
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfur-bridged chromophores for photofunctional materials: using sulfur oxidation state to tune electronic and structural properties
Jennifer
Yuan†
 ,
Zhen
Xu†
and
Michael O.
Wolf
*
,
Zhen
Xu†
and
Michael O.
Wolf
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: mwolf@chem.ubc.ca
First published on 28th April 2022
Abstract
The use of a heteroatom, such as sulfur, as a linker or bridge, in π-conjugated materials has advantages over purely carbon-based ones due to the accessibility of higher oxidation states as a result of hypervalence. Materials containing a sulfide bridge (S) can be systemically oxidized into sulfoxides (SO) and sulfones (SO2), each of which can then influence how a material interacts with light, playing a large role in dictating the photophysical and sometimes photochemical properties. In this perspective, we summarize the progress that our group and others have made, showing how oxidation of a sulfur bridge in symmetric bichromophoric dimers and in diimine ligands can influence the excited state behavior in organic π-conjugated materials and metal complexes.
 Jennifer Yuan | Jennifer Yuan received her BSc in Chemistry at Western University and her PhD at the University of British Columbia with Michael Wolf. Her research was focused on designing new sulfur-bridged chromophore dimers and studying their resulting photophysical and photochemical properties. She is currently a Postdoctoral Associate with Aaron Beeler at Boston University. |
 Zhen Xu | Zhen Xu received his BSc in Chemistry at Wuhan University and his PhD at the University of British Columbia with Michael Wolf. His research was focused on designing new inorganic and organic photochromic and luminescent materials. He is currently a Mitacs Postdoctoral Fellow with Mark MacLachlan at the University of British Columbia. |
 Michael O. Wolf | Michael Wolf received his BSc in Biochemistry and Chemistry at Dalhousie University and his PhD at the Massachusetts Institute of Technology with Mark Wrighton. He then completed a one-and-a-half-year stint as a NSERC Postdoctoral Fellow at the University of Texas-Austin with Marye Anne Fox. He has been on the faculty at the University of British Columbia since 1995 and is now a full Professor and Head of the Department. He was elected a Fellow of the Chemical Institute of Canada in 2015 and his major contributions are to the fields of conjugated polymers and materials, the photochemistry and photophysics of conjugated materials and coordination complexes, and the synthesis of and applications of modified nanomaterials. |
Introduction
Sulfur is an essential element for all living organisms and is utilized biologically in various oxidation states. The oxidation and reduction of sulfur and its compounds occurs in different species of bacteria and plants.1,2Chromatia oxidize sulfides via elemental sulfur to sulfate, whereas sulfur assimilation (reduction of sulfates into sulfides by plants) is a vital metabolic pathway to form the amino acids cysteine and methionine.3 Furthermore, sulfhydryl groups of cysteine residues found in proteins can be oxidized to disulfide bridges, playing an important role in the stability of proteins by maintaining tertiary structures.4,5 The various biological functions of sulfur are related to its hypervalence, which allows for oxidation–reduction transformations to occur. Due to the accessibility and reactivity of sulfur in different oxidation states, sulfur-containing molecules are now widely used in the fields of pharmaceuticals,6 agrochemicals,7,8 and materials.9 Contributions to such diverse fields highlight the importance of design and synthesis of novel organosulfur compounds.3Sulfur-containing heterocycles have been extensively used in organic semiconductors for electronic applications in organic light-emitting devices (OLEDs)10–12 and organic field-effect transistors (OFETs).12–16 Among these, thiophene-based systems are one of the most versatile conjugated materials due to their stability and ease of synthetic modification resulting in controllable optical properties, good charge mobility and low band gaps.11,17,18 As such, methods to increase the fluorescence efficiency and modulate the redox properties of thiophene building blocks have been widely explored to enhance the performance of such devices. Oxidation of the thienyl sulfur to S,S-dioxides was a strategy first proposed by Tanaka to lower the HOMO–LUMO gap in oligothiophenes.19 Barbarella was the first to synthesize a series of oligothiophenes with S,S-dioxide moieties and demonstrate effectiveness of this strategy in enhancing luminescence efficiency, in particular in the solid-state.20–22 Since then, numerous studies on the effect of oxidation on oligothiophenes have been carried out.23–25 While oxidation of heterocyclic sulfur atoms has shown promising results, the incorporation of a sulfur atom as a linker group or bridge provides an interesting design principle for conjugated materials. Electronic interactions between conjugated rings mediated by sulfur bridges may be possible and could result in interesting new electronic properties. Additionally, higher oxidation states are intriguing, and are not available for carbon-based (saturated and unsaturated) linkers where hypervalence is not possible.
To develop efficient solar cells, researchers have taken inspiration from nature, mimicking light harvesting (LH) complexes used in photosynthesis. LH complexes contain an antenna composed of an assembly of hierarchically ordered identical chromophores, which deliver energy (from absorbed photons) efficiently over a large distance, to the reaction center where electron and hole separation occurs. A clear understanding of the excited state properties and their organization is required in fabricating artificial systems. The magnitude of electronic coupling (degree of electronic interactions between neighboring molecules) plays an important role in dictating the rate of the electron and energy transfer processes that occur in these large assemblies.26,27 Efficient coupling can be induced in crystals,28 aggregates29,30 and covalent dimers31,32via through-space or through-bond interactions. In covalently linked systems, rates of energy and electron transfer can be precisely controlled. Through rational design, the linker (or bridge) can bring together the subunits in the correct orientation and distance, inducing characteristic properties compared to the corresponding monomers due to their closely packed π-systems. Additionally, the bridge can participate electronically through through-bond interactions.
In this perspective, we highlight two classes of sulfur-bridged bichromophoric dimers found in (1) organic π-conjugated materials and as (2) ligands for luminescent transition metal complexes (Fig. 1). In addition, we focus on the unique photophysical and photochemical phenomena that arise from different oxidation states. Herein, we review the most recent progress made by our group and others.
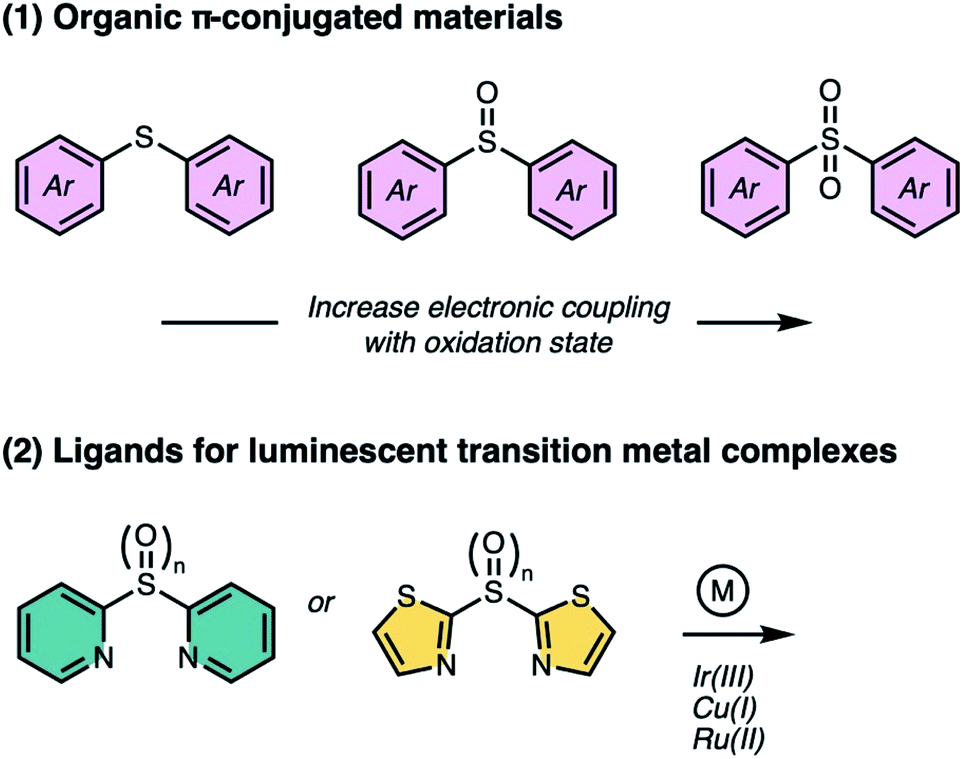 | ||
| Fig. 1 Introduction of sulfur bridges into (1) organic π-conjugated materials and (2) ligands, as a molecular design strategy for controlling electronic coupling and photophysical properties of luminescent transition metal complexes as a function of oxidation state (n = 0–2). | ||
Role of sulfur bridges in bichromophoric systems
Sulfur has been previously used as a non-conjugated bridge to covalently link identical chromophores to induce interchromophoric interactions. Depending on the position and type (sulfide versus disulfide) of linker, the degree of electronic coupling can be controlled by the orientation of the π-systems. For example, the photophysical properties resulting from the dimerization of boron difluoride (BF2) complexes using such bridges have been examined by several groups. In these cases, the dimers were often compared to their monomeric species (a single chromophore with a sulfur substituent) and the effect of oxidation on electronic coupling was not examined.Bröring bridged boron dipyrromethenes (BODIPYs) using sulfur to form dimers 1a–1c (Fig. 2a).29 Regioisomers 1a and 1c (α- versus β-linked, respectively) with sulfur bridges were used to study the effect of the linking position, and the distance between the subunits was varied by increasing the number of sulfur atoms in the bridge using compound 1b (sulfide versus disulfide). Both sulfide- and disulfide-bridged dimers 1a and 1b exhibit exciton coupling with splitting of the S1 level, observed as two peaks in the absorption spectrum (Fig. 2b). This splitting is more pronounced for sulfide 1a than disulfide 1b, due to the closer spatial arrangement of the BODIPY subunits. β-Sulfur-bridged dimer 1c exhibits less exciton splitting than 1a and 1b, which the authors attribute to increased distance between the subunits linked through the β-position compared to the α-position. Both sulfides 1a and 1c exhibit negative solvatochromic behavior in emission, with enhanced quantum yields in nonpolar solvents, suggesting an intramolecular charge transfer (ICT) process in the excited state under solvent-induced symmetry breaking. In contrast, the excited state behavior of disulfide 1b is not influenced by solvent polarity and intramolecular excimer formation is proposed for low quantum yields. Here, the position and length of the sulfur bridge is used to control the relative orientation of the BODIPY subunits which influences the intensity of exciton splitting.
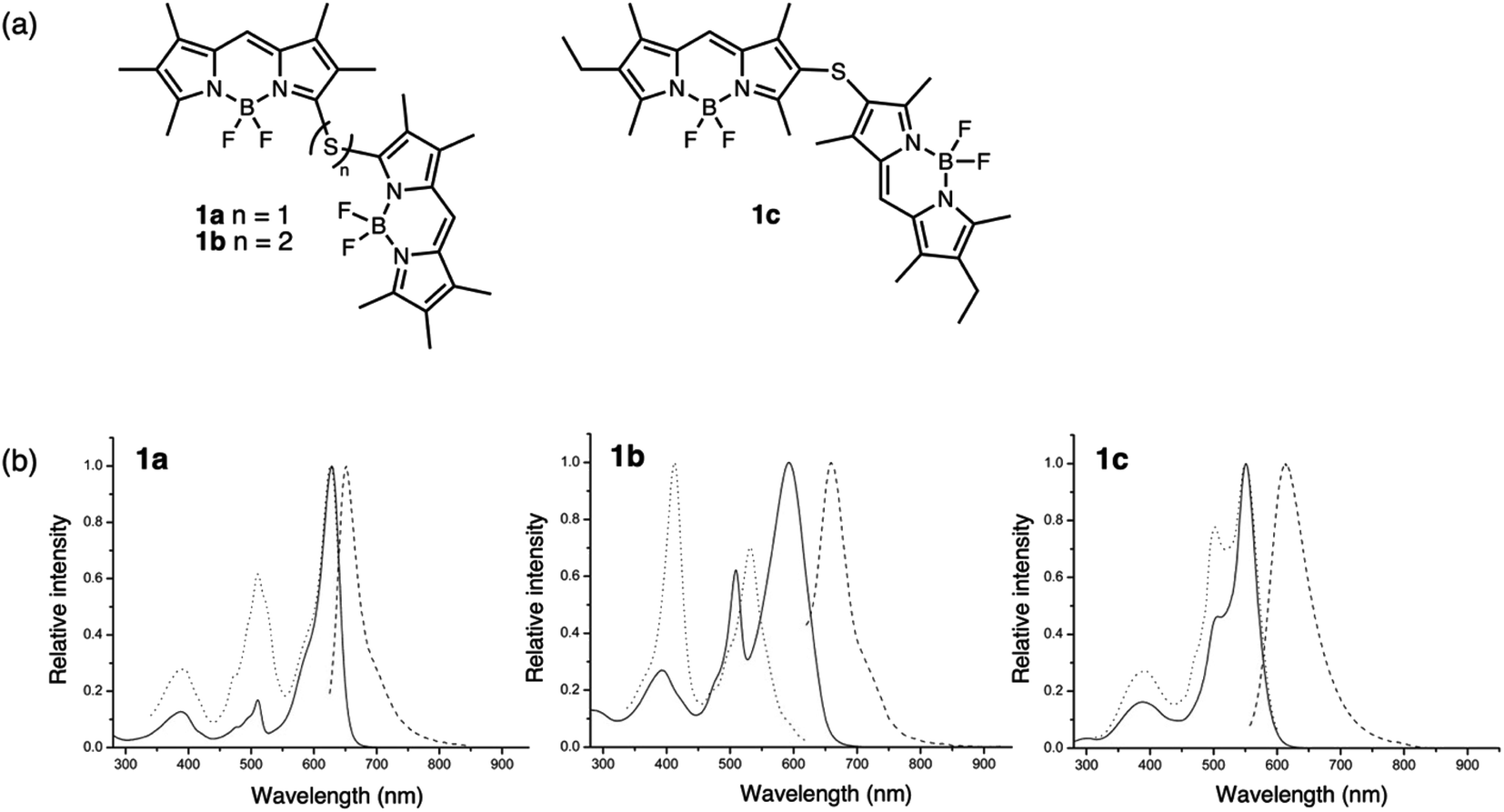 | ||
| Fig. 2 (a) Structures of α- or β-sulfide-bridged 1a and 1c and α-disulfide-bridged 1b BODIPY dimers reported by Bröring. (b) UV-vis absorption (solid line), emission (dashed line) and excitation (dotted line) spectra of the dimers in CH2Cl2. Adapted with permission from ref. 29. Copyright 2013 Wiley. | ||
Boron complexes linked together in the meso-position should show greater overlap of their π-systems than α- or β-linked dimers due to the increased dihedral angle θ which can influence the electronic properties (Fig. 3b). Maeda used a disulfide bridge to bridge two boron dipyrrolyldiketones in the meso-position to form dimers 2a and 2b (Fig. 3a).33 Although disulfide bonds show typical θ of 90° to 100°, smaller θ for more efficient π-overlap can be achieved using intramolecular bonding interactions between the subunits. A blue-shift with the appearance of a shoulder band in the absorption spectrum of dimer 2a (Fig. 3c) compared to its monomer 2c suggests the formation of “oblique H-aggregated” dimers, with dihedral angles of ca. 30° to 40° in solution, promoted by intramolecular π–π and N–H⋯F hydrogen-bonding interactions. A C–S–S–C dihedral angle of 39.4° was also estimated at the B3LYP-GD3BJ/6-31G(d,p) level of theory, in the optimized geometry, consistent with the observed absorption spectrum. Single crystal X-ray analysis of 2a reveals a dihedral angle of 90.3°, contrasting with the results of DFT calculations. However, two pseudopolymorphs were obtained of 2b: plates and needles, exhibiting dihedral angles of 86.7° and 39.1°, respectively. While both 2a and 2b (plate polymorph) showed typical C–S–S–C dihedral angles in the 90° to 100° regime, 2b (needle polymorph) corroborates theoretical studies. In this case, the arrangement of the boron dipyrrolyldiketones subunits was held using a disulfide-bridge in the meso-position and with the help of intramolecular interactions, unusually small C–S–S–C angles were observed.
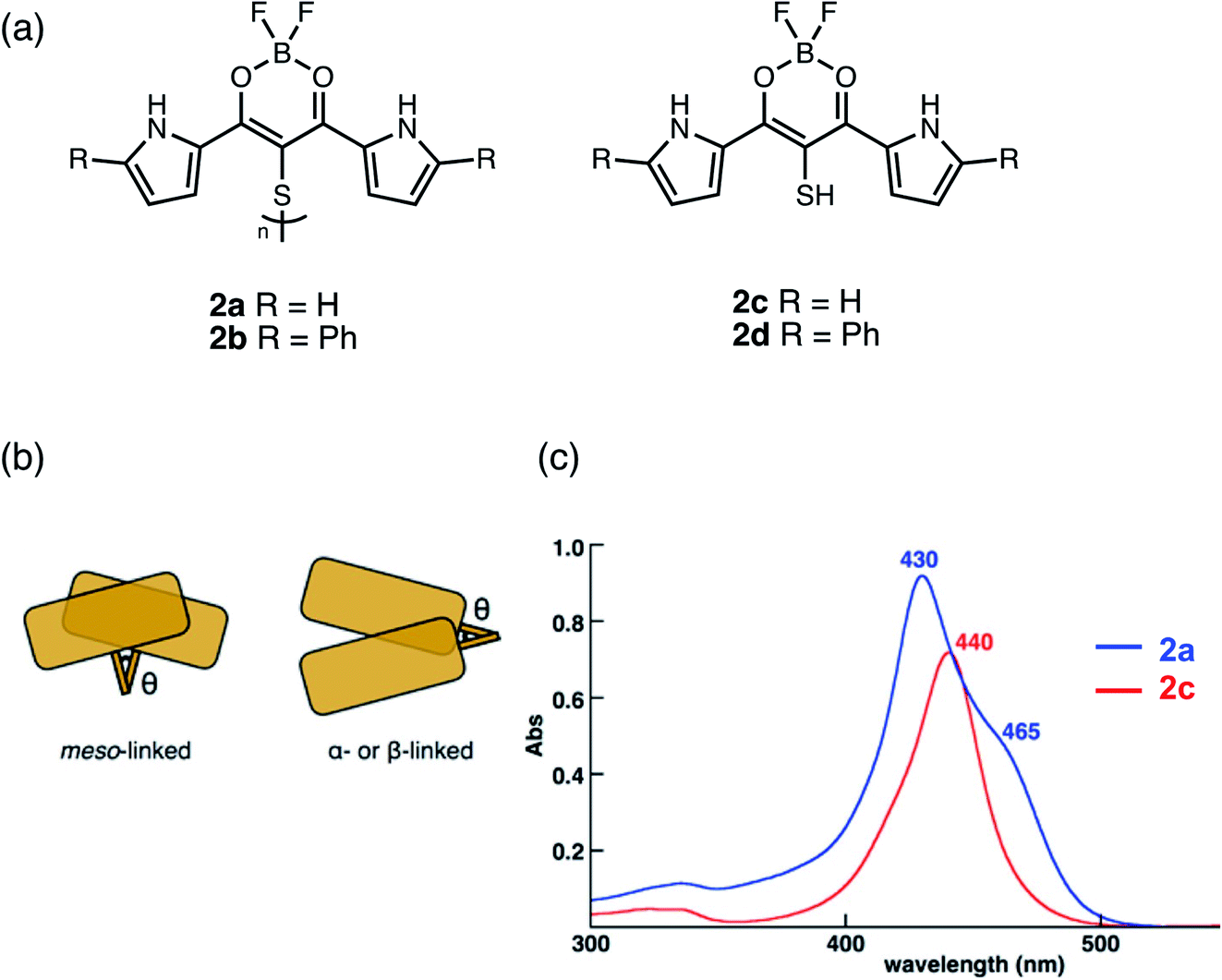 | ||
| Fig. 3 (a) Structures of meso-disulfide-bridged boron dipyrroyldiketone dimers 2a and 2b and monomers 2c and 2d reported by Maeda. (b) Cartoon representation showing how meso-linked chromophores have greater areas of overlap compared to α- or β-linked dimers. (c) UV-vis absorption spectra of dimer 2a (blue) compared to monomer 2c (red) in CH2Cl2. Adapted with permission from ref. 33. Copyright 2017 American Chemical Society. | ||
Sulfur-bridged porphyrins have been synthesized by Senge,34 but their photophysical properties were not examined in detail. Related to the aforementioned BF2 complexes are subporphyrins, a ring-contracted derivative of porphyrins developed by Kim and Osuka.35 Subporphyrin dimers linked by sulfide and disulfide bridges through the meso-position (3a and 3b, respectively) were synthesized and their photophysical properties were examined in detail (Fig. 4a). Sulfide 3a exhibits exciton coupling between the two subunits observed as a split Soret-like band and broad Q-bands in the absorption spectra (Fig. 4b). The red-shifted emission profile of 3a with a large Stokes shift (2670 cm−1) indicates a substantial structural change in the excited state. Comparatively, disulfide 3b does not exhibit exciton splitting and is non emissive (quantum yield of <0.01), suggesting that electronic perturbation by the individual subunits on each other is less than in 3a. Likewise, contrasting transient absorption (TA) profiles were observed for sulfide 3aversus disulfide 3b. The evolution and red-shift of the transient stimulated emission (SE) band in the TA spectrum of 3a (Fig. 4c) suggests effective electronic communication through the sulfur bridge whereas the TA spectrum of 3b (Fig. 4d) shows a rapid decay of the singlet induced absorption, quenching the excited state. The authors suggest a deactivation pathway involving rotation along the S–S axis, originally postulated by Bröring in regard to disulfide-bridged BODIPY dimer 1b.29
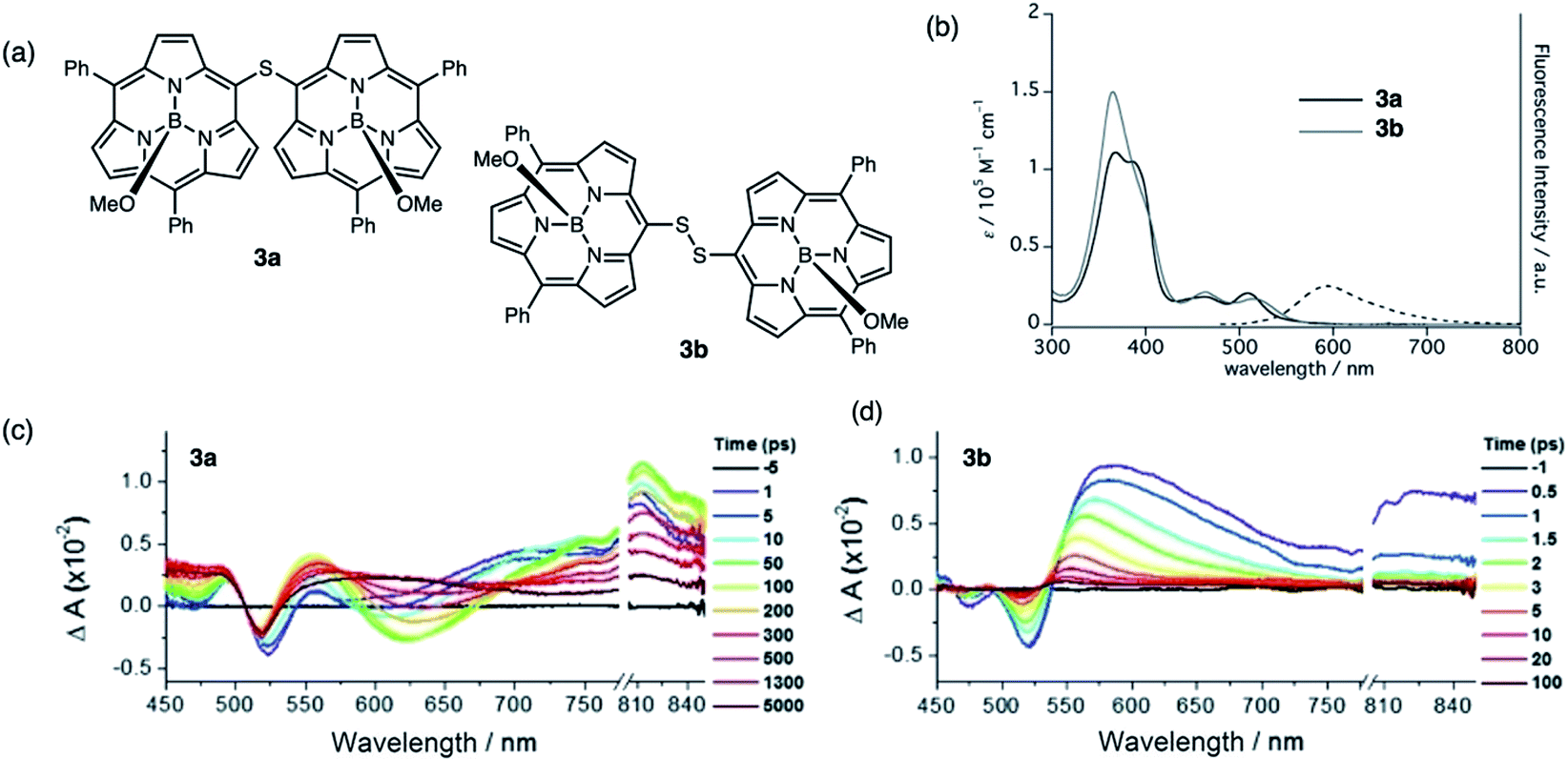 | ||
| Fig. 4 (a) Structures of sulfide-bridged 3a and disulfide-bridged 3b subporphyrin dimers reported by Kim and Osuka. (b) UV-vis absorption (solid) and fluorescence (dashed) spectra of 3a (black) and 3b (gray) in CH2Cl2. (c and d) Transient absorption spectra of 3a and 3b in toluene (λexc = 490 nm). Adapted with permission from ref. 35. Copyright 2016 Wiley. | ||
It has been demonstrated that incorporation of sulfide and disulfide bridges into dimeric systems can impose drastic changes to the electronic properties of the resulting materials. The sulfide bridge can bring the subunits closer together spatially, allowing for strong electronic coupling. While disulfide bridges tend to be more flexible, separating the subunits at greater distance, intramolecular interactions within the dimer can help promote efficient electronic coupling. Additionally, depending on the position of the linkage and thus the conformation of the subunits, the strength of exciton coupling can be controlled. The bridge serves as a non-conjugated linker to enable through-space interactions between the subunits, however the question of whether the sulfur bridge participates in the electronic states was not examined in these works.
Sulfone bridges in luminescent materials
The use of a sulfone (SO2) moiety as the linker has been extensively explored as an acceptor group in the design of luminescent materials exhibiting thermally activated delayed fluorescence (TADF).10,36 TADF is a radiative process from the first singlet excited state (S1). It requires a small singlet-triplet energy gap (ΔEST) to enable reverse intersystem crossing (RISC) from the triplet excited state to the singlet excited state. This approach efficiently harvests triplet excitons through RISC and is beneficial for applications in OLEDs. The tetrahedral geometry of sulfur provides the opportunity to limit the conjugation between donors and acceptors.37 In 2012, Adachi and co-workers prepared three compounds (4a–4c) with donor–acceptor-donor (D–A–D) structures.38 The diphenyl sulfonyl acceptor in the presence of N-containing donors leads to charge transfer (CT) character in the singlet excited state, resulting in a small ΔEST. The introduction of the tert-butyl group enhances the donor strength while the carbazole group raises the energy level of the first triplet excited state (T1). Therefore, 4c shows the smallest ΔEST and highest luminescence efficiency (0.80 at 423 nm) as a film doped into a bis[2-diphenylphosphino)phenyl] ether oxide (DPEPO) host. An OLED device was also fabricated with 4c as the emitting layer with an external quantum efficiency (EQE) of 9.9%. Replacing the tert-butyl group with methoxy substituents (4d) reduces ΔEST from 0.32 eV (4c) to 0.21 eV,39 and the maximum EQE was improved to 14.5% with an emission peak at 460 nm. Since then, various donor groups (Fig. 5) have been chosen to tune ΔEST and TADF efficiency,40–42 and features such as aggregation-induced emission (AIE) have also been imbued within the structure (4g and 4j) to improve efficiency in undoped OLED devices.41,43 In these cases, the sulfone bridge functions as the acceptor and the interaction between monomers (donor groups) was not explored.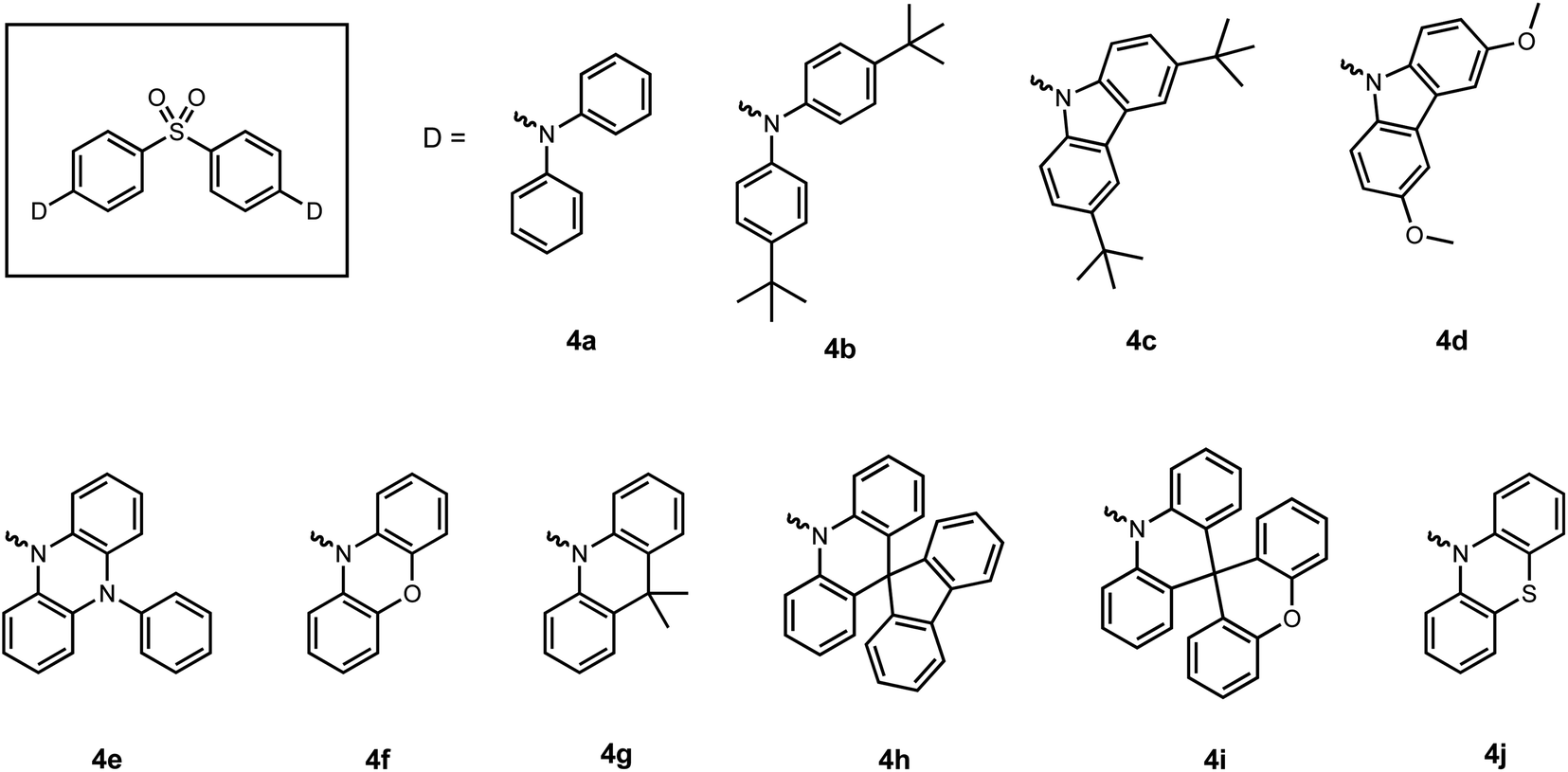 | ||
| Fig. 5 Structures of TADF emitters containing diphenyl sulfonyl acceptors reported previously. | ||
Luminescence enhancement by sulfur oxidation
Although sulfide- and sulfone-bridged systems have been used in novel photofunctional π-conjugated materials, systematic studies on how oxidation of the sulfur bridge alters the structural and electronic properties of these materials are scarce. Rodembusch and Silveira designed a class of asymmetric 5aSOn–5bSOnand symmetric 5cSOn–5eSOn dyes containing a (di)vinylsulfide bridge, which could readily be oxidized to the sulfoxide and sulfone (Fig. 6).44,45 Oxidation increases the electron-withdrawing capacity of the sulfone as an acceptor compared to the sulfide, inducing ICT. No apparent trend in photoluminescence quantum yields (PLQYs) was noted.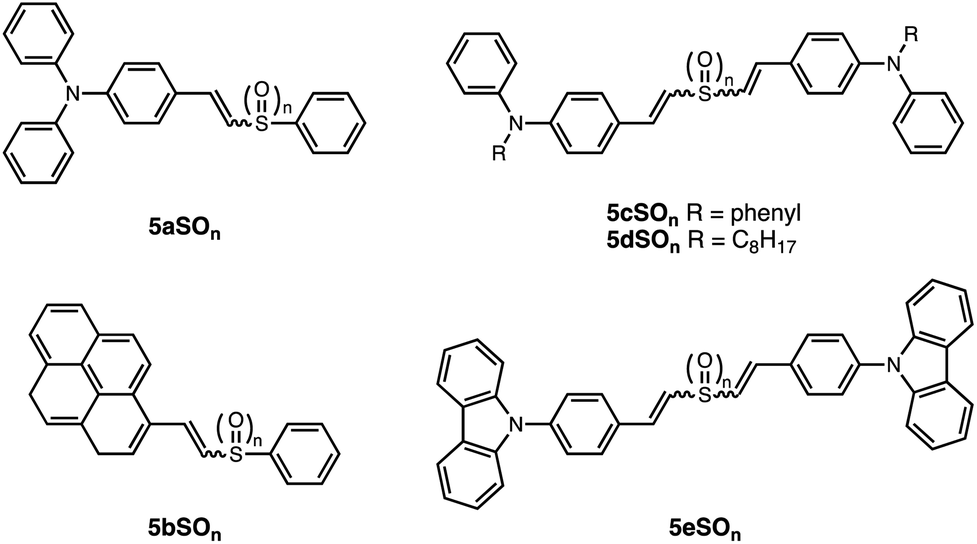 | ||
| Fig. 6 Structures of asymmetric 5aSOn and 5bSOn and symmetric 5cSOn to 5eSOn dyes containing a (di)vinylsulfide bridge (n = 0–2) reported by Rodembusch and Silveira. | ||
In 2013, our group reported how oxidation effects the photophysical properties of symmetric sulfur-bridged chromophore dimers. The chromophores studied were bithiophene (T2), terthiophene (T3), naphthalene (Nap) and pyrene (Pyr) (Fig. 7a).46 All four systems exhibited a systematic increase in their PLQYs upon oxidation of the sulfur (S) to the sulfoxide (SO) and sulfone (SO2) derivatives (Fig. 7). In most cases, the sulfoxide- and sulfone-bridged dimers are more emissive than the parent arenes (Fig. 7c). It was also found that CT character, indicated by solvatochromism in the fluorescence of the compounds, is more prevalent in the sulfoxide- and sulfone-bridged species. This approach provided a general and facile method to improve PLQYs in organic chromophores, which is anticipated to be beneficial for OLED applications.
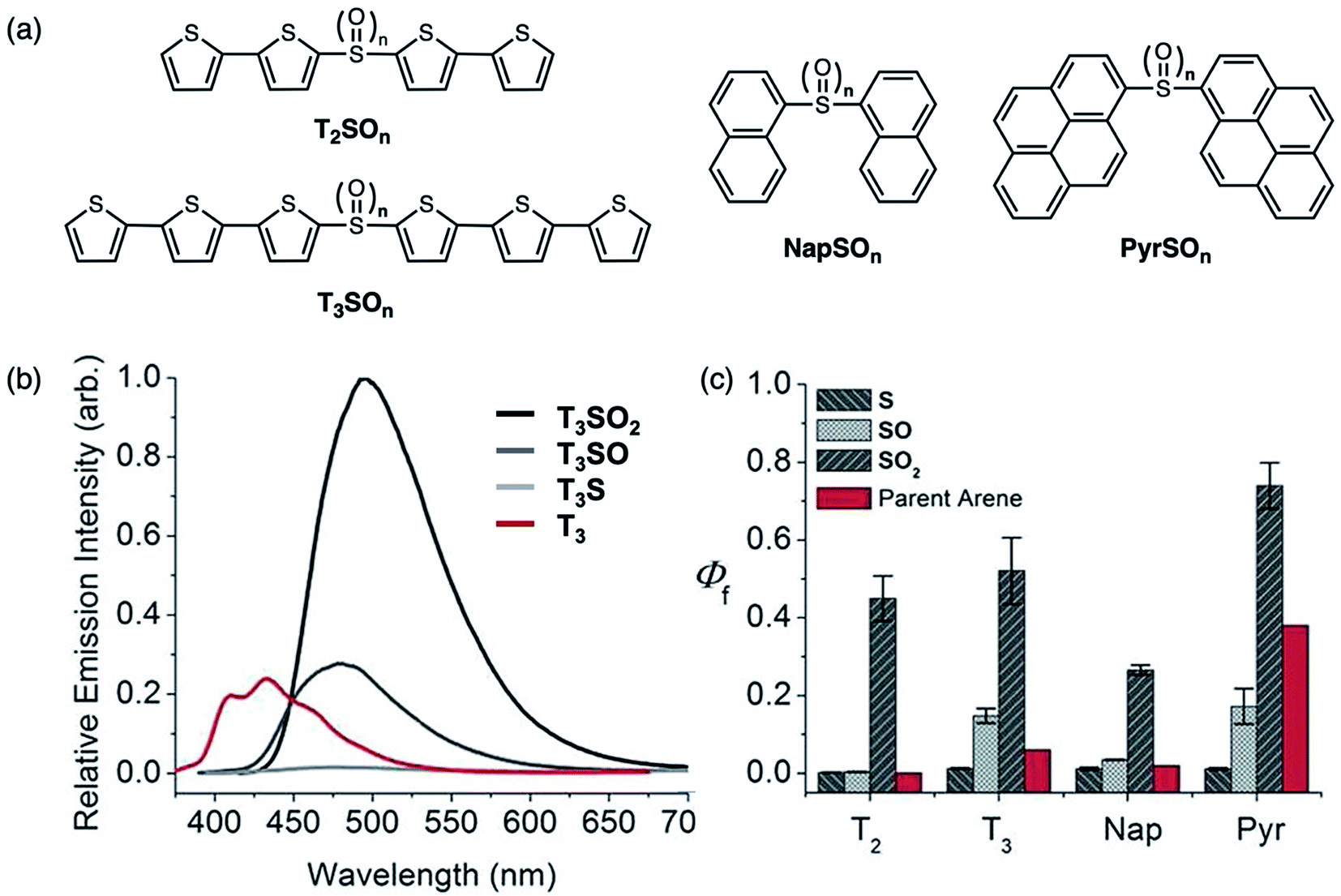 | ||
| Fig. 7 (a) Structures of sulfur-bridged bithiophene T2SOn, terthiophene T3SOn, naphthalene NapSOn and pyrene PyrSOn dimers (n = 0–2) reported by our group. (b) Emission spectra of T3SOn and T3 in CH2Cl2. (c) Photoluminescence quantum yields (Φf) of sulfur-bridged dimers with increasing oxidation states, in comparison to parent arenes. Adapted with permission from ref. 45. Copyright 2013 American Chemical Society. | ||
The mechanism of this photoluminescence enhancement in the terthiophene dimers T3SOn was further investigated using ultrafast TA spectroscopy and computational approaches.47 Methyl-terminated monomers T3SOnMe were synthesized as model compounds (Fig. 8a). Their fluorescence spectra are relatively invariant with increasing solvent polarity and excited state dynamics such as photoluminescence decays resemble those of parent arene T3. These results demonstrate that the CT character originates from the interaction between two T3 monomers and not from the linker itself. On the other hand, fast relaxation (∼5 ps) from the first excited singlet state (S1) to an intermediate singlet excited state 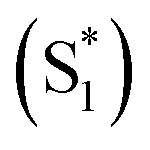 was exclusively observed in T3SOn dimers by femtosecond TA. This excited state exhibits solvent sensitivity and CT character. With the aid of DFT calculations,
was exclusively observed in T3SOn dimers by femtosecond TA. This excited state exhibits solvent sensitivity and CT character. With the aid of DFT calculations, 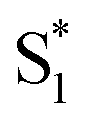 was identified as a symmetry breaking CT state with a net dipole, in which one of the T3 moieties planarizes, localizing the charge (Fig. 8b).
was identified as a symmetry breaking CT state with a net dipole, in which one of the T3 moieties planarizes, localizing the charge (Fig. 8b). 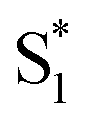 cannot couple efficiently with the triplet excited states due to the one electron nature of the spin–orbit operator. This leads to a decrease in ISC rate resulting in greater fluorescence efficiency compared to T3 (Fig. 8c). Computational studies reveal that the electron lone pairs on the sulfur atom plays a role in mediating intramolecular interactions. Specifically, they “shield” the electronic coupling between monomers by electrostatic screening and suppress relaxation to
cannot couple efficiently with the triplet excited states due to the one electron nature of the spin–orbit operator. This leads to a decrease in ISC rate resulting in greater fluorescence efficiency compared to T3 (Fig. 8c). Computational studies reveal that the electron lone pairs on the sulfur atom plays a role in mediating intramolecular interactions. Specifically, they “shield” the electronic coupling between monomers by electrostatic screening and suppress relaxation to 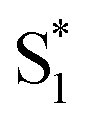 . By oxidizing the sulfur atom, lone pair screening is reduced, and the CT state can be stabilized, which leads to the enhanced PLQYs. Not only do these findings provide insight into the excited state dynamics of sulfur-bridged dimers, but they also open the door to design of efficient organic chromophores for OLEDs and solar energy conversion.
. By oxidizing the sulfur atom, lone pair screening is reduced, and the CT state can be stabilized, which leads to the enhanced PLQYs. Not only do these findings provide insight into the excited state dynamics of sulfur-bridged dimers, but they also open the door to design of efficient organic chromophores for OLEDs and solar energy conversion.
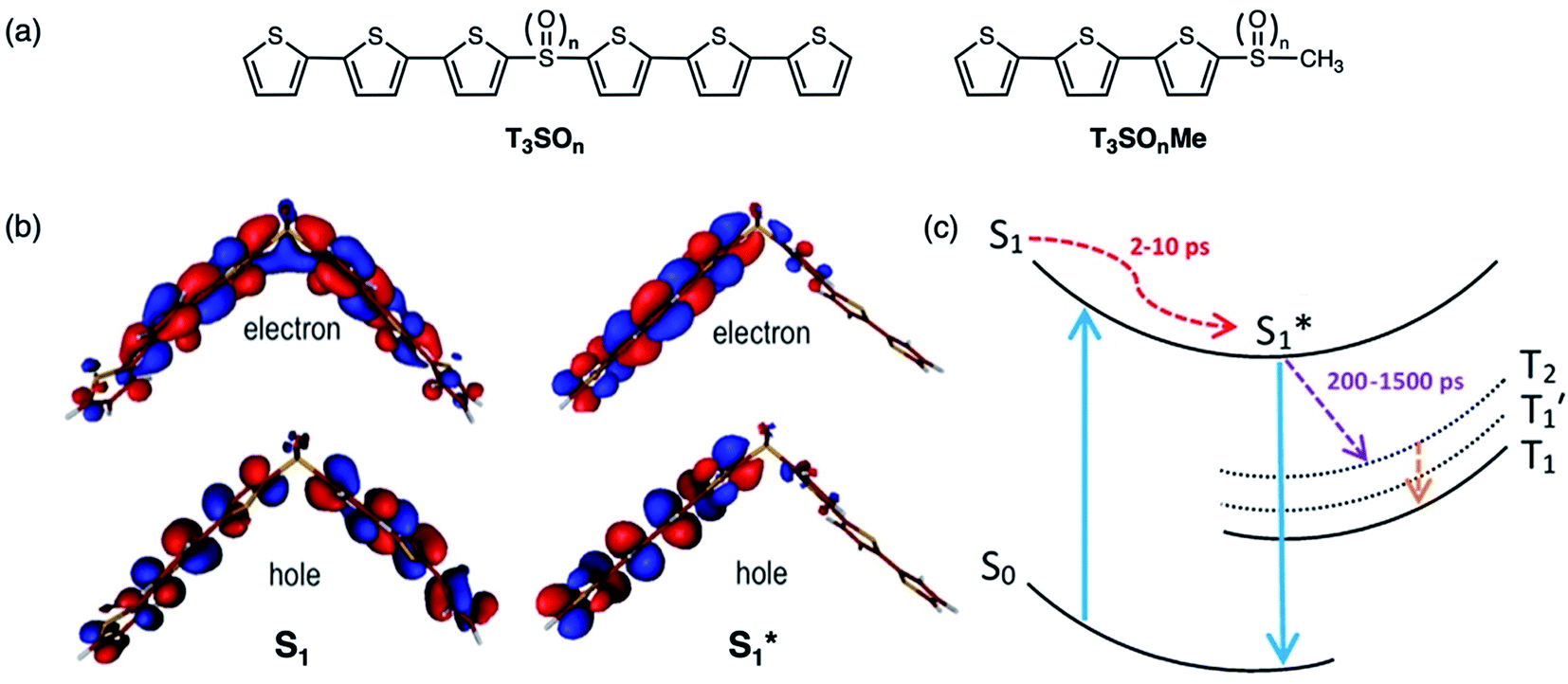 | ||
Fig. 8 (a) Structures of sulfur-bridged terthiophene dimers T3SOn and monomers T3SOnMe (n = 0–2) reported by our group. (b) Electron and hole natural transition orbitals (NTOs) of T3SOn singlet excited states S1 and 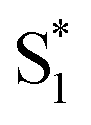 in CH2Cl2. (c) Simplified Jablonski diagram illustrating the excited-state relaxation in T3SOn. Adapted with permission from ref. 46. Copyright 2015 American Chemical Society. in CH2Cl2. (c) Simplified Jablonski diagram illustrating the excited-state relaxation in T3SOn. Adapted with permission from ref. 46. Copyright 2015 American Chemical Society. | ||
Unlike terthiophene, naphthalene does not suffer from ISC as the major non-radiative pathway due to its planarity and lack of thienyl rings. In order to further understand the origin of enhanced emission by sulfur oxidation and to validate the lone-pair screening theory proposed above, naphthalene dimers NapSOn (Fig. 9a) were investigated with computational simulations.48 In this case, the differences in PLQY were shown to be the result of energetically favorable non-radiative decay pathways available in NapS and NapSO, and not NapSO2 (Fig. 9b), indicating that the electron lone pairs on the sulfur atom are largely involved in such transitions. It has previously been proposed that pyramidal inversion of the excited state in aryl sulfoxides results in efficient internal conversion (IC) as the main non-radiative relaxation pathway.49 Calculations show that this inversion is thermally accessible for the NapSO system, but due to a large S0/S1 energy gap of the inversion TS in NapS and NapSO, non-radiative relaxation by IC was ruled out as the main deactivation pathway. We identified a conical intersection (CI) (a S0/S1 state crossing), which is energetically accessible upon photoexcitation, allowing for relaxation back down to the ground state without emission (Fig. 9c). The lack of electron lone pairs on the sulfur in NapSO2 blocks low-lying S0/S1 state crossings, resulting in much higher PLQYs compared to NapS and NapSO. Therefore, it can be concluded that the sulfur electron lone pairs dictate the excited state dynamics in sulfide and sulfoxide dimers, and sulfur oxidation is a general and novel approach for the design of strongly luminescent materials.
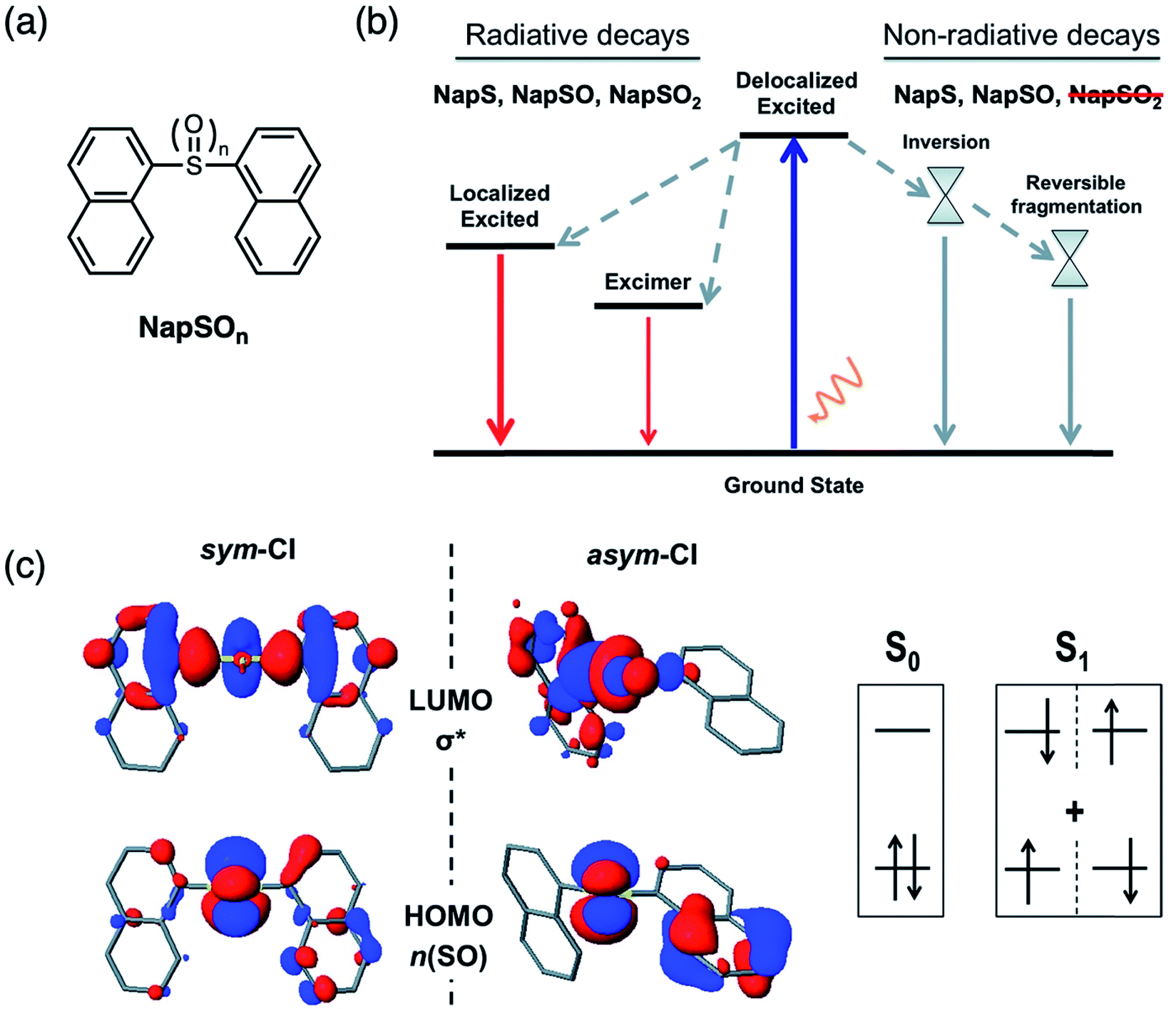 | ||
| Fig. 9 (a) Structures of sulfur-bridged naphthalene dimers NapSOn (n = 0–2) reported by our group. (b) Simplified Jablonski diagram illustrating the deactivation mechanisms in NapSOn. (c) Frontier molecular orbitals (n(SO) and σ*) at the symmetric and asymmetric S0/S1 conical intersection (CI) points for NapSO. Adapted with permission from ref. 47. Copyright 2017 Royal Society of Chemistry. | ||
After demonstrating the mechanism of fluorescence enhancement by sulfur oxidation in small molecules, we further employed this strategy on oligomeric and polymeric substrates (Fig. 10a).50 Other examples of polymeric materials which exhibit enhanced emission focus on the oxidation of thienyl sulfur in polythiophene systems.51–54 We designed thiophene-containing oligomers and polymers in which the main backbone contained alternating (ter)thiophene and sulfide or sulfone moieties. Similar to the trend observed in the small molecules T3SOn, OligoSO2 and PolySO2 exhibited higher PLQYs than OligoS and PolyS. As the number of repeat units increases, the HOMO–LUMO gap gets smaller, and this effect is more prevalent in the sulfide-bridged system compared to the sulfone-bridged system (Fig. 10b). This suggests that the sulfone bridge insulates the interactions between the individual thienyl units. Additionally, while the energy of the HOMO–LUMO gap is lowered upon sulfur oxidation due to the electron withdrawing nature of the sulfone group, the magnitude of the energy gap is also increased upon oxidation.
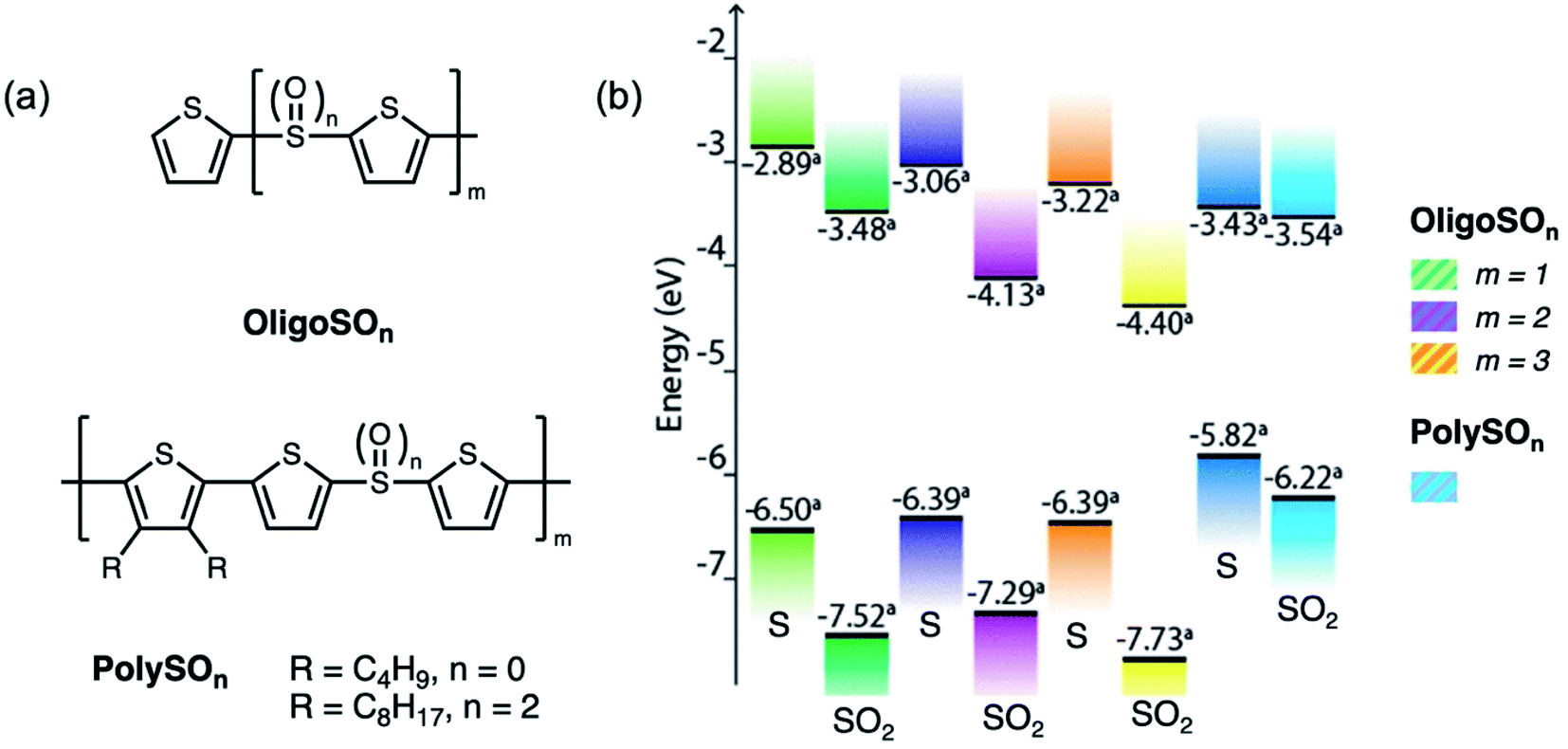 | ||
| Fig. 10 (a) Structures of thiophene-based oligomers OligoSOn and polymers PolySOn containing a sulfur bridge (n = 0, 2) reported by our group. (b) Calculated frontier energy levels obtained from cyclic voltammetry measurements and UV-vis absorption spectra. Adapted with permission from ref. 49. Copyright 2017 American Chemical Society. | ||
Recently, organic room temperature phosphorescence (RTP) has attracted much attention due to the ability to utilize long-lived (>ms) radiative triplet states, yielding potential applications in OLEDs, data encryption, and chemical and biological sensing and imaging.55–57 In order to increase phosphorescence efficiency, functional groups with lone pairs such as carbonyl and sulfone groups are commonly used, due to the capability of facilitating (n, π*) transitions from the first singlet excited (S1) to triplet excited states (Tn) to enhance ISC, according to El Sayed's rule.58,59 On the other hand, RTP lifetimes can be prolonged by utilizing the (π, π*) transition from the first triplet excited state (T1) to the ground state (S0).58,59 These excited state transitions can be controlled by both molecular structure and packing. Having demonstrated that sulfide bridges can enhance intersystem crossing by lone pair screening, we examined the effects of sulfur oxidation state on RTP.60 In the sulfur-bridged carbazole dimers CBZSOn (Fig. 11a), it was found that the single crystal packing modes are almost identical among three compounds with different oxidation states, providing a rare molecular model to study the electronic effects alone without simultaneously altering the intermolecular interactions. All three compounds also exhibit similar RTP lifetimes, largely due to the strong (π, π*) character localized at carbazole groups. Unlike in T3S, sulfur lone pairs in this case can participate in (n, π*) transitions due to the similar energy levels with the HOMO (Fig. 11c). Even though CBZSO has the strongest (n, π*) character indicated by its spin–orbit coupling (SOC) constant, CBZS shows the highest phosphorescence efficiency (Fig. 11b). It can be rationalized that the combination of the (n, π*) transition and electrostatic screening results in the sulfide having the highest phosphorescence. It is also worth noting that the oxygen lone pair orbitals in SO2 lie at much lower energies and do not contribute to SOC between low-lying triplet excited states and the ground state. Sulfone groups have been extensively explored in TADF and RTP materials38,41,61–64 while sulfides have been largely neglected. These findings provide a new perspective for the design of triplet-harvesting luminescent materials.
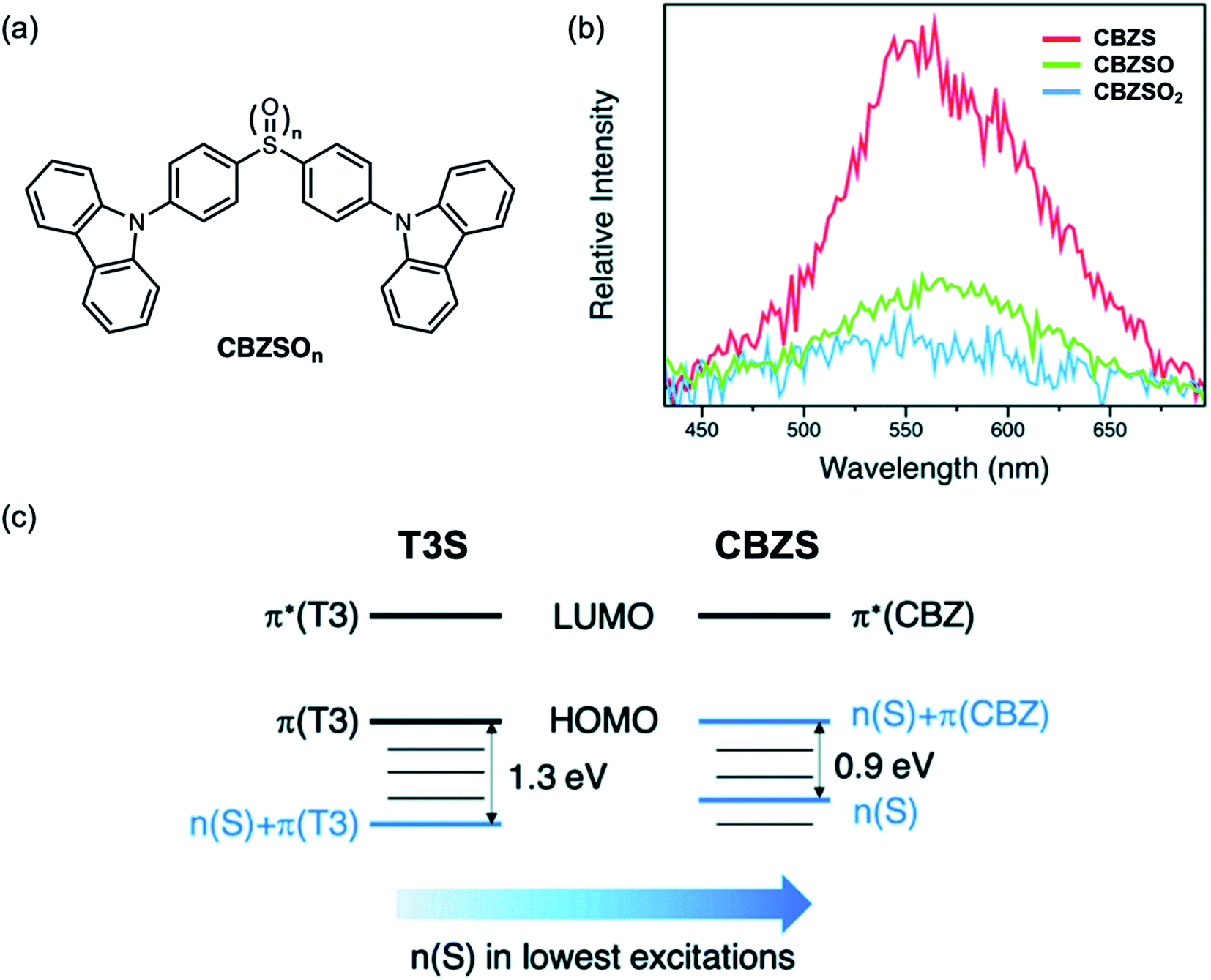 | ||
| Fig. 11 (a) Structures of sulfur-bridged carbazole dimers CBZSOn reported by our group. (b) Time-resolved photoluminescence spectra (60 μs to 110 ms time range) of CBZS, CBZSO and CBZSO2 crystalline samples under ambient conditions at 298 K. The intensity is corrected by fluorescence integration under the same excitation condition as time-resolved measurements and PLQYs. (c) Schematic comparison of frontier orbitals in terthiophene (T3S) and carbazole (CBZS) sulfur-bridged dimers. Adapted with permission from ref. 59. Copyright 2021 Royal Society of Chemistry. | ||
Oxidation-dependent photochemistry
In the search for additional chromophores to examine the effect of oxidation of the bridging sulfur on the photophysical properties, we found that when the substituent was anthracene, sulfur-bridged dimers AnSOn exhibited different photophysical trends than the aforementioned dimers.65 Although anthracene belongs to the same class of molecules (polycyclic aromatic hydrocarbons) as naphthalene and pyrene, AnSO and AnSO2 are less emissive than AnS, contrasting with the behavior previously observed in T2SOn, T3SOn, NapSOn and PyrSOn. The most intriguing aspect is that depending on the oxidation state, AnSOn undergoes different photochemical pathways.The photochemical products of AnSOn are shown in Fig. 12a. Irradiation of AnS with UV light (365 nm) results in no photochemical reaction. Under the same conditions, irradiation of AnSO results in the of loss the sulfoxide bridge, producing 9,9′-bianthracene (BA). The photochemical reaction of AnSO to BA can be monitored using absorption and emission spectroscopies (Fig. 12b and c). Upon conversion to BA, there is a significant increase in the PL intensity. Kinetic studies indicate that the formation of BA is sensitive to the presence of oxygen, and along with triplet sensitization studies using [Ru(bpy)3]2+, suggests the involvement of a triplet excited state. It is not yet known whether sulfur monoxide (SO) is generated from this photolysis reaction. Other examples of sulfur monoxide release from sulfoxides by photolysis have been observed in peri-substituted trisulfide-2-oxides reported by Grainger66 and in dinaphthothiepine bisimide sulfoxide reported by Fukui and Shiokubo.67 On the other hand, the generation of sulfur monoxide by thermal methods has been studied using episulfoxides since the 1960s.68–71 In recent years, both organic and main group precursors72,73 capable of sulfoxide extrusion reactions have been developed.
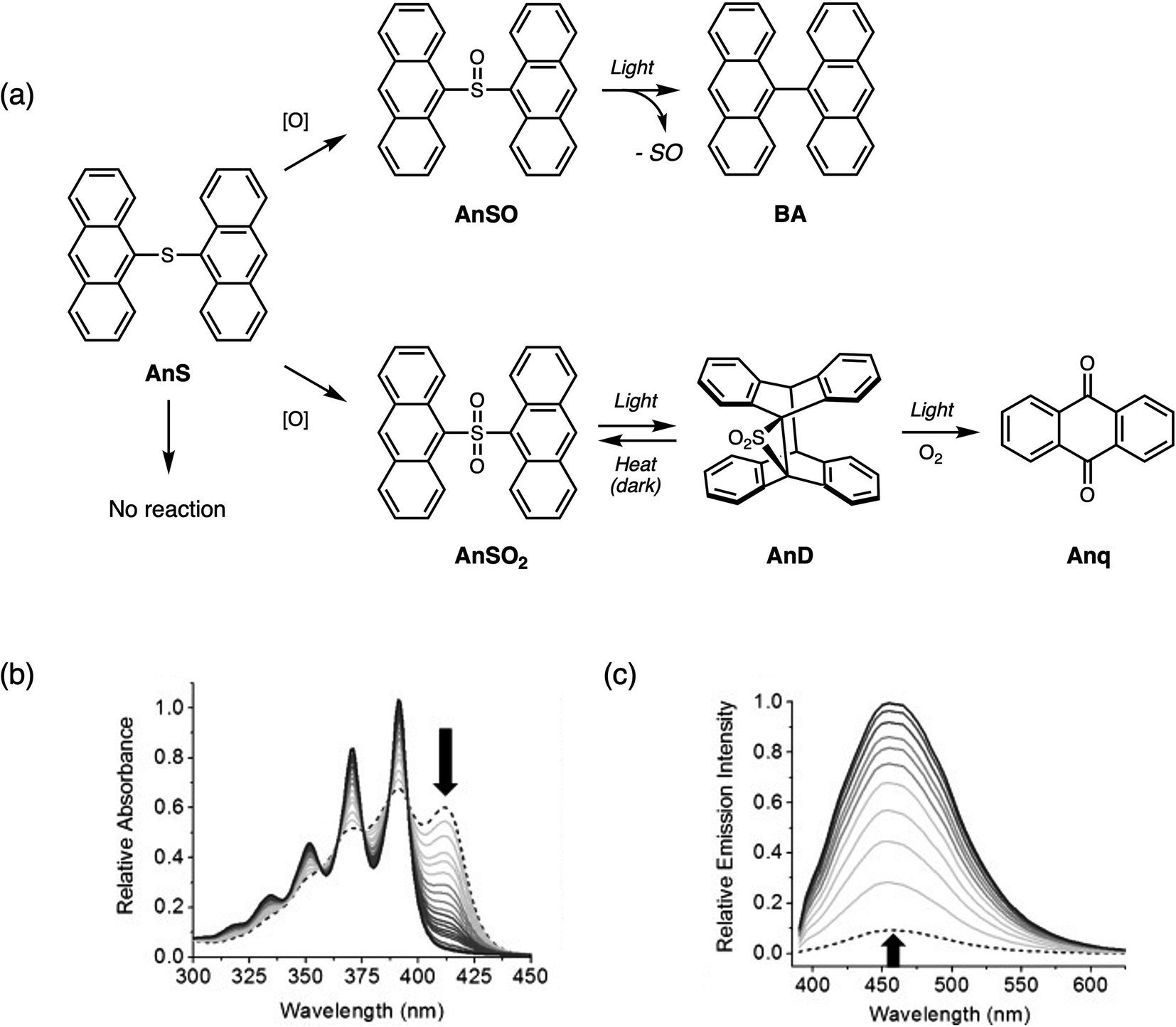 | ||
| Fig. 12 (a) Different photochemical pathways of sulfur-bridged anthracene dimers AnSOn (n = 0–2) depending on the oxidation state. Changes in the (b) UV-vis absorption and (c) emission spectra when AnSO is irradiated with UV light in CH2Cl2. Adapted with permission from ref. 64. Copyright 2013 Wiley. | ||
Irradiation of AnSO2 yields a dimer containing a three-membered episulfone ring AnD, which can thermally revert to the starting bridged species in the dark. The formation of anthracene dimers by [4 + 4] photodimerization (intermolecular) or photocycloisomerization (intramolecular) reactions has been known since 1867 and has been extensively studied and reviewed.74,75 With prolonged irradiation times (overnight) in the presence of oxygen, AnSO2 forms anthraquinone Anq.
We sought to investigate why AnS (unoxidized) is unreactive, whereas AnSO2 (fully oxidized) undergoes a photochemical reaction.76 Phenyl-terminated monomers AnSOnPh were synthesized as model compounds (Fig. 13a). The solvent-dependent PL lifetimes with a monoexponential decay profile of AnS and AnSPh (Fig. 13b) suggests an excited state with CT character, which undergoes rapid non-radiative relaxation, faster than monomeric anthracene An. TA studies identify a population transfer between the singlet and triplet excited states, indicative of ISC. Since the singlet state has appreciable CT character, the energy of the singlet is lowered in solvents of increasing polarity, which brings this state closer in energy to the triplet, facilitating rapid ISC. In the sulfone-bridged systems, phenyl-terminated AnSO2Ph also exhibits solvent-dependent monoexponential PL decay similar to AnS and AnSPh. In this case, there is no observation of a triplet feature in the TA spectrum, suggesting IC and not ISC, as the main nonradiative deactivation pathway, quenching PL. However, AnSO2 exhibits biexponential PL decays in all solvents (Fig. 13c). The TA data suggests that a portion of the singlet population in AnSO2 relaxes back to the ground state via IC similar to AnSO2Ph, but another portion ends up in a long-lived emissive state (>10 ns), which is likely the precursor for the photocycloaddition reaction. Unlike sulfur-bridged terthiophenes T3SOn, the main difference between AnS and AnSO2 is the participation of the sulfur lone pair orbitals. Computational results suggest that the absence of lone pairs in the sulfone-bridge eliminates possible n(S) → σ* and n(S) → π* contributions to ISC and IC. The simplified relaxation dynamics are summarized in the Jablonski diagram in Fig. 13d. Increasing the oxidation state of the sulfur bridge can, in turn, tune the electronic structure of the linker atom which can modulate the relaxation pathways to enable divergent photochemistry.
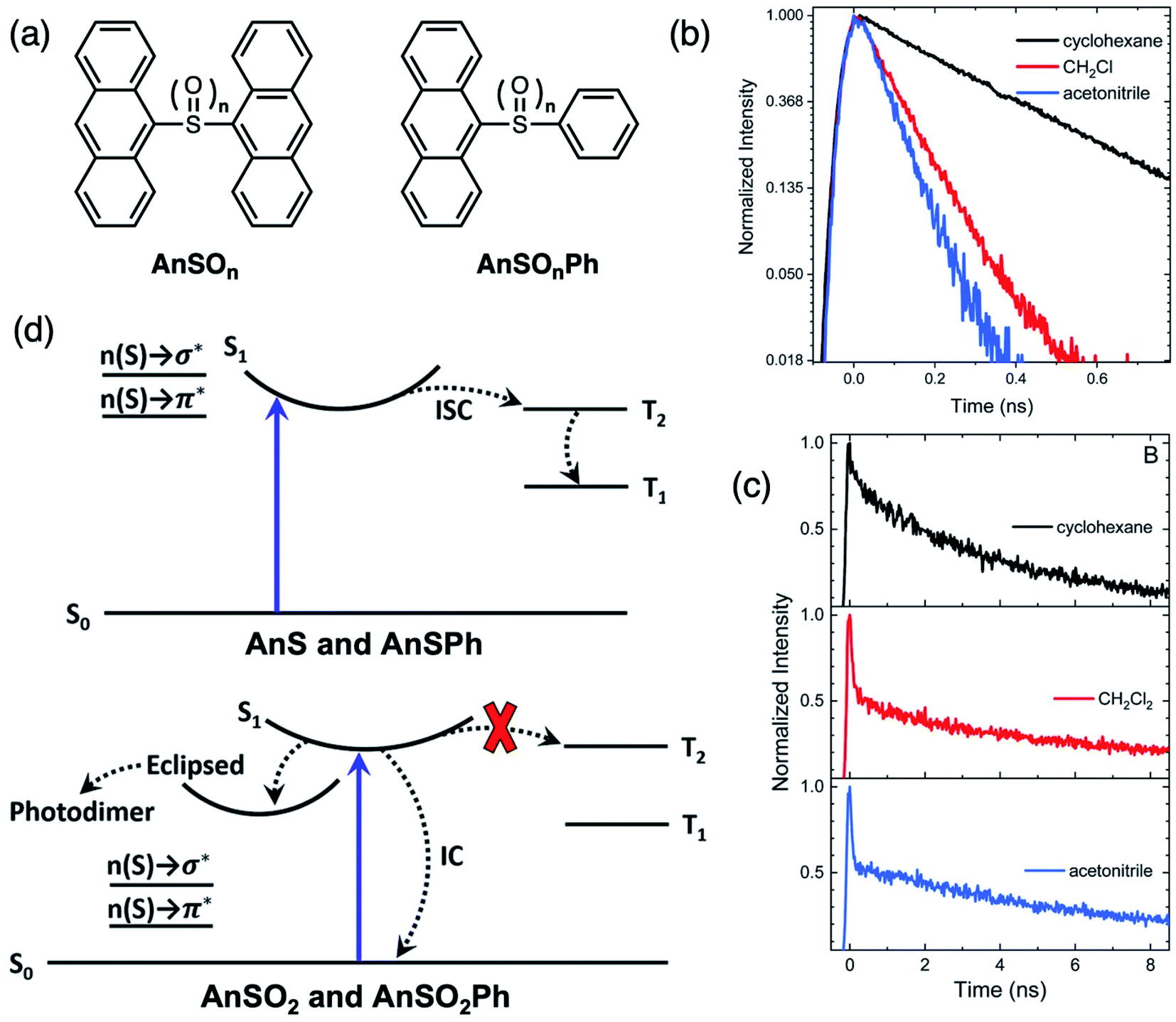 | ||
| Fig. 13 (a) Structures of sulfur-bridged anthracene dimers AnSOn and monomers AnSOnPh (n = 0, 2) reported by our group. (b) Normalized PL time traces on a natural log scale for AnS in cyclohexane (black), CH2Cl2 (red), and acetonitrile (blue) showing that PL lifetime decreases with increasing solvent polarity. (c) Normalized PL time traces integrated over all wavelengths for AnSO2 in cyclohexane (black), CH2Cl2 (red), acetonitrile (blue). (d) Simplified Jablonski diagram illustrating how mixing of the n(S) → σ* and n(S) → π* linker orbitals enable intersystem crossing (ISC) in the sulfide compounds, whereas the lack of mixing in the sulfone prevents ISC from occurring (red X). Adapted with permission from ref. 75. Copyright 2019 Royal Society of Chemistry. | ||
Application of sulfoxide elimination photochemistry
Our group has leveraged the photochemical conversion of sulfoxide-bridged anthracene dimers, which are initially weakly emissive, to strongly emissive bianthracene photoproducts, as potential anti-counterfeiting materials for authentication technology. The dyes were used to formulate photoactive inks that can be used for patterning as the reaction is controlled spatially using light.77 Inks were prepared by doping the dye into a poly(N-vinylcarbazole) (PVK) host at 1 wt%. Additionally, we showed that luminescent patterns can be read using the camera of a smartphone illustrating the ease-of-use for this method, as it reduces the need for expensive and complex instruments required for validation.The first demonstration by our group used AnSO-based inks to design multidimensional QR codes.77 These 2D barcodes technically display 3D information, where barcodes provide two dimensions of positional information and a third dimension corresponding to an intensity of 0 or 1. Thus, by varying the intensity of a specific position from 0 to 1, the data density that can be encoded in a 2D barcode can be increased. Paper substrates were coated with the ink and exposed to 400 nm light through a shadow mask. The shadow mask used comprised of a 2D data matrix encoding the phrase “Hello World” layered on top of a variable transmittance pattern in the form of a radial gradient. After UV exposure, the printed image contained information regarding the data matrix while displaying a background which varies in intensity. The color intensity was extracted using a smartphone to demonstrate the capability of creating multidimensional barcodes.
The main disadvantage of covert luminescent patterns is their ease of reproducibility due to the vast number of dyes with similar emission characteristics (color) available. Thus stimuli-responsive inks, which respond to external factors such as light, have shown to be promising candidates for next-generation materials used in anti-counterfeiting applications. Following our first application of AnSO-based dyes, our group then designed and synthesized both a red and green emissive derivative that could be used in conjunction with AnSO (blue emissive) in order to achieve full color tuning.78 Substitution at the 10,10′-position of the dianthracene core with triarylamine (TAAAnSO) and phenothiazine (PTZAnSO) groups afforded green and red emissive photoproducts, respectively, after irradiation (Fig. 14a). The unique optical properties of the individual sulfoxides allow for different rates of photoconversion, which can be initiated using specific irradiation wavelengths. This approach constitutes a new method for full color tuning since the final emission color of a mixture of the sulfoxides depends on (i) the mixing ratio, (ii) duration of light exposure and (iii) the irradiation wavelength. As a proof-of-concept, an inexpensive, publicly available smartphone application (app) called “Colorimeter” was used to determine the emission color of a mixture containing all three dyes after 1 minute of 400 nm light irradiation, and the corresponding CIELAB coordinates were extracted using the software (Fig. 14c). The final coordinates after light exposure of a specific duration can then be compared against a “true color” to determine the authenticity of a product. Additionally, the ability to visually monitor the “activation process” of this concept is novel to this system, as the mechanism of activation is based on the unique photochemistry of the sulfoxide-bridged dimers.
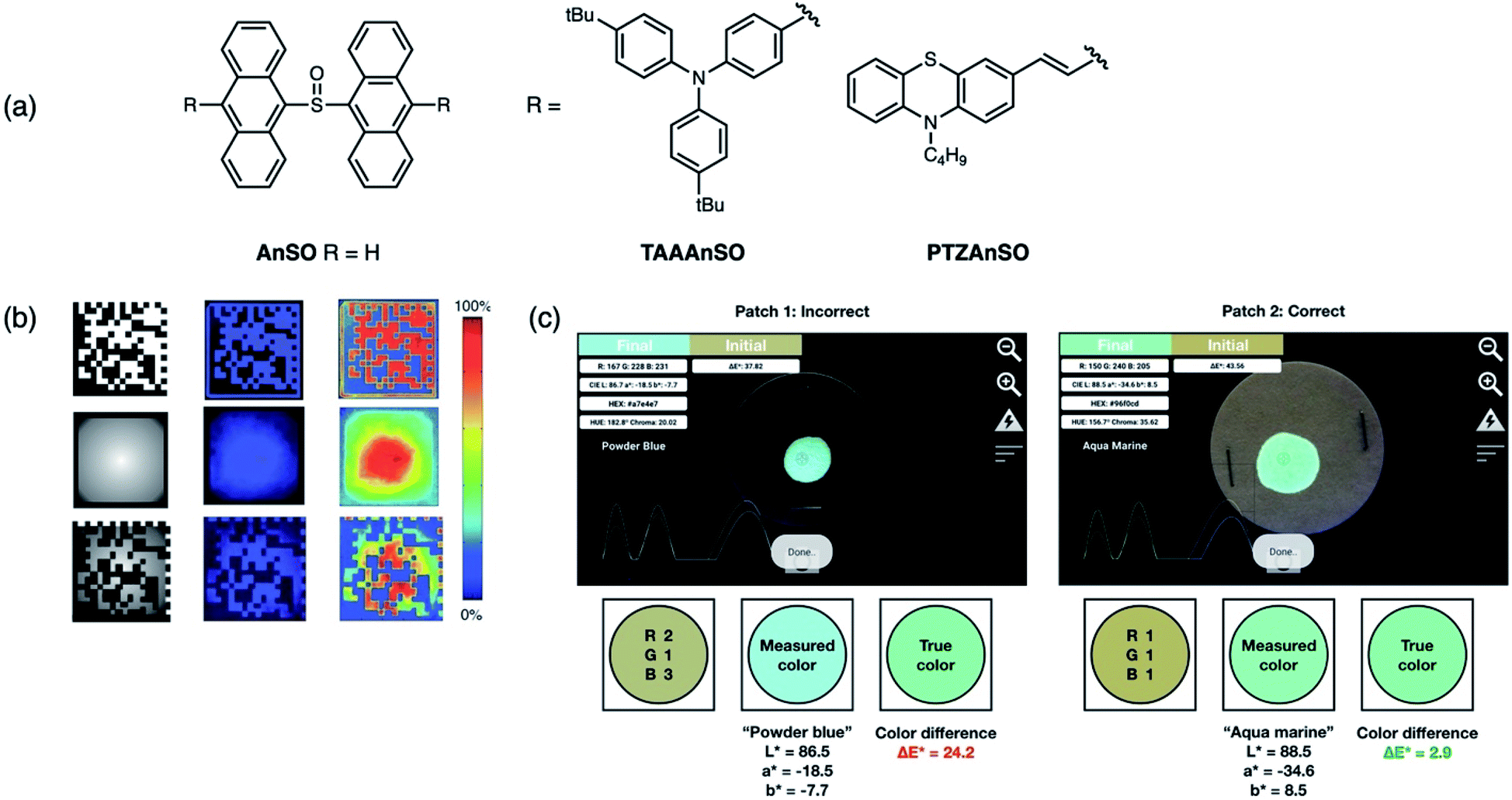 | ||
| Fig. 14 (a) Structures of sulfoxide-bridged anthracene dimers AnSO, TAAAnSO, PTZAnSO reported by our group used to prepare inks for proof-of-concept demonstrations. (b) Using a two-stage mask that contains both a 2D data matrix and variable transmittance pattern (radial gradient to the center), a multidimensional fluorescent barcode can be patterned using UV light. The color intensity is measured using a smartphone camera. Adapted with permission from ref. 76. Copyright 2016 Wiley. (c) Software interface of Colorimeter used to extract CIELAB coordinates. These coordinates are compared to a “true color” to determine authenticity. Adapted with permission from ref. 77. Copyright 2019 Royal Society of Chemistry. | ||
Sulfur-bridged ligands for Ir(III), Cu(I) and Ru(II) complexes
Drawing inspiration from the photophysical trends and emergence of novel photochemical pathways that were dependent on the oxidation state in symmetric sulfur-bridged π-conjugated materials, our group designed new diimine ligands for luminescent metal complexes (Fig. 15). The introduction of substituents such as electron-donating groups (EDGs) or electron-withdrawing groups (EWGs) at, for example, the R1 and/or R2 position of 4,4′-bipyridine (bpy), can be used to modify the photophysical properties of the resulting metal complex. By incorporating a sulfur group in between the diimine groups, we introduced a second degree of tuning. The ability to systematically alter the sulfur centre provides a convenient approach for adjusting electron density on the ligand, which can then be used to fine tune the excited state electronics of luminescent metal complexes. The following section describes the use of sulfur-bridged dipyridyl (DPS) and dithiazolyl (tzS) ligands in Ir(III), Cu(I) and Ru(II) complexes, and how altering the oxidation state of the sulfur bridge on the ligand results in changes in the photophysical and/or electrochemical properties of the metal complex.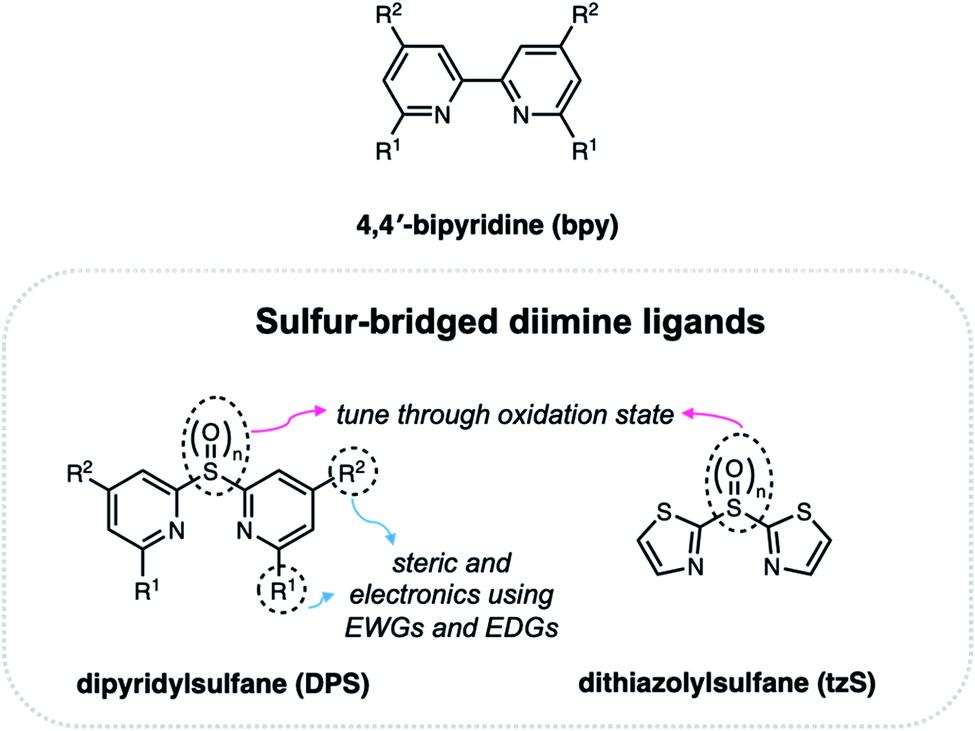 | ||
| Fig. 15 Structure of 4,4′-bipyridine ligand (bpy) compared to sulfur-bridged diimine ligands based on dipyridylsulfane (DPS) and dithiazolylsulfane (tzS). | ||
Since Ir(III) complexes are known for their high efficiency and wide scope of tunability in both OLEDs79,80 and light-emitting electrochemical cells (LEECs),81,82 sulfur-bridged dipyridyl ligands were first installed on Ir(III) metal centers (Fig. 16a).83 Upon oxidizing the bridging sulfur atom from sulfide (Ir-DPS, Ir-Me-DPS) to sulfone (Ir-DPSO2, Ir-Me-DPSO2), the first reduction of the oxidized complex is shifted to a more positive potential (−1.26 and −1.73 V for Ir-DPSO2 and Ir-Me-DPSO2, respectively) compared to the sulfide (−2.16 and −2.40 V for Ir-DPS and Ir-Me-DPS, respectively). This can be attributed to the electron-withdrawing nature of the sulfoxide and sulfone which leads to stabilization of the LUMO. The sulfide and sulfoxide (Ir-DPSO, Ir-Me-DPSO) complexes show almost identical fine-structured spectra with blue-green emission (Fig. 16b), indicative of significant ligand-centered (3LC) contributions to the lowest triplet excited state. This was further confirmed by solvatochromic behavior in the emission spectra and computational studies. Ir-DPSO2 and Ir-Me-DPSO2 on the other hand, exhibit pronounced metal-to-ligand and ligand-to-ligand charge transfer character (3MLCT/3LLCT) with broad and red-shifted emission bands. Complex Ir-Me-DPSO2, with methyl substituents in the 4,4′-positions, shows a slight blue-shift (15 nm) compared to Ir-DPSO2, which is consistent with previous reports.84 PLQYs in deaerated CH2Cl2 were <0.01–0.08, slightly lower than that of the Ir(III) complex with bipyridine ligand (0.14) in deaerated CH3CN, possibly due to increased flexibility of the ligand. Varying the oxidation state of the sulfur centre on these bridged dipyridyl ligands enables control over triplet excited state electronics and demonstrates “two-level” tuning over the emission color.
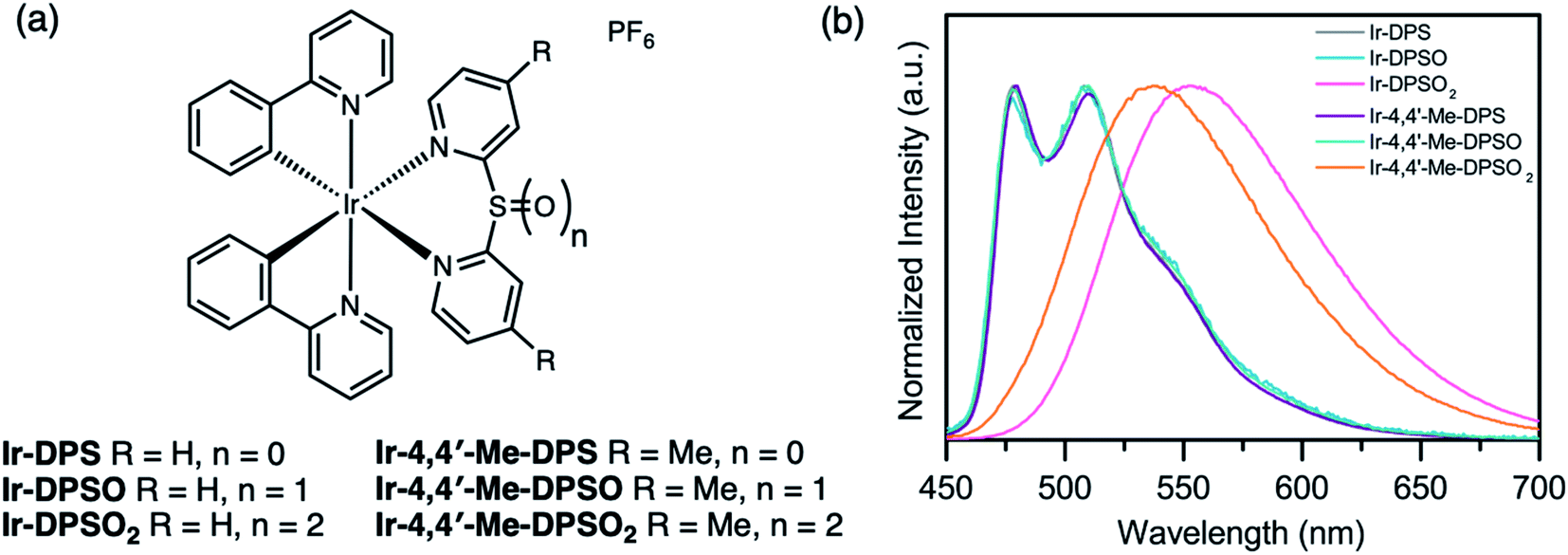 | ||
| Fig. 16 (a) Structures of Ir(III) complexes Ir-DPSOn and Ir-Me-DPSOn (n = 0–2) reported by our group. (b) PL spectra of the Ir(III) complexes in CH2Cl2 solution (λex = 390 nm). Adapted with permission from ref. 82. Copyright 2017 American Chemical Society. | ||
Emissive Cu(I) complexes have attracted great attention in the past decade due to the earth abundance of this metal and lower cost compared to Ir. Furthermore, the capability of Cu(I) complexes to undergo an additional radiative pathway via TADF significantly decreases the photoluminescent lifetime and enhances the quantum efficiency, which is beneficial for device efficiencies in OLEDs and LEECs.85 In the ground-state, the Cu(I) center adopts a pseudotetrahedral geometry with a d10 configuration. Upon photoexcitation, the complex undergoes a fast (<1 ps) Jahn–Teller distortion to a more flattened d9 geometry, opening up nonradiative decay pathways.86–88 Therefore, rational design of the ligand by introducing steric hindrance is crucial to inhibit this undesired distortion in order to maintain a tetrahedral geometry. Surprisingly, not only does sulfur oxidation alter excited state electronics, but it also provides a variety of interesting binding modes and complex geometries with changes in steric constraints (Fig. 17). Without any steric constraints, Cu-DPS and Cu-DPSO2 are mononuclear species with a distorted tetrahedral geometry at the Cu(I) center, retaining the typical N,N binding mode of the diimine ligand. However, with the introduction of methyl substituents on the 6- and 6′-position of the DPS ligand, complexes Cu-Me-DPS and Cu-Me-DPSO2 are bimetallic species with distorted trigonal geometries at the Cu(I) center, bridged by the bi[2-(diphenylphosphino)-phenyl) ether ligand (DPEphos). Upon sulfur oxidation, Cu-Me-DPSO2 was also found to exist as a bimetallic species, in this case with Me-DPSO2 as the bridging ligand through a N,O coordination mode. When the steric constraints are further increased to phenyl groups, monometallic species are formed again. Cu-Ph-DPS adopts a distorted trigonal geometry with the diimine ligand bound through only one pyridyl nitrogen. The sulfone group in Cu-Ph-DPSO2 enables a N,O binding mode, forming a tetrahedral geometry at the Cu(I) metal center. All six complexes are weakly emissive in solution but more strongly emissive in the solid state with PLQYs up to 0.20. Interestingly, complexes Cu-DPS, Cu-DPSO2, Cu-Me-DPS, Cu-Me-DPSO2 and Cu-Ph-DPSO2 all exhibit TADF while Cu-Ph-DPS shows triplet ligand-centered (3LC) emission.
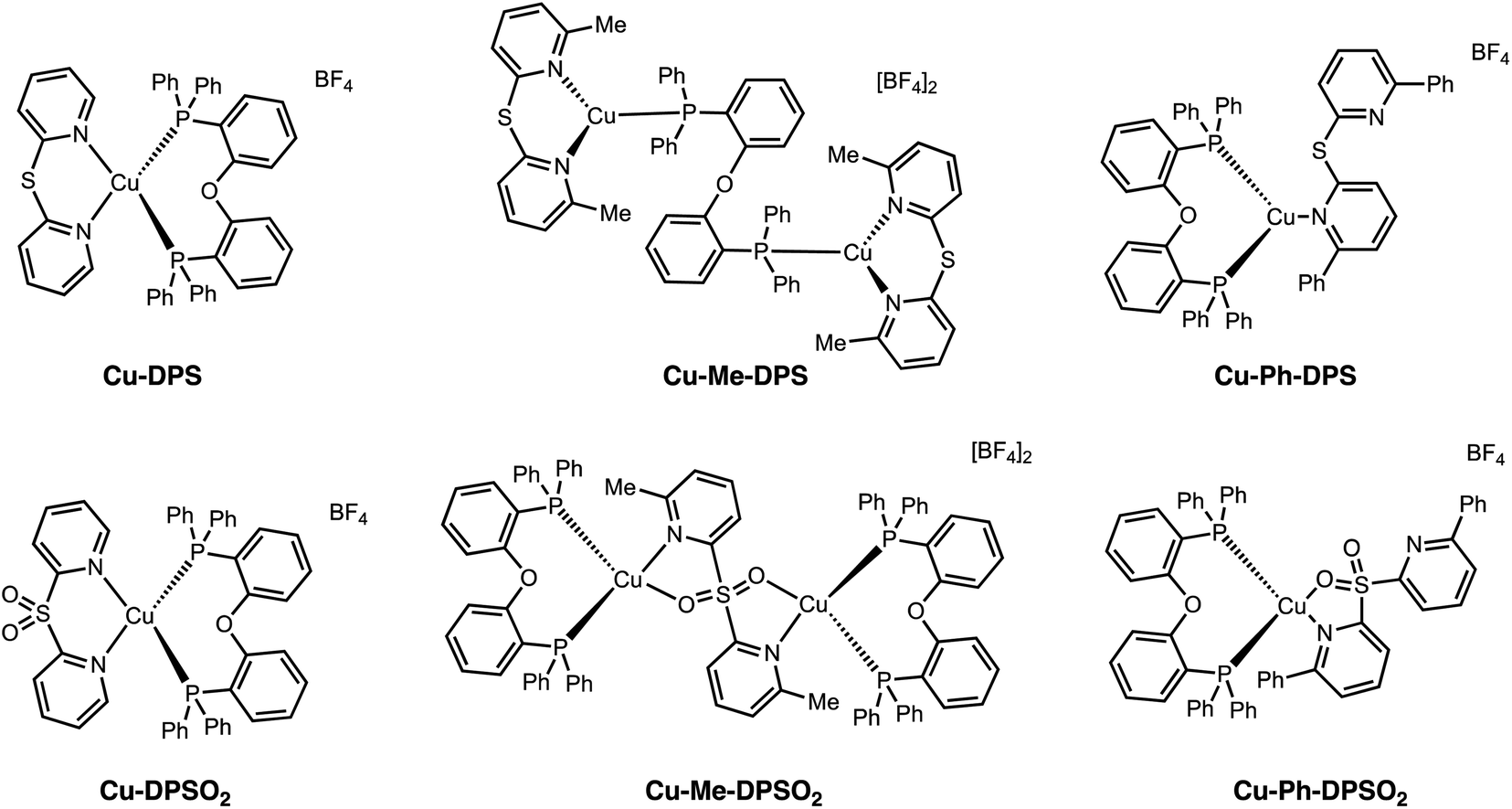 | ||
| Fig. 17 Structures of Cu(I) complexes Cu-DPSOn, Cu-Me-DPSOn and Cu-Ph-DPSOn (where n = 0 or 2) reported by our group. | ||
Interestingly, when sulfoxide-bridged diimine ligands are used, Cu-DPSO gives yellow-green luminescence with the same binding mode as Cu-DPS (Fig. 18a).89 However, Cu-Me-DPSO with methyl substituents at the 6- and 6′-positions on the diimine ligand adopts a N,O binding mode in the single-crystal structure. It is also worth noting that in solution or bulk solid samples of Cu-Me-DPSO, only the typical N,N binding mode can be found, confirmed by NMR spectroscopy and powder X-ray diffraction. Surprisingly, unique phase-dependent and thermochromic emission was also observed (Fig. 18b and c). Cu-Me-DPSO was initially isolated as an amorphous powder (a-Cu-Me-DPSO), which is yellow emissive (542 nm) under a 365 nm UV hand lamp. Upon heating to 180 °C, a crystalline species (c-Cu-Me-DPSO) forms and emits orange fluorescence (658 nm). c-Cu-Me-DPSO also exhibits reversible thermochromic emission, whereby the complex undergoes a large emission color change from orange at room temperature (25 °C) to yellow (556 nm) at −196 °C (Fig. 18d). It is speculated that at room temperature, c-Cu-Me-DPSO adopts a more flattened geometry in the excited state, which gives rise to orange emission (Fig. 18e). However, at low temperatures or in the amorphous phase, the geometry of the excited state is closer to tetrahedral, leading to higher energy emission.90–93 Emissive thermochromic Cu(I) complexes have been largely limited to Cu(I) halide clusters,94,95 and this work provides a new avenue for the design of novel stimuli-responsive Cu(I) complexes.
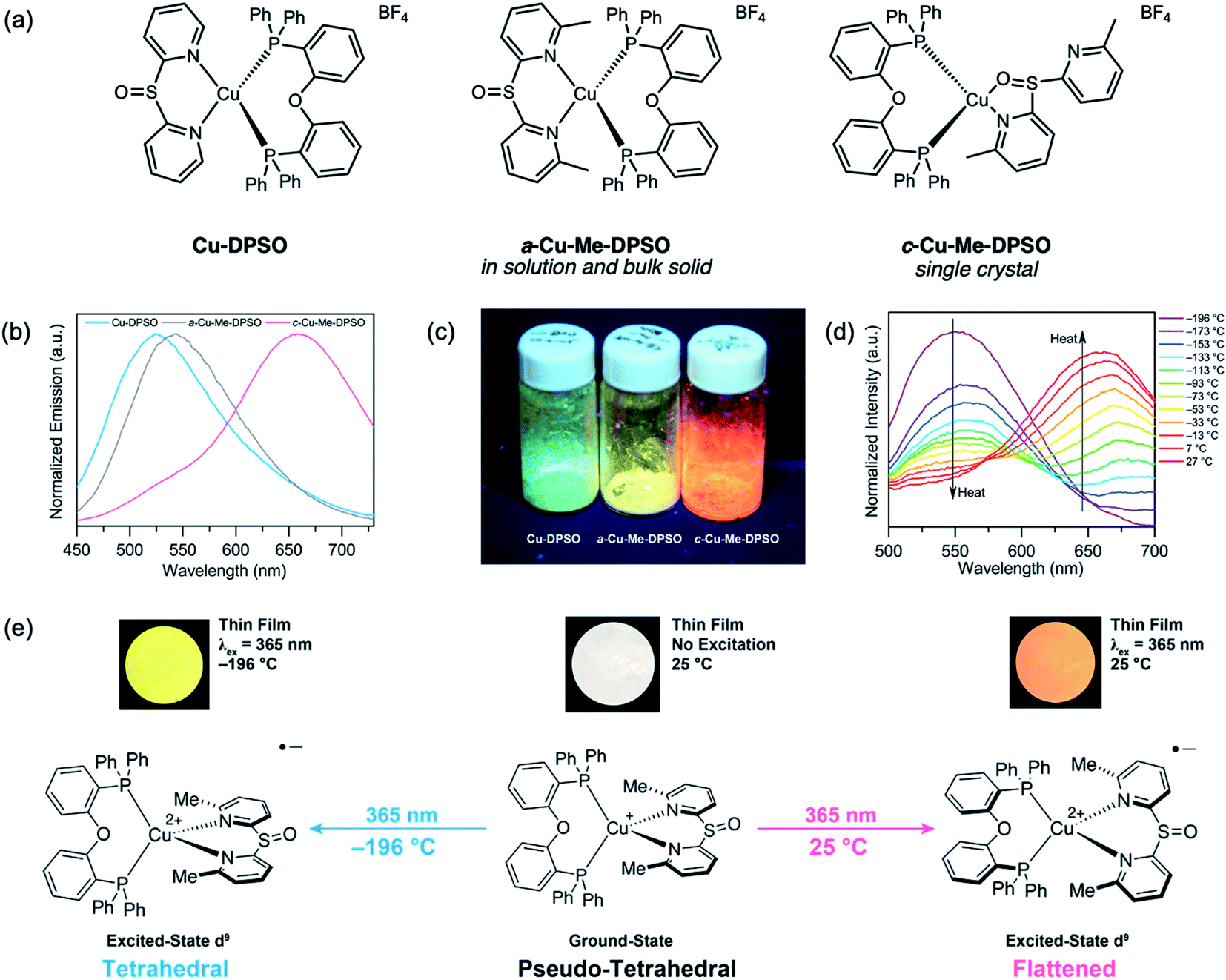 | ||
| Fig. 18 (a) Structures of Cu(I) complexes Cu-DPSO and Cu-Me-DPSO reported by our group. (b) Room-temperature solid-state emission spectra of Cu-DPSO, a-Cu-Me-DPSO and c-Cu-Me-DPSO (drop-cast from MeOH, λex = 390 nm). (c) Photographs of the Cu(I) complexes under UV excitation. (d) Variable-temperature solid-state emission spectra of c-Cu-Me-DPSO from −196 to 27 °C (drop-cast from MeOH, λex = 390 nm). (e) Proposed excited-state conformational changes resulting in the thermochromic emission of c-Cu-Me-DPSO. Adapted with permission from ref. 88. Copyright 2018 American Chemical Society. | ||
More recently, sulfur-bridged bithiazole ligands were installed on both Cu(I) and Ru(II) complexes (Fig. 19a).96 Both Cu-tzS and Cu-tzSOn are weakly emissive in solution as radiative decay is often quenched in Cu(I) complexes by Jahn–Teller distortions, but exhibit strong emission as doped PMMA thin films since the degree of distortion is reduced when confined in a polymer matrix. The oxidation state of the sulfur-bridged bithiazole ligand does not appear to influence the photophysical properties of the Cu(I) complexes in this case. The Ru(II) complexes are non-emissive in solution and exhibit weak emission as both neat solid and doped PMMA thin films at room temperature (293 K). The emission intensity of the Ru(II) complexes as neat thin films and as frozen CH2Cl2 solutions increases when the samples were cooled to 77 K. On the other hand, the emission profiles at 77 K reveal that the nature of the Ru(bpy)2(tzSOn) emitting state depends on the oxidation state. Ru(bpy)2(tzS) has structured emission, representative of a 3MLCT emitting state compared to Ru(bpy)2(tzSO2) which displays a Gaussian-like emission band that is characteristic of a 3MC emitting state. In these examples, the oxidation state of the ligand was shown to tune the electrochemical properties of both the Cu(I) and Ru(II) complexes (Fig. 19b). The redox potentials of complexes bearing sulfur-bridged ligands indicate smaller HOMO–LUMO gaps than for those with sulfone-bridged ligands. This suggests that altering the oxidation state of a bridging sulfur group on diimine ligands not only can influence photophysical properties but can be a useful handle for fine-tuning electrochemical properties of metal complexes as well.
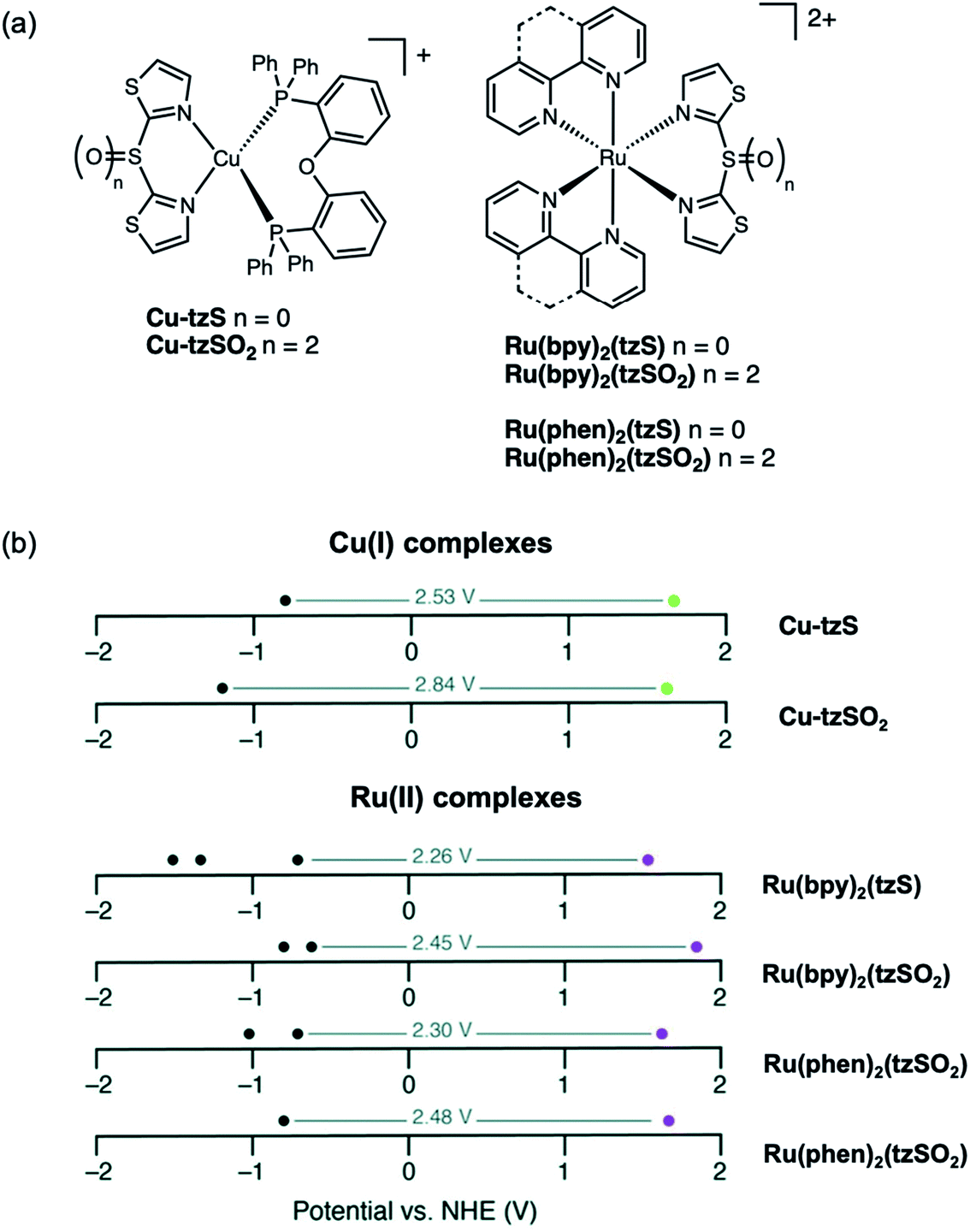 | ||
| Fig. 19 (a) Structures of Cu-tzSOn and Ru(bpy)2(tzSOn) and Ru(phen)2(tzSOn) complexes reported by our group (n = 0, 2). (b) Redox potentials of Cu(I) and Ru(II) complexes with reductive (black dots) and oxidative (green or purple dots) potentials illustrated. Adapted with permission from ref. 95. Copyright 2019 Royal Society of Chemistry. | ||
Conclusions and outlook
Over the past decade, sulfur groups have been used to covalently link chromophores together to prepare symmetric dimers in π-conjugated materials. The identity of the chromophore can be altered to achieve distinct photophysical properties. Sulfur can serve as a passive linker, enabling through-space interactions between the subunits to induce electronic coupling. Perhaps the more intriguing aspect of sulfur as a bridging atom, is the accessibility of higher oxidation states. The lone pair electrons of sulfur can be used to form polarized bonds while maintaining nearly identical geometry. The ability to precisely control the electronic interactions from within the molecule without inducing large conformation changes, or without having to change the external environment (solvent polarity), make these scaffolds particularly interesting to study, and may also be useful for the design of functional materials.The reports summarized have focussed on examining the CT contribution of excited state as a function of oxidation state. Careful selection of the chromophore in both small molecule and macromolecular systems furnished dimers that could exhibit desirable properties such as enhanced PLQYs or room temperature phosphorescence (RTP). The screening effect of the lone pairs on the sulfur was shown to influence the amount of CT character in the singlet excited state. Oxidation to the sulfoxide and sulfone also resulted in a stabilizing effect on the CT state. This in turn affects ISC to the triplet state. Depending on whether it is desired to create or suppress these CT states, the efficiency of the radiative process from either the singlet or triplet state can thus be tuned. This concept of using oxidation states to modulate excited state dynamics in dimeric system can also promote photochemistry in anthracene-based systems, by enabling the formation of long-lived excited states that are presumed to be the precursors for photoproducts.
In addition to organic materials, sulfur bridges were used as a design strategy to prepare “two-level” tunable diimine ligands for luminescent metal complexes. Sulfur-bridged dipyridyl and dithiazoyl ligands were used to synthesize Ir(III), Cu(I) and Ru(II) complexes. In addition to peripheral functionalization of the dipyridyl ligand, oxidation of the sulfur bridge could also result in various complex geometries with the oxygen as the coordinating atom. Oxidation of the bridging sulfur can alter the emission properties such as color and lifetime, as well as give rise to new photophysical phenomena such as thermally activated delayed fluorescence (TADF) or thermochromism.
Beyond the examples presented in this perspective, it is anticipated that other processes that require precise control of chromophore–chromophore interactions should benefit from this design strategy such as photon upconversion97 or intramolecular singlet fission.98 For example, the extent of coupling between chromophores plays a critical role in intramolecular singlet fission in bridged chromophore dimers. The introduction of a sulfur bridge and subsequent oxidation in these systems should allow for modulation of processes including exciton formation and recombination. On the other hand, oxidation of a bridging sulfur in ligands is useful beyond altering the emission characteristics of luminescent metal complexes. These ligands can also be used to control electronic communication between two metal complexes,99 and in turn, effect electron transfer rates, which is crucial for developing systems capable of artificial photosynthesis100 or photocatalytic reactions.101 Likewise, the use of a sulfur bridge can covalently tether the photosensitizer (PS) and catalyst together, and oxidation can potentially optimize these light-stimulated processes. Incorporating sulfur bridges as a molecular design strategy will benefit the discovery of novel organosulfur compounds by offering an opportunity to fine tune optical, electronic and redox properties through systematic oxidation.
Author contributions
J. Y. and Z. X. contributed equally to the writing of this manuscript with input and direction from M. O. W.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council (NSERC) of Canada for funding (Discovery and CREATE NanoMat).References
- G. D. Considine, in Van Nostrand's Scientific Encyclopedia, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2005 Search PubMed.
- R. Hell, M. S. Khan and M. Wirtz, in Plant Cell Monographs, ed. R. Hell and R.-R. Mendel, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 17, pp. 243–279 Search PubMed.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- R. Cecil and J. R. Mcphee, in Advances in Protein Chemistry, ed. C. B. Anfinsen Jr, M. L. Anson, K. Bailey and J. T. Edsall, Academic Press, New York and London, 1959, vol. 14, pp. 255–389 Search PubMed.
- S. F. Betz, Protein Sci., 1993, 2, 1551–1558 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed.
- P. Devendar and G.-F. Yang, in Sulfur Chemistry. Topics in Current Chemistry Collections, ed. X. Jiang, Springer International Publishing, Cham, 2019, pp. 35–78 Search PubMed.
- C. Lamberth, J. Sulfur Chem., 2004, 25, 39–62 CrossRef CAS.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- Y. Li, J.-Y. Liu, Y.-D. Zhao and Y.-C. Cao, Mater. Today, 2017, 20, 258–266 CrossRef.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 RSC.
- T. Baumgartner, J. Inorg. Organomet. Polym. Mater., 2005, 15, 389–409 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- G. Barbarella, M. Melucci and G. Sotgiu, Adv. Mater., 2005, 17, 1581–1593 CrossRef CAS.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- K. Tanaka, S. Wang and T. Yamabe, Synth. Met., 1989, 30, 57–65 CrossRef CAS.
- G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, L. Antolini, O. Pudova and A. Bongini, J. Org. Chem., 1998, 63, 5497–5506 CrossRef CAS.
- G. Barbarella, O. Pudova, C. Arbizzani, M. Mastragostino and A. Bongini, J. Org. Chem., 1998, 63, 1742–1745 CrossRef CAS.
- G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, V. Fattori, M. Cocchi, F. Cacialli, G. Gigli and R. Cingolani, Adv. Mater., 1999, 11, 1375–1379 CrossRef CAS.
- Y. Suzuki, T. Okamoto, A. Wakamiya and S. Yamaguchi, Org. Lett., 2008, 10, 3393–3396 CrossRef CAS PubMed.
- B. Chen, H. Zhang, W. Luo, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 960–968 RSC.
- Q. Yang, D. Li, W. Chi, R. Guo, B. Yan, J. Lan, X. Liu and J. Yin, J. Mater. Chem. C, 2019, 7, 8244–8249 RSC.
- C. P. Hsu, Acc. Chem. Res., 2009, 42, 509–518 CrossRef CAS PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 46, 1290–1299 CrossRef CAS PubMed.
- D. A. da Silva Filho, E.-G. Kim and J.-L. Brédas, Adv. Mater., 2005, 17, 1072–1076 CrossRef CAS.
- J. Ahrens, B. Böker, K. Brandhorst, M. Funk and M. Bröring, Chem. - Eur J., 2013, 19, 11382–11395 CrossRef CAS PubMed.
- T. Eder, T. Stangl, M. Gmelch, K. Remmerssen, D. Laux, S. Höger, J. M. Lupton and J. Vogelsang, Nat. Commun., 2017, 8, 1641 CrossRef PubMed.
- K. Kilså, J. Kajanus, J. Mårtensson and B. Albinsson, J. Phys. Chem. B, 1999, 103, 7329–7339 CrossRef.
- A. Satake and Y. Kobuke, Org. Biomol. Chem., 2007, 5, 1679–1691 RSC.
- Y. Sasano, R. Sato, Y. Shigeta, N. Yasuda and H. Maeda, J. Org. Chem., 2017, 82, 11166–11172 CrossRef CAS PubMed.
- A. A. Ryan, S. Plunkett, A. Casey, T. McCabe and M. O. Senge, Chem. Commun., 2014, 50, 353–355 RSC.
- G. Copley, D. Shimizu, J. Oh, J. Sung, K. Furukawa, D. Kim and A. Osuka, Eur. J. Org. Chem., 2016, 2016, 1977–1981 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2014, 2, 421–424 RSC.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS PubMed.
- G. H. Lee and Y. S. Kim, Polym. Bull., 2016, 73, 2439–2446 CrossRef CAS.
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem. Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- M. Monçalves, D. S. Rampon, P. H. Schneider, F. S. Rodembusch and C. C. Silveira, Dyes Pigments, 2014, 102, 71–78 CrossRef.
- M. Monçalves, G. M. Zanotto, J. M. Toldo, D. S. Rampon, P. H. Schneider, P. F. B. Gonçalves, F. S. Rodembusch and C. C. Silveira, RSC Adv., 2017, 7, 8832–8842 RSC.
- P. R. Christensen, J. K. Nagle, A. Bhatti and M. O. Wolf, J. Am. Chem. Soc., 2013, 135, 8109–8112 CrossRef CAS PubMed.
- C. D. Cruz, P. R. Christensen, E. L. Chronister, D. Casanova, M. O. Wolf and C. J. Bardeen, J. Am. Chem. Soc., 2015, 137, 12552–12564 CrossRef CAS PubMed.
- C. Climent, M. Barbatti, M. O. Wolf, C. J. Bardeen and D. Casanova, Chem. Sci., 2017, 8, 4941–4950 RSC.
- R. S. Kathayat, L. Yang, T. Sattasathuchana, L. Zoppi, K. K. Baldridge, A. Linden and N. S. Finney, J. Am. Chem. Soc., 2016, 138, 15889–15895 CrossRef CAS PubMed.
- E. Caron and M. O. Wolf, Macromolecules, 2017, 50, 7543–7549 CrossRef CAS.
- M. H. M. Cativo, A. C. Kamps, J. Gao, J. K. Grey, G. R. Hutchison and S. J. Park, J. Phys. Chem. B, 2013, 117, 4528–4535 CrossRef CAS PubMed.
- S. Wei, J. Xia, E. J. Dell, Y. Jiang, R. Song, H. Lee, P. Rodenbough, A. L. Briseno and L. M. Campos, Angew. Chem. Int. Ed., 2014, 53, 1832–1836 CrossRef CAS PubMed.
- E. L. Dane, S. B. King and T. M. Swager, J. Am. Chem. Soc., 2010, 132, 7758–7768 CrossRef CAS PubMed.
- Z. Zhou, W. Huang, Y. Long, Y. Chen, Q. Yu, Y. Zhang, S. Liu, Z. Chi, X. Chen and J. Xu, J. Mater. Chem. C, 2017, 5, 8545–8552 RSC.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef PubMed.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- Z. Xu, C. Climent, C. M. Brown, D. Hean, C. J. Bardeen, D. Casanova and M. O. Wolf, Chem. Sci., 2021, 12, 188–195 RSC.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem. Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- Y. Hu, Z. Wang, X. Jiang, X. Cai, S.-J. Su, F. Huang and Y. Cao, Chem. Commun., 2018, 54, 7850–7853 RSC.
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem. Int. Ed., 2019, 58, 17651–17655 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Z. Fan, E. Ubba, W. Li, Y. Li, J. Zhao, Z. Yang, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 179–184 RSC.
- P. R. Christensen, B. O. Patrick, É. Caron and M. O. Wolf, Angew. Chem. Int. Ed., 2013, 52, 12946–12950 CrossRef CAS PubMed.
- R. S. Grainger, B. Patel, B. M. Kariuki, L. Male and N. Spencer, J. Am. Chem. Soc., 2011, 133, 5843–5852 CrossRef CAS PubMed.
- S. Hayakawa, K. Matsuo, H. Yamada, N. Fukui and H. Shinokubo, J. Am. Chem. Soc., 2020, 142, 11663–11668 CrossRef CAS PubMed.
- G. E. Hartzell and J. N. Paige, J. Am. Chem. Soc., 1966, 88, 2616–2617 CrossRef CAS.
- R. M. Dodson and R. F. Sauers, Chem. Commun., 1967, 1189–1190 RSC.
- I. A. Abu-Yousef and D. N. Harpp, Tetrahedron Lett., 1995, 36, 201–204 CrossRef CAS.
- I. A. Abu-Yousef and D. N. Harpp, J. Org. Chem., 1997, 62, 8366–8371 CrossRef CAS PubMed.
- L. E. Longobardi, V. Wolter and D. W. Stephan, Angew. Chem. Int. Ed., 2015, 54, 809–812 CrossRef CAS PubMed.
- M. Joost, M. Nava, W. J. Transue, M. A. Martin-Drumel, M. C. McCarthy, D. Patterson and C. C. Cummins, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5866–5871 CrossRef CAS PubMed.
- H. Bouas-Laurent, A. Castellan, J. P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- H. Bouas-Laurent, J.-P. Desvergne, A. Castellan and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- C. D. Cruz, J. Yuan, C. Climent, N. T. Tierce, P. R. Christensen, E. L. Chronister, D. Casanova, M. O. Wolf and C. J. Bardeen, Chem. Sci., 2019, 10, 7561–7573 RSC.
- P. R. Christensen and M. O. Wolf, Adv. Funct. Mater., 2016, 26, 8471–8477 CrossRef CAS.
- J. Yuan, P. R. Christensen and M. O. Wolf, Chem. Sci., 2019, 10, 10113–10121 RSC.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- G. Zhou, W. Y. Wong and X. Yang, Chem. Asian J., 2011, 6, 1706–1727 CrossRef CAS PubMed.
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206–4215 RSC.
- C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155–177 CrossRef CAS.
- C. M. Brown, M. J. Kitt, Z. Xu, D. Hean, M. B. Ezhova and M. O. Wolf, Inorg. Chem., 2017, 56, 15110–15118 CrossRef CAS PubMed.
- R. D. Costa, E. Ortí, D. Tordera, A. Pertegás, H. J. Bolink, S. Graber, C. E. Housecroft, L. Sachno, M. Neuburger and E. C. Constable, Adv. Energy Mater., 2011, 1, 282–290 CrossRef CAS.
- C. M. Brown, C. Li, V. Carta, W. Li, Z. Xu, P. H. F. Stroppa, I. D. W. Samuel, E. Zysman-Colman and M. O. Wolf, Inorg. Chem., 2019, 58, 7156–7168 CrossRef CAS PubMed.
- Z. A. Siddique, Y. Yamamoto, T. Ohno and K. Nozaki, Inorg. Chem., 2003, 42, 6366–6378 CrossRef CAS PubMed.
- S. Garakyaraghi, E. O. Danilov, C. E. McCusker and F. N. Castellano, J. Phys. Chem. A, 2015, 119, 3181–3193 CrossRef CAS PubMed.
- A. O. Razgoniaev, C. E. McCusker, F. N. Castellano and A. D. Ostrowski, ACS Macro Lett., 2017, 6, 920–924 CrossRef CAS.
- C. M. Brown, V. Carta and M. O. Wolf, Chem. Mater., 2018, 30, 5786–5795 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2007, 129, 5248–5256 CrossRef CAS PubMed.
- A. B. P. Lever, E. Mantovani and J. C. Donini, Inorg. Chem., 1971, 10, 2424–2427 CrossRef CAS.
- A. B. P. Lever and E. Mantovani, Inorg. Chem., 1971, 10, 817–826 CrossRef CAS.
- I. Grenthe, P. Paoletti, M. Sandstrom and S. Glikberg, Inorg. Chem., 1979, 18, 2687–2692 CrossRef CAS.
- S. Perruchas, X. F. Le Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin and J.-P. Boilot, J. Am. Chem. Soc., 2010, 132, 10967–10969 CrossRef CAS PubMed.
- B. Huitorel, Q. Benito, A. Fargues, A. Garcia, T. Gacoin, J.-P. Boilot, S. Perruchas and F. Camerel, Chem. Mater., 2016, 28, 8190–8200 CrossRef CAS.
- E. Caron, C. M. Brown, D. Hean and M. O. Wolf, Dalton Trans., 2019, 48, 1263–1274 RSC.
- A. B. Pun, S. N. Sanders, M. Y. Sfeir, L. M. Campos and D. N. Congreve, Chem. Sci., 2019, 10, 3969–3975 RSC.
- C. Hetzer, D. M. Guldi and R. R. Tykwinski, Chem. - Eur J., 2018, 24, 8245–8257 CrossRef CAS PubMed.
- C. M. Brown, T. Auvray, E. E. Deluca, M. B. Ezhova, G. S. Hanan and M. O. Wolf, Chem. Commun., 2020, 56, 10750–10753 RSC.
- M. D. Kärkäs, E. V Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
Footnote |
| † J. Y. and Z. X. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |