
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShort peptide-based cross-β amyloids exploit dual residues for phosphoesterase like activity†
Chiranjit
Mahato
a,
Sneha
Menon
b,
Abhishek
Singh
a,
Syed Pavel
Afrose
a,
Jagannath
Mondal
b and
Dibyendu
Das
 *a
*a
aDepartment of Chemical Sciences & Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, West Bengal 741246, India. E-mail: dasd@iiserkol.ac.in
bTata Institute of Fundamental Research Hyderabad, Telangana 500046, India
First published on 18th July 2022
Abstract
Herein, we report that short peptides are capable of exploiting their anti-parallel registry to access cross-β stacks to expose more than one catalytic residue, exhibiting the traits of advanced binding pockets of enzymes. Binding pockets decorated with more than one catalytic residue facilitate substrate binding and process kinetically unfavourable chemical transformations. The solvent-exposed guanidinium and imidazole moieties on the cross-β microphases synergistically bind to polarise and hydrolyse diverse kinetically stable model substrates of nucleases and phosphatase. Mutation of either histidine or arginine results in a drastic decline in the rate of hydrolysis. These results not only support the argument of short amyloid peptides as the earliest protein folds but also suggest their interactions with nucleic acid congeners, foreshadowing the mutualistic biopolymer relationships that fueled the chemical emergence of life.
Introduction
Extant enzymes achieve synchronized interactions with specific substrates via the use of multiple residues that are precisely arranged in binding pockets. The native states of enzymes that feature such binding sites result from the folding of chains of hundreds of amino acids. Billions of years of evolution have led to such specific folding pathways to generate the native state, which is an ensemble of closely related, complex three-dimensional structures. The hydrophobicity, pH, polarity, hydrophilicity, and charge of the binding pockets are modulated by the precise yet conformationally flexible spatial arrangements of multiple proximally located amino acid residues. Such a through space arrangement of amino acid residues facilitates proton channeling and stabilization of intermediates required to carry out chemical reactions with high activation barriers. For instance, enzymes such as serine proteases, protein kinases, and phosphatases exploit catalytic dyads or triads of amino acids to facilitate bond-breaking and bond-forming processes involving highly kinetically stable substrates.1–4 The emergence of such remarkable capabilities in modern-day enzymes that can catalyse a myriad of kinetically unfavourable biochemical transformations remains an unresolved puzzle. Recent studies have demonstrated the catalytic competence of amyloid-based peptide assemblies supporting the argument that short amyloid peptides might have been the earliest protein folds.5–13Hydrolysis of activated carboxylic esters is among the most widely explored chemical transformations by using short peptide-based assemblies.14–16 Activated aromatic esters have been extensively targeted due to their kinetic lability, and the presence of histidine residues on the solvent-exposed surface of the assemblies is often sufficient for this hydrolytic step which has a relatively lower activation barrier.17–20 In contrast, natural hydrolases use a combination of multiple residues (e.g., serine, histidine, and aspartate) and hence, reports on the hydrolysis of substrates with extreme kinetic stability (e.g., phosphate ester bonds, amide bonds, and so forth) by short amyloid peptides remain scarce. This is due to the fact that the catalytic amyloid microphases reported thus far mostly expose a single catalytic residue on the surface limited by the short length of the sequence which is insufficient to fold in ways that can expose multiple residues required for binding and catalysis.
In this regard, extant DNase, RNase, and phosphatase utilize their long peptide chains to expose multiple residues and cofactors for specific binding and lower the activation energies required to cleave the kinetically stable phosphate ester bonds of diverse origins.4,21 The molecular recognition of nucleic acids seen in such DNA and RNA binding proteins and their decorative relationships even in ribosomes suggest a biopolymer mutualism that is argued to be necessary for the emergence of cellular life.22,23 It will be intriguing to probe whether short peptide-based assemblies can expose multiple residues on the surface to bind and lower the activation barrier of phosphoester hydrolysis. Herein, we report that the paracrystalline stacks of cross-β sheets accessed from short amyloid peptides can expose and importantly, utilize more than one catalytic residue for cleaving stable phosphate esters in the absence of any co-factors. Utilizing antiparallel β-sheet stacks to proximally locate specific amino acid residues, the amyloid microphases fostered the hydrolytic cleavage of phosphate bonds, expanding the catalytic diversity of primitive short peptide-based folds.
Results and discussion
The short fragment17LVFF21A of the Aβ(1–42) sequence associated with the fibrillar protein deposits seen in Alzheimer's disease was used. At first, we started with the question whether a single catalytic residue can catalyse the hydrolysis of phosphate ester at all. Histidine and arginine were chosen as catalytic residues for checking phosphoester hydrolysis.So, arginine or histidine was installed separately at the N-terminal of the sequence while the C-terminal was coupled to leucine, yielding the peptide sequences Ac-RLVFFAL (RL) and Ac-HLVFFAL (HL) respectively (Scheme 1). These peptides self-assembled to form nanotubular morphologies with surface-exposed cationic guanidinium (RL) or imidazole (HL) moieties (Fig. S1†).11,18 The nuclease activities of these peptide assemblies were probed with an anionic RNA model substrate 2-hydroxypropyl-4-nitrophenylphosphate (HPNPP, Scheme 1). The cationic guanidinium groups were expected to bind to the anionic HPNPP and facilitate the cleavage of the phosphoester bond.24–32 The imidazole moiety of histidine is known to facilitate phosphoester hydrolysis by acting as a general acid/base catalyst or by acting as a nucleophile.33,34 The rate of hydrolysis was monitored with the help of UV-vis spectroscopy by the change of absorbance at 405 nm. Despite the presence of catalytic residues on the surface of the morphologies, addition of the substrate did not show any noticeable enhancement of hydrolytic rates compared to the background (Fig. S2†). In this context, it is important to note that a combination of arginine and histidine with other amino acids and suitable co-factors is used by contemporary nucleases for cleaving the phosphoester bonds.35 Hence, arginine and imidazole were installed at the N-terminal and leucine was installed at the C-terminal of the core sequence 17LVFF21A, yielding Im-RLVFFAL (Im-RL, Scheme 1, see the ESI for details†). Im-RL formed nanotubular morphologies with surface-exposed arrays of imidazole and guanidinium residues (Fig. S1 and S3,† FTIR suggested antiparallel β-sheet arrangement).15 Indeed, close to a three-fold enhancement of the hydrolytic rate was registered compared to HL and RL (Fig. S2†). At this point, we speculated that if the peptide can utilize its antiparallel β-sheet packing to locate the two residues proximally, catalysis might be facilitated (Scheme 1).
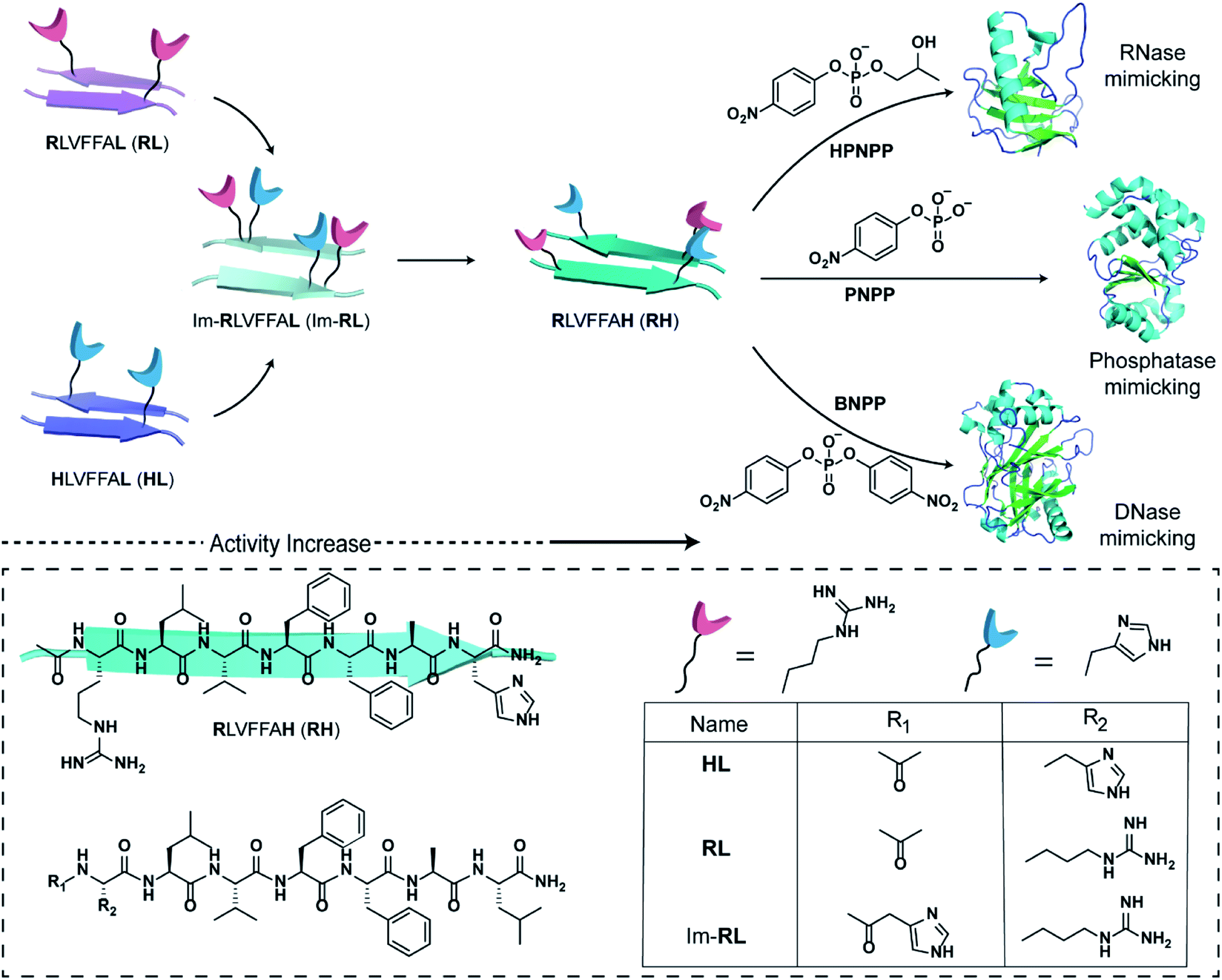 | ||
| Scheme 1 Schematic representation of the design of peptide sequences for mimicking RNase, phosphatase and DNase activity. Chemical structures of the amyloid sequences used are shown in the box. | ||
Specifically, if the sequence features guanidinium and imidazole at the extreme termini, interstrand arrangement would possibly provide more degrees of freedom and plasticity to the catalytic moieties as opposed to intrastrand arrangement in Im-RL (Scheme 1). Hence, the sequence was modified by incorporating arginine and histidine at the N and C terminals of the sequence 17LVFF21A to access Ac-RLVFFAH-NH2 (RH, Scheme 1). RH self-assembled into sheet-like morphology, characterized by TEM (Fig. 1a) and atomic force microscopy (AFM, Fig. 1b; the average height of the assemblies was 4.94 ± 0.8 nm). Confocal microscopy performed in the presence of coumarin-343 dye revealed fluorescent structures which suggested the capability of the RH microphases to bind small molecular guests (Fig. 1c). The antiparallel β-sheet conformation of the peptide assembly was indicated by FTIR and circular dichroism (CD, Fig. S4†). Powder X-ray diffraction (PXRD) showed d-spacings of 10.3 Å and 4.78 Å, corresponding to H-bonded β-sheet laminate and interstrand distances respectively (Fig. 1d). In order to investigate the structural features of RH assembly, molecular dynamics (MD) simulations were performed. The simulations revealed that both imidazole and guanidinium moieties are surface exposed via antiparallel packing (Fig. 1e). The mean laminate distance varied between 10.0 to 10.5 Å and the mean interstrand distance was close to 5.3 Å which corroborated the experimental PXRD findings (d-spacings of 10.3 Å and 4.78 Å respectively, for details see ESI, Fig. S5 and S6†). The studies done with negatively charged AuNPs revealed an ordered array of AuNPs bound to the assemblies of RH at pH = 7, indicating the presence of positively charged residues on the surface (Fig. 1f and S7†). To confirm the presence of both imidazole and guanidinium residues on the solvent-exposed surface, AuNPs were incubated with the assembly at pH 5.5, which is lower than the pKa of both moieties. The higher density of AuNPs observed in the case of pH 5.5 on the surface of assemblies compared to pH 7 indicated the presence of both the catalytic moieties on the surface of the microphases (Fig. 1g and S7†).
 | ||
| Fig. 1 TEM micrographs (a) and AFM topographic image (b) of RH (inset shows the height profile). (c) Confocal microscopy image of coumarin-bound-RH, (d) PXRD of RH assemblies. (e) van der Waals surface of the RH assembly displaying solvent-exposed arginine (pink) and histidine (blue). TEM micrographs of negatively charged gold nanoparticles with RH at (f) pH 7 and (g) pH 5.5. (h) Bar diagram of initial hydrolysis rates of HPNPP in different self-assembled systems, [catalyst] = 100 μM, and [substrate] = 500 μM. | ||
After exploring the self-assembling properties of the designed peptide RH, we investigated its phosphodiester cleaving ability. The catalytic nature of the RH assembly was assessed by UV-vis spectroscopy [vi= (23.76 ± 7.63) × 10−10 M s−1, Fig. 1h and S2†]. Kinetics at different substrate concentrations typically followed the Michaelis–Menten equation and allowed the determination of the Michaelis–Menten parameters; KM = 0.70 ± 0.20 mM and kcat = (4.45 ± 0.54) × 10−5 s−1 (Fig. S8, 2a and Table S1†). The catalytic rate was ∼2.22 × 102 fold higher compared to the uncatalyzed reaction (kuncat = 2.0 × 10−7 s−1, Fig. 2a, Table S1†). Interestingly, the rate of hydrolysis of HPNPP was six-fold higher compared to Im-RL (Fig. 1h and S2†) despite the presence of guanidinium and imidazole in both sequences. The significant difference in rates, along with the results of the simulations, suggested that the antiparallel packing in RH facilitated the closer proximity of imidazole and guanidinium moieties which is exigent for catalysis. The stability of the microphases at the experimental temperature was probed by incubating the assemblies at 40 °C for more than 24 hours. TEM of the incubated sample revealed that the morphology was retained (Fig. S9†). Furthermore, the role of a self-assembled surface was probed by using an HFIP treated RH assembly (see details in the ESI†). Hydrolytic activities decreased drastically following HFIP mediated disassembly (Fig. 1h). These results suggested that the co-localization of the catalytic moieties on the surface of the microphases helped in facilitating RNase activity in a cooperative manner.
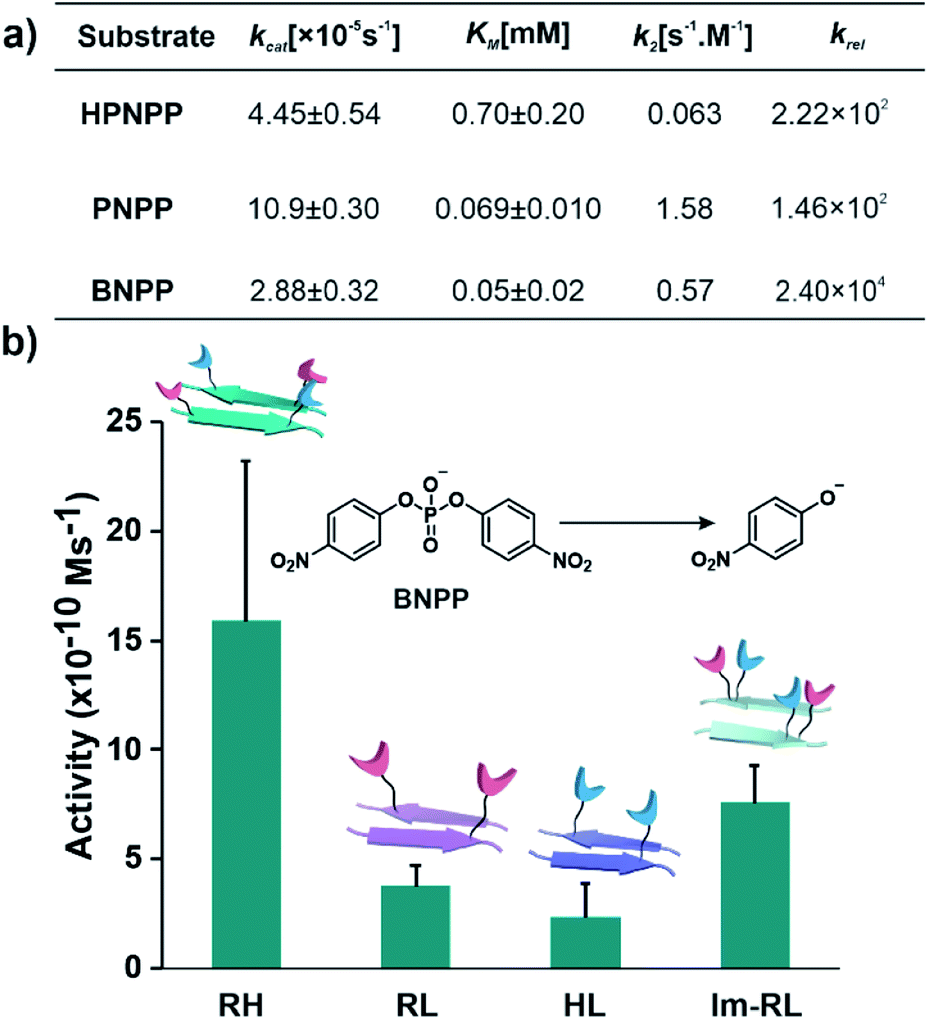 | ||
| Fig. 2 (a) Summary of kinetic parameters of RH with different substrates. (b) Bar diagram of initial rates of hydrolysis of BNPP in different self-assembled systems, [catalyst] = 100 μM, and [substrate] = 500 μM (see the ESI for details†). | ||
After demonstrating the RNase activity by RH, the catalytic versatility was monitored by exposing it to different substrates of varying reactivity. 4-Nitrophenyl phosphate (PNPP, Scheme 1), a well-known model substrate to probe the phosphatase activity, was used. The double negative charge on the substrate was expected to lead to improved binding with the positively charged surface of the assemblies to facilitate hydrolysis. When mixed with the substrate PNPP, the RH assemblies showed appreciable rates of hydrolysis. The catalytic rate constant (kcat) was found to be (10.9 ± 0.30) × 10−5 s−1 (kuncat = 7.5 × 10−7 s−1 at 40 °C), indicating an ∼1.46 × 102 fold rate acceleration in the presence of the self-assemblies (Fig. 2a, S10 and Table S2†). Controls with RL and HL displayed significantly lower hydrolytic rates compared to RH, suggesting the importance of both guanidinium and imidazole side chains (Fig. S11†). However, in contrast to HPNPP, the activity with Im-RL was only ∼1.2-fold lower compared to that with RH (Fig. S11†). The HFIP treated assemblies showed no significant hydrolysis due to the absence of the assemblies (Fig. S12†).
At this point, we sought to explore DNase-like activity by using cofactor-free short peptide-based amyloid assemblies. Bis(4-nitrophenyl)phosphate (BNPP), a well-known DNA model substrate was selected. Unlike HPNPP, BNPP lacked a hydroxyl group and was comparatively more stable towards hydrolysis. The rate constant (kcat) of the RH catalysed reaction was found to be (2.88 ± 0.32) × 10−5 s−1 and the Michaelis–Menten constant was 0.05 ± 0.02 mM (Fig. 2a, S13 and Table S3†). Similar controls, as done before, showed a decrease in activities, again underpinning the role of the self-assembled surface with the exposed imidazole and arginine (Fig. 2b and S14†). A comparison of the kinetic parameters of RH with other peptide-based self-assembled systems reported in the literature for the hydrolysis of PNPP is shown in Table 1. To the best of our knowledge, no report thus far shows short peptide-based self-assembled catalysts that can hydrolyze three different phosphoester based substrates.
An equilibrated model of the assembly from the MD simulation trajectory revealed the presence of the catalytic residues guanidinium and imidazole in close proximity, resulting from the antiparallel β-sheet packing (Fig. 3). To gain insights into the catalytic proficiency of RH assembly, we performed molecular docking of the substrate molecules HPNPP, BNPP and PNPP on the surface of the assembly (Fig. 3d and S15†). It is notable that the predominant docking mode of all the substrates was in the cross-β grooves between the imidazole and guanidinium groups that are exposed on the fibril surface owing to the anti-parallel packing. Redocking of the substrates to the dominant binding sites of the RH assembly yielded binding affinities of −5.6 kcal mol−1, −5.3 kcal mol−1 and −6.5 kcal mol−1 for HPNPP, PNPP and BNPP, respectively. Docking of phosphate esters on the catalytic microphases shows the favourable proximity of the ester and the imidazole and guanidinium moieties in the binding grooves. Such proximity has been argued to be critically important for catalysis.36 As depicted in Fig. 3a–c, these substrates form a variety of interactions such as salt-bridges, hydrogen bonds, π-stacking interactions and hydrophobic interactions with the imidazole and guanidinium side chains of histidine and arginine. These observations, along with the fact that disassembled peptides show diminished activities, underpins the crucial role of the antiparallel arrangement for the interaction with the substrates leading to hydrolysis.
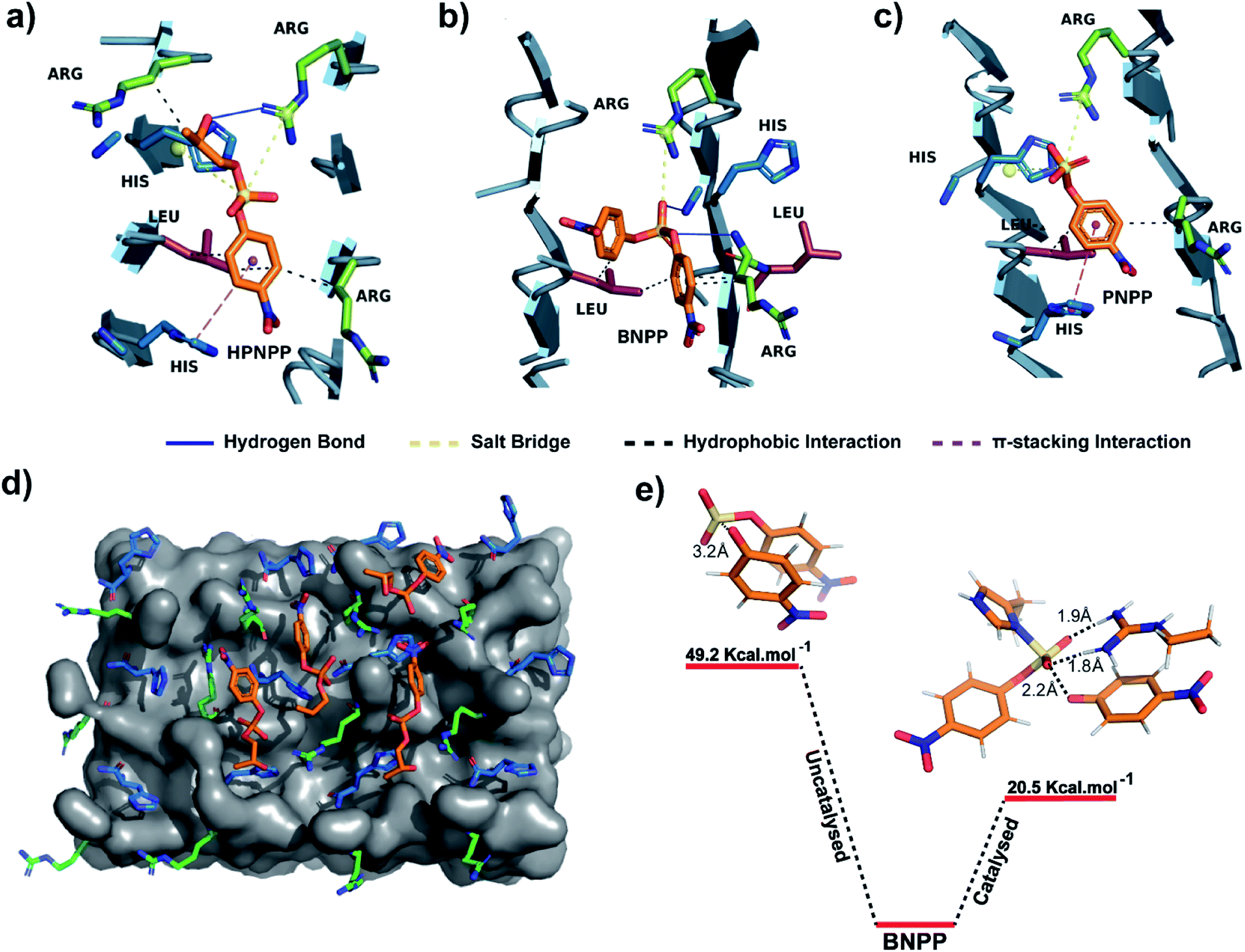 | ||
| Fig. 3 (a–c) Binding interactions of HPNPP, BNPP and PNPP with catalytic moieties on the surface of the RH assembly. (d) Molecular docking of HPNPP substrate molecules on the surface of the RH assembly modelled via MD simulations. (e) Comparison of transition state energies of catalyzed and uncatalyzed reactions of BNPP. | ||
To probe the role of the catalytic moieties in phosphoester hydrolysis, we mapped the reaction pathways using density functional theory (DFT) with BNPP and HPNPP substrates in the presence and absence of RH assemblies (see the ESI†). Bond dissociation enthalpies (BDE) of BNPP (93.8 kcal mol−1) and HPNPP (98.7 kcal mol−1) calculated as the standard enthalpy change resulting from homolytic cleavage of the phosphoester bond were found to be only marginally different (Fig. S16†). To understand the low rates of hydrolysis of BNPP compared to HPNPP, we discerned the energetics of the uncatalyzed reaction pathway by following the transition states of phosphoester bond cleavage. Free energy of activation for BNPP was found to be 50 kcal mol−1 while that for the –OH mediated transition state for HPNPP for phosphoester bond cleavage was only 18.4 kcal mol−1 which explains the enhanced rates of hydrolysis in HPNPP over BNPP despite the marginal difference in BDE (4.9 kcal mol−1, Fig. S17†).37,38
Next, a substrate along with truncated peptide binding pockets (MD simulations, Fig. 3a–d) were used to investigate the underlying energetics in the catalytic systems by modelling the transition state (see the ESI for details†). Optimised structures revealed several points of contact between BNPP and the catalytic guanidinium and imidazole moieties (Fig. S18†). We performed an analysis of the closest guanidinium and imidazole residues to the substrate as potential catalytic centres. The analysis suggested that the guanidinium moiety facilitated the nucleophilic attack of imidazole on the phosphorous centre by forming H-bonds with phosphate anions.42 The transition state corresponding to phosphoester bond cleavage and imidazole–phosphorus bond formation (1.9 Å) gained stabilisation by forming multiple H-bonds between the dissociated p-nitrophenolate anion and positively charged guanidinium (Fig. S18†). It showed a significantly lower free energy activation barrier of 20.5 kcal Mol−1, i.e., 28.7 kcal Mol−1 lower than that of the uncatalyzed reaction which explains the higher hydrolytic rates in the presence of a catalyst (Fig. 3e). We could not obtain the transition states of HPNPP due to the complex nature of the potential energy surface (Fig. S19†).
Conclusions
The collaborative mutualism between nucleic acids and short peptide assemblies has been argued to initiate the dynamic evolving network in cellular life that catalyses and carries information based on specific recognitions via non-covalent interactions. The present study demonstrates the capability of short peptides to capitalize the paracrystalline packing and the antiparallel arrangement to bind diverse phosphate esters and subsequently utilize more than one catalytic residue for cleavage. This work is among the first examples where the registry and consequently the plasticity of a paracrystalline amyloid assembly are exploited to access advanced binding pockets to interact with nucleic acid congeners, which eventually leads to the cleaving of phosphodiester bonds with high kinetic stability. The versatility of the minimal self-assembled catalyst towards kinetically unfavourable chemical transformations, supports the argument of their role as primitive catalytic folds and also hints towards their involvement in biopolymer co-evolution.43–45 Furthermore, within the limits of a short sequence such flexibility along with the utilisation of a peptide registry can, in principle, process more challenging chemical transformations which were thus far believed to be achievable only by extant enzymes.Data availability
All experimental and computational data associated with this work are available in the ESI.†Author contributions
C. M., A. S., S. P. A., and D. D. concieved the project and designed the experiments. S. M. and J. M. performed molecular dynamic simulations and docking analyses. All authors co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DD and AS are thankful to Swarnajayanti (SB/SJF/2020-21/08), GOI for financial assistance. DD acknowledges CSIR (02(0406)/21/EMR-II. CM and SPA acknowledge CSIR and UGC for fellowships. JM and SM acknowledge support of the Department of Atomic Energy, Government of India, under Project Identification No. RTI 4007. JM acknowledges Core Research grants provided by the Department of Science and Technology (DST) of India (CRG/2019/001219).References
- Z. Wang, P. A. Cole, Z. Wang and P. A. Cole, Methods Enzymol., 2014, 548, 1–21 CAS.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4523 CrossRef CAS PubMed.
- M. Diez-Castellnou, F. Mancin and P. Scrimin, J. Am. Chem. Soc., 2014, 136, 1158–1161 CrossRef CAS PubMed.
- E. E. Kim and H. W. Wyckoff, J. Mol. Biol., 1991, 218, 449–464 CrossRef CAS PubMed.
- J. Greenwald and R. Riek, J. Mol. Biol., 2012, 421, 417–426 CrossRef CAS PubMed.
- N. Singh, M. Kumar, J. F. Miravet, R. V Ulijn and B. Escuder, Chem. – Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- F. Rodriguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478–11484 CrossRef CAS PubMed.
- A. Reja, S. P. Afrose and D. Das, Angew. Chem., Int. Ed., 2020, 59, 4329–4334 CrossRef CAS PubMed.
- J. Ottelé, A. S. Hussain, C. Mayer and S. Otto, Nat. Catal., 2020, 3, 547–553 CrossRef.
- O. Carny and E. Gazit, FASEB J., 2005, 19, 1051–1055 CrossRef CAS PubMed.
- T. O. Omosun, M. C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 CrossRef CAS PubMed.
- A. Chatterjee, C. Mahato and D. Das, Angew. Chem., Int. Ed., 2021, 60, 202–207 CrossRef CAS PubMed.
- J. Greenwald, W. Kwiatkowski and R. Riek, J. Mol. Biol., 2018, 430, 3735–3750 CrossRef CAS PubMed.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. Degrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- B. Sarkhel, A. Chatterjee and D. Das, J. Am. Chem. Soc., 2020, 142, 4098–4103 CrossRef CAS PubMed.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977–985 CrossRef CAS PubMed.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS PubMed.
- A. Chatterjee, S. P. Afrose, S. Ahmed, A. Venugopal and D. Das, Chem. Commun., 2020, 56, 7869–7872 RSC.
- C. Zhang, R. Shafi, A. Lampel, D. Macpherson, C. G. Pappas, V. Narang, T. Wang, C. Maldarelli and R. V Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, A. A. Orr, P. Makam, B. Redko, E. Haimov, Y. Wang, L. J. W. Shimon, S. Rencus-Lazar, M. Ju, P. Tamamis, H. Dong and E. Gazit, Angew. Chem., Int. Ed., 2021, 60, 17164–17170 CrossRef CAS PubMed.
- W. Yang, Q. Rev. Biophys., 2011, 44, 1–93 CrossRef CAS PubMed.
- C. R. Woese, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8742–8747 CrossRef CAS PubMed.
- J. E. Smith, A. K. Mowles, A. K. Mehta and D. G. Lynn, Life, 2014, 4, 887–902 CrossRef PubMed.
- L. Baldini, R. Cacciapaglia, A. Casnati, L. Mandolini, R. Salvio, F. Sansone and R. Ungaro, J. Org. Chem., 2012, 77, 3381–3389 CrossRef CAS PubMed.
- P. S. Muñana, G. Ragazzon, J. Dupont, C. Z.-J. Ren, L. J. Prins and J. L.-Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 16469–16474 CrossRef PubMed.
- R. Salvio, S. Volpi, T. Folcarelli, A. Casnati and R. Cacciapaglia, Org. Biomol. Chem., 2019, 17, 7482–7492 RSC.
- J. L. Y. Chen, C. Pezzato, P. Scrimin and L. J. Prins, Chem. – Eur. J., 2016, 22, 7028–7032 CrossRef CAS PubMed.
- A. Pecina, D. R. Gastaldo, L. Riccardi, S. F. Ulloa, E. Milan, P. Scrimin, F. Mancin and M. D. Vivo, ACS Catal., 2021, 11, 8736–8748 CrossRef CAS PubMed.
- R. Chen, K. Das, M. A. Cardona, L. Gabrielli and L. J. Prins, J. Am. Chem. Soc., 2022, 144, 2010–2018 CrossRef CAS PubMed.
- C. Z.-J. Ren, P. S. Muñana, G. G. Warr and J. L.-Y. Chen, ACS Catal., 2020, 10, 8395–8401 CrossRef CAS.
- S. Neri, S. G. Martin, C. Pezzato and L. J. Prins, J. Am. Chem. Soc., 2017, 139, 1794–1797 CrossRef CAS PubMed.
- R. Salvio, S. Volpi, R. Cacciapaglia, F. Sansone, L. Mandolini and A. Casnati, J. Org. Chem., 2016, 81, 9012–9019 CrossRef CAS PubMed.
- J. G. L. Ferreira, L. M. Ramos, A. L. de Oliveira, E. S. Orth and B. A. D. Neto, J. Org. Chem., 2015, 80, 5979–5983 CrossRef CAS PubMed.
- E. S. Orth, T. A. S. Brandão, B. S. Souza, J. R. Pliego, B. G. Vaz, M. N. Eberlin, A. J. Kirby and F. Nome, J. Am. Chem. Soc., 2010, 132, 8513–8523 CrossRef CAS PubMed.
- M. Ghosh, G. Meiss, A. Pingoud, R. E. London and L. C. Pedersen, J. Biol. Chem., 2005, 280, 27990–27997 CrossRef CAS PubMed.
- F. M. Menger and F. Nome, ACS Chem. Biol., 2019, 14, 1386–1392 CrossRef CAS PubMed.
- S. Mikkola, T. Lönnberg and H. Lönnberg, Beilstein J. Org. Chem., 2018, 14, 803–837 CrossRef CAS PubMed.
- J. Czescik, F. Mancin, R. Strömberg and P. Scrimin, Chem. – Eur. J., 2021, 27, 8143–8148 CrossRef CAS PubMed.
- Y. Wang, L. Yang, M. Wang, J. Zhang, W. Qi, R. Su and Z. He, ACS Catal., 2021, 11, 5839–5849 CrossRef CAS.
- A. S. Pina, L. Morgado, K. L. Duncan, S. Carvalho, H. F. Carvalho, A. J. M. Barbosa, B. de P. Mariz, I. P. Moreira, D. Kalafatovic, B. M. M. Faustino, V. Narang, T. Wang, C. G. Pappas, I. Ferreira, A. C. A. Roque and R. V. Ulijn, Chem. Sci., 2022, 13, 210–217 RSC.
- G. Gulseren, I. C. Yasa, O. Ustahuseyin, E. D. Tekin, A. B. Tekinay and M. O. Guler, Biomacromolecules, 2015, 16, 2198–2208 CrossRef CAS PubMed.
- D. G. Liz, A. M. Manfredi, M. Medeiros, R. Montecinos, B. Gómez-González, L. Garcia-Rio and F. Nome, Chem. Commun., 2016, 52, 3167–3170 RSC.
- J. T. Goodwin, A. K. Mehta and D. G. Lynn, Acc. Chem. Res., 2012, 45, 2189–2199 CrossRef CAS PubMed.
- A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 51, 3047–3070 RSC.
- A. Harish and G. Caetano-Anollés, PLoS One, 2012, 7, e32776 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03205h |
| This journal is © The Royal Society of Chemistry 2022 |