
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImaging strategies for monitoring the immune response
Xiaohan
Liu†
 ,
Limeng
Pan†
,
Kaiye
Wang
,
Wei
Pan
,
Na
Li
* and
Bo
Tang
*
,
Limeng
Pan†
,
Kaiye
Wang
,
Wei
Pan
,
Na
Li
* and
Bo
Tang
*
College of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Institute of Molecular and Nano Science, Shandong Normal University, Jinan, 250014, P. R. China. E-mail: lina@sdnu.edu.cn; tangb@sdnu.edu.cn
First published on 7th October 2022
Abstract
Real-time monitoring of the immune response can be used to evaluate the immune status of the body and to distinguish immune responders and non-responders, so as to better guide immunotherapy. Through direct labelling of immune cells and imaging specific biomarkers of different cells, the activation status of immune cells and immunosuppressive status of tumor cells can be visualized. The immunotherapeutic regimen can then be adjusted accordingly in a timely manner to improve the efficacy of immunotherapy. In this review, various imaging methods, immune-related imaging probes, current challenges and opportunities are summarized and discussed.
1. Introduction
Tumor immunotherapy has opened a new era of cancer therapy, offering new hope to many cancer patients, especially those achieving poor responses to traditional treatments.1–4 The body's immune system is the most effective weapon to defend against invasion by pathogens. Cancer vaccine, immune checkpoint blockade therapy (such as the PD-1/PD-L1 antibody and CTLA-4 antibody) and adoptive cell immunotherapy (such as CAR-T cell therapy), as the main means of immunotherapy, have attracted significant attention.5–7 These immunotherapeutic methods have been successfully applied in tumor therapy. However, there are many frustrating experiences, which are caused by several reasons. Cancer cells may establish a defense network against the immune system.8–11 They will reduce the antigen expression to avoid being recognized by immune cells.12 The complexity of immune cell composition and heterogeneity of tumor antigens have reduced the universality of immunotherapy.13,14 Immune-related adverse events also hinder the clinical application of immunotherapy.15,16 Moreover, drug resistance17 and the failure of T lymphocyte infiltration18 need to be addressed. Given a series of problems of immunotherapy, it is important to assess the immune response in real-time.Immunological assessment is to monitor and evaluate the immune status of the body before and after immunotherapy using invasive or non-invasive methods.19–23 The level and status of immune cells in each person are different. Real-time immunological assessment can fully reflect the current status of immune function.24 Before treatment, the basic immune status of patients should be evaluated to comprehensively understand the level of PD-L1, NK cells, killing T cells and other immune cells in the body, so as to master the immune reserve and response. Immunological assessment can help distinguish immune responders and non-responders.20,25–28 On this basis, the types and programs of cell therapy can be more accurate and effective. After treatment, the immune reconstitution status of the patients needs to be regularly evaluated to determine whether the patients are relieved from immunosuppression.
Various drugs have diverse effects on different tumor cell lines. Applying real-time immunosurveillance can be used to screen drugs in preclinical models. It is significant to horizontally and vertically evaluate the effects of different drugs on tumors.29,30 In addition, immunoimaging technology can also provide insight into the pharmacokinetic physiological and pathological mechanisms of immunotherapeutic drugs, which could provide certain support for the formulation of strategies and timely adjustment of drugs.
Many imaging strategies have been developed to predict or track the immune response, providing a basis for the implementation of immunotherapy.28,31 The immune response can be evaluated by immune markers secreted or expressed by immune cells in activated or suppressed status.32–34 Appropriate labelling strategies can track the dynamic changes of immune cells in real time.35–37 This review introduces different imaging modalities and summarizes the designs of probes according to the different cell types (Fig. 1 and Table 1). The current problems and the future development direction of immune-related imaging are also discussed.
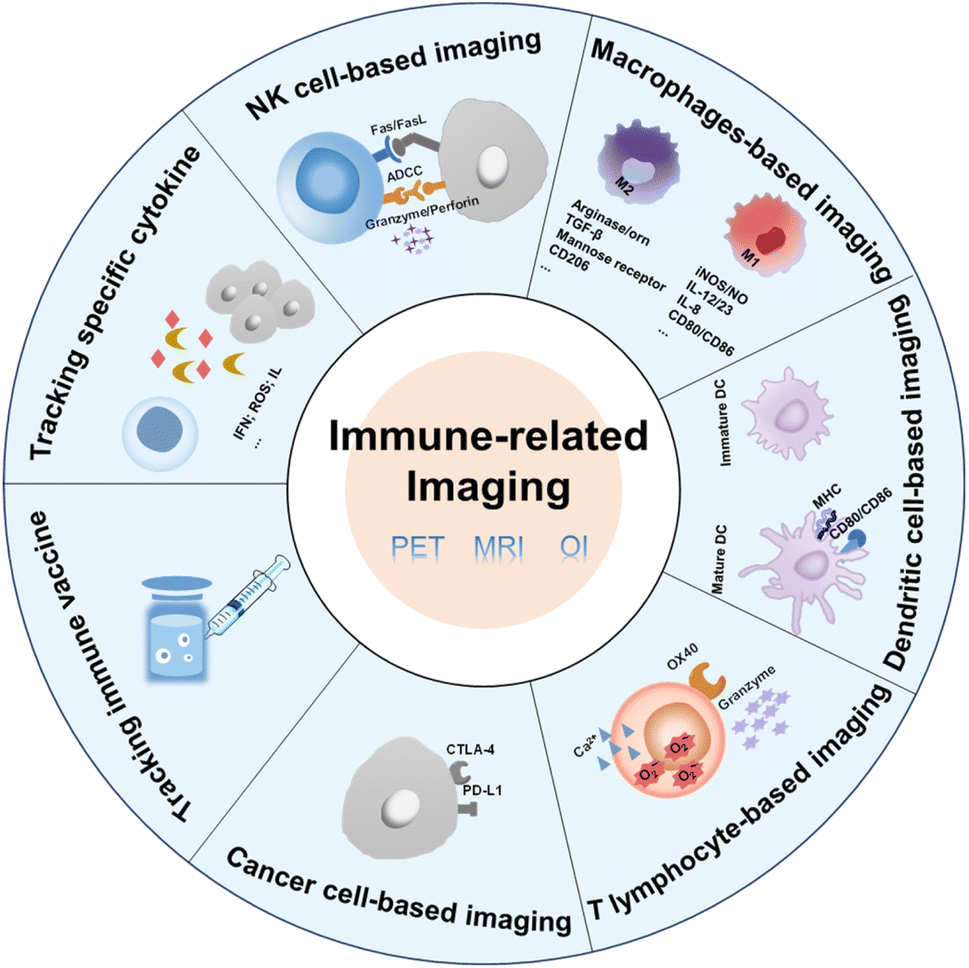 | ||
| Fig. 1 Factors associated with immune cells and cancer cells that can be used in immune-related imaging. | ||
| Cell type | Labelling cells or imaging markers | Imaging method | Designs |
|---|---|---|---|
| NK cells | Labelling NK cells | MRI | A magnetic nanocomposite including hyaluronic acid (HA), protamine (P), and ferumoxtol (F)35 |
| ROS | OI | Down-conversion nanoparticles (DCNPs) coated with a ROS-sensitive dye60 | |
| Granzyme B | OI | Granzyme B-elicited chemiluminescence probes61 | |
| Macrophages | Labelling macrophages | PETI | 64Cu-labelled polyglucose nanoparticles70 |
| 18F-labelled macrophages71 | |||
| MRI | Mannose-modified iron oxide nanoparticles (IONPs)72 | ||
| OI | Dextran modified by ICG75 | ||
| Aggregation-induced emission (AIE) molecules encapsulated in glucan particles81 | |||
| OI/MRI | MARCO antibody-conjugated upconversion nanoparticles (UCNPs)36 | ||
| ROS | OI | NO imaging probes encapsulated in liposomes30 | |
| Dendritic cell | Labelling dendritic cells | PETI | The nanoprobe formed by PPa-conjugated short-chain PEG and FITC-modified Trp2 peptide antigens88 |
| T lymphocyte | Labelling T lymphocyte | PETI | 64Cu-labelled OVA-CTLs37 |
| 89Zr-PEGylated anti-CD8 VHH91 | |||
| 99mTc-labelled CD4 monoclonal antibodies93 | |||
| OI | NIR fluorescent dye-labelled OX40 antibody for imaging T lymphocytes97 | ||
| OI/PETI | NIR dye and 89Zr dual-labelled silica nanoparticles98 | ||
| Granzyme B | PETI | Granzyme B-cleavable peptide chain conjugated with a chelating agent containing 68Ga33 | |
| 64Cu-chelated and membrane-anchored probes101 | |||
| OI | NIR dye-conjugated peptide substrate of granzyme B29 | ||
| Semiconducting polymer (SP) conjugated with IR800-labelled granzyme B-cleavable peptides102 | |||
| Polymer-loaded peptide substrates simultaneously linked with fluorophores and quench groups103 | |||
| Granzyme B-responsive platform transformed from molecules to nanoparticles104 | |||
| O2˙− | OI | The nanoprobe constructed with an O2˙−-activable chemiluminescence substrate, SP, and triblock polymer via a nano-precipitation method105 | |
| Ca2+ | CT/OI | G5 PAMAM dendrimers loaded with the indicator of Ca2+and Au NPs108 | |
| Tumor cells | CTLA-4 | PETI | 64Cu-labelled ipilimumab for specifically binding to CTLA-4 expressed in human non-small cell lung cancer cells110 |
| PD-L1 | PETI | 64Cu-labelled αPD-L1 Fab incorporated with one UAA, p-azidophenylalanine (pAzF)112 | |
| 68Ga-NOTA-Nb109 (a nonblocking single-domain antibody)48 | |||
| OI | PD-L1 mAb connected with NIR-II fluorophores115 | ||
| PD-L1-functionalized ErNPs with NIR-IIb luminescence properties116 | |||
| NIR-II nanoprobe based on a boron-dipyrromethene (BDP) photosensitizer encapsulated in nanomicelles117 | |||
| Simultaneously labelling internal and external antigens for multispectral fluorescence imaging to track the programmed delivery of combined nanovaccines118 | |||
| Tracking immune vaccine | — | OI | 4-[18F]fluoro-1-naphthol ([18F]4FN)119 |
| Tracking specific cytokine markers | High-energy free radicals | PET/CT | 89Zr-anti-IFNγ122 |
| IFN | PET |
2. Immune-related imaging strategies
2.1. Imaging methods
2.2. Imaging strategies based on different cells
2.2.1.1. Labelling NK cells. For the direct labelling of NK cells, a magnetic nanocomposite (denoted as HAPF) consisting of hyaluronic acid (HA), protamine (P), and ferumoxtol (F) was developed by Kim et al., which achieved effective treatment of MRI-guided adoptive NK cell therapy.35 Under a magnetic field, HAPF-labelled NK cells were activated and cell lysis was enhanced. Furthermore, MRI was used to monitor NK cell transport and aggregation in tumor tissue, providing an effective tool for controlling NK cell infusion. Song et al. employed a ratiometric imaging method to monitor the status of NK cells in adoptive cell immunotherapy.60 The ROS-sensitive dye IR786s and F127 were coated on down-conversion nanoparticles (DCNP) to form the nanoprobes (denoted as DCNP@786s) (Fig. 2A). They were used to directly mark NK cells and track the viability of NK cells in real time. Excessive ROS could be produced by NK cells during the process of cell death, which resulted in the degradation of IR786s. The fluorescent signal at 1550 nm of the dye excited by an 808 nm laser was increased, while the signal of DNCP excited by a 980 nm laser remained unchanged. Therefore, the signal ratio of F1550Em,980Ex/F1550Em,808Ex could convey information about NK cell activity. Under experimental conditions, the nanoprobe was non-toxic to NK cells and had no effect on cell phenotype. In vitro experiments demonstrated that various ROS could stimulate different degrees of near-infrared region II (NIR-II) fluorescence recovery under laser irradiation at 808 nm, while the fluorescent signal under 980 nm showed no obvious change (Fig. 2B). Under a series of harsh conditions, the level of ROS in NK cells increased significantly. These results demonstrated that the nanoprobe could assess the activity of NK cells by monitoring the changes of ROS. With the imaging of this nanoprobe, the ratio increased with the simultaneous treatment of NK cells with different cytokines, suggesting the effectiveness of the cytokine-assisted NK cell immunotherapy in the HCC tumor-bearing mice.
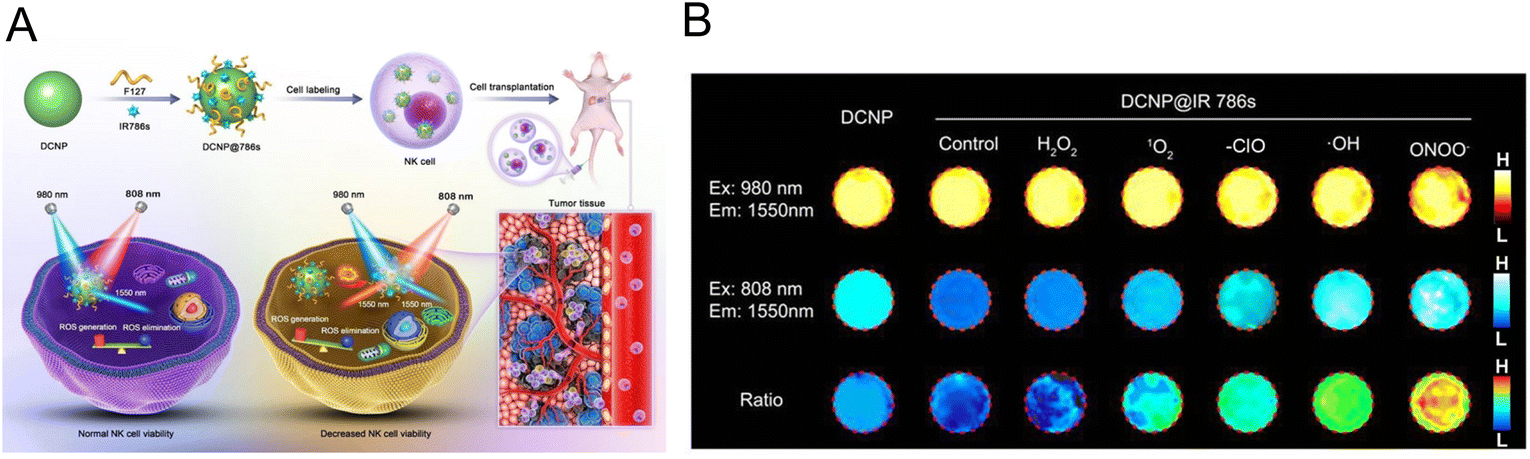 | ||
| Fig. 2 (A) The preparation of DCNP@786s and the schematic diagram of labelling and ratiometrically imaging NK cells in the NIR-II window. (B) Fluorescence images of DCNP@786s treated with various ROS. Reproduced from ref. 60 with permission from Wiley-VCH GmbH, Copyright 2021. | ||
2.2.1.2. Imaging specific biomarkers of NK cells. Regarding monitoring the specific biomarkers of NK cells, Vendrell et al. used a granzyme B-elicited chemiluminescence probe to assess the activity of NK cells.61 The peptide substrate (Ile-Glu-Pro-Asp) of granzyme B was connected to the phenoxydioxetane moiety via a self-eliminating linker (Fig. 3A). In the presence of granzyme B, a bright chemiluminescence signal with a higher signal-to-noise ratio (SNR) was recovered as a signal of immune activation. In the tumor-bearing mice model for adoptive NK cell transfer, the chemiluminescence signal was only collected from the tumor that had been injected with NK cells (Fig. 3B).
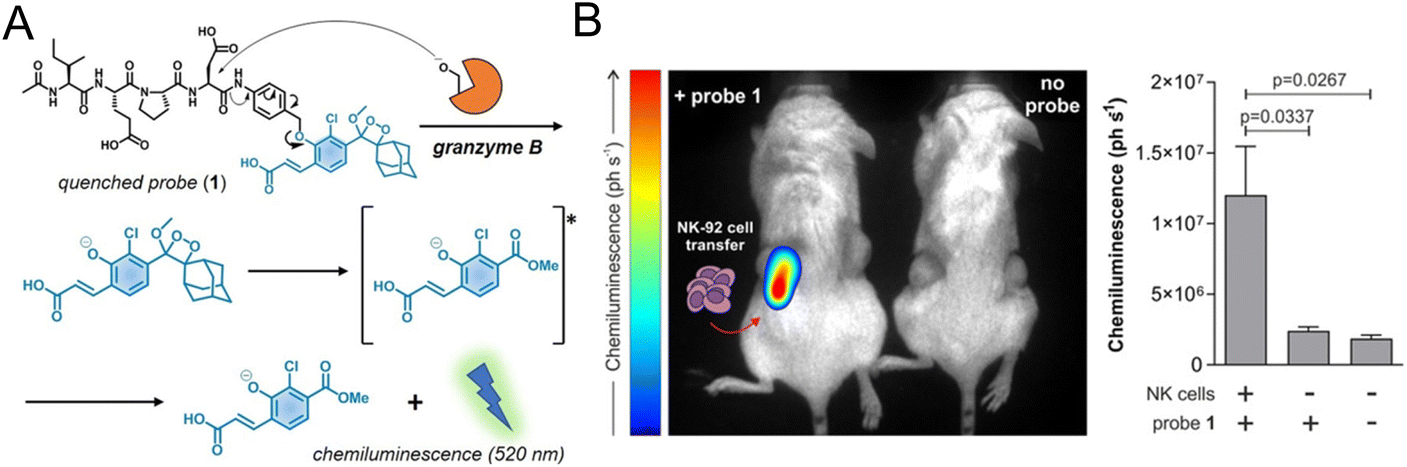 | ||
| Fig. 3 (A) Structure of the granzyme B-activated chemiluminescence probe and the mechanism of fluorescence recovery. (B) In vivo imaging and quantification of the chemiluminescence signal of tumors with or without NK-92 cell transfer. Reproduced from ref. 61 with permission from Wiley-VCH GmbH, Copyright 2021. | ||
Adoptive NK cell immunotherapy has broad prospects for improving treatment of malignant tumors. Therefore, imaging adoptive NK cells is conducive to visualizing the activities and immune status of NK cells, which will provide a basis for further improvement of adoptive NK cells. The efficiency of in vitro labelling and its effect on NK cell activity should be fully evaluated to ensure nondestructive labelling.
2.2.2.1. Labelling macrophages. To directly label macrophages, multiple imaging methods have been used to track the accumulation of macrophages in tumor tissue and specifically image the changes of a specific type of macrophage. For PET imaging of macrophages, the 64Cu-containing radioactive chelating agent was linked to the biodegradable polyglucose nanoparticles (Macrin) viaL-lysine to quantify tumor-associated macrophages (TAMs) in tumor tissue.70 Compared to other cells, Macrin could be selectively taken up by macrophages. From the imaging results, enhanced permeability and retention effect could be found in the tumor tissue due to the massive accumulation of macrophages. Macrin could help to select suitable nanomaterials for tumor therapy and predict the immune properties of drugs. In the strategy designed by Kim et al., macrophages were tracked by PET imaging using a biorthogonal reaction (Fig. 4).71 First, dibenzocyclooctyne (DBCO)-modified mesoporous silica nanoparticles (MSNs) were prepared and used to incubate with RAW 264.7 cells. Modified macrophages were injected into mice and then the 18F-labelled azide was used to react with DBCO to real-timely locate RAW 264.7 cells in tumor-bearing and ApoE-/- mouse models. By means of the migration property of macrophages to tumor tissue and atherosclerosis, PET images of tumor tissue and atherosclerosis were obtained. This system was expected to be used for other cell-tracking experiments with the in vivo alkyne–azide cycloaddition.
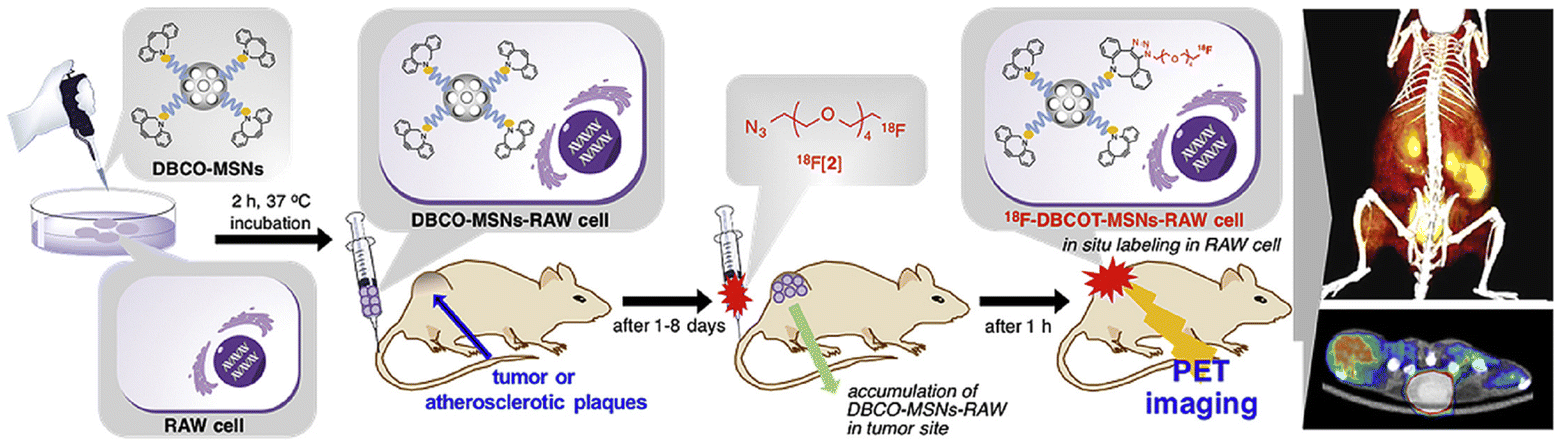 | ||
| Fig. 4 Schematic of DBCO-MSN-incubated macrophages and further bonding with an 18F-labelled azide synthon to track the macrophages via PETI. Reproduced from ref. 71 with permission from Elsevier Ltd, Copyright 2019. | ||
In the strategy designed by Mao et al.,72 specific MRI was performed for M2 macrophages (Fig. 5A). Magnetic iron oxide nanoparticles (IONPs) were coated with the polyethylene glycol-block-allyl glycidyl ether (PEG-b-AGE) polymer to prevent nonspecific adsorption. Then mannose was modified on the surface to specifically target M2 macrophages. Confocal imaging experiments showed that mannose-modified magnetic nanoparticles could be effectively taken up by M2-type macrophages, and the uptake was significantly reduced after CD206 was blocked, which demonstrated that CD206 mediated the uptake of the probe by M2-type macrophages. In vivo imaging experiments (Fig. 5B) demonstrated that the T2 signal was significantly reduced in the tumor tissue injected with mannose-modified nanoparticles, while no significant change was observed in the nontargeted group. The potent M2 macrophage-targeting property enables the nanoprobe to be used for indicating changes in M2 macrophages, providing an effective evaluation tool for the development of M2 macrophage-related drugs.
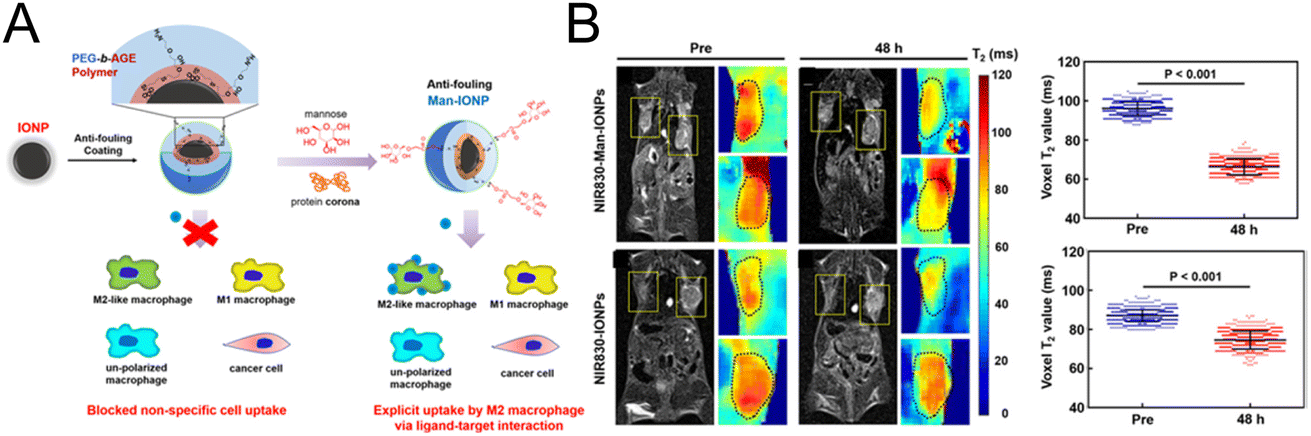 | ||
| Fig. 5 (A) The preparation of mannose-modified IONPs with a reduced nonspecific adsorption effect. (B) MRI imaging and the quantification of T2 signals in tumor tissue before and after the injection of IONPs with or without mannose modification. Reproduced from ref. 72 with permission from American Chemical Society, Copyright 2020. | ||
For optical imaging, fluorescent dyes in the NIR II region (1000–1700 nm) with a deeper penetration depth have better spatial resolution and can greatly reduce light damage to biological tissues.73,74 Sheng et al. designed a probe with NIR II emission property to image macrophages in deep tumor tissue (∼0.5 cm).75 Dextran was cross-linked with lysine to obtain the amino-functionalized dextran. The carboxylated ICG was then connected to synthesize the final nanoprobe (DN-ICG) with biocompatible and metabolizable properties (Fig. 6A). The interaction between dextran and specific ICAM-3-grabbing nonintegrin related 1 (SIGN-R1) mediated nanoprobe uptake by macrophages. High signal-to-background ratio (SBR) imaging was observed in the NIR II region, suggesting the potential of this probe to image macrophages in tumor tissue with high quality. Subsequently, the probe was applied to measure the macrophage-related response after different treatments. The increased fluorescence intensity in the low dose X-ray-treated group and the decreased intensity in the zoledronic acid-treated group (Fig. 6B) were consistent with the results of the characterization of macrophage number using a flow cytometer (Fig. 6C), which demonstrated the potential of this probe for macrophage-related imaging. The immune detection of macrophages can also be applied in the postoperative evaluation of transplantation. Extensive macrophage infiltration will damage the transplanted organ and cause a strong immune rejection response.76 Therefore, monitoring macrophages after transplantation can provide the status of transplantation in a timely manner. Fluorescent molecules with aggregation-induced emission (AIE) properties possess a large Stokes shift and excellent optical stability.77,78 AIE molecules can solve the aggregation-induced fluorescence quenching of traditional fluorescence dyes.79,80 The AIE material is regarded as a potential contrast agent. In the design of Xie et al., AIE molecules were encapsulated in glucan nanoparticles to trace the infiltration of macrophages after transplantation.81 This nanoprobe could be selectively internalized by macrophages via mimicking the intestinal infection behavior of yeast and could be retained in macrophages for a prolonged period. No obvious innate immune response was elicited with this nanoparticle, suggesting that this system was compatible with transplantation. In the skin graft model, fluorescence imaging results showed that the macrophages gradually gathered at the skin graft site with time, and the fluorescence intensity was higher in the xenograft than in the allograft model. This was consistent with the in vitro validation of macrophage infiltration, which confirmed the feasibility of this nanoprobe for detecting the immune response after transplantation.
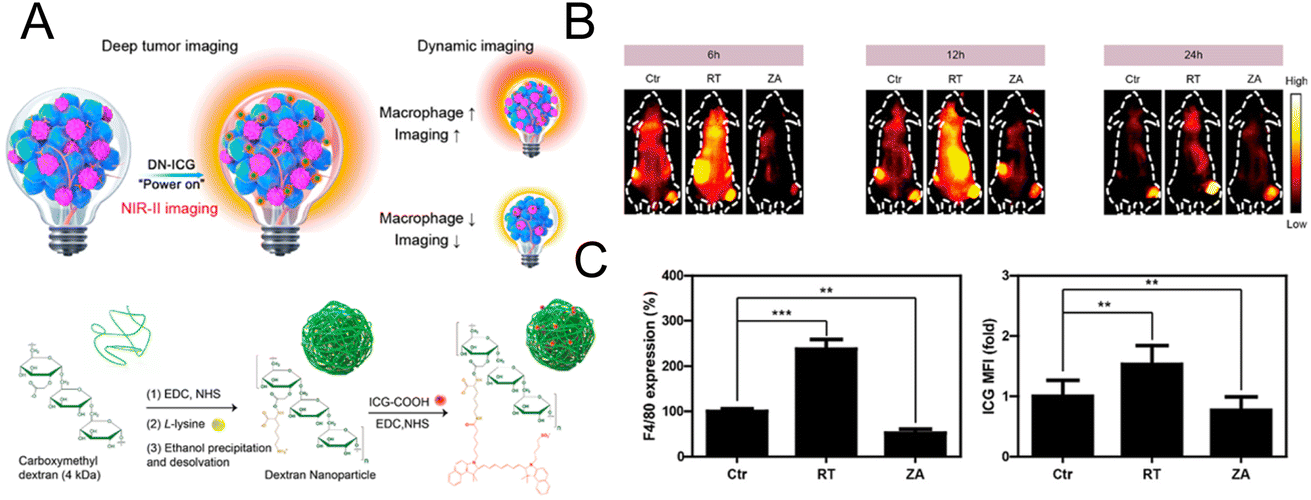 | ||
| Fig. 6 (A) Schematic diagram of the mechanism and synthesis of DN-ICG. (B) NIR-II fluorescence images of macrophages in tumor tissue after treatment with X-ray and zoledronic acid. (C) Flow cytometric analysis of the number of macrophages in tumor tissue. Reproduced from ref. 75 with permission from American Chemical Society, Copyright 2021. | ||
Combining multiple imaging methods is mutually complementary for directly locating cells. Changes of activated M1-type macrophages can reflect the plaque progression in atherosclerosis. Therefore, imaging M1-type macrophages can provide a basis for the diagnosis of atherosclerosis. Gao et al. designed and synthesized a Gd-doped and MARCO antibody-conjugated upconversion nanoparticle (UCNP) system to image M1-type macrophages in an atherosclerotic mice model.36 MARCO antibody enabled the probe to actively target M1 macrophages. UCNPs transmitted the fluorescence signal, and the Gd element achieved MRI. The fluorescence imaging results showed that the nanoparticles could actively target M1 macrophages and foam macrophages, and preferentially accumulate in vulnerable plaques of the carotid artery. The same phenomenon could also be seen in the MRI results. The dual-mode imaging capability of the nanoprobe could be used for the diagnosis of vulnerable plaques.
2.2.2.2. Imaging specific biomarker of macrophage. For imaging macrophage-related marker to reflect the immune status, Kulkarni et al.30 constructed a platform for monitoring M1 macrophages by OI. Considering that NO can be seen as a marker of antitumoral M1 macrophages, NO imaging probes were encapsulated in liposomes as the nanoreporter (Fig. 7A). In the 4T1 tumor-bearing mice model, macrophage activation was successfully imaged in mice treated with different drugs with this nanoprobe. Experimental results demonstrated that the most significant and prolonged optimal activation signal was observed in the group with liposomes simultaneously encapsulating CSF1R inhibiting amphiphile and the NO imaging reagent (Fig. 7B). The excellent tumor inhibition rate and relatively high content of M1 macrophages in this group were consistent with the fluorescence imaging results, demonstrating that this NO nanoreporter could potentially be a tool for detecting the immune response of M1 macrophages with various treatments.
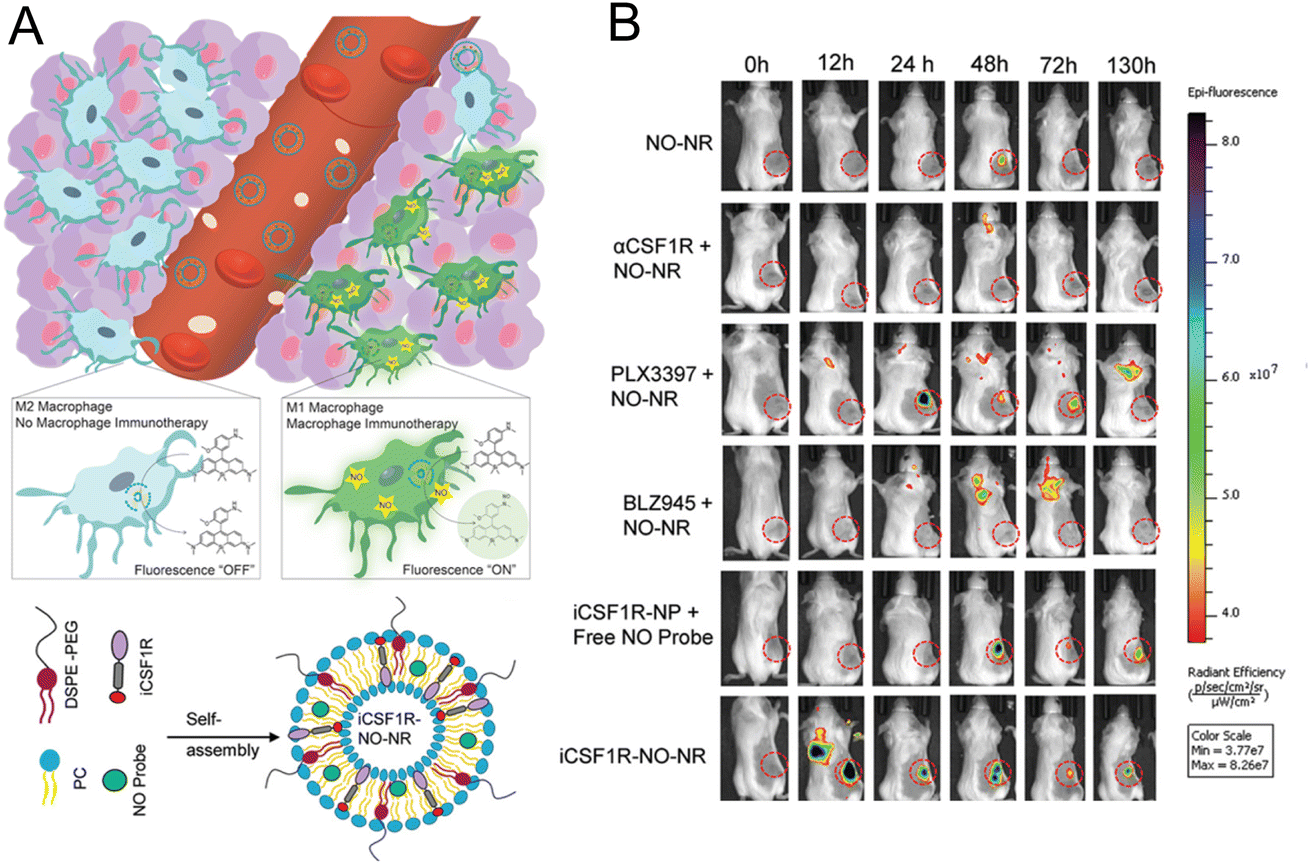 | ||
| Fig. 7 (A) Principle of the NO nanoreporter for specifically responding to M1 macrophages and the schematic illustration of the simultaneous encapsulation of CSF1R inhibiting amphiphile and NO imaging probes in liposomes. (B) Fluorescence images of the mice with different treatments. Tumor tissues were marked by circles. Reproduced from ref. 30 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Copyright 2020. | ||
Macrophages can not only directly engulf pathogens but also act as antigen-presenting cells to participate in the cellular immune response.82,83 Imaging macrophages can help distinguish the phenotypes in the tumor tissue and further stimulate the change of cellular phenotype according to the requirements of treatment. In addition, it is significant to fully verify and improve the relationship between macrophage phenotype and the development of disease.
In studies by Gao et al., short-chain PEG was conjugated with photosensitizer PPa, and then the FITC-modified Trp2 peptide antigen was self-assembled with this conjugate to form the nanoprobe.88 No significant cytotoxicity to dendritic cells was detected. The tetrapyrrole ring of PPa can be labelled with 64Cu for PET imaging of the migration of dendritic cells. The in vivo PET imaging result demonstrated that the signal gradually migrated from the injection site toward draining lymph nodes.
2.2.4.1. Labelling T lymphocytes. Liu et al. used PETI to record the accumulation of adoptive T cells in tumor tissues to further evaluate the therapeutic effects of adoptive T cell therapy. They used metabolic glycoengineering to label azide groups on the surface of OVA-specific cytotoxic T lymphocytes (OVA-CTLs) (Fig. 8A).37 Then 64Cu was successfully labelled on CTLs by the reaction between DBCO and azide groups to noninvasively track OVA-CTLs. PET imaging results demonstrated that 64Cu-labelled OVA-CTLs accumulated more in tumor tissue than 64Cu-labelled control-CTLs in B16-OVA tumor-bearing mice (Fig. 8B). Moreover, an increased accumulation of 64Cu-labelled OVA-CTLs could be detected in PET images after the treatment of PLGA-FAKi (focal adhesion kinase inhibitor) nanoparticles. The excellent antitumor efficiency of OVA-CTLs plus the PLGA-FAKi group also confirmed the PET imaging results. Ploegh et al. designed 89Zr-labelled PEGylated single-domain antibody fragments (nanobodies or VHHs) to evaluate the therapeutic effect of PD-1 blockade.91 The immuno-PET imaging mediated by the 89Zr-PEGylated anti-CD8 VHH probe demonstrated that more CD8+ T cells infiltrated into the tumor tissue in responders. In addition, 89Zr-labelled PEGylated anti-CD11b VHH was utilized to study the changes of the CD11b+ population. The results showed that CD11b+ cells were already present in the tumor tissue before treatment and were more evenly distributed in tumor tissue after anti-PD-1 treatment.
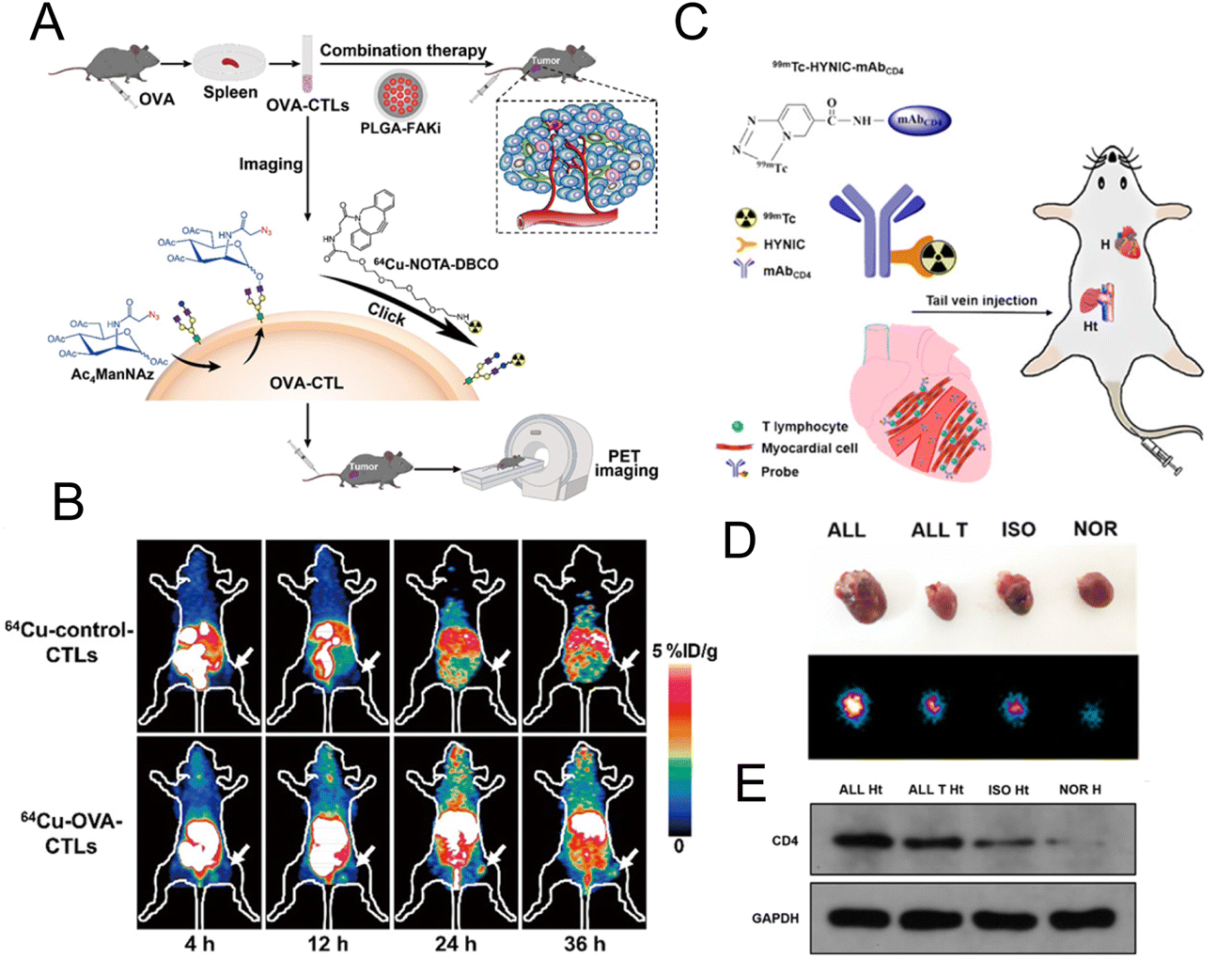 | ||
| Fig. 8 (A) Schematic illustration of 64Cu-labelled OVA-CTL for PET imaging and synergistic treatment with PLGA-FAKi. (B) PET imaging for tumor tissue treated with 64Cu-control-CTLs and 64Cu-OVA-CTLs. (C) Construction of the 99mTc-HYNIC-mAbCD4 probe for SPECT imaging. (D) The SPECT images of graft heart from different groups. (E) Western blot analysis of CD4 expression. (A and B) Reproduced from ref. 37 with permission from Springer Nature, Copyright 2021. (C, D and E) Reproduced from ref. 93 with permission from American Chemical Society, Copyright 2021. | ||
CD4+ T lymphocytes are the main T lymphocytes that mediate the acute rejection (AR) after heart transplantation (HT).92 Therefore, the specific imaging of CD4+ T lymphocytes can provide an accurate diagnostic tool for AR. Xie et al. developed a radionuclide 99mTc-labelled CD4 monoclonal antibody (mAbCD4) as the imaging probe (denoted as 99mTc-HYNIC-mAbCD4) to bind with CD4+ T lymphocytes and accumulate in the graft heart (Fig. 8C).93 In the allograft (ALL) model, the clear position of graft heart could be visualized at 1 h after injection with this probe. Compared with the other three groups, relatively high radioactivity occurred in the heart of the ALL group with SPECT/CT imaging (Fig. 8D). Western blotting evaluating the expression of CD4 (Fig. 8E) further confirmed that CD4+ T lymphocytes were significantly increased in the AR after HT and this probe could effectively reflect the expression of CD4+ T lymphocytes.
OX40 is expressed primarily on the surface of activated T cells and can induce the expression of proteins with antiapoptotic and cell cycle progression properties.94–96 Furthermore, OX40 can counteract the inhibitory effect of other factors on immune cells and directly stimulate effector T cells. In the design of Shan et al., targeted OX40 imaging was used to predict the early immune response triggered by the vaccine.97 The NIR fluorescent dye was connected to the OX40 antibody to form the imaging probe (denoted as IRDye800-AbOX40). The experimental results showed that probe uptake in the CpG-treated tumor was significantly increased compared to the control group. In addition, the intensity of the fluorescence signal in vivo showed a strong correlation with the expression of OX40. The IRDye800-AbOX40 probe had excellent performance in predicting the early immune response of hepatocellular carcinoma.
Aras et al. applied NIR dye and 89Zr dual-labelled silica nanoparticles as the tracer agents for monitoring the dynamics of immune cells in adoptive cell transfer therapy.98 The NIRF/PET nanotag-labelled human carcinoembryonic antigen (hCEA)-redirected CAR T cells were studied in the peritoneal carcinomatosis model. As shown in the fluorescence imaging result, the signal of cancer cells rapidly overlapped with the double signal of CART cells, achieving the real-time tracing of CAR T cells.
2.2.4.2. Imaging specific biomarkers of T lymphocytes. T lymphocytes can release perforin and granzyme B to mediate the killing of tumor cells.99,100 The activation of T cells involves the cooperation of many cytokines. Thus, imaging specific biomarkers can reflect the activation status of T lymphocytes and provide support for the diagnosis of diseases.
Many strategies have been applied to the imaging of granzyme B to assess the immune activation of T lymphocytes. Mahmood et al. have confirmed that granzyme B could be an early biomarker of immune activation using a PETI probe.33 The granzyme B-cleavable peptide chain was modified to achieve irreversible connection and then conjugated with a chelating agent containing 68Ga to form a probe for PETI. The PETI results showed that significantly increased probe uptake was observed in the tumor regression group after treatment, while there was a relatively low uptake rate in nonresponsive tumors. For the analysis of biopsy specimens from melanoma patients, responders and non-responders showed significant differences in the expression of granzyme B and could be distinguished with this probe. The results demonstrated that PETI for granzyme B could serve as a quantitative and useful predictive method to evaluate immunotherapy. Craik et al. designed a 64Cu-chelated tracer for PETI to explore the secretion and biochemical activity of granzyme B in vivo.101 The peptide which can bind tightly to the phospholipid bilayer was incorporated with the radioactive element. The peptide masking domain was linked with the granzyme B-cleavable peptide. Extracellular granzyme B cleaved the linker and the membrane-binding peptide inserted into the adjacent cell membrane to fix the sign for detection. The experimental results demonstrated that the cut off and tissue uptake changes were driven by immune cell activation and the secretion of granzyme B. This probe can be applied for the early detection of the immunotherapeutic response.
The “always on” signals from imaging reagents reflect a poor correlation with biomarkers related to immune activation. In view of this, Pu et al.29 designed a granzyme B-activatable NIR fluorescent probe for the real-time monitoring of immune activation. The granzyme B-cleavable peptide was linked to a NIR hemicyanine dye and quenched the NIR fluorescence. Subsequently, the hydrophilic poly(ethylene glycol) (PEG) chain was connected to form the macromolecule fluorescence reporter, which could passively target the tumor tissue (Fig. 9A). The fluorescence recovery induced by granzyme B had a good correlation with the level of immune activation, which could perform immunological assessment in a non-invasive manner. The probe was used to image the immune-activation level in the tumor-bearing mice treated with five immune-drugs, respectively. Compared to other groups, a stronger fluorescence signal emerged in the group treated with S-(2-boronoethyl)-L-cysteine hydrochloride (BEC) (Fig. 9B). The expression of CD8 antibodies was also relatively high (Fig. 9C). In another study, a ratiometric imaging nanoprobe with granzyme B-responsive property was synthesized to assess the immune activation level of T lymphocytes.102 Granzyme B-cleavable peptide substrates were labelled with the NIR dye IR800 and further conjugated on the semiconducting polymer (SP) (Fig. 10A). IR800 and SP could produce both NIR fluorescence (NIRF) and photoacoustic (PA) signals. When encountering granzyme B, the NIRF820 and PA760 signals from IR800 decreased. Meanwhile, these two signals (740 nm for NIRF and 700 nm for PA) from SP remained unchanged. In mice treated with BEC, the ratios of NIRF740/NIRF820 and PA700/PA760 were higher compared to the control groups (Fig. 10B and C). These results suggested that this nanoprobe with high sensitivity and specificity for granzyme B could be used for in vivo imaging of T lymphocytes.
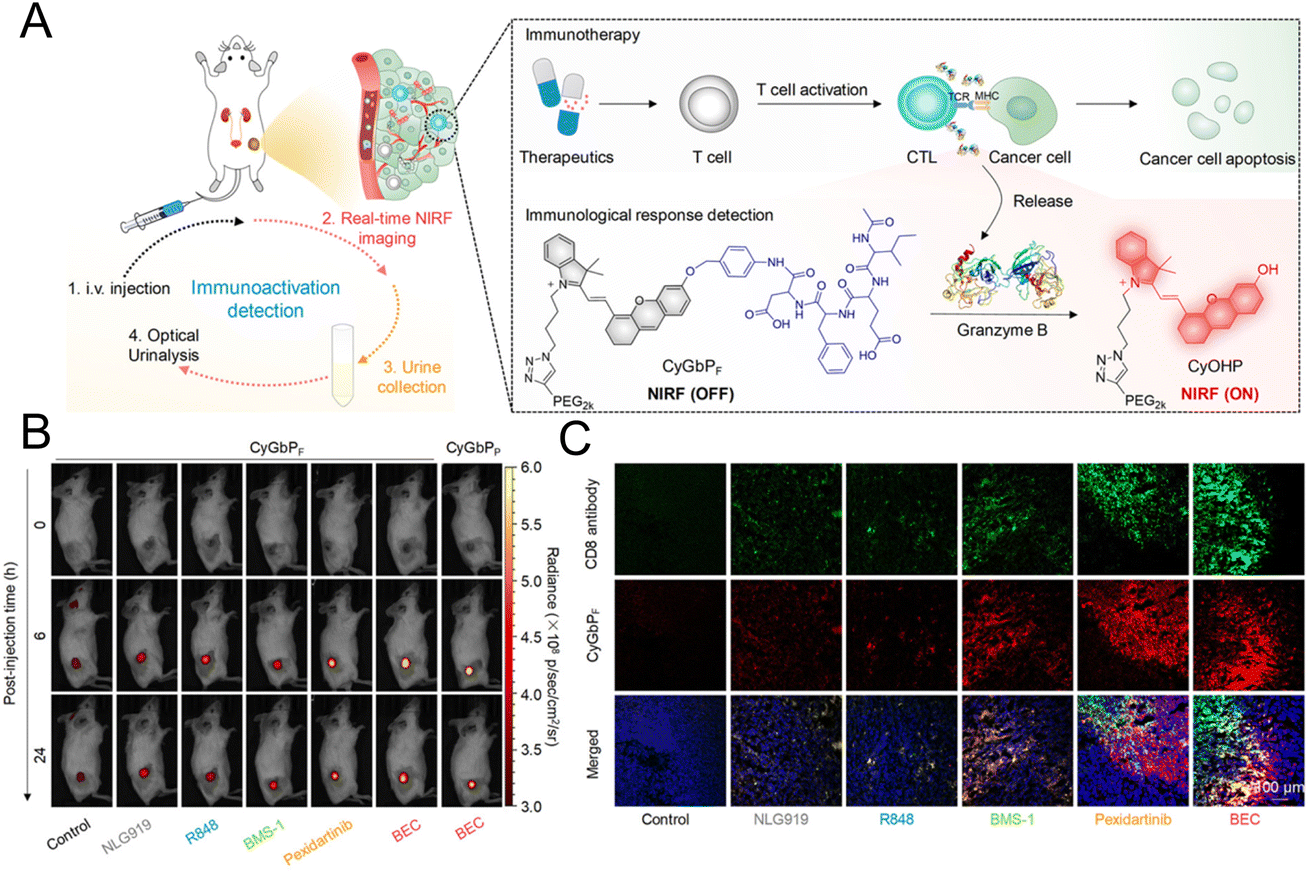 | ||
| Fig. 9 (A) Schematic diagram of the granzyme B-responsive imaging probe for detecting the activation status of T lymphocytes. (B) The fluorescent signals of tumor tissue treated with different immunotherapeutic drugs. (C) Immunofluorescence staining images of the CD8 antibody. Reproduced from ref. 29 with permission from American Chemical Society, Copyright 2020. | ||
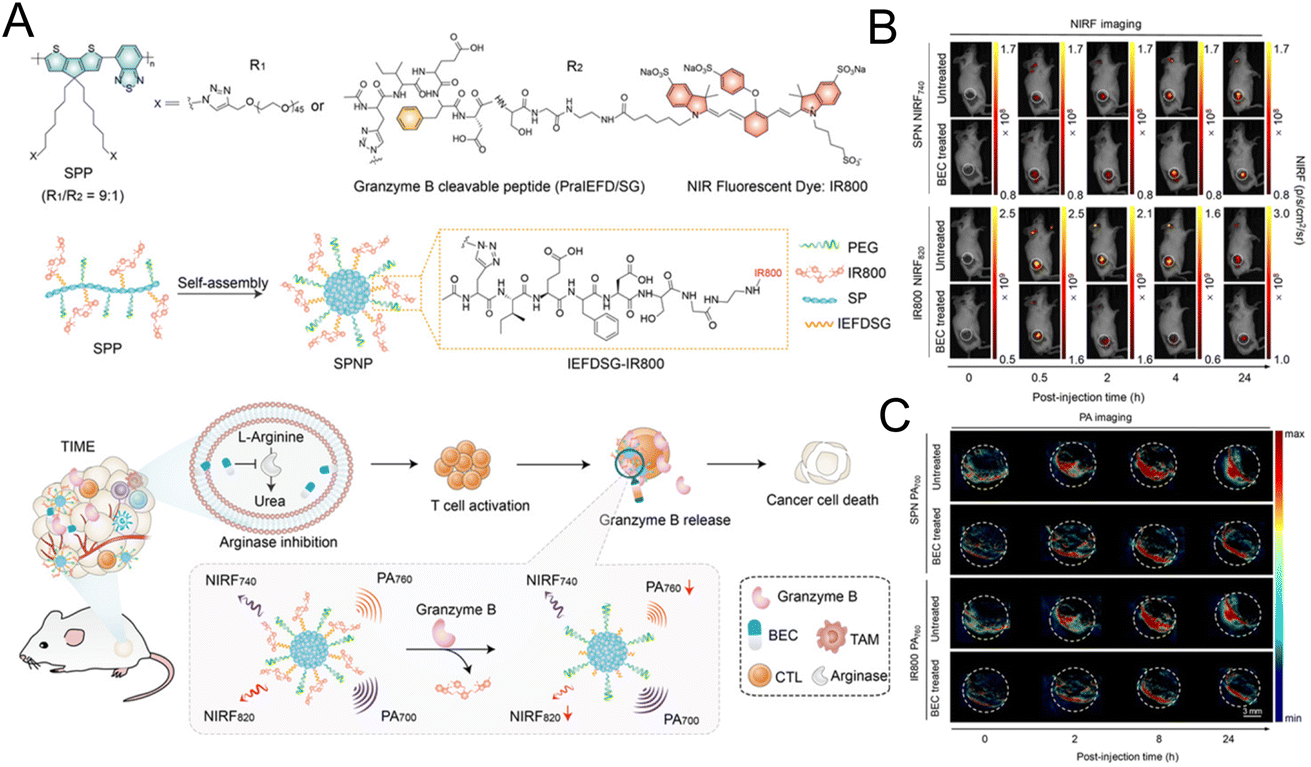 | ||
| Fig. 10 (A) The synthesis of the SP nanoprobe (SPNP) and schematic diagram of granzyme B-triggered ratiometric imaging. Fluorescence imaging (B) and PA imaging (C) of tumor tissue under different excitation lasers. Reproduced from ref. 102 with permission from Wiley-VCH GmbH, Copyright 2021. | ||
Kulkarni et al. constructed a nanoreporter (GNR) simultaneously carrying therapeutic components and the dye for imaging granzyme B. The quenching group and the fluorescent dye are simultaneously attached to the granzyme B-cleaved peptide substrate. The peptide was then conjugated to the poly(isobutylene-alt-maleic anhydride) (PIMA) skeleton (Fig. 11A).103 Due to the high reactivity of the polymer framework, PD-L1 antibodies were conjugated to form an integrated platform for diagnosis and treatment. The interception of the PD-1/PD-L1 pathway stimulated the T lymphocyte to release granzyme B. Granzyme B cleaved the peptide, leading to the release of the quencher and the recovery of the fluorescence signal. This nanoreporter could help identify tumors with a high response or low response to the immunotherapy. A bilateral tumor model was constructed with a poorly responsive WT-B16/F10 tumor and highly responsive WT-MC38 colon adenocarcinoma tumor (Fig. 11B). As shown in Fig. 11C, an obvious fluorescence signal could be seen in the MC38 tumor treated with αPDL1-GNR. The experimental results of the flow cytometry and immunofluorescence staining assay have also confirmed that a highest level of tumor-infiltrating CD8+ T lymphocytes and granzyme B could be detected in αPDL1-GNR-treated MC38 tumors.
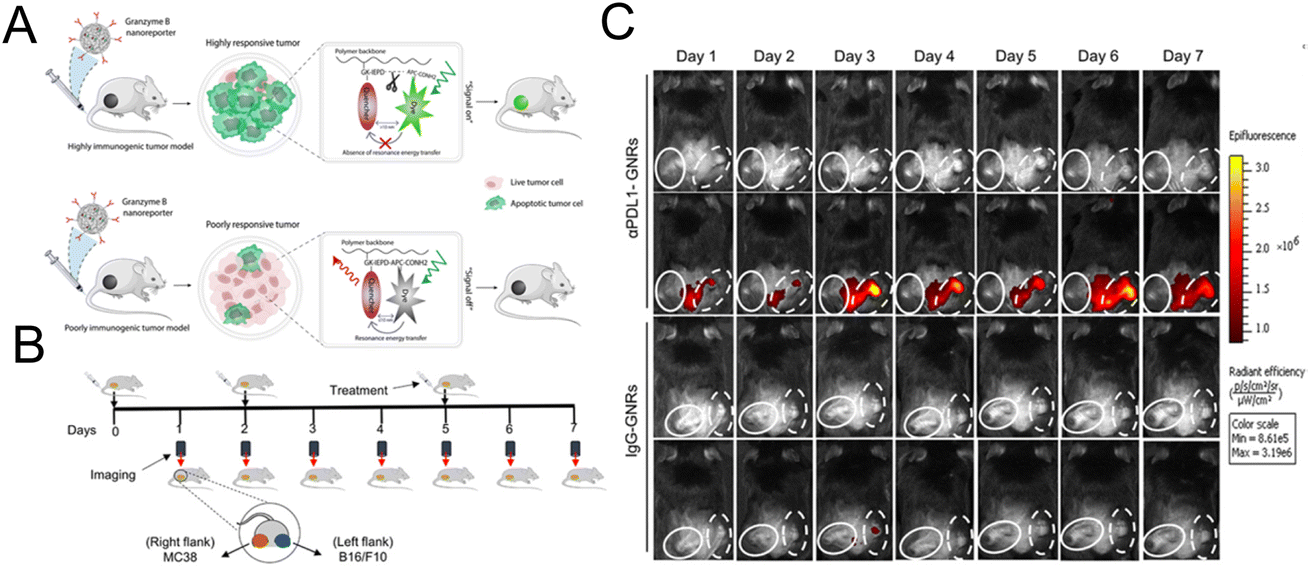 | ||
| Fig. 11 (A) The construction of GNRs and the mechanism of granzyme B-mediated fluorescence signal recovery. (B) Establishment of a bilateral tumor model. (C) Fluorescence imaging of tumor tissue treated with αPDL1-GNRs or lgG-GNRs. Reproduced from ref. 103 with permission from Science, Copyright 2020. | ||
In the design of Rao et al., an intelligent platform which could respond to granzyme B and be transformed from a molecule to a nanoparticle was developed for imaging the immune response.104 In the presence of granzyme B and GSH, the cysteine residues were exposed, and could react with the aromatic nitrile group to form large aggregates. Labelling with Cy5 enabled fluorescence imaging of granzyme B, and thus the efficiencies of CAR T cells and checkpoint blockade therapies were successfully evaluated with this probe.
Superoxide anion (O2˙−) plays an important role in T lymphocyte activation and cytokine secretion, which can also be regarded as a marker of immune activation.105 Zhang et al. synthesized a SP probe for chemiluminescence imaging of O2˙− to evaluate the immune activation.105 O2˙−-activable group-modified dioxethane derivatives were used as chemiluminescence precursors. The nanoprobe was then constructed with a chemiluminescence substrate, semiconducting polymer and triblock polymer via a nano-precipitation method. In the presence of O2˙−, the chemiluminescence of the dioxetane derivative excited the SP and the emission light at 700 nm was produced. The enhanced chemiluminescence signal in vivo after immunotherapy was associated with an increase in the number of T lymphocytes in the tumor, demonstrating its feasibility in tracking T lymphocyte activation.
The concentration of Ca2+ in T cells can obviously increase along with T cell activation and can regulate downstream signals, which allows it to act as another signaling molecule for T cell activation.106,107 Shi et al. used a dual-mode Ca2+ imaging probe to track and monitor T cells via computed tomography (CT) and fluorescence imaging.108 The Fluo-4 probe, as an indicator of the Ca2+ level, was connected on the surface of G5 PAMAM dendrimers through the functionalized PEG. In addition, Au NPs were encapsulated in the internal cavities for the CT imaging. Intracellular esterase could break the ester bond to release Fluo-4 for Ca2+-related fluorescence imaging.
Cai et al. labelled ipilimumab with 64Cu to specifically bind to CTLA-4 expressed in human non-small cell lung cancer cells.110 PETI was used in three cell line models with different expression levels of CTLA-4. Western blotting and ELISA analysis suggested that the A549 cell line had a relatively higher level of CTLA-4 compared to the H460 and H358 cell lines. PET images also exhibited a similar tendency. After blocking CTLA-4, the uptake of tracers decreased significantly, suggesting the specificity of the probe to CTLA-4. This probe was expected to monitor the CTLA-4-expressing tissue in a non-invasive manner and be a useful tool for the further understanding of the activity of immune checkpoint blocking therapy.
The PD-1/PD-L1 pathway in cancer cells and their mechanisms that confuse T cells have been extensively studied. PD-L1 receptors expressed by cancer cells can bind to PD-1 on the surface of T cells, thus resulting in the inactivation of T cells.13,111 Imaging PD-L1 can map tumor cell profiles to guide surgical resection and predict the responder to PD-L1 immune checkpoint blocking therapy. In the design of Wang et al., 64Cu-NOTA connected with αPD-L1 Fab was used to perform the PETI of PD-L1. The αPD-L1 Fab was incorporated with one UAA, p-azidophenylalanine (pAzF).112 The 64Cu-NOTA-αPD-L1 could specifically bind to PD-L1, which could be validated by the reduced intake after the blocking of PD-L1. With the reduced nonspecific accumulation and rapid clearance properties, Fab fragment-based PET agents possessed higher SNR imaging capability compared with intact full-length antibody counterparts.
In the design of Lin et al., Nb109, a nonblocking single-domain antibody, was attached to the 68Ga-labeled chelator NOTA (68Ga-NOTA-Nb109).48 This imaging probe did not have a histidine label and thus a lower renal permeability was obtained. Significant accumulation could be seen in A375-hPD-L1 cells with high expression of PD-L1. Rapid clearance of blood and muscle resulted in a high SNR. Importantly, the tracer did not influence the PD-1 and PD-L1 binding, which may be an effective platform for predicting and adjusting PD-L1/PD-1 blockade therapy by evaluating PD-L1 expression. Subsequently, various cancer cells with different expression levels of PD-L1 were used to validate the ability of 68Ga-NOTA-Nb109 to recognize PD-L1.113 Compared to HCT 116 and NCI-H1299 tumors with low expression of PD-L1, the accumulation of this probe was significantly increased in U87 tumors with high expression of PD-L1. Furthermore, it could be seen from PET images that PD-L1 expression in NCI-H1299 cells was increased following treatment with a certain amount of CDDP. In the comparison experiment with the 18F-FDG probe, 68Ga-NOTA-Nb109 had a higher SNR for detecting PD-L1 expression. Tumors with high or low expression of PD-L1 could be distinguished with this probe. To expand and evaluate its clinical application, Sun et al. constructed several mouse models for a series of validation studies.114 The same species tumor with different PD-L1 expression levels in the same mouse were constructed to exclude the interference of different tumors. According to the PETI result, a higher PET signal was observed in the tumor with a high level of PD-L1.
In the work of Dai et al., a PD-L1 monoclonal antibody (PD-L1 mAb) was connected with a NIR-II fluorophore to image PD-L1.115 The length of side chains on the shielding unit was adjusted to obtain the fluorophore with high fluorescence quantum yield and renal clearance ability. This probe could distinguish various tumors with different PD-L1 expression levels by fluorescence signals and could be used for surgical guidance. Moreover, the same group also developed erbium-based rare-earth nanoparticles (ErNPs) with near-infrared-IIb (NIR-IIb) luminescence characteristics (∼1600 nm) to visualize PD-L1.116 A PD-L1-functionalized hydrophilic polymer cross-linking network endowed ErNPs with water solubility and biocompatibility. The tumor was found to have a signal ratio of about 40 compared to normal tissue. In vivo imaging of PD-L1 and CD8 revealed the presence of cytotoxic T lymphocytes in the tumor microenvironment after immunotherapy. In the design of Jiang et al., a NIR-II nanoprobe based on the boron-dipyrromethene (BDP) photosensitizer was encapsulated in nanomicelles.117 The interaction between PD-L1 and PD-1 mediated the aggregation of nanomicelles in tumor tissues. The signal in the tumor was up to 14 times higher than that in non-tumor tissue.
Lv et al. used a multispectral fluorescence imaging strategy to track the programmed delivery of a combined nanovaccine.118 Genipin was cross-linked with the amino groups of PEI and OVA proteins to produce the fluorescence signal for labelling the internal antigens. The external antigen as the initial exposure antigen was labeled with the fluorescent dye RB (magenta). The transfer of the components of the composite vaccine could be monitored by collecting different fluorescence signals. The results of the dynamic fluorescence imaging showed that the combined vaccine could effectively delay antigen degradation and stimulate a stronger immune response.
High-energy free radicals are a hallmark of focal innate immune activation. Piwnica-worms et al. identified a new molecule 4-[18F]fluoro-1-naphthol ([18F]4FN) that could selectively react with high-energy free radicals and be used for whole-body imaging by PET/CT to detect multiorgan inflammation in real time.119 When oxidized by myeloperoxidase plus H2O2, [18F]4FN selectively binds to nearby proteins and cells. Studies have shown that [18F]4FN is superior to [18F]FDG in detecting inflammation.
IFN is a kind of lymphokine with extensive immunomodulatory effect.120,121 Directly imaging the effector molecule can provide a better indication of the immune response to treatment. Viola-Villegas et al. developed a 89Zr-labelled probe (89Zr-anti-IFNγ) utilizing the IFN-targeting mAb to perform PETI of IFNγ.122 A TUBO tumor-bearing mouse model was established and treated with HER2 vaccine. The results demonstrated that the tumor with higher tracer uptake achieved slower growth rates.
3. Conclusions and perspectives
In this review, we have introduced immune-related imaging strategies based on various immune cells and tumor cells to monitor the immune response. Labelling cells and imaging specific biomarkers can reflect the activation level of the immune cells and the immunosuppressive state of tumor cells. A variety of imaging strategies have achieved preliminary results. However, to further improve their application, a series of problems should be addressed. In this section, we provide the following suggestions, as a reference for achieving new advancements in the field.(I) Enhancing the sensitivity and specificity of probes. Highly sensitive and specific probes can be used to assess the immune status more accurately. Developing signal amplification strategies through the design of molecular structures can effectively improve the SNR. Ratiometric measurement is not affected by the local concentration of the probe and other factors. Designing ratiometric probes is also an effective strategy to improve the SNR. Rationally designing the structure of substrates is necessary to enhance the response efficiency and specific recognition to the target.
(II) As for the imaging methods, OI presents high temporal and spatial resolution and sensitivity. However, poor tissue penetration limits the development of OI. Fluorescence imaging in the NIR-II (1000–1700 nm) window and mediated by two photons can reduce the tissue absorption and scattering of light, thus enhancing the depth of penetration. Hence, probes with NIR-II and two-photon excitation properties should be extensively developed. For PETI, chelating agents with high stability and biocompatibility should be widely explored to prevent damage from radionuclides. For MRI, deep tissue penetration can be achieved. However, it lacks molecular sensitivity and specificity. It is necessary to develop probes that release imaging agents in response to the tumor microenvironment to improve molecular sensitivity.
(III) Based on the advantages and disadvantages of various imaging methods, multi-mode imaging systems are expected to be developed. It is feasible to combine OI with PETI or MRI, which can enhance the depth of penetration while enhancing the molecular sensitivity.
(IV) The immunogenicity of the probe itself needs to be studied. Biocompatible and nonimmunogenic probes should be explored. Probe metabolism should be intensively studied to ensure that there are no long-term effects on the body. In addition, it is also necessary to verify the effects of tags on the secretion function of immune cells, while characterizing the effects on cell survival.
In general, monitoring the immune response in real time plays a guiding role during the course of immunotherapy. With the efforts of many researchers, more innovative immune-related monitoring strategies will further improve the efficiency of immunotherapy.
Author contributions
X. Liu, L. Pan, N. Li and B. Tang proposed and designed the review. X. Liu and L. Pan collected references. K. Wang and W. Pan designed the figures. X. Liu, L. Pan, K. Wang, W. Pan, N. Li and B. Tang drafted the manuscript and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFA0210100) and Jinan Innovation Team (2021GXRC034).Notes and references
- J. C. Del Paggio, Nat. Rev. Clin. Oncol., 2018, 15, 268–270 CrossRef PubMed.
- L. Milling, Y. Zhang and D. J. Irvine, Adv. Drug Delivery Rev., 2017, 114, 79–101 CrossRef PubMed.
- C. W. Shields, L. L. Wang, M. A. Evans and S. Mitragotri, Adv. Mater., 2020, 32, 1901633 CrossRef PubMed.
- S. Yan, Z. Luo, Z. Li, Y. Wang, J. Tao, C. Gong and X. Liu, Angew. Chem., Int. Ed., 2020, 59, 17332–17343 CrossRef PubMed.
- L. Liu, E. Bi, X. Ma, W. Xiong, J. Qian, L. Ye, P. Su, Q. Wang, L. Xiao, M. Yang, Y. Lu and Q. Yi, Nat. Commun., 2020, 11, 5902 CrossRef CAS PubMed.
- J. T. Schiller, D. R. Lowy, I. H. Frazer, O. J. Finn, E. Vilar, H. K. Lyerly, S. Gnjatic, N. Zaidi, P. A. Ott, V. P. Balachandran, P. Y. Dietrich, D. Migliorini, R. H. Vonderheide and S. M. Domchek, Cancer Cell, 2022, 40, 559–564 CrossRef CAS.
- T. Wieder, T. Eigentler, E. Brenner and M. Rocken, J. Allergy Clin. Immunol., 2018, 142, 1403–1414 CrossRef CAS.
- N. E. Donlon, R. Power, C. Hayes, J. V. Reynolds and J. Lysaght, Cancer Lett., 2021, 502, 84–96 CrossRef CAS PubMed.
- B. Feng, F. Zhou, B. Hou, D. Wang, T. Wang, Y. Fu, Y. Ma, H. Yu and Y. Li, Adv. Mater., 2018, 30, 1803001 CrossRef.
- Y. Li, S. Gong, W. Pan, Y. Chen, B. Liu, N. Li and B. Tang, Chem. Sci., 2020, 11, 7429–7437 RSC.
- P. Zhao, Y. Wang, X. Kang, A. Wu, W. Yin, Y. Tang, J. Wang, M. Zhang, Y. Duan and Y. Huang, Chem. Sci., 2018, 9, 2674–2689 RSC.
- N. Ahmed, V. S. Salsman, E. Yvon, C. U. Louis, L. Perlaky, W. S. Wels, M. K. Dishop, E. E. Kleinerman, M. Pule, C. M. Rooney, H. E. Heslop and S. Gottschalk, Mol. Ther., 2009, 17, 1779–1787 CrossRef CAS.
- T. F. Gajewski, H. Schreiber and Y. X. Fu, Nat. Immunol., 2013, 14, 1014–1022 CrossRef CAS PubMed.
- M. Benvenuto, C. Focaccetti, V. Izzi, L. Masuelli, A. Modesti and R. Bei, Semin. Cancer Biol., 2021, 72, 65–75 CrossRef CAS.
- C. Connolly, K. Bambhania and J. Naidoo, Front. Oncol., 2019, 9, 530 CrossRef.
- M. Conroy and J. Naidoo, Nat. Commun., 2022, 13, 392 CrossRef PubMed.
- B. L. Russell, S. A. Sooklal, S. T. Malindisa, L. J. Daka and M. Ntwasa, Front. Oncol., 2021, 11, 641428 CrossRef.
- M. B. Schaaf, A. D. Garg and P. Agostinis, Cell Death Dis., 2018, 9, 115 CrossRef.
- K. E. de Visser, A. Eichten and L. M. Coussens, Nat. Rev. Cancer, 2006, 6, 24–37 CrossRef CAS.
- A. Gruneboom, O. Aust, Z. Cibir, F. Weber, D. M. Hermann and M. Gunzer, Immunol. Rev., 2022, 306, 293–303 CrossRef.
- J. Shi, J. Liu, X. Tu, B. Li, Z. Tong, T. Wang, Y. Zheng, H. Shi, X. Zeng, W. Chen, W. Yin and W. Fang, J. Immunother. Cancer, 2022, 10, e003133 CrossRef.
- P. Wang, T. Kim, M. Harada, C. Contag, X. Huang and B. R. Smith, Nanoscale Horiz., 2020, 5, 628–653 RSC.
- A. L. Wilson, K. L. Wilson, M. Bilandzic, L. R. Moffitt, M. Makanji, M. D. Gorrell, M. K. Oehler, A. Rainczuk, A. N. Stephens and M. Plebanski, Cancers, 2018, 11, 32 CrossRef.
- L. Li and Z. Zeng, Front. Immunol., 2020, 11, 564768 CrossRef CAS.
- L. Goldfarb, B. Duchemann, K. Chouahnia, L. Zelek and M. Soussan, EJNMMI Res., 2019, 9, 8 CrossRef PubMed.
- L. Tian, M. Shao, Y. Gong, Y. Chao, T. Wei, K. Yang, Q. Chen and Z. Liu, Sci. China: Chem., 2022, 65, 574–583 CrossRef CAS.
- P. Wang, Y. Chen and C. Wang, Front. Oncol., 2021, 11, 672677 CrossRef PubMed.
- L. Xu, Y. Wang, Y. Ma, S. Huan and G. Song, ChemMedChem, 2021, 16, 2547–2557 CrossRef PubMed.
- S. He, J. Li, Y. Lyu, J. Huang and K. Pu, J. Am. Chem. Soc., 2020, 142, 7075–7082 CrossRef.
- A. Ramesh, S. Kumar, A. Brouillard, D. Nandi and A. Kulkarni, Adv. Mater., 2020, 32, 2000648 CrossRef.
- S. Nisar, A. A. Bhat, S. Hashem, S. K. Yadav, A. Rizwan, M. Singh, P. Bagga, M. A. Macha, M. P. Frenneaux, R. Reddy and M. Haris, J. Transl. Med., 2020, 18, 471 CrossRef PubMed.
- F. K. Krebs, E. R. Trzeciak, S. Zimmer, D. Ozistanbullu, H. Mitzel-Rink, M. Meissner, S. Grabbe, C. Loquai and A. Tuettenberg, Cancer Med., 2021, 10, 1562–1575 CrossRef CAS PubMed.
- B. M. Larimer, E. Wehrenberg-Klee, F. Dubois, A. Mehta, T. Kalomeris, K. Flaherty, G. Boland and U. Mahmood, Cancer Res., 2017, 77, 2318–2327 CrossRef CAS.
- R. Tavare, H. Escuin-Ordinas, S. Mok, M. N. McCracken, K. A. Zettlitz, F. B. Salazar, O. N. Witte, A. Ribas and A. M. Wu, Cancer Res., 2016, 76, 73–82 CrossRef CAS PubMed.
- T. Sim, B. Choi, S. W. Kwon, K. S. Kim, H. Choi, A. Ross and D. H. Kim, ACS Nano, 2021, 15, 12780–12793 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, Z. Wang, J. Zhang, R. R. Qiao, M. Xu, N. Yang, L. Gao, H. Qiao, M. Gao and F. Cao, Biomaterials, 2019, 219, 119378 CrossRef CAS PubMed.
- D. Lu, Y. Wang, T. Zhang, F. Wang, K. Li, S. Zhou, H. Zhu, Z. Yang and Z. Liu, J. Nanobiotechnol., 2021, 19, 175 CrossRef CAS.
- S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS.
- Y. Jiang and K. Pu, Chem. Rev., 2021, 121, 13086–13131 CrossRef CAS PubMed.
- V. Ntziachristos, Annu. Rev. Biomed. Eng., 2006, 8, 1–33 CrossRef CAS.
- X. Chen, W. Niu, Z. Du, Y. Zhang, D. Su and X. Gao, Chin. Chem. Lett., 2022, 33, 3349–3360 CrossRef.
- R. F. Gamache, K. A. Zettlitz, W. K. Tsai, J. Collins, A. M. Wu and J. M. Murphy, Chem. Sci., 2020, 11, 1832–1838 RSC.
- K. Heinzmann, L. M. Carter, J. S. Lewis and E. O. Aboagye, Nat. Biomed. Eng., 2017, 1, 697–713 CrossRef PubMed.
- Y. Sun, M. Yu, S. Liang, Y. Zhang, C. Li, T. Mou, W. Yang, X. Zhang, B. Li, C. Huang and F. Li, Biomaterials, 2011, 32, 2999–3007 CrossRef.
- Y. Xing, J. Zhao, P. S. Conti and K. Chen, Theranostics, 2014, 4, 290–306 CrossRef PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- P. Chen, H. Wang, H. Wu, P. Zou, C. Wang, X. Liu, Y. Pan, Y. Liu and G. Liang, Anal. Chem., 2021, 93, 6329–6334 CrossRef CAS.
- G. Lv, X. Sun, L. Qiu, Y. Sun, K. Li, Q. Liu, Q. Zhao, S. Qin and J. Lin, J. Nucl. Med., 2020, 61, 117–122 CrossRef CAS.
- J. Cui, R. Jiang, C. Guo, X. Bai, S. Xu and L. Wang, J. Am. Chem. Soc., 2018, 140, 5890–5894 CrossRef CAS PubMed.
- Y. Zhang, Q. Ma, Y. Yan, C. Guo, S. Xu and L. Wang, Anal. Chem., 2020, 92, 15679–15684 CrossRef CAS.
- H. Zhong, Y. X. Wu, S. Yu, X. Wang, K. He, D. Li, Y. Cao and N. Gan, Anal. Chem., 2021, 93, 5691–5699 CrossRef CAS PubMed.
- E. T. Ahrens and J. W. Bulte, Nat. Rev. Immunol., 2013, 13, 755–763 CrossRef CAS PubMed.
- N. Shimasaki, A. Jain and D. Campana, Nat. Rev. Drug Discovery, 2020, 19, 200–218 CrossRef CAS PubMed.
- L. Chiossone, M. Vienne, Y. M. Kerdiles and E. Vivier, Semin. Immunol., 2017, 31, 55–63 CrossRef CAS.
- T. A. Fehniger, S. F. Cai, X. Cao, A. J. Bredemeyer, R. M. Presti, A. R. French and T. J. Ley, Immunity, 2007, 26, 798–811 CrossRef CAS.
- I. Prager, C. Liesche, H. van Ooijen, D. Urlaub, Q. Verron, N. Sandstrom, F. Fasbender, M. Claus, R. Eils, J. Beaudouin, B. Onfelt and C. Watzl, J. Exp. Med., 2019, 216, 2113–2127 CrossRef PubMed.
- E. Vivier, D. H. Raulet, A. Moretta, M. A. Caligiuri, L. Zitvogel, L. L. Lanier, W. M. Yokoyama and S. Ugolini, Science, 2011, 331, 44–49 CrossRef PubMed.
- S. Paul and G. Lal, Front. Immunol., 2017, 8, 1124 CrossRef.
- J. A. Myers and J. S. Miller, Nat. Rev. Clin. Oncol., 2021, 18, 85–100 CrossRef PubMed.
- N. Liao, L. Su, Y. Zheng, B. Zhao, M. Wu, D. Zhang, H. Yang, X. Liu and J. Song, Angew. Chem., Int. Ed., 2021, 60, 20888–20896 CrossRef PubMed.
- J. I. Scott, S. Gutkin, O. Green, E. J. Thompson, T. Kitamura, D. Shabat and M. Vendrell, Angew. Chem., Int. Ed., 2021, 60, 5699–5703 CrossRef.
- S. J. Jenkins, D. Ruckerl, P. C. Cook, L. H. Jones, F. D. Finkelman, N. van Rooijen, A. S. MacDonald and J. E. Allen, Science, 2011, 332, 1284–1288 CrossRef PubMed.
- J. W. Pollard, Nat. Rev. Immunol., 2009, 9, 259–270 CrossRef PubMed.
- K. S. Siveen and G. Kuttan, Immunol. Lett., 2009, 123, 97–102 CrossRef PubMed.
- B. Ruffell and L. M. Coussens, Cancer Cell, 2015, 27, 462–472 CrossRef PubMed.
- C. E. Lewis and J. W. Pollard, Cancer Res., 2006, 66, 605–612 CrossRef PubMed.
- C. D. Mills and K. Ley, J. Innate Immun., 2014, 6, 716–726 CrossRef PubMed.
- M. Chittezhath, M. K. Dhillon, J. Y. Lim, D. Laoui, I. N. Shalova, Y. L. Teo, J. Chen, R. Kamaraj, L. Raman, J. Lum, T. P. Thamboo, E. Chiong, F. Zolezzi, H. Yang, J. A. Van Ginderachter, M. Poidinger, A. S. Wong and S. K. Biswas, Immunity, 2014, 41, 815–829 CrossRef CAS PubMed.
- K. K. Goswami, T. Ghosh, S. Ghosh, M. Sarkar, A. Bose and R. Baral, Cell. Immunol., 2017, 316, 1–10 CrossRef CAS.
- H. Y. Kim, R. Li, T. S. C. Ng, G. Courties, C. B. Rodell, M. Prytyskach, R. H. Kohler, M. J. Pittet, M. Nahrendorf, R. Weissleder and M. A. Miller, ACS Nano, 2018, 12, 12015–12029 CrossRef CAS PubMed.
- H. J. Jeong, R. J. Yoo, J. K. Kim, M. H. Kim, S. H. Park, H. Kim, J. W. Lim, S. H. Do, K. C. Lee, Y. J. Lee and D. W. Kim, Biomaterials, 2019, 199, 32–39 CrossRef.
- Y. Li, H. Wu, B. Ji, W. Qian, S. Xia, L. Wang, Y. Xu, J. Chen, L. Yang and H. Mao, ACS Appl. Bio Mater., 2020, 3, 4335–4347 CrossRef PubMed.
- A. L. Antaris, J. T. Robinson, O. K. Yaghi, G. Hong, S. Diao, R. Luong and H. Dai, ACS Nano, 2013, 7, 3644–3652 CrossRef PubMed.
- Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16294–16308 CrossRef PubMed.
- X. Luo, D. Hu, D. Gao, Y. Wang, X. Chen, X. Liu, H. Zheng, M. Sun and Z. Sheng, ACS Nano, 2021, 15, 10010–10024 CrossRef PubMed.
- C. S. Leung, J. Li, F. Xu, A. S. L. Wong and K. O. Lui, FASEB J., 2019, 33, 484–493 CrossRef CAS.
- J. Qi, C. Sun, A. Zebibula, H. Zhang, R. T. K. Kwok, X. Zhao, W. Xi, J. W. Y. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2018, 30, 1706856 CrossRef.
- Z. Zheng, T. Zhang, H. Liu, Y. Chen, R. T. K. Kwok, C. Ma, P. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, K. S. Wong and B. Z. Tang, ACS Nano, 2018, 12, 8145–8159 CrossRef CAS.
- S. Suzuki, S. Sasaki, A. S. Sairi, R. Iwai, B. Z. Tang and G. I. Konishi, Angew. Chem., Int. Ed., 2020, 59, 9856–9867 CrossRef CAS PubMed.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS PubMed.
- T. Gao, Y. Wu, W. Wang, C. Deng, Y. Chen, L. Yi, Y. Song, W. Li, L. Xu, Y. Xie, L. Fang, Q. Jin, L. Zhang, B. Z. Tang and M. Xie, ACS Nano, 2021, 15, 11908–11928 CrossRef CAS.
- F. Geissmann, M. G. Manz, S. Jung, M. H. Sieweke, M. Merad and K. Ley, Science, 2010, 327, 656–661 CrossRef CAS.
- M. Benoit, B. Desnues and J. L. Mege, J. Immunol., 2008, 181, 3733–3739 CrossRef CAS PubMed.
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Nat. Rev. Immunol., 2020, 20, 7–24 CrossRef PubMed.
- J. Banchereau, F. Briere, C. Caux, J. Davoust, S. Lebecque, Y. J. Liu, B. Pulendran and K. Palucka, Annu. Rev. Immunol., 2000, 18, 767–811 CrossRef.
- K. Hoebe, E. Janssen and B. Beutler, Nat. Immunol., 2004, 5, 971–974 CrossRef.
- F. C. Hartgers, C. G. Figdor and G. J. Adema, Immunol. Today, 2000, 21, 542–545 CrossRef.
- Z. He, H. Jia, M. Zheng, H. Wang, W. Yang, L. Gao, Z. Zhang, J. Xue, B. Xu, W. Yang, G. Xing, X. Gao and F. Gao, ACS Appl. Bio Mater., 2021, 4, 5707–5716 CrossRef CAS PubMed.
- L. Chen and D. B. Flies, Nat. Rev. Immunol., 2013, 13, 227–242 CrossRef.
- J. A. Siller-Farfan and O. Dushek, Immunol. Rev., 2018, 285, 194–205 CrossRef CAS.
- M. Rashidian, M. W. LaFleur, V. L. Verschoor, A. Dongre, Y. Zhang, T. H. Nguyen, S. Kolifrath, A. R. Aref, C. J. Lau, C. P. Paweletz, X. Bu, G. J. Freeman, M. I. Barrasa, R. A. Weinberg, A. H. Sharpe and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16971–16980 CrossRef CAS.
- T. J. Grazia, R. J. Plenter, S. M. Weber, H. M. Lepper, F. Victorino, M. R. Zamora, B. A. Pietra and R. G. Gill, Transplantation, 2010, 89, 33–39 CrossRef PubMed.
- H. Li, Y. Chen, Q. Jin, Y. Wu, C. Deng, Y. Gai, Z. Sun, Y. Li, J. Wang, Y. Yang, Q. Lv, Y. Zhang, R. An, X. Lan, L. Zhang and M. Xie, Mol. Pharm., 2021, 18, 1317–1326 CrossRef CAS.
- M. Croft, Nat. Rev. Immunol., 2009, 9, 271–285 CrossRef CAS.
- B. D. Curti, M. Kovacsovics-Bankowski, N. Morris, E. Walker, L. Chisholm, K. Floyd, J. Walker, I. Gonzalez, T. Meeuwsen, B. A. Fox, T. Moudgil, W. Miller, D. Haley, T. Coffey, B. Fisher, L. Delanty-Miller, N. Rymarchyk, T. Kelly, T. Crocenzi, E. Bernstein, R. Sanborn, W. J. Urba and A. D. Weinberg, Cancer Res., 2013, 73, 7189–7198 CrossRef CAS.
- N. Kitamura, S. Murata, T. Ueki, E. Mekata, R. T. Reilly, E. M. Jaffee and T. Tani, Int. J. Cancer, 2009, 125, 630–638 CrossRef CAS PubMed.
- X. Chen, D. Li, Y. Cao, J. Gao, H. Jin and H. Shan, Mol. Pharm., 2019, 16, 4252–4259 CrossRef CAS.
- S. Harmsen, E. I. Medine, M. Moroz, F. Nurili, J. Lobo, Y. Dong, M. Turkekul, N. V. K. Pillarsetty, R. Ting, V. Ponomarev, O. Akin and O. Aras, Biomaterials, 2021, 269, 120630 CrossRef PubMed.
- E. R. Podack, Curr. Opin. Immunol., 1995, 7, 11–16 CrossRef.
- P. R. Hiebert and D. J. Granville, Trends Mol. Med., 2012, 18, 732–741 CrossRef.
- N. Zhao, C. Bardine, A. L. Lourenco, Y. H. Wang, Y. Huang, S. J. Cleary, D. M. Wilson, D. Y. Oh, L. Fong, M. R. Looney, M. J. Evans and C. S. Craik, ACS Cent. Sci., 2021, 7, 1638–1649 CrossRef PubMed.
- Y. Zhang, S. He, W. Chen, Y. Liu, X. Zhang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 5921–5927 CrossRef.
- A. Nguyen, A. Ramesh, S. Kumar, D. Nandi, A. Brouillard, A. Wells, L. Pobezinsky, B. Osborne and A. A. Kulkarni, Sci. Adv., 2020, 6, eabc2777 CrossRef.
- J. Xie, F. El Rami, K. Zhou, F. Simonetta, Z. Chen, X. Zheng, M. Chen, P. B. Balakrishnan, S. Y. Dai, S. Murty, I. S. Alam, J. Baker, R. S. Negrin, S. S. Gambhir and J. Rao, ACS Cent. Sci., 2022, 8, 590–602 CrossRef PubMed.
- D. Cui, J. Li, X. Zhao, K. Pu and R. Zhang, Adv. Mater., 2020, 32, 1906314 CrossRef PubMed.
- E. C. Schwarz, B. Qu and M. Hoth, Biochim. Biophys. Acta, 2013, 1833, 1603–1611 CrossRef CAS PubMed.
- S. N. Christo, K. R. Diener and J. D. Hayball, Immunol. Cell Biol., 2015, 93, 694–704 CrossRef CAS.
- M. Chen, O. Betzer, Y. Fan, Y. Gao, M. Shen, T. Sadan, R. Popovtzer and X. Shi, Biomacromolecules, 2020, 21, 1587–1595 CrossRef CAS PubMed.
- Y. Li, X. Liu, X. Zhang, W. Pan, N. Li and B. Tang, Adv. Funct. Mater., 2021, 31, 2107540 CrossRef CAS.
- E. B. Ehlerding, C. G. England, R. L. Majewski, H. F. Valdovinos, D. Jiang, G. Liu, D. G. McNeel, R. J. Nickles and W. Cai, Mol. Pharm., 2017, 14, 1782–1789 CrossRef CAS.
- S. Maleki Vareki, C. Garrigos and I. Duran, Crit. Rev. Oncol. Hematol., 2017, 116, 116–124 CrossRef.
- H. L. Wissler, E. B. Ehlerding, Z. Lyu, Y. Zhao, S. Zhang, A. Eshraghi, Z. Y. Buuh, J. C. McGuth, Y. Guan, J. W. Engle, S. J. Bartlett, V. A. Voelz, W. Cai and R. E. Wang, Mol. Pharm., 2019, 16, 2028–2036 CrossRef CAS.
- Q. Liu, L. Jiang, K. Li, H. Li, G. Lv, J. Lin and L. Qiu, Cancer Immunol. Immunother., 2021, 70, 1721–1733 CrossRef CAS PubMed.
- S. Qin, Y. Yu, H. Guan, Y. Yang, F. Sun, Y. Sun, J. Zhu, L. Xing, J. Yu and X. Sun, Aging, 2021, 13, 13006–13022 CrossRef.
- H. Wan, H. Ma, S. Zhu, F. Wang, Y. Tian, R. Ma, Q. Yang, Z. Hu, T. Zhu, W. Wang, Z. Ma, M. Zhang, Y. Zhong, H. Sun, Y. Liang and H. Dai, Adv. Funct. Mater., 2018, 28, 1804956 CrossRef PubMed.
- Y. Zhong, Z. Ma, F. Wang, X. Wang, Y. Yang, Y. Liu, X. Zhao, J. Li, H. Du, M. Zhang, Q. Cui, S. Zhu, Q. Sun, H. Wan, Y. Tian, Q. Liu, W. Wang, K. C. Garcia and H. Dai, Nat. Biotechnol., 2019, 37, 1322–1331 CrossRef.
- Q. Liu, J. Tian, Y. Tian, Q. Sun, D. Sun, F. Wang, H. Xu, G. Ying, J. Wang, A. K. Yetisen and N. Jiang, ACS Nano, 2021, 15, 515–525 CrossRef PubMed.
- X. Dong, J. Liang, A. Yang, C. Wang, D. Kong and F. Lv, ACS Appl. Mater. Interfaces, 2018, 10, 21861–21875 CrossRef CAS PubMed.
- F. Pisaneschi, S. T. Gammon, V. Paolillo, S. A. Qureshy and D. Piwnica-Worms, Nat. Biotechnol., 2022, 40, 965–973 CrossRef CAS.
- S. E. Fenton, D. Saleiro and L. C. Platanias, Cancers, 2021, 13, 1037 CrossRef CAS PubMed.
- E. Alspach, D. M. Lussier and R. D. Schreiber, Cold Spring Harbor Perspect. Biol., 2019, 11, a028480 CrossRef CAS.
- H. M. Gibson, B. N. McKnight, A. Malysa, G. Dyson, W. N. Wiesend, C. E. McCarthy, J. Reyes, W. Z. Wei and N. T. Viola-Villegas, Cancer Res., 2018, 78, 5706–5717 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |