
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
19F-centred NMR analysis of mono-fluorinated compounds†
Alan J. R. Smith,
Richard York,
Dušan Uhrín and
Nicholle G. A. Bell*
and
Nicholle G. A. Bell*
EaStCHEM School of Chemistry, University of Edinburgh, David Brewster Rd, Edinburgh, EH9 3FJ, UK. E-mail: Nicholle.Bell@ed.ac.uk
First published on 30th March 2022
Abstract
Addressing limitations of the existing NMR techniques for the structure determination of mono-fluorinated compounds, we have developed methodology that uses 19F as the focal point of this process. The proposed 19F-centred NMR analysis consists of a complementary set of broadband, phase-sensitive NMR experiments that utilise the substantial sensitivity of 19F and its far reaching couplings with 1H and 13C to obtain a large number of NMR parameters. The assembled 1H, 13C and 19F chemical shifts, values of JHF, JHH, and JFC coupling constants and the size of 13C induced 19F isotopic shifts constitute a rich source of information that enables structure elucidation of fluorinated moieties and even complete structures of molecules. Here we introduce the methodology, provide a detailed description of each NMR experiment and illustrate their interpretation using 3-fluoro-3-deoxy-D-glucose. This novel approach performs particularly well in the structure elucidation of fluorinated compounds embedded in complex mixtures, eliminating the need for compound separation or use of standards to confirm the structures. It represents a major contribution towards the analysis of fluorinated agrochemicals and (radio)pharmaceuticals at any point during their lifetime, including preparation, use, biotransformation and biodegradation in the environment. The developed methodology can also assist with the investigations of the stability of fluoroorganics and their pharmacokinetics. Studies of reaction mechanisms using fluorinated molecules as convenient reporters of these processes, will also benefit.
Introduction
Fluorine's unique properties, such as high electronegativity, strength of a single fluorine–carbon bond and small atomic radius, impart significant benefits to fluorinated organic molecules.1 Fluorination has been shown to enhance potency and/or specificity of molecular interactions, increase membrane permeability, modulate metabolism, moderate the pKa of proximal functionalities, influence conformation, stabilise inherently reactive functionalities and produce viable bioisosteres.2,3 Currently, about 20% of the commercial pharmaceuticals contain fluorine and the proportion of newly approved fluoro-pharmaceuticals is rising steadily.4,5 The proportion of fluoro-agrochemicals is even larger; 53% of all active agrochemicals registered during 1998–2020 belong to this category.6 Similarly, 18F is the most frequently used radioisotope in positron emission tomography radiopharmaceuticals.7 Fluorination also has the potential to become a useful tool for improving properties of fragrance and semiochemical molecules.8To capitalise on the ability of fluorine to improve molecular properties, there is a drive to design efficient and environmentally-safe chemical,9,10 enzymatic11 and chemo-enzymatic12–14 fluorination methods. To assist these efforts, efficient analytical methods for the characterisation of fluorinated molecules are required. 19F NMR spectroscopy plays a prominent role in this area due to the favourable properties of 19F, such as its high sensitivity, 100% natural abundance, large chemical shift dispersion, large and far-reaching spin–spin interactions and 13C induced 19F isotopic shifts.
The lack of background 19F signals, due to the scarcity of fluorinated endogenous compounds, makes 19F NMR perfect for the analysis of mixtures produced by chemical or chemoenzymatic reactions with minimum clean-up steps or compound separation required. 1D 19F NMR spectroscopy has been widely used in studies of biodegradation and biotransformation of fluorinated compounds15–21 mostly relying on the use of known standards15 or tabulated 19F chemical shifts. In a similar manner, 19F NMR has also been used for probing the mechanism and kinetics of chemical reactions, were fluorine is a convenient reporter of the processes taking place.22,23
In support of such wide ranging activities, we have developed a 19F-centred NMR approach for the analysis of mono-fluorinated compounds, taking 19F NMR beyond recording simple 1D NMR spectra. Put together, the information obtained allows the structure elucidation of fluorine-containing molecular moieties and complete structure determination of small fluorine-containing molecules. It is well suited for the studies of complex mixtures. The 19F-centred NMR shares similarities to the “NMR spy” approach developed for the analysis of complex mixtures of soil organic matter, where –O13CH3 tags are introduced to a subset of molecules.24–26 Nevertheless, there are significant differences between the two approaches. Firstly, fluorinated molecules already contain 19F and therefore do not require additional chemical modifications. Secondly, the fluorine atom is typically closer to the protons and carbons of an organic molecule than are the nuclei of the –O13CH3 group which, when combined with far reaching 19F couplings, allows to inspect parts of the molecule that are more remote from the 19F “tag.” The FESTA family of NMR experiments27–29 that relies on selective manipulation of individual 1H and 19F resonances illustrated this approach and provided 1H–19F chemical shift correlations and 1H–19F coupling constants when such spin manipulations were possible.
Our methodology utilises the far reaching 1H–19F and 19F–13C couplings to obtain 1H and 13C chemical shifts of nuclei multiple bonds away from the 19F atom, provides accurate values of numerous JHF, JFC, and JHH coupling constants and 13C induced 19F isotopic shifts from several purposely designed nonselective 2D NMR experiments. Their advantages over similar existing NMR experiments are highlighted. The 19F-centered approach is illustrated using 3-fluoro-3-deoxy-D-glucose, 1, which can be characterized as a simple mixture of two 19F-containing molecules. Application of this methodology to a very complex mixture of compounds produced by chloramination of a single fluorinated molecule is presented elsewhere.30
Experimental
The sample of 3-fluoro-3-deoxy-D-glucose (30 mg), 1, was dissolved in 600 μL of D2O (Merck, 99.9 atom% D) and placed into a 5 mm NMR tube. Spectra involving 19F were acquired at 300 K on a 400 MHz Bruker Avance III NMR spectrometers equipped with a TBO BB-H/F-D probe. A 1D 1H spectrum was acquired on an 800 MHz Bruker Avance III NMR spectrometer equipped with a TCI 5 mm probe. Parameters of the performed NMR experiments are presented in Table S1† and Bruker pulse sequences compatible with TopSpin 3 can be found in the ESI† (pp. 1–6).The following symbols are used to depict the pulse sequences in Fig. 1–6: the thin and thick filled rectangles represent high power 90° (1H, p1 or 19F, p3) and 180° (1H, p2) pulses, respectively. 1 ms adiabatic CHIRP pulses with a peak power of 10.3 kHz (p44, shaded trapezoid with an inclined arrow) were applied to 19F. A 20 ms 60 kHz CHIRP 1H pulse with a peak power of 2294 Hz (p32, trapezoid with inclined arrow) was used as part of the z-filter. A 500 μs CHIRP pulse (p14) and 2 ms composite CHIRP pulse (p24) were applied to 13C with a peak power of 9800 Hz. Unless stated otherwise, the r.f. pulses were applied from the x-axis. The 100% pulsed field gradient strength corresponds to 53.5 G cm−1.
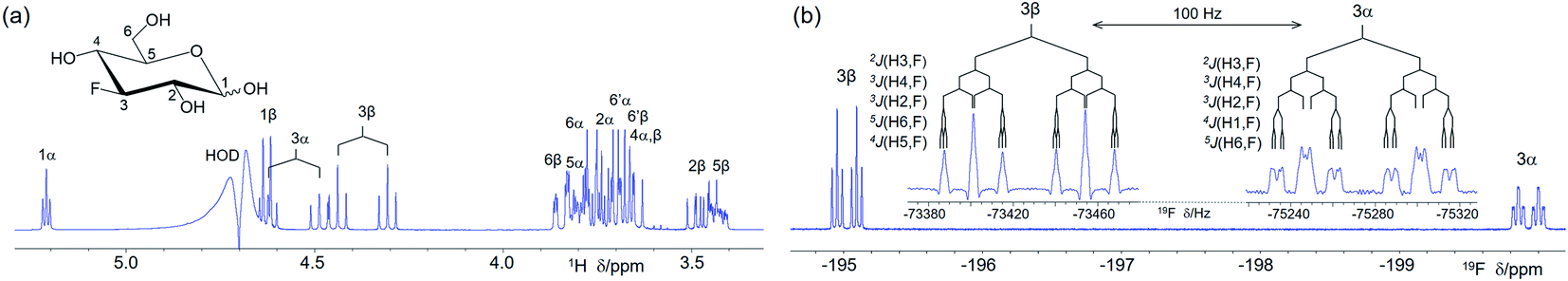 | ||
| Fig. 1 (a) 400 MHz 1D 1H spectrum of 1 (structure in the inset) with HOD suppression and resonance assignments; (b) 1H-coupled 1D 19F spectrum of 1. Expansions of 19F multiplets of both anomeric forms from resolution-enhanced spectra produced using Lorentzian to Gaussian line shape conversion (LB = −2.0 Hz, GB = 0.5) are given in the inset. | ||
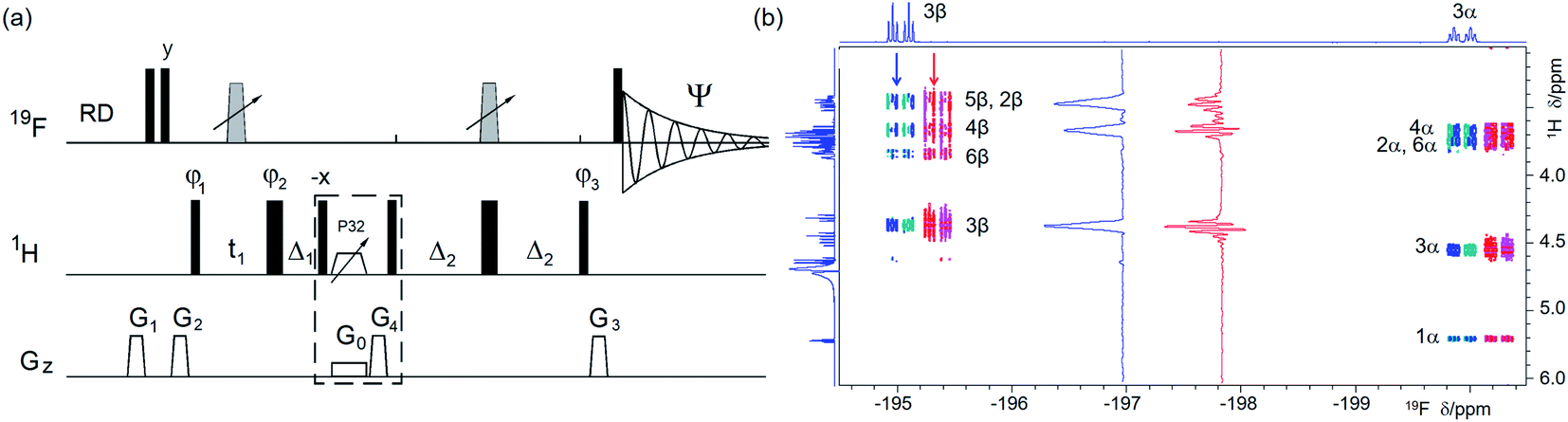 | ||
| Fig. 2 (a) Pulse sequence of a 19F-detected z-filtered 2D 1H, 19F HETCOR. In a non z-filtered experiment, the part within the dashed rectangle is not included. For explanation of symbols used for pulses see Experimental. The NMR parameters used are given in Table S1.† The delays used were as follows: Δ1 = p44; Δ2 = one half of the JHF evolution; t1(0), the initial t1 evolution delay time = 0.5 × in0, where in0 is the t1 increment. The gradient strengths were as follows: G0 = 3%; G1 = 17%; G2 = 31%; G3 = 24%; G4 = 10.0%. The following phase cycling was used: φ1 = x, −x; φ2 = 4x, 4(−x); φ3 = 2y, 2(−y); Ψ = x, 2(−x), x. States-TPPI protocol was used for sign discrimination in F1 with the phase φ1 incremented by 90°. Purging of 19F magnetisation at the beginning of the pulse sequence by a composite 90° 19F pulse and pulsed field gradients (PFGs) minimises the cancellation artefacts. (b) An overlay of the 19F-detected 2D 1H, 19F HETCOR spectra with (blue/turquoise) and without the z-filter (red/magenta). For clarity, the spectrum acquired without a z-filter was offset horizontally to the right. Insets show 1D F1 traces taken at positions indicated by arrows. 1D 1H and 19F spectra are shown along the left and top, respectively. | ||
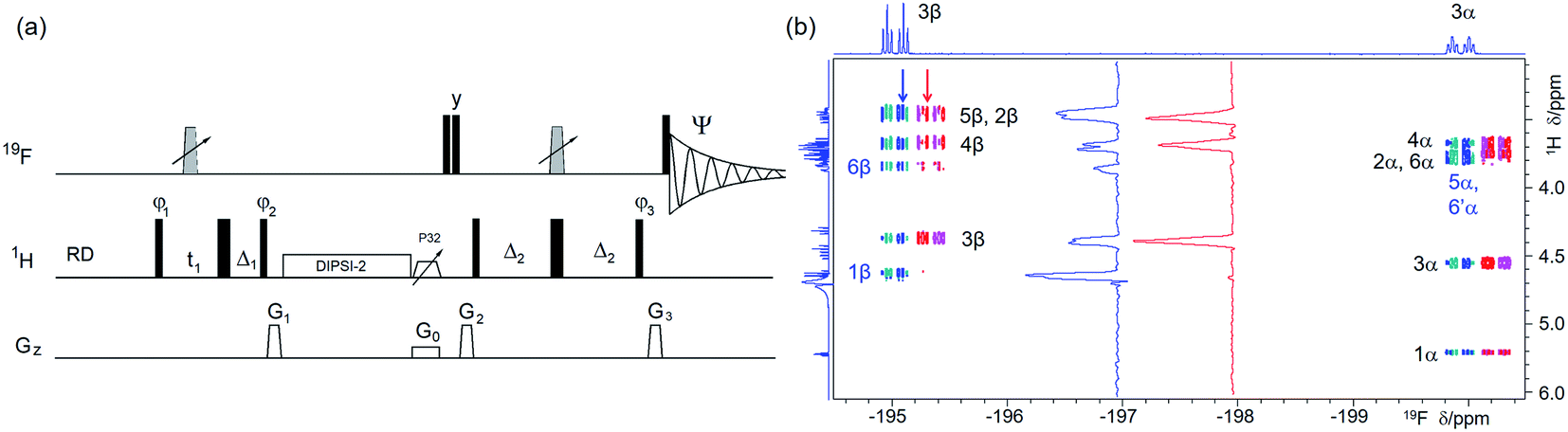 | ||
| Fig. 3 (a) Pulse sequence of a 2D 1H, 19F TOCSY–HETCOR. For explanation of symbols used for pulses see Experimental. The NMR parameters used are given in Table S1.†The delays were as follows: Δ1 = p44; Δ2 = one half of the JHF evolution; t1(0) is the initial t1 evolution delay time = 0.5 × in0, where in0 is the t1 increment. The gradient strengths were are follows: G0 = 5%; G1 = 17%; G2 = 31%; G3 = 24%. The following phase cycling was used: φ1 = x, −x; φ2 = 4x, 4(−x); φ3 = 2y, 2(−y); Ψ = x, 2(−x), x, −x, 2x, −x. States-TPPI protocol was used for sign discrimination in F1 with the phase φ1 incremented by 90°. Purging of 19F magnetisation after the z-filter by a composite 90° 19F pulse followed by the G2 PFG minimises the cancellation artefacts. (b) An overlay of the 19F-detected 2D 1H,19F TOCSY–HETCOR spectrum (blue/turquoise) and a z-filtered VT 19F-detected 2D 1H, 19F HETCOR spectrum (red/magenta, horizontally offset to the right) of 1 acquired with the pulse sequence shown in (a) and Fig. 2a, respectively. Vertical traces of the two spectra as indicated by arrows are shown in the inset. Exclusive/stronger TOCSY cross peaks are labelled in blue. 1D 1H and 19F spectra are shown along the left and the top, respectively. | ||
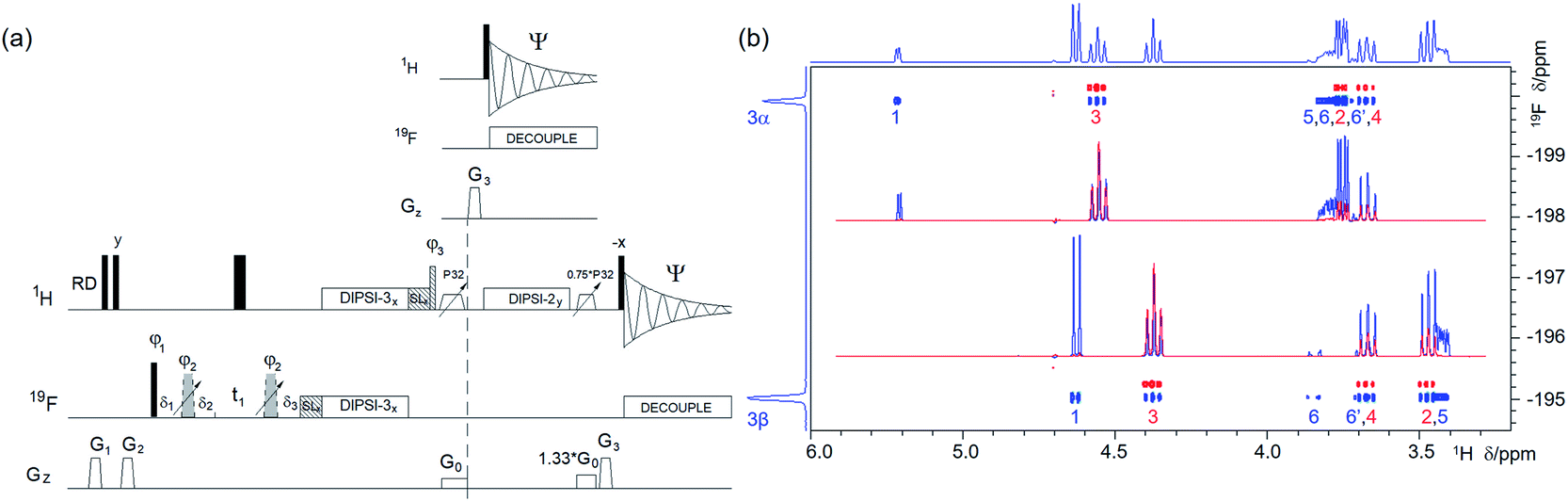 | ||
| Fig. 4 (a) Pulse sequence of a 2D 19F, 1H CP-DIPSI3–DIPSI2. For explanation of symbols used for pulses see Experimental. The NMR parameters used are given in Table S1.†The dashed line indicates signal acquisition before an optional 1H–1H spin-lock. For description of pulses see Experimental. The delays were as follows: δ1 = 20 μs; δ2 = δ1 + (2/π) × p3; δ3 = p2; t1(0) is the initial t1 evolution delay time = 0.5 × in0, where in0 is the t1 increment. The gradient strengths were as follows: G0 = 5%; G1 = 17%; G2 = 31%; G3 = 66%. The following phase cycling was used: φ1 = y, −y; φ2 = 4x, 4(−x); φ3 = 2y, 2(−y); Ψ = x, 2(−x), x. The states-TPPI protocol was used for sign discrimination in F1 with the phase φ1 incremented by 90°. Purging of 19F magnetisation at the beginning of the pulse sequence by a composite 90° 19F pulse and PFGs minimises the cancellation artefacts. (b) An overlay of two 2D 19F, 1H CP-DIPSI3–DIPSI2 spectra acquired with 20 ms 19F → 1H cross-polarisation (CP) only (red) and an additional 50 ms 1H → 1H spin-lock (blue) using the pulse sequence shown in (a). The red spectrum was offset vertically to facilitate visualisation of the cross peaks. The two insets show overlaid 1D F2 traces through 19F resonances of α- and β-D-glucose from both spectra. Twice as many scans were acquired for the blue spectrum as for the red spectrum. 1D 19F and 1H projections of the blue spectrum are shown along the left and top, respectively. | ||
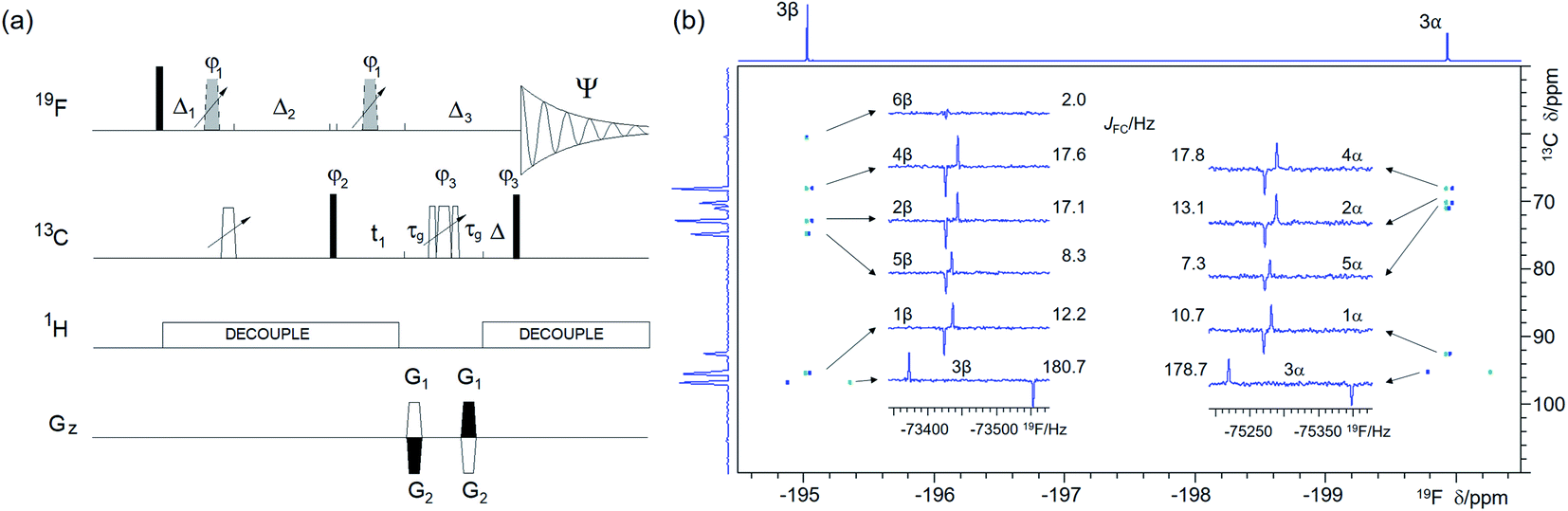 | ||
| Fig. 5 (a) Pulse sequence of the 2D 19F, 13C HMBC optimised for nJFC correlations. For explanation of symbols used for pulses see Experimental. The NMR parameters used are given in Table S1.† The delays were as follows: d6 = 0.25/nJFC; Δ = p44; Δ3 = 2 × p16 + 2 × d16 + p24 + Δ + 8 μs; Δ1 = d6 − Δ3/2; Δ2 = d6 + Δ3/2 − p14 + (2/π) × p1; t1(0) is the initial t1 evolution delay time = 0.5 × in0, where in0 is the t1 increment. The gradient strengths were are follows: G1 = 80%; G2 = cnst30 × G1, where cnst30 = (1 − sfo2/sfo1)/(1 + sfo2/sfo1) and sfo1 and sfo2 are 19F and 13C frequencies, respectively. The following phase cycling was used: φ1 = 2x, 2(−x); φ2 = x, −x; φ3 = 4x, 4(−x); Ψ = 2(x, −x), 2(−x, x). The echo-antiecho protocol was used with PFGs changing sign between real and imaginary increments. Phases φ2 and Ψ were incremented by 180° together with the PFG sign change, (b) A 2D 19F, 13C HMBC spectrum of 1 optimised for nJFC of 20 Hz acquired using the pulse sequence of shown in (a). The two insets show 1D F2 traces for individual 13C resonances of the α- and β-forms of 1. 1D 1H-decoupled 19F NMR spectrum and the 13C projection are shown on the top and along the left of the spectrum, respectively. | ||
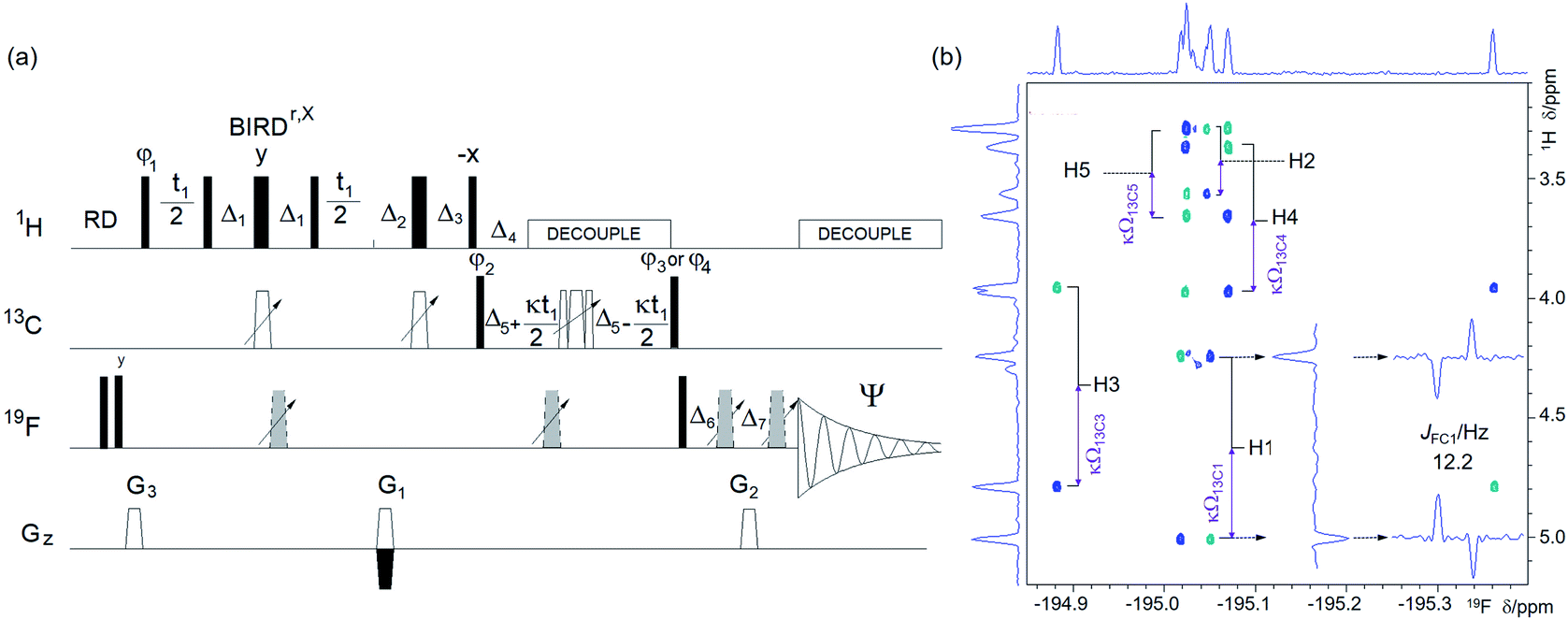 | ||
| Fig. 6 (a) Pulse sequence of the (3, 2)D H1CnF correlation experiment. For explanation of symbols used for pulses see Experimental. The NMR parameters used are given in Table S1.†The delays were as follows: d2 = 0.25/1JHC; d3 = 0.5/1JHC; d4 = 0.25/nJFC; d6 = cnst1/1JHC, where cnst1 = 0.5 for CH and 0.25 for CH2 groups; Δ1 = d3 − p14/2; Δ2 = d2 − p14/2 − p16 − d16; Δ3 = d2 − p14/2 − 2t1(0); Δ4 = d6; Δ5 = d4; Δ6 = p16 + d16 − (2/π)p1 + 4 μs; Δ7 = p16 + d16 + 4 μs, where p16 and d16 are the PFG length and the recovery time, respectively. The gradient strengths were as follows: G1 = 40%; G2 = 42.51%; G3 = 13%. The following phase cycling was used: φ1 = y, −y; φ2 = 4x, 4(−x); φ3 = 2x, 2(−x); φ4 = 2y, 2(−y); Ψ = x, 2(−x), x, −x, 2x, −x. The echo-antiecho protocol was used with G1 changing sign between real and imaginary increments. Phases φ1 and Ψ were incremented by 180° together with the sign change. Two interleaved experiments were acquired applying either the φ3 or φ4 phase to the last 90° 13C pulse, (b) an F1 antiphase (3, 2)D H1CnF spectra of 1 acquired using the pulse sequence shown in (a) showing the cross peaks of the β-anomer of 1. Positive and negative cross peaks are shown in blue and turquoise, respectively. The insets contain vertical and horizontal traces through the H1, F cross peaks. The 1H chemical shift of protons directly attached to 13C atoms and the associated κΩ13C frequencies are indicated. Antiphase doublets in F2 show nJFC coupling constants. Horizontal and vertical internal projections are shown on the top and along the left side of all spectra, respectively. The editing process that simplifies this spectrum is explained in the text and shown in Fig. S5.† | ||
Results and discussion
1D 1H and 19F spectra of 3-fluoro-3-deoxy-D-glucose, 1
A 400 MHz 1D 1H spectrum of 1 with the suppression of the HOD signal shows considerable overlap of 1H resonances (Fig. 1a). A 1H-coupled 1D 19F spectrum 1 (Fig. 1b) contains two 19F signals belonging to α- and β-anomeric forms of 1. The insets highlight numerous 1H–19F coupling constants of 1. NMR parameters of 1 obtained using the developed experiments, including those involving 13C, are presented in Table S2.†19F-centred NMR experiments – novelty and hardware requirements
Although a number of NMR experiments exist that correlate 19F chemical shifts with those of other nuclei,31 the majority of existing techniques yield magnitude mode spectra.32,33 Acquisition of pure-phase absorption signals in a phase-sensitive manner is much preferred, as it provides higher sensitivity and allows for accurate determination of coupling constants, including identification of the active coupling constants. Some existing phase-sensitive experiments yield complicated cross peak structures that lower their sensitivity.34,35The optimal performance of experiments constituting the 19F-centred NMR approach across a range of 19F frequencies, is ensured by the use of adiabatic inversion pulses.36,37 The experiments provide pure phase multiplets with simple structure afforded by 1H or 19F decoupling and were designed to minimise the effect of passive spins; they do not use refocusing intervals, which maximises their sensitivity. NMR hardware capable of pulsing simultaneously on 1H and 19F frequencies is required; fortunately, such systems are more widespread now. To access the rich information provided by 13C–19F interactions, a three-channel NMR spectrometer is necessary. Maximum benefits are realised on systems equipped with highly sensitive low temperature probes. These have also become more widely available, mainly due to their use in binding studies of biomacromolecules with fluorinated ligands.
Fluorine–proton and proton–proton correlation
Following the acquisition of 1H-decoupled and 1H-coupled 1D 19F spectra, mapping of the 1H–19F correlations is the natural next step in investigating the structure of fluorinated compounds. For this task a choice of three types of experiment exist: hetero-COSY, HETCOR or HMBC.31,32 Most of these can be implemented using 19F or 1H as the directly detected nucleus. Using 19F as the directly detected nucleus, the 2D 1H, 19F HMBC has the highest sensitivity, but yields mixed-phased multiplets. 2D 1H, 19F hetero-COSY can be implemented with either nucleus being sampled in the directly detected (F2) dimension. Nevertheless, sampling 19F in the F2 dimension has a distinct advantage of acquiring spectra with the high digital resolution required for the identification of active and passive JHF coupling constants and potentially also for their measurements. A disadvantage of COSY type spectra is the antiphase nature of their cross peaks (particularly in F1) and their large footprint.Choosing to obtain the 1H–19F correlations using a phase-sensitive 19F-detected 2D 1H, 19F HETCOR experiment (Fig. 2a) retains the advantages of 19F detection. Its uniform performance across a large 19F chemical shift range is guaranteed by the use of broadband inversion CHIRP pulses38 arranged in a double inversion adiabatic sweep (Fig. S1†), a feature applied in several experiments presented here to eliminate phase evolution of the transverse magnetisation during pulses.39–41 This allows the use of such pulses not only for spin inversion but also refocusing.
The structure of cross peaks in HETCOR spectra is simplified by the application of a 180° 19F pulse in the middle of the t1 interval, reducing the probability of signal overlap in spectra of complex mixtures. A drawback of this experiment is the evolution of 1H–1H couplings during the defocusing interval 2Δ2, which competes with the evolution of 1H–19F couplings, decreasing its sensitivity. This decrease can often be tolerated because of the 100% natural abundance of both nuclei.
Due to diverse sizes of JHF coupling constants, no attempt was made to refocus 19F magnetisation prior to detection and 1H decoupling was not applied during t2. Preserving the antiphase character of cross peaks is important, as it allows the identification of active couplings. Nevertheless, if a 1H-coupled 19F 1D spectrum is overlap free, it is advised to read the coupling constants from this spectrum, where accurate values are readily obtained (see Fig. 1b).
In a basic HETCOR experiment,32 the evolution of 1H–1H couplings during the 1H–19F defocusing interval, 2Δ2, leads to the appearance of mixed phase proton multiplets in F1 – a feature that is masked by the magnitude mode presentation of spectra. This issue was resolved in the proposed phase-sensitive experiment by inserting a z-filter42 after the t1 period, which separates the evolution of 1H–1H couplings during the t1 and the 2Δ2 defocusing interval. Providing the t1max is kept short (<30 ms), the cross peaks appear as singlets in F1. The described features of the experiment are illustrated on a 2D 1H, 19F HETCOR spectrum of 1 (Fig. 2b), where correlations with many 19F coupled protons are observed.
Protons not coupled by a sizable (>1.0 Hz) coupling constant to a 19F, but which are part of a spin system containing at least one 1H coupled to a 19F, are detected in a 2D 1H, 19F TOCSY–HETCOR experiment (Fig. 3a). Here, the 1H chemical shifts are labelled before their magnetisation is spread through the network of JHH coupled spins by a DIPSI-2 spin-lock.43 Part of the magnetisation that has reached the 19F-coupled protons is then transferred to 19F for detection in a subsequent HETCOR step. An overlay of the 2D 1H, 19F HETCOR and 2D 1H, 19F TOCSY–HETCOR spectra (Fig. 3b) revealed several protons with a JHF close to zero, which were not detected by the HETCOR experiment. Other protons of both anomeric forms of 1 coupled with small coupling constants to 19F showed increased intensities.
In addition to JHF coupling constants, JHH coupling constants provide important structural information that for complex mixtures is inaccessible by standard 2D experiments, but can be retrieved when some form of 19F editing is used. In principle, 1H–1H couplings modulate cross peaks in the F1 dimension of the 2D (TOCSY–)HETCOR experiments discussed above but in practice, typical t1 acquisition times used to record such spectra are too short to resolve them. The 1H–1H couplings are more likely to be resolved in the F2 dimension of 1H-detected experiments considering a non-refocused 2D 1H-detected 19F, 1H HETCOR, this experiment shows F2 multiplets with JHF and JHH coupling constants as anti-phase and inphase splitting, respectively, complicating access to JHH coupling constants (data not shown).
The JHH coupling constants can be measured more effectively from inphase proton multiplets acquired in the presence of 19F decoupling. Developed for simple mixtures of fluorinated compounds, this reasoning has led to the design of FESTA experiments.27–29 These 1D selective experiments require that both 19F and 1H multiplets are amenable to selective inversion, which is rarely the case for complex mixtures; experiments that do not rely on selective manipulations of spins are more robust.
A suitable alternative involving the use of 19F → 1H cross-polarisation (CP) that produces inphase 1H multiplets was already proposed in the form of a 3D CP 19F, 1H heteronuclear TOCSY experiment.44 We did not find it necessary to label the 1H chemical shifts after the initial 19F → 1H magnetisation transfer and present here a 2D version of this experiment in the form of a 2D 19F, 1H CP-DIPSI3–DIPSI2 (Fig. 4a). Here, the signal acquisition can start immediately after the z-filter42 that follows the CP step. Note that signals of protons not coupled to 19F can appear in the spectrum even at this point due to the 1H–1H TOCSY transfer that takes place simultaneously with the heteronuclear CP step.
This pulse sequence can be extended by a dedicated 1H–1H DIPSI-2 spin-lock propagating the magnetisation transfer to more remote parts of the spin system. Application of two z-filters and 19F decoupling ensures that pure inphase 1H multiplets are eventually acquired. DIPSI-3,45 using 40 μs 19F/1H pulses, was applied for the CP step covering a ±4 kHz frequency range with >75% efficiency. A slight improvement was achieved with the FLOPSY-16 mixing scheme46 covering ±4.7 kHz, i.e. 25 ppm of 19F resonances on a 400 MHz NMR spectrometer with >65% efficiency relative to the on-resonance signal (Fig. S2†). Further improvements, not explored here, can be achieved by using broadband pulses during the CP step.47
An overlay of two 400 MHz 2D 19F, 1H CP-DIPSI3–DIPSI2 spectra acquired with a 20 ms 19F → 1H cross-polarisation (red) and an additional 50 ms 1H → 1H spin-lock (blue) using the pulse sequence of Fig. 4a is presented in Fig. 4b. Both spectra are suitable for the determination of the JHH coupling constants. The former spectrum contains pure in phase multiplets of protons H2, 3 and 4 of 1, while the latter spectrum also shows all their other protons. Note the dominance of the H3 signals in the red spectra caused by an effective CP via large JH3F3 (∼50 Hz).
Fluorine–carbon correlation
Structure determination of sparsely protonated fluorinated molecules, such as heavily substituted aromatic rings, based only on 1H and 19F chemical shifts and coupling constants could be problematic. Thanks to the far-reaching 19F–13C couplings (nJFC, n = 1–5), many 19F-coupled 13C atoms can be identified by 2D 19F, 13C correlated experiments such as HMBC or HSQC, making structure determination of such molecules possible. A 2D 19F, 13C HSQC experiment33,41 was not considered in this study mainly because of a larger complexity of the double INEPT transfer. For small molecules, the slower relaxation of single-quantum (HSQC) relative to multiple-quantum (HMBC) coherences does not make a substantial difference to their sensitivity and for mono-fluorinated compounds F1 singlets are produced by both experiments.As the 1JFC coupling constants are large (∼150–250 Hz), while the n>1JFC typically range from 0 to 50 Hz,48 the one-bond (Fig. S3†) and long-range correlation (Fig. 5a) experiments are best performed separately. A single long-range optimised experiment can also yield one-bond correlations if multiple rotations of the 19F magnetisation vectors during the evolution interval fall outside of even multiples of 0.5/1JFC. This approach can only be used when values of 1JFC coupling constants are known, and if dealing with mixtures, their spread is narrow. Values of 1JFC coupling constants required for such optimisation can be obtained from 1D 1H-decoupled 19F spectra acquired with a sufficient S/N ratio. Alternatively, accordion optimisation49 can be used to obtain simultaneously both types of correlations. Both experiments perform best when 1H decoupling is applied during most of the pulse sequence. Such decoupling removes splitting of cross peaks by 1H–13C couplings in F1 and by JHF in F2. Resulting F1 singlets and F2 anti-phase doublets split by 19F–13C interactions (Fig. 5b) allow accurate measurement of JFC coupling constants that provide valuable structural information.
A comparison of 19F chemical shifts of 13C isotopomers obtained from 2D 19F, 13C HMBC spectra with the 19F signal in a 1D 1H-decoupled 19F spectrum yields 13C induced 19F isotopic shifts (see a large isotopic shift of C3 resonances in Fig. 5b). In aliphatic systems these decrease with the number of bonds separating the two atoms and are generally measurable to up to four bonds separating 13C and 19F. A careful alignment of the one-bond correlation trace from the pure phase HMBC spectrum and the satellites from the 1D 19F spectrum is required to obtain accurate values of these isotopic shifts.
Proton–carbon–fluorine correlation
1H–1H and 1H–13C interactions are the cornerstone of NMR structure determination of small molecules. For fluorinated compounds, the existence of 1H–19F and 19F–13C couplings makes this process even more robust. However, for complex mixtures, mapping of these interactions separately, can compromise identification of the nuclei belonging to individual molecules.This ambiguity can be avoided by correlating all three spin types in a dedicated HCF experiment. There are numerous possibilities for how such an experiment can be designed. Inspired by the 3D HNCA, a pulse sequence for assigning protein backbone resonances,50 a 1H-detected 3D triple-resonance 1H, 13C, 19F experiment has been proposed previously.51,52 This out-and-back 3D experiment contains JFC defocusing and refocusing intervals, samples 19F and 13C chemical shifts indirectly and applies simultaneous 13C and 19F decoupling during the direct detection of 1H. We prefer to use a unidirectional polarisation transfer pathway and direct detection of 19F; both of these features are well suited for molecules with a large spread of coupling constants, as is typical for 19F–13C interactions. The pulse sequence of such an experiment starts with a one-bond 1H–13C correlation step followed by a 13C, 19F long-range transfer step. It incorporates a reduced dimensionality approach53–55 and samples 13C chemical shifts simultaneously with the indirect labelling of 1H resonances. The resulting 2D experiment is referred to as (3, 2)D H1CnF, where the superscripts indicate the type of 13C and 19F interactions (1-one-bond, n-long-range) mediating the polarisation transfer (Fig. 6a).
In the (3, 2)D H1CnF experiment, the 1H chemical shifts are recorded first, while suppressing the evolution of 1H–1H and 1H–19F couplings by a BIRDr,X pulse56,57 and a 180° 19F pulse applied in the middle of the t1 period, respectively.
The magnetisation is then transferred in an INEPT step to 13C via one-bond 1H–13C couplings, where it is refocused before starting 1H decoupling. During the subsequent evolution interval, the 19F–13C anti-phase magnetisation is developed while the central 180° 13C and 19F pulses move simultaneously with the t1 incrementation. This causes modulation of 1H chemical shifts by 13C offsets, Ω13C (=δ(13C) − 13C r.f. carrier frequency) of their directly bonded 13C, splitting the signals into doublets centred at the 1H chemical shift. The size of 13C doublets can be scaled down relative to the t1 evolution (κ factor), keeping the F1 spectral width small and without any limitations for setting the length of the constant-time 19F–13C coupling evolution interval, 2Δ5.
The signal is finally transferred to 19F, where it is detected during t2 under 1H decoupling as a pure phase doublet in anti-phase with regard to JFC (Fig. 6b).
Interleaved acquisition of two spectra, differing by 90° in the phase of the last 90° 13C pulse of the pulse sequence, generates inphase and anti-phase F1 doublets, respectively, allowing spectra to be simplified by spectral editing58,59 as illustrated in Fig. S5.† A pulsed field gradient assisted echo-antiecho protocol is used to obtain pure phase signals in F1.
Overall, the reduced dimensionality experiment retains the full information content of 3D spectra with substantially increased digital resolution. Due to the use of a single nJFC evolution interval, sensitivity is also improved relative to the original 3D HCF experiment.51 Detecting 19F under 1H decoupling during t2 further increases sensitivity of this experiment, while providing values of JFC coupling constants. The (3, 2)D H1CnF experiment thus complements the 2D 19F, 13C HMBC technique discussed above and for protonated carbons correlates unambiguously three atom types, HCF, instead of aiming to achieve the same through a combined interpretation of 2D 1H, 13C HSQC and 2D 19F, 13C HMBC spectra, which for complex mixtures, is problematic.
Structure determination process in 19F-centred NMR
This process is briefly summarised with the help of a graphical representation in Fig. 7, using the β-anomeric form of 1 as an example. The 19F–1H correlations experiments, 2D 1H, 19F HETCOR and 2D 1H, 19F TOCSY–HETCOR spectra, together with 1D 1H-coupled/decoupled 19F spectra provided the parameters summarised in Fig. 7a, while 2D 19F, 1H CP-DIPSI3–DIPSI2 experiments extended the identified spin system to protons not directly coupled to fluorine (Fig. 7b).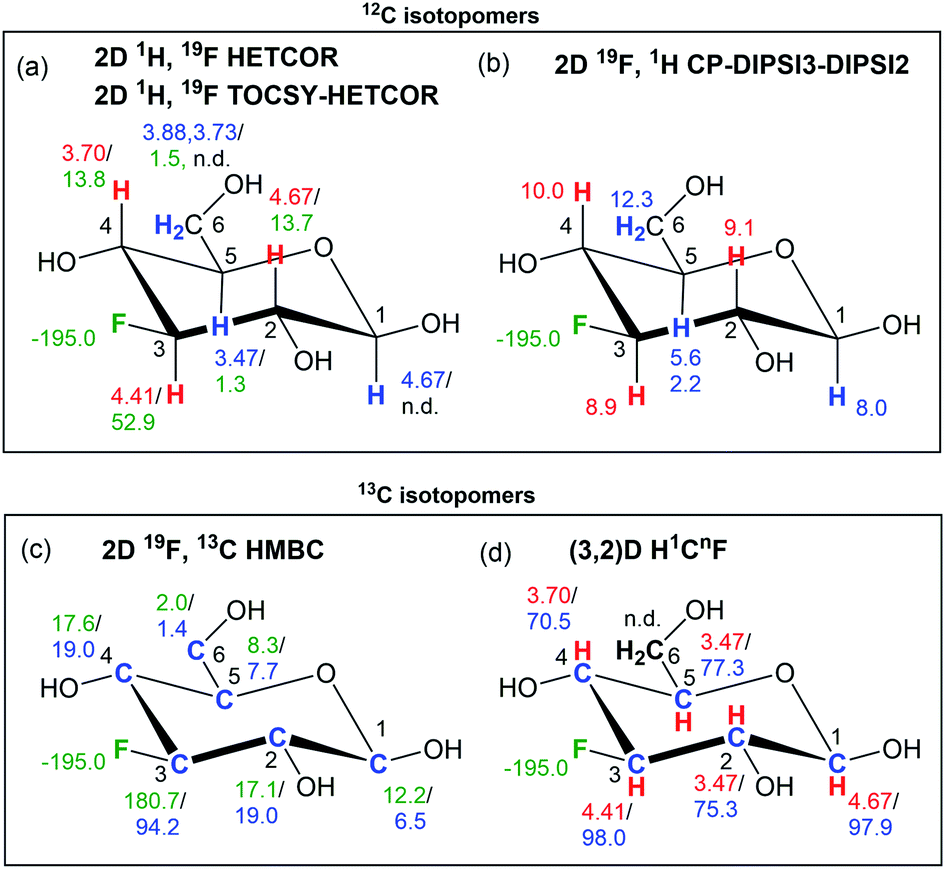 | ||
| Fig. 7 NMR parameters obtained by 19F-centred NMR for the β-anomeric form of 1. Chemical shifts, coupling constants and 13C isotopic shifts are given in ppm, Hz and ppb, respectively. (a) δ1H/nJHF; red and blue colours indicate correlations obtained from 2D 1H, 19F HETCOR and 2D 1H, 19F TOCSY–HETCOR spectra, respectively; (b) JHi,H(i+1), red and blue colour indicates correlation from 2D 19F, 1H CP-DIPSI3–DIPSI2 without and with the DIPSI-2 extension; (c) nJFC/Δ19F(13C); (d) δ1H/δ13C; n.d. – not detected. | ||
These experiments thus provide 19F and 1H chemical shift correlations together with nJHF (n = 2–4)60 and nJHH (n = 2–3) coupling constants, enabling the start of a structure determination process.
Experiments involving 19F–13C correlations are very informative. Central to these is the 2D 19F, 13C HMBC experiment, which provides long-range 19F–13C correlations and nJFC coupling constants and in conjunction with a 1D 1H decoupled 19F spectrum also the 13C induced 19F isotopic chemical shifts (Fig. 7c). The subsequent (3, 2)D H1CnF experiment provides correlations of HC pairs, in which the carbon is coupled to 19F, and if present, a distinction between non-protonated and protonated carbons (Fig. 7d).
Occasionally, a 2D 1H, 19F HOESY experiment31,61 can be used to identify protons not accessible by exploring J coupled networks of spins. In general, at this point, the chemical shift assignment and of numerous 1H, 13C and 19F resonances, values of JHF, JHH and JFC coupling constants and 13C induced 19F isotopic shifts are known and the structure determination of fluorine containing moieties can be completed.
For larger molecules, which contain spin systems isolated from those containing 19F, the 19F-centered approach provides a starting point by identifying protons and carbons that appear in both the 19F-centered and the standard 1H–1H and 1H–13C 2D chemical shift correlated spectra. These resonances can then be used to extend the structures and connect the fluorinated and non-fluorinated parts of molecules, e.g. via 1H–1H NOESY experiments or 1H–13C HMBC experiments, which can bridge such spin-systems. This approach is particularly beneficial for analyses of mixtures, where the identity of cross peaks belonging to the non-fluorinated parts of the molecule could be difficult to establish.
Although the discussed NMR experiments were developed for mono-fluorinated compounds, they can also be applied to compounds bearing more than one fluorine atom. Nevertheless, the presence of multiple 19F atoms should be taken into account when setting up some of the experiments, as the existence of passive 1H–19F (or 19F–13C) couplings need to be reflected in the parameters used as outlined in Table S3.†
It should be emphasised, that the 19F-centered approach takes full advantage of the high sensitivity of 19F to its environment and minute differences in the 19F chemical shift of the order of few Hz are sufficient to obtain the kind of information illustrated here on a very simple mixture provided by 1. Application of the 19F-centered approach to a very complex mixture of chloramination by-products is presented elsewhere.30
Conclusions
The described methodology is based on a concerted use of several NMR experiments, nevertheless, these can also be used in their own right. Collectively, these experiments represent the most effective NMR approach for the structure determination of mono-fluorinated compounds, particularly those contained in mixtures.The 19F-centred approach developed here is applicable at any point during the lifetime of fluorinated compounds, e.g. in analysing reaction mixtures during their production, performing mechanistic studies to understand reaction mechanisms and to optimise chemical reactions, investigating their stability, pharmacokinetics, biodegradation and biotransformation and ultimately to follow their fate in the environment.62
Data availability
The spectra obtained in this study are available here: https://doi.org/10.7488/ds/3422.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NGAB would like to acknowledge NERC soil security programme NE/N020227/1 for funding. AJRS was supported by Scottish Water and EPSRC grant EP/N509644/1 and RY by the NERC Centre for Doctoral Training, E4 (NE/S007407/1). Instrument support was in part provided by the EPSRC grant EP/R030065/1. The authors would like to thank Juraj Bella and Dr Lorna Murray for maintenance of the NMR spectrometers.References
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- B. M. Johnson, Y. Z. Shu, X. L. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- J. L. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O’Hagan, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- O. Jacobson, D. O. Kiesewetter and X. Y. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- P. T. Lowe and D. O’Hagan, J. Fluorine Chem., 2020, 230, 109420 CrossRef CAS.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- J. Fang, D. Hait, M. Head-Gordon and M. C. Y. Chang, Angew. Chem., Int. Ed., 2019, 58, 11841–11845 CrossRef CAS PubMed.
- T. Hayashi, G. Kehr, K. Bergander and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 3814–3818 CrossRef CAS PubMed.
- A. Rentmeister, F. H. Arnold and R. Fasan, Nat. Chem. Biol., 2009, 5, 26–28 CrossRef CAS PubMed.
- C. D. Murphy, Biotechnol. Lett., 2010, 32, 351–359 CrossRef CAS PubMed.
- B. D. Key, R. D. Howell and C. S. Criddle, Environ. Sci. Technol., 1997, 31, 2445–2454 CrossRef CAS.
- X. J. Zhang, T. B. Lai and R. Y. C. Kong, in Fluorous Chemistry, ed. I. T. Horvath, 2012, vol. 308, pp. 365–404 Search PubMed.
- M. G. Boersma, T. Y. Dinarieva, W. J. Middelhoven, W. J. H. van Berkel, J. Doran, J. Vervoort and I. Rietjens, Appl. Environ. Microbiol., 1998, 64, 1256–1263 CrossRef CAS PubMed.
- V. S. Bondar, M. G. Boersma, E. L. Golovlev, J. Vervoort, W. J. H. Van Berkel, Z. I. Finkelstein, I. P. Solyanikova, L. A. Golovleva and I. Rietjens, Biodegradation, 1998, 9, 475–486 CrossRef CAS PubMed.
- M. Kiel and K. H. Engesser, Appl. Microbiol. Biotechnol., 2015, 99, 7433–7464 CrossRef CAS PubMed.
- R. Natarajan, R. Azerad, B. Badet and E. Copin, J. Fluorine Chem., 2005, 126, 425–436 CrossRef.
- E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS PubMed.
- R. Wei, A. M. R. Hall, R. Behrens, M. S. Pritchard, E. J. King and G. C. Lloyd-Jones, Eur. J. Org. Chem., 2021, 2021, 2331–2342 CrossRef CAS.
- N. G. A. Bell, L. Murray, M. C. Graham and D. Uhrin, Chem. Commun., 2014, 50, 1694–1697 RSC.
- N. G. A. Bell, M. C. Graham and D. Uhrin, Analyst, 2016, 141, 4614–4624 RSC.
- G. A. Bell, A. A. L. Michalchuk, J. W. T. Blackburn, M. C. Graham and D. Uhrin, Angew. Chem., Int. Ed., 2015, 54, 8382–8385 CrossRef PubMed.
- L. Castanar, P. Moutzouri, T. M. Barbosa, C. F. Tormena, R. Rittner, A. R. Phillips, S. R. Coombes, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 5445–5450 CrossRef CAS PubMed.
- T. M. Barbosa, L. Castanar, P. Moutzouri, M. Nilsson, G. A. Morris, R. Rittner and C. F. Tormena, Anal. Chem., 2020, 92, 2224–2228 CrossRef CAS PubMed.
- G. Dal Poggetto, J. V. Soares and C. F. Tormena, Anal. Chem., 2020, 92, 14047–14053 CrossRef CAS PubMed.
- A. J. R. Smith, R. York, D. Uhrin and N. G. A. Bell, Chem. Sci., 2021 10.1039/D1SC06057K.
- J. Battiste and R. A. Newmark, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 1–23 CrossRef CAS.
- A. A. Marchione, R. J. Dooley and B. Conklin, Magn. Reson. Chem., 2014, 52, 183–189 CrossRef CAS PubMed.
- R. A. Newmark and R. J. Webb, J. Fluorine Chem., 2005, 126, 355–360 CrossRef CAS.
- K. A. M. Ampt, R. Aspers, P. Dvortsak, R. M. van der Werf, S. S. Wijmenga and M. Jaeger, J. Magn. Reson., 2012, 215, 27–33 CrossRef CAS PubMed.
- R. Aspers, K. A. M. Ampt, P. Dvortsak, M. Jaeger and S. S. Wijmenga, J. Magn. Reson., 2013, 231, 79–89 CrossRef CAS PubMed.
- A. A. Marchione and B. Conklin, Appl. Magn. Reson., 2017, 48, 485–499 CrossRef CAS.
- J. E. Power, M. Foroozandeh, P. Moutzouri, R. W. Adams, M. Nilsson, S. R. Coombes, A. R. Phillips and G. A. Morris, Chem. Commun., 2016, 52, 6892–6894 RSC.
- J. M. Bohlen, I. Burghardt, M. Rey and G. Bodenhausen, J. Magn. Reson., 1990, 90, 183–191 Search PubMed.
- T. L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- M. H. Levitt and R. Freeman, J. Magn. Reson., 1981, 43, 65–80 CAS.
- B. Adams, Magn. Reson. Chem., 2008, 46, 377–380 CrossRef CAS PubMed.
- M. J. Thrippleton and J. Keeler, Angew. Chem., Int. Ed., 2003, 42, 3938–3941 CrossRef CAS PubMed.
- S. P. Rucker and A. J. Shaka, Mol. Phys., 1989, 68, 509–517 CrossRef CAS.
- H. Hu, P. Kulanthaivel and K. Krishnamurthy, J. Org. Chem., 2007, 72, 6259–6262 CrossRef CAS PubMed.
- A. J. Shaka, C. J. Lee and A. Pines, J. Magn. Reson., 1988, 77, 274–293 Search PubMed.
- M. Kadkhodaie, O. Rivas, M. Tan, A. Mohebbi and A. J. Shaka, J. Magn. Reson., 1991, 91, 437–443 CAS.
- A. A. Marchione and E. L. Diaz, J. Magn. Reson., 2018, 286, 143–147 CrossRef CAS PubMed.
- H. Duddeck and M. R. Islam, Tetrahedron, 1981, 37, 1193–1197 CrossRef CAS.
- G. Bodenhausen and R. R. Ernst, J. Am. Chem. Soc., 1982, 104, 1304–1309 CrossRef CAS.
- L. E. Kay, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1990, 89, 496–514 CAS.
- L. Li and P. L. Rinaldi, Macromolecules, 1996, 29, 4808–4810 CrossRef CAS.
- L. L. Li, B. Zhang, F. Wyzgoski, X. H. Li, E. F. McCord and P. L. Rinaldi, ACS Macro Lett., 2013, 2, 141–145 CrossRef CAS.
- G. Bodenhausen and R. R. Ernst, J. Magn. Reson., 1981, 45, 367–373 CAS.
- Y. Shen, H. S. Atreya, G. H. Liu and T. Szyperski, J. Am. Chem. Soc., 2005, 127, 9085–9099 CrossRef CAS PubMed.
- W. Kozminski and I. Zhukov, J. Biomol. NMR, 2003, 26, 157–166 CrossRef CAS PubMed.
- J. R. Garbow, D. P. Weitekamp and A. Pines, Chem. Phys. Lett., 1982, 93, 504–509 CrossRef CAS.
- D. Uhrin, T. Liptaj and K. E. Kover, J. Magn. Reson., Ser. A, 1993, 101, 41–46 CrossRef CAS.
- N. Brodaczewska, Z. Kostalova and D. Uhrin, J. Biomol. NMR, 2018, 70, 115–122 CrossRef CAS PubMed.
- J. Sakas and N. G. A. Bell, Faraday Discuss., 2019, 218, 191–201 RSC.
- Hans Reich's Collection, 19F NMR Spectroscopy,https://organicchemistrydata.org/hansreich/resources/nmr/?index=nmr_index%2F19F_coupling#f-data08.
- P. L. Rinaldi, J. Am. Chem. Soc., 1983, 105, 5167–5168 CrossRef CAS.
- M. T. Anaraki, D. H. Lysak, K. Downey, F. V. C. Kock, X. You, R. D. Majumdar, A. Barison, L. M. Liao, A. G. Ferreira, V. Decker, B. Goerling, M. Spraul, M. Godejohann, P. A. Helm, S. Kleywegt, K. Jobst, R. Soong, M. J. Simpson and A. J. Simpson, Prog. Nucl. Magn. Reson. Spectrosc., 2021, 126–127, 121–180 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08046f |
| This journal is © The Royal Society of Chemistry 2022 |