scheme 12).34 A variety of branched acyclic α-allyl β-keto esters were accessed in high levels of diastereo- and enantioselectivities under mild conditions. The introduction of an ester group provided an extra coordination site for the chiral-nickel(II) catalyst to achieve better enantiocontrol. To minimize epimerization of 1,3-dicarbonyl products, purification was performed by flash chromatography on silica gel at −20 °C. Control experiments confirmed that the cooperative catalysis combining both gold and nickel catalysts was essential for this transformation. Of note, one limitation is that the less electron-rich aliphatic alkynyl ester failed to give the target product. Based on this work, the concept of relay catalysis via bimetallic catalysts has rapidly emerged as a promising strategy for other asymmetric tandem sigmatropic rearrangements.
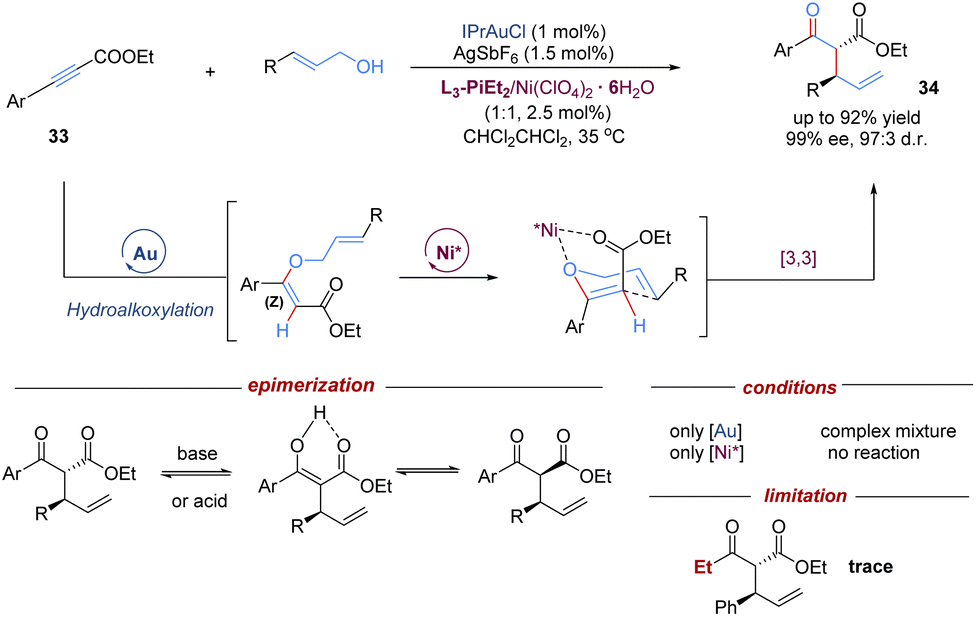 | ||
| Scheme 12 Gold/nickel relay catalysis for tandem hydroalkoxylation/Claisen rearrangement. | ||
Another important variant of tandem asymmetric [3,3]-rearrangements involves the utilization of highly reactive metal carbene intermediates, enabling numerous useful synthetic transformations. An impressive study in this chemistry is the tandem reaction between vinyldiazoacetates and allylic C–H bonds, called “combined C–H activation/Cope rearrangement” (CHCR), which was originally explored by the Davies group.35 Many examples based on this concept have been reported in an enantioselective version, confirming its potential value in organic synthesis. Here, we choose the CHCR reaction of vinyl ethers as the model for a brief introduction, not to summarize all relevant work in this field (Scheme 13).36 With a chiral dirhodium catalyst Rh2(S-DOSP)4, vinyl ethers 35 and vinyldiazoacetates 36 underwent the combined asymmetric C–H functionalization/Cope rearrangement reaction to generate homoallylic alcohol products 37 in a highly anti-selective and enantioselective version. According to computational studies, the author proposed that the reaction proceeded through a s-cis-boat transition state, following a concerted but asynchronous hydride transfer/C–C bond formation process. Notably, much lower amounts of byproducts from the competing direct C–H insertion were detected even in the acyclic vinyl ethers, which further broadened the substrate scope (37c).
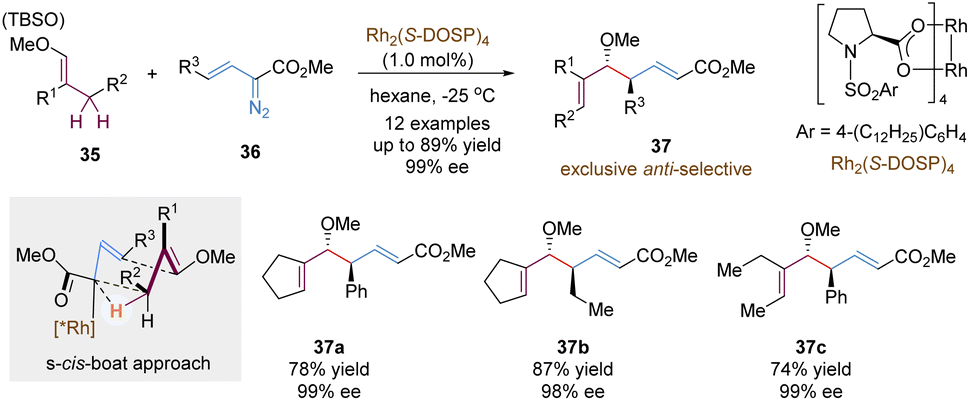 | ||
| Scheme 13 Rhodium-catalysed C–H insertion/Cope rearrangement. | ||
In addition to C–H insertion, metal carbenes can also undergo O–H insertion to form allyl vinyl ethers in situ, followed by a tandem Claisen rearrangement. For example, the Feng group reported a catalytic tandem O–H insertion/asymmetric Claisen rearrangement reaction of N-sulfonyl-1,2,3-triazoles with allylic alcohols through the relay catalysis of achiral rhodium(II) salt and a chiral N,N′-dioxide–indium(III) complex (Scheme 14a).37N-Sulfonyl-1,2,3-triazoles 38 are convenient precursors of carbenoid species that are readily converted into metal-bound imino-carbene intermediates in the presence of transition-metals, such as rhodium catalysts.38 Subsequently, following the O–H insertion of allylic alcohols, proton transfer and release of the Rh(II)-catalyst, the free (Z)-allyl vinyl ethers 43 are generated, thereby resulting in no efficient enantiocontrol for the subsequent Claisen rearrangement reaction even under the catalysis of a chiral Rh(II) catalyst. To address this challenging issue, Feng and co-workers developed a strategy combining chiral Lewis acid catalysis and transition metal catalysis. Through the installation of an ester substitution group, the readily formed intermediate 43 could be bonded and activated by a chiral Lewis acid catalyst, i.e. indium(III) complex. Then the Claisen-type [3,3]-rearrangement process proceeded in an enantioselective manner. Benefiting from the good compatibility of rhodium and indium catalysts, various valuable γ-oxo-β-amino acid esters 40 were directly obtained in high yields with good to excellent selectivities (up to 20:1 d.r., 96% ee).
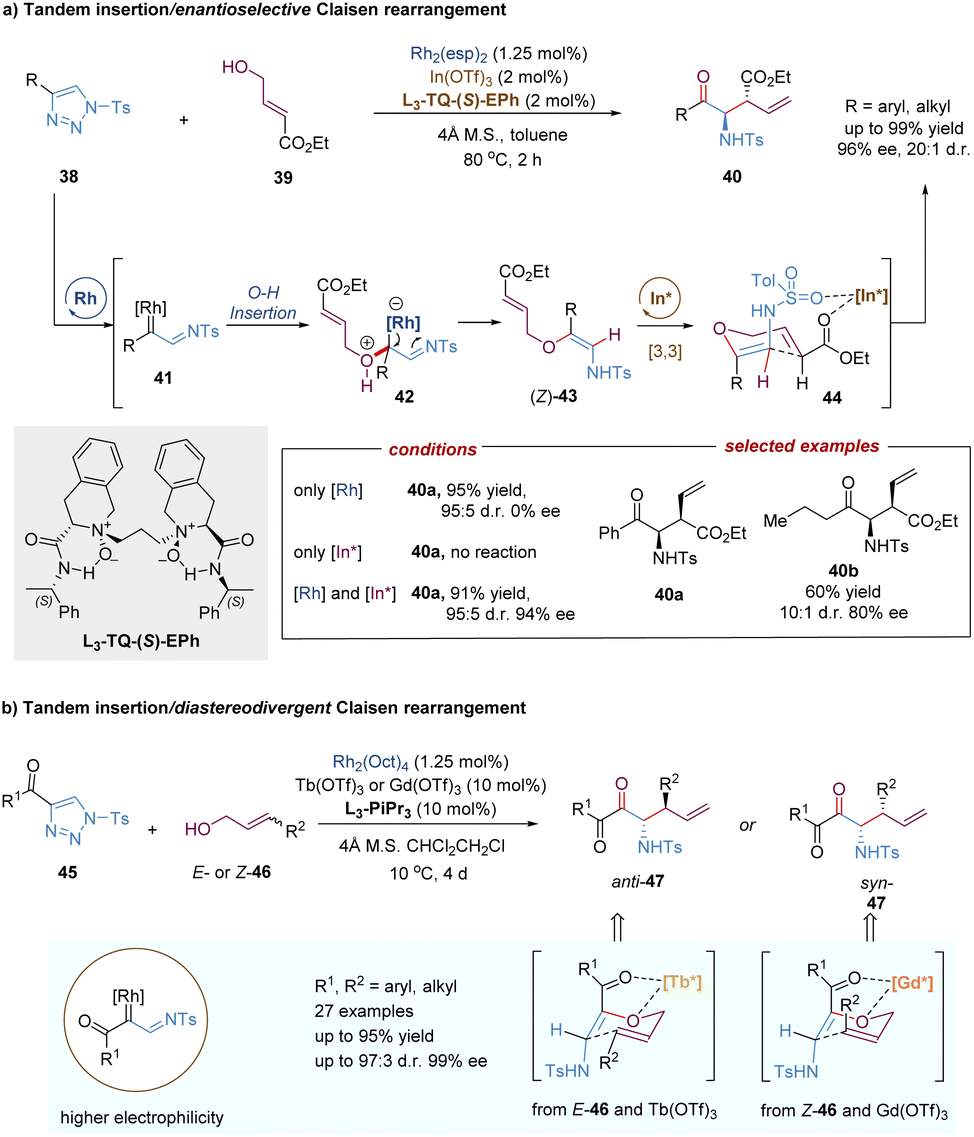 | ||
| Scheme 14 Rhodium/indium relay catalysis for tandem O–H insertion/Claisen rearrangement. | ||
Recently, Feng, Lin and co-workers described a similar tandem asymmetric catalytic O–H insertion/Barnes–Claisen rearrangement reaction from 4-acyl-1-sulfonyl-1,2,3-triazoles (45) with allylic alcohols (Scheme 14b).39 In comparison to 4-aryl 1-sulfonyl-1,2,3-triazoles 38, rhodium carbenoids generated in situ here are the accepter/accepter type due to the presence of acyl-imino groups, exhibiting higher electrophilicity under milder conditions. The introduction of a carbonyl group also affords a new two-point binding coordination model for the N,N′-dioxide–metal-promoted asymmetric Barnes–Claisen rearrangement. The unique advantage of this transformation is that both chiral anti- and syn-α-aminoketones were obtained in high yields with good stereoselectivities by tuning the source of Lewis acids and the configuration of allylic alcohols.
Similarly, allyl vinyl sulfonium ylides generated from the nucleophilic attack of sulfides and metal-carbenes can undergo tandem charge-accelerated thio-Claisen rearrangements with a faster reaction rate. In 2020, the Feng group developed such an enantioselective transformation with 2-thioindoles and α-diazo pyrazoleamides, providing an efficient route to access 3-allyl substituted indole-sulfide derivatives with good yields and high ee values (Scheme 15).40 The authors proposed a possibly mechanistic pathway: initially, the L2-Pi(OiBu)2/Ni(OTf)2 complex interacts with α-diazo pyrazoleamides to form a chiral nickel(II) carbene with the loss of nitrogen gas. The key step is that 2-thioindoles attack the nickel carbenoids in an enantioselective manner, generating sulfonium ylides 51 with an R configuration at the sulfur-center. The subsequent proton transfer and [3,3]-rearrangement reaction are still promoted by the chiral nickel(II) catalyst, thus leading to the generation of (R,E)-configured products 49 with high enantioselectivities. It is to be noted that an additional coordination site from the α-diazo pyrazoleamide plays a crucial role for the formation of a chiral N,N′-dioxide–nickel carbenoid. Other related asymmetric transformations based on the above carbenes have been well developed, such as the stereoselective [2,3]-sigmatropic rearrangement depicted later.
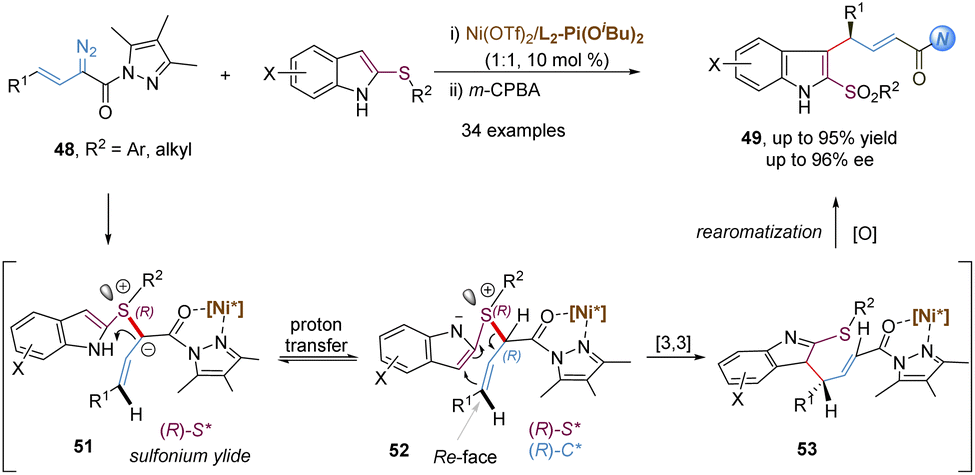 | ||
| Scheme 15 Nickel-catalysed thio-Claisen rearrangement via in situ generated sulfonium ylides. | ||
3. [2,3]-Sigmatropic rearrangements
Stereoselective [2,3]-sigmatropic rearrangements are of great importance in organic synthesis and have received tremendous attention from chemists.41 Particularly, heteroatoms (such as O, N, S, Se, and I etc.) are readily involved in [2,3]-rearrangements, providing a straightforward approach to access the C–X bond in an enantioselective manner for the synthesis of complex targets. [2,3]-Sigmatropic rearrangements can be broadly generalized into three types according to the characteristics of the substrate precursors: allyl/propargyl ethers, allylic N-oxides, and onium ylides (Scheme 16). Mechanistically, [2,3]-rearrangements proceed through a concerted process via a five-membered, envelope-like transition state in which the substituents are pseudo-equatorial. Likewise, stereospecificity is also a distinct feature of [2,3]-sigmatropic rearrangements: (E)-substrates prefer to afford anti-products, while (Z)-substrates generally give syn-products with high selectivity (Scheme 17). However, compared with [3,3]-sigmatropic rearrangements, the greater conformational flexibility of transition states for the [2,3]-rearrangement often makes it more susceptible to the substituent effects, and it becomes more difficult to achieve complete stereocontrol.42 Previously, extensive efforts had been devoted to the development of metal-catalysed diastereoselective and/or enantioselective [2,3]-rearrangements, but the main strategy was based on the chiral auxiliaries or enantiomerically enriched starting materials. A truly chiral metal complex-controlled highly enantioselective [2,3]-rearrangement has been more challenging. As such, engineered enzymes have been investigated to achieve catalytic enantioselective [2,3]-rearrangements of allylic sulfides and diazo reagents, only giving poor asymmetric induction.43 In the last decade, benefiting from the refined substrate design, rapid development of new chiral ligands, and a better understanding of the stereochemical mechanism, many important milestones in metal-catalysed enantioselective [2,3]-rearrangements have been achieved.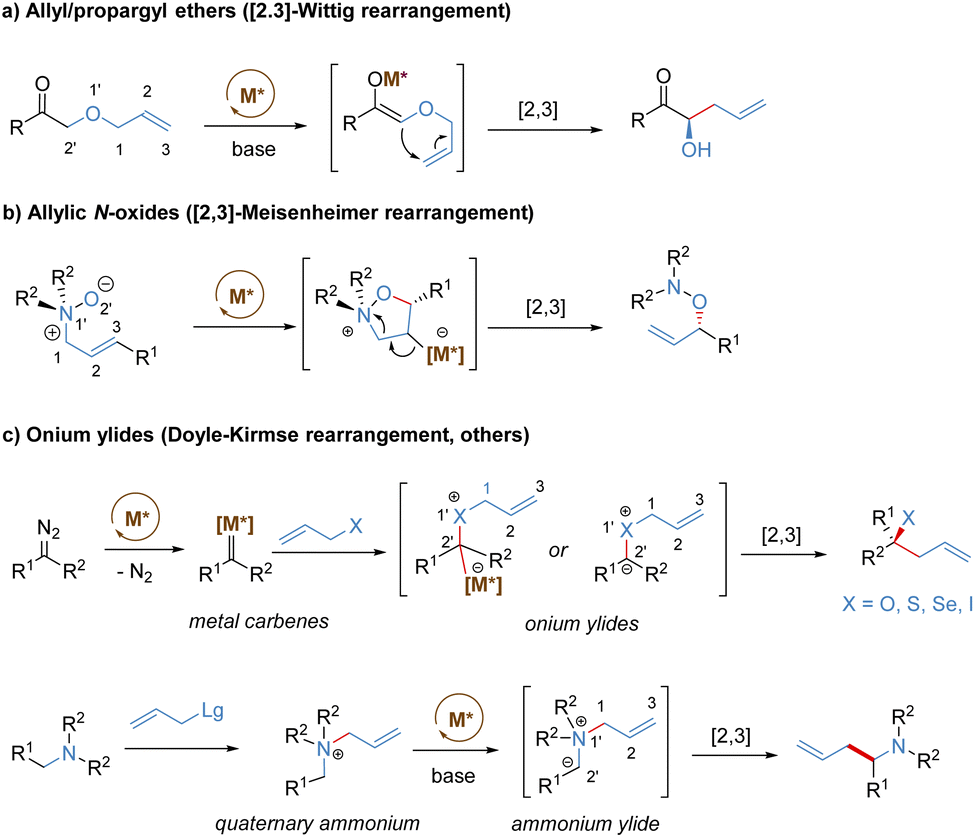 | ||
| Scheme 16 Metal-catalysed asymmetric [2,3]-sigmatropic rearrangements. | ||
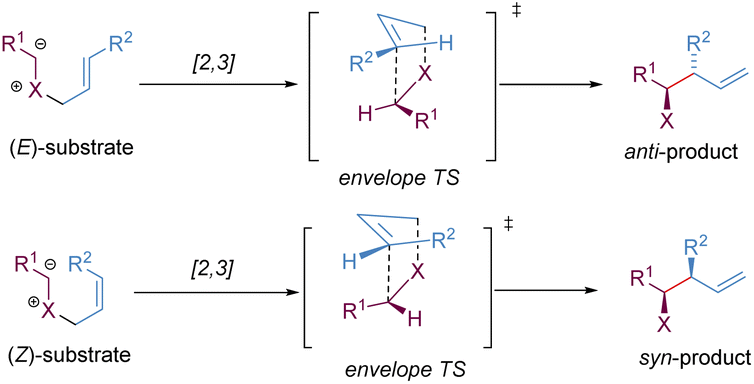 | ||
| Scheme 17 Diastereoselective [2,3]-rearrangement from (E)- and (Z)-substrates. | ||
3.1. [2,3]-Wittig rearrangement of allyl/propargyl ethers
In 1942, G. Wittig found that allylic ethers underwent deprotonation in the conditions of strong bases and followed a [2,3]-sigmatropic shift to afford sterically hindered homoallyl alcohols.44 In this context, the anion-promoted [2,3]-Wittig rearrangements have been extensively studied in organic synthesis and natural product total synthesis. In view of enantioselective versions, the examples of catalytic asymmetric [2,3]-Wittig rearrangements remain scarce,45 probably because strong bases are typically required to generate anionic species. Most previously catalytic enantioselective examples were realized through asymmetric organocatalysis,45c–f such as aminocatalysis, phase-transfer catalysis, thiourea-based catalysis and chiral Lewis base catalysis. Recently, some progress has also been made in the field of chiral Lewis acid-catalysed [2,3]-Wittig rearrangements. As shown in Scheme 18, Kanger and co-workers reported the first example of a chiral calcium(II) complex-catalysed enantioselective [2,3]-Wittig rearrangement of α-allyloxymalonate derivatives.46 The calcium salt Ca(NTf2)2 combined with tridentate Inda-Pybox to form a strong chiral Lewis acid complex, which tightly coordinated with two oxygen atoms of the allyloxy and enolate of ester. Optically active homoallyl alcohols were prepared in good yields, but only moderate enantioselectivities (32%–85% ee). This methodology was quite sensitive to the steric hindrance of ester groups, and no products were formed with bulky isopropyl or tert-butyl derivatives. Diketone substrates also showed poor reactivity for the [2,3]-rearrangement. Additionally, the selection of imidazole as a base additive was crucial for achieving higher conversion and enantioselectivity than the other bases, indicating that an appropriate base could promote the formation of anionic species and also suppress strong racemic background reactions.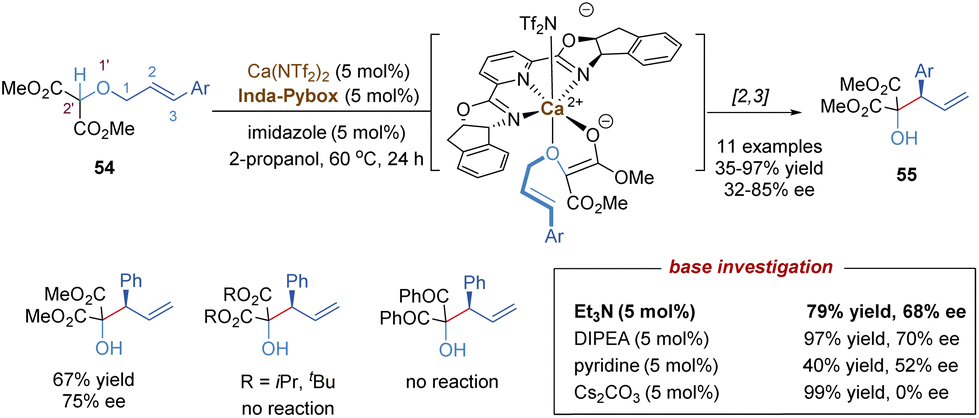 | ||
| Scheme 18 Calcium-catalysed asymmetric [2,3]-Wittig rearrangement of allyloxymalonate. | ||
In a similar fashion to the alkyne Claisen rearrangement, the [2,3]-Wittig rearrangement of propargylic ethers also provides a direct route to versatile functionalized allenes. However, owing to the low rearrangement reactivity of the triple bond and the competitive nucleophilic attack on the alkyne moiety, such propargyl [2,3]-Wittig rearrangement has been less studied. In 2008, the Feng group disclosed an enantioselective propargyl [2,3]-Wittig rearrangement of oxindole derivatives that relied on chiral N,N′-dioxide–nickel(II) as the catalyst and Et3N as the base (Scheme 19a).47 The coordination of the chiral Lewis acid complex to propargylic oxindole ethers results in the decrease of the pKa value of the α-C–H of carbonyl group, and accelerates the enolization of amide to generate a carbanion intermediate, which undergoes an enantioselective [2,3]-Wittig rearrangement under relatively mild reaction conditions. This novel approach affords a direct route to access chiral 3-hydroxyl oxindoles 57 bearing functionalized allenes in high yields and excellent enantioselectivities (up to 99% ee). The challenging aliphatic alkynes (such as cyclohexyl, methyl) are also well tolerated, giving the desired products with excellent enantioselectivities (up to 98% ee) and moderate yields (43–50%). It is worth pointing out that a strong positive non-linear effect was observed in this system, allowing access to enantioenriched rearrangement products (92% ee) with only 15% ee ligand.
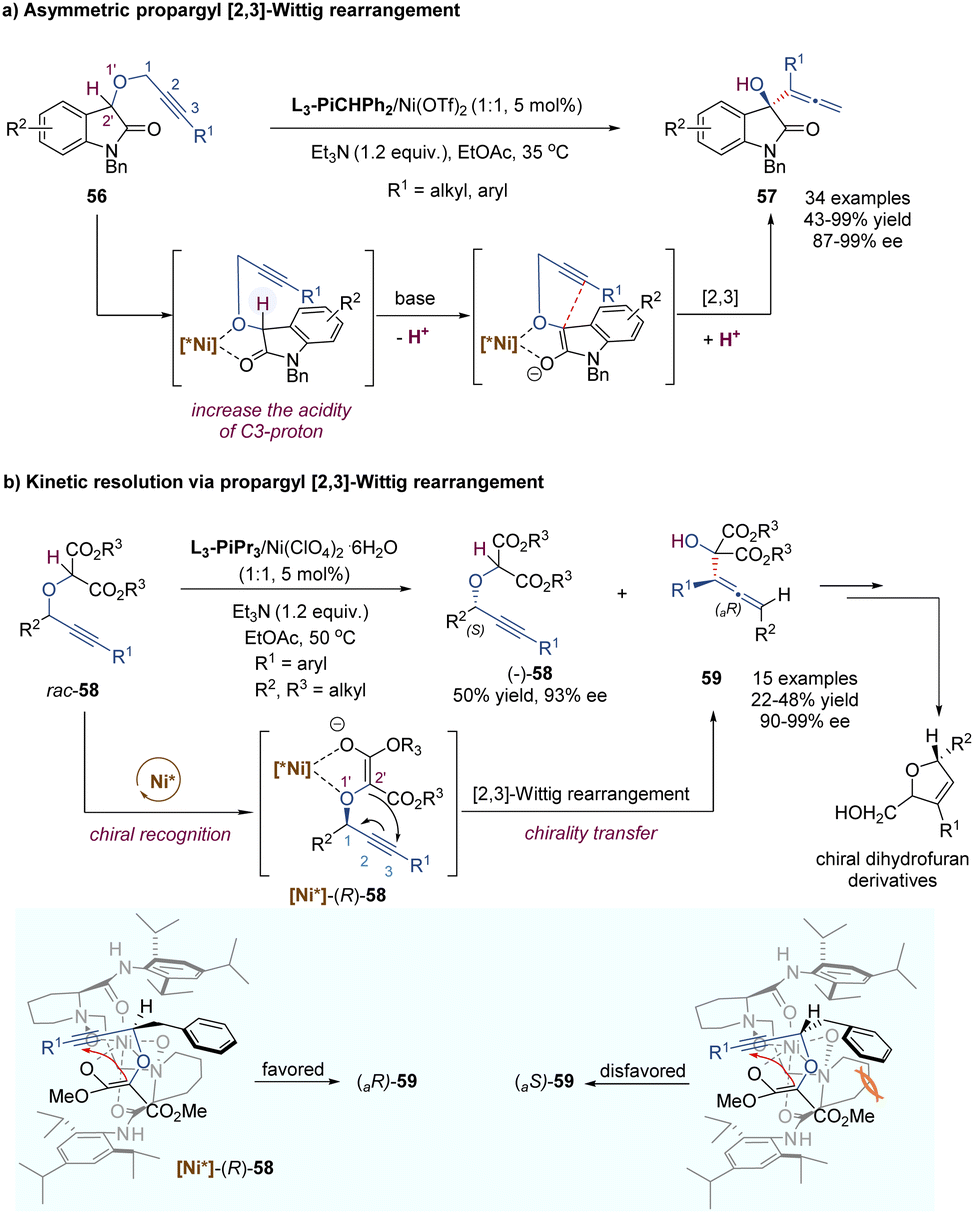 | ||
| Scheme 19 Nickel-catalysed asymmetric [2,3]-Wittig rearrangement of propargylic ethers. | ||
Furthermore, they developed a kinetic resolution process of racemic propargyloxy dicarbonyl compounds via [2,3]-Wittig rearrangement by using a systemic chiral Lewis acid catalyst as above (Scheme 19b).48 A chiral nickel(II) complex can recognize the (R)-enantiomer-58 and promote its rearrangement into an axially chiral α-hydroxyallene product 59 through a chirality transfer process, accompanied by an enantioenriched (S)-enantiomer 58 left. As depicted in the proposed asymmetric induction models, with the assistance of a base additive, the enolization intermediate coordinates tightly to the Lewis acid catalyst through the ether oxygen atom and the enolate ion of the auxiliary ester group. Due to the steric hindrance between the Bn group (R2) and amino acid skeleton unit, the adduct of [Ni*]-(S)-58 was disfavored to undergo rearrangement. Of note, the resulting chiral α-allenyl alcohols can serve as useful synthetic intermediates in organic synthesis, which could be easily transformed into chiral dihydrofuran derivatives widely existing in natural products.
3.2. [2,3]-Meisenheimer rearrangement of allylic N-oxides
The [2,3]-rearrangement of allylic N-oxides into hydroxylamine derivatives was named [2,3]-Meisenheimer rearrangement.49 Since its initial discovery in 1919, the catalytic asymmetric version of this reaction remains elusive for many years,50 presumably because of thermally spontaneous rearrangement of the reactive substrates even without the assistance of an external catalyst. Tambar and co-workers made a breakthrough in this area and reported the first catalytic asymmetric [2,3]-Meisenheimer rearrangement catalysed by the phosphoramidite/Pd(OAc)2 complex (Scheme 20a).51 The relatively stable allylic amine N-oxides 60 were transformed into chiral secondary allylic hydroxylamines 62, tolerating various reactive functional groups such as free alcohols, aldehydes, and phosphate esters. Based on the experimental data, the author proposed that the chiral palladium(II) catalyst might act as a π-acid to activate the double bond of allylic N-oxide substrates, followed by intramolecular attack to generate the heterocyclic intermediates 64. C2-substituted substrates cannot form a sterically hindered fully substituted carbon bond to Pd, resulting in poor reactivity (65c, trace). Subsequently, Grob-type fragmentation delivered the formal [2,3]-rearrangement products in excellent outcomes (87%–97% ee). After the reductive cleavage of the N–O bond in hydroxylamine products, optically active secondary allylic alcohols could be conveniently achieved.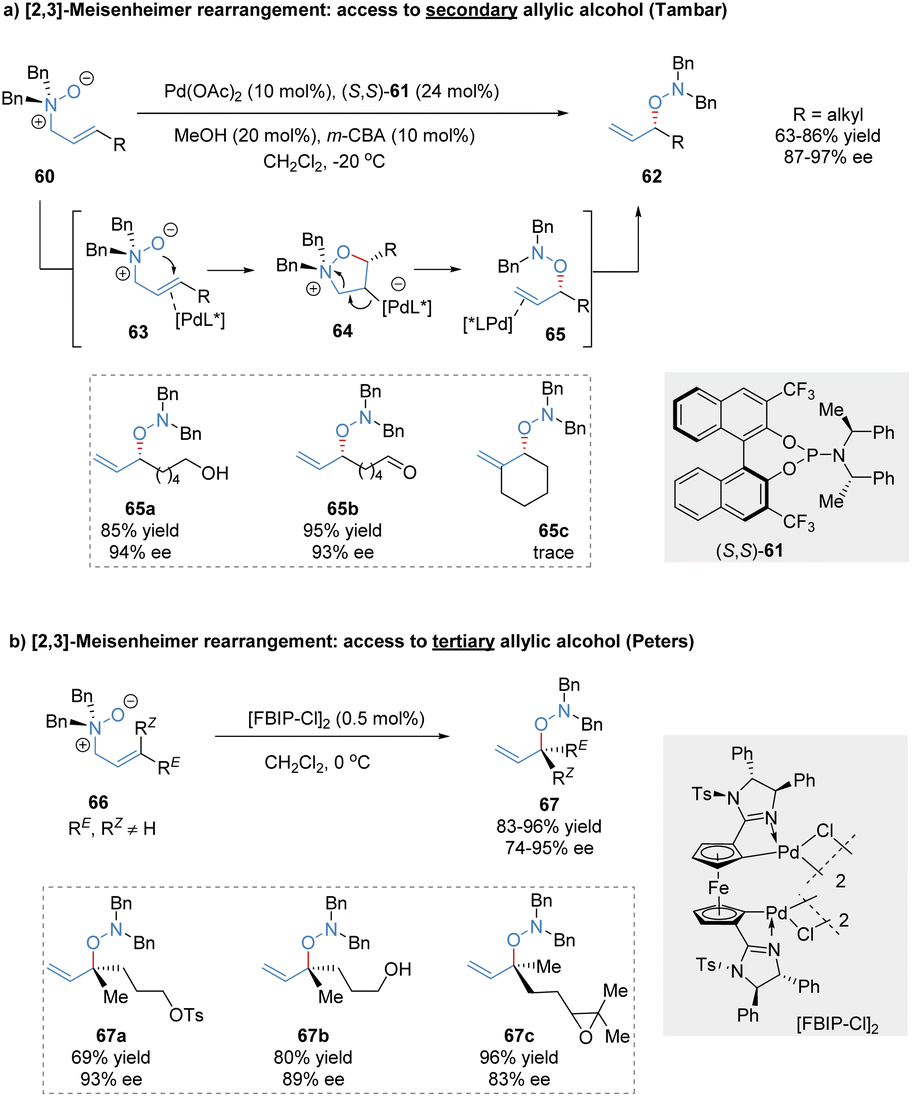 | ||
| Scheme 20 Palladium-catalysed [2,3]-rearrangement of allylic N-oxides. | ||
In 2020, a similar approach for the synthesis of chiral tertiary allylic alcohols through catalytic [2,3]-Meisenheimer rearrangement was described by Peters and co-workers (Scheme 20b).52 The allylic amine N-oxides 66 possessing trisubstituted olefin moieties were prepared by meta-chloroperbenzoic acid oxidation at −20 °C. The planar chiral ferrocene-based bispalladacycle catalyst [FBIP-Cl]2 displayed superior catalytic activity, enabling the enantioselective [2,3]-Meisenheimer rearrangement at reduced catalyst loading (0.5 mol%). Various trisubstituted allylic N-oxides bearing highly reactive functional groups even with similar RE and RZ residues were well tolerated, producing chiral acyclic tertiary alcohol precursors 67 at high levels of enantioselectivity (74%–95% ee).
3.3. [2,3]-Rearrangement of onium ylides
Transition metal-carbenoids generated from diazo compounds can readily react with allylic heteroatom-nucleophiles (such as O, S, Se, and halides) to form the corresponding allylic onium ylides, followed by a facile in situ [2,3]-rearrangement to access complex heteroatom-containing molecules (Scheme 21). The enantioselective versions of the above process have been widely explored via chiral metal complexes. The degree of association between the chiral metal complex and the ylide can lead to two differentiated pathways to achieve stereocontrol. In the first case, the chiral metal-complex catalysts may prefer to tightly interact with the ylide intermediate based on the inherent stereoelectronic properties of the ylide or with the assistance of an additional coordination group. Subsequently, an enantioselective [2,3]-rearrangement of the allylic group occurs with the aid of the chiral metal-complex catalysts (path a). In the second case, the metal-complex catalysts dissociate prior to the [2,3]-rearrangement to release a chiral free ylide, for instance, the relatively stable sulfonium ylides, which may undergo the chirality transfer process to deliver enantiomeric enriched rearrangement products (path b). The equilibrium between the metal-bonded ylide and the chiral free ylide also makes enantiocontrol more challenging.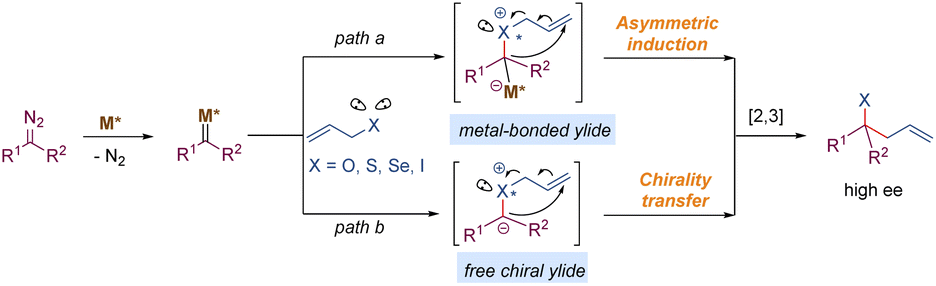 | ||
| Scheme 21 Asymmetric [2,3]-rearrangement of onium ylides. | ||
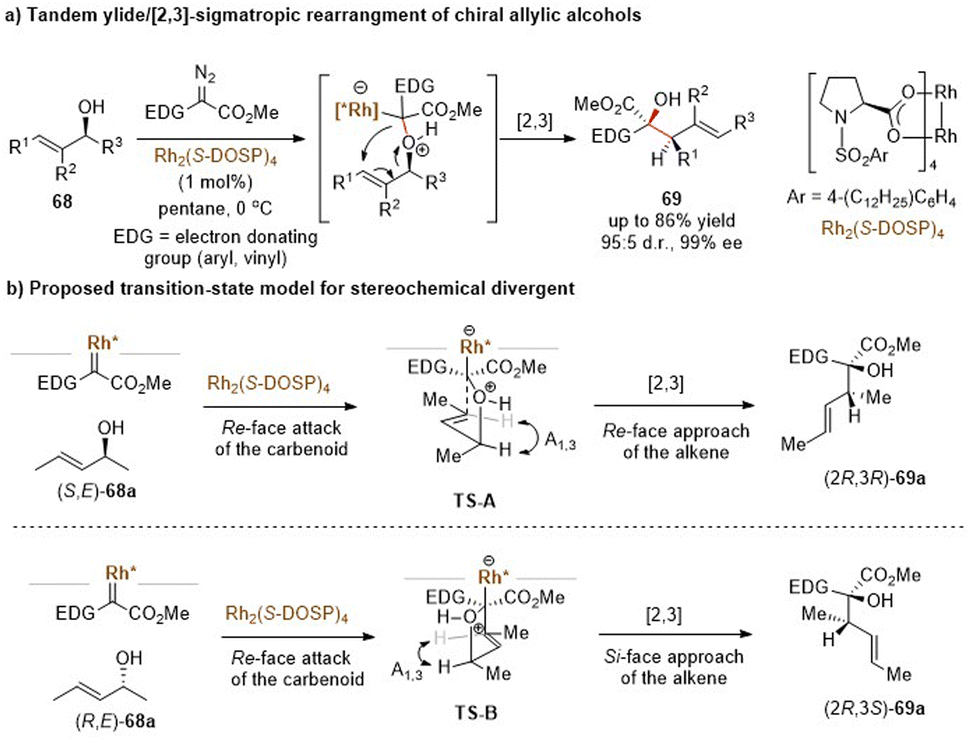 | ||
| Scheme 22 Rhodium-catalysed [2,3]-rearrangement of chiral allylic alcohols. | ||
The Davies group also reported a similar tandem oxonium ylide formation/[2,3]-rearrangement of α-styryl diazoacetates with propargylic alcohols to generate chiral α-hydroxy allenes (Scheme 23a).57 They found that when donor/acceptor carbenoids and highly functionalized tertiary propargylic alcohols were used as substrates, the [2,3]-rearrangement reaction of the in situ oxonium ylide became predominantly favored over the standard O–H insertion reaction. In the presence of the same catalytic system Rh2(S-DOSP)4, most of the enantioenriched allene derivatives 71 were produced in high yields and with excellent enantioselectivities, except for bulky substituents (tBu, TMS) on alkyne to give only moderate yields of 71b and 71c. Notably, a kinetic resolution transformation of racemic tertiary propargylic alcohols was also smoothly furnished under mild conditions (Scheme 23b). The rearranged allenic alcohols bearing two stereogenic centers 73 were obtained in good stereoselectivities, and the unreacted propargylic alcohols (R)-72 were isolated in good results.
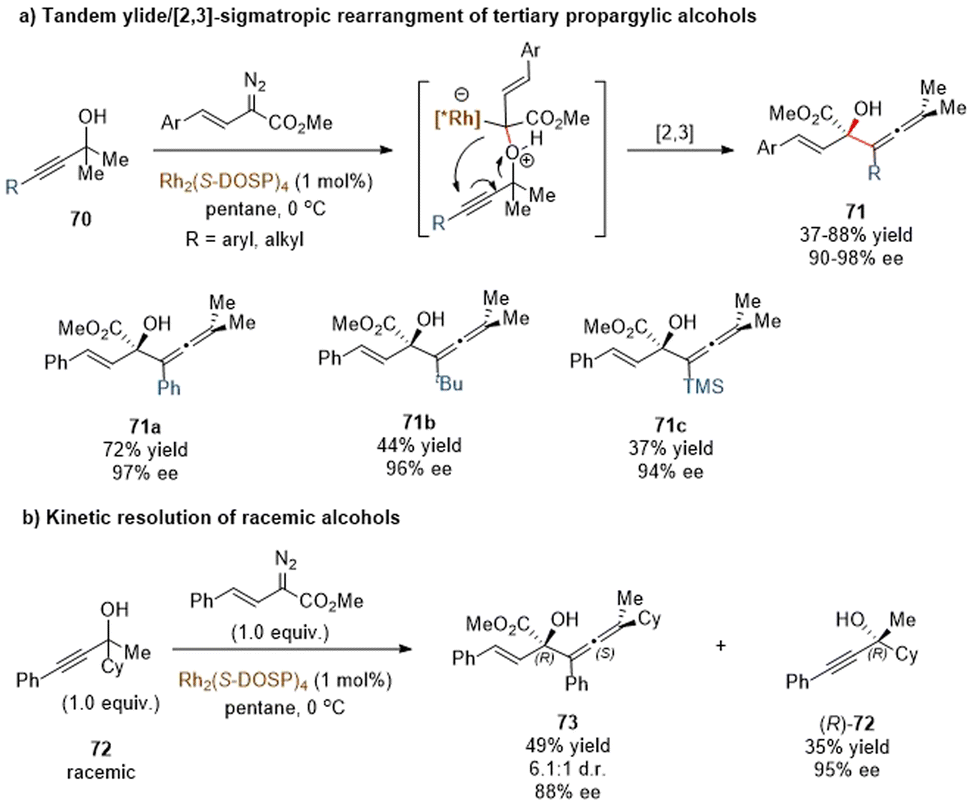 | ||
| Scheme 23 Rhodium-catalysed [2,3]-rearrangement of tertiary propargylic alcohols. | ||
Until 2017, the Wang group made a great advance in the enantioselective [2,3]-sigmatropic rearrangement of α-diazoacetates with trifluoromethyl allyl or propargyl sulfides in the presence of chiral Rh(II) or Cu(I) catalysts (Scheme 24).62 The desired products 74 containing a valuable trifluoromethylthio (SCF3) functional group were obtained with high yields and excellent enantioselectivities (up to 98% yield and 98% ee). In control experiments, the present catalytic system lacked enantiocontrol in the reaction of the symmetric diallylsulfide. Therefore, the author proposed that the enantio-discriminating step might be the nucleophilic addition of the sulfide with one of its two heterotopic free electron pairs to the metal carbene complex. The rhodium complex dissociated to form a sulfur-centered chiral free ylide intermediate, followed by a chirality transfer process from sulfur to carbon. In addition, the author also investigated the same reaction with low-cost and tunable Cu(I)-catalysis. It was found that good yields and enantioselectivities could be obtained for aryl α-diazoesters in general. However, the reaction with vinyl α-diazoacetates only gave poor enantioselectivities, and the reaction with propargyl trifluoromethyl thioethers was sluggish in the presence of the copper(I) complex catalyst.
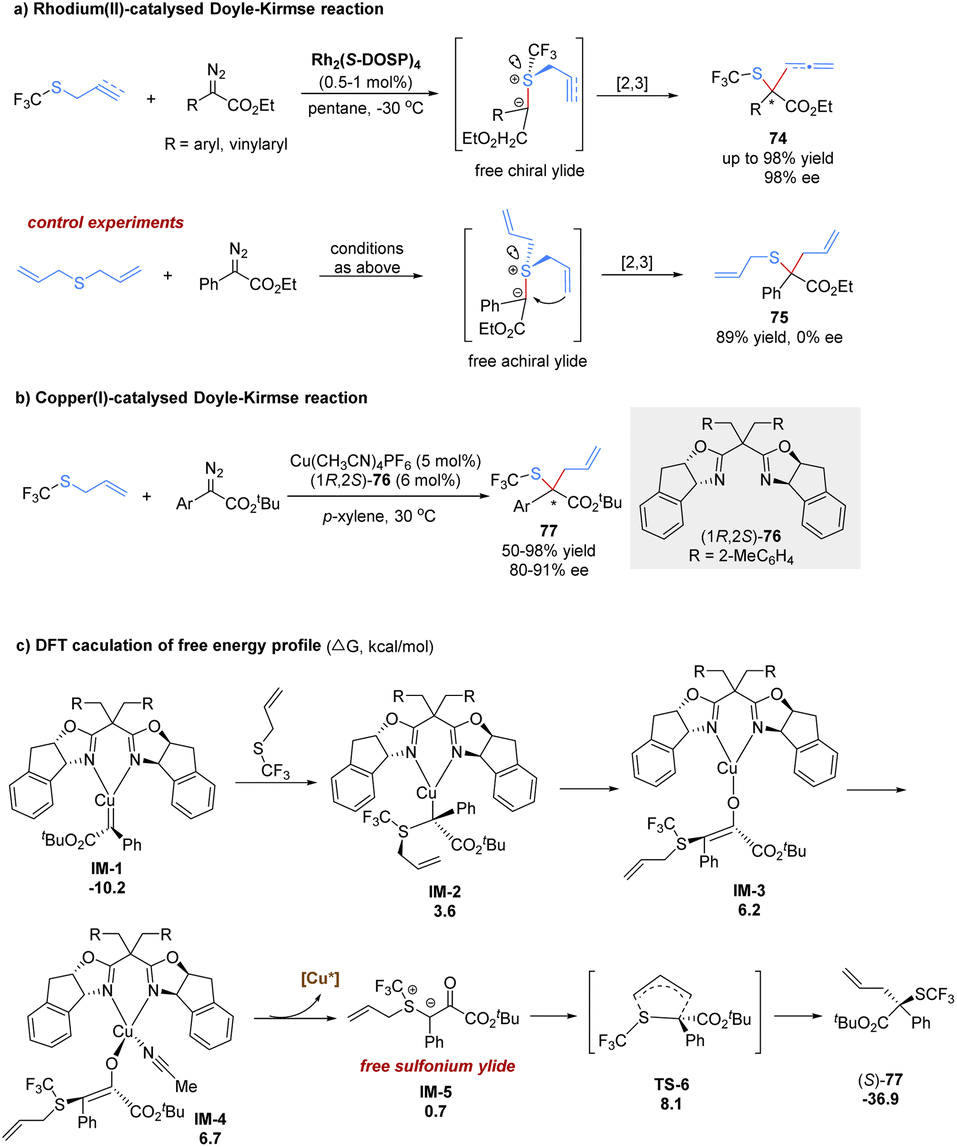 | ||
| Scheme 24 Doyle–Kirmse reaction of trifluoromethyl thioethers and DFT mechanistic study. | ||
Recently, Dang and Tantillo have investigated density functional theory calculations for the asymmetric [2,3]-rearrangements of α-diazoesters with allylic sulfides or iodides to reveal the mechanism and origin of regio- and stereoselectivity (Scheme 24).63 Initially, nucleophilic attack to the Cu-carbene delivers the metal-bound ylide IM-2, following a keto–enol tautomerism at the copper(I) center to offer enolate tautomer IM-3. The free sulfonium ylide IM-5 is generated through a ligand exchange process involving the acetonitrile coordination and ylide dissociation. Finally, the [2,3]-rearrangement of IM-5 occurs in a five membered electrophilic transition state (TS-6) with a strong exergonic effect (37.6 kcal mol−1). The author proposes that the stereoselectivity is mainly controlled by the substrate–ligand steric interactions. A stereochemical transition state that avoids severe steric exclusion between the bulky substituents on the carbene/allyl moieties and the bulky groups on the bisoxazoline ligands is more favorable.
Later, the Feng group described a different strategy to address the difficulties of the asymmetric Doyle–Kirmse reaction by using a new type of α-diazo pyrazoleamides and developing a dual-task paradigm of the chiral N,N′-dioxide–metal complex (Scheme 25).64 Mechanistically, the chiral Ni(II) complex reacted with α-diazo pyrazoleamides 78 to generate a metal-carbene intermediate with the release of N2. Subsequently, the attack of allyl sulfides produced the sulfonium ylide species. The pyrazoleamide substituent played an important role in the overall process. On the one hand, it adjusted the reactivity of the α-diazo compound for the formation of nickel-carbenoid. Switching α-diazo pyrazoleamide into α-diazoester resulted in poor results (35% yield, 6% ee after 10 h). On the other hand, it served as a bidentate coordination group in favor of the formation of chiral nickel(II)-bound ylide intermediate A. The reaction of symmetric diallylic sulfides delivered achiral sulfonium ylides, which still could provide the desired products 79a and 79b with good enantioselectivities, indicating the discrimination of the heterotopic lone electron pairs of sulfur was not the key step for enantio-determination in this catalytic system. Most likely, the [2,3]-rearrangement step of the chiral catalyst-bound ylide A was the enantioselective-determining step (Scheme 25b).
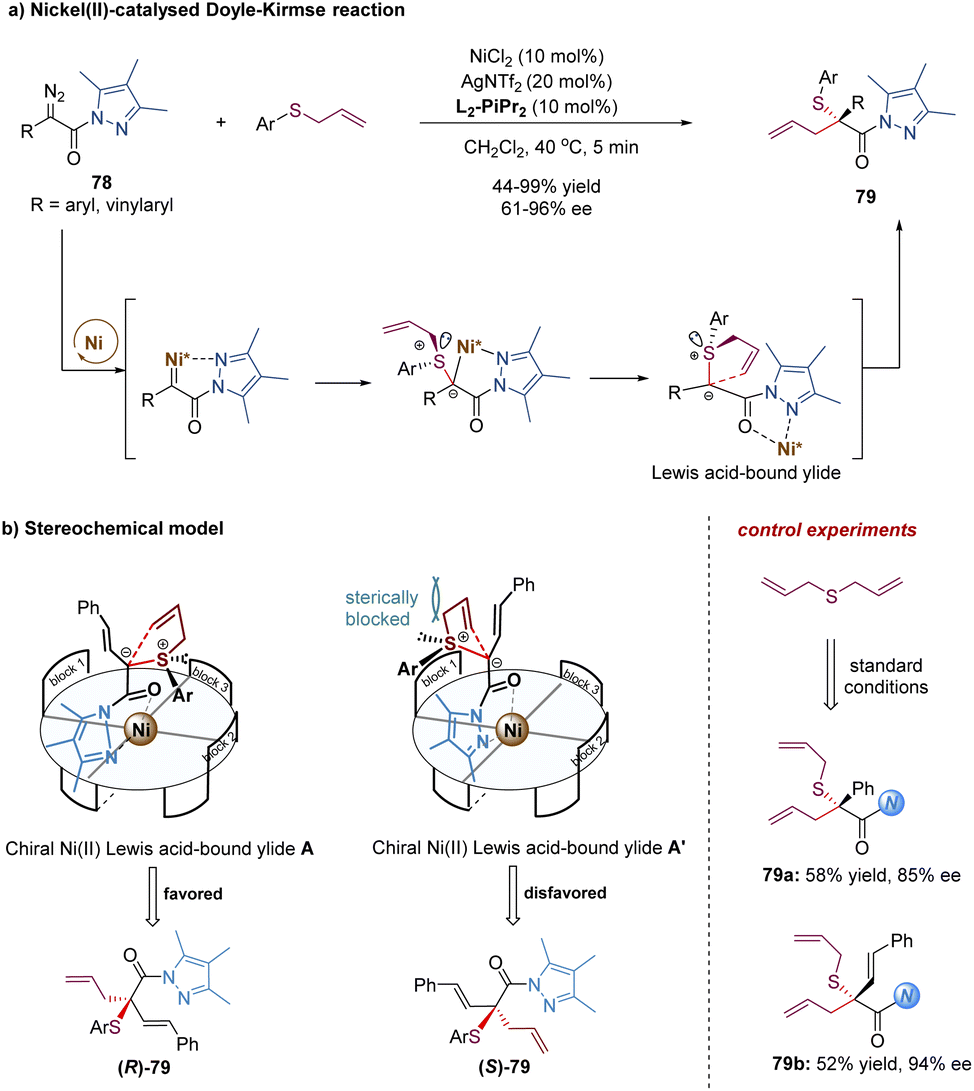 | ||
| Scheme 25 Doyle–Kirmse reaction of allylic sulfides. | ||
Similarly, Feng and co-workers also described the application of α-diazo pyrazoleamides in asymmetric [2,3]-Stevens and Sommelet–Hauser rearrangements catalysed by chiral N,N′-dioxide–nickel(II) complexes, providing a direct and elegant route to convert the C–S bond into a C–C bond (Scheme 26).65 These transformations generally involve the sulfonium ylide formation, 1,3-proton transfer and [2,3]-rearrangement. Ni(OTf)2/L2-PiPr2 as the catalyst was able to activate the vinyl or aryl substituted α-diazo pyrazoleamides to react with sulfides, forming chiral sulfonium ylide species with S-stereocenters (TS-1 and TS-4). A diastereoselective 1,3-proton transfer with the assistance of the external carbonic acid additive or a trace amount of water in the system yielded tautomeric chiral new ylides, bearing S- and C-stereocenters (TS-3 and TS-5). For the asymmetric [2,3]-Stevens rearrangement, various functionalized 1,6-dicarbonyl products 81 were readily accessed in modest to good yields and dr values with high enantiomeric excesses (Scheme 26a). For the asymmetric Sommelet–Hauser rearrangement, the chiral nickel-bound ylide TS-5 underwent the [2,3]-rearrangement at the ortho-position of the aryl group to generate the dearomative intermediate TS-6, followed by another 1,3-proton shift to deliver the multi-substituted arene 83 bearing a C–S bond stereocenter in good outcomes (Scheme 26b). Different from the above Doyle–Kirmse reaction of symmetric diallylsulfide, symmetric diethyl 2,2′-thiodiacetate only gave racemic rearranged products 81a and 83a (Scheme 26c), indicating that the discrimination of heterotopic lone pairs of sulfur reagents was essential to improve the stereoselectivity of the whole transformations. The author proposed that this [2,3]-rearrangement process occurred in an enantioselective version due to the combined effect of chirality transfer from the chiral sulfonium ylide intermediate and asymmetric induction of the chiral Lewis acid complex.
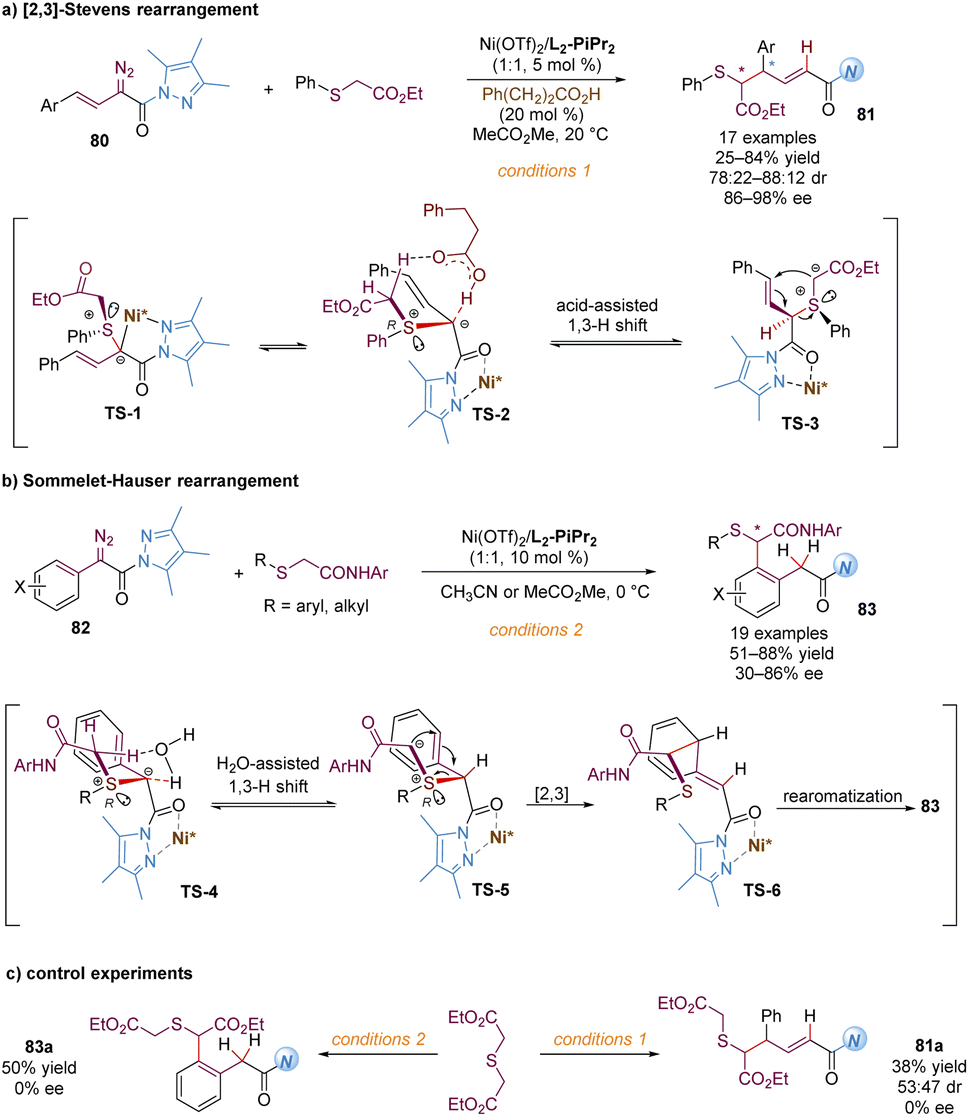 | ||
| Scheme 26 [2,3]-Stevens rearrangement and Sommelet–Hauser rearrangement. | ||
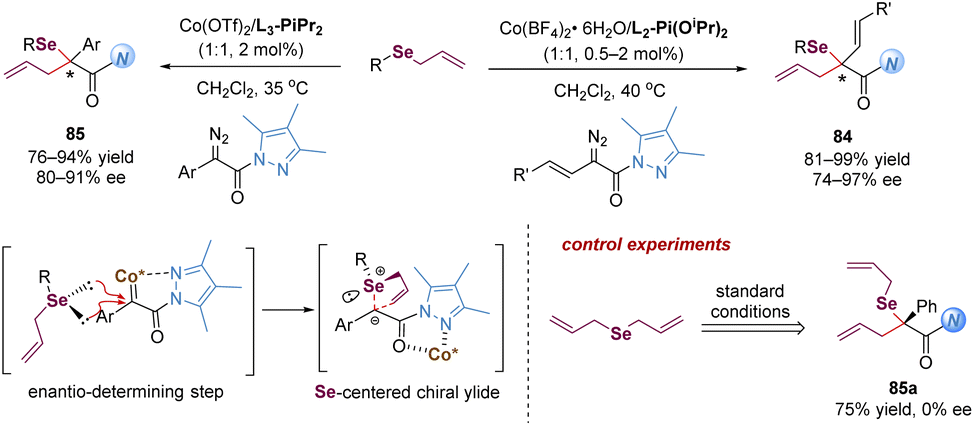 | ||
| Scheme 27 Cobalt-catalysed [2,3]-rearrangement of allylic selenides. | ||
The significant breakthrough in catalytic asymmetric [2,3]-rearrangements of halonium ylides was disclosed by the Tambar group, who achieved the first highly enantioselective, diastereoselective and regioselective [2,3]-iodonium rearrangement (Scheme 28a).69 In the presence of a chiral bisoxazoline–copper(I) catalyst, various substituted (Z)-allylic iodides coupled with metal-carbenoids to generate metal-bonded iodonium ylides with an equilibrium to the enolate form. Following the subsequent asymmetric [2,3]-rearrangement, the synthetically versatile chiral α-iodoester products were accessed with excellent stereoselectivities (up to 20:1 r.r., 20:1 d.r., and 97% ee). Notably, the bisoxazoline ligands preferentially yielded [2,3]-rearrangement products over [1,2]-rearrangement products. The use of (Z)-allylic iodides greatly enhanced diastereoselectivity without the loss of regioselectivity, and the bulkier tert-butyl α-diazoesters further improved the enantiomeric excess. Moreover, the potential utility was demonstrated by a series of stereospecific transformations of the C–I bond into other C–X (S, N, C, and H) bonds.
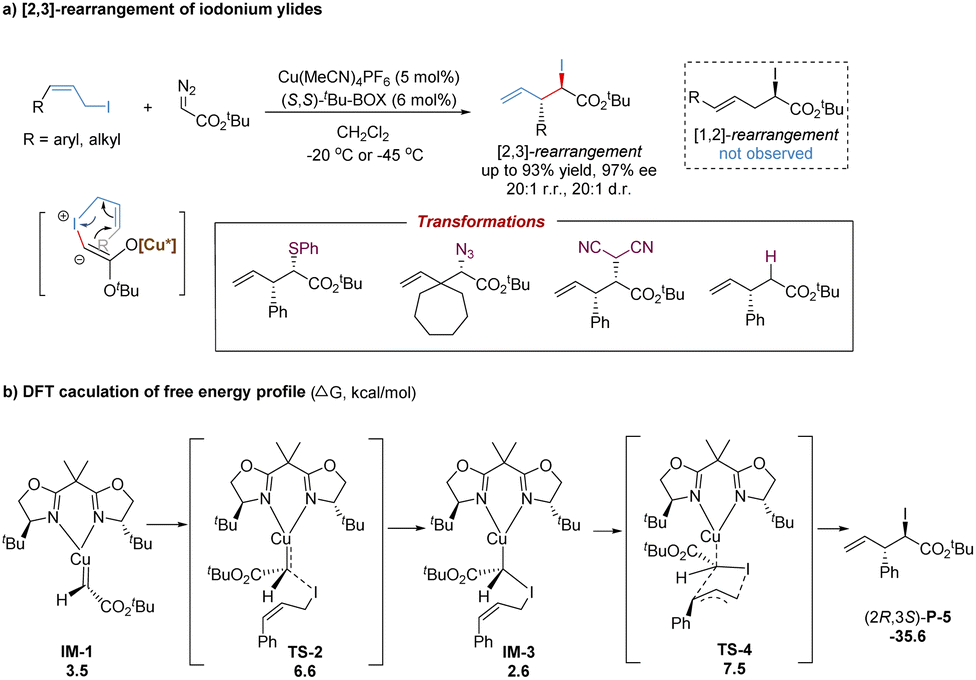 | ||
| Scheme 28 Copper-catalysed [2,3]-rearrangement of iodonium ylides. | ||
As shown in Scheme 28b, a possible reaction pathway has been proposed: firstly, the metal-bound ylide IM-3 is formed from the nucleophilic attack of (Z)-allylic iodide to the metal-carbene (IM-1) with an energy barrier of 6.6 kcal mol−1.63 Subsequently, the [2,3]-sigmatropic rearrangement occurs via a five-membered envelope transition state TS-4 with a barrier of 7.5 kcal mol−1. The major product (2R,3S)-P-5 is produced by releasing an energy of 35.6 kcal mol−1. In comparison, the free sulfonium ylide in [2,3]-rearrangements is more stable owing to the strong electronegativity of sulfur. But the free iodonium ylide with a carbanion and an iodine cation becomes unstable due to the low electronegativity of iodine. Therefore, it is necessary to introduce the copper(I) center to stabilize the iodonium ylide. Based on the DFT calculations, the nucleophilic addition of allylic iodide to the Cu(I)-carbene prominently controls the stereoselectivity, following a favored transition state to circumvent substrate–ligand and substrate–substrate steric influences.
In 2020, the group of Feng reported a chiral N,N′-dioxide–Mg(II) complex-catalysed enantioselective [2,3]-rearrangement of allylic ammonium salts generated in situ from glycine pyrazoleamides and allyl bromides (Scheme 29).71 Such allylic ammonium salts possessed a pyrazoleamide group as the bidentate coordination site, which were readily subjected to deprotonation with external base iPr2NH to form chiral Lewis acid complex bonded ammonium ylides. Then, these ammonium ylides would undergo a stereoselective [2,3]-rearrangement to deliver anti-configured α-amino acid derivatives 86 in high yields and excellent diastereo- and enantioselectivities. By employing the chiral magnesium(II) catalyst, the π-allyl cation unit approached the pyrazoleamide anion unit via an exo-transition state to deliver highly anti-selective rearranged products, which were in contrast to the previous organocatalysis giving exclusive syn-selective products reported by Smith.72
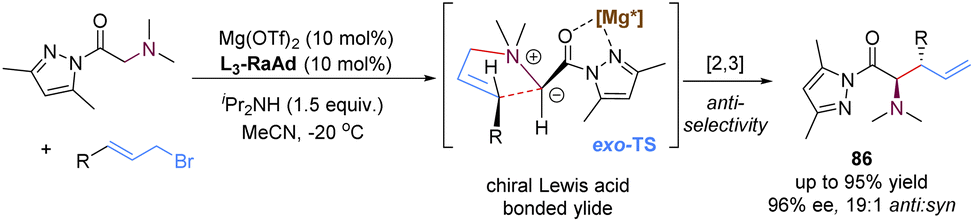 | ||
| Scheme 29 Magnesium-catalysed [2,3]-rearrangement of ammonium ylides. | ||
4. [1,3]-Sigmatropic rearrangements
[1,3] O-to-C rearrangements were reported more than 100 years ago to be investigated as an alternative strategy for the construction of C–C bonds in organic synthesis (Scheme 30).73 In comparison with its [3,3]-rearrangement counterpart, the development of [1,3] O-to-C rearrangement has gained much less attention, especially for enantioselective versions.74 Due to being orbital symmetry-forbidden,75 suprafacial [1,3]-rearrangement is an unfavorable process, and harsh reaction conditions, such as high temperatures, are generally required to alter the pathway through a non-concerted diradical intermediate. By relying on a transition metal catalyst, organocatalyst, and Lewis acid catalyst, the [1,3] O-to-C rearrangements have been achieved under relatively mild conditions. As such, when treating with a suitable Lewis acid catalyst, the heterolytic cleavage of the C–O bond of an enol ether would generate a zwitterion pair intermediate. The subsequent recombination of the carbocation and enolate anion furnishes the [1,3] O-to-C rearrangement. Consequently, by judiciously choosing the chiral Lewis acid catalytic system, the stereochemical process of the [1,3]-rearrangement is controllable and tunable.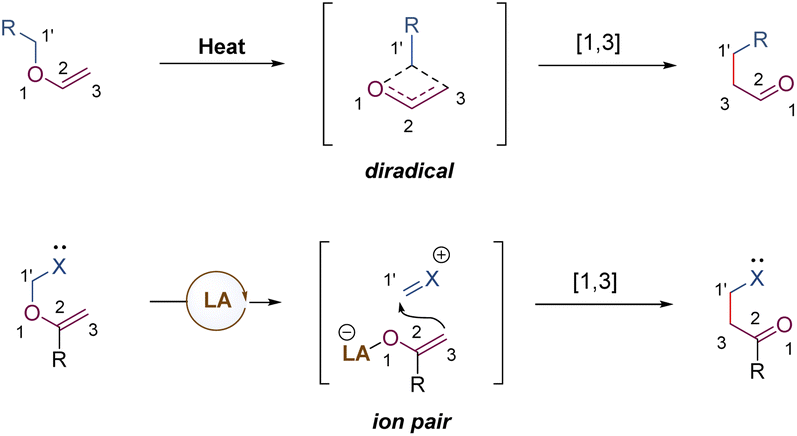 | ||
| Scheme 30 Lewis acid promoted [1,3]-rearrangement. | ||
In 2019, Ishihara demonstrated an unprecedented catalytic asymmetric dearomative [1,3]-rearrangement of allyl naphthyl ethers catalysed by a chiral π–Cu(II) complex (Scheme 31).76 A series of highly functionalized β-naphthalenone derivatives 88 bearing an all-carbon quaternary stereocenter was prepared in high yields with excellent enantioselectivities. Most of the allyl moieties should contain at least one aryl group, otherwise the dialkyl substituted substrate only gave the product 88c in modest outcomes (58% yield, 68% ee). In addition, benzyl naphthyl ethers with strong electron-donating substituents also smoothly underwent enantioselective [1,3]-rearrangement. Mechanistic studies showed that the coordination of the Cu(II) center with substrate 87 would promote the heterolytic C–O cleavage to form a reactive and tight ion pair intermediate. The anion was further stabilized by the α-electron-withdrawing substituent (ester group), as well as by the coordination of the chiral copper(II) complex. Then the intramolecular allyl cation approached the Re-face of the counter anion to afford the rearranged product 88 in S-configuration. These results illustrate that the success of catalytic asymmetric [1,3]-rearrangement is feasible by a well-designed substrate, along with a carefully selected Lewis acid catalyst.
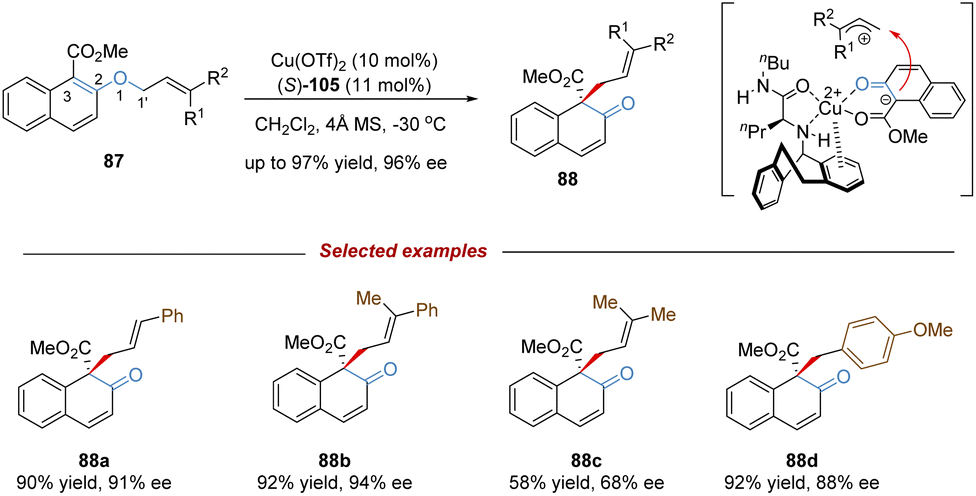 | ||
| Scheme 31 Copper-catalysed enantioselective [1,3]-rearrangement. | ||
In 2020, the Feng group discovered an enantioselective catalysis in [1,3] O-to-C rearrangement of unsubstituted racemic benzyl vinyl ethers catalysed by a chiral N,N′-dioxide–iron(II) complex (Scheme 32).77 The introduction of a phenoxy group provided an additional binding site to the iron(II) center, which was favorable to form a stable chelated transition state. This intermediate followed a heterolytic cleavage of the benzyl C–O bond to generate an in situ enolate and o-quinone methide cation. The subsequent formal 1,4-conjugate addition/cyclization cascade reaction afforded a wide range of chromanols with high yields and excellent enantioselectivities. In most cases, the overall conversion efficiency was very high with catalyst loadings as low as 0.1 mol%. As an important structural motif in many natural products and pharmaceuticals, the product chromanol was utilized to prepare urological drug (R)-tolterodine via reductive amination in the presence of diisopropylamine and sodium cyanoborohydride.
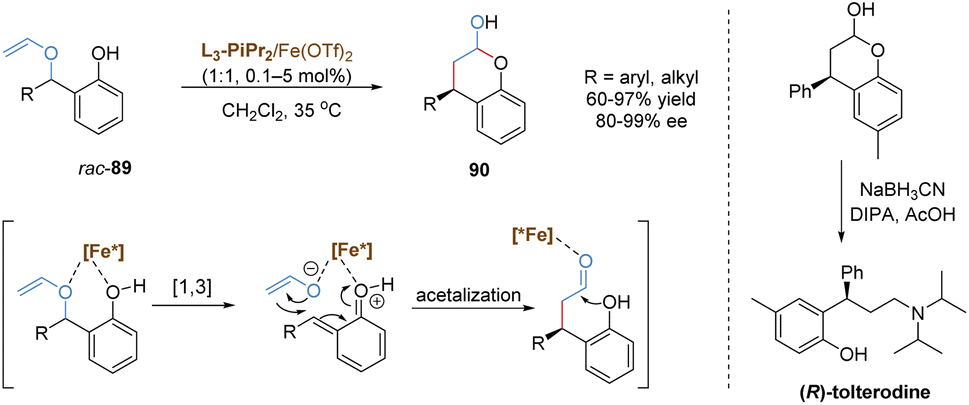 | ||
| Scheme 32 Iron-catalysed asymmetric [1,3]-rearrangement. | ||
Feng and co-workers recently reported a relay catalysis of the tandem O–H insertion/[1,3]-rearrangement reaction of N-sulfonyl-1,2,3-triazoles and benzyl alcohols by the use of a bimetallic system composed of a dirhodium catalyst and a chiral N,N′-dioxide–indium(III) complex (Scheme 33).78 Various synthetically useful chiral α-aminoketone derivatives 91 were readily prepared in excellent yields and enantioselectivities even at 90 °C. Initially, Rh(II)-stabilized imino carbene, in situ generated from N-sulfonyl-1,2,3-triazole, inserted into the O–H bond of the benzylic alcohol to form a (Z)-amino enol ether, which served as the intermediate for the following [1,3]-rearrangement. Based on the control experiments, it was found that the configuration at the C3 position was governed by the starting material (S)-phenylethanol, and the configuration at the C2 position was affected by both (S)-phenylethanol and the chiral catalyst. Therefore, the author speculated that an ion-pair mechanism for the [1,3]-rearrangement step was more likely. The O–Bn bond could be weakened by the chiral N,N′-dioxide–indium(III) complex to facilitate its heterolytic cleavage, leading to the generation of a tight ion pair species. Eventually, the chiral Lewis acid-bonded enolate counterpart attacked the benzylic carbocation in an enantioselective manner, delivering the [1,3]-rearrangement product in excellent stereoselectivity. The bimetallic catalysis combining transition metal and Lewis acid demonstrated a great potential in catalytic asymmetric [1,3]-rearrangement.
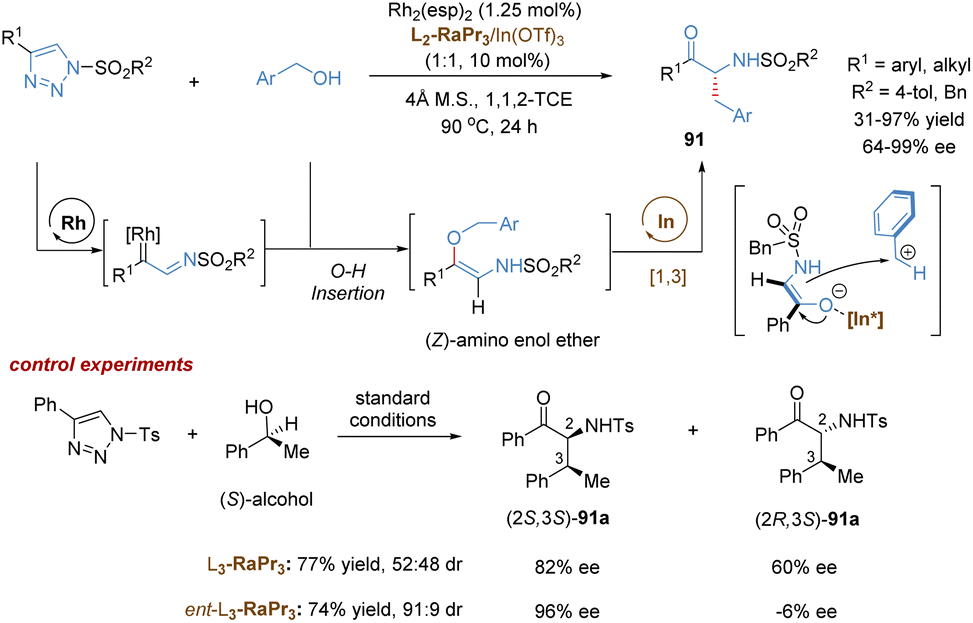 | ||
| Scheme 33 Relay catalysis for tandem O–H insertion/[1,3]-rearrangement. | ||
5. Conclusions
Metal-catalysed asymmetric sigmatropic rearrangements have achieved significant advances in the past decade. This review listed some significant contributions in this field, categorized by types of [3,3], [2,3], and [1,3]-rearrangements. A broad substrate scope, excellent diastereo- and enantioselectivity, diverse tandem process and new application in target-oriented synthesis have been demonstrated through the efficient protocols as summarized in this review. Generally, the close association between the chiral metal-complex and substrate via a tight bidentate-bonding provides an excellent control over reactivity and stereoselectivity. And some intrinsic limitations of traditional sigmatropic rearrangements, such as stoichiometric catalyst loading, harsh reaction conditions or low enantioselectivities, have been overcome to some extent with the assistance of well-designed substrates and carefully selected chiral metal-complex catalysts. Specifically, we are intrigued that the tandem process involving the cooperation of sigmatropic rearrangements and other catalytic reactions (such as bimetallic relay catalysis), has emerged as a remarkable advance in creating intricate structures with extreme efficiency. These promising tandem sigmatropic rearrangement transformations will overcome the limitations in either single process and provide more versatility in the synthesis of valuable molecules. At present, it is still worthy to explore the potential in metal-catalysed asymmetric sigmatropic rearrangements. We hope that this concise review will inspire more scientists to work in this field and promote development in the future.Author contributions
Dr Yangbin Liu wrote the manuscript-original draft. Prof. Xiaohua Liu guided and edited the manuscript. Prof. Xiaoming Feng conceived the project. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from National Natural Science Foundation of China (22001177, 22188101) and Shenzhen Bay Laboratory (S201100003). We also appreciate Mr F. Q. Zhang for helpful discussions and manuscript proofreading.Notes and references
-
(a) R. Hoffmann and R. B. Woodward, Acc. Chem. Res., 1968, 1, 17–22 CrossRef CAS
; (b) C. M. Rojas, Molecular Rearrangement in Organic Synthesis, Wiley, Hoboken, 2015 CrossRef
-
(a) A. M. M. Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef CAS PubMed
-
(a) H. Wu, Q. Wang and J. Zhu, Eur. J. Org. Chem., 2019, 1964–1980 CrossRef CAS
-
Fundamentals of Asymmetric Catalysis, ed. P. J. Walsh and M. C. Kozlowski, University Science Books, Sausalito, California, 2008 Search PubMed
- R. M. Romero, M. F. Roberts and J. D. Phillipson, Phytochemistry, 1995, 40, 1015–1025 CrossRef CAS
- For selected examples, see:
(a) C. Uyeda and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 9228–9229 CrossRef CAS PubMed
- T. C. A. F. Rodrigues, W. A. Silva and A. H. L. Machado, Curr. Org. Synth., 2015, 12, 795–805 CrossRef CAS
- L. Claisen, Chem. Ber., 1912, 45, 3157–3166 CrossRef CAS
-
(a)
M. Hiersemann and U. Nubbemeyer, The Claisen Rearrangement: Methods and Applications, 1st edn, Wiley-VCH, 2007 CrossRef
- H. Ito and T. Taguchi, Chem. Soc. Rev., 1999, 28, 43–50 RSC
-
(a) K. N. Houk, J. González and Y. Li, Acc. Chem. Res., 1995, 28, 81–90 CrossRef CAS
-
(a) W. v. E. Doering and W. R. Roth, Tetrahedron, 1962, 18, 67–74 CrossRef CAS
- J. S. Cannon and L. E. Overman, Acc. Chem. Res., 2016, 49, 2220–2231 CrossRef CAS PubMed
- L. Abraham, R. Czerwonka and M. Hiersemann, Angew. Chem., Int. Ed., 2001, 40, 4700–4703 CrossRef CAS PubMed
-
(a) J. Becker, L. Butt, V. von Kiedrowski, E. Mischler, F. Quentin and M. Hiersemann, Org. Lett., 2013, 15, 5982–5985 CrossRef CAS PubMed
- J. Tan, C.-H. Cheon and H. Yamamoto, Angew. Chem., Int. Ed., 2012, 51, 8264–8267 CrossRef CAS PubMed
- C. E. McKenna and B. A. Kashemirov, Top. Curr. Chem., 2002, 220, 201–238 CrossRef CAS
-
(a) B. M. Trost and M. K. Brennan, Synthesis, 2009, 3003–3025 CrossRef CAS
- E. C. Linton and M. C. Kozlowski, J. Am. Chem. Soc., 2008, 130, 16162–16163 CrossRef CAS PubMed
- O. Gutierrez, C. E. Hendrick and M. C. Kozlowski, Org. Lett., 2018, 20, 6539–6543 CrossRef CAS PubMed
- T. Cao, J. Deitch, E. C. Linton and M. C. Kozlowski, Angew. Chem., Int. Ed., 2012, 51, 2448–2451 CrossRef CAS PubMed
- Z.-W. Lai, C. Liu, H. Sun and S.-L. You, Chin. J. Chem., 2017, 35, 1512–1516 CrossRef CAS
-
(a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed
- Y. B. Liu, H. P. Hu, H. F. Zheng, Y. Xia, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2014, 53, 11579–11582 CrossRef CAS PubMed
- Y. B. Liu, H. P. Hu, L. L. Lin, X. Y. Hao, X. H. Liu and X. M. Feng, Chem. Commun., 2016, 52, 11963–11966 RSC
- B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978–15979 CrossRef CAS PubMed
- Y. B. Liu, X. H. Liu, H. P. Hu, J. Guo, Y. Xia, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 4054–4058 CrossRef CAS PubMed
- H. F. Zheng, Y. Wang, C. R. Xu, X. Xu, L. L. Lin, X. H. Liu and X. M. Feng, Nat. Commun., 2018, 9, 1968–1974 CrossRef PubMed
-
(a) Y. Aramaki, K. Chiba and M. Tada, Phytochemistry, 1995, 38, 1419–1421 CrossRef CAS
- A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 2556–2591 CrossRef CAS PubMed
- For iridium-catalysed tandem [3,3]-rearrangement, see:
(a) W.-B. Liu, N. Okamoto, E. J. Alexy, A. Y. Hong, K. Tran and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 5234–5237 CrossRef CAS PubMed
- H. Wu, W. Zi, G. Li, H. Lu and F. D. Toste, Angew. Chem., Int. Ed., 2015, 54, 8529–8532 CrossRef CAS PubMed
- H. Kim, J. Jang and S. Shin, J. Am. Chem. Soc., 2020, 142, 20788–20795 CrossRef CAS PubMed
- J. Li, L. L. Lin, B. W. Hu, P. F. Zhou, T. Y. Huang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2017, 56, 885–888 CrossRef CAS PubMed
-
(a) H. M. L. Davies and Q. Jin, J. Am. Chem. Soc., 2004, 126, 10862–10863 CrossRef CAS PubMed
- Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 11940–11943 CrossRef CAS PubMed
- Y. S. Chen, S. X. Dong, X. Xu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 16554–16558 CrossRef CAS PubMed
- T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed
- Y. Liu, Y. S. Chen, A. Yihuo, Y. Q. Zhou, X. H. Liu, L. L. Lin and X. M. Feng, ACS Catal., 2022, 12, 1784–1790 CrossRef CAS
- W. Yang, X. B. Lin, Y. Y. Zhang, W. D. Cao, X. H. Liu and X. M. Feng, Chem. Commun., 2020, 56, 10002–10005 RSC
-
(a)
Nitrogen, Oxygen and Sulfur Ylide Chemistry: A Practical Approach in Chemistry, ed. J. S. Clark, Oxford University Press, New York, 2002 Search PubMed
- R. W. Hoffmann, Angew. Chem., Int. Ed. Engl., 1979, 18, 563–572 CrossRef
- V. Tyagi, G. Sreenilayam, P. Bajaj, A. Tinoco and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 13562–13566 CrossRef CAS PubMed
- T. Nakai and K. Mikami, Chem. Rev., 1986, 86, 885–902 CrossRef CAS
-
(a) J. A. Workman, N. P. Garrido, J. Sançon, E. Roberts, H. P. Wessel and J. B. Sweeney, J. Am. Chem. Soc., 2005, 127, 1066–1067 CrossRef CAS PubMed
- M. Ošeka, M. Kimm, I. Järving, K. Lippur and T. Kanger, J. Org. Chem., 2017, 82, 2889–2897 CrossRef PubMed
- X. Xu, J. L. Zhang, S. X. Dong, L. L. Lin, X. B. Lin, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 8734–8738 CrossRef CAS PubMed
- X. Xu, S. X. Dong, L. L. Feng, S. J. Wang, X. H. Liu and X. M. Feng, Org. Lett., 2020, 22, 2692–2696 CrossRef CAS PubMed
- R. F. Kleinschmidt and A. C. Cope, J. Am. Chem. Soc., 1944, 66, 1929–1933 CrossRef CAS
-
(a) A. Guarna, E. G. Occhiato, M. Pizzetti, D. Scarpi, S. Sisi and M. van Sterkenburg, Tetrahedron: Asymmetry, 2000, 11, 4227–4238 CrossRef CAS
- H. Bao, X. Qi and U. K. Tambar, J. Am. Chem. Soc., 2011, 133, 1206–1208 CrossRef CAS PubMed
- X. Yu, N. Wannenmacher and R. Peters, Angew. Chem., Int. Ed., 2020, 59, 10944–10948 CrossRef CAS PubMed
-
(a) M. P. Doyle, V. Bagheri and N. K. Harn, Tetrahedron Lett., 1988, 29, 5119–5122 CrossRef CAS
- For selected examples, see:
(a) M. P. Doyle, D. C. Forbes, M. M. Vasbinder and C. S. Peterson, J. Am. Chem. Soc., 1998, 120, 7653–7654 CrossRef CAS
-
(a) B. T. Parr, Z. Li and H. M. L. Davies, Chem. Sci., 2011, 2, 2378–2382 RSC
- Z. Li, B. T. Parr and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 10942–10946 CrossRef CAS PubMed
- Z. Li, V. Boyarskikh, J. H. Hansen, J. Autschbach, D. G. Musaev and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 15497–15504 CrossRef CAS PubMed
-
(a) W. Kirmse and M. Kapps, Chem. Ber., 1968, 101, 994–1003 CrossRef CAS
- X. Zhang, Z. Qu, Z. Ma, W. Shi, X. Jin and J. Wang, J. Org. Chem., 2002, 67, 5621–5625 CrossRef CAS PubMed
- K. J. Hock and R. M. Koenigs, Angew. Chem., Int. Ed., 2017, 56, 13566–13568 CrossRef CAS PubMed
-
(a) M. Ma, L. Peng, C. Li, X. Zhang and J. Wang, J. Am. Chem. Soc., 2005, 127, 15016–15107 CrossRef CAS PubMed
- Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W.-D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970–976 CrossRef CAS PubMed
-
(a) Z. Liu, X. Jin and Y. Dang, ACS Catal., 2021, 11, 691–702 CrossRef CAS
- X. B. Lin, Y. Tang, W. Yang, F. Tan, L. L. Lin, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2018, 140, 3299–3305 CrossRef CAS PubMed
- X. B. Lin, W. Yang, W. K. Yang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 13492–13498 CrossRef CAS PubMed
- For selected examples, see:
(a) T. Wirth, Angew. Chem., Int. Ed., 2000, 39, 3740–3749 CrossRef CAS
- X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1423–1433 Search PubMed
-
(a) M. P. Doyle, W. H. Tamblyn and V. Bagheri, J. Org. Chem., 1981, 46, 5094–5102 CrossRef CAS
- B. Xu and U. K. Tambar, Angew. Chem., Int. Ed., 2017, 56, 9868–9871 CrossRef CAS PubMed
-
(a) E. Tayama, N. Naganuma, H. Iwamoto and E. Hasegawa, Chem. Commun., 2014, 50, 6860–6862 RSC
- Q. C. Lin, B. W. Hu, X. Xu, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Sci., 2020, 11, 3068–3073 RSC
-
(a) T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H.-Y. Cheong, G. C. Lloyd-Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366–4375 CrossRef CAS PubMed
- S. J. Meek and J. P. A. Harrity, Tetrahedron, 2007, 63, 3081–3092 CrossRef CAS
-
(a) A. B. Gade, P. N. Bagle, P. S. Shinde, V. Bhardwaj, S. Banerjee, A. Chande and N. T. Patil, Angew. Chem., Int. Ed., 2018, 57, 5735–5739 CrossRef CAS PubMed
-
(a)
R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Verlag Chemie, Weinheim, 1970 Search PubMed
- L. Yao and K. Ishihara, Chem. Sci., 2019, 10, 2259–2263 RSC
- L. F. Wang, P. F. Zhou, Q. C. Lin, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Sci., 2020, 11, 10101–10106 RSC
- Y. S. Chen, Y. Liu, Z. J. Li, S. X. Dong, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2020, 59, 8052–8056 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2022 |