
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeep learning in single-molecule imaging and analysis: recent advances and prospects
Xiaolong
Liu
 ab,
Yifei
Jiang
c,
Yutong
Cui
ab,
Jinghe
Yuan
*a and
Xiaohong
Fang
*abc
ab,
Yifei
Jiang
c,
Yutong
Cui
ab,
Jinghe
Yuan
*a and
Xiaohong
Fang
*abc
aKey Laboratory of Molecular Nanostructure and Nanotechnology, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: xfang@iccas.ac.cn; jhyuan@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cInstitute of Basic Medicine and Cancer, Chinese Academy of Sciences, Hangzhou 310022, Zhejiang, China
First published on 22nd September 2022
Abstract
Single-molecule microscopy is advantageous in characterizing heterogeneous dynamics at the molecular level. However, there are several challenges that currently hinder the wide application of single molecule imaging in bio-chemical studies, including how to perform single-molecule measurements efficiently with minimal run-to-run variations, how to analyze weak single-molecule signals efficiently and accurately without the influence of human bias, and how to extract complete information about dynamics of interest from single-molecule data. As a new class of computer algorithms that simulate the human brain to extract data features, deep learning networks excel in task parallelism and model generalization, and are well-suited for handling nonlinear functions and extracting weak features, which provide a promising approach for single-molecule experiment automation and data processing. In this perspective, we will highlight recent advances in the application of deep learning to single-molecule studies, discuss how deep learning has been used to address the challenges in the field as well as the pitfalls of existing applications, and outline the directions for future development.
 Xiaolong Liu | Xiaolong Liu received his B. S. degree in Chemistry in 2019 from Harbin Institute of Technology. He is currently pursuing his PhD degree in chemistry at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), conducting single-molecule imaging research under the supervision of Dr Xiaohong Fang. His research interests focus on single molecule imaging for membrane protein interaction analysis. |
 Yifei Jiang | Dr. Yifei Jiang earned a Bachelor's degree in Chemical Physics from the University of Science and Technology of China and a PhD in Physical Chemistry from Clemson University. He did his post-doc with Prof. Daniel Chiu at the University of Washington, focusing on developing fluorescent probes and spectroscopy methods for single exosome characterization. He is currently a professor at the Institute of Basic Medicine and Cancer (IBMC), Chinese Academy of Sciences (CAS). His current research focuses on superresolution microscopy and its biomedical applications. |
 Yutong Cui | Yutong Cui studied at the University of Chinese Academy of Sciences as an undergraduate and joined Xiaohong Fang's research group in 2021 to participate in single-molecule imaging research. She will continue her doctoral studies at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). |
 Jinghe Yuan | Dr. Jinghe Yuan obtained his doctorate degree in Optics Engineering (2002) at the Institute of Modern Optics, Nankai University, China. Then he joined the State Key Laboratory of high field laser physics, Shanghai Institute of Optics and Fine Mechanics, Chinese Academy of Sciences as a postdoctoral researcher. In 2009, he joined the Key Laboratory of Molecular Nanostructure and Nanotechnology as an associate research fellow and a research fellow. His research interests include super-resolution microscopy and data processing with deep-learning. |
 Xiaohong Fang | Dr. Xiaohong Fang obtained her PhD degree in Analytical Chemistry from Peking University in 1996. After one-year postdoc work at the Univ. of Waterloo (Canada), she worked as a research associate at the Univ. of Florida (USA) from 1998 to 2001. She was recruited to the Chinese Academy of Sciences (CAS) in 2001 and became a professor of chemistry at the Institute of Chemistry, CAS. In 2021, she was appointed as a professor at the Institute of Basic Medicine and Cancer (IBMC), CAS. Her major research interest is the development of new bioanalytical and biomedical methods for protein detection and interaction studies at the single molecule level, as well as the discovery and diagnosis of cancer biomarkers. |
1 Introduction
In recent years, driven by the interest to study cellular processes at the molecular level, a variety of single-molecule microscopic methods have been developed, including single-molecule localization microscopy (SMLM),1 single-particle tracking (SPT),2 single-molecule fluorescence resonance energy transfer (smFRET),3 single-molecule polarization imaging,4etc. By observing and analyzing the behavior of individual molecules directly, including their aggregation states,5 kinetic characteristics, and conformation changes,6,7 single molecule imaging can unveil structural and kinetic heterogeneities that are not accessible to conventional ensemble measurements.8–11 Due to these unique advantages, single-molecule imaging has also attracted research interest from other fields, including electrochemistry,12,13 materials science,14 and pharmaceutical science.15However, there are several barriers that hinder the wide application of single-molecule imaging in biochemical studies. Firstly, single-molecule imaging is a generally sensitive and time/labor-consuming process, which requires high stability of the instrument and extensive experience from the researcher. Run-to-run variation increases measurement errors and makes the result hard to interpret. Secondly, a single-molecule signal is often weak and heterogeneous with various types of dynamics. The event of interest is also convolved with noise and photo-physical kinetics, as well as instrument fluctuation, which results in highly complex data.16 Traditional algorithms that assume the data to follow a certain distribution might not work well with single-molecule data.17 Thirdly, single-molecule imaging typically generates a large amount of data. Its data analysis method requires lots of time/effort from experienced users and the procedures are easily affected by human subjective factors, which affects the accuracy and the consistency of analysis.
Recently, as a new class of computer algorithms that simulate the human brain to extract data features, deep learning has been applied to a wide range of research fields with excellent performance.18 Deep learning networks excel in task parallelism and model generalization and are well-suited for handling nonlinear functions and extracting weak features, which provide a promising approach for single-molecule experiment automation and data processing.19 Recent published studies that apply deep learning to single-molecule imaging and analysis have shown that, compared to previous algorithms, deep learning provides superior performances in terms of sensitivity, accuracy, and processing speed.
In this perspective, we will first introduce the basic principles of single-molecule microscopy, in particular, current challenges in experiment automation and data processing. Then we will review recent advances in deep learning in single-molecule studies and highlight how deep learning has been used to address the challenges in the field. Finally, we will conclude with the current stage of deep learning in single-molecule imaging and data analysis, discuss the pitfalls of the existing applications, and outline the directions for future development. It should be noted that deep-learning-assisted SMLM, including the single molecule localization method,20–22 image reconstruction,23,24 background estimation,25 and point spread function (PSF) engineering,26–30 has received broad attention and been reviewed extensively.1,31–33 Equally important but often neglected areas are single-molecule imaging automation and single-molecule feature recognition, which will be the focus of this review.
2 Basics of single-molecule fluorescence imaging and analysis
Single-molecule microscopy is a powerful tool to characterize structural and dynamic heterogeneities in biological systems, which mainly contains two tasks: imaging and data analysis. In the imaging process, researchers would like to fully preserve the dynamics of interest, which involves measuring the behavior of single molecules with high sensitivity and precision. In the data analysis process, researchers would like to extract as much information from the images as possible, which requires the use of highly specialized and carefully optimized algorithms. In this section, we will briefly introduce the basics of single-molecule fluorescence imaging and analysis methods, and discuss the recent progress in this field.2.1 Single-molecule fluorescence imaging
Single-molecule imaging relies on the efficient collection and detection of limited fluorescence photons emitted by individual molecules. Under ideal conditions, such as labeled dyes immobilized on a clean coverslip or diffusing in a solution with a low fluorescence background, a combination of a high numerical aperture objective and a highly sensitive detector can readily provide the signal-to-noise-ratio (SNR) required to detect single molecules. However, applying single-molecule imaging to more complex systems, such as live cells and thick tissues, places additional requirements on the imaging SNR and imaging depths.7 In addition, in order to study biological processes that occur on a shorter time and length scales, it is also necessary to further improve the spatial and temporal resolutions of single-molecule imaging.In general, single-molecule imaging methods enhance the imaging SNR and detection sensitivity by reducing the excitation/detection volumes using different types of fluorescence microscopes. For example, total internal reflection fluorescence microscopy (TIRFM)34–38 exploits evanescent waves to selectively excite molecules near the interface; confocal microscopy uses pinholes to filter out non-focal fluorescence signals; light-sheet fluorescence microscopy (LSFM) uses a 2D light sheet to illuminate and image samples in thin slices, etc. Among these methods, TIRFM has very shallow imaging depth and is most suitable for studying lateral structures/dynamics; confocal microscopy, as a point-scanning technique, offers 3D resolution but suffers from low imaging efficiency; LSFM,39,40 on the other hand, combines wide-field planar excitation with axial optical-sectioning, which offers a balanced performance between axial resolution and imaging speed.41–43
In addition to the various excitation schemes, single-molecule detection schemes can also be modified to extend the imaging depth and obtain additional information. For example, PSF engineering methods use conventional epi excitation schemes and modify the shape of the PSF to reflect the axial position of the fluorophore. By introducing cylindrical optics or phase plates into the detection light path, the conventional Gaussian-like PSF can be transformed into ellipse, double helix, and tetrapod shapes.44–47 PSF engineering methods offer very good temporal resolution and extend the imaging depth to as deep as 20 μm, which is particularly useful for 3D imaging and particle tracking.48 Hyperspectral imaging determines the spectra of individual molecules through dispersion of fluorescence photons, which can provide information about structure and dynamic heterogeneities.49–52 In addition, the emission polarization of fluorescent probes can be used to study the orientation and the rotational movements of biomolecules.53–58
The spatial resolution of conventional optical microscopy is limited by the diffraction of light.59 Depending on the numerical aperture of the objective and imaging wavelength, the lateral and axial resolutions of fluorescence microscopy are typically 200–300 nm and 500–600 nm, respectively. Driven by the interest in studying biological structures/processes below the diffraction limit, a variety of methods have been developed to further improve the spatial resolution of fluorescence imaging, including single-molecule localization methods, such as stochastic optical reconstruction microscopy (STORM)60 and photoactivated localization microscopy (PALM),61 methods that exploit fluorophores' non-linear response to excitation, such as stimulated emission depletion microscopy (STED)62–64 and ground state depletion microscopy (GSD),65 and post-acquisition processing methods, such as super-resolution optical fluctuation imaging (SOFI),66etc. Among these techniques, SMLM67 has attracted particular research interest, as it offers high spatial resolution while using relatively simple instrumentation.66,68,69 By combining SMLM with point-function engineering, 3D super-resolution imaging with a lateral and an axial resolution of 5 nm and 10 nm has been demonstrated, which greatly improves the level of detail provided by single-molecule imaging.70,71 SMLM and its deep learning applications have been extensively reviewed.1,31–33,72–77 Due to the limited space, we will focus on single-molecule imaging and only mention SMLM briefly in the review.
Overall, advances in imaging techniques have increased the detection sensitivity, imaging depth, and spatial and temporal resolution of single-molecule imaging. Due to the high sensitivity of the measurement, maintaining focus and minimizing sample drift are crucial for reducing measurement variations and obtaining reliable results. In addition, advanced applications, such as deep particle tracking and 3D single-molecule imaging, require careful optimization and calibration of the instruments. To address these challenges, we will discuss how deep learning has been used to set up single-molecule experiments, optimize imaging conditions, and improve the quality of the results in Sections 4.1 and 4.2.
2.2 Single-molecule imaging data analysis
Single-molecule imaging is well-suited to address heterogeneity and characterize unsynchronized sequences of events. Single-molecule data contain a large amount of information, which can be used to reveal aggregation states, kinetic characteristics, and conformation changes of individual molecules. However, a single-molecule signal is typically weak and highly complex. The dynamics of interest can be easily confused with various types of noises, as well as photo-physical kinetics. Extracting information from single-molecule data is a challenging process that requires specialized algorithms and careful optimization of the analysis procedures.Localization of single molecules in the images is the first step of single-molecule data processing. Single-molecule localization allows obtaining basic information such as location, intensity, and orientation of single molecules, which can be used to visualize the subcellular structure and to construct the fluorescence intensity and position traces. The conventional approach is to use a two-dimensional Gaussian distribution to fit the PSF. Multiple iterations are performed using maximum likelihood estimation (MLE)78 or nonlinear least squares (NLLS)79 until the best Gaussian model is found. Such an iterative approach is usually time-consuming. There is also the wavelet segmentation algorithm, which converts the raw data into wave maps and performs single-molecule localization using a wavefront to accelerate the process.80
After initial localization, analysis of the fluorescence intensity traces can be used to obtain a variety of valuable information on biomolecule structures and functions (Fig. 1). For example, counting the number of steps in a photobleaching trajectory can be used to determine the single-molecule aggregation state (Fig. 1b), smFRET analysis can be used to study protein interactions (Fig. 1c), single-molecule recognition through equilibrium Poisson sampling (SiMREPS) can be used to characterize the binding dynamics of bio-molecules (Fig. 1d), etc.5 For diffusing molecules, the trajectory is constructed by linking localized positions between sequential frames,81,82 which can be used to characterize the state of single molecules and their interactions with the microenvironment (Fig. 1a).83,84 Many physical parameters associated with biological processes can be extracted from the analysis of the trajectories, such as total displacement, furthest distance from the starting point, confinement ratio, local orientation, directional change, instantaneous velocity, mean curve rate, root mean square displacement (RMSD) and diffusion coefficient (D).85,86 These parameters reflect the state of single molecules and their interactions with the surroundings. For example, molecular diffusion models, such as Brownian motion, directional diffusion, confined diffusion, etc., are extensively used to analyze the interactions of proteins on the membrane.7
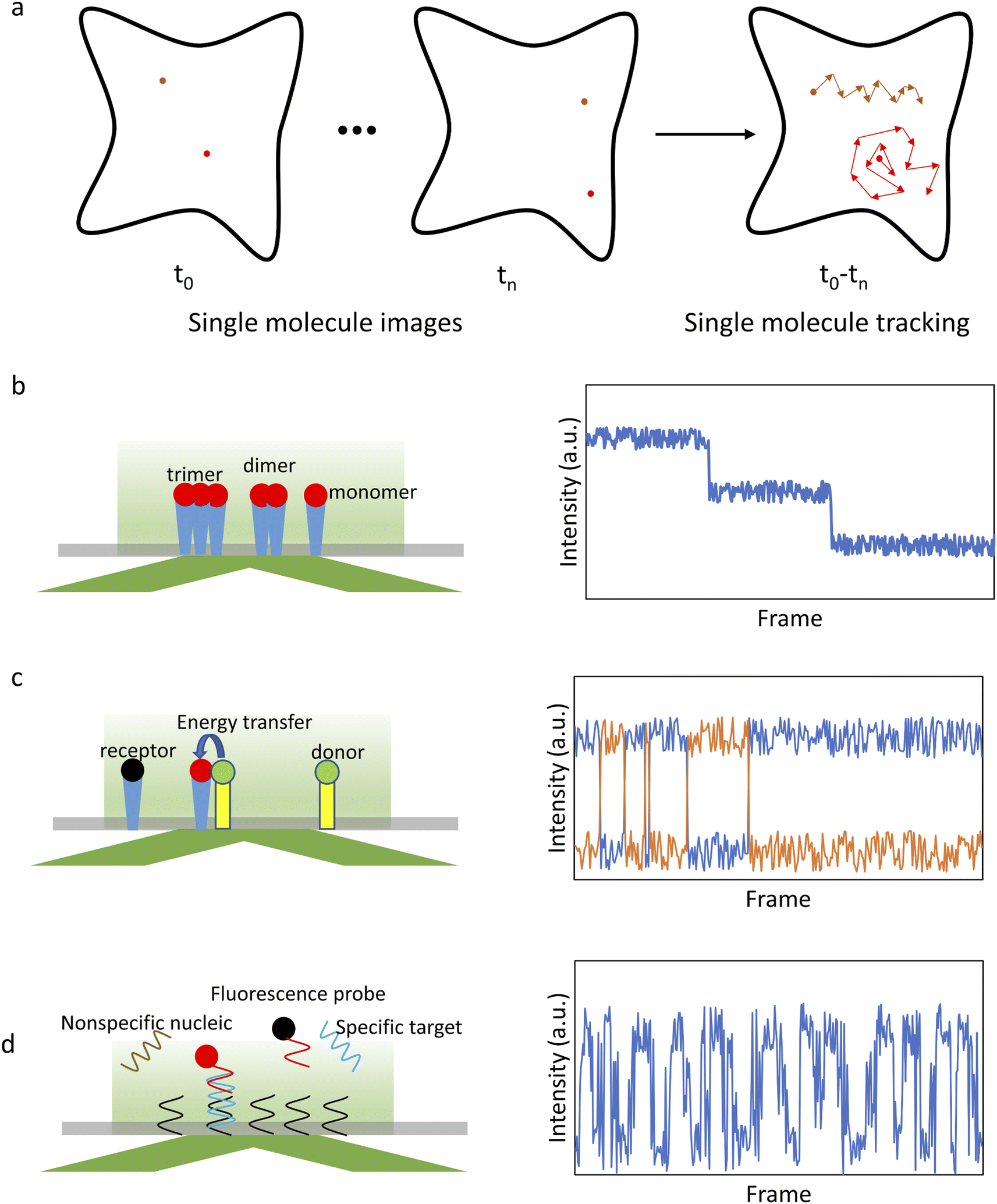 | ||
| Fig. 1 Schematic of single-molecule imaging methods. (a) Imaging and tracking of single molecules. (b) Principle of single-molecule photobleaching step-counting analysis (smPSCA). (c) Principle of smFRET. (d) Principle of SiMREPS. Red/green and dark spots represent the excited and non-excited molecules, respectively. | ||
Single-molecule data analysis is a challenging process by traditional methods. On one hand, single-molecule data often contains a variety of dynamics, and do not follow a certain distribution. On the other hand, single-molecule imaging typically generates a large amount of data, which are easily influenced by human bias with reduced accuracy and consistency. We will discuss how deep learning algorithms have been used to address these problems and facilitate single-molecule data analysis in Sections 4.3 and 4.4.
3 Deep learning algorithms
In recent years, with the development of computational hardware and algorithms, tremendous progress has been made in deep learning neural networks (DNNs).87 Deep learning has been applied in various fields and plays an important role in high-throughput data processing.18 Here, we will introduce the basic concepts of deep learning and hardware requirements.The basic units of DNNs are neurons.18 Each neuron is a simple operator that yields an output from multiple inputs. Multiple neurons in parallel form a layer of neurons, and the output of the neurons in one layer is used as the input of the neurons in the next layer, thus forming a neural network. The number of layers, the number of neurons in each layer, and the weights of neurons are all adjustable parameters in the model. The parameters are determined by learning a large amount of training data. Due to the advantages of task parallelism and model generalization, DNNs can be used to fit nonlinear functions and simulate feature extraction functions of the human brain.
Deep neural networks can be divided into two main categories in terms of training methods:88 supervised learning networks and unsupervised learning networks. Supervised learning feeds the model with already labeled data for training. The output targets of the training data are already known in advance, and the model only needs to iterate continuously so that the objective function converges to minimum error. The advantage of supervised learning is the high accuracy of the trained model. However, the data needs to be labeled in advance, which is difficult for some applications due to the lack of a priori knowledge. In contrast, an unsupervised learning network is a type of learning in which the training data does not need to be labeled in advance and the model automatically finds features and classifies all the data. As a result, unsupervised learning performs well in cluster analysis and is able to find small classes that traditional methods cannot find.
The most widely used deep neural network is convolutional neural networks (CNNs).89 CNNs are suitable for processing multidimensional data, such as images, audio signals, etc. CNNs generally consist of several types of network layers: input layer, convolutional layer, activation layer, pooling layer, and fully connected layer. The input layer feeds the raw or pre-processed data into the convolutional neural network. As the core layer in CNNs, the convolutional layer performs a sliding window operation using smaller convolutional kernels to detect different features, which is similar to the receptive field in a biological visual system. The activation layer converts linear mapping into a nonlinear mapping using a nonlinear activation function, such as a rectified linear activation function or sigmoid function. The pooling layer is a down-sampling layer, sandwiched between successive convolutional layers, used to compress the number of parameters and reduce overfitting. In a fully connected layer, all the neurons between two successive layers are interconnected with weights to map the learned features into the sample labeling space.
Recurrent neural networks (RNNs) are often used for time series data, such as speech signals.89 The RNN is a recursion of a neural network, which uses the previous output as well as a hidden layer, in which the information about all the past processed elements is preserved, as the input.90 The memory layer adds a layer of weights with each propagation, which results in a reduced amount of previous information in the later memory (vanishing gradiant). To solve the problem of gradient disappearance, long-short-term memory (LSTM) networks have been developed.91 By adding a forgetting gate, LSTMs chose which memory to remember or forget and can preserve long-term information. Long-term memory is propagated by linear summation operations so that gradient disappearance does not occur in back propagation. LSTMs have been shown to perform better than conventional RNNs in most problems.91
A generative adversarial network (GAN), contains two sequential networks: the generative network and the discriminative network.92 The generative network is used to generate data based on a probability distribution and the discriminative network is used to extract features from the generated data. The two models are trained to promote each other. As a type of GAN, the discriminator-generator network (DGN) uses two bi-directional long-short term memory networks (biLSTMs) as a generator and a discriminator respectively.93,94 BiLSTMs can access both past and future contexts to improve the prediction results. The discriminator is used to map the input sequence to a hidden state vector, and then the generator recovers the input time sequence from this hidden state vector. The discriminator and generator are jointly trained to optimize the prediction accuracy, thus uncovering the hidden state behind a time series.
Considering that single-molecule imaging data is mainly images and time-series, CNN and RNN-based networks are well-suited for single-molecule data analysis. In addition, in the case of single-molecule data with unknown features or with features that cannot be labeled, a GAN-based unsupervised network is particularly useful.
4 Applications of deep learning in single-molecule imaging and analysis
Deep learning has been applied to almost every stage of single-molecule imaging and analysis, including single-molecule imaging automation, single-molecule localization, fluorescence intensity and position trace analysis. By replacing human labor in single-molecule experiments and data processing, deep learning algorithms have reduced run-to-run variations induced by bias and human error, thus improving the accuracy of the measurement and the analysis. In addition, deep learning excels in handling nonlinear functions and extracting weak features that cannot be detected using conventional algorithms.The application of deep learning in single molecule data analysis includes two stages: model training and experimental data analysis. Model training is time- and power-consuming. Once the model is trained, the analysis of experimental data only takes seconds to minutes. For most deep learning tasks, a personal computer with an appropriate configuration is enough. There are three parts that need to be considered: central processing unit (CPU), graphics processing unit (GPU), and random access memory (RAM). A deep learning model always processes a large amount of data. The performance of the CPU mainly limits the speed of data loading and pre-processing. Most mainstream CPUs can meet the requirements. Most deep learning models are trained on the GPU. An excellent GPU with a memory of no less than 8 GB can accelerate the speed of training, e.g., Nvidia RTX 1080 – 3080 series and Titan series. Insufficient RAM will limit the processing speed of the CPU and GPU. The RAM should be larger than the GPU memory. We recommend RAM greater than 32 GB. There are several cloud computing platforms providing free GPUs, which facilitate the use of deep learning and project sharing, e.g., Amazon Web Service (AWS), Microsoft Azure, and Google Colaboratory.
In Table 1, we have listed recent representative applications of deep learning in single-molecule imaging/analysis and summarized the key information, including network type, input/output of the model, training hardware and training time. In this part, we will review these applications in detail and compare the performances of the various deep learning algorithms.
| Applications | Network type | Input | Output | GPU | Train time | Ref. |
|---|---|---|---|---|---|---|
| Autofocus for SMLM | CNNs | Defocus image | Defocus degree | GeForce RTX 2080 SUPER VENTUS XS OC GPU | 3 h | Lightley95 |
| Offline autofocus for fluorescence microscopy | GAN | Defocus image | Focused image | GeForce RTX 2080Ti | 30 h | Luo96 |
| Single-shot autofocus for fluorescence microscopy | Fully connected Fourier neural network (FCFNN) | Defocus image | Defocus degree | GTX 1080Ti | 30 min (GPU) | Pinkard97 |
| 15 h (CPU) | ||||||
| Automated single-molecule imaging | CNN | Image | Classified image based on expression level | NVIDIA Quadro 4000 | — | Yasui98,99 |
| Protein stoichiometry analysis for epidermal growth factor receptors (EGFRs) | CNN, LSTM | Single molecule intensity-time series | Aggregation state | GeForce 1080Ti GPU | — | Xu16 |
| Protein stoichiometry analysis for transforming the growth factor-β type II receptor (TβRII) | biLSTM | Single molecule intensity-time series | Aggregation state and state change dynamics | — | — | Yuan100 |
| Protein stoichiometry analysis for the chemokine receptor (CXCR4) | CNN | Single molecule images | Aggregation state and state change dynamics | GeForce 2080Ti | — | Wang101 |
| Protein stoichiometry analysis for CXCR4 | CNN, LSTM | Single molecule blinking intensity time series | Aggregation state | GeForce RTX 2080Ti | — | Wang102 |
| FRET trace classification | LSTM | SmFRET intensity time series | Classified intensity time series based on FRET activity | — | — | Thomsen103 |
| DNA point mutation recognition by SiMREPS and FRET traces classification | LSTM | SiMREPS or smFRET intensity time series | Classified intensity time series based on binding activity | NVIDIA's TESLA V100 | 1 h (GPU) | Li104 |
| 5–10 h (CPU) | ||||||
| Diffusion model classification | CNN | Single molecule position traces | Diffusion type | NVIDIA GeForce Titan GTX | — | Granik105 |
4.1 Deep-learning-assisted single-molecule autofocus
A single-molecule data acquisition process requires a high level of instrument stability and a high level of experience from the researcher. On one hand, single-molecule imaging typically takes hours of continuous experiments to acquire enough data for statistical analysis. Focus variation and sample drift during the acquisition greatly affect the measurement accuracy. On the other hand, experimental procedures, such as searching for cells in the desired state or focusing onto the structure of interest, are prone to human error and require extensive practice. Deep learning can improve the performance of single-molecule imaging by automating the experimental process and reducing the amount of human labor involved.Autofocus is a very useful function in microscopic imaging. It can quickly find the focal plane without human judgment and, in addition, prevent samples from defocusing during long-time imaging. Traditional real-time autofocus includes two main types: hardware-based autofocus and image-based autofocus. Hardware-based autofocus relies on an additional sensor that detects the back-reflection signal from the coverslip to determine the focus drift and then performs re-focus. Lightley et al.95 recently improved the working distance of the hardware-based autofocus system by developing a CNN-based algorithm. A diode laser with a wavelength of 830 nm is focused onto a coverslip. The detector is located on the conjugate plane of the coverslip. The reflected NIR laser is detected by using a camera and the spatial distribution of intensity is recorded (Fig. 2a). The shape of the distribution is influenced by the focal condition. A CNN model is trained with the acquired images of various out-of-focus depths. The off-focus distance can be quickly calculated and corrected by analyzing the distribution shape during the imaging process (Fig. 2b). This method has been applied in SMLM and works well over a range of ±100 μm. The image-based auto-focusing takes a series of images along the Z-axis and determines the off-focus distance by calculating the sharpness of the feature edges.
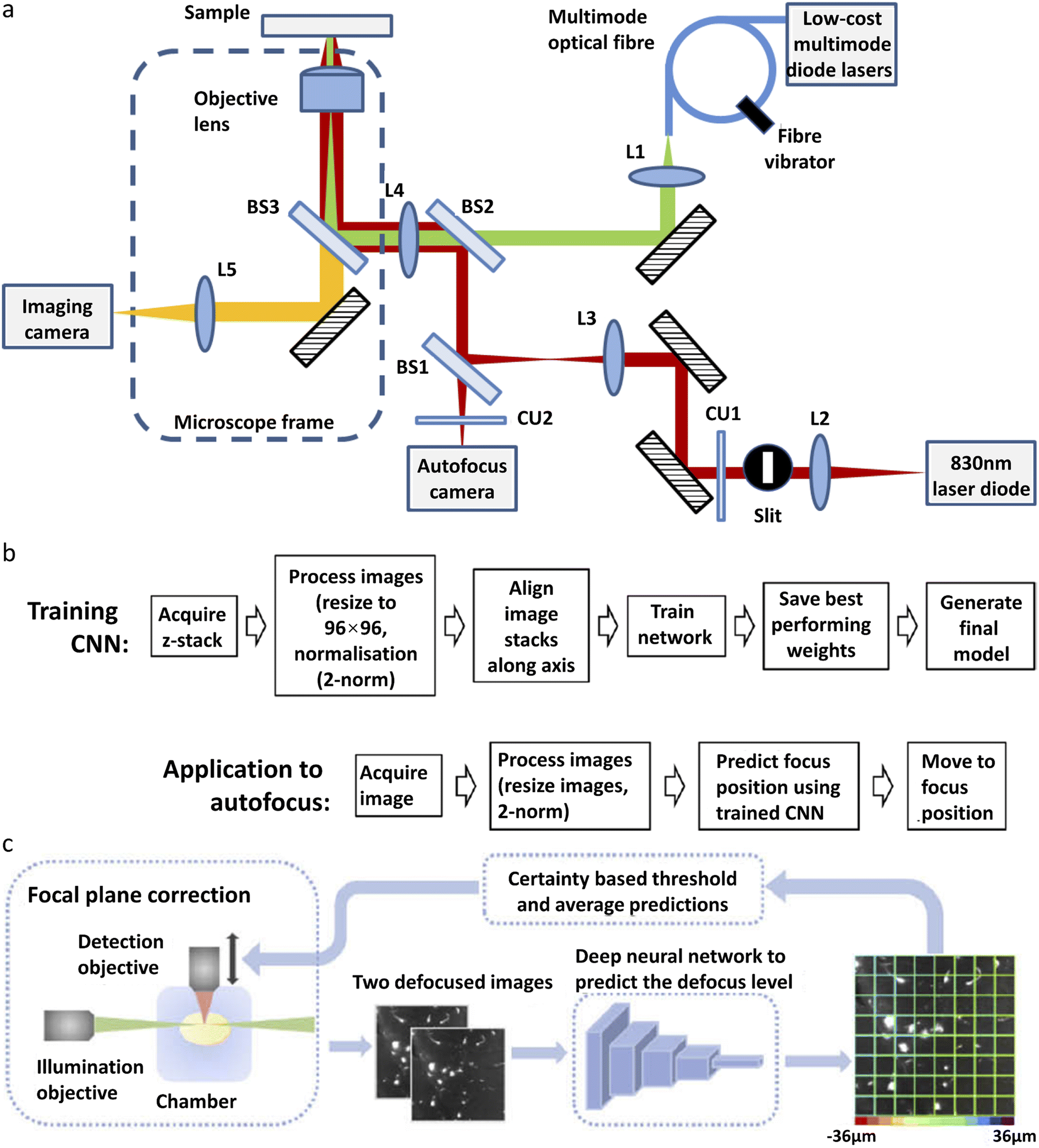 | ||
| Fig. 2 Different types of deep-learning assisted autofocus system. (a) Instrumentation for a CNN-assisted hardware-based online autofocus system. (b) The process of CNN training for hardware-based autofocus application.95 Copyright © 2021, The Authors. (c) Overview of the integration of the deep-learning-assisted image-based autofocus method with a custom-built LSFM.106 Copyright © 2021, Optica Publishing Group. | ||
Henry et al.97 reported a single-shot focusing method based on deep learning, which relies on one or more off-axis illumination sources to find the correct focal plane. While the idea of single-shot focusing is compelling, the requirement for an extra illumination source could limit its application in single-molecule imaging. Li et al.106 developed a deep learning model for autofocus of LSFM (Fig. 2c). Hundreds of defocused image stacks are acquired, each containing a series of images with various off-focus distances. For every image stack, two defocused images with a known off-focus distance are fed into the network for training. The known defocus distance served as the ground truth. After training, this model can determine the off-focus distance according to two defocused images in LSFM. This model has been demonstrated in the imaging of mouse forebrain and pig cochleae samples.
More recently, a new off-line autofocus method has been developed. Luo et al.96 developed a post-imaging autofocus system called Deep-R based on the GAN. Different levels of out-of-focus and in-focus images are used for training. The generative network takes the out-of-focus images as input data and outputs the in-focus images, and then the discriminative network takes the output of the generative network as the input and generates the out-of-focus images. The two networks are trained jointly. The in-focus image generated by the model is compared with the actual in-focus image to filter out the wrong models. After the training, with an out-of-focus image as the input, the generative network is able to generate the corresponding in-focus image quickly and accurately.
4.2 Deep-learning-assisted single-molecule image acquisition
The data acquisition process of single-molecule imaging is time-consuming and complex. To simplify this process and reduce the possibility of human error, Yasui et al.98,99 built an automated single-molecule imaging platform AiSIS, based on deep learning. It consists of three key modules: an oil immersion feedback system, an autofocus system, and an automated cell search system. The autofocus system is hardware-based with the assistance of DNNs, which is briefly described as follows. An iris is conjugated to the upper surface of the coverslip and the image of the iris is captured by a surface reflection interference contrast filter (SRIC). When the image is out-of-focus, the image of the iris is blurred. Pre-acquired in-focus and out-of-focus images are used to train the neural network. This neural network is used to determine whether it is out-of-focus and to perform focusing according to the image. Automated search for cells with suitable single-molecule density (1–3 molecules per μm2) is also achieved by using a deep learning method. The model is pre-trained with single-molecule images of suitable density. Fluorescent spots generated by cell fragments are also excluded by the deep learning model. Combined with their custom-built mechanics, they are able to fully automate the processes of well plate placement, dosing stimulation, and multi-well imaging without the need for human labor, which provides great advantages for high-throughput single-molecule imaging. The system is capable of imaging and analyzing 1600 cells in 96-well plates within one day.Baddeley developed the Python-Microscopy Environment (PYME)107 which is an integrated platform for high-throughput SMLM. Deep learning neural network Mask R-CNN is trained to detect nuclei for ROI selection automatically. Mask R-CNN is a flexible framework for object instance segmentation, which has been applied in human pose estimation, tumor identification, artifact detection, etc. A dataset BBBC038v1, which contains a large number of segmented nuclei images, is used as training data. The system exploits data compression, distributed storage, and distributed analysis for automatic real-time localization analysis, which massively increases the throughput of SMLM to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells a day.
000 cells a day.
4.3 Deep-learning-assisted single molecule fluorescence intensity trace analysis
Here, we will focus on three types of single-molecule fluorescence intensity trace analysis, which are single-molecule photobleaching step-counting analysis (smPSCA), smFRET, and SiMREPS, and discuss how deep-learning was applied to improve the performance of these methods.Cellular proteins generally function as multimers, aggregates or protein complexes. SmPSCA has become a common method to count the number of fluorescent proteins within a diffraction-limited-spot and determine the stoichiometry and aggregation state of the proteins. In photobleaching trajectories, the dynamics of interest are easily confused with various types of noise and photophysical kinetics, such as photoblinking, which are not accounted for in conventional analysis methods, such as the filter method,108 threshold method, multiscale product analysis, motion t-test method, and step fitting method.109 Taking the temporal information into account, the hidden Markov model (HMM) can partially eliminate the interference of photoblinking.110 However, HMM methods show a weak ability to correlate long-term events and require users to preset parameters such as initial states, state numbers and a transition matrix. All the methods above require the input of parameters based on prior knowledge of the biological system as well as the algorithms, which could be challenging for users and might affect the accuracy of the analysis. Xu et al.16 reported the first deep learning model to solve these problems in smPSCA, which is referred to as the convolutional and long-short-term memory deep learning neural network (CLDNN) (Fig. 3a). This model consists of both a convolutional layer and LSTM layer. Single-molecule photobleaching traces are used as input data, and the output is the number of steps. The convolutional layer is introduced to accurately extract features of steplike photobleaching events, and the LSTM layer is to remember the previous fluorescence intensity for photoblinking identification. Manually labeled experimental data and artificially synthesized data are used as the training sets. Once the model is trained, it can analyze a large amount of data quickly without setting any parameters. The CLDNN model effectively removes the interference of photoblinking and noise on bleaching step recognition. Compared to the previously reported algorithms for smPSCA, the CLDNN shows higher accuracy with even over 90% at a low SNR value (SNR = 1.9), and higher computational efficiency with 2–3 orders of faster speed.
 | ||
| Fig. 3 Deep learning for single molecule stoichiometry studies. (a) Architecture of the CLDNN for SMPSCA.16 Copyright © 2019, American Chemical Society. (b) The training and performances of the DGN on both SMPSCA and dynamic finding with fluorescence intensity traces.100 Copyright © 2020, The Author(s). | ||
The CLDNN is a supervised-learning network. Training data need to be labeled manually, which is often difficult to realize without human bias. Yuan et al.100 developed an unsupervised neural network, DGN, which can be used not only for protein stoichiometry determination but also for the kinetic characteristics of protein aggregation state changes in live cells (Fig. 3b). The DGN model consists of two biLSTMs. Each biLSTM consists of two inverse LSTMs. The LSTM is suitable to analyze the change of the aggregation state, in which the previous state affects the prediction of the later data. In the traditional LSTM, the model predicts the current time point according to previous information. However, there is very little reference information for the first data point, which leads to less accurate prediction. Therefore, a bi-directional LSTM is used so that there is data to refer to for both front and back feature extraction. In order to achieve unsupervised learning, two biLSTMs are used as a generator and discriminator, respectively. The discriminator identifies the hidden state behind the input fluorescence intensity traces, and then the generator generates fluorescence intensity traces using the hidden state sequence from the discriminator. The generator and the discriminator are trained jointly. After training, the DGN exhibits excellent accuracy in counting photobleaching steps. At SNR = 1.40, the DGN is able to achieve 79.6% accuracy, while conventional methods such as HMM can only achieve 30.9% accuracy. In addition, the DGN can recover the state path, which allows the dynamic information to be obtained from the analysis of fluorescence intensity traces of live cells, including durations of protein association, transition rates during protein interactions and state occupancies of different protein aggregation states. The authors used the model to investigate the TGF-β receptor monomer and dimeric/oligomeric state change under different conditions. They found that while the ligand TGF-β can drive the balance forward to receptor oligomer formation, disruption of lipid-rafts by nystatin can make TGF-β receptor association or disassociation more active, and oligomers are difficult to stably exist.
Wang et al.101 developed a deep learning convolutional neutral network (DLCNN) to recognize receptor monomers and complexes. When receptors form a complex, multiple fluorophores are integrated into a diffraction volume to create an overly bright or abnormal spot, which can be used to identify the complex state. This model was trained with images of single quantum dot (QD) particles and aggregates. After training, it can visualize the complex formation of chemokine receptor CXCR4 in real time and reach an accuracy of >98% for identifying monomers and complexes. They also developed deep-blinking fingerprint recognition (BFR) for identification of oligomeric states.102 They labeled the CXCR4 receptor with carbon dots (CDs). According to the different aggregation states of the receptor, CD blinking creates different intensity fingerprints. Deep learning models extract the fingerprints and classify the receptor aggregation states. They demonstrate that the heterogeneous organizations of CXCR4 can be regulated by various stimuli at different degrees. For 42-residue amyloid-β peptide (Aβ42), it is difficult to probe individual aggregation pathways in a mixture because existing fibrils grow and new fibrils appear. A deep neural network (FNet)111 was developed to split highly overlapping fibrils into single fibrils, which enables tracking of the changes of individual fibrils.
smFRET can be used to analyze protein interactions and achieve highly sensitive detection of targets. Performing smFRET requires preprocessing of images, and extracting, classifying and segmenting smFRET traces.112 Traditionally, the selection of traces requires a lot of subjective judgment and is time-consuming. The two-color intensity trajectories of smFRET need to match the inverse relationship such that as one falls, the other rises. One of the major advantages of deep learning lies in fast feature recognition. Therefore, classification and analysis of smFRET trajectories using DNNs has been reported. Thomsen et al.103 developed software for smFRET data analysis based on DNNs: DeepFRET. This model includes the whole process from image pre-processing, extraction of trajectories, and selection of trajectories, to data analysis. The LSTM is used to learn the temporality of the data and propagate the learned information to the later frames. The introduction of the LSTM eliminates the interference of noise. The model was pre-trained using 150000 simulated data that included all possible FRET states, inter-state transition probabilities, state dwell time etc. On the real data, the model was able to achieve an accuracy of over 95%, while using 1% of the time required by the traditional method.
SiMREPS uses fluorescence to detect the specific binding and dissociation of the labeled molecules with fixed targets (Fig. 4a).113 The binding and dissociation of the molecule are reflected as an increase or a decrease in the fluorescence intensity (Fig. 4b). The rate constant of dissociation represents the strength of the binding, and the frequency of binding represents the concentration of the diffusing molecules. SiMREPS requires classification of intensity traces based on resident (“on”) time, which is easily influenced by photobleaching, protein aggregation, and noise variation when analysed using traditional algorithms. Li et al.104 developed a LSTM-based single-molecule fluorescence trace classification model, i.e., an automatic SMFM trace selector (AutoSiM) (Fig. 4c). The authors applied this model to analyze DNA sequences with point mutations. A solution containing a DNA sequence with a pre-known proportion that exhibits point mutations, which randomly bind to complementary strands was immobilized on the surface of a glass plate. The DNA with a specific base mutation shows a shorter residence time in the fluorescence state due to the reduced binding between the mutated DNA and the target. When used for the analysis of experimental data, the recognition specificity increased by 4.25 times compared to that by the conventional HMM method. The number of layers of the LSTM is adjusted in this model, with 7 layers for classification and 8 layers for segmenting useful trajectories. The model was trained with real experimental and synthetic FRET data. The FRET data classification network was able to achieve an accuracy of 90.2 ± 0.9%. To extend the applicability of the model, transfer learning was introduced. Only 559 manually analyzed FRET trajectories of a Mn2+ sensor were used to complete the model training, which took less than 15 min. A classification accuracy of 91% was achieved for the experimental data using the transfer learning model.
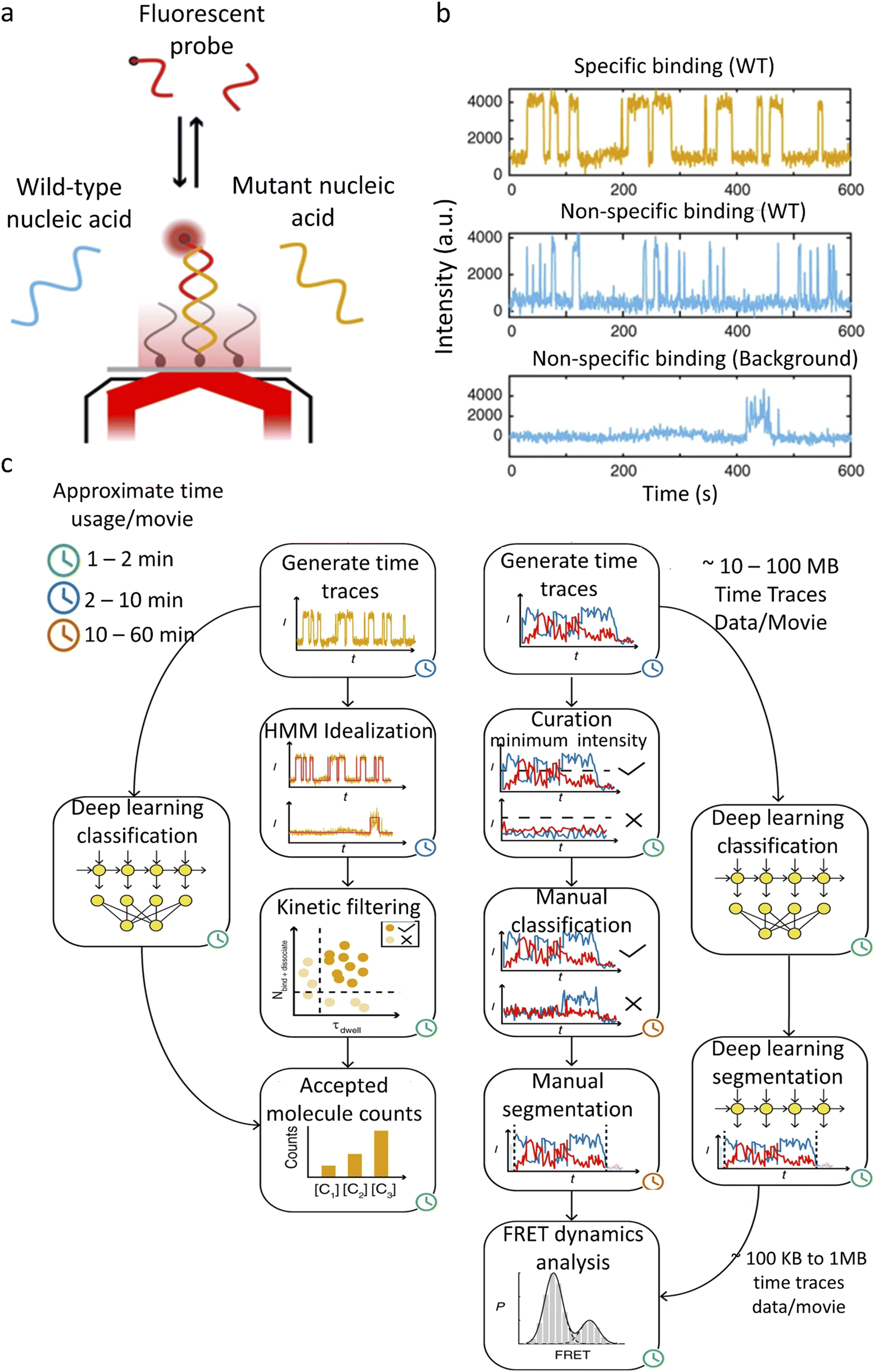 | ||
| Fig. 4 AutoSIM for SiMREPS and smFRET data classification. (a) Schematic of an experimental system for detection of a mutant DNA sequence by SiMREPS. (b) Representative experimental fluorescence intensity traces showing repeated binding of mutant and wild type DNAs to the complementary strands, as well as a typical trace showing non-repetitive nonspecific binding. (c) SiMREPS and smFRET data analysis steps bypassed by the deep learning methods in AutoSIM.104 Copyright © 2020, The Author(s). | ||
4.4 Deep-learning-assisted single molecule position trace analysis
The traditional methods for extracting diffusion features are the mean square displacement (MSD) method114,115 and HMM method.116 In the MSD analysis, the slope and curvature of the MSD–Δt curve reflect the D and diffusion mode of the molecule, respectively. Segmented MSD analysis can be used to detect the change of molecule diffusion states over time. The HMM is commonly used as a machine learning method to extract hidden state changes from noisy time series data. The HMM is typically combined with other parameter estimation methods, such as maximum likelihood estimation, Bayesian estimation,86 and Rayleigh mixture distribution17 for model selection. As discussed in the previous section, HMM methods require the input of parameters which are often difficult to obtain in advance. In addition, both the MSD and the HMM methods assume that there is a clear mathematical relationship between MSD and Δt, which allows the diffusion parameters to be obtained by mathematical fitting. However, sometimes, the diffusion model is not known in advance or the dynamics do not follow a simple diffusion pattern, which prevents such methods from accurately extracting information from the position traces. Recently, deep learning has been applied to the analysis of single molecule position traces, which can be classified into two categories, i.e., classification of single-molecule diffusion mode and construction of single-molecule diffusion fingerprints.Granik et al.105 used CNNs to classify diffusion trajectories. Three diffusion modes were analyzed: Brownian motion, continuous time random walk (CTRW) and fractional Brownian motion (FBM). FBM and CTRW have similar motion characteristics for shorter trajectories, but they obey different physical laws: FBM is associated with crowded cellular environments, while CTRW motion mainly occurs in trap-containing environments. The neural network was trained with 300000 trajectories. The accuracy of the model was evaluated using real experiment trajectories. The diffusion of fluorescent beads follows FBM in gel networks of different densities, and pure Brownian motion in water and glycerol solutions. The diffusion of proteins across the cell membrane is a combination of FBM and CTRW. Based on 100-step tracks of beads with two sizes, the network can distinguish two different populations, and the mean values are similar to predicted theoretical diffusion coefficients; however, the existence of two populations cannot be distinguished by TAMSD with 100 steps.
Due to the complexity of the cellular environment, single-molecule diffusion is often a combination of multiple models and varies over time. So far there is no unified model that can completely describe all the kinetic characteristics of single-molecule diffusion. To address this problem, deep learning has been recently used to construct diffusion fingerprints for single molecule position traces. Pinholt et al.117 proposed a single-molecule diffusion fingerprinting method that integrates 17 single-molecule diffusion characteristics (Fig. 5). This approach creates an exclusive diffusion fingerprint for each type of single-molecule diffusion, which allows better classification of different diffusion entities. The 17 characteristics include 8 features from HMM estimation: D of the four states and the respective residence times, two features from classical RMSD analysis: the diffusion constants describing irregular diffusion, four features based on trajectory shape: kurtosis, dimension, efficiency, and trappedness, and three features describing the general trend: the average speed, track duration, and MSD parameters. These features are partially overlapped and can be used to distinguish subtle differences between trajectories. A logistic regression classifier is used to predict the experimental environment that generates such data. A linear discriminant analysis (LDA) is used to rank the most relevant features. Single molecule diffusion fingerprinting was applied to identify Thermomyces lanuginosus lipase (TLL) and L3 mutants. The mutant and wild type have almost the same catalytic rate. The step length distributions among the single-molecule trajectories were very similar and difficult to differentiate using conventional methods. The analysis of the diffusion fingerprints identified the feature that distinguishes the two enzymes: the residence time of the HMM diffusion state. The L3 mutant diffuses away from the generated product region in larger steps and spends more time in faster states. This allows the L3 mutant to have less end-product inhibition. This is also in agreement with the available experimental results. This approach of diffusion fingerprinting combined with multiple traditional characterization methods provides a more comprehensive understanding of different diffusion patterns. However, when the feature selection is not optimal, the accuracy of the classification is not very high. Replacing the simple logistic regression model with a CNN or LSTM could potentially improve the classification accuracy.
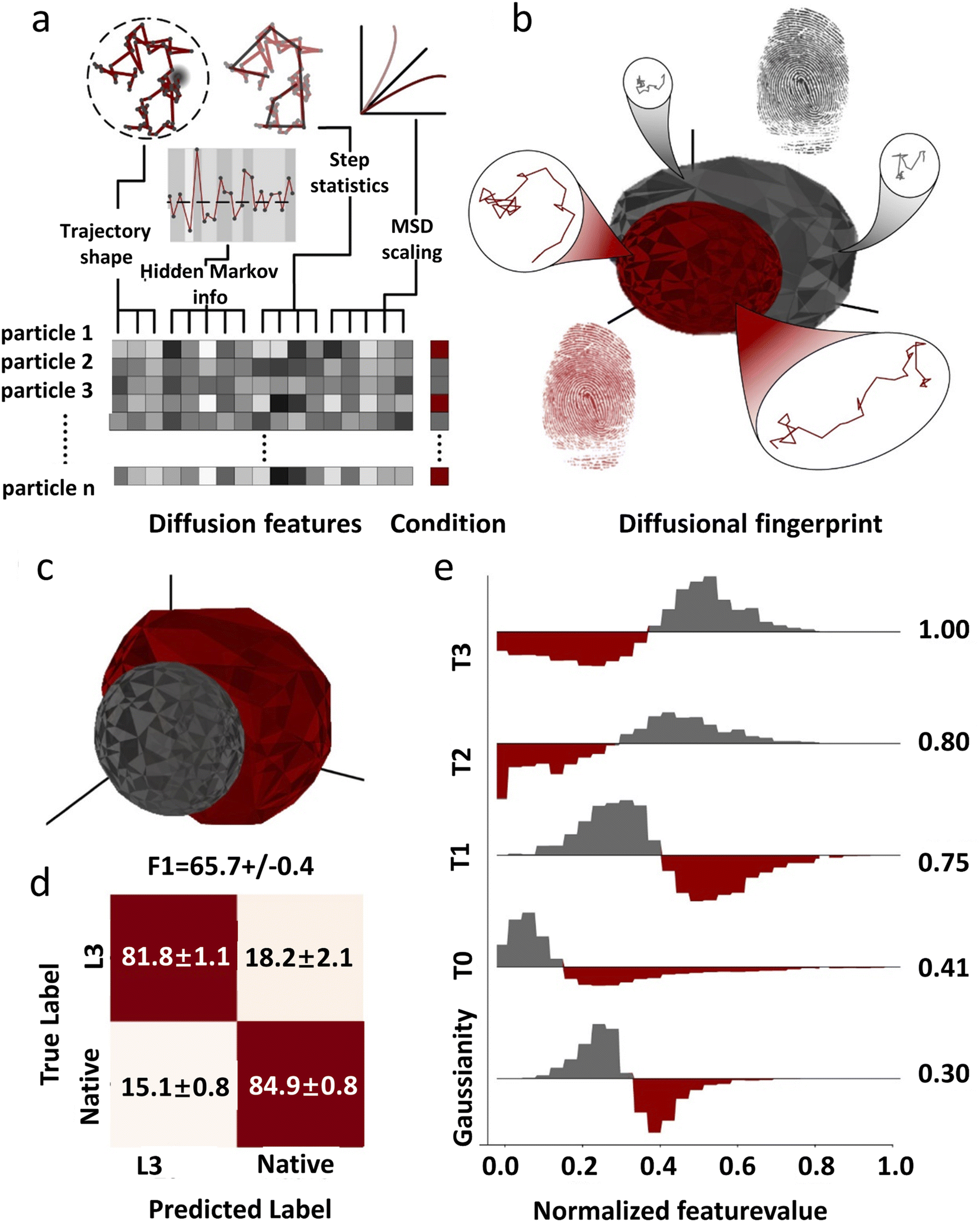 | ||
| Fig. 5 The concept of diffusional fingerprinting for classifying molecular identity based on SPT data. (a) Analysis of the trajectory and extraction of 17 descriptive features. (b) The diffusional fingerprint is composed of the feature distributions for each particle type. (c) Diffusional fingerprinting of SPT data for two functionally similar TLL variants, L3 and native. (d) Confusion matrix for classifying two kinds of TLL. (e) Differential histograms of the five highest-ranked features.117 Copyright © 2021, National Academy of Science. | ||
Overall, traditional feature-based methods of single-molecule diffusion analysis assume that the diffusion of particles obeys some basic physical diffusion patterns, which in reality is often more complex. Analyzing a single feature of diffusion cannot reflect the full information of single-molecule motion. Compared to intensity trajectories, the single-molecule diffusion trajectory has higher dimensions and therefore contains more features, which may not obey existing physical laws or models. Deep learning can be used to discover these weak features, achieve a comprehensive description of single-molecule diffusion, and truly establish a single-molecule diffusion fingerprint.
5 Summary and outlook
In summary, deep learning has performed well in single-molecule experiment automation and data analysis. Automated data acquisition assisted by deep learning can greatly reduce variations induced by human error and improves the reliability of the measurement. Analysis by deep learning is objective, accurate and fast compared with that by conventional analytical methods. Most conventional analytical methods need to pre-know the mathematical model of variables. However, in reality, biomolecule activities often do not strictly follow a specific mathematical distribution. By training, a deep learning method can find the most appropriate, exact and nonlinear function for each variable, which cannot be achieved by conventional algorithms.There are still some pitfalls to be solved for the application of deep learning in single molecule studies. (1) In order to obtain a good model, a large amount of data is required for training. Acquisition of such data is time- and labor-demanding. (2) Deep learning methods often suffer from the problem of overfitting. A model that learns well on training data may not be able to accurately handle unfamiliar data. Some methods have been developed to mitigate this problem, but it is still a tricky situation. (3) Deep learning is a black box; the distribution of features has no analytical form and steps in the algorithm cannot be correlated to the features, which makes it impossible to get an exact interpretation of the algorithm. (4) Deep learning is not easy to get started with. Performing deep learning requires extensive knowledge of the related algorithms and programming skills. Skilled scientists often had difficulties tuning parameters and fixing bugs, let alone the freshman. These problems limit the application of deep learning in single-molecule imaging and analysis.
In the future, development of more advanced algorithms will reduce the requirement for training data volume and makes deep learning more user friendly. Construction of an authoritative single-molecule database can facilitate the generalization of deep learning methods. Not only does it help scientists to verify the accuracy of the methods, but it also contributes to building deep-learning models that are applicable to different instruments and experimental conditions. The standardization of instruments allows for the comparison of different research studies. For home-built microscopy systems, scientists should add more imaging parameters such as: SNR, laser power, TIRFM angle, etc. We should develop more convenient deep learning platforms and modularize different deep learning methods so that even inexperienced users can invoke them easily with a mouse click. Cloud computation platforms have dramatically lowered the barrier for deep learning applications, but more efforts are needed. Scientists who use deep learning for single-molecule data processing should share the code and package their models into easy-to-use applications. In addition, adding more single-molecule parameters (polarization, spectrum, phase, etc) to the deep learning model can help it extract less-obvious features with enhanced accuracy. By adding these advantages, deep learning can further improve the performance of single-molecule microscopy in a low SNR environment, providing a truly powerful tool set for biochemical applications. Further development of deep learning-aided single molecule imaging should also contribute to clinical studies, including disease diagnoses, pathological investigations, and drug discovery.
Author contributions
X.-F. and J.-Y. set the scope of the perspective and supervised the completion of the manuscript. X.-L. wrote the outline and first draft of the perspective. Y.-C participated in the literature research. X.-L., Y.-J., Y.-C., X.-F. and J.-Y. participated in the revision. All authors contributed to the editing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos. 21735006, 21890742, and 22077124) and the Chinese Academy of Science.References
- L. Möckl and W. E. Moerner, J. Am. Chem. Soc., 2020, 142, 17828–17844 CrossRef PubMed.
- A. Kusumi, T. A. Tsunoyama, K. M. Hirosawa, R. S. Kasai and T. K. Fujiwara, Nat. Chem. Biol., 2014, 10, 524–532 CrossRef CAS PubMed.
- D. K. Sasmal, L. E. Pulido, S. Kasal and J. Huang, Nanoscale, 2016, 8, 19928–19944 RSC.
- K. Zhanghao, L. Chen, X.-S. Yang, M.-Y. Wang, Z.-L. Jing, H.-B. Han, M. Q. Zhang, D. Jin, J.-T. Gao and P. Xi, Light: Sci. Appl., 2016, 5, e16166 CrossRef CAS PubMed.
- W. Zhang, Y. Jiang, Q. Wang, X. Ma, Z. Xiao, W. Zuo, X. Fang and Y.-G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15679–15683 CrossRef CAS PubMed.
- T. Xia, N. Li and X. Fang, Annu. Rev. Phys. Chem., 2013, 64, 459–480 CrossRef CAS PubMed.
- F. Luo, G. Qin, T. Xia and X. Fang, Annu. Rev. Anal. Chem., 2020, 13, 337–361 CrossRef CAS PubMed.
- J. Pi, H. Jin, F. Yang, Z. W. Chen and J. Cai, Nanoscale, 2014, 6, 12229–12249 RSC.
- T. Sungkaworn, M.-L. Jobin, K. Burnecki, A. Weron, M. J. Lohse and D. Calebiro, Nature, 2017, 550, 543–547 CrossRef CAS PubMed.
- K. Eichel, D. Jullié, B. Barsi-Rhyne, N. R. Latorraca, M. Masureel, J.-B. Sibarita, R. O. Dror and M. von Zastrow, Nature, 2018, 557, 381–386 CrossRef CAS PubMed.
- W. T. C. Lee, Y. Yin, M. J. Morten, P. Tonzi, P. P. Gwo, D. C. Odermatt, M. Modesti, S. B. Cantor, K. Gari, T. T. Huang and E. Rothenberg, Nat. Commun., 2021, 12, 2525 CrossRef CAS PubMed.
- J. Dong, Y. Lu, Y. Xu, F. Chen, J. Yang, Y. Chen and J. Feng, Nature, 2021, 596, 244–249 CrossRef CAS PubMed.
- R. Hao, Z. Peng and B. Zhang, ACS Omega, 2020, 5, 89–97 CrossRef CAS PubMed.
- B. Dong, N. Mansour, T.-X. Huang, W. Huang and N. Fang, Chem. Soc. Rev., 2021, 50, 6483–6506 RSC.
- S. Okay, Assay Drug Dev. Technol., 2020, 18, 56–63 CrossRef CAS PubMed.
- J. Xu, G. Qin, F. Luo, L. Wang, R. Zhao, N. Li, J. Yuan and X. Fang, J. Am. Chem. Soc., 2019, 141, 6976–6985 CrossRef CAS PubMed.
- R. Zhao, J. Yuan, N. Li, Y. Sun, T. Xia and X. Fang, Anal. Chem., 2019, 91, 13390–13397 CrossRef CAS PubMed.
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436–444 CrossRef CAS PubMed.
- H. Ismail Fawaz, G. Forestier, J. Weber, L. Idoumghar and P.-A. Muller, Data. Min. Knowl. Discov., 2019, 33, 917–963 CrossRef.
- P. Cascarano, M. C. Comes, A. Sebastiani, A. Mencattini, E. Loli Piccolomini and E. Martinelli, Bioinformatics, 2022, 38, 1411–1419 CrossRef CAS PubMed.
- E. Nehme, L. E. Weiss, T. Michaeli and Y. Shechtman, Optica, 2018, 5, 458 CrossRef CAS.
- E. Hershko, L. E. Weiss, T. Michaeli and Y. Shechtman, Opt. Express, 2019, 27, 6158 CrossRef CAS PubMed.
- L. von Chamier, R. F. Laine, J. Jukkala, C. Spahn, D. Krentzel, E. Nehme, M. Lerche, S. Hernández-Pérez, P. K. Mattila, E. Karinou, S. Holden, A. C. Solak, A. Krull, T.-O. Buchholz, M. L. Jones, L. A. Royer, C. Leterrier, Y. Shechtman, F. Jug, M. Heilemann, G. Jacquemet and R. Henriques, Nat. Commun., 2021, 12, 2276 CrossRef CAS PubMed.
- A. Speiser, L.-R. Müller, P. Hoess, U. Matti, C. J. Obara, W. R. Legant, A. Kreshuk, J. H. Macke, J. Ries and S. C. Turaga, Nat. Methods, 2021, 18, 1082–1090 CrossRef CAS PubMed.
- L. Möckl, A. R. Roy, P. N. Petrov and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 60–67 CrossRef PubMed.
- E. Nehme, D. Freedman, R. Gordon, B. Ferdman, L. E. Weiss, O. Alalouf, T. Naor, R. Orange, T. Michaeli and Y. Shechtman, Nat. Methods, 2020, 17, 734–740 CrossRef CAS PubMed.
- M. J. Mlodzianoski, P. J. Cheng-Hathaway, S. M. Bemiller, T. J. McCray, S. Liu, D. A. Miller, B. T. Lamb, G. E. Landreth and F. Huang, Nat. Methods, 2018, 15, 583–586 CrossRef CAS PubMed.
- L. Möckl, P. N. Petrov and W. E. Moerner, Appl. Phys. Lett., 2019, 115, 251106 CrossRef PubMed.
- F. Xu, D. Ma, K. P. MacPherson, S. Liu, Y. Bu, Y. Wang, Y. Tang, C. Bi, T. Kwok, A. A. Chubykin, P. Yin, S. Calve, G. E. Landreth and F. Huang, Nat. Methods, 2020, 17, 531–540 CrossRef CAS PubMed.
- P. Zhang, S. Liu, A. Chaurasia, D. Ma, M. J. Mlodzianoski, E. Culurciello and F. Huang, Biophys. J., 2019, 116, 281a CrossRef.
- C. Belthangady and L. A. Royer, Nat. Methods, 2019, 16, 1215–1225 CrossRef CAS PubMed.
- L. Möckl, A. R. Roy and W. E. Moerner, Biomed. Opt. Express, 2020, 11, 1633 CrossRef PubMed.
- Y. Shechtman, Biophys. Rev., 2020, 12, 1303–1309 CrossRef PubMed.
- D. Axelrod, J. Cell Biol., 1981, 89, 141–145 CrossRef CAS PubMed.
- M. Tokunaga, N. Imamoto and K. Sakata-Sogawa, Nat. Methods, 2008, 5, 159–161 CrossRef CAS PubMed.
- W. Luo, T. Xia, L. Xu, Y.-G. Chen and X. Fang, J. Biophotonics, 2014, 7, 788–798 CrossRef CAS PubMed.
- J. Tang, C.-H. Weng, J. B. Oleske and K. Y. Han, J. Visualized Exp., 2019, 59360 Search PubMed.
- T. Chen, D. Ji and S. Tian, BMC Plant Biol., 2018, 18, 43 CrossRef PubMed.
- Y. Wan, K. McDole and P. J. Keller, Annu. Rev. Cell Dev. Biol., 2019, 35, 655–681 CrossRef CAS PubMed.
- E. M. C. Hillman, V. Voleti, W. Li and H. Yu, Annu. Rev. Neurosci., 2019, 42, 295–313 CrossRef CAS PubMed.
- F. Cella Zanacchi, Z. Lavagnino, M. Perrone Donnorso, A. Del Bue, L. Furia, M. Faretta and A. Diaspro, Nat. Methods, 2011, 8, 1047–1049 CrossRef PubMed.
- T. A. Planchon, L. Gao, D. E. Milkie, M. W. Davidson, J. A. Galbraith, C. G. Galbraith and E. Betzig, Nat. Methods, 2011, 8, 417–423 CrossRef CAS PubMed.
- L. Gao, L. Shao, B.-C. Chen and E. Betzig, Nat. Protoc., 2014, 9, 1083–1101 CrossRef CAS PubMed.
- S. R. P. Pavani, M. A. Thompson, J. S. Biteen, S. J. Lord, N. Liu, R. J. Twieg, R. Piestun and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2995–2999 CrossRef CAS PubMed.
- M. Badieirostami, M. D. Lew, M. A. Thompson and W. E. Moerner, Appl. Phys. Lett., 2010, 97, 161103 CrossRef PubMed.
- Y. Shechtman, L. E. Weiss, A. S. Backer, M. Y. Lee and W. E. Moerner, Nat. Photonics, 2016, 10, 590–594 CrossRef CAS PubMed.
- P. Zhang, S. Liu, A. Chaurasia, D. Ma, M. J. Mlodzianoski, E. Culurciello and F. Huang, Nat. Methods, 2018, 15, 913–916 CrossRef CAS PubMed.
- Y. Shechtman, L. E. Weiss, A. S. Backer, S. J. Sahl and W. E. Moerner, Nano Lett., 2015, 15, 4194–4199 CrossRef CAS PubMed.
- K.-H. Song, B. Brenner, W.-H. Yeo, J. Kweon, Z. Cai, Y. Zhang, Y. Lee, X. Yang, C. Sun and H. F. Zhang, Nanophotonics, 2022, 11, 1527–1535 CrossRef CAS PubMed.
- Y. Jiang and J. McNeill, Nat. Commun., 2018, 9, 4314 CrossRef PubMed.
- Y. Jiang, H. Chen, X. Men, Z. Sun, Z. Yuan, X. Zhang, D. T. Chiu, C. Wu and J. McNeill, Nano Lett., 2021, 21, 4255–4261 CrossRef CAS PubMed.
- R. Yan, K. Chen and K. Xu, J. Am. Chem. Soc., 2020, 142, 18866–18873 CrossRef CAS PubMed.
- M. P. Backlund, M. D. Lew, A. S. Backer, S. J. Sahl, G. Grover, A. Agrawal, R. Piestun and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19087–19092 CrossRef CAS PubMed.
- K. Zhanghao, L. Chen, X.-S. Yang, M.-Y. Wang, Z.-L. Jing, H.-B. Han, M. Q. Zhang, D. Jin, J.-T. Gao and P. Xi, Light: Sci. Appl., 2016, 5, e16166 CrossRef CAS PubMed.
- J. Lu, H. Mazidi, T. Ding, O. Zhang and M. D. Lew, Angew. Chem., Int. Ed., 2020, 59, 17572–17579 CrossRef CAS PubMed.
- V. Curcio, L. A. Alemán-Castañeda, T. G. Brown, S. Brasselet and M. A. Alonso, Nat. Commun., 2020, 11, 5307 CrossRef CAS PubMed.
- O. Zhang, W. Zhou, J. Lu, T. Wu and M. D. Lew, Nano Lett., 2022, 22, 1024–1031 CrossRef CAS PubMed.
- T. Wu, J. Lu and M. D. Lew, Optica, 2022, 9, 505 CrossRef CAS PubMed.
- E. Abbe, Archiv f. mikrosk. Anatomie, 1873, 9, 413–468 CrossRef.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- H. Blom and J. Widengren, Chem. Rev., 2017, 117, 7377–7427 CrossRef CAS PubMed.
- H. Yang, Y. Wu, H. Ruan, F. Guo, Y. Liang, G. Qin, X. Liu, Z. Zhang, J. Yuan and X. Fang, Anal. Chem., 2022, 94, 3056–3064 CrossRef CAS PubMed.
- Y. Wu, H. Ruan, R. Zhao, Z. Dong, W. Li, X. Tang, J. Yuan and X. Fang, Adv. Opt. Mater., 2018, 6, 1800333 CrossRef.
- S. Bretschneider, C. Eggeling and S. W. Hell, Phys. Rev. Lett., 2007, 98, 218103 CrossRef PubMed.
- T. Dertinger, R. Colyer, G. Iyer, S. Weiss and J. Enderlein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22287–22292 CrossRef CAS PubMed.
- L. von Diezmann, Y. Shechtman and W. E. Moerner, Chem. Rev., 2017, 117, 7244–7275 CrossRef CAS PubMed.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Nano Lett., 2010, 10, 4756–4761 CrossRef CAS PubMed.
- K. Xu, H. P. Babcock and X. Zhuang, Nat. Methods, 2012, 9, 185–188 CrossRef CAS PubMed.
- Y. Jiang, Q. Hu, H. Chen, J. Zhang, D. T. Chiu and J. McNeill, Angew. Chem., Int. Ed. Engl., 2020, 59, 16173–16180 CrossRef PubMed.
- I. M. Khater, I. R. Nabi and G. Hamarneh, Patterns, 2020, 1, 100038 CrossRef CAS PubMed.
- Z. Liu, L. Jin, J. Chen, Q. Fang, S. Ablameyko, Z. Yin and Y. Xu, Comput. Biol. Med., 2021, 134, 104523 CrossRef PubMed.
- T. Yang, Y. Luo, W. Ji and G. Yang, Biophys. Rep., 2021, 7(4), 253–266 Search PubMed.
- Y. Hyun and D. Kim, Int. J. Mol. Sci., 2022, 23, 6896 CrossRef PubMed.
- M. Fazel and M. J. Wester, AIP Adv., 2022, 12, 010701 CrossRef CAS.
- L. von Chamier, R. F. Laine and R. Henriques, Biochem. Soc. Trans., 2019, 47, 1029–1040 CrossRef CAS PubMed.
- K. I. Mortensen, L. S. Churchman, J. A. Spudich and H. Flyvbjerg, Nat. Methods, 2010, 7, 377–381 CrossRef CAS PubMed.
- A. V. Abraham, S. Ram, J. Chao, E. S. Ward and R. J. Ober, Opt. Express, 2009, 17, 23352 CrossRef CAS PubMed.
- J.-C. Olivo-Marin, Pattern Recognit., 2002, 35, 1989–1996 CrossRef.
- K. Jaqaman, D. Loerke, M. Mettlen, H. Kuwata, S. Grinstein, S. L. Schmid and G. Danuser, Nat. Methods, 2008, 5, 695–702 CrossRef CAS PubMed.
- J.-Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, Methods, 2017, 115, 80–90 CrossRef CAS PubMed.
- N. Chenouard, I. Smal, F. de Chaumont, M. Maška, I. F. Sbalzarini, Y. Gong, J. Cardinale, C. Carthel, S. Coraluppi, M. Winter, A. R. Cohen, W. J. Godinez, K. Rohr, Y. Kalaidzidis, L. Liang, J. Duncan, H. Shen, Y. Xu, K. E. G. Magnusson, J. Jaldén, H. M. Blau, P. Paul-Gilloteaux, P. Roudot, C. Kervrann, F. Waharte, J.-Y. Tinevez, S. L. Shorte, J. Willemse, K. Celler, G. P. van Wezel, H.-W. Dan, Y.-S. Tsai, C. O. de Solórzano, J.-C. Olivo-Marin and E. Meijering, Nat. Methods, 2014, 11, 281–289 CrossRef CAS PubMed.
- L. Xiang, K. Chen, R. Yan, W. Li and K. Xu, Nat. Methods, 2020, 17, 524–530 CrossRef CAS PubMed.
- H. Shen, L. J. Tauzin, R. Baiyasi, W. Wang, N. Moringo, B. Shuang and C. F. Landes, Chem. Rev., 2017, 117, 7331–7376 CrossRef CAS PubMed.
- F. Persson, M. Lindén, C. Unoson and J. Elf, Nat. Methods, 2013, 10, 265–269 CrossRef PubMed.
- L. Alzubaidi, J. Zhang, A. J. Humaidi, A. Al-Dujaili, Y. Duan, O. Al-Shamma, J. Santamaría, M. A. Fadhel, M. Al-Amidie and L. Farhan, J. Big Data, 2021, 8, 53 CrossRef PubMed.
- M. R. Minar and J. Naher, arXiv, 2018, preprint, arXiv:1807.08169 [cs, stat], DOI:10.13140/RG.2.2.24831.10403.
- J. Schmidhuber, Scholarpedia, 2015, 10, 32832 CrossRef.
- G. Koppe, S. Guloksuz, U. Reininghaus and D. Durstewitz, Schizophr. Bull., 2019, 45, 272–276 CrossRef PubMed.
- Y. Yu, X. Si, C. Hu and J. Zhang, Neural Comput., 2019, 31, 1235–1270 CrossRef PubMed.
- I. J. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, arXiv, 2014, preprint, arXiv:1406.2661 [cs, stat], DOI:10.48550/arXiv.1406.2661.93.
- M. Schuster and K. K. Paliwal, IEEE Trans. Signal Process., 1997, 45, 2673–2681 Search PubMed.
- A. Graves and J. Schmidhuber, Neural Networks, 2005, 18, 602–610 Search PubMed.
- J. Lightley, F. Görlitz, S. Kumar, R. Kalita, A. Kolbeinsson, E. Garcia, Y. Alexandrov, V. Bousgouni, R. Wysoczanski, P. Barnes, L. Donnelly, C. Bakal, C. Dunsby, M. A. A. Neil, S. Flaxman and P. M. W. French, J. Microsc., 2021, 13020 CrossRef PubMed.
- Y. Luo, L. Huang, Y. Rivenson and A. Ozcan, ACS Photonics, 2021, 8, 625–638 CrossRef CAS.
- H. Pinkard, Z. Phillips, A. Babakhani, D. A. Fletcher and L. Waller, Optica, 2019, 6, 794 CrossRef.
- M. Hiroshima, M. Yasui and M. Ueda, Microscopy, 2020, 69, 69–78 CrossRef CAS PubMed.
- M. Yasui, M. Hiroshima, J. Kozuka, Y. Sako and M. Ueda, Nat. Commun., 2018, 9, 3061 CrossRef PubMed.
- J. Yuan, R. Zhao, J. Xu, M. Cheng, Z. Qin, X. Kou and X. Fang, Commun. Biol., 2020, 3, 669 CrossRef CAS PubMed.
- Q. Wang, H. He, Q. Zhang, Z. Feng, J. Li, X. Chen, L. Liu, X. Wang, B. Ge, D. Yu, H. Ren and F. Huang, Anal. Chem., 2021, 93, 8810–8816 CrossRef CAS PubMed.
- Q. Wang, Q. Zhang, H. He, Z. Feng, J. Mao, X. Hu, X. Wei, S. Bi, G. Qin, X. Wang, B. Ge, D. Yu, H. Ren and F. Huang, Anal. Chem., 2022,(94), 3914–3921 CrossRef CAS PubMed.
- J. Thomsen, M. B. Sletfjerding, S. B. Jensen, S. Stella, B. Paul, M. G. Malle, G. Montoya, T. C. Petersen and N. S. Hatzakis, eLife, 2020, 9, e60404 CAS.
- J. Li, L. Zhang, A. Johnson-Buck and N. G. Walter, Nat. Commun., 2020, 11, 5833 CrossRef CAS PubMed.
- N. Granik, L. E. Weiss, E. Nehme, M. Levin, M. Chein, E. Perlson, Y. Roichman and Y. Shechtman, Biophys. J., 2019, 117, 185–192 CrossRef CAS PubMed.
- C. Li, A. Moatti, X. Zhang, H. Troy Ghashghaei and A. Greenbaum, Biomed. Opt. Express, 2021, 12, 5214 CrossRef PubMed.
- A. E. S. Barentine, Y. Lin, E. M. Courvan, P. Kidd, M. Liu, L. Balduf, T. Phan, F. Rivera-Molina, M. R. Grace, Z. Marin, M. Lessard, J. R. Chen, S. Wang, K. M. Neugebauer, J. Bewersdorf and D. Baddeley, BioRixv, 2019, 606954, DOI:10.1101/606954.
- K. Nakajo, M. H. Ulbrich, Y. Kubo and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18862–18867 CrossRef CAS PubMed.
- J. W. J. Kerssemakers, E. Laura Munteanu, L. Laan, T. L. Noetzel, M. E. Janson and M. Dogterom, Nature, 2006, 442, 709–712 CrossRef CAS PubMed.
- T. C. Messina, H. Kim, J. T. Giurleo and D. S. Talaga, J. Phys. Chem. B, 2006, 110, 16366–16376 CrossRef CAS PubMed.
- F. Meng, J. Yoo and H. S. Chung, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116736119 CrossRef CAS PubMed.
- S. Hohng, S. Lee, J. Lee and M. H. Jo, Chem. Soc. Rev., 2014, 43, 1007–1013 RSC.
- A. Johnson-Buck, X. Su, M. D. Giraldez, M. Zhao, M. Tewari and N. G. Walter, Nat. Biotechnol., 2015, 33, 730–732 CrossRef CAS PubMed.
- D. Calebiro, F. Rieken, J. Wagner, T. Sungkaworn, U. Zabel, A. Borzi, E. Cocucci, A. Zurn and M. J. Lohse, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 743–748 CrossRef CAS PubMed.
- M. J. Saxton and K. Jacobson, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 373–399 CrossRef CAS PubMed.
- I. Chung, R. Akita, R. Vandlen, D. Toomre, J. Schlessinger and I. Mellman, Nature, 2010, 464, 783–787 CrossRef CAS PubMed.
- H. D. Pinholt, S. S.-R. Bohr, J. F. Iversen, W. Boomsma and N. S. Hatzakis, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104624118 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |