
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneration of organo-alkaline earth metal complexes from non-polar unsaturated molecules and their synthetic applications
Kohei
Watanabe†
 b,
Jia Hao
Pang†
a,
Ryo
Takita
*b and
Shunsuke
Chiba
*a
b,
Jia Hao
Pang†
a,
Ryo
Takita
*b and
Shunsuke
Chiba
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: shunsuke@ntu.edu.sg
bGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: takita@mol.f.u-tokyo.ac.jp
First published on 19th November 2021
Abstract
Organomagnesium compounds, represented by the Grignard reagents, are one of the most classical yet versatile carbanion species which have widely been utilized in synthetic chemistry. These reagents are typically prepared via oxidative addition of organic halides to magnesium metals, via halogen–magnesium exchange between halo(hetero)arenes and organomagnesium reagents or via deprotonative magnesiation of prefunctionalized (hetero)arenes. On the other hand, recent studies have demonstrated that the organo-alkaline earth metal complexes including those based on heavier alkaline earth metals such as calcium, strontium and barium could be generated from readily available non-polar unsaturated molecules such as alkenes, alkynes, 1,3-enynes and arenes through unique metallation processes. Nonetheless, the resulting organo-alkaline earth metal complexes could be further functionalized with a variety of electrophiles in various reaction modes. In particular, organocalcium, strontium and barium species have shown unprecedented reactivity in the downstream functionalization, which could not be observed in the reactivity of organomagnesium complexes. This perspective will focus on the newly emerging protocols for the generation of organo-alkaline earth metal complexes from non-polar unsaturated molecules and their applications in chemical synthesis and catalysis.
 Kohei Watanabe | Kohei Watanabe earned his PhD degree from Chiba University in 2018. He engaged in research on main group chemistry under the supervision of Prof. Gerhard Erker at Westfälische Wilhelms-Universität as a postdoc, and then started to work at the University of Tokyo as Assistant Professor with Prof. Ryo Takita from 2019. He focuses on organometallic chemistry for the development of methodologies in synthetic organic chemistry. |
 Jia Hao Pang | Jia Hao Pang completed his undergraduate studies at Nanyang Technological University (NTU), Singapore in 2016 before beginning his PhD work in the laboratory of Shunsuke Chiba at NTU. He is currently focusing on synthetic chemistry of main group metal hydrides. |
 Ryo Takita | Ryo Takita got his PhD degree in 2006 under the supervision of Prof. Masakatsu Shibasaki at the University of Tokyo. After a postdoc with Prof. Timothy M. Swager at MIT, he became an Assistant Professor at Kyoto University and then moved to the University of Tokyo and RIKEN. He is currently Associate Professor at the University of Tokyo. His research group focuses on the development of reactions and molecular functions featuring element-based characteristics. |
 Shunsuke Chiba | Shunsuke Chiba earned his PhD degree in 2006 under the supervision of Prof. Koichi Narasaka at the University of Tokyo. In 2007, he embarked on his independent career as the faculty of Nanyang Technological University (NTU), Singapore, where he is currently Professor of Chemistry. His research group focuses on methodology development in the area of synthetic chemistry and catalysis. |
1. Introduction
As part of the larger movement towards sustainable and green chemistry, transition-metal free methodologies have attracted much attention in the fields of synthetic chemistry and catalysis. In this context, leveraging of alkali/alkaline earth metals to drive desired synthetic processes is extremely attractive due to their abundance in nature and lower toxicity. Organomagnesium reagents, typified by the Grignard reagents, are one of the most classical yet versatile carbanion species, which have widely been utilized in synthetic chemistry.1,2 These reagents are typically prepared via oxidative addition of organic halides to magnesium metals (Scheme 1A)3 or via halogen–magnesium exchange between halo(hetero)arenes and alkylmagnesium reagents (Scheme 1B).4–8 Deprotonative magnesiation of (hetero)arenes has also been developed for the direct preparation of arylmagnesium reagents, while it commonly needs prefunctionalization at a suitable position of the (hetero)arene substrates (Scheme 1C).9,10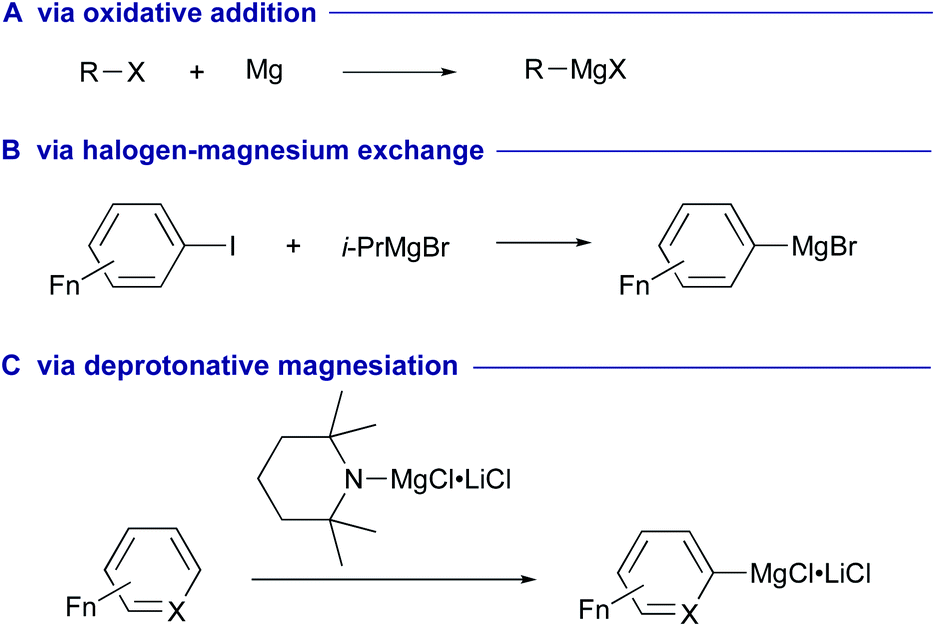 | ||
| Scheme 1 Conventional preparation methods of organomagnesium reagents. | ||
Recently, synthesis and structural characterization of various molecular magnesium(II) hydrides has successfully been achieved.11 Employment of sterically hindered ligands (L) is the key to stabilize molecular magnesium(II) hydrides [L–Mg–H] kinetically from their Schlenk equilibrium to homoleptic magnesium-anionic ligand complexes [MgL2] and insoluble magnesium hydride [MgH2]n of the bulk lattice due to its higher lattice energy.12 Thermally stable lower valent magnesium(I) dimers [L–Mg–Mg–L] could also be designed and synthesized with the aid of bulky anionic ligands.13–15 The reactivity assessments of these magnesium(II) hydride and magnesium(I) dimer complexes have led to the discovery of unprecedented magnesiation processes of non-polar unsaturated carbon systems such as alkenes, alkynes and arenes, which could be applied in chemical synthesis and catalysis mainly via downstream functionalization of the resulting organomagnesium intermediates with various electrophiles. On the other hand, in situ generation of active magnesium hydride species could be mediated by σ-bond metathesis of hydrosilanes or pinacolborane with organomagnesium complexes or counter ion metathesis between sodium hydride (NaH)16 and magnesium iodide (MgI2) without the use of ligands, facilitating unique molecular transformations of non-polar unsaturated compounds. Moreover, heavier alkaline earth metal hydride complexes based on calcium, strontium and barium have also been designed and prepared.17,18 These hydride complexes commonly display more hydridic reactivity for hydrometallation to non-polar unsaturated molecules to generate organo-heavier alkaline earth metal intermediates.19,20 These species further perform unprecedented molecular transformations and catalysis, which are not often observed in the reactivities of organomagnesium complexes. The purpose of this perspective is to highlight recent advances in the development of new metallation methods of non-polar unsaturated systems with alkaline earth metal reagents and the applications of the resulting organo-alkaline earth metal intermediates in chemical synthesis and catalysis.21 It should be noted that this perspective does not include the preparation methods of the key alkaline earth metal complexes that are used for the metallation of non-polar unsaturated systems. Readers can find the protocols in the corresponding references.
2. Organomagnesium complexes
2.1. Transformation of alkenes
The terminal magnesium hydride carbatrane complex 1 having a tris[(1-isopropylbenzimidazol-2-yl)dimethylsilyl]methyl ligand was recently synthesized by Parkin and it was found to serve as a catalyst for the Markovnikov hydrosilylation and hydroboration of styrene (2) through hydromagnesiation and ensuing σ-bond metathesis of the resulting alkylmagnesium intermediate with hydrosilane 3 (PhSiH3) or pinacolborane (4) [HB(pin)] (Scheme 2).22 The turnover frequency of the hydrosilylation was identified as 0.9 h−1, whereas that of the hydroboration was 0.3 h−1.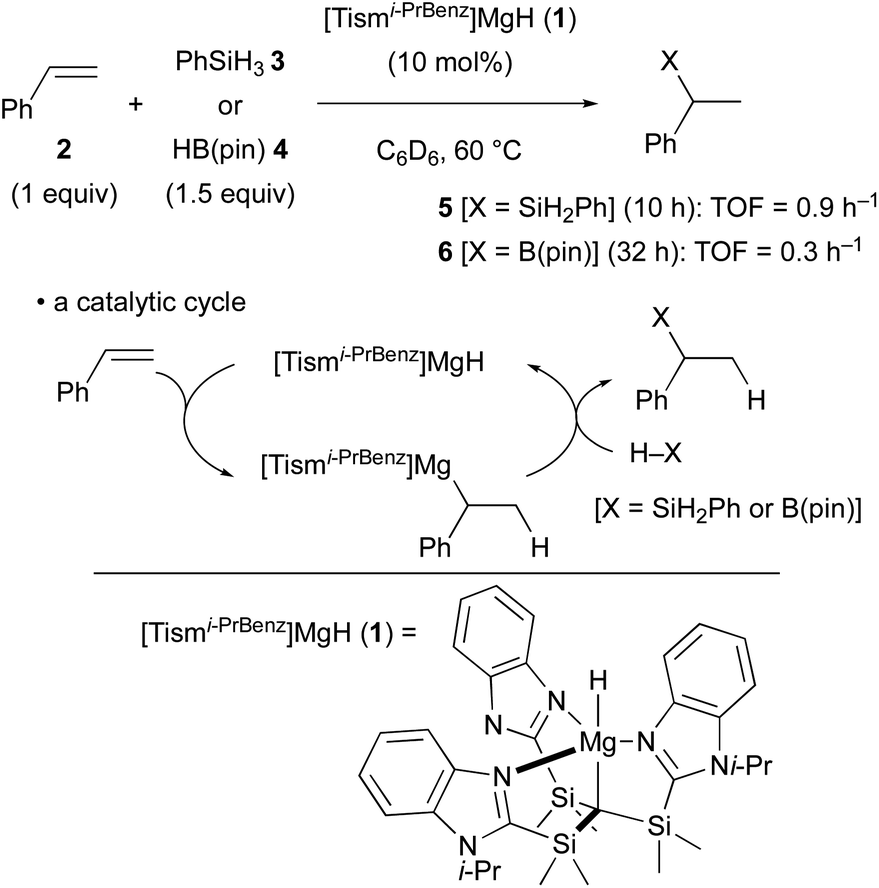 | ||
| Scheme 2 Catalytic hydrosilylation and hydroboration of styrene (Parkin, 2017). | ||
In contrast, β-diketiminato magnesium hydride dimer 7 (ref. 23) performed anti-Markovnikov hydromagnesiation of unactivated aliphatic terminal alkenes in good efficiency to afford the corresponding organomagnesium complexes 8–10 (Scheme 3A).24 The anti-Markovnikov regioselectivity and the reaction rate are primarily governed by the steric effect. In turn, the hydromagnesiation of styrene (2) gave a 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45 mixture of linear and branched organomagnesium species 11 and 12 (Scheme 3B). Although 1,2-disubstituted alkenes were generally inert toward hydromagnesiation, strained bicyclic alkene, norbornene (13) showed good reactivity with magnesium hydride 7 to afford 2-norbornylmagnesium 14 (Scheme 3C). A domino sequence of hydromagnesiation and 5-exo carbomagnesiation was observed in the reaction of 1,5-hexadiene (15), affording cyclopentylmethylmagnesium 17via 5-hexenylmagnesium 16 (Scheme 3D).
:45 mixture of linear and branched organomagnesium species 11 and 12 (Scheme 3B). Although 1,2-disubstituted alkenes were generally inert toward hydromagnesiation, strained bicyclic alkene, norbornene (13) showed good reactivity with magnesium hydride 7 to afford 2-norbornylmagnesium 14 (Scheme 3C). A domino sequence of hydromagnesiation and 5-exo carbomagnesiation was observed in the reaction of 1,5-hexadiene (15), affording cyclopentylmethylmagnesium 17via 5-hexenylmagnesium 16 (Scheme 3D).
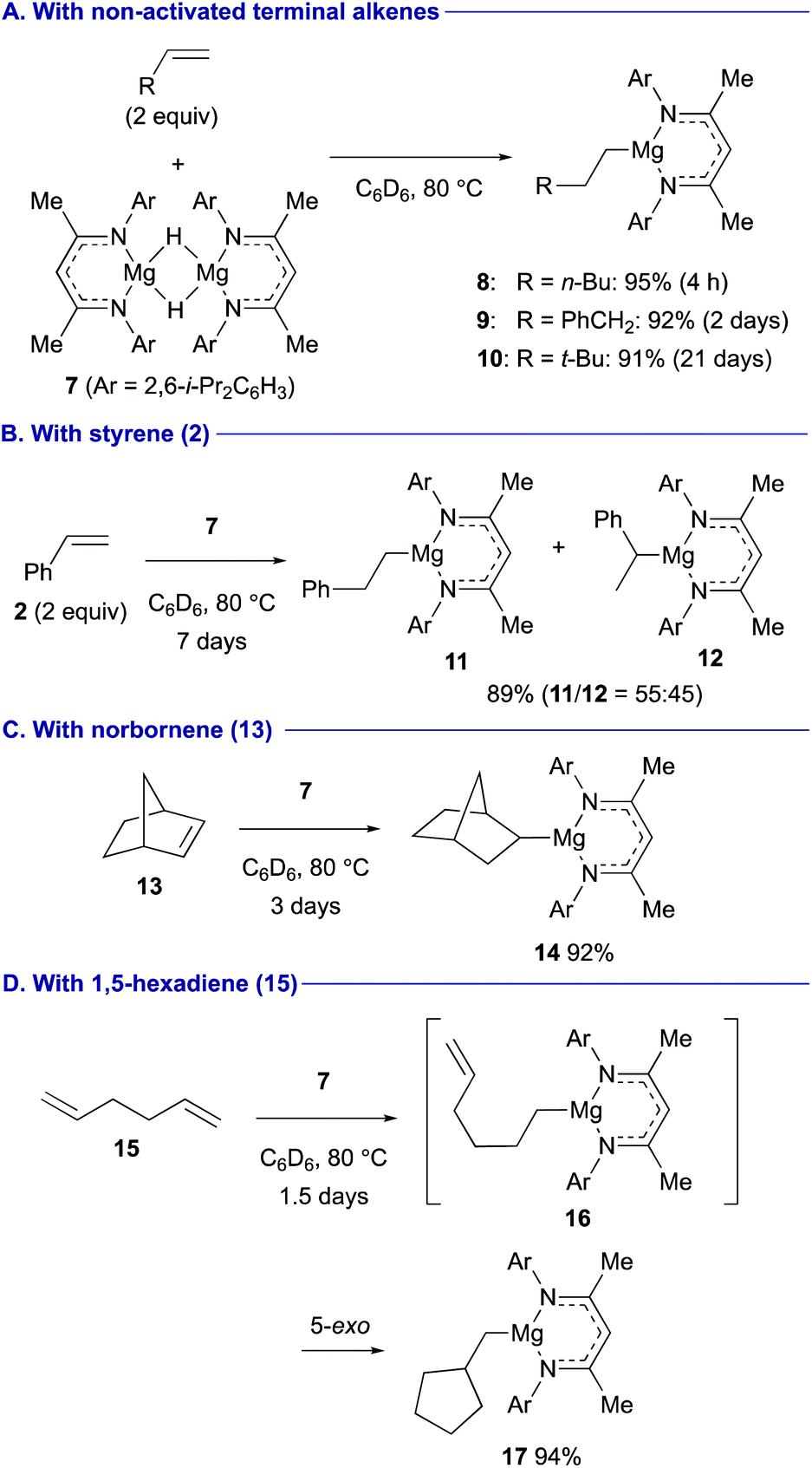 | ||
| Scheme 3 Hydromagnesiation of alkenes with magnesium hydride 7 (Maron and Hill, 2019). | ||
Thus, this hydromagnesiation of non-activated alkenes with magnesium hydride 7 could be applied for the catalytic hydrosilylation using PhSiH33, which is responsible to undergo rate-determining σ-bond metathesis with the organomagnesium intermediates generated by hydromagnesiation to maintain the catalytic turnover (Scheme 4).
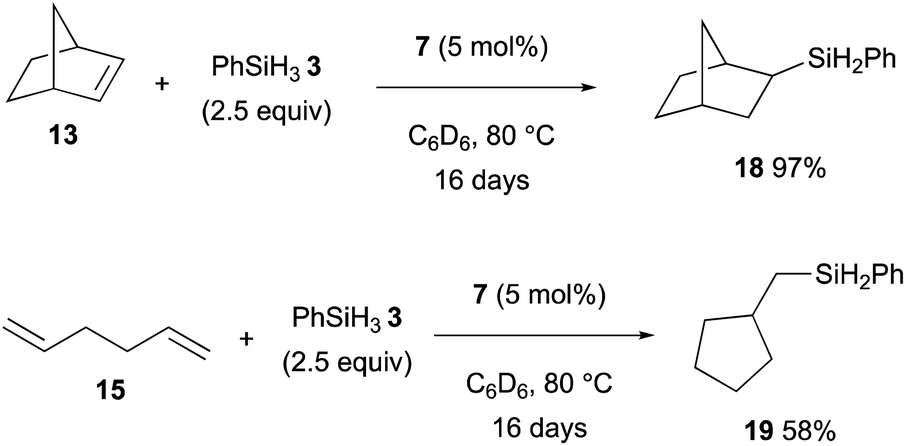 | ||
| Scheme 4 Catalytic hydrosilylation of alkenes with 7 (Maron and Hill, 2019). | ||
Jones and Maron revealed that magnesium(I) dimers 20 (ref. 25) and 21 (ref. 26) supported by β-diketiminato ligands showed unprecedented reactivity toward alkenes. For example, 1,1-diphenylethylene (22) underwent oxidative insertion into the Mg(I)–Mg(I) bond of 20 and 21 to form 1,2-dimagnesioethane complexes 23 and 24, respectively. At ambient temperature, this process was found reversible via reductive elimination of 1,1-diphenylethylene (22) (Scheme 5).27
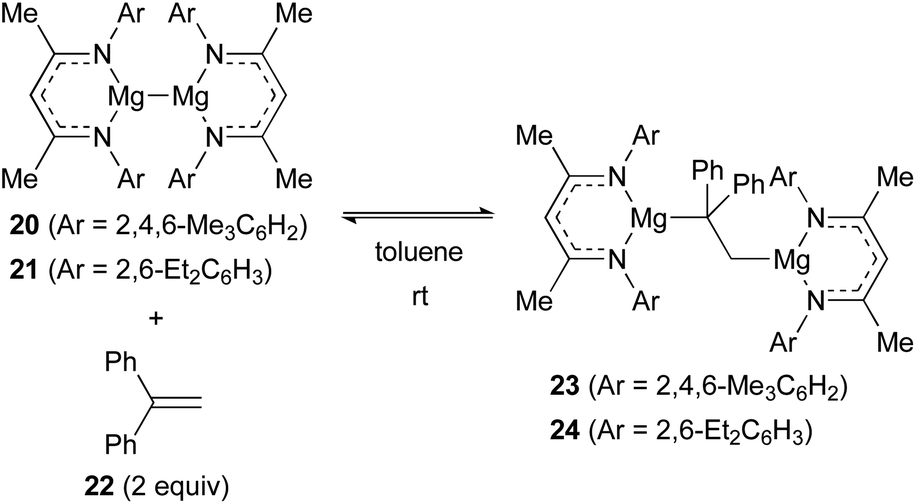 | ||
| Scheme 5 Reversible insertion of 1,1-diphenylethylene into the Mg(I)–Mg(I) bond (Jones and Maron, 2017). | ||
Treatment of the 1,2-dimagnesioethane complex 23 with H2 resulted in regioselective hydrogenation at the Mg–CPh2 moiety to liberate alkylmagnesium 25 and magnesium hydride dimer 26, while the reaction of 23 with ethylene induced its insertion into the Mg–CPh2 bond, providing 1,4-dimagnesiobutane 27 (Scheme 6A). Interestingly, in the reaction of 24 with CO, cyclobutenediolate 28 was formed via sequential incorporation of two molecules of CO and cyclization (Scheme 6B).
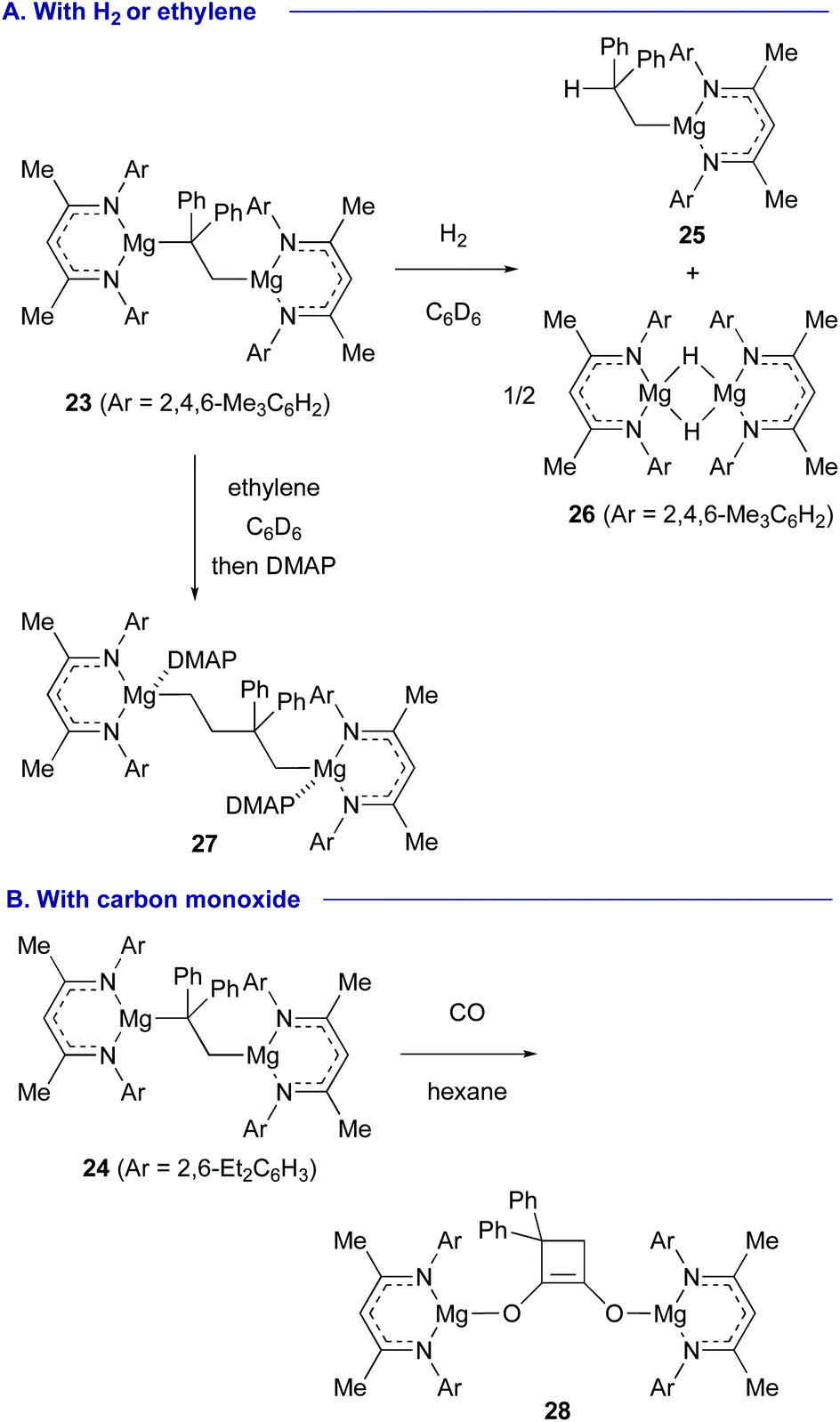 | ||
| Scheme 6 Unique reactivity of 1,2-dimagnesioethane complexes 23 and 24 (Jones and Maron, 2017). | ||
In the presence of an N-heterocyclic carbene ligand that can coordinate with Mg(I) centers as a Lewis base, the magnesium(I) dimer 20 could reductively activate even inert ethylene, forming 1,4-dimagnesiobutane 29 in 43% yield (Scheme 7).28
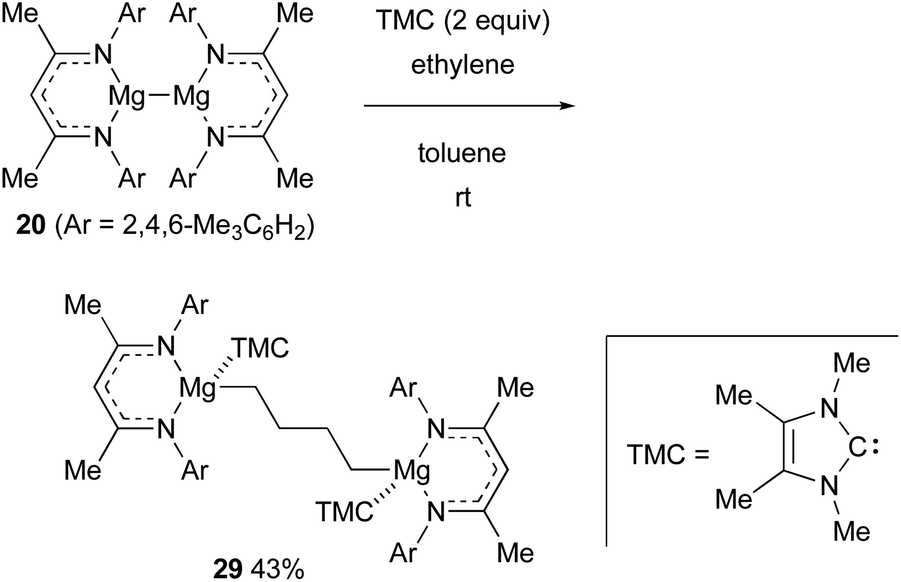 | ||
| Scheme 7 Reductive activation of ethylene with Mg(I) dimer 20 (Jones and Maron, 2020). | ||
Treatment of alkylidene cyclopropane 30 with magnesium(I) dimer 20 resulted in the formation of 1,3-dimagnesio-3-butene 31via 1,2-dimagnesiation of the alkene moiety of 30 followed by β-alkyl migration associated with the ring-opening of the cyclopropane ring (Scheme 8A).29 A combination of hydromagnesiation of alkenes and this β-alkyl migration led to the development of catalytic hydrosilylation of alkylidene cyclopropane 30 using magnesium hydride 7 and hydrosilane 3, affording homoallylsilane 32 in high yield (Scheme 8B).
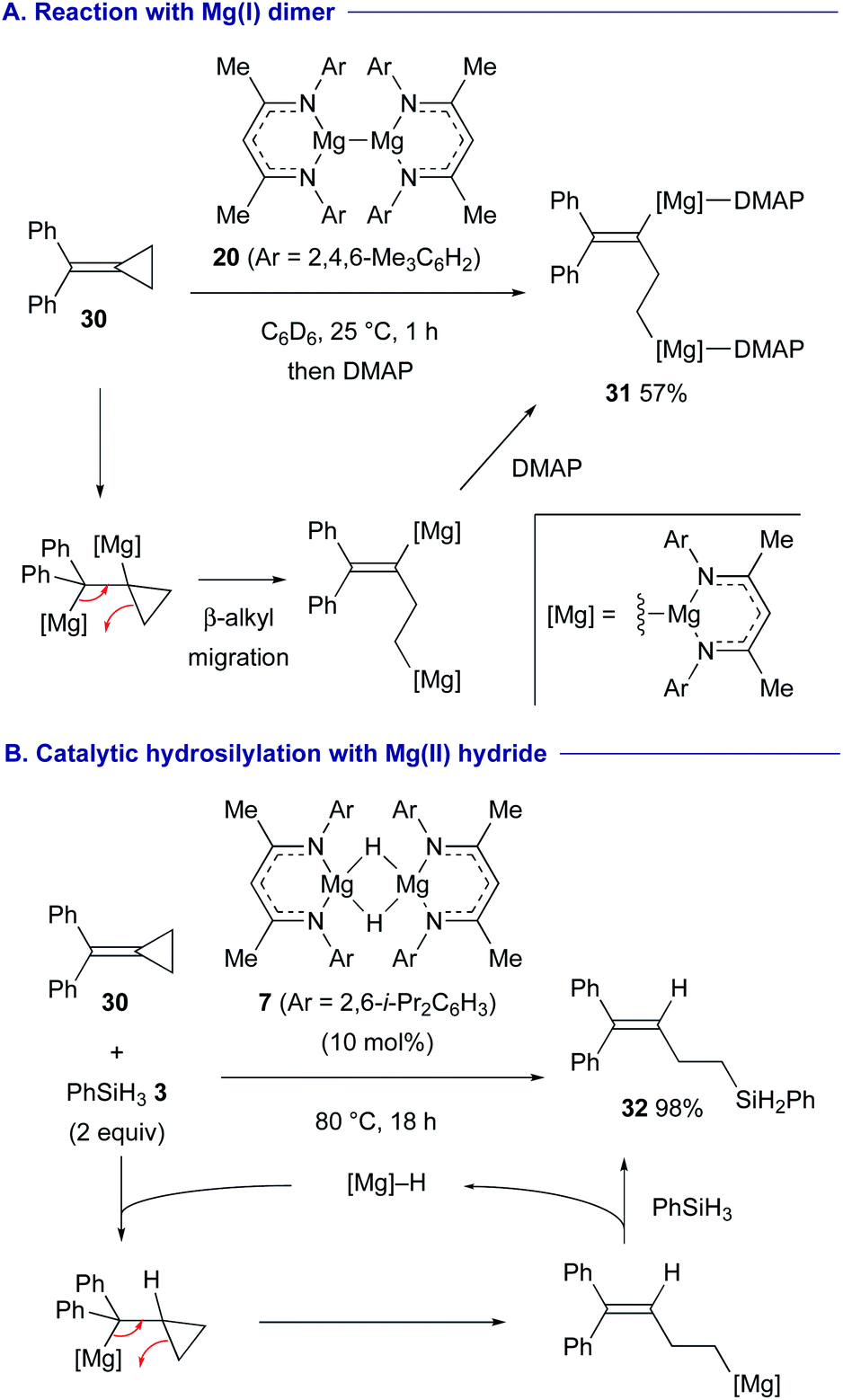 | ||
| Scheme 8 1,2-Dimagnesiation of alkylidene cyclopropane 30 with Mg(I) dimer 20 followed by β-alkyl migration and its catalytic variant with Mg(II) hydride 7 (Crimmin, 2020). | ||
2.2. Transformation of alkynes
The magnesium(I) dimer 33 containing unsymmetrical β-diketiminate ligands was found to be an effective pre-catalyst for the anti-Markovnikov hydroboration of terminal alkynes such as 34 and 35 with pinacolborane (4) [HB(pin)] (Scheme 9).30 In these processes, it was proposed that the magnesium(I) dimer precatalyst 33 is converted into magnesium hydride via the reaction with 4, although the precise mechanism is unclear. The resulting magnesium hydride underwent syn-hydromagnesiation of the alkyne to form an alkenylmagnesium intermediate. Finally, σ-bond metathesis of alkenylmagnesium with HB(pin) 4 liberated the alkenylborane products such as 36 and 37 along with the regeneration of the magnesium hydride.31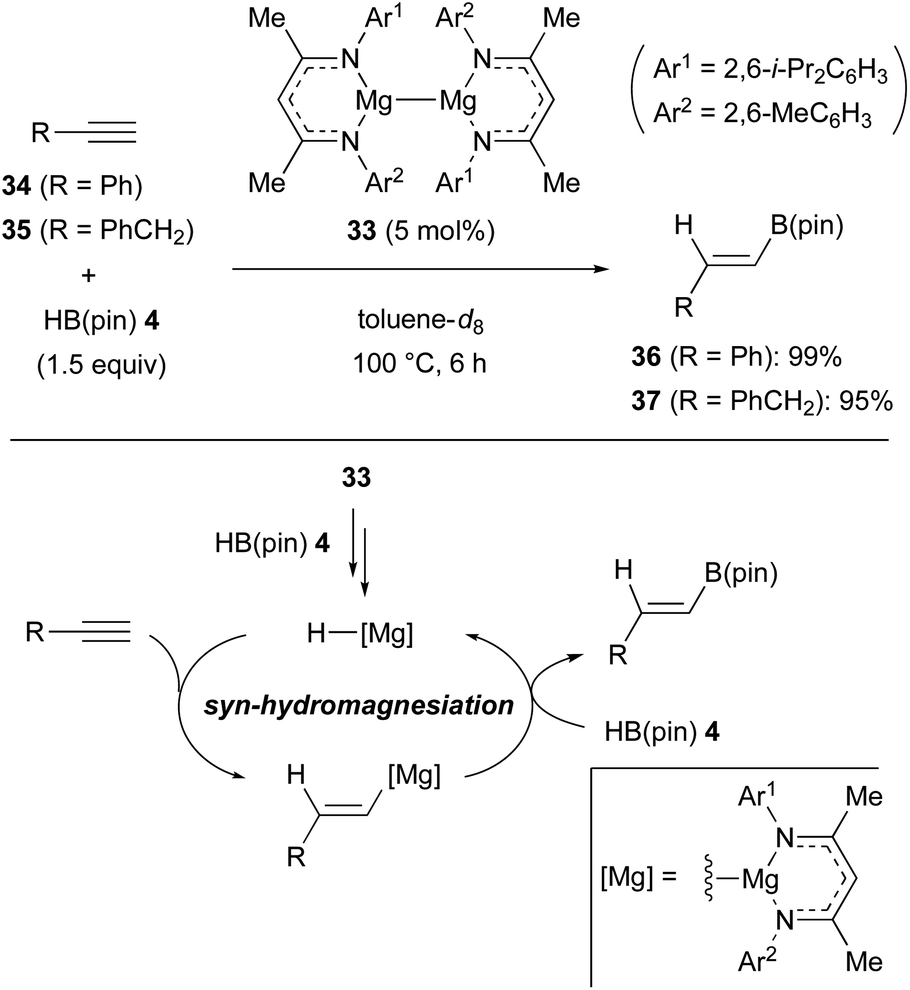 | ||
| Scheme 9 Catalytic hydroboration of alkynes with unsymmetrical Mg(I) dimer 33 (Ma, 2018). | ||
Rueping and Cavallo discovered that a catalytic amount of dibutylmagnesium (38) (Bu2Mg), in the presence of HB(pin) 4, is able to effect the syn-selective hydroboration of both terminal and internal alkynes in good yields (Scheme 10A).32 In the case of unsymmetrical internal alkynes such as (cyclohexylethynyl)benzene (39), the regioselectivity was controlled by the steric difference of the substituents (see Scheme 10B for the regioselective formation of 40 over 41). The active catalytic species was estimated as butylmagnesium hydride coordinated with HB(pin) 4, which could be generated in situ through σ-bond metathesis between Bu2Mg (38) and HB(pin) 4 (Scheme 10C). The syn-hydromagnesiation of the alkyne generated the vinyl magnesium intermediate, which underwent another σ-bond metathesis with HB(pin) 4, to regenerate butylmagnesium hydride species with the release of the hydroborated product.33
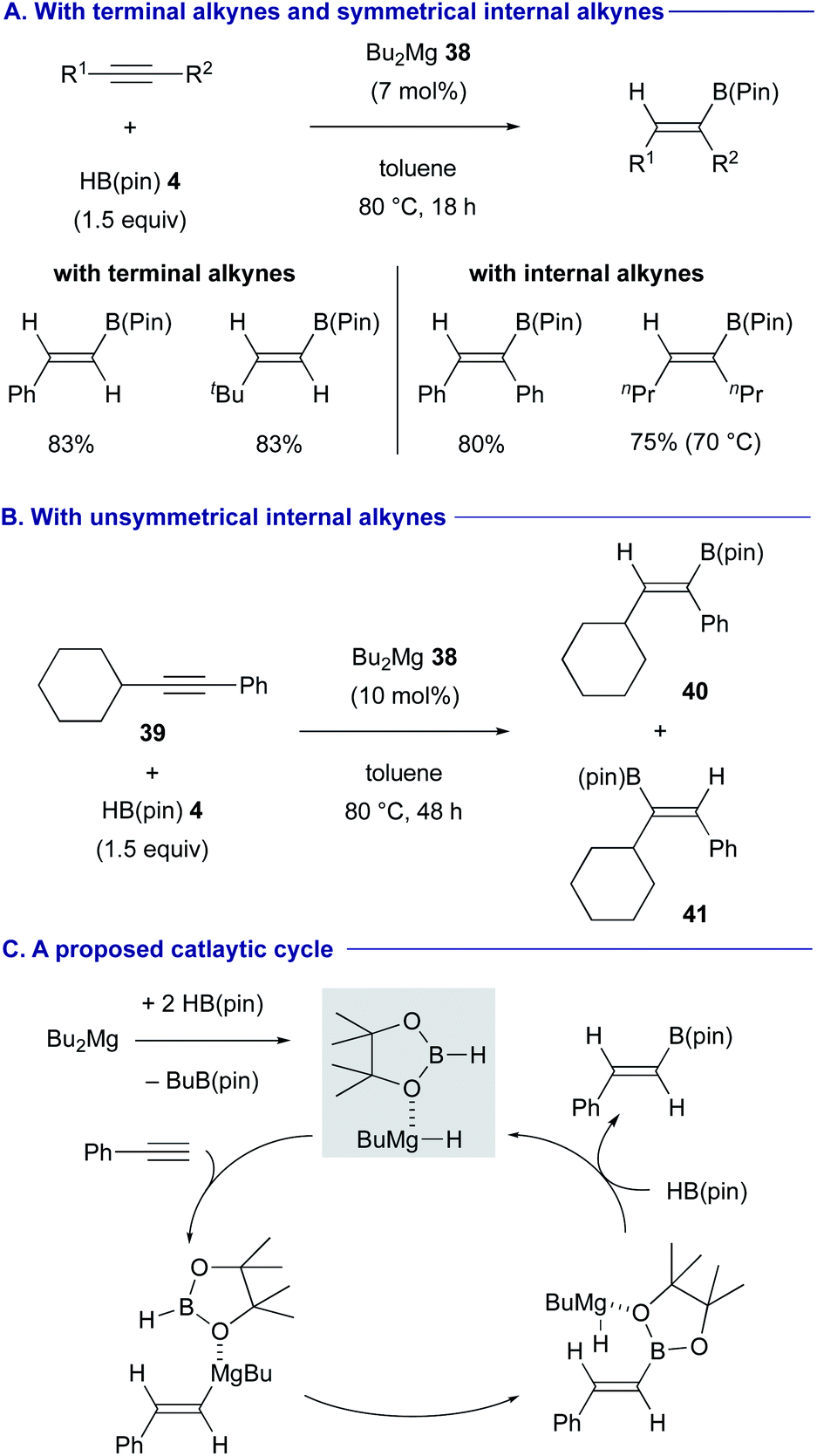 | ||
| Scheme 10 Catalytic hydroboration of terminal and internal alkynes with Bu2Mg (Rueping and Cavallo, 2019). | ||
Solvothermal treatment of sodium hydride (42) with magnesium iodide (43) in THF was found to allow for counter ion metathesis, generating highly reactive magnesium hydride (44) [MgH2]n, which could induce unprecedented anti-hydromagnesiation of arylalkynes such as diphenylacetylene (45) (Scheme 11A).34 The resulting alkenylmagnesium intermediate 46 could be trapped with a series of electrophiles such as water (for 47), HB(pin) 4 (for 48), 1,2-dibromotetrachloroethane (for 49) and dimethylformamide (DMF) (for 50) to afford stereochemically well defined functionalized alkenes. In turn, the reactions of propargyl alcohols such as 51 underwent anti-hydromagnesiation with perfect diastereoselectivity via 5-membered ring magnesiocycles such as 52, and ensuing treatment with electrophiles allowed for further downstream functionalization (see Scheme 11B for the synthesis of 53–55). The DFT calculation using a MgH2 dimer as a model active species suggested that the reaction via transition state TSanti is the favored process, where the polar hydride transfer mechanism is involved, to afford the anti-alkenylmagnesium species (Scheme 11C). On the other hand, the large distortion of diphenylacetylene is required in TSsyn, making the syn-hydromagnesiation unfavorable.
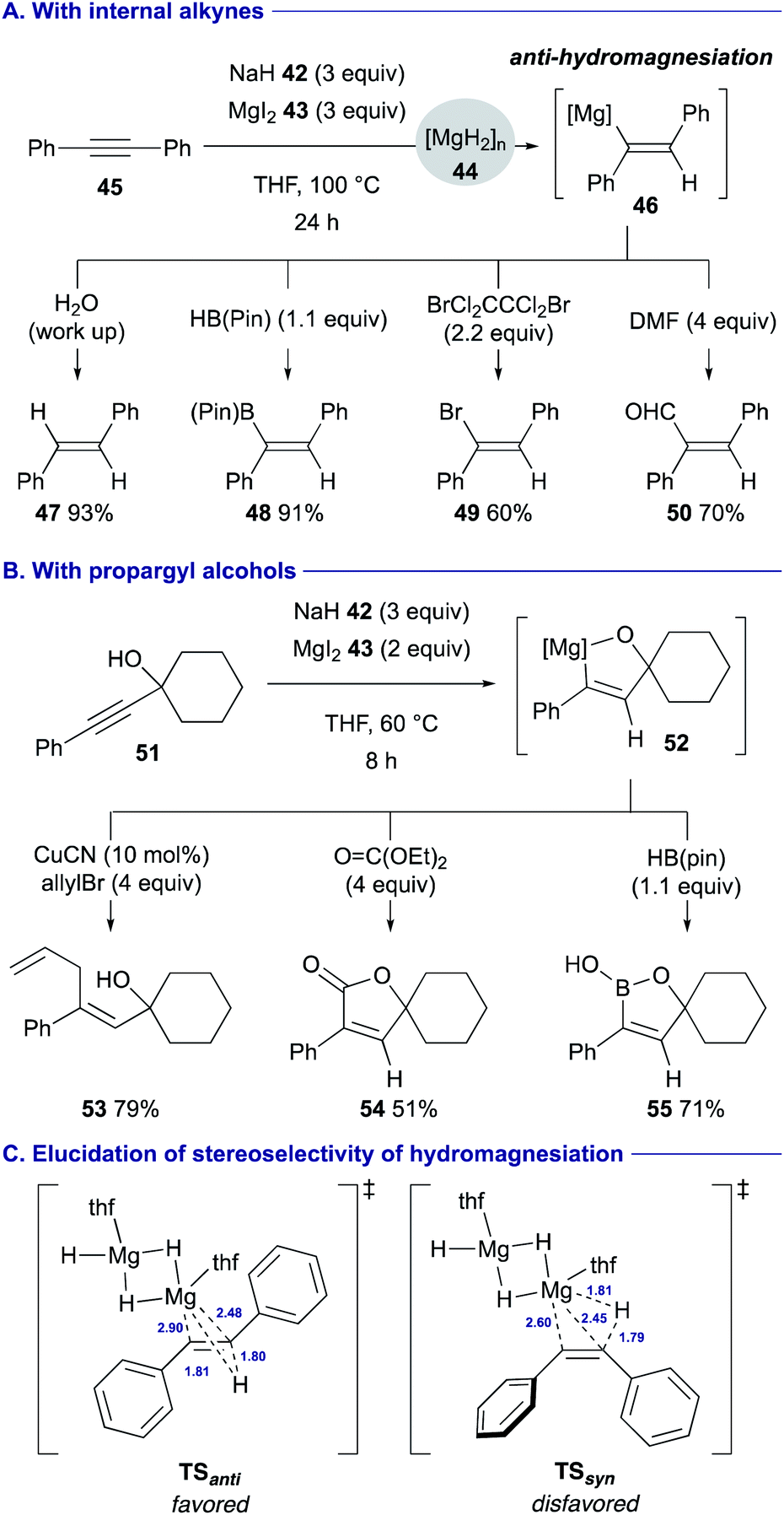 | ||
| Scheme 11 Anti-hydromagnesiation of arylalkynes by in situ generated MgH2 from NaH and MgI2 (Chiba and Takita, 2020). | ||
2.3. Transformation of 1,3-enynes
The unique hydridic reactivity of magnesium hydride (44) generated in situ from sodium hydride (42) and magnesium iodide (43) was extended further to regioselective hydromagnesiation of 1,3-enynes 56 to form an equilibrium mixture of allenylmagnesium 57 and propargylmagnesium 58 (Scheme 12).35 Downstream functionalization of the resulting organomagnesium intermediates 57 and 58 was demonstrated by subsequent treatment with a series of alkyl and silyl halides in the presence of CuCN as a catalyst, affording polysubstituted allenes 59–62.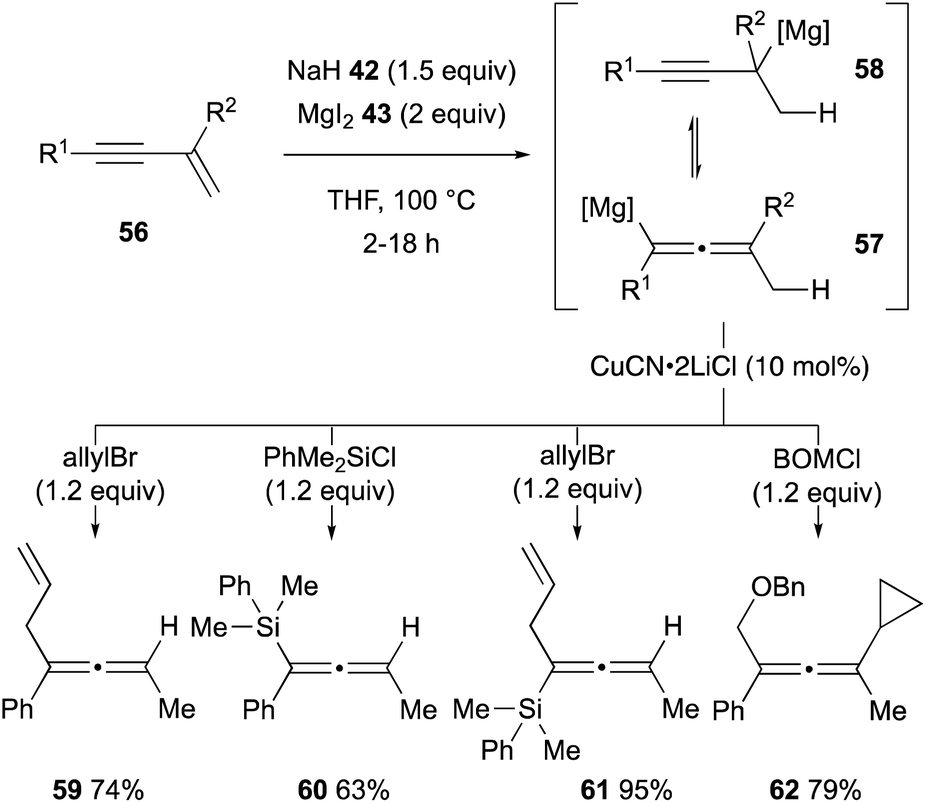 | ||
| Scheme 12 Hydromagnesiation of 1,3-enynes with MgH2 and downstream functionalization for the synthesis of substituted allenes (Chiba and Takita, 2021). | ||
In turn, downstream treatment of the allenyl/propargylmagnesium intermediates derived from hydromagnesiation of 1,3-enynes with organo nitro compounds36,37 enabled engagement of propargylmagnesium species for the amination, resulting in the formation of α-alkynylnitrones (Scheme 13).38 The α-alkynylnitrone 65 derived from 1,3-enyne 63 and 1-allyl-2-nitrobenzene (64) underwent intramolecular 1,3-dipolar cycloaddition to afford tetrahydro-1,4-epoxybenzo[b]azepine 66 as the major product and the ensuing reductive N–O bond cleavage delivered diastereomerically pure tetrahydro-1H-benzo[b]azepin-4-ol 67.
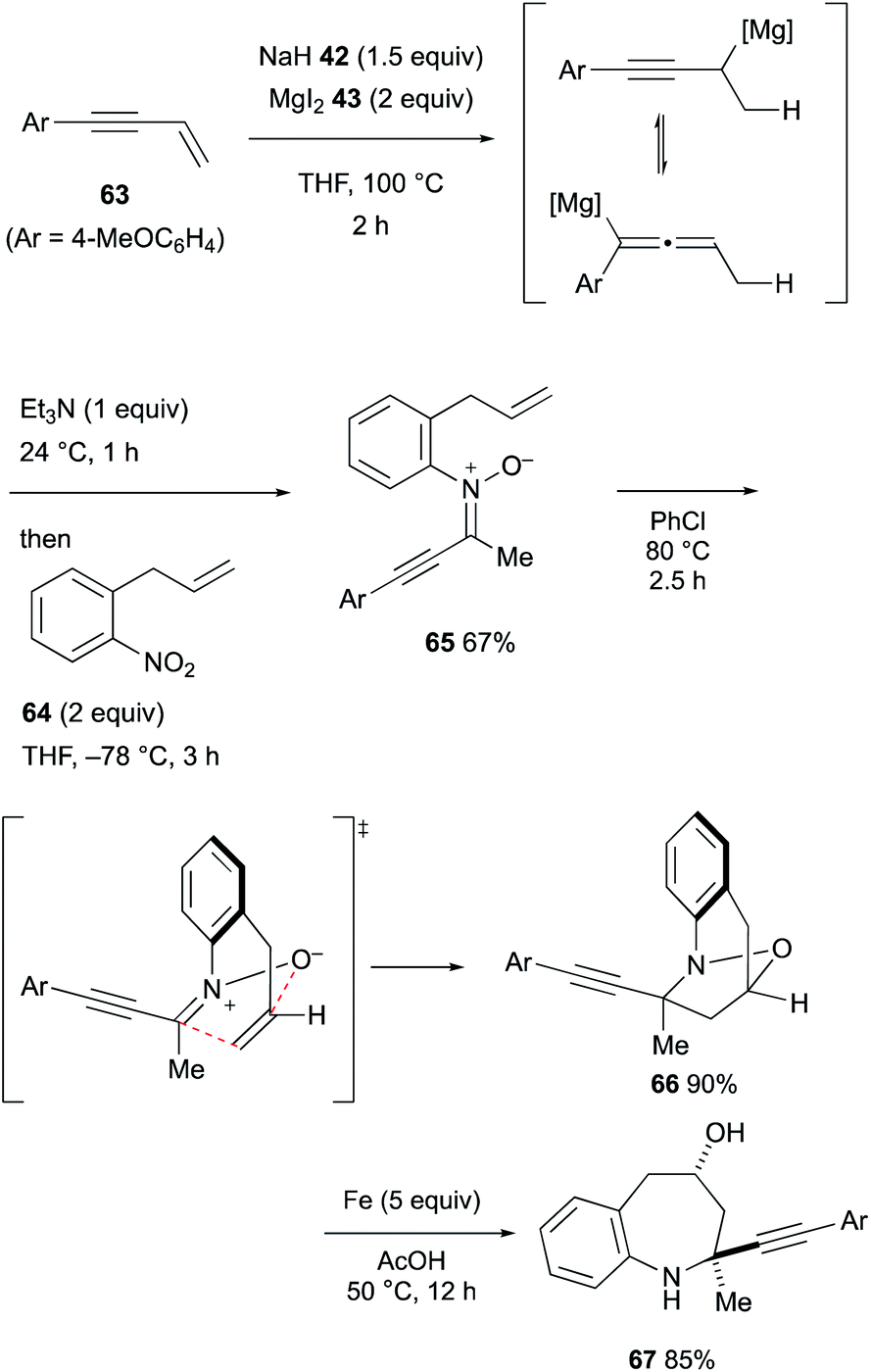 | ||
| Scheme 13 Hydromagnesiation of 1,3-enynes with MgH2 and downstream functionalization for the synthesis of nitrones (Chiba, 2021). | ||
2.4. Transformation of arenes
Two new strategies have recently emerged to convert inert arenes into the corresponding arylmagnesium complexes with in situ generated magnesium(I) radical species.Reduction of magnesium iodide complex 68 with a super bulky β-diketiminate ligand having 2,6-diisopentylphenyl groups on the imine nitrogen by sodium (Na) in the presence of tetramethylethylenediamine (TMEDA) in benzene generated cyclohexandienediyl bridged magnesium complex 69 (dearomatized C6H6 dianion sandwiched between divalent magnesium cations) (Scheme 14A).39 The reaction was initiated by the generation of doublet magnesium(I) radical 70 and its subsequent addition to benzene to form cyclohexadienyl anion radical 71, which was further reduced by 70 to liberate 69. The use of the bulky ligand and bidentate TMEDA is the key to kinetically stabilize the magnesium(I) radical 70 and prevent its dimerization. Addition of THF to 69 led to the formation of centrosymmetric flat C6H6 dianion THF adduct 72. The reaction of this dearomatized C6H6 dianion complex 69 in toluene at 120 °C for 3 days gave phenylmagnesium 73 and magnesium hydride species 74 (Scheme 14B).
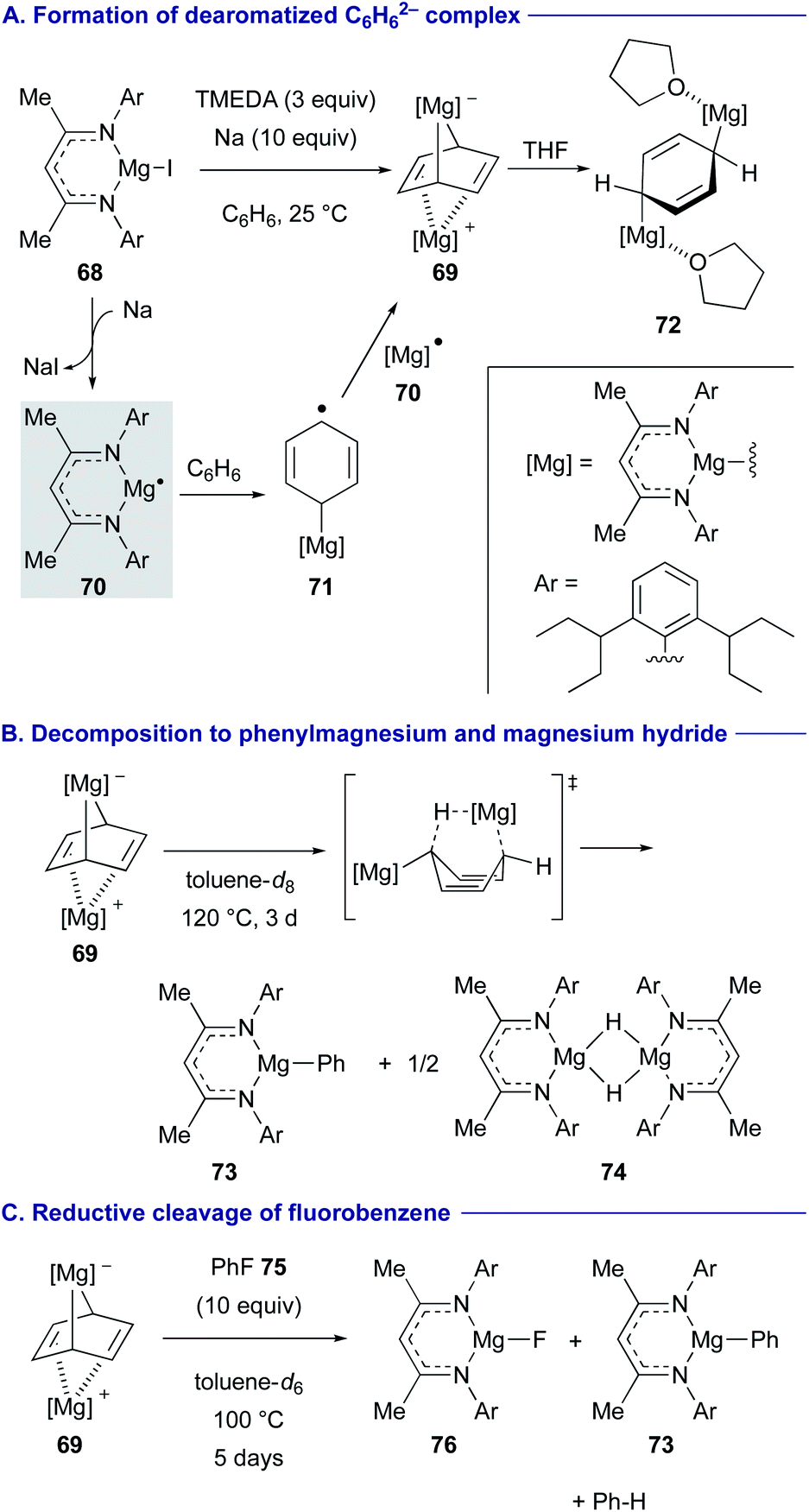 | ||
| Scheme 14 Reductive magnesiation of benzene with magnesium(I) radical 70 (Harder, 2019). | ||
The C6H6 dianion complex 69 also displayed highly reducing reactivity. For example, addition of fluorobenzene (75) to 69 induced reductive cleavage of the C–F bond to give magnesium fluoride 76 and phenylmagnesium 73 with the release of benzene (Scheme 14C).
On the other hand, Maron and Jones discovered that photoexcitation of magnesium(I) dimer 77 induced homolysis of the Mg–Mg bond to generate a highly reactive magnesium(I) radical 78, which could be used for reductive dearomatization of benzene to form cyclohexadienediyl bridged magnesium complex 79 (Scheme 15A).40 Similarly, 79 could be readily converted into phenylmagnesium 80 and magnesium hydride complex 7 upon gentle heating at 60 °C (Scheme 15A). Interestingly, the photoinduced magnesiation reactions of substituted arenes with 77 were observed to be regioselective. For example, the magnesiation of toluene (81) was observed at the meta-position to form 82, whereas that of o-xylene (83) afforded ortho-magnesiated product 84 as a single product (Scheme 15B).
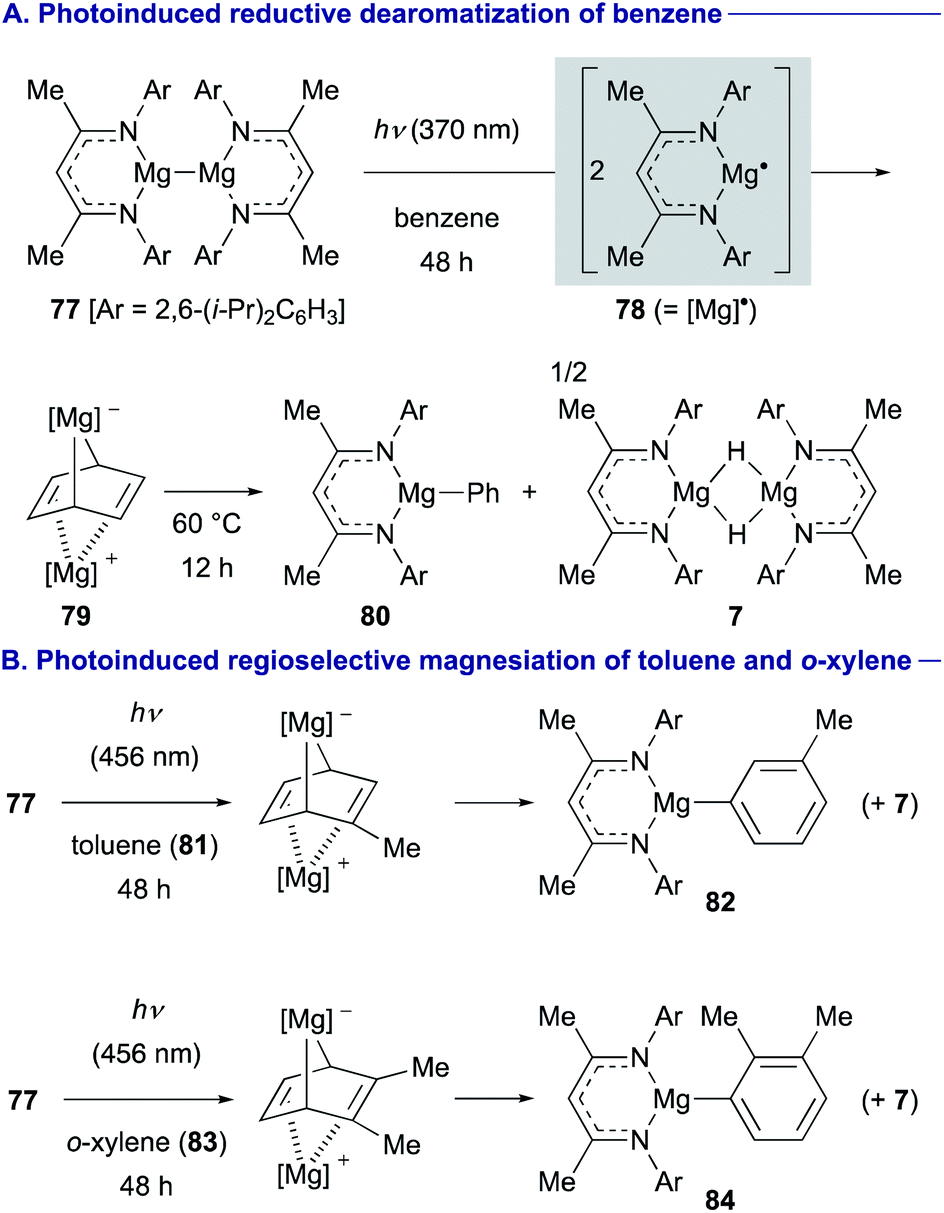 | ||
| Scheme 15 Reduction and C–H magnesiation of inert arenes using photochemically generated magnesium(I) radical 78 (Maron and Jones, 2021). | ||
3. Organo-heavier alkaline earth metal complexes
This section highlights recent selected examples for the generation of organo-heavier alkaline earth metal complexes from non-polar unsaturated molecules and their exotic reactivities in chemical synthesis and catalysis.Maron and Okuda recently developed cationic dinuclear calcium hydride complex 85 supported by 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane as the neutral tetradentate macrocyclic nitrogen ligand and tetraarylborate as the counter anion.41 This calcium hydride complex functioned as a catalyst for the hydrogenation of terminal alkenes such as styrene (2) and 1-hexene (87) under a hydrogen atmosphere, whereas internal alkenes could be kept intact (Scheme 16). Thus, in the reaction of skipped diene 89, selective hydrogenation of the terminal alkene moiety was observed to afford 90 as the sole product. It was speculated that the process is initiated by hydrometallation of terminal alkenes by the cationic calcium hydride complex 85 to form organocalcium intermediates, which are subsequently protonated with molecular hydrogen via heterolysis of the H–H bond to give the hydrogenation products along with the regeneration of the calcium hydride complex 85 to maintain the catalytic turnover.
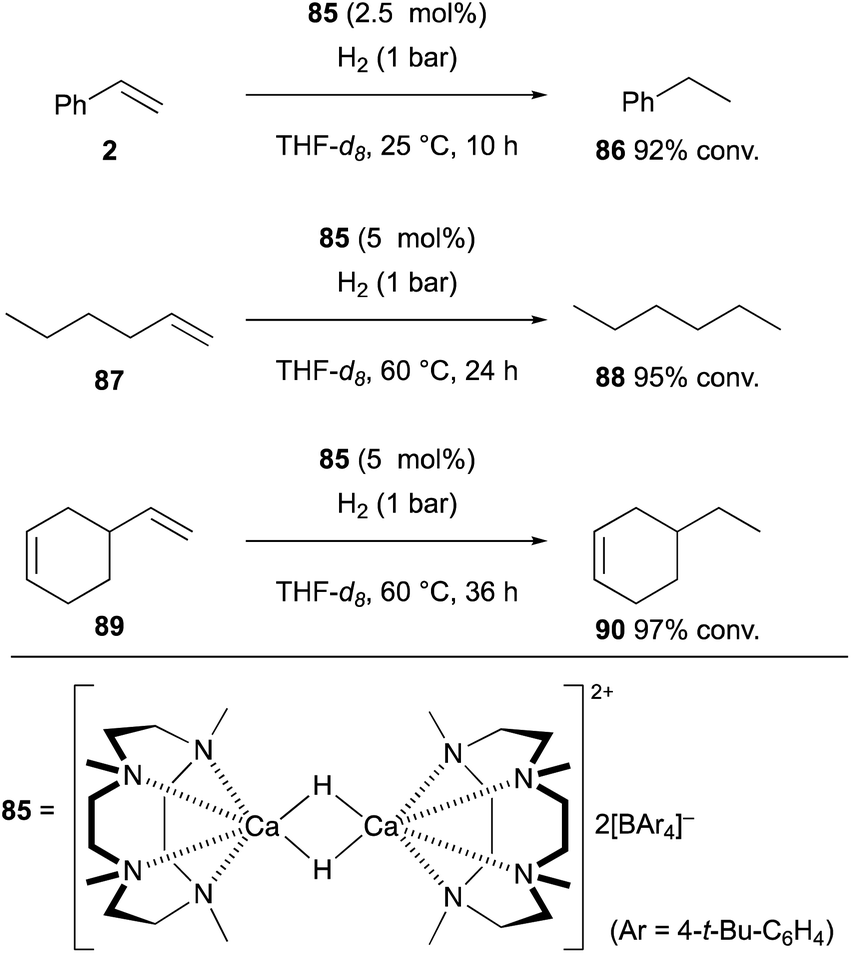 | ||
| Scheme 16 Hydrogenation of alkenes catalyzed by cationic calcium hydride dimer 85 (Maron and Okuda, 2017). | ||
The regioselectivity of the hydrometallation of terminal alkenes is determined by the electronic nature of the alkenes. Okuda demonstrated hydrosilylation of terminal alkenes using the cationic dinuclear calcium hydride complex 91 as the catalyst in the presence of hydrosilanes (Scheme 17). The reaction with styrene (2) proceeded in the Markovnikov manner to form branched benzylsilane 92 as the product (Scheme 17A), whereas anti-Markovnikov selectivity was observed in the hydrosilylation of non-activated terminal alkenes such as 1-hexene (87) (Scheme 17B).42 It should also be noted that this catalytic system is amenable to the hydrosilylation of ethylene (Scheme 17C). A more recent study revealed that the employment of 1,4,7,10,13-pentamethyl–1,4,7,10,13-pentaazacyclopentadecane as the pentadentate macrocyclic ligand could enhance the catalytic activity of the calcium hydride complex toward the hydrogenation and hydrosilylation.43
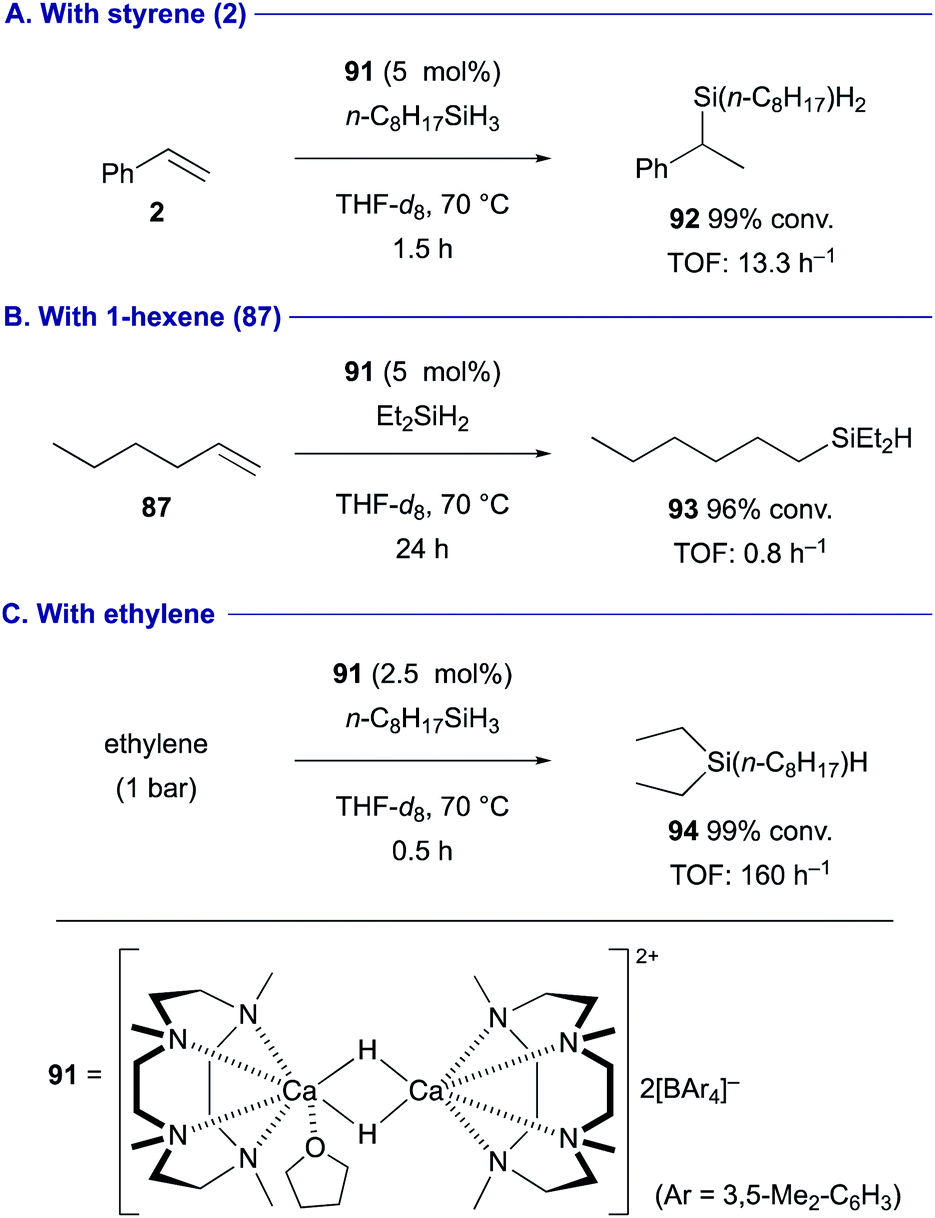 | ||
| Scheme 17 Hydrosilylation of alkenes catalyzed by cationic calcium hydride dimer 91 (Okuda, 2020). | ||
Maron discovered that β-diketiminato calcium hydride dimer 95 with no THF solvation on the calcium center undergoes hydrometallation of non-activated terminal alkenes such as 1-butene (96) in C6D6 at ambient temperature to form alkylcalcium dimer 97 (Scheme 18).44 Further treatment of 97 at 60 °C liberated n-butylbenzene 99 along with the regeneration of the calcium deuteride dimer 95-d2. This unusual alkylation of benzene was characterized as the concerted nucleophilic aromatic substitution reaction where an alkyl carbanion is installed with concurrent elimination of a hydride on sp2 hybridized carbon via the transition state 98.45
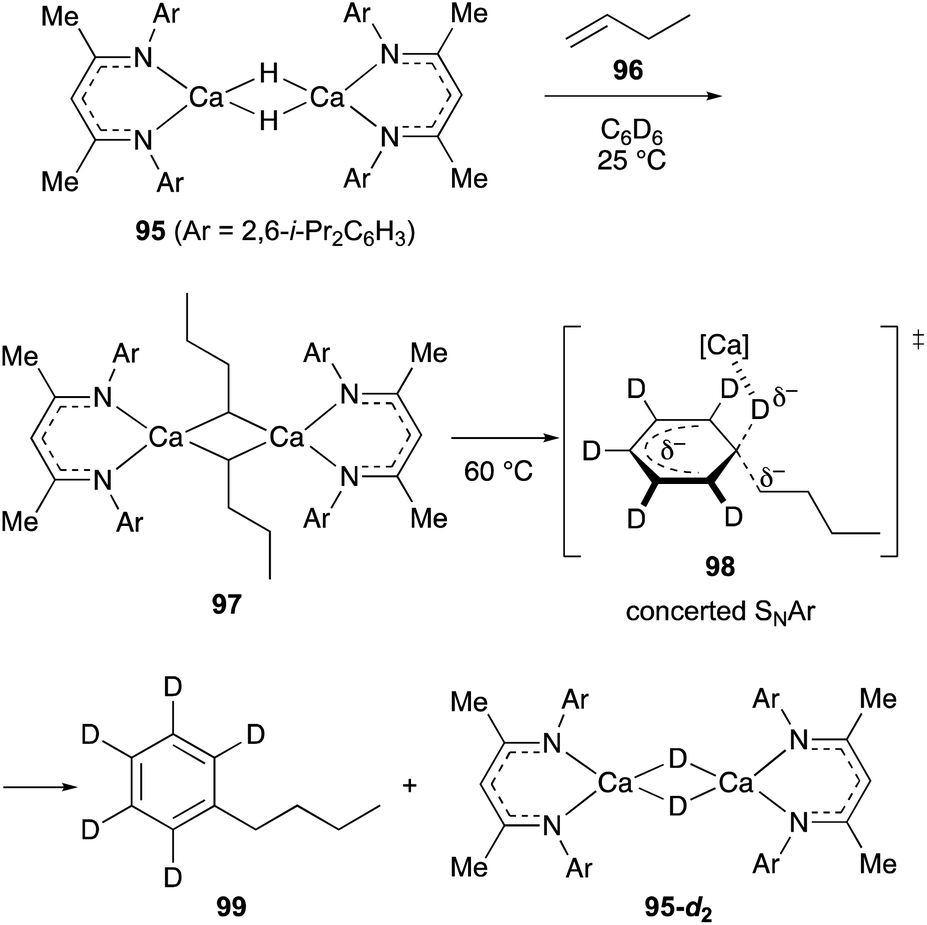 | ||
| Scheme 18 Hydrometallation of non-activated terminal alkenes and the following SNAr alkylation of benzene promoted by calcium hydride dimer 95 (Maron, 2017). | ||
Hill and Maron demonstrated that under a hydrogen atmosphere, β-diketiminato calcium hydride dimer 95 could function as the catalyst for the hydrogenation of terminal alkenes such as allylbenzene (100) as well as some activated internal alkenes such norbornene (13) (Scheme 19A).46 Based on the experimental observations and the DFT calculations, it was proposed that the dimeric structure of the calcium complex is likely maintained during the catalytic cycle (Scheme 19B). Upon insertion of one molecule of alkene to calcium hydride dimer 95, the resulting alkyl hydride dicalcium complex 103 liberates the reduced alkane via subsequent deprotonation of H2. Alternatively, the alkyl hydride dicalcium complex 103 can also undergo the second alkene insertion to generate the dialkyl complex 104, prior to deprotonation of H2 to release the reduced alkane with the regeneration of the dimeric calcium hydride species 95.
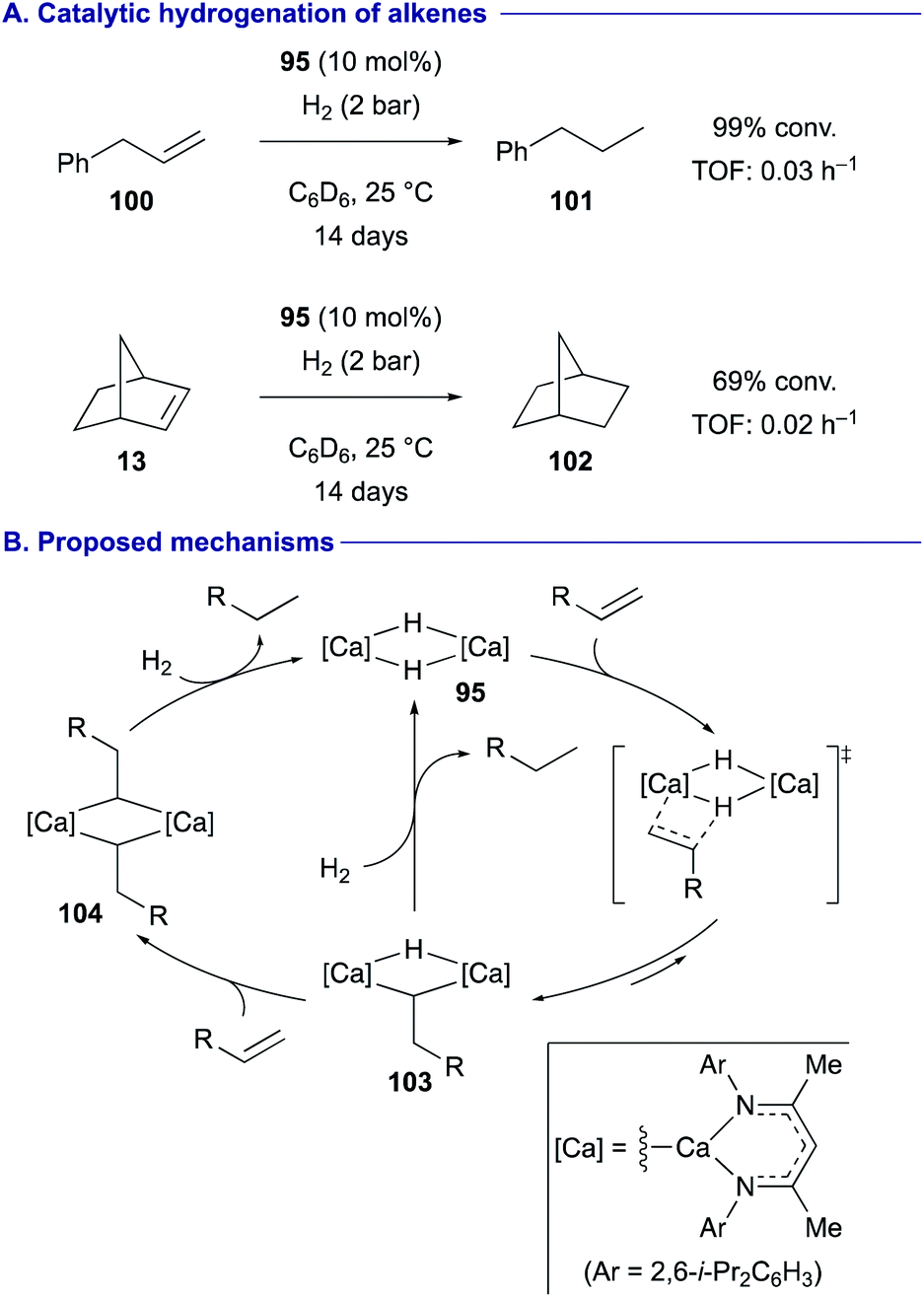 | ||
| Scheme 19 Hydrogenation of alkenes catalyzed by calcium hydride dimer 95 (Hill and Maron, 2018). | ||
Amidinato calcium hydride dimer complex 105 developed by Harder47 was found to react with diphenylacetylene (45) at 80 °C in benzene to form organocalcium complex 106 symmetrically bridged by a stilbene dianion (Scheme 20A).48 Oxidation of 106 with I2 gave trans-stilbene (47) with calcium iodide dimer 107. In turn, the treatment of 106 under a H2 atmosphere (6 bar) resulted in the formation of 1,2-diphenylethane (108) along with the regeneration of the calcium hydride dimer complex 105via deprotonation of H2. Thus, the organocalcium complex 105 could be employed as the catalyst for hydrogenation of diphenylacetylene (45) to 1,2-diphenylethane (108) under a H2 atmosphere (6 bar) (Scheme 20B).
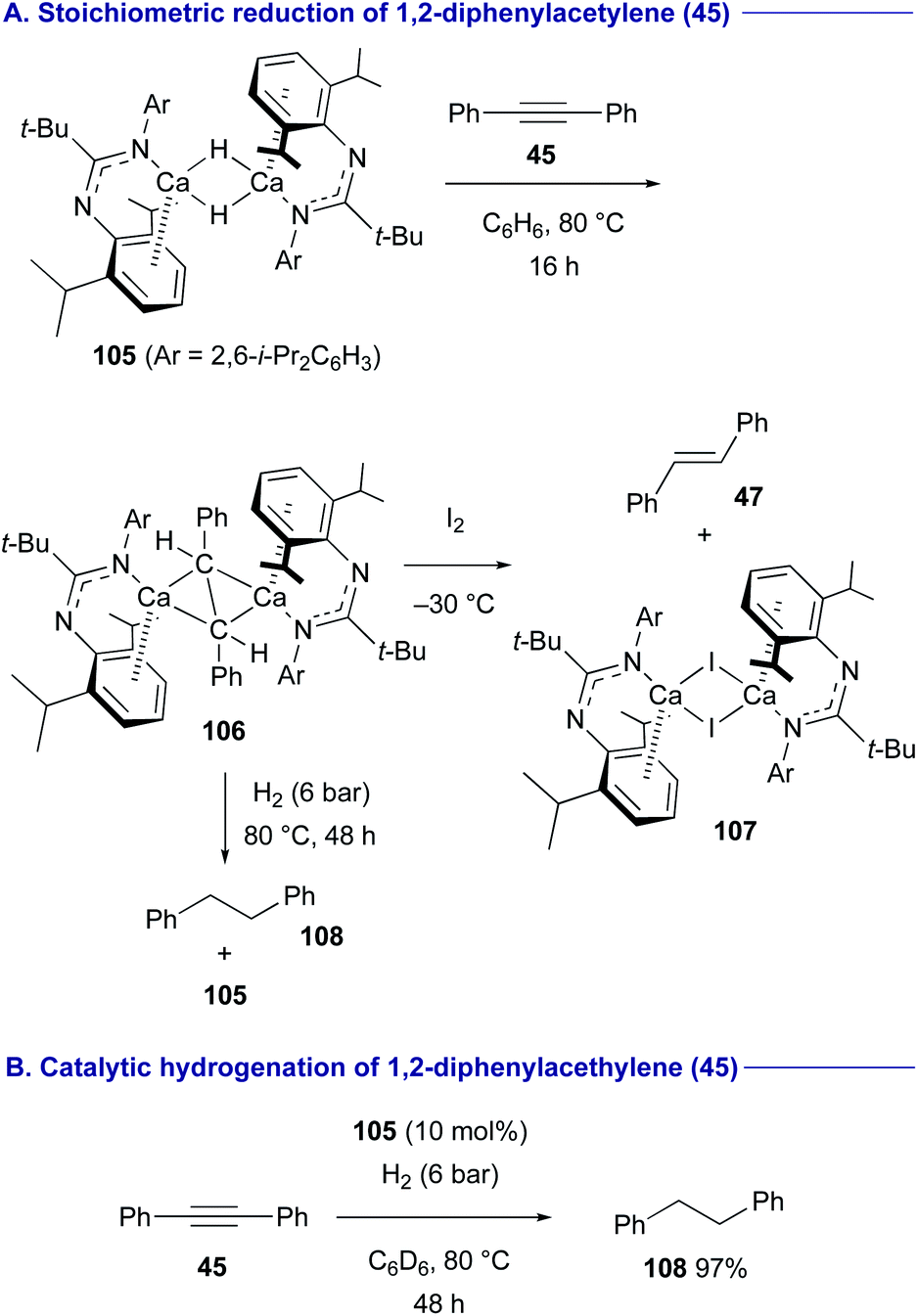 | ||
| Scheme 20 Hydrogenation of alkynes with amidinato calcium hydride dimer 105 (Harder, 2017). | ||
Cheng revealed that benzylcalcium complex 109 with a trispyrazolyl borate ligand is converted into the mononuclear calcium hydride complex 110 under a H2 atmosphere (Scheme 21A).49 The resulting calcium hydride 110 could serve as an active catalyst for the hydrogenation of alkenes including non-activated internal alkenes such as trans-2-octene (111) and cyclooctene (113) (Scheme 21B).
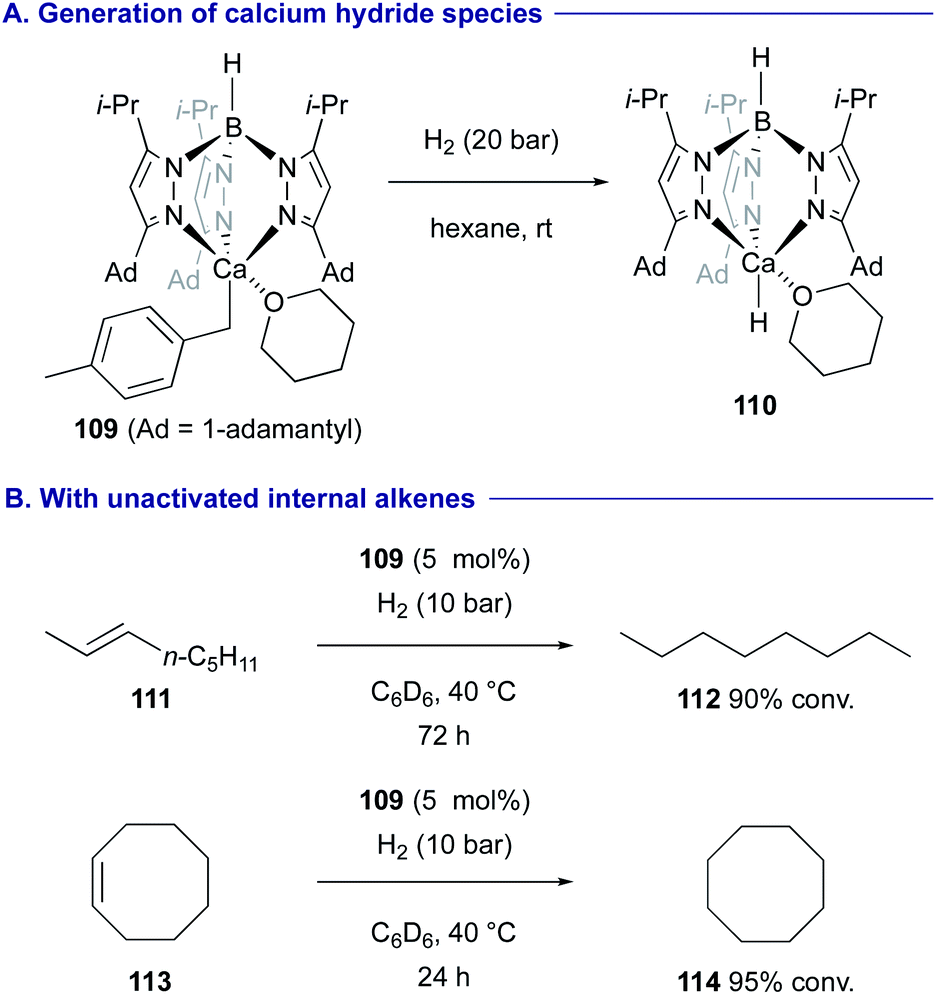 | ||
| Scheme 21 Hydrogenation of alkenes with mononuclear calcium hydride 110 (Cheng, 2020). | ||
A strontium hydride dimer 115 having extremely bulky β-diketiminato ligands was found to display higher hydridic reactivity toward benzene by Harder (Scheme 22).50 The stoichiometric reaction of 115 with C6D6 induces unprecedented hydride addition to C6D6 to form dearomatized anionic intermediate 116, which undergoes re-aromatization via elimination of deuteride to form strontium deuteride dimer 115-d2 and C6D5H 117 (Scheme 22A). On the other hand, the strontium hydride dimer 115 performed fast deprotonative H–D exchange under a D2 atmosphere (1.5 bar) at ambient temperature. Thus, these reactivities could be combined to develop catalytic deuteration of benzene under a D2 atmosphere by 115 (Scheme 22B). In turn, under an ethylene atmosphere (1 bar) in C6D6 at ambient temperature, the strontium hydride dimer 115 is quickly converted into alkylstrontium complexes 118 having ethyl, butyl, hexyl and higher ethylene oligomers, which could be confirmed by the detection of the corresponding alkanes 119 by GC-MS analysis. Furthermore, these alkylstrontium complexes 118 undergo nucleophilic aromatic substitution with C6D6 to form alkylated arenes 120 and 115-d2 (Scheme 22C).
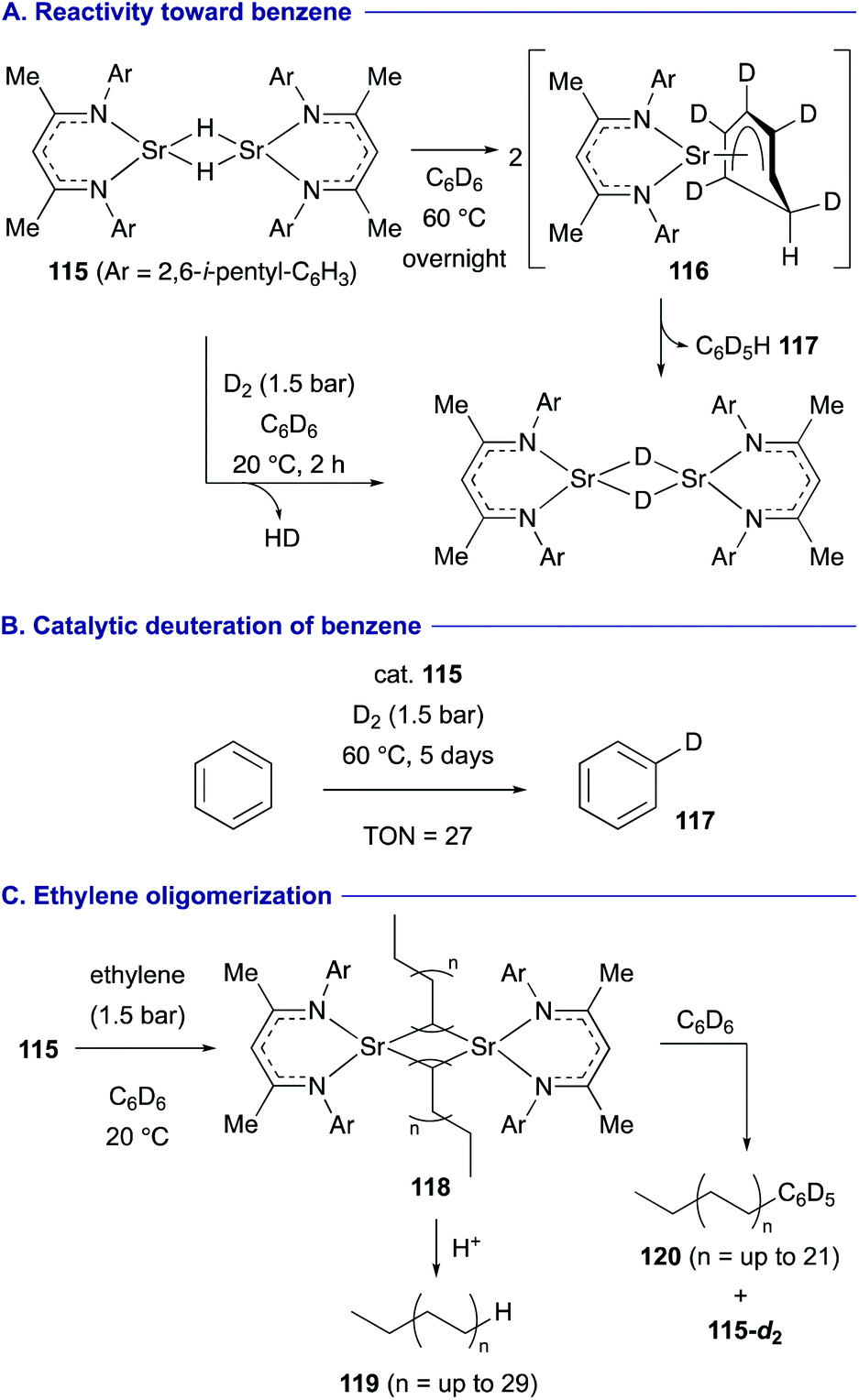 | ||
| Scheme 22 Nucleophilic hydrogenation and alkylation of benzene promoted by strontium hydride dimer 115 (Harder, 2019). | ||
Harder recently exploited alkaline earth metal complexes 121–124 having an extremely bulky bis(triisopropylsilyl)amide ligand as the precatalyst for the alkaline earth metal hydrides capable of catalytic hydrogenation of alkenes under a H2 atmosphere (Scheme 23A).51 The barium complex 124 was found to be most reactive, performing hydrogenation of even unactivated internal alkenes such as trans-3-hexene (125) (Scheme 23B). Moreover, the barium amide 124 complex could perform hydrogenation of conjugated arenes such as naphthalene (126) and biphenyl (128) to tetralin (127) and phenylcyclohexane (129), respectively (Scheme 23C). This catalytic system was found to be amenable to the hydrogenation of benzene to cyclohexane (130) (Scheme 23D). With 2.5 mol% of 124 under higher H2 pressure (50 bar) at 140 °C, 18% conversion of benzene to cyclohexane (130) was attained after 3 days. On the other hand, treatment of benzene with 1 mol% of 124 under milder pressure of D2 (10 bar) at 120 °C allowed for H/D exchange of benzene at 37% incorporation rate within 4 h.52
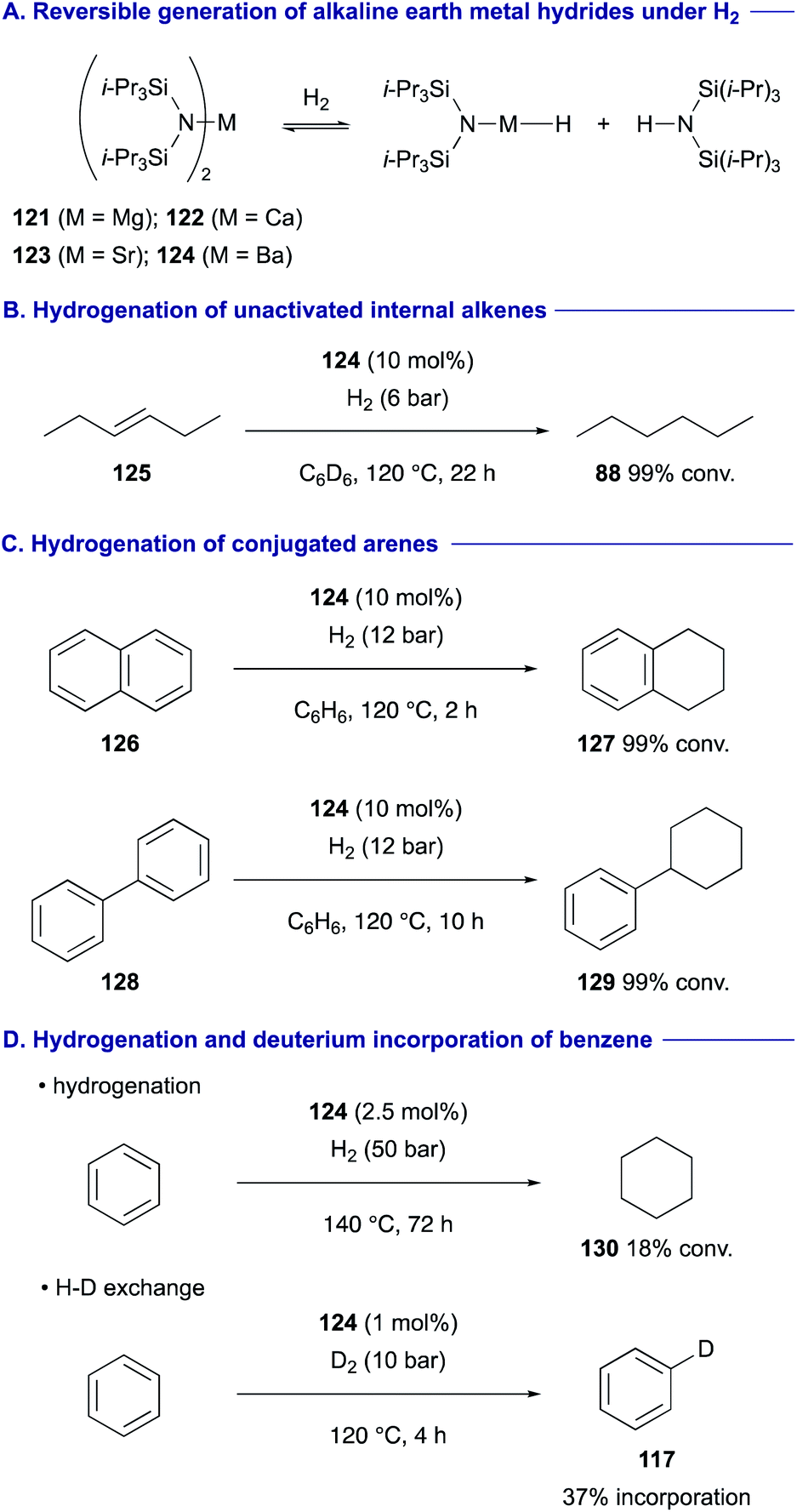 | ||
| Scheme 23 Hydrogenation of non-activated alkenes and arenes with a barium hydride complex having a bis(triisopropylsilyl)amide ligand (Harder, 2020). | ||
4. Conclusions
In this perspective, we have highlighted the protocols for the generation of organomagnesium complexes from non-polar unsaturated compounds and their unique reactivities in chemical synthesis and catalysis. The key enabling advance in these transformations takes advantage of well-defined molecular magnesium(II) hydrides or magnesium(I) dimer complexes, which are supported by the sophisticated sterically hindered anionic ligands. The method for in situ generation of active magnesium hydrides via either σ-bond metathesis between pinacolborane and dibutylmagnesium or counter ion metathesis between sodium hydride and magnesium iodide without the use of any supporting ligands also enabled concise transformation of alkynes and 1,3-enynes into various scaffolds via hydromagnesiation and ensuing downstream functionalization with electrophiles. Engagement of heavier alkaline earth metal hydride complexes having well-defined bulky ligands allowed for metallation of non-activated internal alkenes and arenes including benzene. The resulting organo-heavier alkaline earth metal complexes have shown exotic and unique reactivities in the downstream functionalization. Given the versatile reactivities of organo-alkaline earth metal complexes to drive the unprecedented chemical processes which have been dominated by transition-metal catalysts, we view that more unique and capable synthetic methods, especially catalysis, that leverage organo-alkaline earth metal complexes as the key components, will be devised and engaged in various synthetic endeavours.Author contributions
S. C. and R. T. conceived the contents of the perspective. All the authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the Nanyang Technological University (NTU) and the Singapore Ministry of Education (Academic Research Fund Tier 2: MOE2019-T2-1-089) (for S. C.) as well as JSPS KAKENHI grant JP19K06992 (for R. T.).Notes and references
- D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- The Chemistry of Organomagnesium Compounds, ed. Z. Rappaport and I. Marek, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- J. F. Garst and M. P. Soriaga, Coord. Chem. Rev., 2004, 248, 623–652 CrossRef CAS.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2014, 114, 1207–1257 CrossRef CAS.
- T. Klatt, J. T. Markiewicz, C. Sämann and P. Knochel, J. Org. Chem., 2014, 79, 4253–4269 CrossRef CAS.
- H. Ila, O. Baron, A. J. Wagner and P. Knochel, Chem. Lett., 2006, 35, 2–7 CrossRef CAS.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- M. Balkenhohl and P. Knochel, SynOpen, 2018, 2, 78–95 CrossRef CAS.
- P. Knochel, M. A. Schade, S. Bernhardt, G. Manolikakes, A. Matzger, F. M. Piller, C. J. Rohbogner and M. Mosrin, Beilstein J. Org. Chem., 2011, 7, 1261–1277 CrossRef CAS.
- D. Mukherjee and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 1458–1473 CrossRef CAS.
- Physical Constants of Organic Compounds, in CRC Handbook of Chemistry and Physics, ed. J. R. Rumble, CRC Press/Taylor & Francis, Boca Raton, FL, 102nd edn (Internet Version), 2021 Search PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- M. Westerhausen, Angew. Chem., Int. Ed., 2008, 47, 2185–2187 CrossRef CAS.
- S. P. Green, G. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS.
- For the generation of zinc hydrides through counter ion metathesis between NaH and zinc halides, see: D. Y. Ong, Z. Yen, A. Yoshii, J. R. Imbernon, R. Takita and S. Chiba, Angew. Chem., Int. Ed., 2019, 58, 4992–4997 CrossRef CAS.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- M. Westerhausen, A. Koch, H. Görls and S. Krieck, Chem.–Eur. J., 2017, 23, 1456–1483 CrossRef CAS.
- M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516–1531 CrossRef CAS.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- M. Rauch, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2017, 139, 13264–13267 CrossRef CAS.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed.
- L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Chem. Sci., 2019, 10, 8108–8118 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS.
- A. J. Boutland, A. Carroll, C. A. Lamsfus, A. Stasch, L. Maron and C. Jones, J. Am. Chem. Soc., 2017, 139, 18190–18193 CrossRef CAS PubMed.
- K. Yuvaraj, I. Douair, L. Maron and C. Jones, Chem.–Eur. J., 2020, 26, 14665–14670 CrossRef CAS.
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2020, 142, 11967–11971 CrossRef CAS PubMed.
- J. Li, M. Luo, X. Sheng, H. Hua, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, Org. Chem. Front., 2018, 5, 3538–3547 RSC.
- J. W. Clary, T. J. Rettenmaier, R. Snelling, W. Bryks, J. Banwell, W. T. Wipke and B. Singaram, J. Org. Chem., 2011, 76, 9602–9610 CrossRef CAS PubMed.
- M. Magre, B. Maity, A. Falconnet, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 7025–7029 CrossRef CAS PubMed.
- Dibutylmagnesium was also found to function as a catalyst to induce a regio- and stereoselective hydrostannylation of both internal and terminal alkynes, while this process unlikely involves magnesium hydride species, see: M. Magre, M. Szewczyk and M. Rueping, Org. Lett., 2020, 22, 1594–1598 CrossRef CAS.
- B. Wang, D. Y. Ong, Y. Li, J. H. Pang, K. Watanabe, R. Takita and S. Chiba, Chem. Sci., 2020, 11, 5267–5272 RSC.
- B. Wang, Y. Li, J. H. Pang, K. Watanabe, R. Takita and S. Chiba, Angew. Chem., Int. Ed., 2021, 60, 217–221 CrossRef CAS PubMed.
- G. Bartoli, E. Marcantoni and M. Petrini, J. Org. Chem., 1992, 57, 5834–5840 CrossRef CAS.
- G. Bartoli, E. Marcantoni, M. Petrini and R. Dalpozzo, J. Org. Chem., 1990, 55, 4456–4459 CrossRef CAS.
- Y. Li, J. S. Ng, B. Wang and S. Chiba, Org. Lett., 2021, 23, 5060–5064 CrossRef CAS.
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed.
- D. D. L. Jones, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 7087–7092 CrossRef CAS.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS.
- D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 59, 310–314 CrossRef CAS.
- T. Höllerhage, D. Schuhknecht, A. Mistry, T. P. Spaniol, Y. Yang, L. Maron and J. Okuda, Chem.–Eur. J., 2021, 27, 3002–3007 CrossRef.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS.
- S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 16368–16388 CrossRef CAS.
- A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed.
- A. Causero, G. Ballmann, J. Pahl, H. Zijlstra, C. Färber and S. Harder, Organometallics, 2016, 35, 3350–3360 CrossRef CAS.
- A. Causero, H. Elsen, G. Ballmann, A. Escalona and S. Harder, Chem. Commun., 2017, 53, 10386–10389 RSC.
- X. Shi, C. Hou, L. Zhao, P. Deng and J. Cheng, Chem. Commun., 2020, 56, 5162–5165 RSC.
- B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef.
- J. Martin, C. Knüpfer, J. Eyselein, C. Färber, S. Grams, J. Langer, K. Thum, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 9102–9112 CrossRef CAS.
- J. Martin, J. Eyselein, S. Grams and S. Harder, ACS Catal., 2020, 10, 7792–7799 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |