DOI:
10.1039/D1QO01341F
(Review Article)
Org. Chem. Front., 2022,
9, 517-571
A brief overview of classical natural product drug synthesis and bioactivity
Received
8th September 2021
, Accepted 18th November 2021
First published on 20th November 2021
Abstract
Traditional medicines consisting of compounds derived from natural organisms have been used for human health care worldwide since ancient times. Since the last century, huge numbers of bioactive natural entities with diverse chemical scaffolds have been discovered, and some have been explored as clinical medications to treat various diseases. The advent of modern technologies has promoted the discovery of natural product-based pharmaceutical agents. The synthesis of natural products not only paves the way to confirm their molecular structures but also offers the structural modification opportunity to rationally optimize the drug-likeness parameters and evaluate the bioactivity of analogs. By providing a brief overview of a miscellaneous collection of complex natural products synthesis and the efforts of the structure–activity relationship, the present report aims to highlight the impact of chemical synthesis in natural product generation, diversification, bioactivity evaluation, and natural product-based drug development.
 Gen Li | Gen Li was born in Xinjiang, China. He received his B.Sc. (2004) in Chemistry from Xinjiang University and his Ph.D. (2011) degree in Chemical Biology from Peking University under the supervision of Professor Xin-Shan Ye. After a seven year research stage at Xinjiang Technical Institute of Physics and Chemistry, CAS, as an assistant professor and associate professor, he joined Qi's group at the National Institute of Biological Science (NIBS) at the beginning of 2019. His research interests focus on the total synthesis of natural products, and medicinal chemistry. |
 Mingliang Lou | Mingliang Lou was born in Anhui Province, China. He received his BS degree in Biology from Nankai University in 2014. In 2020, he received his Ph.D. degree in Medicinal Chemistry from Peking Union Medical College under the direction of Professor Xiangbing Qi. Currently, he works in the National Institute of Biological Sciences (NIBS) and focuses on natural product synthesis and drug development. |
 Xiangbing Qi | Xiangbing Qi was born in Shandong, China, and received his Ph.D. in chemistry and biochemistry from UT Southwestern Medical Center, Dallas, in 2005. After postdoctoral training at the University of Illinois Urbana-Champaign and medicinal chemistry research at UT Southwestern Medical Center, Dr Qi joined the faculty at the National Institute of Biological Sciences, Beijing, in 2013 and he is currently the Director of the Chemistry Center. The central theme of the research program is focused on the interface of synthetic chemistry, chemical biology, natural product total synthesis, and medicinal chemistry. |
1. Introduction
For millennia, nature has provided an invaluable source of traditional medicines from a variety of plants, animals, microorganisms, and minerals for humans to treat a wide spectrum of diseases. Since around 2600 BCE the successful use of natural medicines and remedies, particularly plant-originated substances, has been well documented in a large number of countries.1 The World Health Organization (WHO) estimated in 1985 that more than 4000 million people in the world relied mainly on traditional medicines for their primary health care.2 Since the 19th century, developments in the fields of phytochemistry and analytical chemistry have enhanced humans’ ability to examine natural materials more deeply and to identify pure bioactive constitutes to further leverage the application potential of natural products.
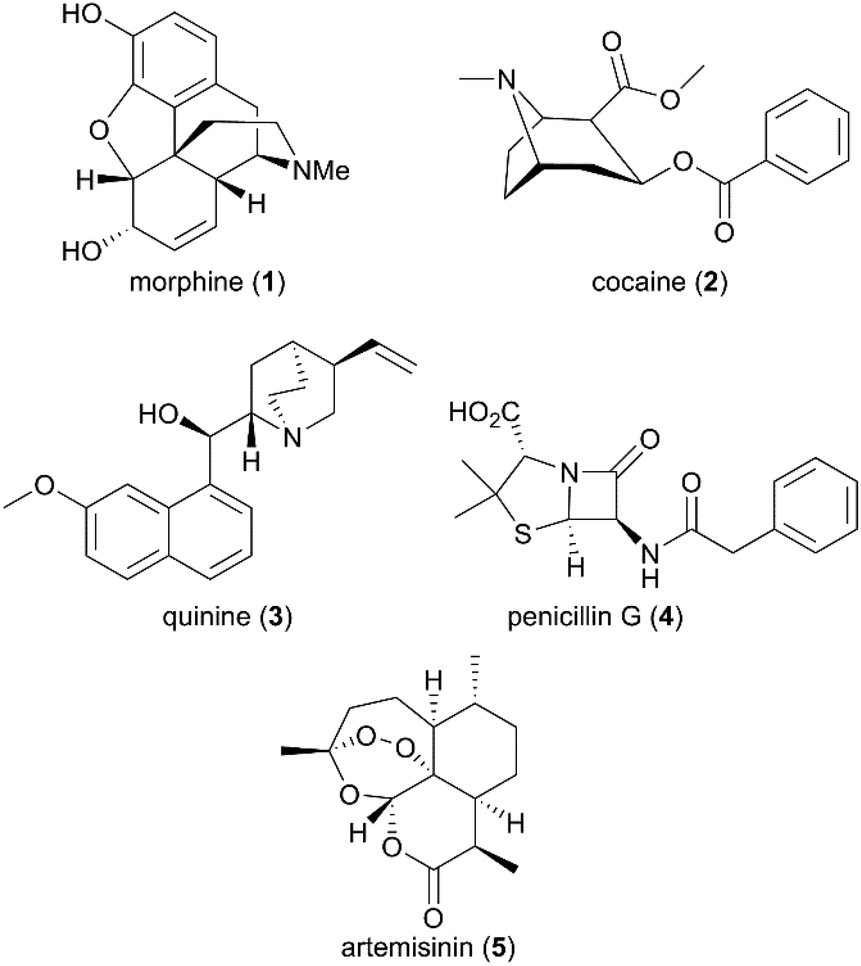
Beginning with the isolation of morphine, the first active principle from the opium poppy, by German pharmacist Friedrich Sertürner in 1806, and followed by its commercialization as an analgesic and sedative by Merck in 1827,3 several active natural products were subsequently purified, such as cocaine and quinine from plant coca and cinchona, respectively (Fig. 1).4 However, not until the last century did drug discovery-based natural products come of age due to the scientific breakthroughs in modern chemistry and medicine. The natural products pool was enriched with broader chemical structures and diverse spectrum bioactivities. Compared with conventional synthetic molecules, naturally generated products are characterized by more complex skeletons with a higher molecular mass and a larger number of sp3-hybridized atoms, which can be advantageous for the exploration of small molecule and biomolecule interactions.
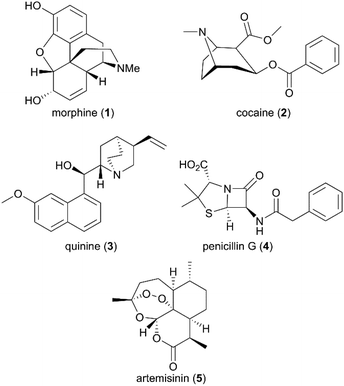 |
| | Fig. 1 Structures of morphine, cocaine, quinine, penicillin G, and artemisinin. | |
Despite the successful discovery of multiple drugs based on original natural products, such as morphine and artemisinin (Fig. 1), they still suffer from source and availability challenges. More importantly, the low success rate of the natural entity in approved drugs emphasizes the importance of structural optimization to improve natural products’ drug-like characteristics during drug development. With the development of new synthetic strategies and methodologies, more complex natural products have been assembled by efficient total synthesis or semisynthesis. The complementation of total synthesis strategies with semisynthetic transformations has been applied widely in the diversity- and bioactivity-oriented synthesis of drug-like entities.
Natural products and their synthetically modified analogs have historically made a remarkable contribution to the development of new drugs. A number of reports and books have reviewed the development of natural product-based drugs,1,5–13 including antibiotic drugs,14–16 antitumor agents,17–20 analgesics,21,22 and antiparasitic drugs.23–25 All the reviews mentioned above emphasize that original natural products play significant roles in the drug discovery and development process. This is also confirmed further in the recent survey provided in serial reviews from Newman and Cragg,5,26–29 which reported that only 463 purely synthetic drugs accounted for 24.6% of 1881 approved new chemical entities between 1981 and 2019. The rest of the new drugs on the market were either derived from the original natural structure or inspired by the rigidly complex skeletons of natural entities; 427 naturally occurring products (N, Newman and Cragg's category) and their structural derivatives (ND) were approved over almost 39 years.
Even though these reviews summarized nicely the natural product-based new drug development, druggability-related synthesis was not intensively tackled. We proposed to provide a brief overview of a miscellaneous collection of complex natural products synthesis and the endeavors of structure–activity relationships (SAR) studies to highlight the impact of chemical synthesis in natural product medicinal chemistry. Given the large number of approved natural product drugs in history, only the representative complex natural products whose chemical synthesis played an important role during the related drugs development are selected here. We admit that some natural products drugs, such as penicillin, vancomycin, ciclosporin, and rapamycin, are also classical and complex, but these are not covered due to limited space. Herein, the total synthesis of eight natural entities that were approved as drugs is presented according to the structural features, and the roles that total synthesis played in drug discovery and development are overviewed. The SAR studies are also discussed extensively.
2. Morphine and morphine derivatives
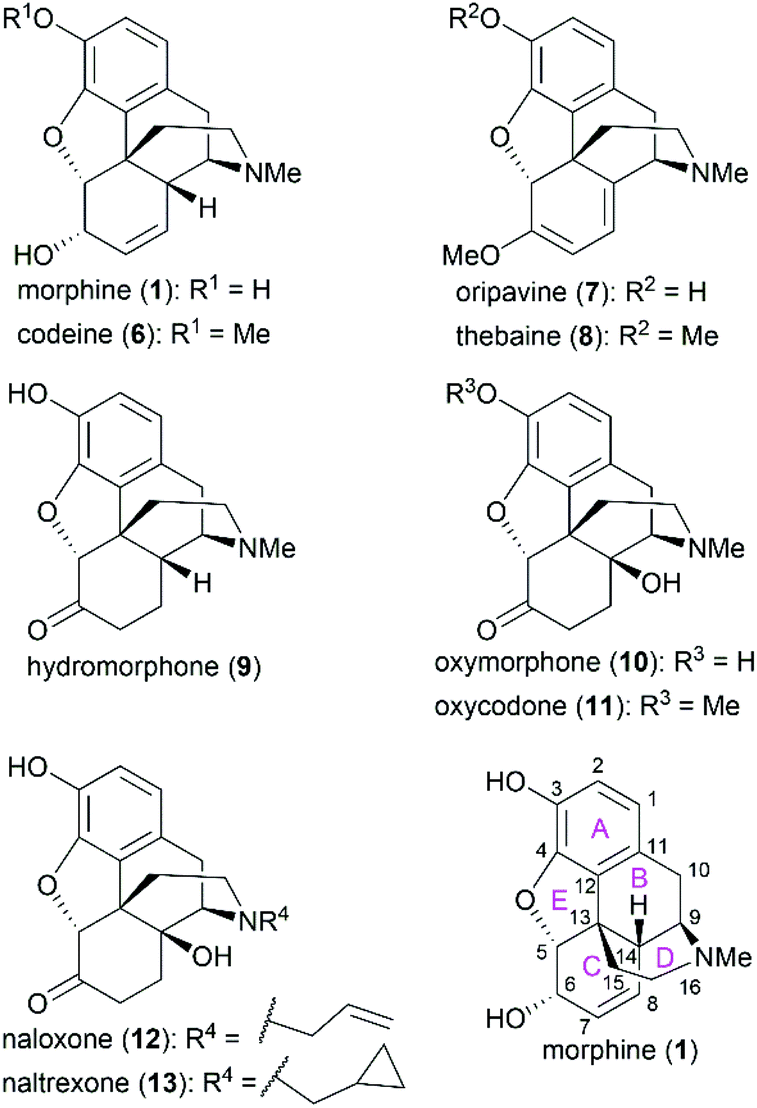
Morphine (1), isolated in 1806 by a German pharmacist, Friedrich Sertürner, is used primarily to treat both acute and chronic moderate-to-severe pain.30–34 It functions through selective binding to the μ-type opioid receptor in the brain and spinal cord.35,36 Many natural analogs and semisynthetic derivatives of morphine have been isolated or synthesized until now, some of which are presented in Fig. 2. The primary source of morphine and its congeners is isolation from the opium poppy, which is greatly affected by climate and political instability.32,37 Although the morphine biosynthesis pathway has been elucidated and the laboratory scale production of morphine in yeast has been realized, major challenges remain to be addressed before the realization of practical strategies for industrial-scale production.38,39 Therefore, total synthesis may be a plausible surrogate for extraction in the production of morphine and its derivatives.
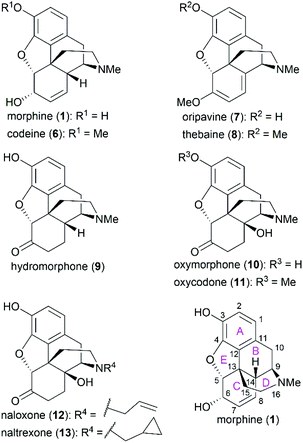 |
| | Fig. 2 Morphine and related compounds, and their ring lettering and carbon numbering system. | |
The structure of morphine was first proposed by Robinson in 1925, and this structure was supported by Gates’ first total synthesis of morphine in 1952 and later by X-ray analysis in 1954.40–43 As shown in Fig. 2, there are several intriguing structural features of morphine: a rigid pentacyclic bridged/fused ring structure (designated A, B, C, D, and E), five contiguous stereocenters (C5, C6, C9, C13, and C14) including one quaternary stereocenter (C13), an ether linkage between C4 and C5, two hydroxy groups (C3 and C6), a basic tertiary amine moiety, and a double bond between C7 and C8, which make its total synthesis a challenging task. The numerous studies on the synthesis of morphine alkaloids have led to over 40 synthesis routes, which have been summarized in several recent reviews.31,33,34,37,44,45 Selected representative routes for catalytic asymmetric total synthesis of morphine alkaloids will be discussed in the following section.
2.1 Catalytic asymmetric total synthesis of morphine alkaloids
2.1.1 Overman's total synthesis of (−)-morphine.
Overman and co-workers completed the first catalytic asymmetric synthesis of (−)-morphine (1).46 In their synthesis, enantioselective reduction of ketone 14 with catecholborane in the presence of (R)-oxazaborolidine catalyst (15) and subsequent condensation of the alcohol with phenyl isocyanate produced 16 (Scheme 1). The latter was transformed into 18 through two key operations: catalytic dihydroxylation of the terminal double bond and introduction of an allylsilane group, in which the desired C14 (following the numbering in morphine) stereocenter was established. Condensation of 18 with a tetrasubstituted A ring aldehyde 19 through the Mannich reaction generated ring D in 20. An intramolecular Heck reaction within 20 and subsequent removal of the benzyl group gave the tetracyclic compound 21 concomitant with the construction of the crucial quaternary C13 stereocenter. Ring E was constructed through epoxidation of the double bond and an epoxide ring-opening cyclization sequence. The pentacyclic compound 22 was transformed into (−)-morphine (1) following Rice's method.47
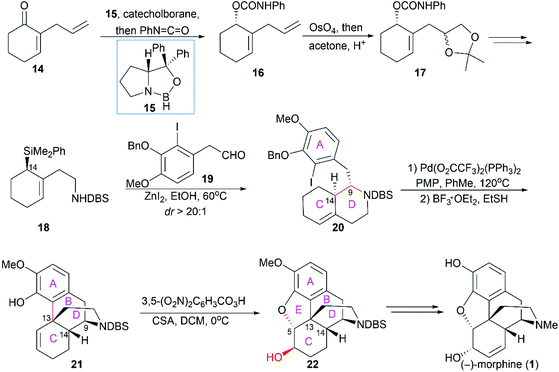 |
| | Scheme 1 Overman's synthetic route. | |
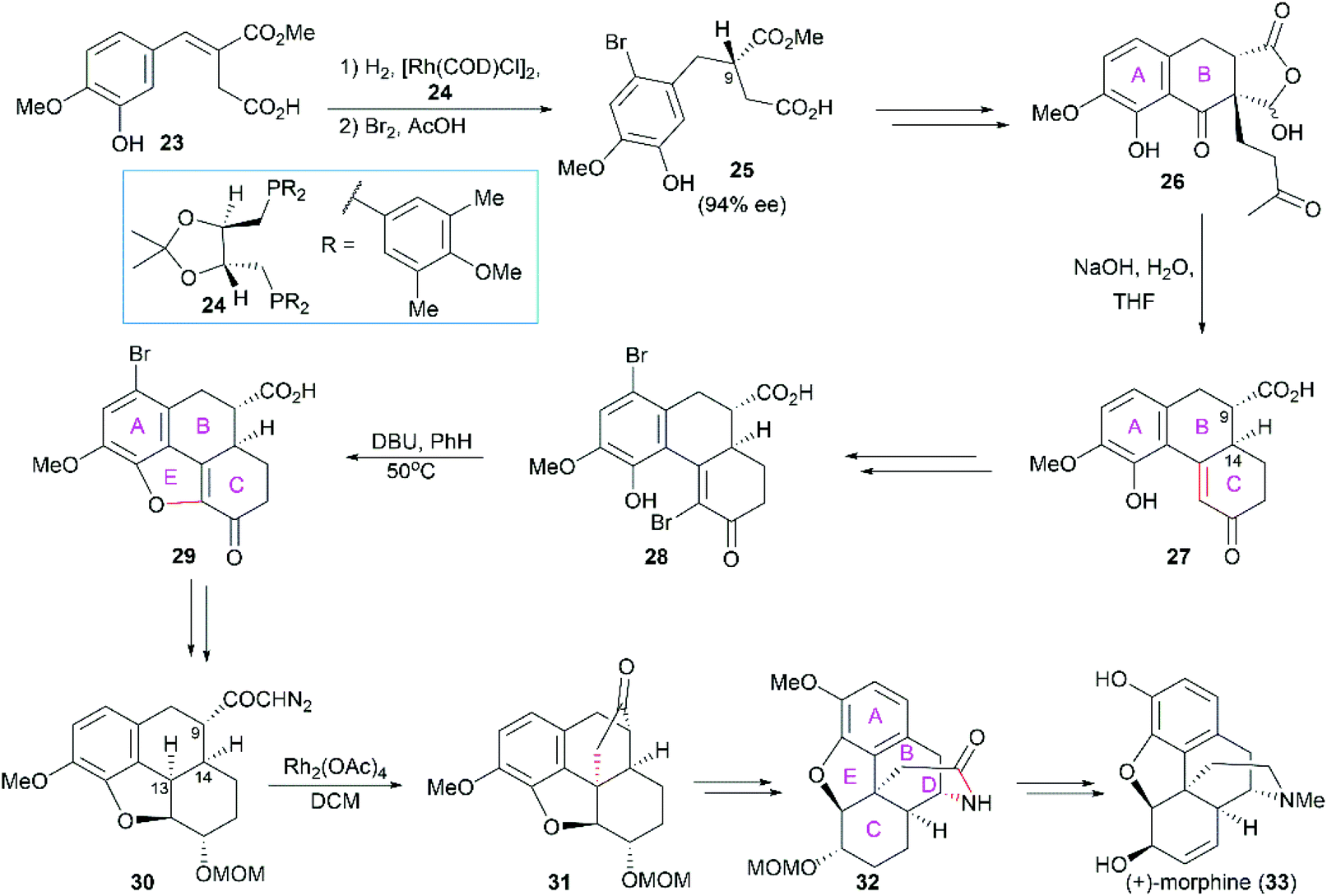
2.1.2 White's total synthesis of (+)-morphine.
In White's synthesis,48 the phenanthrene ring system (ring A/B/C) was constructed first, and this ring system was used as a platform for the sequential introduction of the dihydrofuran ring E and piperidine ring D. The first chiral center (C9) was established through Rh-catalyzed enantioselective hydrogenation of the α,β-unsaturated ester 23 in the presence of (4R,5R)-MOD-DIOP 24 (Scheme 2). The following intramolecular Friedel–Crafts reaction and Robinson annulation provided the phenanthrene nucleus 28. DBU-promoted cyclization yielded the tetracyclic compound 29, which was transformed into the diazo ketone 30. A Rh(II)-catalyzed carbenoid C–H insertion reaction was used to construct the challenging C13 all-carbon stereocenter in 31. The cyclopentanone was transformed into the piperidine ring through Beckmann rearrangement, which completed the synthesis of the pentacyclic core skeleton 32. Several functional group manipulations successfully led to the synthesis of (+)-morphine.
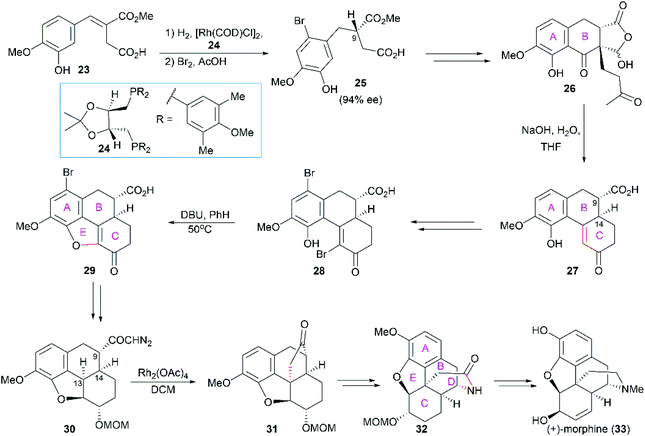 |
| | Scheme 2 White's synthetic route. | |
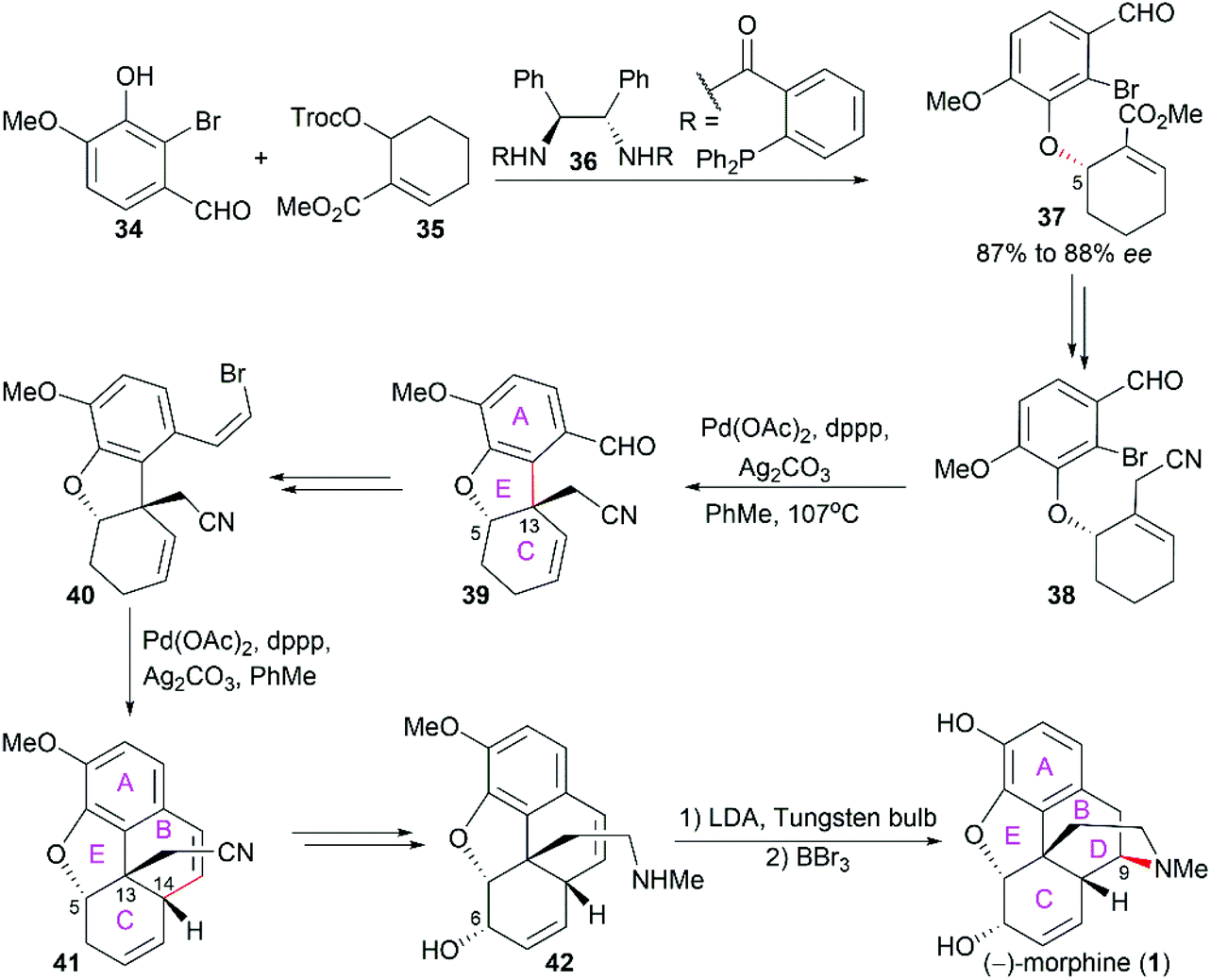
2.1.3 Trost's total synthesis of (−)-morphine.
Trost's asymmetric total synthesis of (−)-morphine (1) features a series of Pd-catalyzed reactions.49–51 By employing Pd-catalyzed asymmetric allylic alkylation of allylic ester 35 with 2-bromovanillin 34 in the presence of chiral ligand 36, the aryl ether 37 with 87% to 88% ee and the desired stereo configuration at C5 was obtained (Scheme 3). Then, a Pd-catalyzed intramolecular Heck reaction was applied to construct the ring E accompanied by the establishment of the quaternary stereocenter C13 (38 to 39). Another intramolecular Heck reaction on 40 smoothly led to tetracyclic compound 41, which was transformed into allyl alcohol 42. The final piperidine ring D was constructed through an intriguing visible light-promoted hydroamination reaction. It should be noted that intramolecular Heck reactions were also used in Hudlicky's asymmetric total synthesis of (−)-codeine (6) to construct ring E and ring B, wherein the chiral starting material was derived from enzyme-catalyzed enantioselective dihydroxylation of β-bromoethylbenzene.52,53
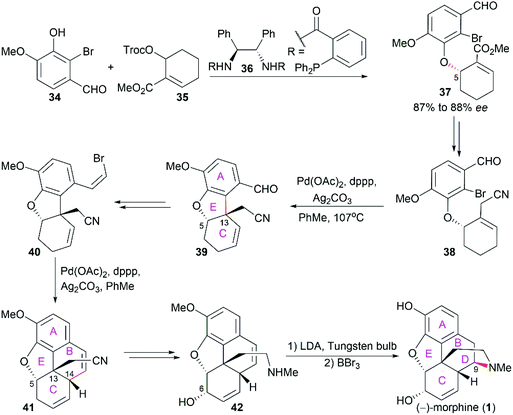 |
| | Scheme 3 Trost's synthetic route. | |
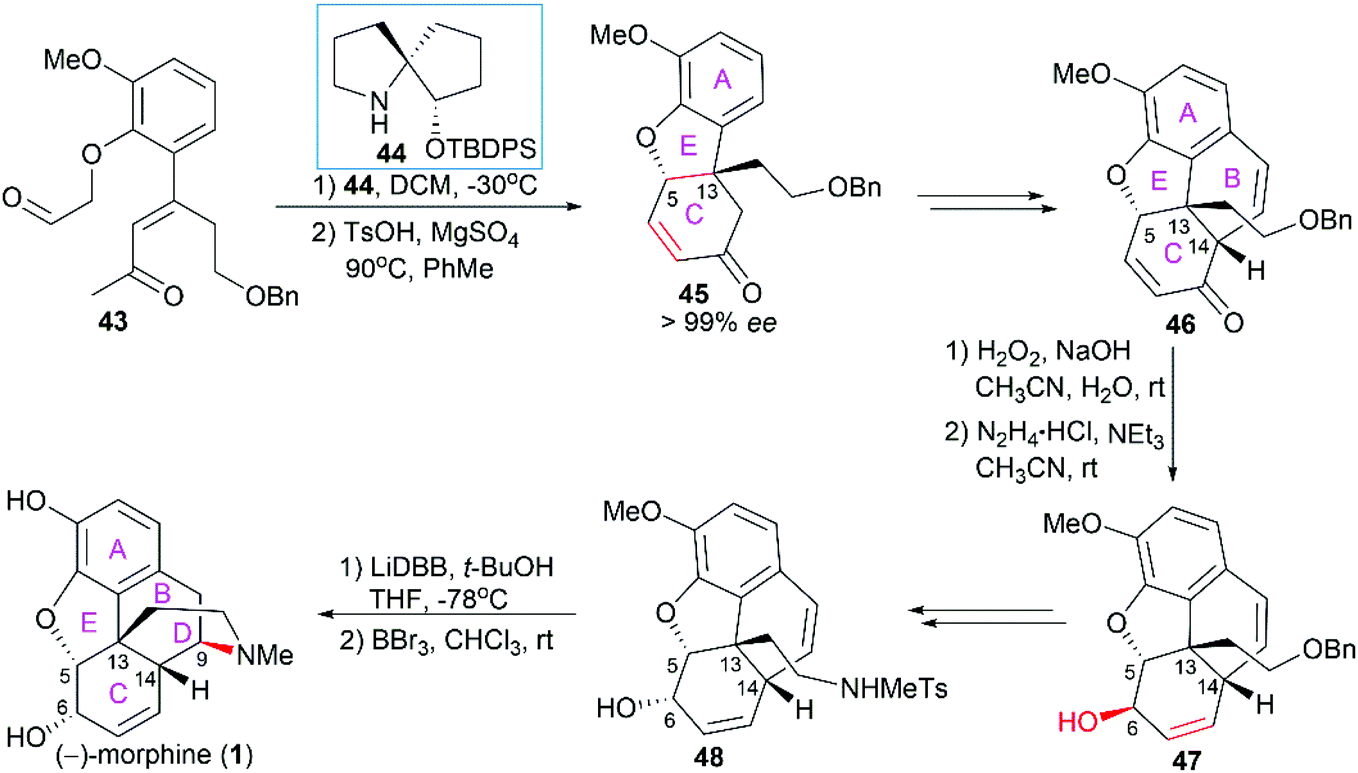
2.1.4 Tu's total synthesis of (−)-morphine.
In Tu's total synthesis of (−)-morphine (1),54 chiral spiropyrrolidine (SPD) organocatalyst 44-catalyzed asymmetric Michael addition within 43 and subsequent p-toluenesulfonic acid-promoted Robinson annulation were utilized to construct the A/C/E ring system with the establishment of the quaternary stereocenter C13 (Scheme 4). Introduction of an aldehyde group on 45 and subsequent polyphosphoric acid (PPA)-catalyzed Friedel–Crafts type cyclization led to the tetracyclic compound 46. A Wharton reaction was used to transform α,β-unsaturated ketone 46 to allylic alcohol 47. Introduction of the sulfonamide through a regioselective Mitsunobu reaction and configuration inversion of C6 hydroxy group via an oxidation/reduction process led to 48, which was ready for the introduction of the final piperidine ring D. A lithium 4,4′-di-tert-butylbiphenylide (LiDBB)-promoted hydroamination reaction and a subsequent demethylation operation successfully led to (−)-morphine (1).
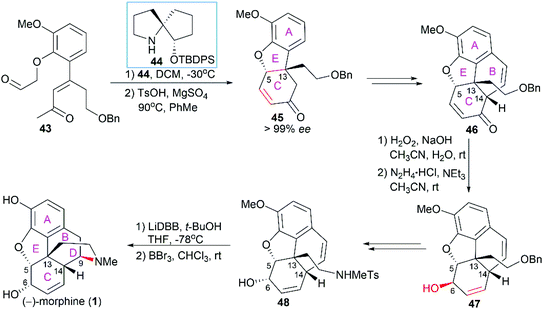 |
| | Scheme 4 Tu's synthetic route. | |
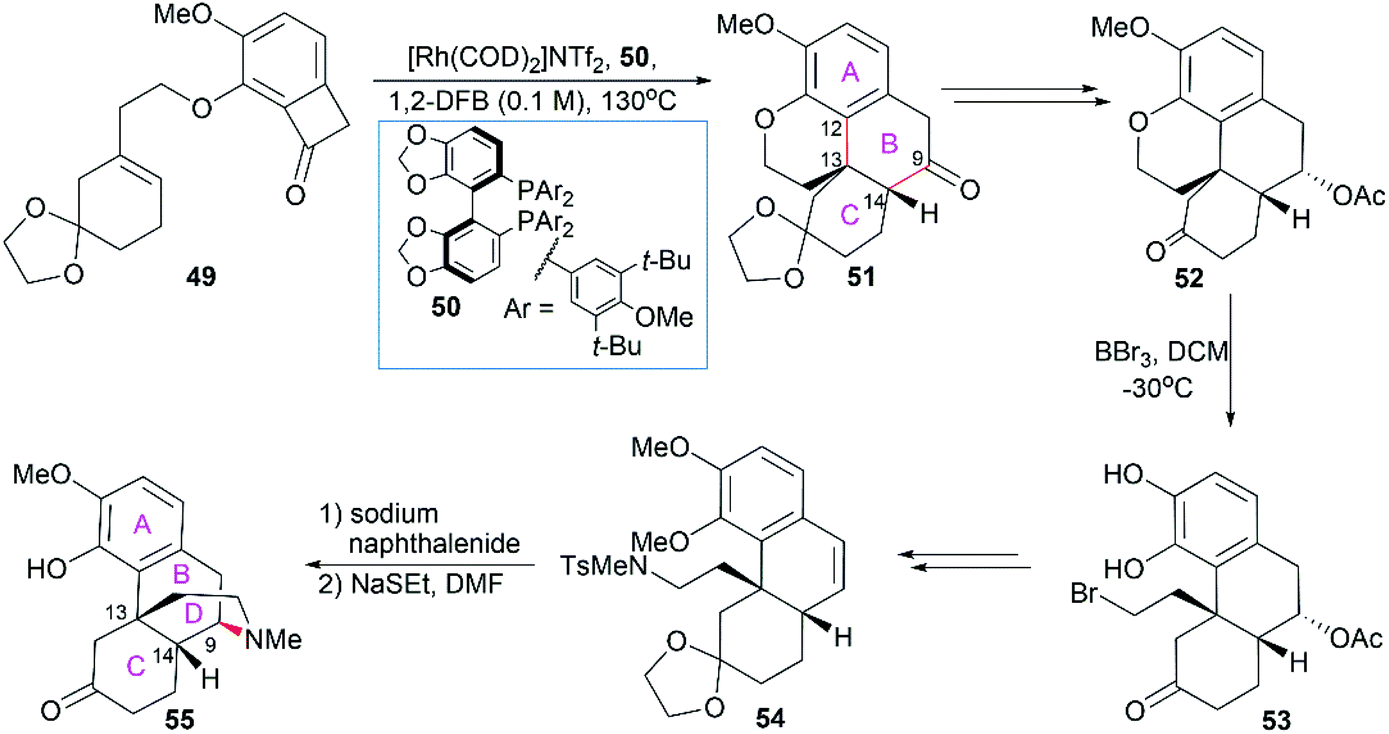
2.1.5 Dong's total synthesis of morphine alkaloids.
Dong and co-workers developed a deconstructive strategy for the asymmetric total synthesis of morphine alkaloids (Scheme 5).55 In their synthesis, the phenanthrene ring system in 51 was efficiently constructed through Rh-catalyzed “cut-and-sew” transformation between a sterically hindered trisubstituted alkene and benzocyclobutenone within 49 in the presence of the chiral bidentate phosphine ligand 50. The chiral C13 and C14 centers with the desired stereo configuration were also set up. Then, BBr3-mediated ether bond cleavage successfully yielded compound 53, which was a suitable substrate for the introduction of the sulfonamide group in 54. The piperidine ring was constructed through sodium naphthalenide-mediated removal of the Ts group and subsequent radical cyclization. Selective demethylation on one of the two MeO ethers using NaSEt led to compound 55, which was a common precursor for the synthesis of morphine (1), codeine (6), and thebainone A.55
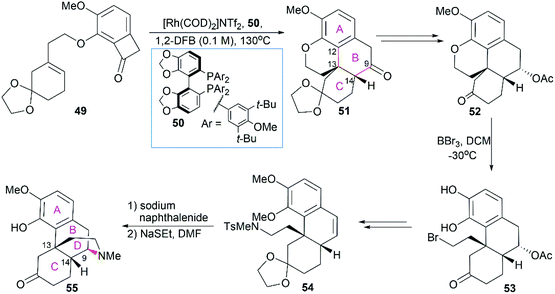 |
| | Scheme 5 Dong's synthetic route. | |
2.2 Structure–activity relationships of morphine derivatives
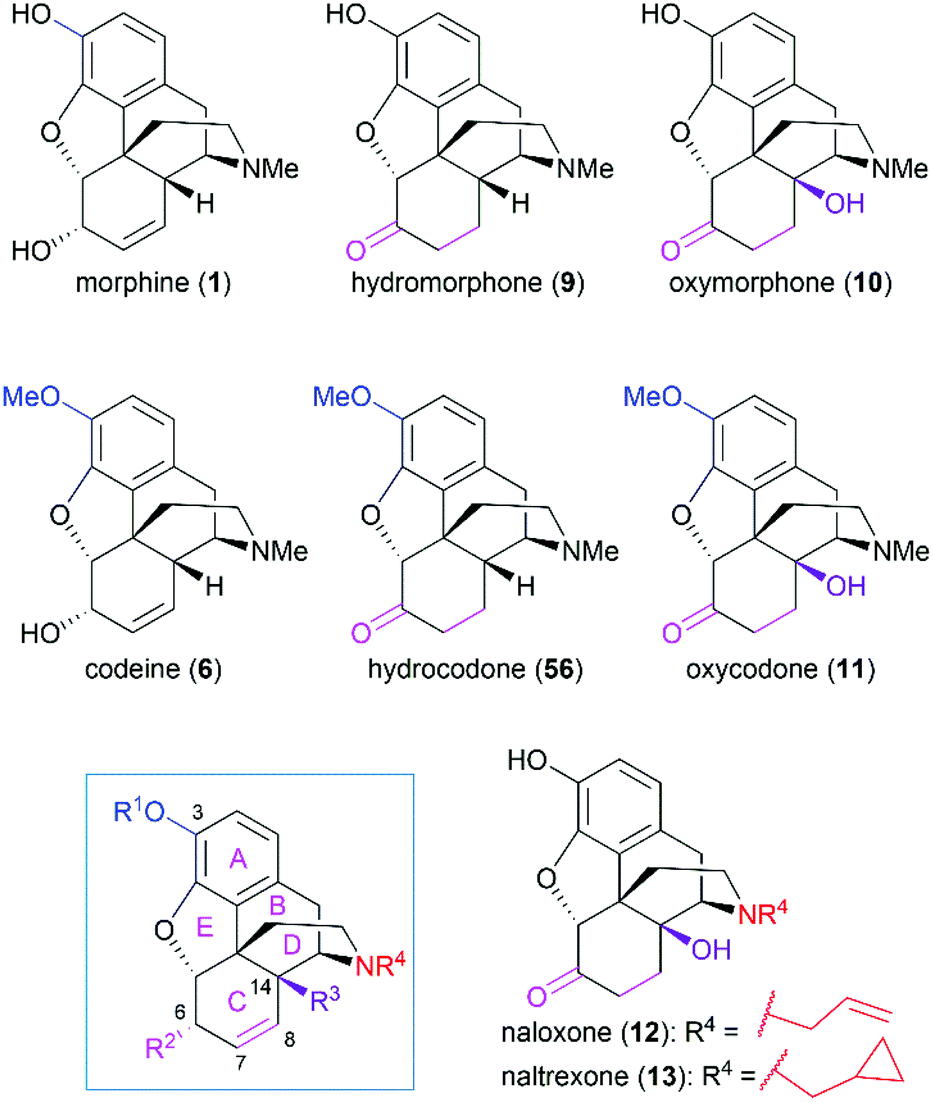
As the prototypical opioid agonist, morphine (1) has been intensively studied, which led to various types of morphine-related drugs. Representative pentacyclic morphine-based drugs are listed in Fig. 3. There are several structural features that are important for morphine's biological function: the C3 phenolic hydroxy group, C6 allylic hydroxy group and C7–C8 double bond, the B/C cis-fused ring system, the substitution groups at C14, and the amine group. Detailed SAR studies have been summarized previously56 and a brief discussion is provided in the following paragraph.
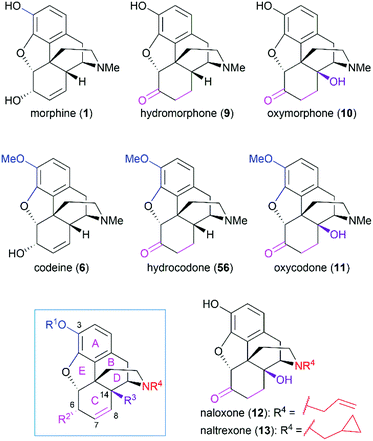 |
| | Fig. 3 Representative morphine-related drugs. | |
The C3 phenolic hydroxy group is essential for the binding of morphine with the μ receptor. Therefore, codeine (6), the C3 methoxy ether derivative of morphine, shows 10-fold lower potency than morphine.57 However, codeine possesses greatly enhanced oral bioavailability. A similar relationship is observed between codeine-derivatized hydrocodone (56) and morphine-derivatized hydromorphone (9), which are the same as oxycodone (11) and oxymorphone (10).56 These codeine-derivatives function as prodrugs and must undergo CYP2D6-mediated demethylation of their corresponding phenolic methyl ethers before binding to μ receptors.56
The C6 allylic hydroxy group is engaged in a weak hydrogen bond between morphine and its receptor.58 However, removal of the hydroxy group increases the molecular lipophilicity and therefore leads to a 10-fold increase in activity.59 If the alcohol is oxidized to ketone, a 3-fold decrease in activity is observed; further hydrogenation of the C7–C8 double bond led to hydromorphone (9) with more flexibility in ring C, which shows a 6-fold increase in potency compared with morphine.56 Introduction of a β-hydroxy group at C14 transforms hydromorphone (9) into oxymorphone (10), which shows a 2- to 3-fold increase in receptor affinity when compared with hydromorphone (9).
The essential amino nitrogen functions in a protonated form by forming an electrostatic bond with the receptor. Substituents at this nitrogen atom have a great influence on its biological function. A methyl group is commonly observed in morphine-derived agonists; however, naloxone (12) and naltrexone (13), with allyl or cyclopropylmethyl group, respectively, are pure antagonists.
3. Tetracyclines
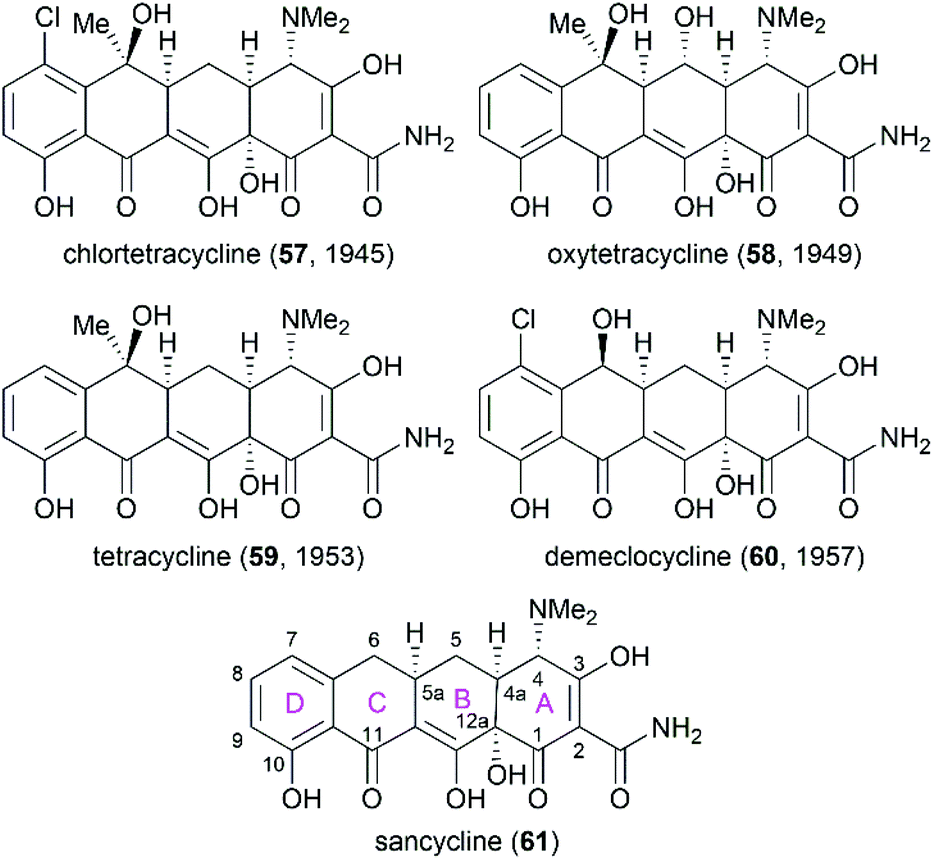
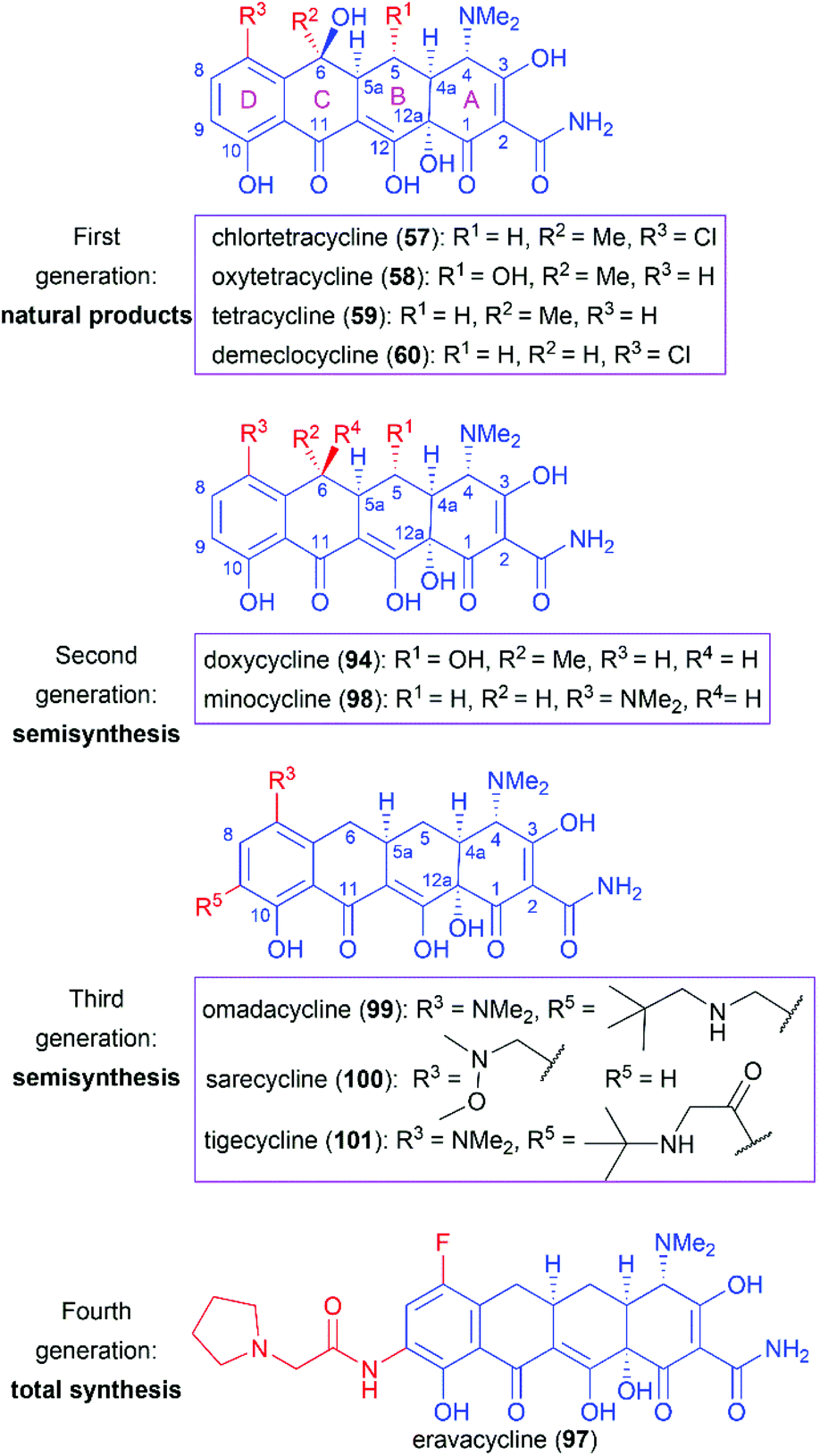
Following the discovery of chlortetracycline (57) by Duggar in 1945,60 several other natural tetracyclines—oxytetracycline (58),61 tetracycline (59),62 and demeclocycline (60),63—were discovered in the following years (Fig. 4). The broad-spectrum activity and low side effects of tetracyclines make them one of the most important classes of antibiotics.64 It is well established that tetracyclines function by binding to the 30S ribosomal subunit, which prevents the association of aminoacyl-tRNA with the bacterial ribosome and therefore inhibits bacterial protein synthesis.65 Over 70 years of widespread use has resulted in the emergence of bacterial resistance to tetracyclines. There are three main mechanisms responsible for tetracycline resistance: (1) efflux pumps reduce the intracellular tetracycline concentration by exporting tetracycline from the cell; (2) ribosomal protection proteins protect ribosomes from tetracycline inhibition via various mechanisms; and (3) protection enzymes inactivate tetracycline through modification and degradation.64,66–68 The growing problem of tetracycline resistance calls for much more investigation on the synthesis of tetracycline derivatives for the development of higher potency antibiotic drugs.
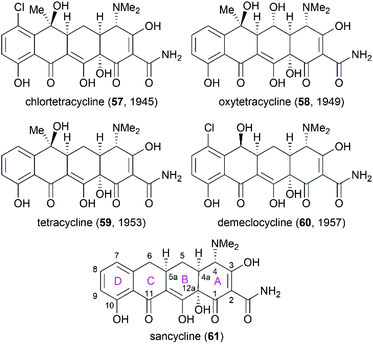 |
| | Fig. 4 Structures of natural tetracyclines and the core structure of tetracyclines. | |
In the early 1950s, the structures of chlortetracycline (57) and oxytetracycline (58) were first determined by scientists from Pfizer with the help of Woodward.69–71 Subsequent studies showed that the tetracyclines share one common nucleus, sancycline (61), which is composed of four linearly fused 6-membered rings (designated A, B, C, and D) with four stereocenters. A combination of different substitution groups, for example, chloride, hydroxy, and methyl, with the different substitution positions on the nucleus could lead to various tetracyclines with more structural complexity. The tetracyclines are frangible under an array of conditions: (1) the C6 hydroxy group undergoes dehydration in acidic media; (2) the C4 dimethylamino group epimerizes in mildly acidic conditions; and (3) the B and C rings undergo a retro-Dieckmann reaction in basic media.72,73 The important biological functions and the complicated structural features of tetracyclines have attracted much attention of synthetic chemists.
3.1 Total synthesis of tetracyclines
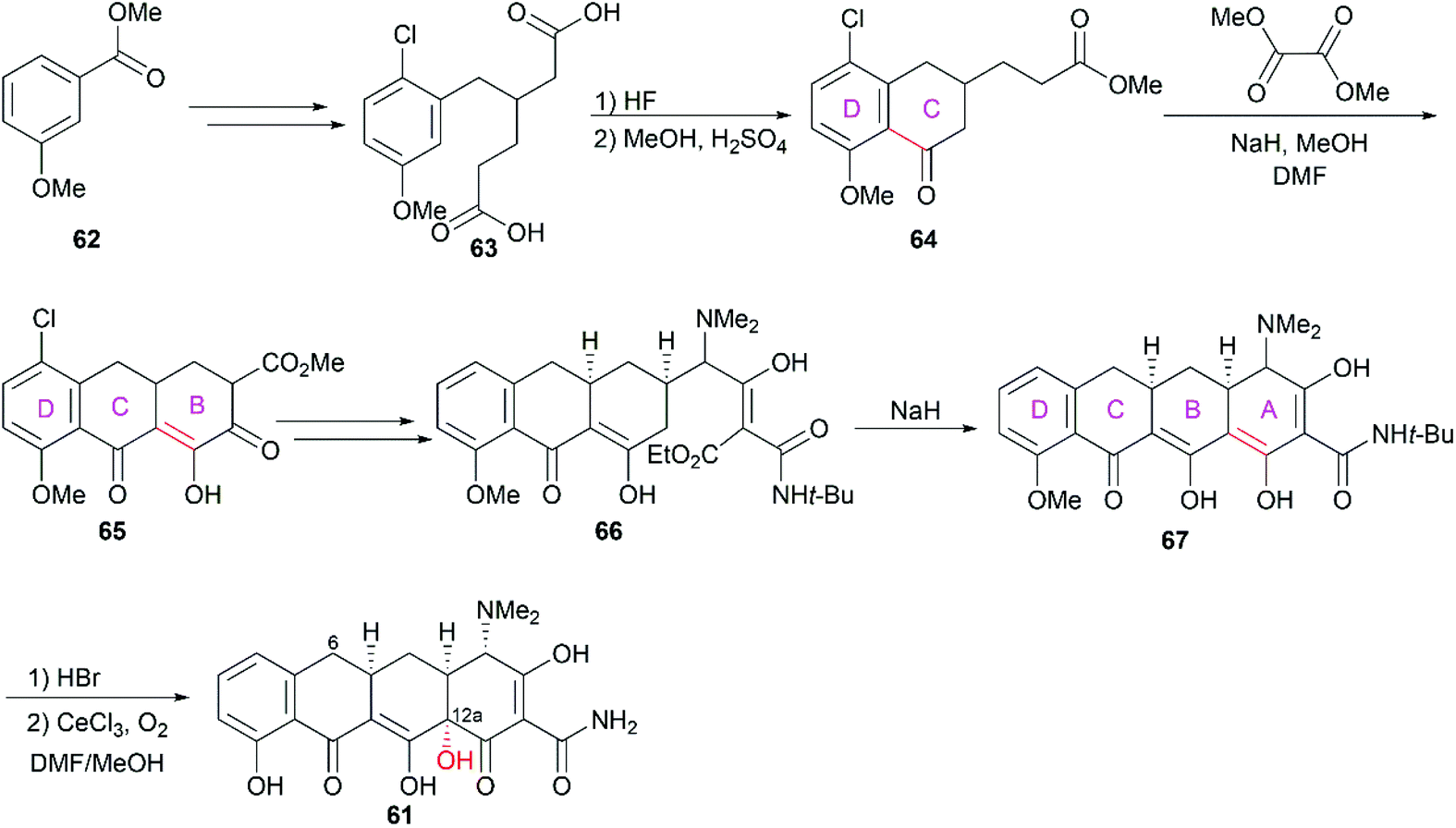
3.1.1 Woodward and Shemyakin's total synthesis.
One decade after they elucidated the structures of chlortetracycline (57) and oxytetracycline (58), Woodward and co-workers completed the first total synthesis of the prototypic tetracycline antibiotic, 6-demethyl-6-deoxytetracycline (61, sancycline).74–76 Starting from methyl 3-methoxybenzoate (62), they constructed the four fused rings one by one in a D to A direction through sequential Friedel–Crafts acylation and Claisen-/Dieckmann-type condensation reactions (Scheme 6). At the final stage of their synthesis, C12a was selectively oxidized with O2 in the presence of CeCl3 to construct the hydroxy group, which gave the desired product sancycline.
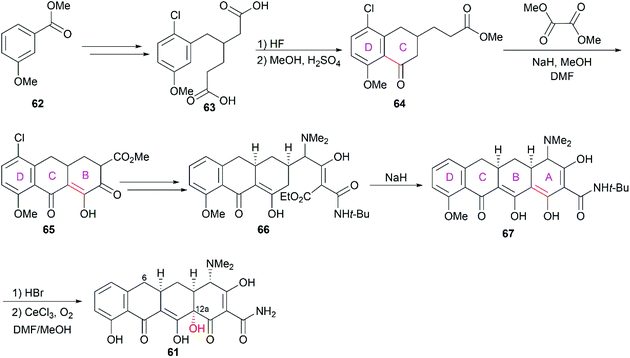 |
| | Scheme 6 Woodward's total synthesis of sancycline. | |
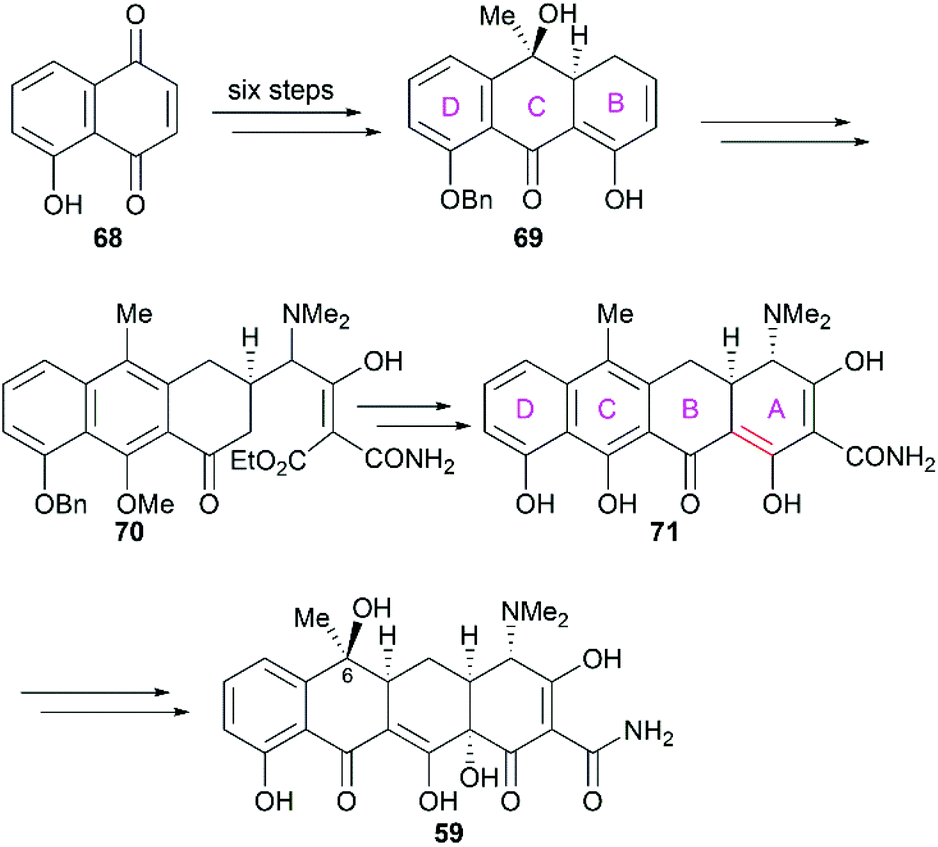
Not long after Woodward's synthesis of sancycline, Shemyakin reported their synthesis of tetracycline.77 Starting from juglone (68), they developed a synthetic strategy similar to that of Woodward (Scheme 7). It should be noted that the C6 hydroxy group was installed using a photooxidation method.78
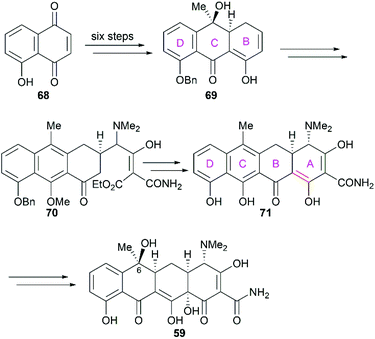 |
| | Scheme 7 Shemyakin's total synthesis of tetracycline. | |
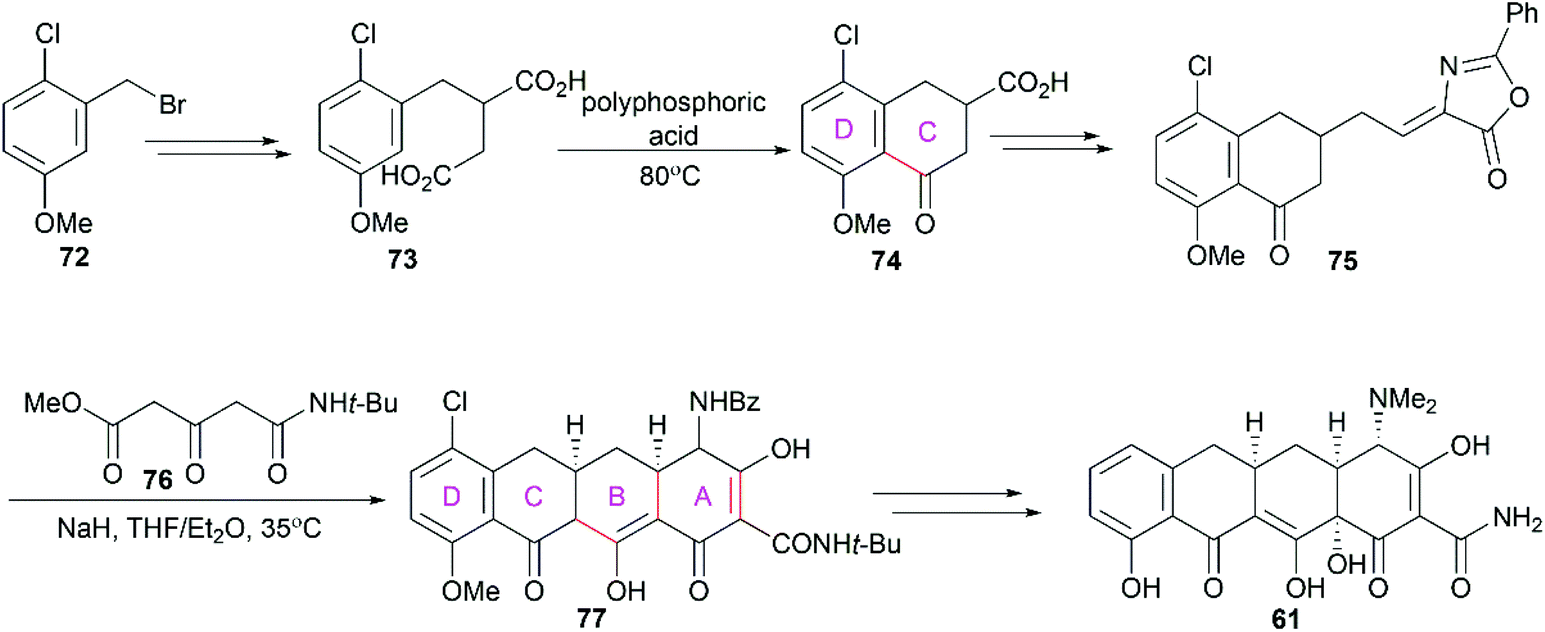
3.1.2 Muxfeldt's total synthesis.
In 1965, Muxfeldt and co-workers reported their total synthesis of sancycline (61).79 Still performed in the D to A direction, their synthesis utilized 1-chloro-2-bromomethyl-4-methoxybenzene (72) as a starting material and the D/C ring fused compound 75 was synthesized (Scheme 8). Condensation of 75 with methyl N-t-butyl-3-oxoglutaramate (76) in the presence of sodium hydride led to the tetracyclic product 77. Notably, their strategy constructed the A and B rings in one step utilizing a cascade reaction consisting of intramolecular Michael addition and Dieckmann condensations. Further exploration of this strategy led to their achievement of the total synthesis of oxytetracycline (58).80
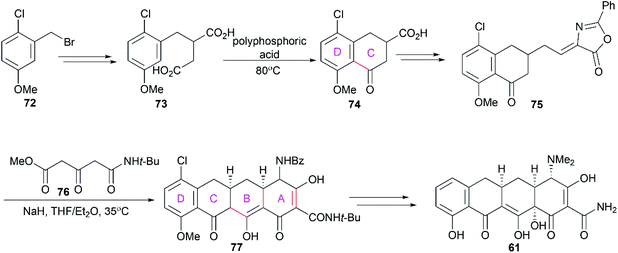 |
| | Scheme 8 Muxfeldt's total synthesis of sancycline. | |
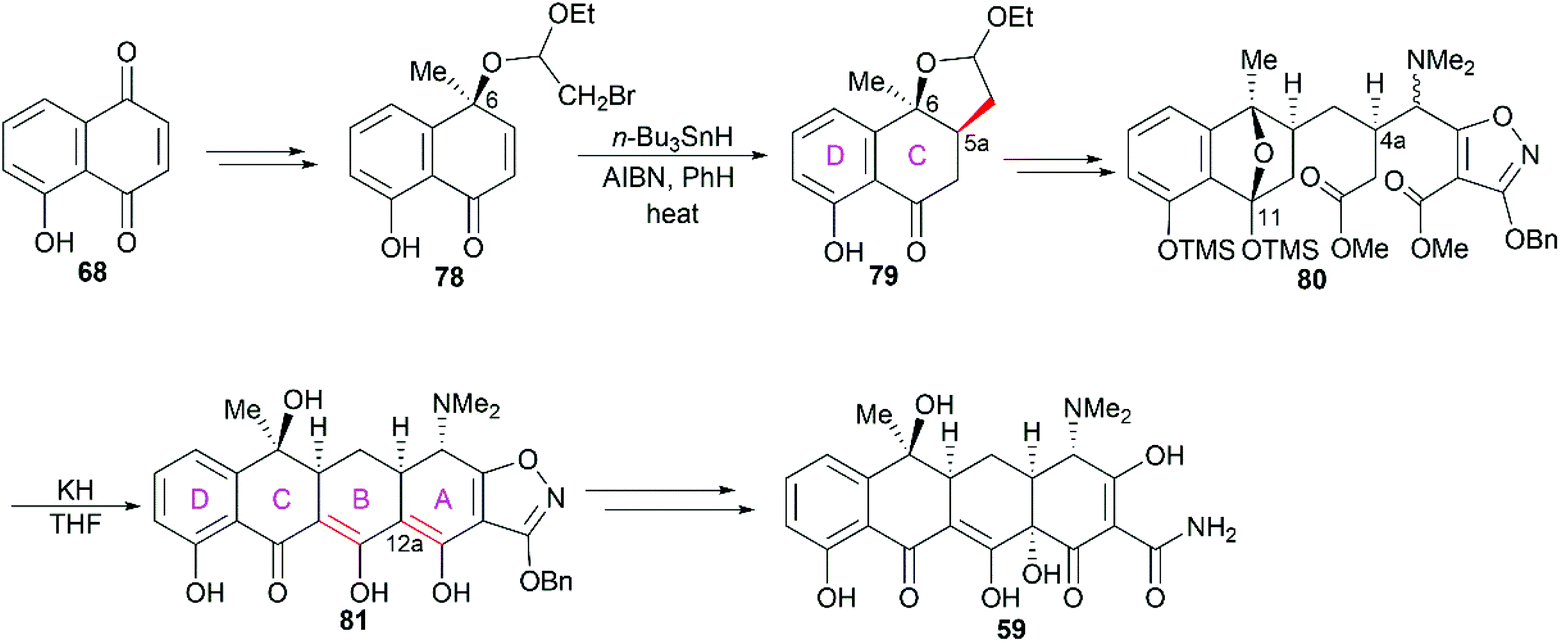
3.1.3 Stork's total synthesis.
In Stork's total synthesis of tetracycline,81 the C5a stereocenter was established using C6 hydroxy group-directed radical cyclization (78 to 79, Scheme 9), and the C4a stereocenter was correctly constructed in a substrate-controlled stereoselective Michael reaction to generate 80. Dieckmann cyclization of 80 in the presence of potassium hydride smoothly led to the pentacyclic product 81, which could be transformed into tetracycline (59) with hydrogenolytic opening of the isoxazole ring and incorporation of the C12a hydroxy group. It should be pointed out that when ring B was constructed first, trials to synthesize ring A were unsuccessful. Therefore, the authors proposed that ring A was formed prior to ring B in the transformation of 80 into 81.
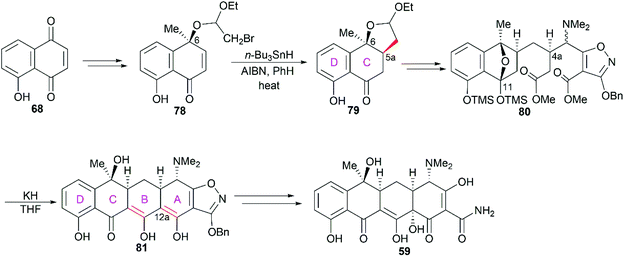 |
| | Scheme 9 Stork's total synthesis of tetracycline. | |
3.1.4 Tatsuta's total synthesis.
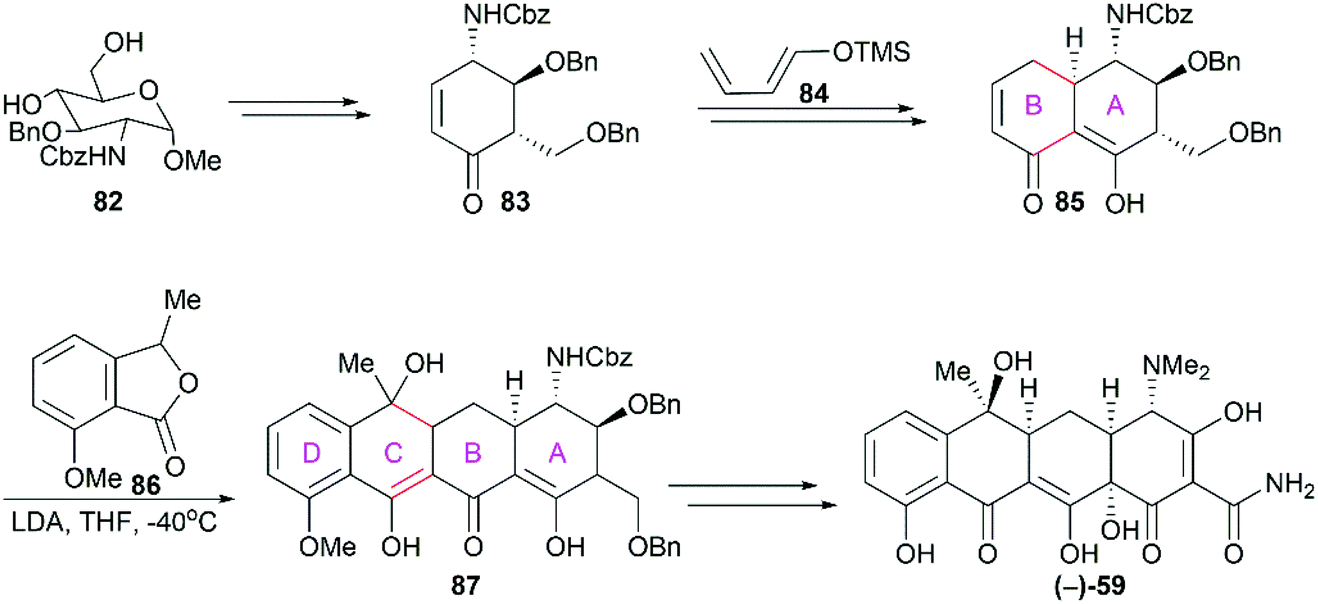
There was no asymmetric total synthesis of tetracyclines until Tatsuta's seminal report on the completion of (−)-tetracycline in 2000.82 Starting from 82, which was derivatized from D-glucosamine, the functionalized ring A (83) was synthesized first (Scheme 10). The subsequent regio- and stereo-selective [4 + 2] cycloaddition reaction between 83 and the diene 84 successfully led to the A/B ring-fused bicyclic intermediate. The following tandem Michael-Dieckmann-type reaction between 85 and 86 constructed the ring C with the concomitant introduction of the ring D. In contrast to previous synthetic strategies, Tatsuta's synthesis builds the tetracyclic framework in the ring A to ring D direction.
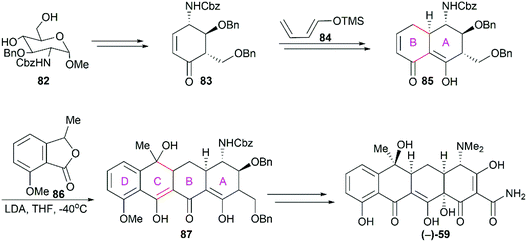 |
| | Scheme 10 Tatsuta's total synthesis of (−)-tetracycline. | |
3.1.5 Myers’ total synthesis.
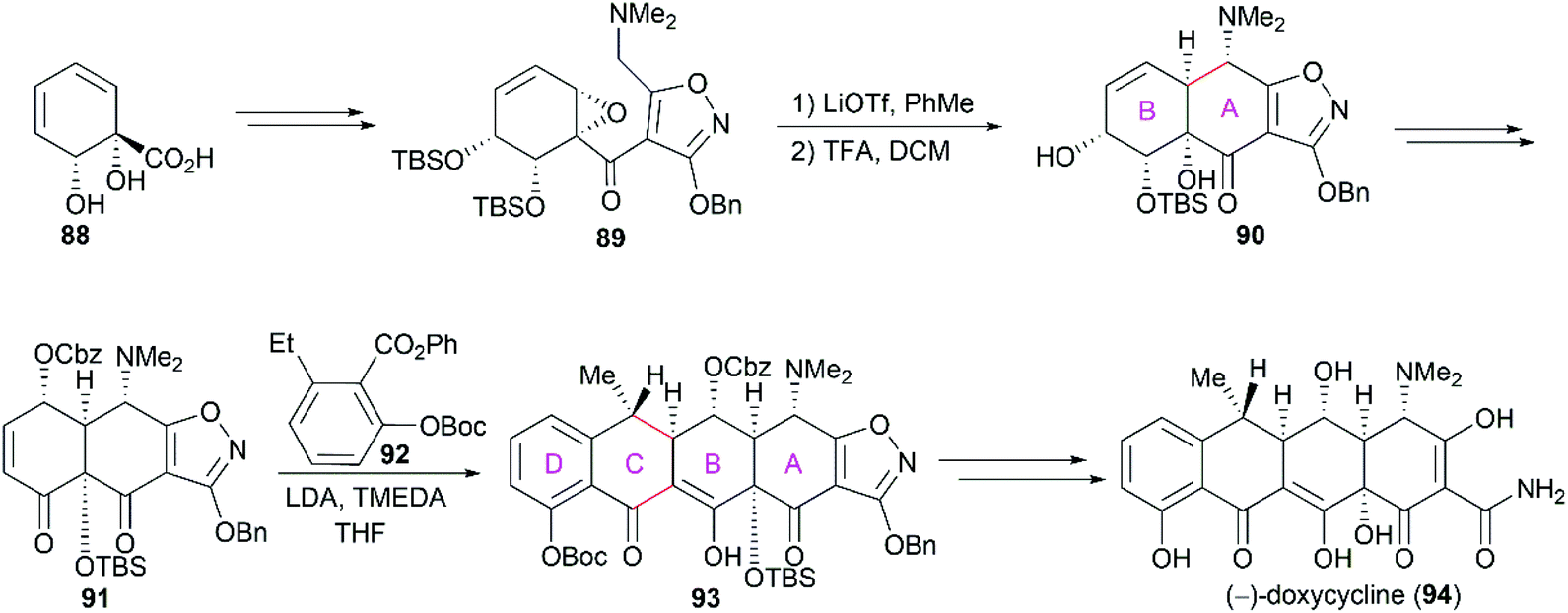
In Myers’ total synthesis of the FDA-approved drug (−)-doxycycline (94), the functionalized ring B 88,83 derivatized from benzoic acid, was transformed into compound 89, which was a precursor of the tricyclic compound 90 with A/B-fused rings (Scheme 11). Like that in Stork's synthesis,81 Myers and co-workers used a functionalized isoxazole as a precursor for the sensitive groups on ring A. Following a strategy similar to that used in Tasuta's total synthesis of (−)-tetracycline,82 the cascade Michael-Dieckmann reaction between 91 and 92 successfully yielded the pentacyclic compound 93, which could be efficiently transformed into (−)-doxycycline (94).
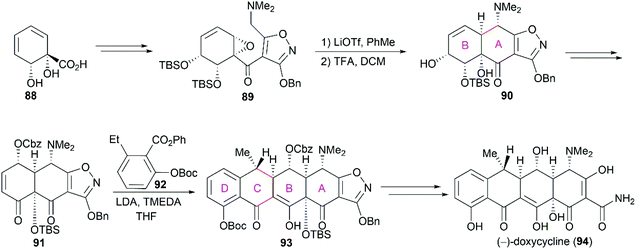 |
| | Scheme 11 Myers’ total synthesis of (−)-doxycycline. | |
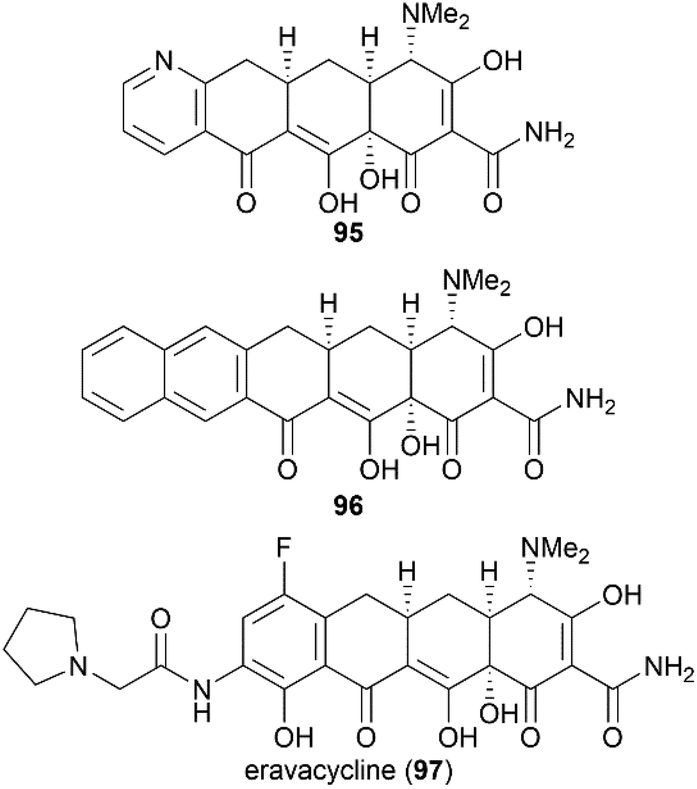
Previous X-ray crystallography studies on the interaction between tetracycline (59) and the 30S ribosomal subunit indicated that the D ring is suitable for modifications to generate more potent antibiotics.65 Therefore, Myers’ late-stage construction of ring C with the concomitant introduction of structurally varied forms of ring D is a promising strategy to synthesize tetracycline derivatives with modified D ring. With continuing optimization, this synthetic strategy is now suitable for industrial-scale application.84–88 What's more, utilizing Myers’ method, countless tetracycline derivatives, which are difficult to synthesize or are inaccessible by semisynthesis, can be constructed. Fig. 5 shows three representative tetracycline derivatives with different substitutions, the 7-azatetracycline 95, the pentacycline 96, and the fluorocycline 97, among which 97, the first fully synthetic tetracycline antibiotic, was approved in 2018.89 To date, more than 3000 tetracycline derivatives have been synthesized based on Myers’ strategy by a company named Tetraphase.90 These tetracycline derivatives with diverse modifications make more detailed SAR studies possible.
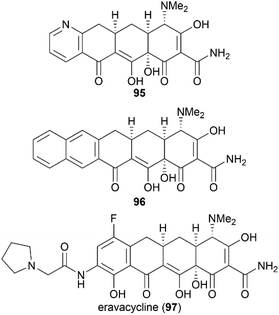 |
| | Fig. 5 Representative tetracycline derivatives synthesized based on Myers’ strategy. | |
3.2 Structure–activity relationships of tetracycline derivatives
Since the application of chlortetracycline (1) as the first broad-spectrum antibiotic, extensive efforts have been devoted to the SAR studies of tetracyclines, through natural tetracyclines to semisynthetic derivatives and then to fully total synthetic candidates. The tetracycline drugs can be roughly divided into four generations and representative members are presented in Fig. 6.
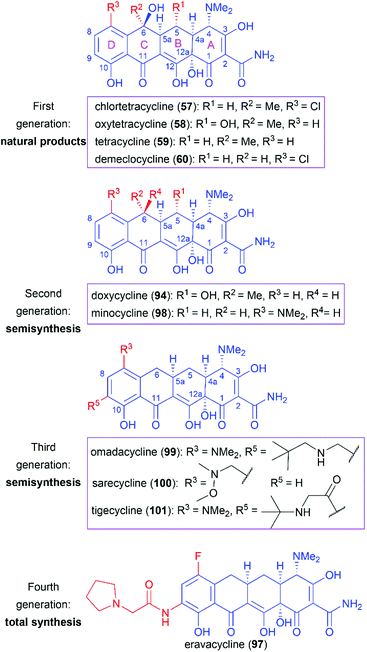 |
| | Fig. 6 Representative members of approved tetracycline drugs. | |
Naturally isolated tetracyclines are the main members of the first-generation drugs. These natural drugs share a common pharmacophore and comprise different substitution groups at C5, C6, and C7. Several structural features are important for their bioactivities. Maintenance of the four linearly fused six-membered carbocyclic skeleton and the α stereochemical configurations of C4, C4a, C5a, and C12a are essential for their high antibacterial activity.64,91,92 Chelation with metal ions has a great influence on tetracyclines’ activity, and the keto–enol systems (C1–C3 and C11–C12) and the carboxamide (C2), which are the sites of interaction with ions, are necessary.64,91 Alkylation of the carboxamide (C2) generally decreases activity, whereas suitable substituents can increase the water solubility, as in the case of the prodrug lymecycline.64,92 Removal of the C4 dimethylamino or its replacement with higher alkylamino groups reduces activity.91 Furthermore, esterification of the C12a hydroxy group diminishes the activity.
However, as mentioned above, the C6 hydroxy group in these natural products is labile and undergoes dehydration in acidic media to produce anhydrotetracycline, which shows nephrotoxicity.93 Therefore, removal of the C6 hydroxy group and further modifications through semisynthesis led to two second-generation drugs, doxycycline (94) and minocycline (98), which are more stable while maintaining similar or even higher antibacterial activity.94,95
The widespread use of tetracycline antibiotics has brought about serious resistance problems, which has led to the call for more-effective drugs. Investigations on C9-substituted tetracyclines led to the discovery of one glycylcycline named tigecycline (101), which is a derivative of minocycline (98).96 Tigecycline (101) shows high potency on tetracycline-resistant strains that utilize the efflux and ribosomal protection mechanisms of resistance.97 However, tigecycline (101) is only available in an injectable formulation.91 Further modifications led to the discovery of omadacycline (99), which is a C9-aminomethylcycline. Compared with tigecycline (101), omadacycline (99) shows improved oral bioavailability while maintaining activity against tetracycline-resistant bacteria.98 Another C7-aminomethylcycline, sarecycline (100), was approved as a narrow-spectrum tetracycline for the treatment of acne.
The success of the second- and third-generation tetracycline drugs indicates the potential role of the tetracycline pharmacophore in developing new antibiotics. However, the use of semisynthesis, as opposed to total synthesis, for the modification of tetracyclines has inherent limitations. The power of total synthesis is demonstrated by the approval of eravacycline (97),89,99 the first fully synthetic tetracycline drug. We believe that total synthesis will play a much more important role in the campaign against growing antibiotic resistance.
4. Macrolides
The macrolides, a series of macrocyclic lactones with 14-, 15- and 16-membered rings and decorated with one or two sugar moieties are primarily isolated from different Streptomyces spp. The first macrolide and wide-spread antimicrobial agent, erythromycin (102), was isolated in 1952 from the culture broth of Saccharopolyspora erythera, and its structure features, a highly substituted 14-membered lactone bearing 10 chiral carbon atoms, and configuration were successively defined by chemical and X-ray diffraction studies.100–102 Inspired by its excellent and broad-spectrum activity against Gram-positive pathogens, the mechanism by which erythromycin binds to the 23S RNA of the 50S ribosomal subunit to hamper the exit peptide tunnel and inhibit protein synthesis was elucidated and confirmed further by determining the crystal structure of erythromycin bound to the 50S ribosomal subunit.103,104 Since erythromycin was launched as Ilosone to treat bacterial infections of the respiratory tract, skin, and soft tissues, the problem of varying levels of resistance to macrolides has been uncovered; resistance was characterized as resulting from the expression of efflux proteins in Gram-positive pathogens,105,106 a change in erythromycin binding site in mutated ribosomal proteins,107 and bio-modifications of macrolides by methylase108,109 and esterases.110,111 In addition, erythromycin is unstable, especially in acidic environments, due to ketal formation between the C6 and C12 hydroxy groups and the C9 ketone; this undesired side reaction alters the structural properties and leads to low safety and bioavailability.112 To effectively address these problems and improve the drug-likeness potential of natural macrolides, total synthesis and semisynthesis strategies for the macrolides and their structural analogs have been explored to develop novel lead structures with new molecular features and mechanisms of actions.
4.1 Total synthesis of macrolides
4.1.1 Woodward's total synthesis of erythromycin A.
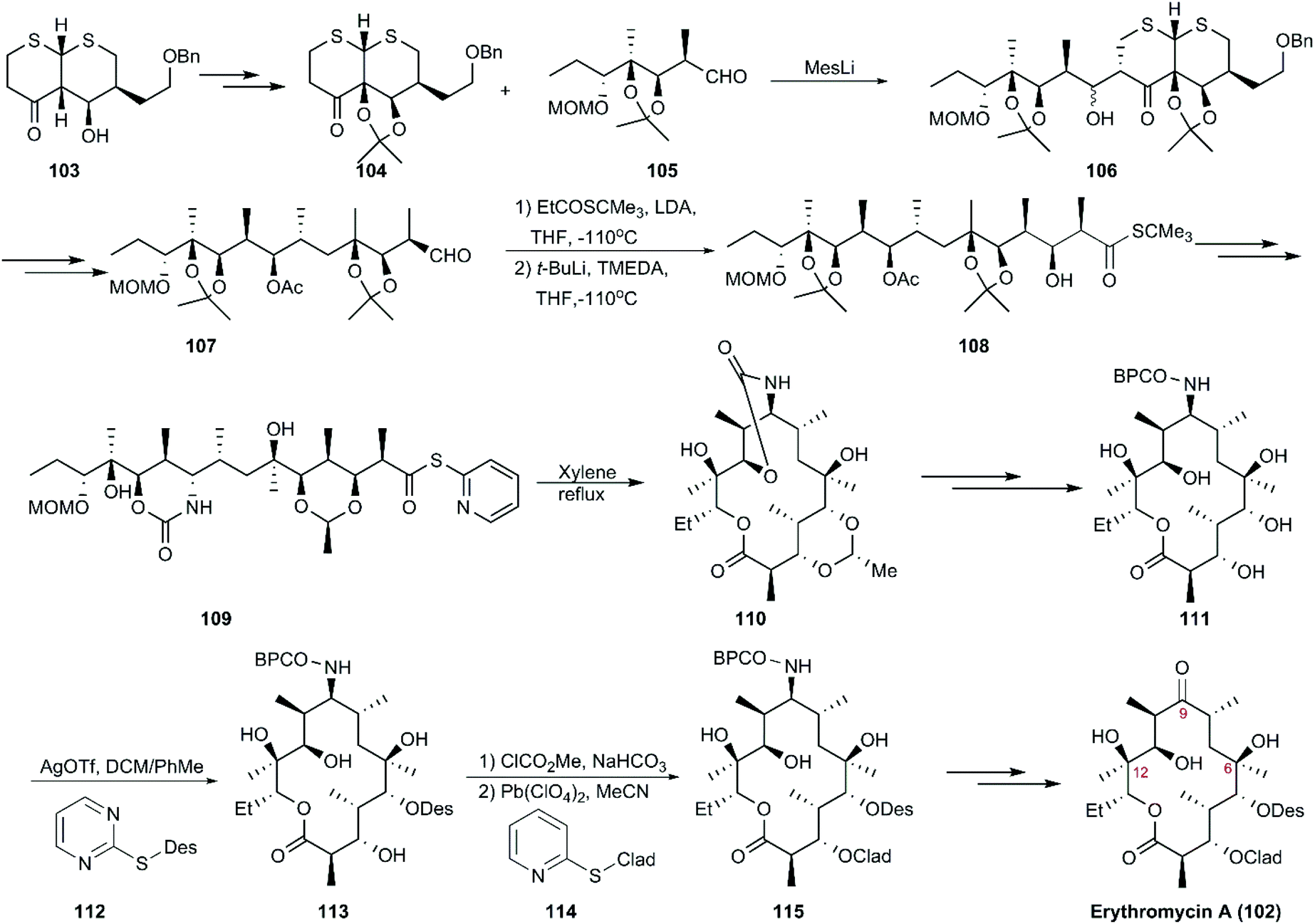
Only three groups in the world, including Woodward and Martin's teams, have completed the total synthesis of erythromycin A and B. The first route to erythromycin A (102) was reported by Woodward and co-workers using dithiohemiacetal as starting material, which was followed by 12 conversion steps to generate a common intermediate (103). Then, 103 was converted to intermediate 104 and aldehyde 105, followed by aldolization in the presence of mesityllithium as a base, yielding diastereomeric aldols (106), which underwent a series of reactions, including deprotection, protection, and oxidation, to generate aldehyde 107. The coupling of 107 with the enolate tert-butyl thiopropionate exclusively generated the “Cram” product with an undesired stereochemistry at C2. The desired stereochemistry at C2 was subsequently obtained in the presence of t-BuLi, to yield 108, which was transformed into a single seco-acid derivative (109) decorated with 2-pyridyl-thioester as a precursor of macrocyclization. Next, 109 was subjected to Corey's method of lactonization to afford 110, and this was followed by ten steps including two innovative glycosidation manipulations using D-desosaminide 112 and L-cladinoside 114 as Königs–Knorr glycosyl donors, which finally led to the production of erythromycin A (102) in 48 steps (Scheme 12).113–115
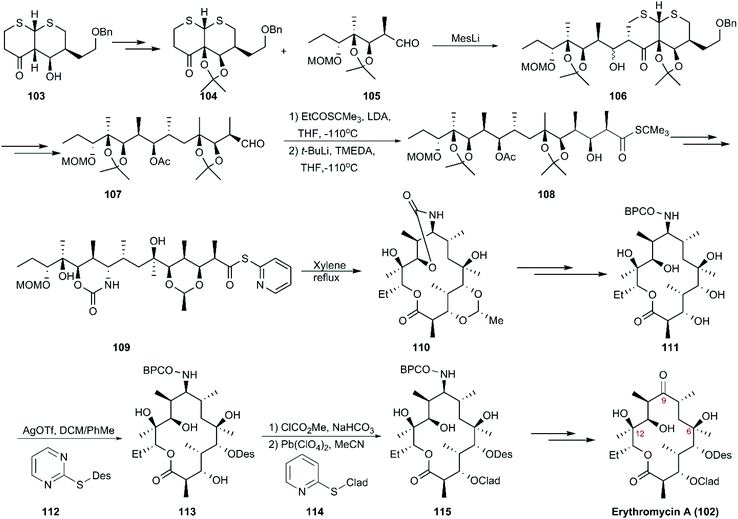 |
| | Scheme 12 Woodward's synthesis of erythromycin A. | |
4.1.2 Martin's total synthesis of erythromycin B.
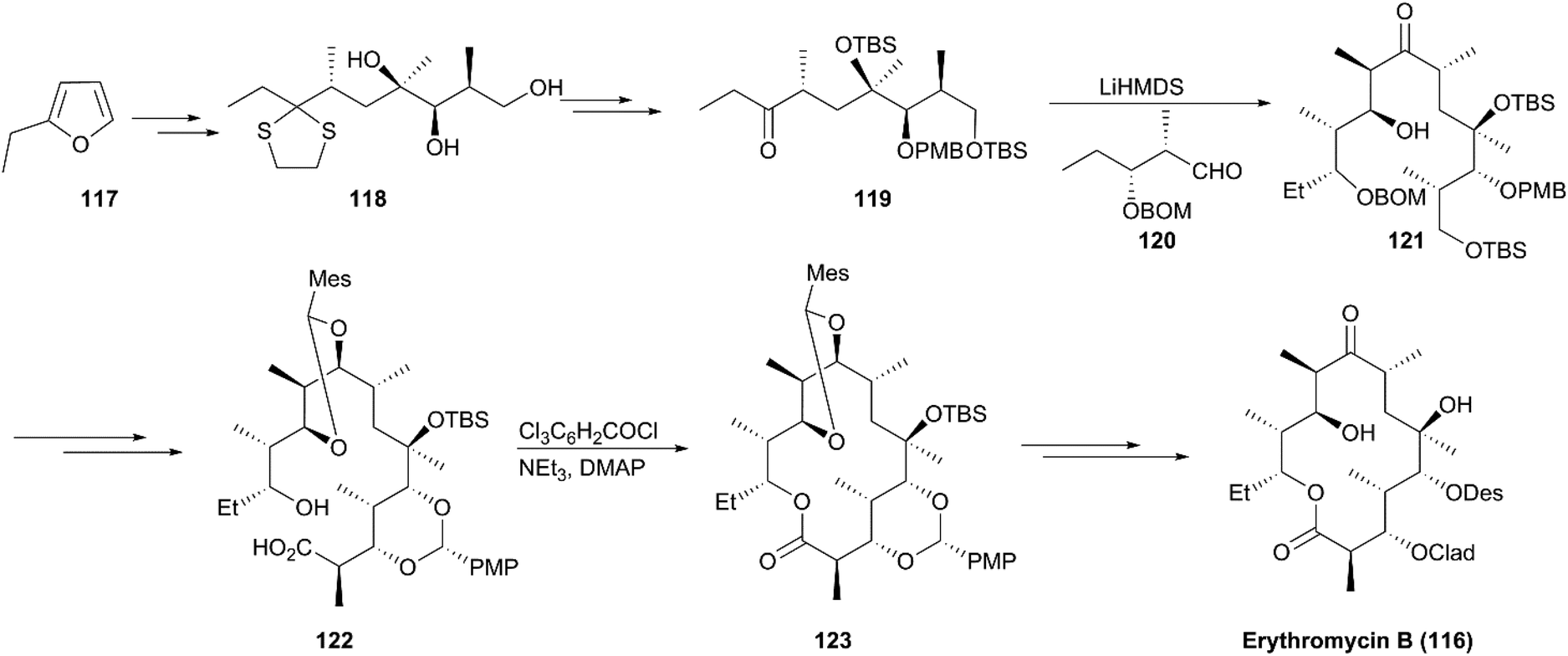
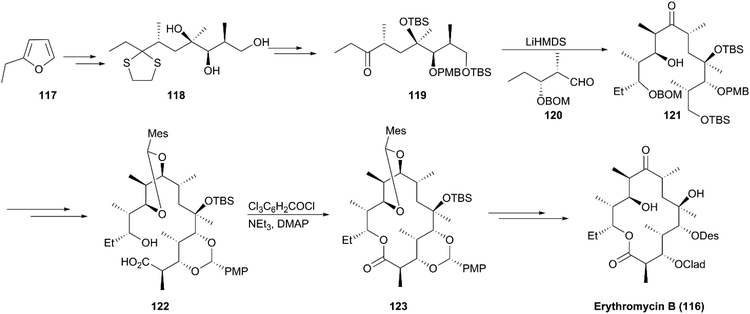
A decade after Woodward's approach to erythromycin A synthesis was reported, the formal synthesis of erythromycin A was reported by Oishi's group and a highly efficient strategy for semisynthesis of erythromycin A from its natural aglycon was accomplished by Tatsuta using altered glycosidation strategy. In 1997, Martin completed the total synthesis of erythromycin B (116), a congener of erythromycin A lacking a hydroxy group at C12 (Scheme 13).116 Based on known trihydroxy intermediate 118 prepared according to a reported method, synthesis of ketone 119 was achieved via interconversions of protecting groups and oxidation. This step was followed by aldolization with aldehyde 120 to generate intermediate 121. A macrocyclization precursor, seco-acid (122), was prepared from 121 with highly stereoselective aldolization as a critical conversion step. Using excellent Yamaguchi's macrolactonization, synthesis of the macrolide core skeleton (123) was achieved with a high yield. After multiple steps of protecting group transformations accompanied by two steps of glycosidation using Woodward's protocol, erythromycin B (116) was obtained in 23 steps.
 |
| | Scheme 13 Martin's synthesis of erythromycin B. | |
4.1.3 Kang's total synthesis of azithromycin.
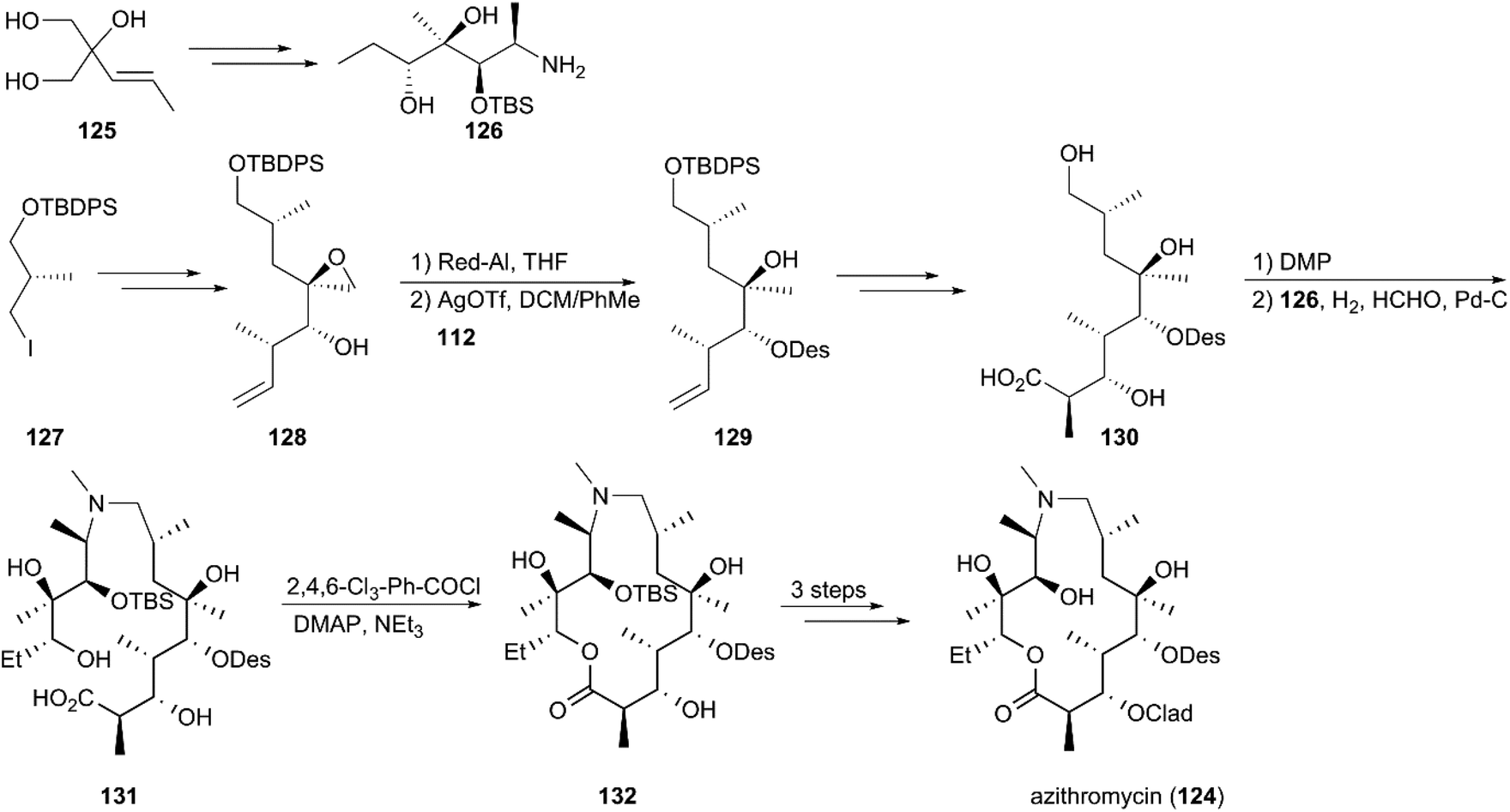
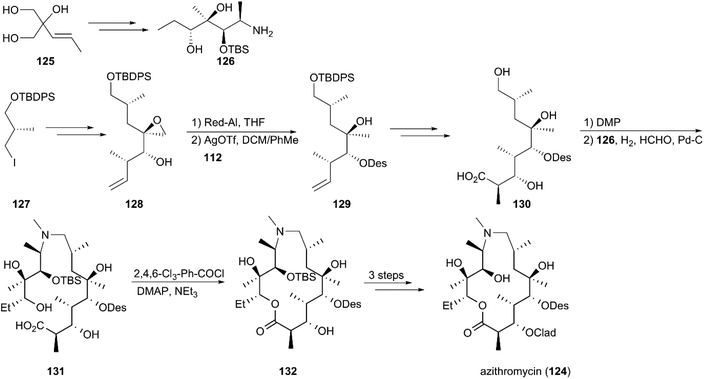
The approved drug azithromycin (124), a semisynthetic analog, is prepared from erythromycin A in four steps. Considering its clinical use as a first-line antibiotic, Kang and co-workers used a novel synthetic plan instead of using erythromycin A as the precursor to construct azithromycin (124) (Scheme 14).117 According to the strategy, the triol (125) was converted to alkanolamine intermediate (126) in 11 steps using desymmetric mono-benzoylation and epoxide chemistry as key reactions. The eastern building block (130) with a carboxylic acid, was obtained by homologation, desymmerization, crotylation, and reduction and glycosidation using the known chiral moiety (127) as starting material. The alkanolamine (126) was coupled with an aldehyde by reductive amination under hydrogenation conditions, and amino methylation was completed in the presence of formaldehyde to give seco-acid (131). These and the following steps, including Yamaguchi macrocyclization, glycosidation, and deprotection, yielded azithromycin (124) in a total of 18 steps.
 |
| | Scheme 14 Kang's total synthesis of azithromycin. | |
4.2 Structure–activity relationships of macrolide derivatives
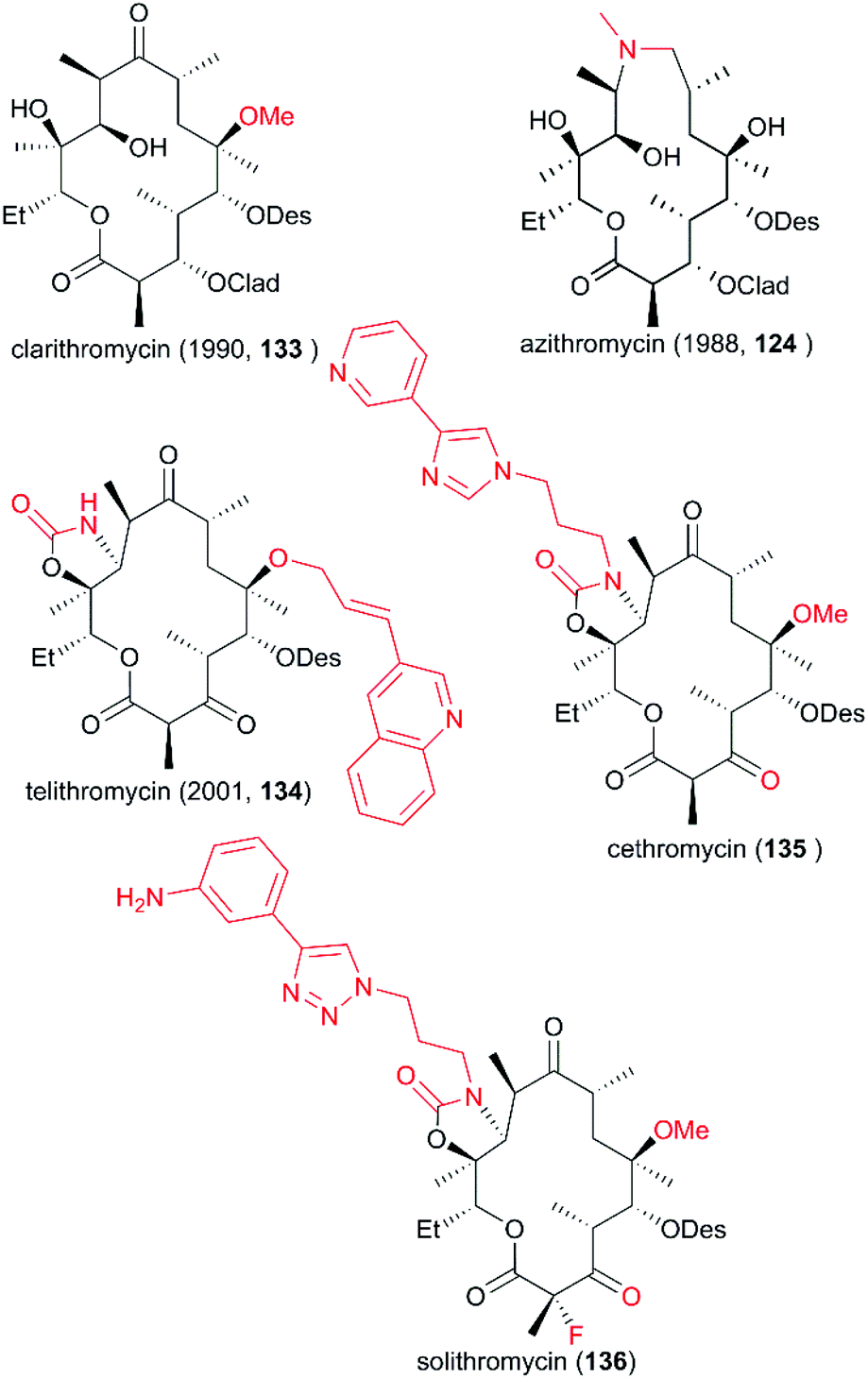
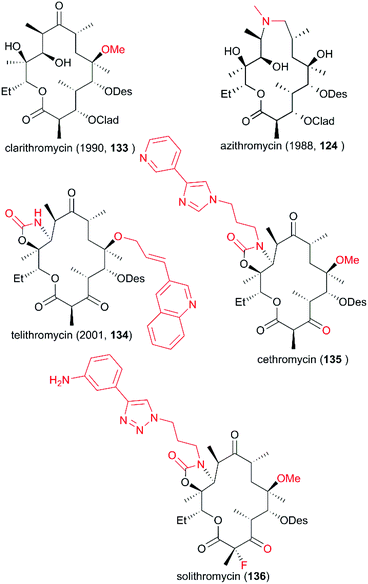
Since the discovery of erythromycin as the first macrolide with broad-spectrum antibacterial activity, the development of more potential novel antibiotic candidates has been spurred, leading to the preparation and testing of hundreds of macrolide derivatives. As mentioned above, spiroketal formation between the ketone at C9 with hydroxy groups at C6 and C11 under acidic conditions contributes to the low stability of erythromycin. To avoid this unpleasant side effect, a more acid-resistant and powerful derivative with a 6-O-methyl group was developed by Ōmura and co-workers in 1984; six years later this derivative, clarithromycin (133), was approved for clinical use (Fig. 7).118 Meanwhile, a 15-membered ring of azamacrolide was furnished through Beckmann rearrangement of erythromycin A oxime at C9. This new strategy from Lazarevski's group led to a novel chemical scaffold with excellent stability and activity; this chemical was approved as a new drug in 1988, named azithromycin (124). In addition, the C3-cladinose moiety was shown to be unnecessary for protein contact.119 The success of the marketed drug telithromycin (134) showed that its ability to bind to the ribosome was improved by the introduction of a C11/C12 carbamate ring with an extended aryl substituted alkyl chain, and binding was also improved by oxidation of the C3 hydroxy group to give C3 ketone without sugar.120–122 However, a more effective antibiotic is urgently needed because of the serious drug resistance problem induced by the widespread use of the antibiotic. The development of more drug-like candidates for clinical applications, such as cethromycin (135) and solithromycin (136), is currently underway.
 |
| | Fig. 7 Derivatives of erythromycin A. | |
5. Vinca alkaloids
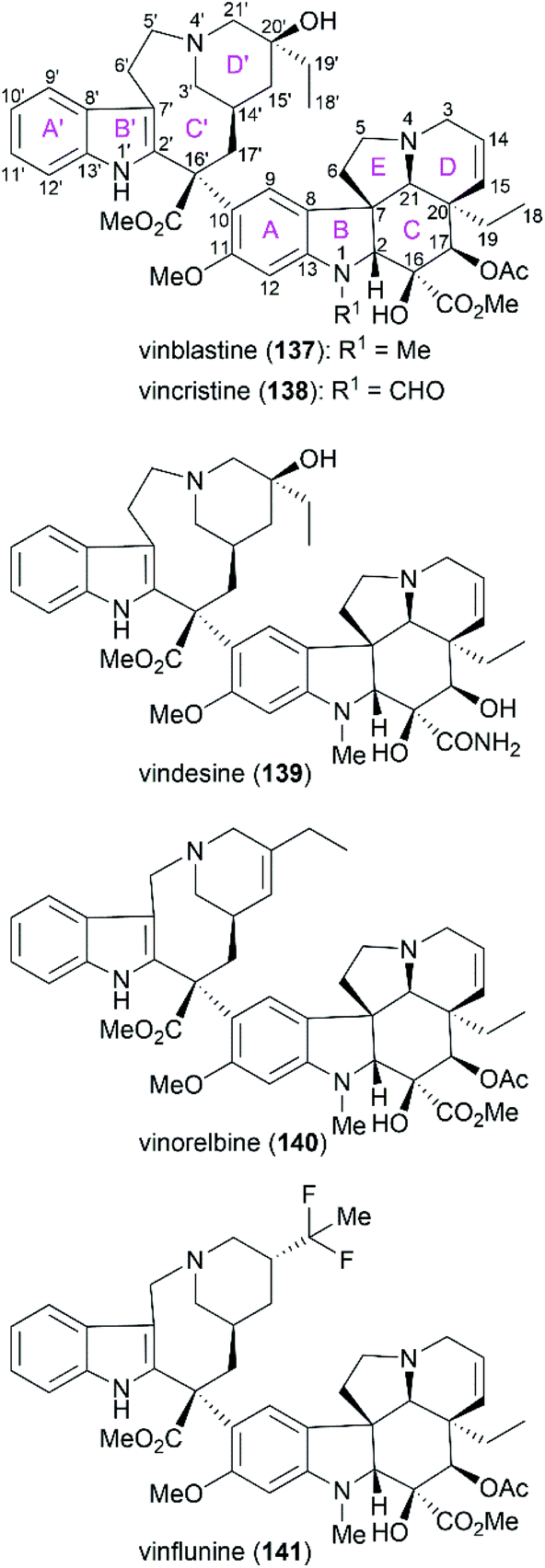
Vinblastine (137) and vincristine (138) are characteristic members of the biologically important vinca alkaloids (Fig. 8). Along with these two alkaloids, derivatives of these alkaloids, including vindesine (139), vinorelbine (140), and vinflunine (141), have been widely applied as well-known antitumor drugs. These molecules function through binding to tubulin, which disrupts microtubule function and results in mitotic arrest and apoptosis.123–127
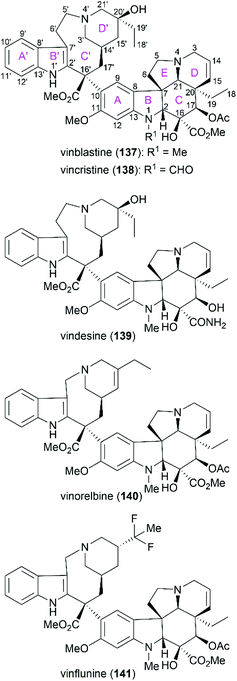 |
| | Fig. 8 Representative vinca alkaloids and related derivatives used as antitumor drugs and their ring lettering and carbon numbering system. | |
Vinblastine and vincristine were isolated from the leaves of Catharanthus roseus in 1958 and 1961,128–130 respectively, and their absolute configuration was determined by X-ray crystallography in 1965.131 Both vinblastine and vincristine possess an upper velbanamine subunit and a lower vindoline or vindoline-derived subunit. For vinblastine, the lower vindoline subunit contains a pentacyclic skeleton (ABCDE rings), which bears six contiguous stereocenters on ring C with two quaternary chiral centers. The upper velbanamine subunit is composed of four rings, including one 9-membered ring C′. There are three stereocenters in the upper subunit, including the C16′ quaternary chiral center resulted from the linkage between the two subunits. The pharmaceutical importance combined with their intriguing structural features make this kind of alkaloid attractive synthetic targets, and great endeavors from the synthetic community in the past several decades have resulted in several elegant total syntheses of vinblastine and vincristine. These strategies are discussed in the following section.
5.1 Total synthesis of vinca alkaloids
5.1.1 Potier and Kutney's total synthesis of (+)-vinblastine.
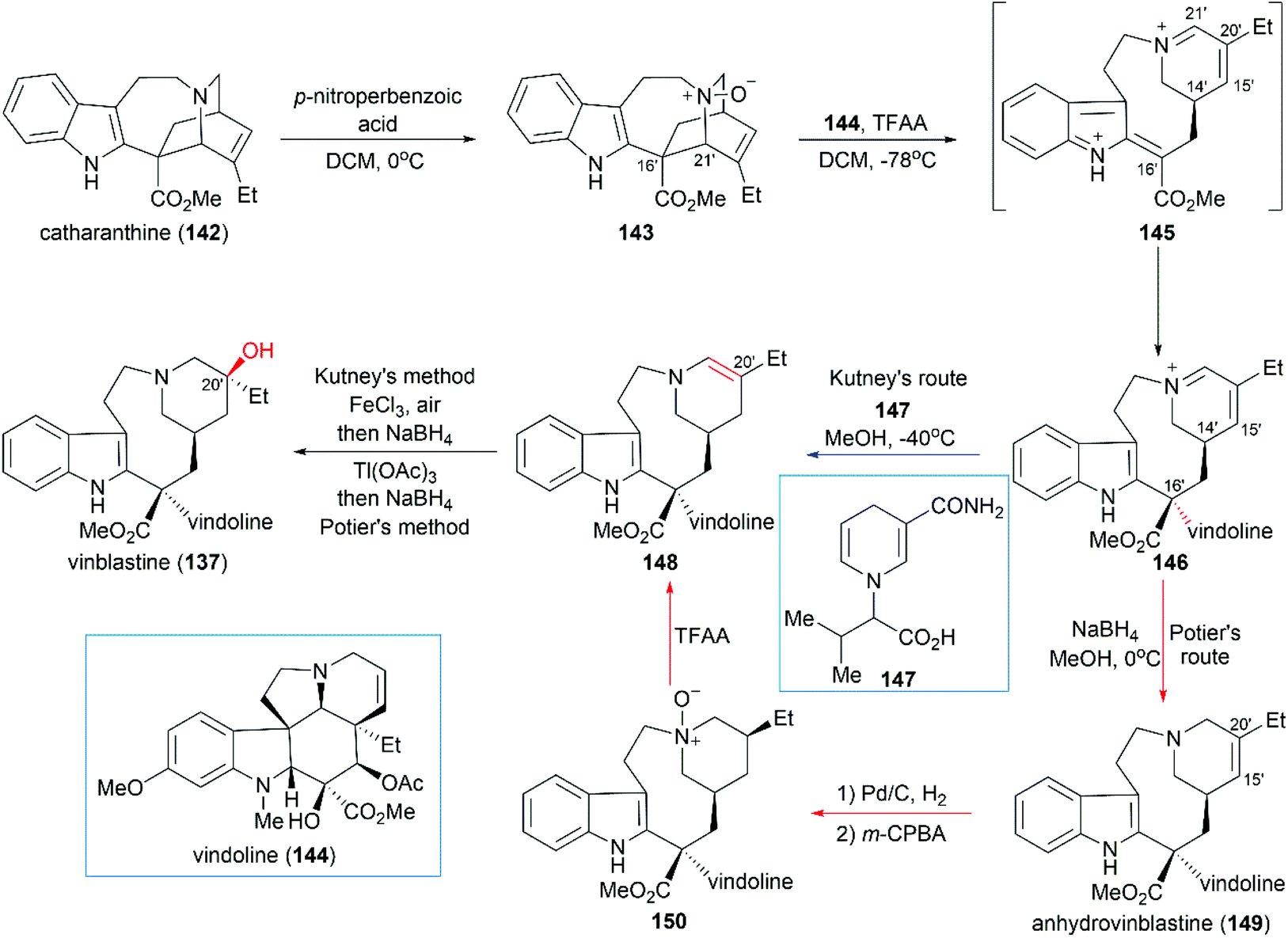
Inspired by the proposed biogenetic pathway for the synthesis of vinblastine, Potier and Kutney developed a pioneering strategy for direct coupling of catharanthine (142) with vindoline (144), which are the precursors of vinblastine (Scheme 15).132–134 In their synthetic design, catharanthine (142) was first oxidized to N-oxide 143, which underwent fragmentation to give bis-iminiumion 145 through a TFAA-promoted Polonovski reaction. Then, the coupling of 145 with vindoline (144) successfully produced iminiumion 146 with the desired stereoconfiguration at C16′. Careful examination of reaction parameters showed that the C16′ diastereoselectivity was temperature- and concentration-dependent: low temperature and high concentration gave better diastereoselectivity.132
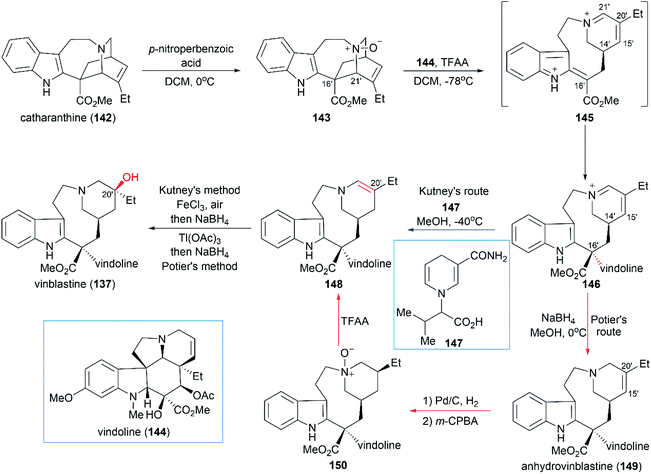 |
| | Scheme 15 Potier and Kutney's synthetic route. | |
Potier and Kutney applied different strategies for the transformation of iminiumion 146 into vinblastine (137). In Kutney's one-pot operation, a 1,4-reduction of 146 with the carefully designed reductant 147 yielded enamine 148, which was successfully oxidized with O2 in the presence of FeCl3 to establish the C20′ hydroxy group.134 Potier's 1,2-reduction of iminiumion 146 with NaBH4 produced anhydrovinblastine 149, which is a naturally occurring vinca alkaloid. Hydrogenation of the C15′–C20′ double bond and oxidation gave 150. The following Polonovski reaction transformed 150 into enamine 148. Installation of the C20′ hydroxy group was achieved through Tl(OAc)3 mediated oxidation followed by reduction with NaBH4.133
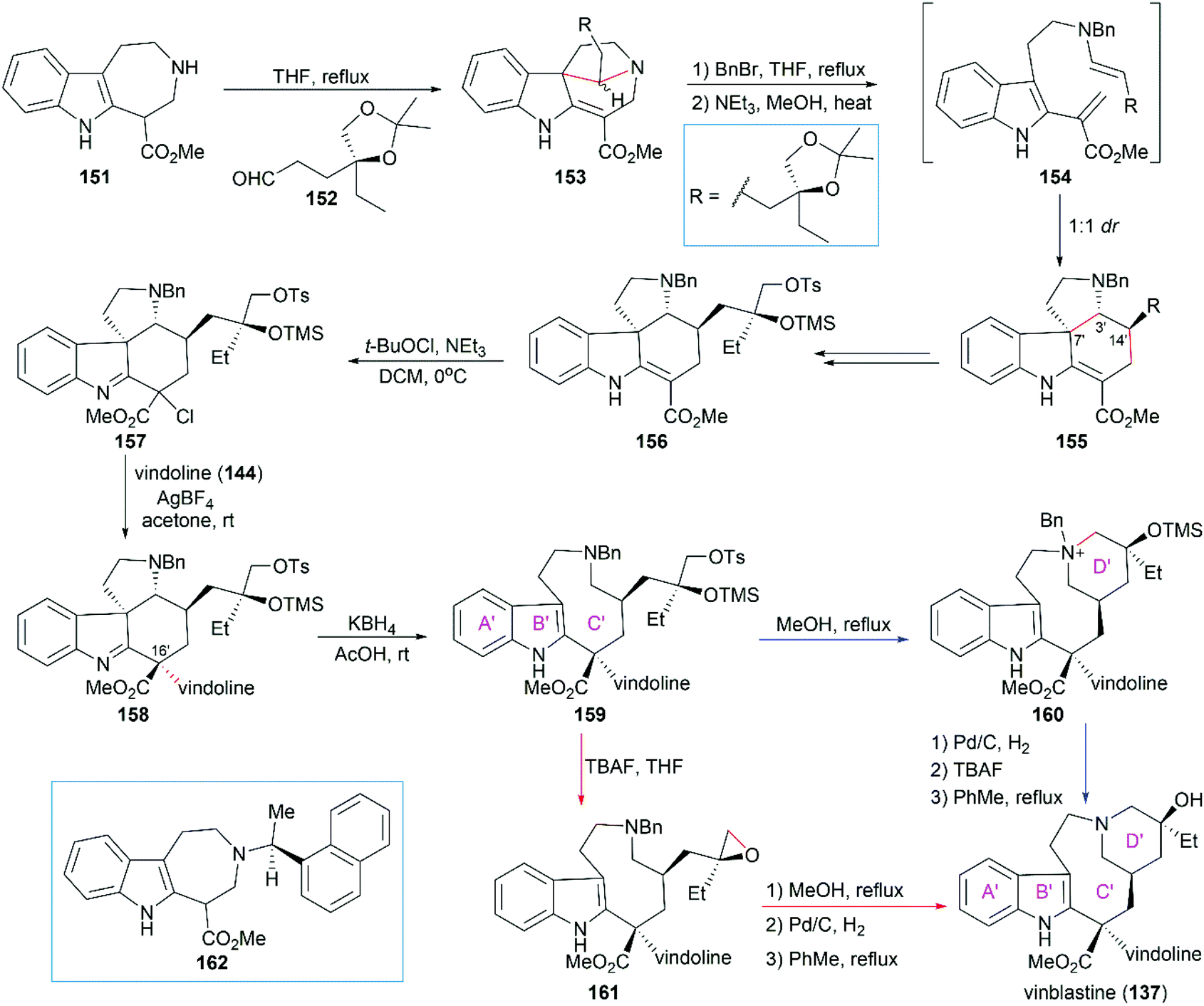
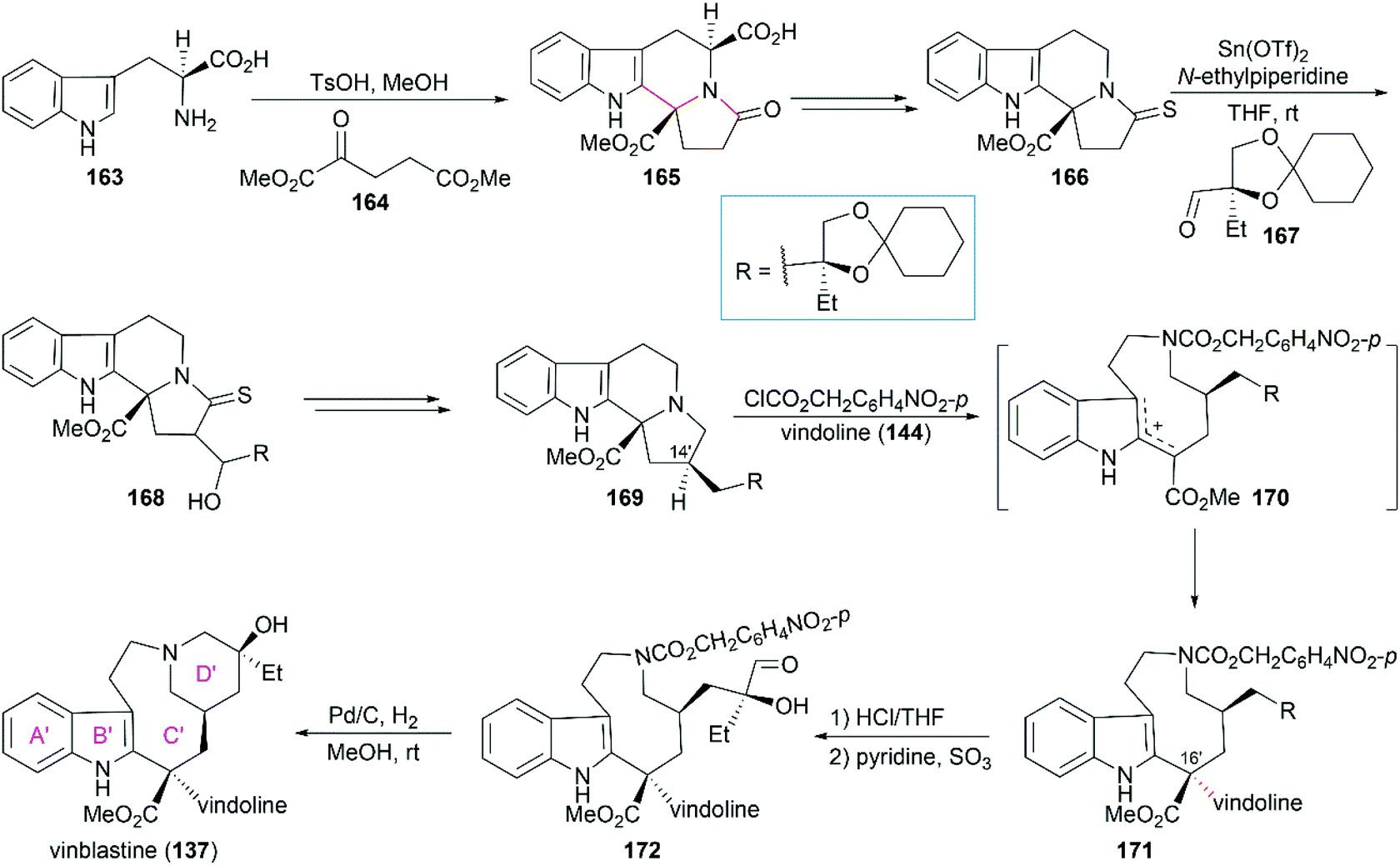
5.1.3 Magnus’ total synthesis of (+)-vinblastine.
In Magnus’ total synthesis of vinblastine (137), condensation of L-tryptophan (163) with diester ketone 164 produced the tetracyclic lactam 165 through Pictet–Spengler reaction (Scheme 17).137 Aldol condensation of 166 with chiral aldehyde 167 provided 168, which could be converted to 169 with the required C14′ stereo configuration after dehydration, desulfurization, and hydrogenation. When 169 was treated with ClCO2CH2C6H4NO2-p, carbamate formation followed by fragmentation led to delocalized carbocation 170, which was trapped by vindoline (144) to give the coupled product 171. Their study showed that the stereochemistry of the newly generated C16′ chiral center was influenced by temperature, reaction solvent, and nucleophilicity of the aromatic partner. The best diastereoselectivity could be achieved in a ratio of 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 (C16′ S/R) when the reaction was run in CH3CN/H2O (15:1) at −15 °C. Hydrolysis of 171 followed by mono-oxidation of the diol and subsequent cyclization through reductive amination provided vinblastine (137).
:16 (C16′ S/R) when the reaction was run in CH3CN/H2O (15:1) at −15 °C. Hydrolysis of 171 followed by mono-oxidation of the diol and subsequent cyclization through reductive amination provided vinblastine (137).
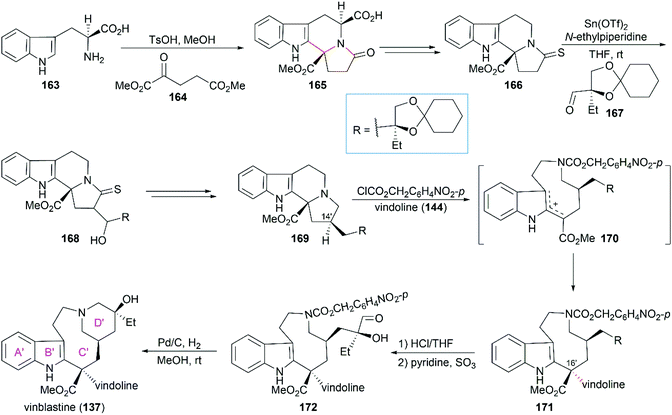 |
| | Scheme 17 Magnus’ synthetic route. | |
5.1.4 Fukuyama's total synthesis of (+)-vinblastine.
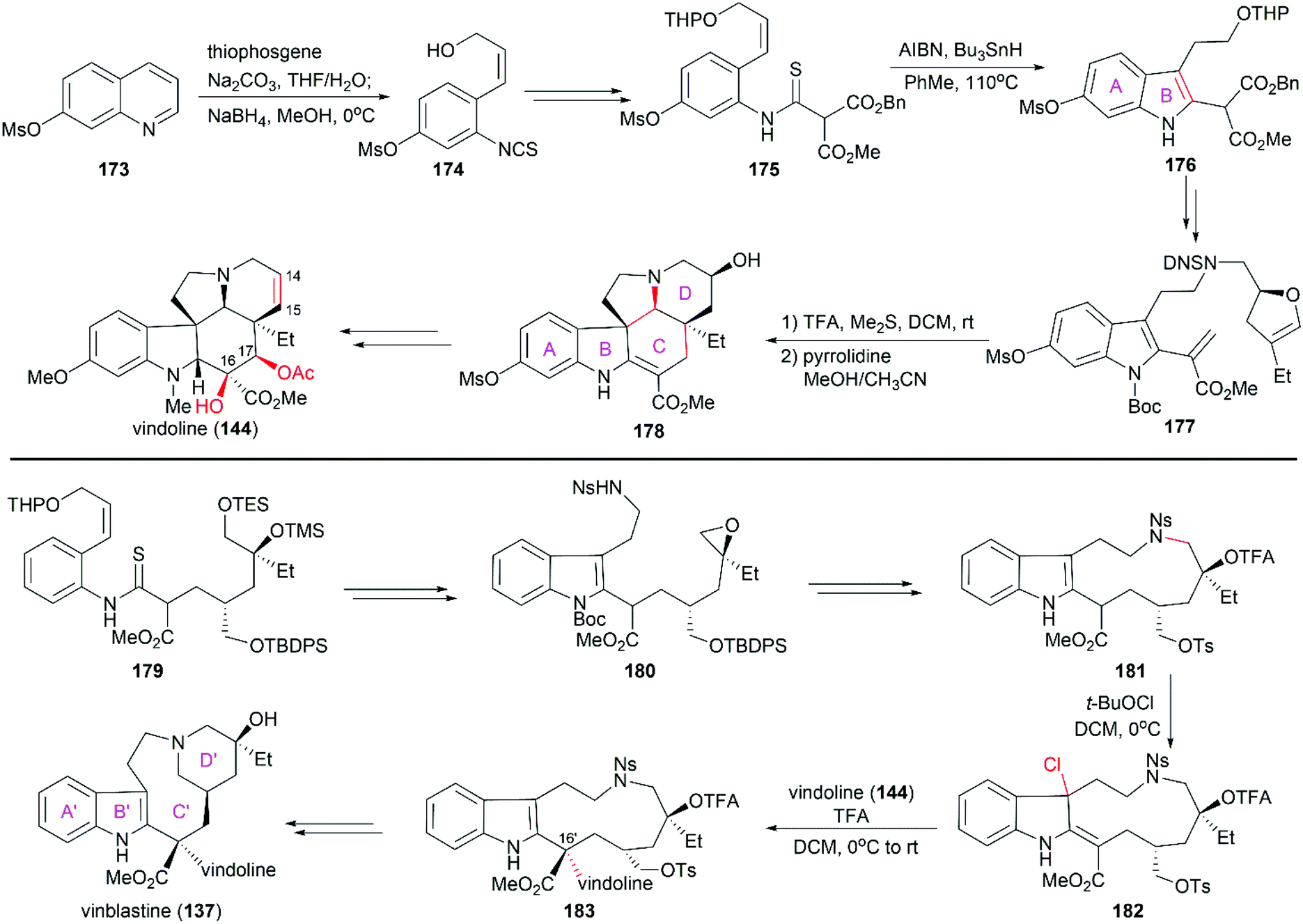
Fukuyama's total synthesis of vinblastine started with the enantioselective synthesis of the lower (−)-vindoline (144) subunit (Scheme 18).138,139 Ring-opening of 7-mesyloxyquinoline (173) with thiophosgene followed by reduction with NaBH4 provided isothiocyanate 174, which was transformed into thioanilide 175. Following the protocol developed in their laboratory, 175 was converted to indole compound 176 through radical-mediated cyclization. Further elaboration of 176 led to 177, which was transformed into pentacyclic compound 178 upon hydrolysis in the presence of TFA followed by removal of DNS protection and subsequent cyclization. Further manipulations including dehydration to construct the C14–C15 double bond, and introduction of the C16 and C17 hydroxy groups successfully provided (−)-vindoline (144). The synthesis of the upper subunit also relied on a radical-type indole synthesis method developed by Fukuyama and co-workers to provide epoxide 180, which was a precursor of the eleven-membered-ring compound 181. When treated with tert-butyl hypochlorite, 181 was smoothly converted to chloroindolenine 182, which was coupled with (−)-vindoline (144) in the presence of TFA to yield 183 with the desired stereoconfiguration at C16′. It is worth noting that the diastereoselectivity of this transformation was very high and no other stereoisomer was found. The final D′ ring was synthesized through deprotection of the tertial alcohol, removal of the Ns protection, and subsequent cyclization. This strategy was also applied to the synthesis of (+)-vincristine and vinblastine derivatives in the Fukuyama laboratory.140,141
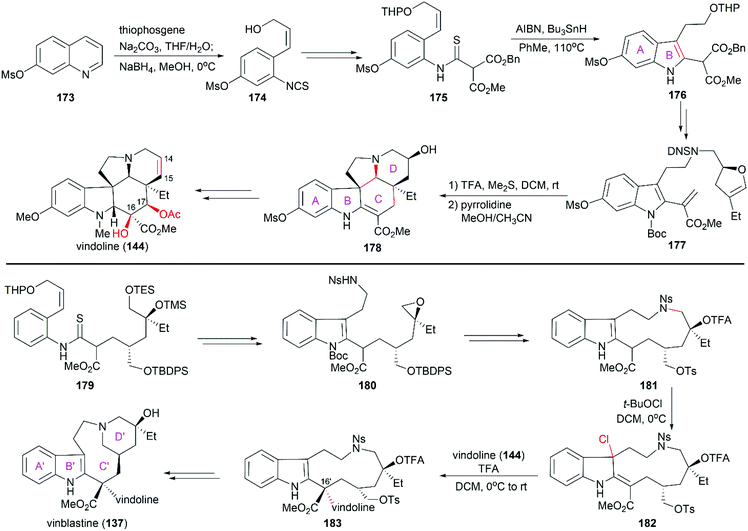 |
| | Scheme 18 Fukuyama's synthetic route. | |
5.1.5 Boger's total synthesis of (+)-vinblastine.
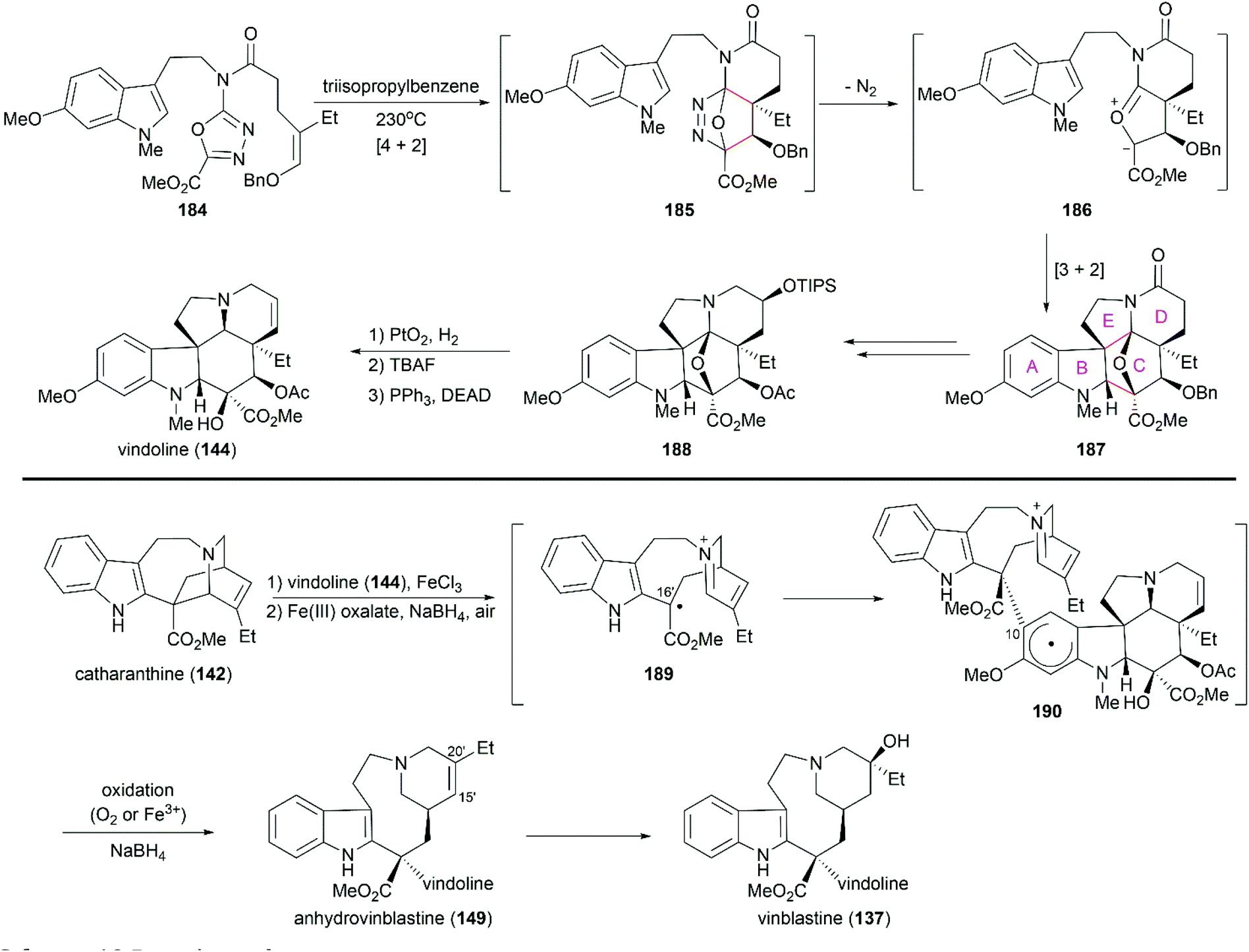
Boger's concise synthesis of vinblastine utilized a powerful cascade cycloaddition reaction developed in their laboratory.142,143 When heated at a high temperature, the advanced intermediate oxadiazole 184 underwent an inverse electron demand Diels–Alder reaction to give 185, which was transformed into the bridged cycloadduct 187 after the loss of N2 and subsequent 1,3-dipolar cycloaddition (Scheme 19). It should be noted that this elegant cascade cycloaddition reaction constructed four C–C bonds and three rings concomitant with the establishment of the six stereocenters with high diastereoselectivity. Hydrogenolytic cleavage of the oxygen bridge in 188 followed by silyl ether cleavage and subsequent dehydration under Mitsunobu reaction condition successfully produced (−)-vindoline (144). The following Fe(III)-mediated coupling of (−)-vindoline (144) with catharanthine (142) and installation of C20′ alcohol successfully produced vinblastine (137). According to their preliminary mechanistic studies, a plausible radical pathway was proposed for the coupling reaction: in the presence of FeCl3, 142 was oxidized to radical cation 189, which underwent radical addition to the aromatic ring of vindoline at C10 to yield 190.144 Further oxidation for rearomatization and subsequent reduction of the iminiumion provided anhydrovinblastine (149). They also proposed another Fe(III)-mediated radical pathway for the installation of a hydroxy group at C20′.145 They applied this elegant strategy to the syntheses of vinblastine-related natural products and analogs.144,145
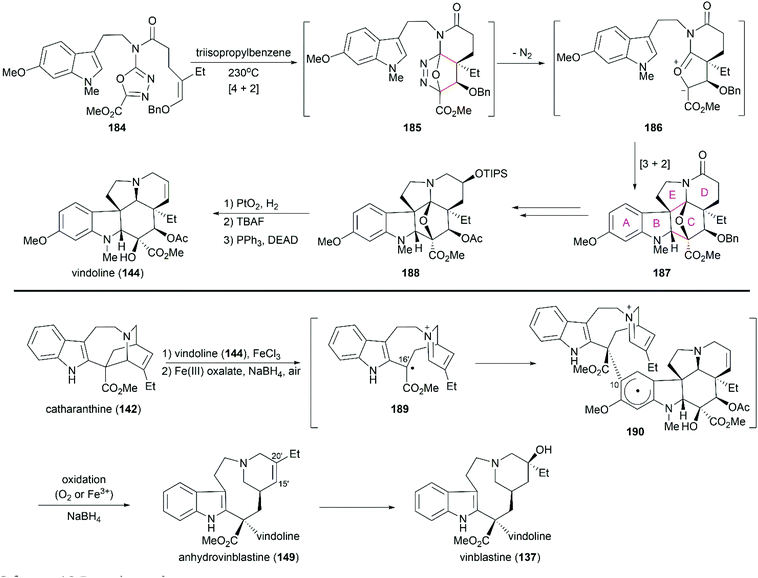 |
| | Scheme 19 Boger's synthetic route. | |
5.1.6 Jiang's total synthesis of (+)-vinblastine.
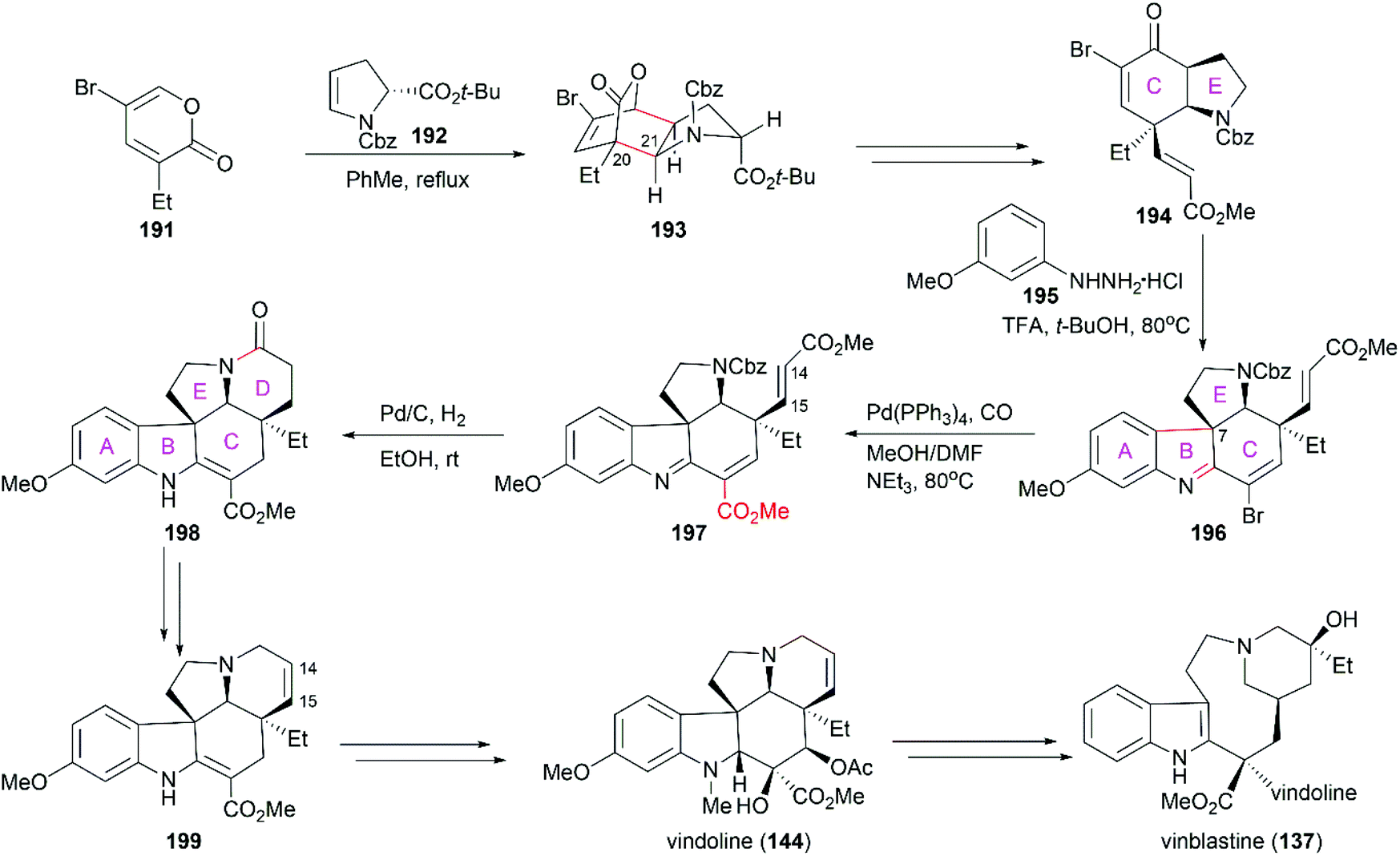
Jiang's total synthesis of vinblastine started with inverse-electron-demand Diels–Alder cycloaddition of pyrone 191 with chiral enamide 192 to give bridged lactone 193, which constructed the C20 and C21 chiral centers (Scheme 20).146 Further manipulations on 193, including decarboxylation, reductive ring-opening, installation of the ester group, and oxidation, led to α,β-unsaturated ketone 194. A challenging Fischer indolization on 194 for the introduction of the A/B rings and the C7 quaternary chiral center was optimized to give tetracyclic compound 196 with excellent diastereoselectivity and regioselectivity. Pd(0)-catalyzed installation of the ester group provided 197, which was transformed into pentacyclic compound 198 through a Pd/C-catalyzed one-pot cascade reaction including lactamization to construct ring D. Reintroduction of the C14–C15 double bond and reduction of the amide produced 199, which was an advanced intermediate in Fukuyama's total synthesis of (−)-vindoline (144).138 Coupling of vindoline with catharanthine (142) following Boger's protocol successfully provided vinblastine.143
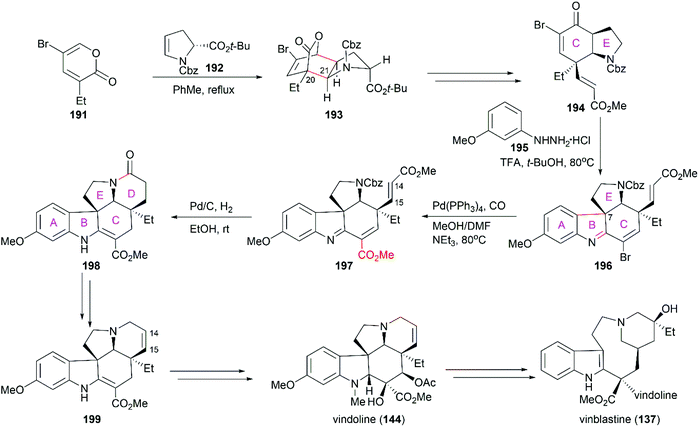 |
| | Scheme 20 Jiang's synthetic route. | |
5.2 Structure–activity relationships of vinblastine derivatives
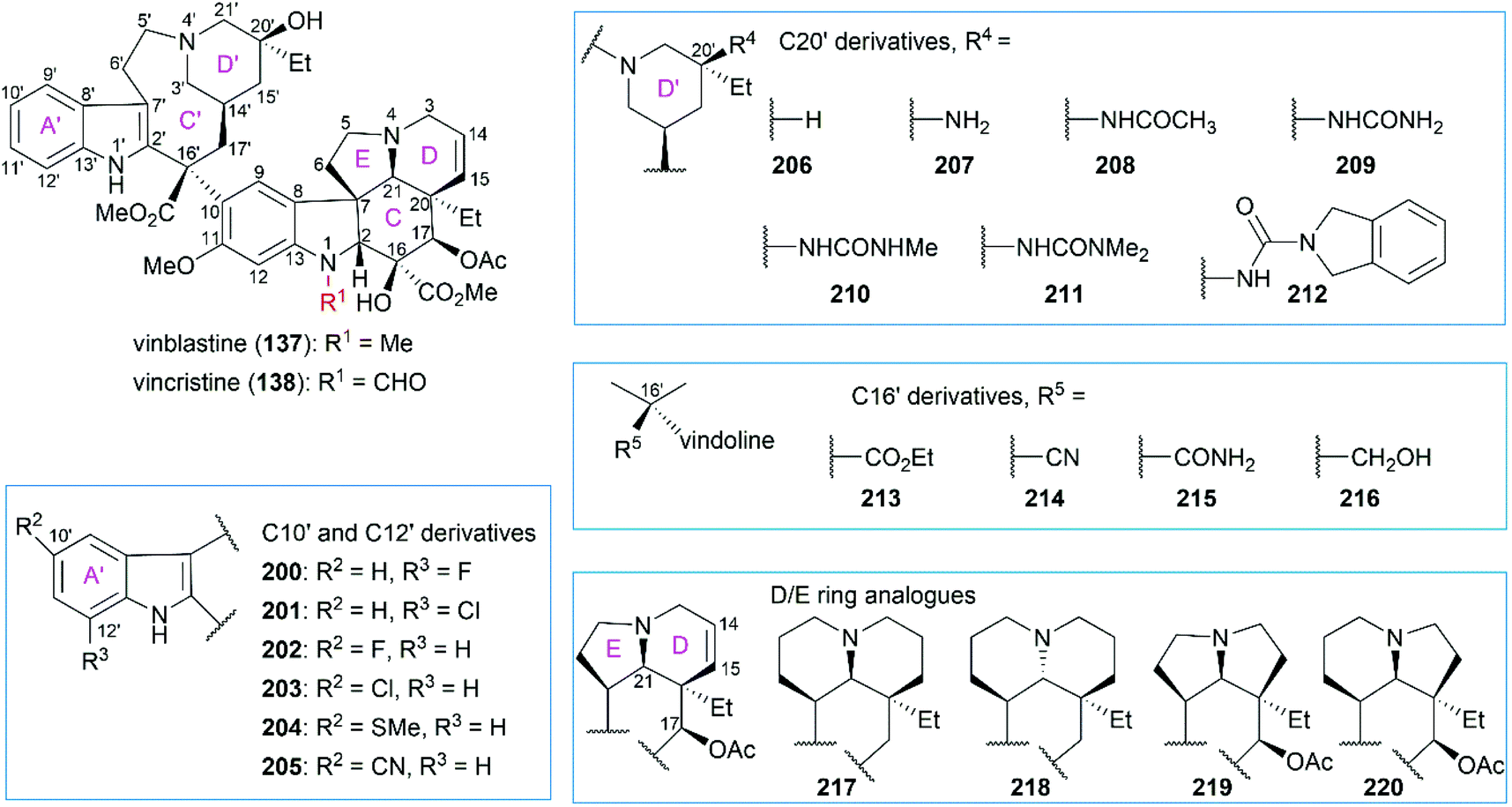
Extensive SAR studies on vinblastine and related natural or synthesized derivatives led to the approval of several important antitumor drugs, some of which are listed in Fig. 8. However, the emergence of drug resistance caused by overexpression of the drug efflux pump phosphoglycoprotein (Pgp) calls for more potent vinblastine derivatives to be synthesized and evaluated in the future. Since several review papers have given a detailed summary of the SAR results,147–150 we briefly discuss research progress achieved in recent years, especially those in Boger's laboratory (Fig. 9 and Table 1).
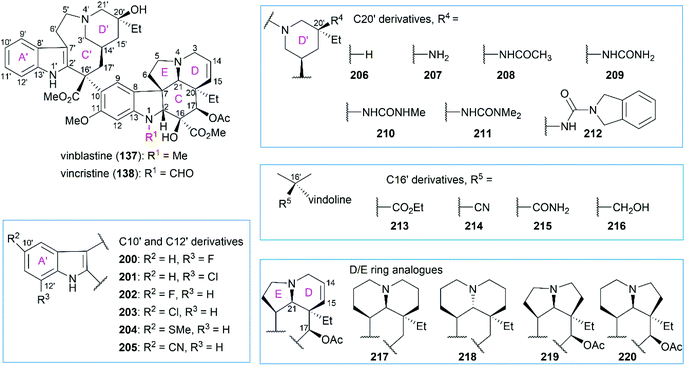 |
| | Fig. 9 SAR studies of vinblastine-related compounds. | |
Table 1 Cytotoxicities of vinblastine and related derivatives to three carcinoma cell linesa
| Entry |
Compound |
IC50 (nM) |
| L1210b |
HCT116b |
HCT116/VM46b |
|
Values given are IC50 in nM and original activity data can be found in ref. 150–155.
L1210: murine leukemia cell line; HCT116: human colon cancer cell line; HCT116/VM46: resistant human colon cancer cell line, Pgp overexpression.
|
| 1 |
Vinblastine (137) |
6.0 |
6.8 |
6.0 × 102 |
| 2 |
200
|
50 |
65 |
7.3 × 102 |
| 3 |
201
|
3.3 × 102 |
2.2 × 102 |
5.0 × 103 |
| 4 |
202
|
0.70 |
0.80 |
80 |
| 5 |
203
|
6.2 |
7.6 |
7.2 × 102 |
| 6 |
204
|
50 |
50 |
7.8 × 102 |
| 7 |
205
|
6.3 × 102 |
6.2 × 102 |
>1.0 × 104 |
| 8 |
206
|
— |
60 |
6.0 × 102 |
| 9 |
207
|
— |
6.0 × 102 |
>1.0 × 104 |
| 10 |
208
|
— |
90 |
7.5 × 103 |
| 11 |
209
|
40 |
7.5 |
4.4 × 103 |
| 12 |
210
|
— |
0.82 |
5.3 × 102 |
| 13 |
211
|
5.9 |
2.8 |
80 |
| 14 |
212
|
0.51 |
0.60 |
7.5 |
| 15 |
213
|
60 |
70 |
8.3 × 102 |
| 16 |
214
|
6.3 × 102 |
6.7 × 102 |
7.4 × 103 |
| 17 |
215
|
>1.0 × 104 |
>1.0 × 104 |
>1.0 × 104 |
| 18 |
216
|
6.5 × 103 |
5.8 × 103 |
>1.0 × 104 |
| 19 |
217
|
5.7 × 103 |
7.4 × 103 |
>1.0 × 104 |
| 20 |
218
|
55 |
80 |
9.0 × 102 |
| 21 |
219
|
5.6 × 102 |
80 |
7.8 × 103 |
| 22 |
220
|
7.2 |
6.5 |
4.5 × 102 |
Although substitution at the C12′ position of ring A′ in the upper velbanamine subunit (200 and 201) often led to reduced activity, substitution at the C10′ gave promising candidates. Applying their one-pot Fe(III)-mediated coupling and oxidation reaction, Boger and co-workers synthesized a series of vinblastine derivatives including the C10′-substituted ones.151 These derivatives exhibited activity that correlated with the size and shape of the substituents: 10′-chlorovinblastine 203 matched the potency of vinblastine; 10′-fluorovinblastine 202 exhibited an 8-fold increase in activity against both a sensitive (HCT116) and a vinblastine-resistant tumor cell line (HCT116/VM46); in contrast, derivatives bearing the larger (SMe, 204) or rigidly extended (CN, 205) substituents were 10–100-fold less potent. According to the X-ray structure of vinblastine bound to tubulin127 and the results of activity assays, the authors proposed that the 10′-fluorine substituent interacts with the protein at a hydrophobic site uniquely sensitive to steric interactions.151
Initial substitution of the C20′ hydroxy group with a hydrogen atom (206) or free amine (207) gave derivatives with less potency.152,153 Acetylation of the free amine (208) resulted in activity improvement, and a derivative with the urea group (209) showed comparable potency with vinblastine. However, these derivatives exhibited a further decrease in activity against a resistant HCT116/VM46 cell line compared with vinblastine. Further substitution of the terminal nitrogen led to improved activity against both sensitive and resistant cell lines (210 and 211). Finally, they found that the isoindoline-substituted compound 212 exhibited a 10-fold increase in activity against the sensitive HCT116 cell line and an 80-fold increase in activity against the resistant HCT116/VM46 cell line.
Replacement of the C16′ methyl ester with an ethyl ester (213), a cyano group (214), a primary carboxamide (215), or a hydroxy group (216) dramatically reduced the activity against both sensitive and resistant cell lines.154 Derivatives (217–220) containing 6,6-, 5,5-, and 5,6-membered DE ring systems, instead of the vindoline 6,5-DE ring, were synthesized.155 Among these analogs, 220 and 218 matched the potency of vinblastine and C17-deacetoxyvinblastine, respectively. Studies also showed that the N1-methyl group, C20 ethyl group, C14–C15 double bond, and C11 methoxy group were all essential to vinblastine's cytotoxic activity.150
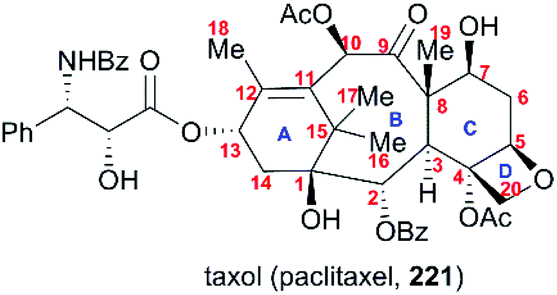
6. Taxol
One of the most famous natural taxane products, taxol (221, paclitaxel) was first isolated from the stem bark of Pacific yew by Wall and Wani in 1966, and in 1971 its complex structure was elucidated by NMR techniques and X-ray crystal structural analysis as a diterpenoid with a unique 6-8-6 tricyclic carbon skeleton bearing 11 stereocenters, an unusual oxetane ring, and a β-phenylisoserine chain ester at C13 (Fig. 10).156 Because of its potent anticancer activity, the worldwide academic and industrial research communities have developed different taxane agents for clinical use in the treatment of various cancers, including the marketed Taxol® and Anzatax®, as well as the synthetic structural derivatives under the tradenames Taxotere®, Jevtana®, and Abraxane®. In addition to its unique structure, taxol also demonstrates modes of action different from those of known anticancer agents. Mechanistic studies showed that during cancer cell mitosis, taxol diffuses through nanopores in the microtubule to bind the β-tubulin subunit on the inner surface of the microtubule, thereby inhibiting the microtubule depolymerization process and arresting the cell in the late G2/M phase, which blocks the cell division and leads to apoptosis. Cancer cells are believed to have a higher cell division rate than “normal” cells; therefore, they are more sensitive to taxol.157–159 As observed for other target-specific anticancer drugs, the mechanism of action discussed above and the long-term use as a first-line treatment have given rise to resistance against taxane anticancer agents. The actual resistance mechanisms are unclear, even though many of these mechanisms have been uncovered in different cell lines. For example, the over-expression of the mdr1 gene gives rise to the drug efflux of the membrane-bound P-glycoprotein. The abnormal expression of microtubule-associated proteins leads to drug resistance.
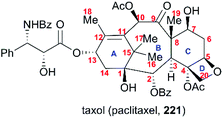 |
| | Fig. 10 The structure of taxol. | |
Scarcity of the natural resources of taxane entities hampers the development of taxane-derived anticancer agents, and scientists from academia and industry never stop procuring the taxane products on an industrial scale. Research groups around the world have been working on the synthesis of taxanes and testing accessible synthetic strategies for the construction of the 20-carbon skeleton of the taxane family.
6.1 Total synthesis of taxol
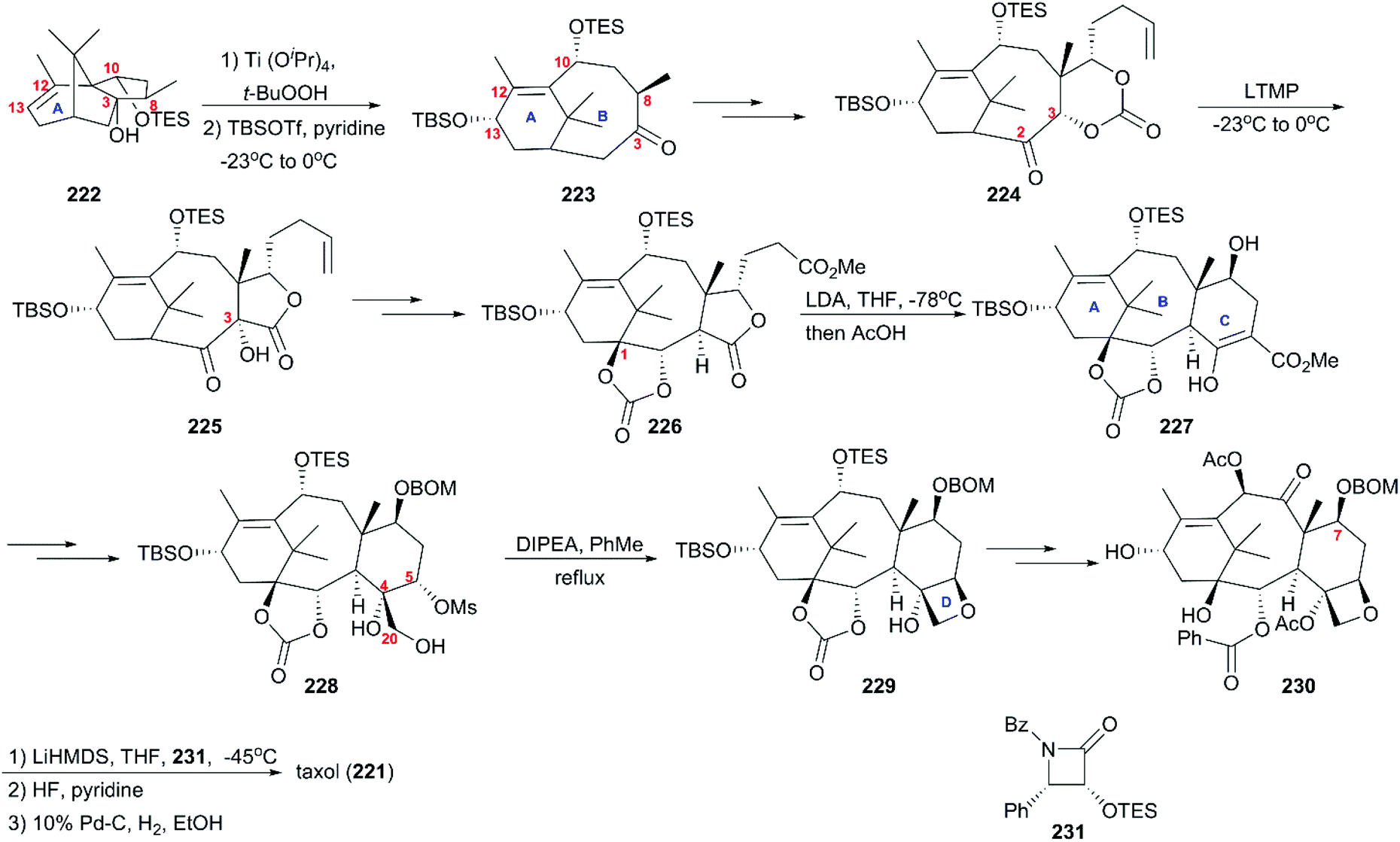
6.1.1 Holton's total synthesis of taxol.
In 1994, nearly three decades after the discovery of taxol (221), Holton's group reported the first total synthesis strategy using the readily available camphor derivative 222 as a starting material (Scheme 21).160,161 This derivative underwent epoxidation and alcohol fragmentation followed by protection at C13 to construct the B ring of taxusin intermediate 223, which was subjected to the aldol condensation and a series of oxidations and reduction to give 224 with a ketocarbonyl at C2. With ketone 224 in hand, Chan's rearrangement in the cyclic system was applied to produce trans-fused hydroxy lactone 225 in the presence of LTMP at low temperatures. Then, multi-step transformations gave rise to intermediate 226, which was converted to the C ring-fused enol ester 227 through Dieckmann cyclization. Subsequent decarbomethoxylation and oxidation at C5 and the introduction of C4/C20 dihydroxys led to 228, which was further converted to the D ring-attached precursor 229. Additional functionality interconversions produced 7-BOM baccatin III 230, which was lithium alkoxided and treated with β-lactam 231 to successfully produce taxol (221) after deprotection of C7 BOM.
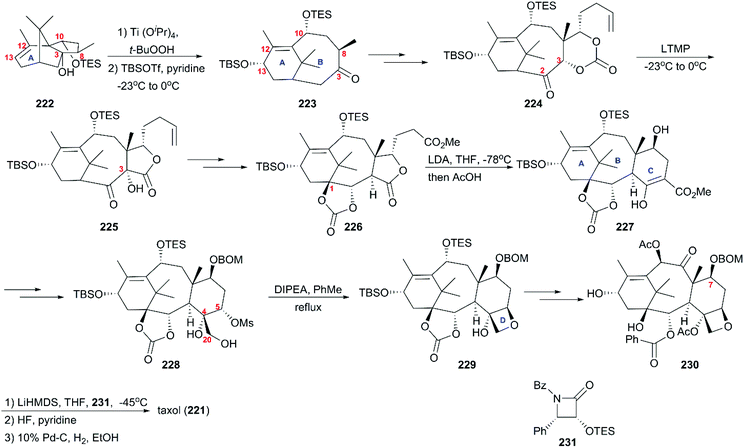 |
| | Scheme 21 Total synthesis of taxol by Holton. | |
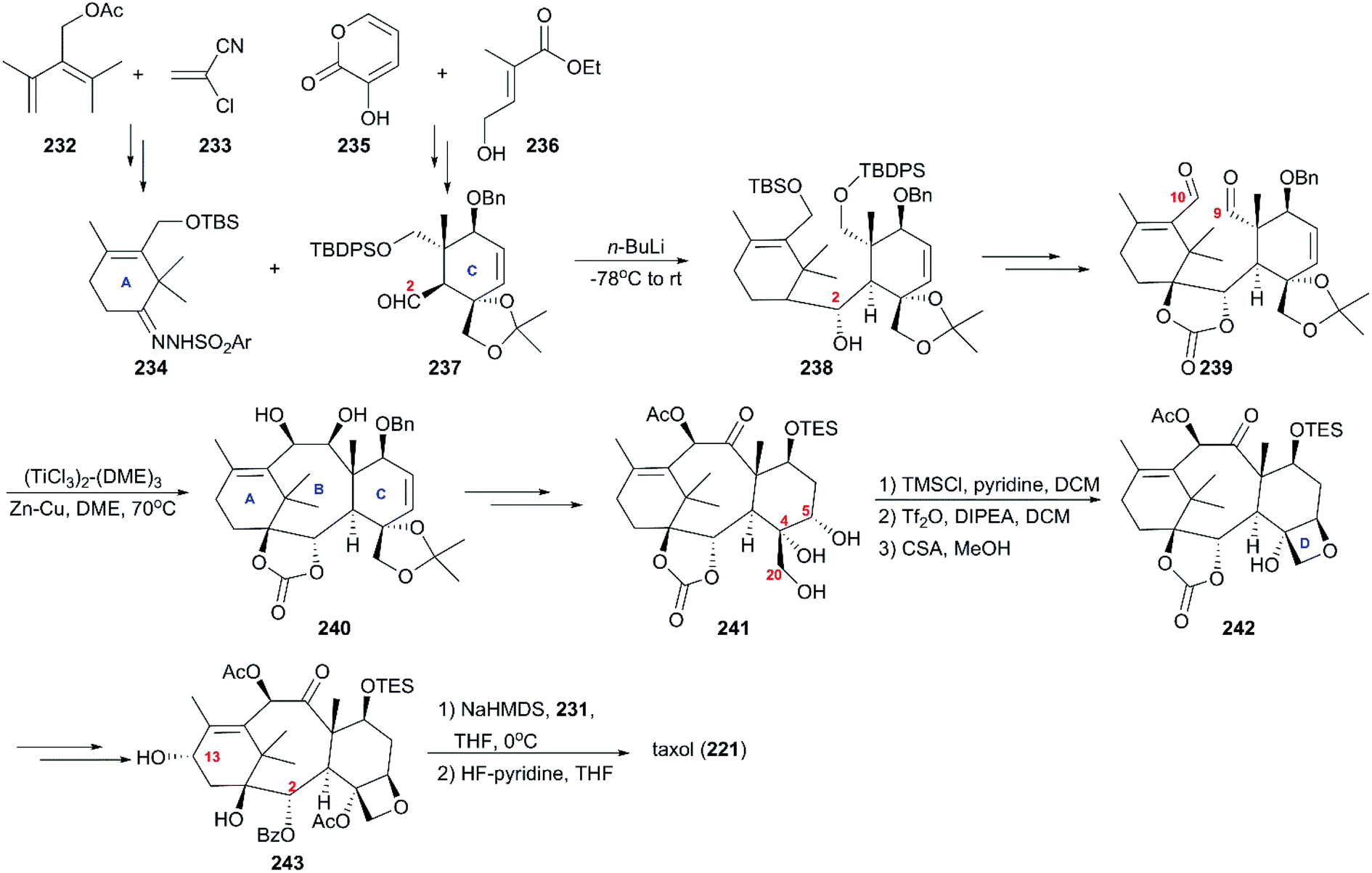
6.1.2 Nicolaou's total synthesis of taxol.
Soon after Holton's report, significant work on the total synthesis of taxol from Nicolaou and co-workers was published.162 In their strategy (Scheme 22), two pivotal intermediates, 234 and 237, with the ring A and ring B present in taxol, were both prepared by Diels–Alder cyclization between commercially available 232 and 233 and between 235 and 236, respectively; 234 and 237 were further decorated with functional groups. Then, they were subjected to Shapiro coupling to obtain a single diastereoisomer of hydroxy compound 238, which underwent epoxidation, regioselective reduction, deprotection, and oxidations to create dialdehyde 239. Subsequent optimal McMurry coupling resulted in the construction of the taxoid 6-8-6 ring skeleton 240 with 23% yield, and the configurations of the newly generated centers were confirmed by X-ray crystallographic analysis. After hydroboration-oxidation and interconversion of protecting groups, the important intermediate 241 with trihydroxy installed at C4, C5, and C20 was produced. During the next stage, the oxetane ring was built by sequential monosilylation of the primary hydroxy, triflation of the second hydroxy at C5, and acid treatment to produce a tetracyclic ring system. The allylic oxidation at C13 and benzoylation of C2-OH generated 7-TES-baccatin III 243, which was subjected to the protocol they previously reported using NaHMDS for C13 alkoxidation and consecutive treatment with β-lactam 231 to finally produce taxol (221).163
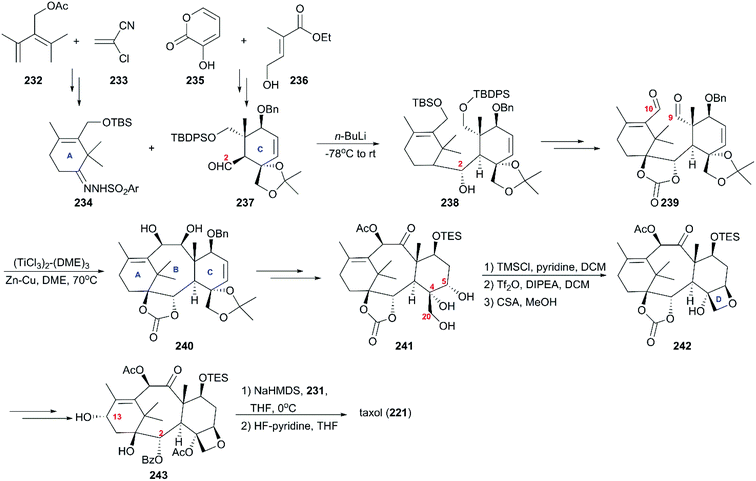 |
| | Scheme 22 Total synthesis of taxol by Nicolaou. | |
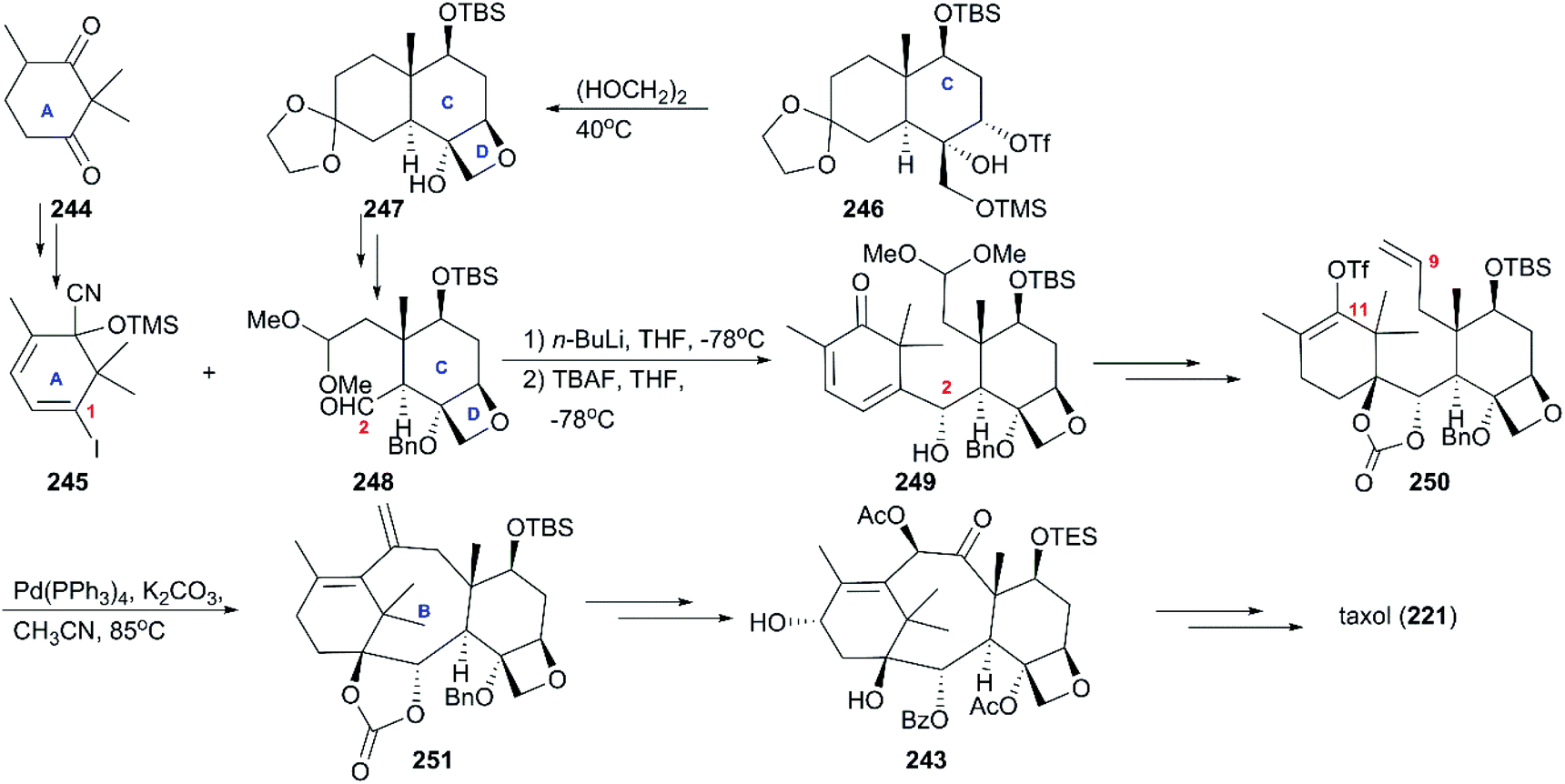
6.1.3 Danishefsky's total synthesis of taxol.
Different from the approaches previously reported, in which oxetane was constructed at a later stage, Danishefsky and co-workers prepared it at the beginning of their strategy using partially protected triol 246, which was obtained from a Wieland-Miescher ketone (Scheme 23).164 With the intermediate 247 in hand, additional functionalization led to the B ring precursor 248, which coupled with 245 derived from dione 244 to give rise to a single carbinol 249. After a series of conversions including epoxidation, hydrogenation, deprotection, and Wittig olefination, the precursor of cyclization 250 was obtained. In the next phase of synthesis, the intramolecular Heck reaction was employed to construct the tetracyclic skeleton 251, which was converted to the building block 243 used by Nicolaou through successive oxidation and protection reactions. By following previously described protocols for the final steps, the total synthesis of taxol was completed.
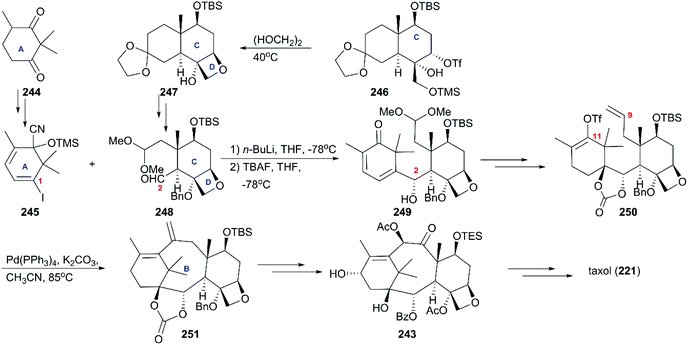 |
| | Scheme 23 Total synthesis of taxol by Danishefsky. | |
6.1.4 Wender's total synthesis of taxol.
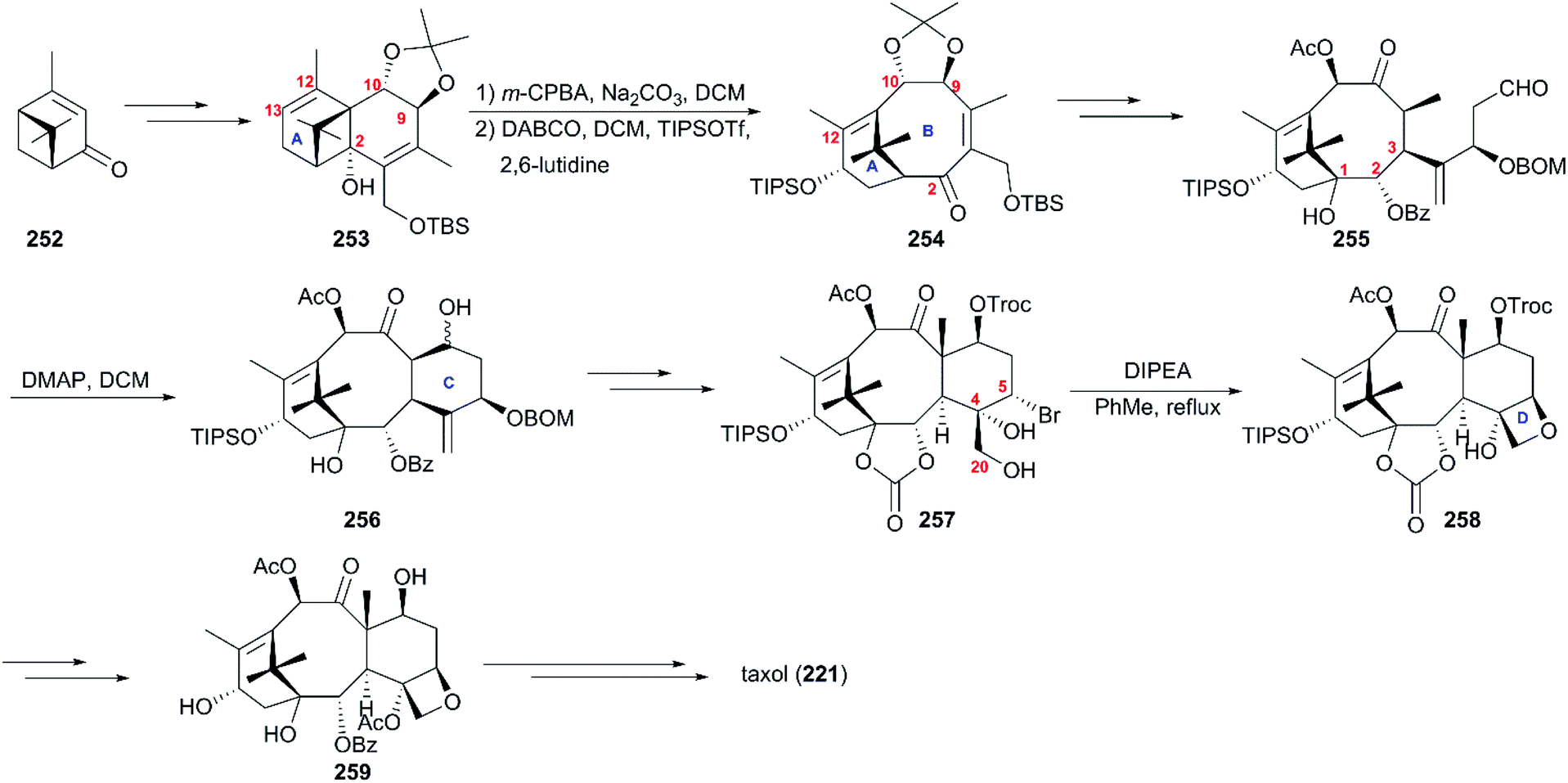
The synthesis strategy from the Wender team used verbenone 252, the air oxidation product of pinene, as the starting material (Scheme 24).165,166 Reactions consisting of the major transformations of alkylation, photorearrangement, and nucleophilic addition produced the subunit tricyclic 253 as a supplier of the A–B ring system and source of chirality of the taxol core. Then, 253 was successfully converted to the A–B bicyclic ketone 254via hydroxy-epoxide fragmentation induced by DABCO. After installation of the hydroxy at C1 and the alkyl with aldehyde group at C4, the intermediate 255 was subjected to intramolecular aldol cyclization in the presence of DMAP to construct the C ring. In the next phase, compound 256 underwent stereoselective bromination at C5 and dihydroxylation at C4/C20 to give rise to precursor 257, which directly closed to afford compound 258 with the D ring. After the protecting group interconversions, baccatin III (259) reacted with β-lactam 231 following previously reported procedures to produce the target taxol (221).
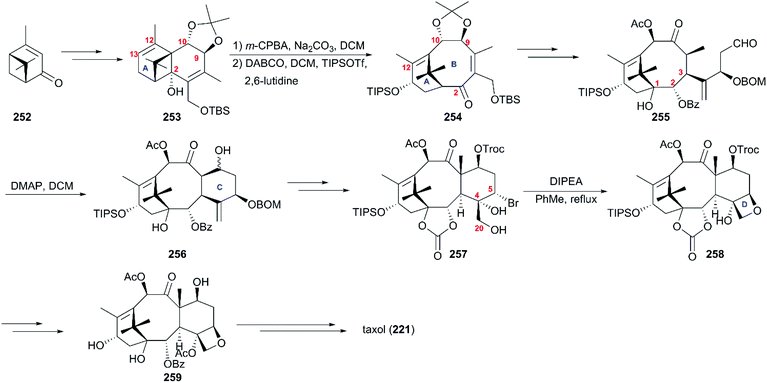 |
| | Scheme 24 Total synthesis of taxol by Wender. | |
6.1.5 Kuwajima's total synthesis of taxol.
In the strategy developed by Kuwajima's group (Scheme 25),167,168 the starting materials 261 and 263, which were used as suppliers of the A ring/C2/C10 and C ring/C9, were prepared from compounds 260 and 262, respectively. In the presence of Mg(II) ions, the coupling reaction of 261 with the lithiated 263 yielded a single diastereoisomer, which was converted to the cyclization precursor 264 through the protection of the newly produced vicinal diol as a boronate. Several Lewis acids were optimized to induce cyclization, and, finally, TiCl2(OiPr)2 was selected as the most powerful one for the construction of the B ring. This step was followed by the removal of a protecting group, generating the tricarbocycle 265. After generation of hydroxy at C7 and ketocarbonyl at C4, cyclopropanation of the double bond between C3 and C8 was induced to give cyclopropyl ketone 266, which was exposed to the SmI2/HMPA/methanol reductive system to smoothly produce 267 with a newly angled methyl group at C8. Functionalizations of 267 generated 268 as the precursor of oxetane, and in the presence of DBU, the tetracycle 269 was generated. In the final phase, Kuwajima and colleagues also applied previously reported protocols with some modifications to finish the total synthesis of taxol (221).
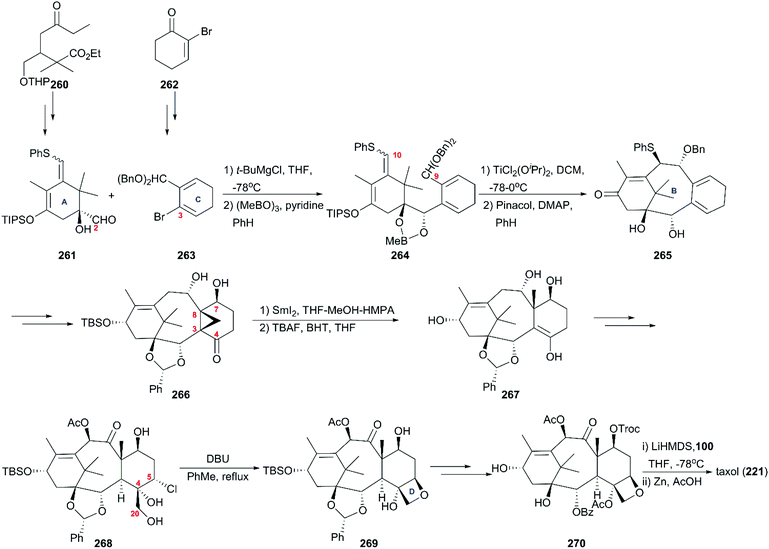 |
| | Scheme 25 Total synthesis of taxol by Kuwajima. | |
6.1.6 Mukaiyama's total synthesis of taxol.
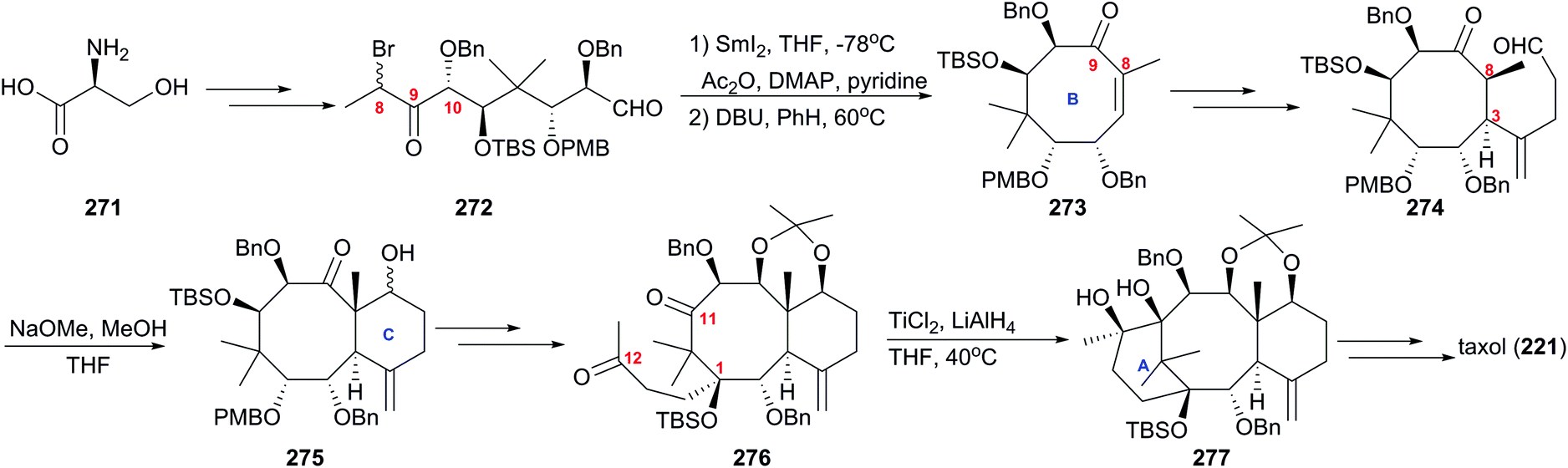
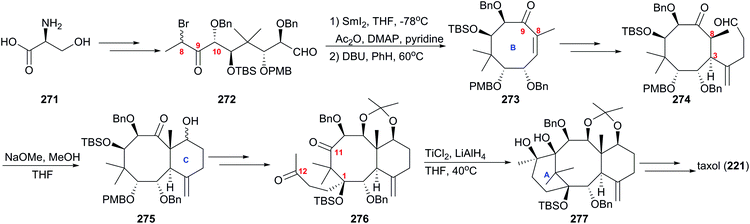
In 1999, Mukaiyama's group applied a novel approach to taxol using optically active polyoxy linear unit 272 to prepare the B ring of taxol at the early stage of the plan (Scheme 26).169 The cyclic precursor 272 prepared from amino acid 271 by repeated intermolecular aldol reactions, was converted to the 8-membered ring smoothly in the presence of SmI2, and followed by dehydration, α,β-unsaturated cyclooctanone 273 was provided. Introduction of the alkyl aldehyde at C3 produced 274, which was converted into the desired bicyclic compound 275 by the intramolecular aldol reaction in the presence of NaOMe. After transformations of the functional groups and protections of newly formed hydroxy groups, diketone 276 was given and it subsequently was subjected to the intramolecular pinacol coupling reaction to produce the desired pinacol 277 in the presence of low valent titanium. At the next stage, the protocols used previously were applied here to get taxol (221).
 |
| | Scheme 26 Total synthesis of taxol by Mukaiyama. | |
6.1.7 Baran's total synthesis of taxol.
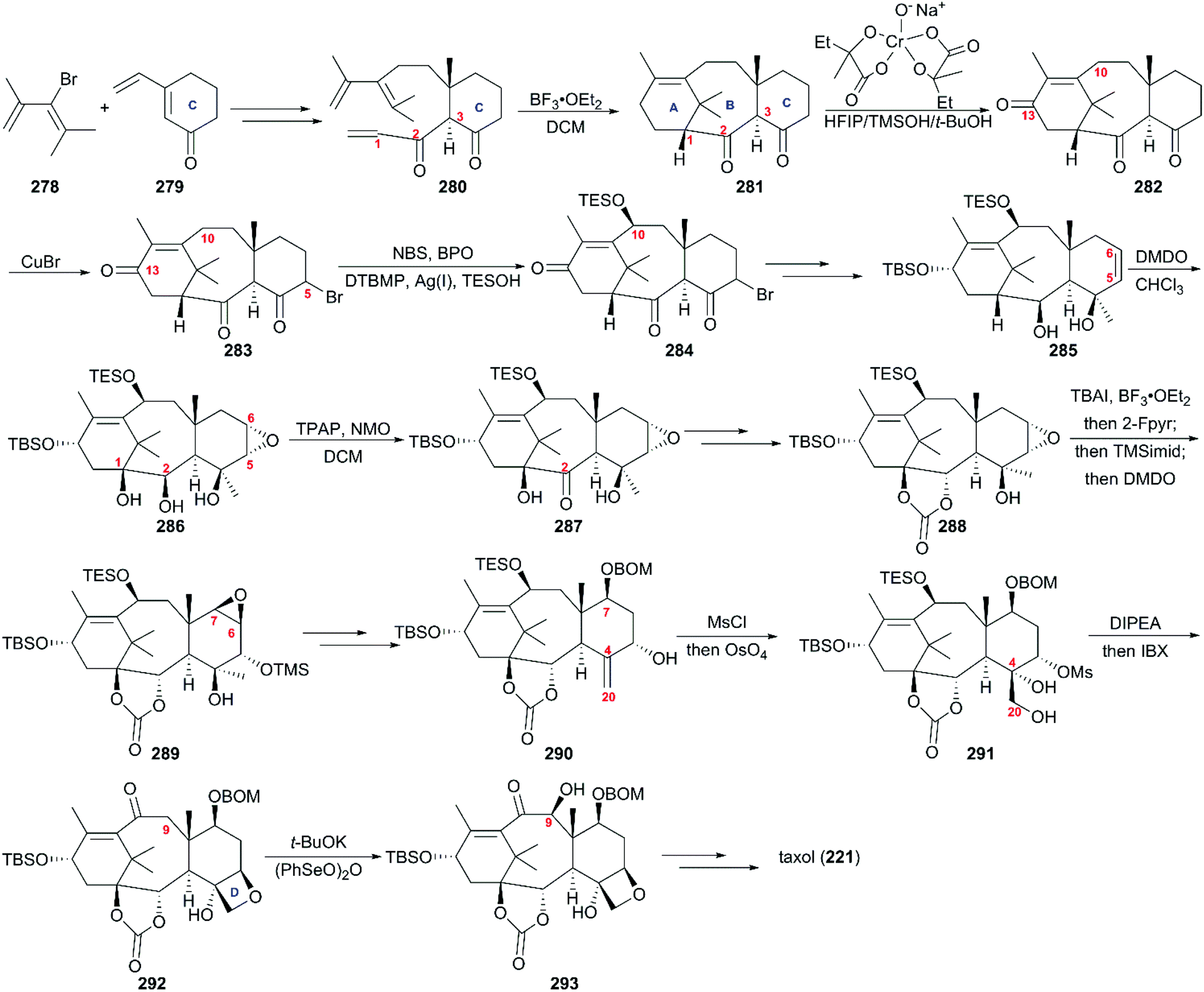
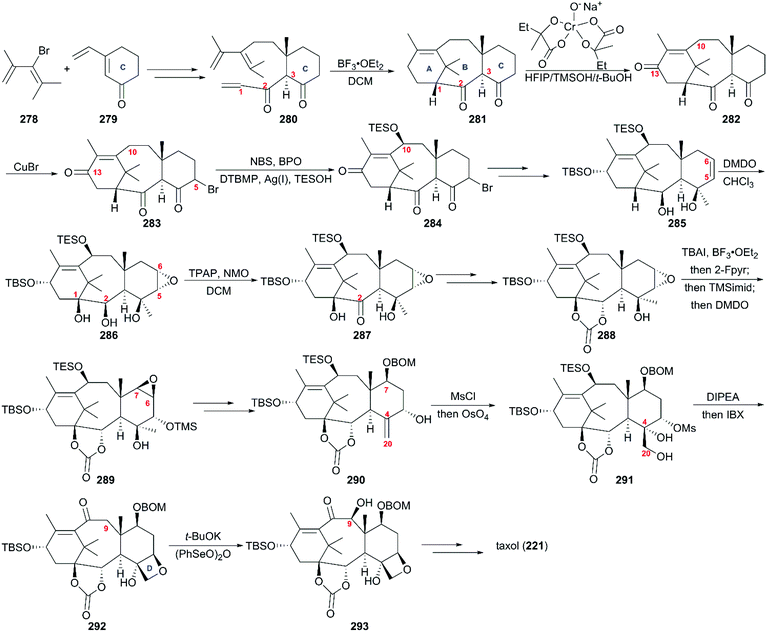
Except for the formal syntheses of taxol reported by Takahashi,170 Nakada,171 and Chida,172,173 no synthetic approach was reported in the past two decades before Baran's work. The conventional convergent approaches to taxol proved not to be practical in the pharmaceutical industry; on the contrary, the relatively linear natural route used to synthesize taxol is efficient and the oxidation sequence of taxol biosynthesis could produce more natural analogs of taxol.174,175 Inspired by the taxol biosynthetic approaches, including the generation of C–C bonds by the first type of enzymes in a “cyclase-phase” and installation of C–O bonds by the second type of enzyme in an “oxidase-phase”, Baran's team presented a two-phase synthesis scheme for taxol.176,177 As shown in Scheme 27, in the first “cyclase phase”,178 the cyclization precursor 280 was prepared on a decagram scale by the Lewis acid-modified organocopper 1,6-addition of known compounds 278, 279, and a subsequent aldol reaction and oxidation. With mixture 280 in hand, the tricyclic intermediate 281 was generated in the presence of BF3·OEt2. At the beginning of the “oxidase phase”, regioselective allylic oxidation was more feasible at C13 than at C10 in the presence of the Cr(V)-based oxidant. After bromination at C5, compound 283 experienced radical oxidation and nucleophilic substitution at C10 to generate 284, which was subjected to sequential elimination of C5 bromide, the addition of MeMgBr at C4, and reduction of the C2 and C13 ketone groups accompanied by TBS installation to give the oxidation precursor 285. The oxidant DMDO was employed to produce the epoxy-triol 286 using CHCl3 as an optimal solvent, followed by TPAP-mediated oxidation of C2 to afford 287, which was reduced using Na/i-PrOH and exposed to triphosgene to produce carbonate 288. After sequential iodine-oxidation in the presence of TBAI/BF3·OEt2 and elimination and epoxidation by DMDO, 288 was converted to epoxy-taxane 289. After the reductive opening of epoxide, BOM protection, and Burgess dehydration of C4, allylic alcohol 290 was generated in moderate yield. The intermediate with a hydroxy group activated by MsCl at C5 was unstable and immediately exposed to OsO4 to obtain the oxetane precursor 291, which was subjected to oxetane ring formation to generate tetracyclic carbonate 292. Then, (PhSeO)2O was applied to introduce a hydroxy group at C9 in the present of t-BuOK to deliver 293. Finally, synthesis of the target molecule 221 was achieved using previously reported protocols.
 |
| | Scheme 27 Total synthesis of taxol by Baran. | |
6.2 Structure–activity relationships of taxol derivatives
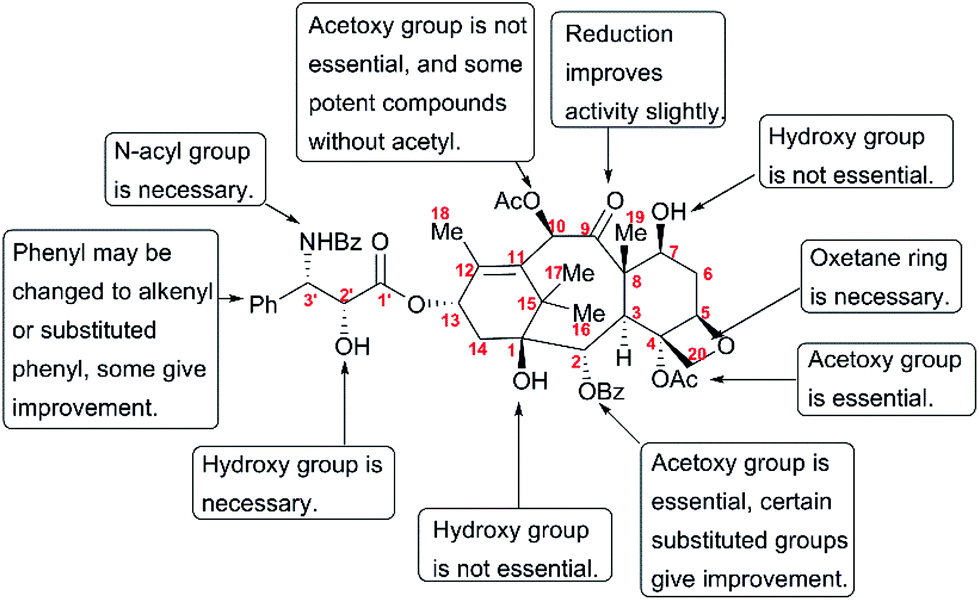
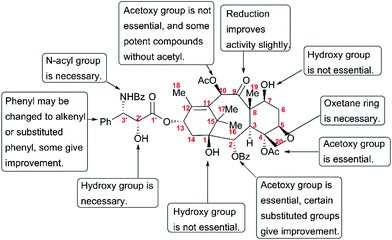
The discovery of taxol as one of the most potent anticancer agents spurred a large number of academic and industrial researchers to investigate the SAR for identifying lead compounds with drug-likeness potential. However, the complex structure of taxol, a heavily oxygenated polycyclic diterpenoid, presents a huge challenge for pharmaceutical studies that call for a substance-rich library. A number of studies have uncovered the still-incomplete SAR information about taxol. This information has been described by several reviews,179–183 and is summarized here in Fig. 11.
 |
| | Fig. 11 Structure–activity relationships of taxol. | |
Studies on the modifications at C1, such as removal and esterification, revealed that the C1-OH is not essential for the activity of taxol.184 In addition, C7-OH, C9-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C10-OAc play insignificant roles in the anticancer activity of taxol.185–188 SAR studies have shown that the benzoyl group at C2 is necessary for activity; 2-deoxy-taxol displayed decreased antitumor activity,189 and 2-benzoate analogs did not lead to any remarkable improvement in biological activity in a microtubule assay or cytotoxicity test.189 The acetoxy group at C4 was also demonstrated to be essential for activity, and the analog 4-deoxytaxol was inactive on several tumor cell lines.190,191 However, oxetane plays a critical role in the bioactivity of taxol, and analogs in which the oxygen atom was replaced by nitrogen, sulfur, or other heteroatoms did not significantly enhance antitumor activity.192,193
O, and C10-OAc play insignificant roles in the anticancer activity of taxol.185–188 SAR studies have shown that the benzoyl group at C2 is necessary for activity; 2-deoxy-taxol displayed decreased antitumor activity,189 and 2-benzoate analogs did not lead to any remarkable improvement in biological activity in a microtubule assay or cytotoxicity test.189 The acetoxy group at C4 was also demonstrated to be essential for activity, and the analog 4-deoxytaxol was inactive on several tumor cell lines.190,191 However, oxetane plays a critical role in the bioactivity of taxol, and analogs in which the oxygen atom was replaced by nitrogen, sulfur, or other heteroatoms did not significantly enhance antitumor activity.192,193
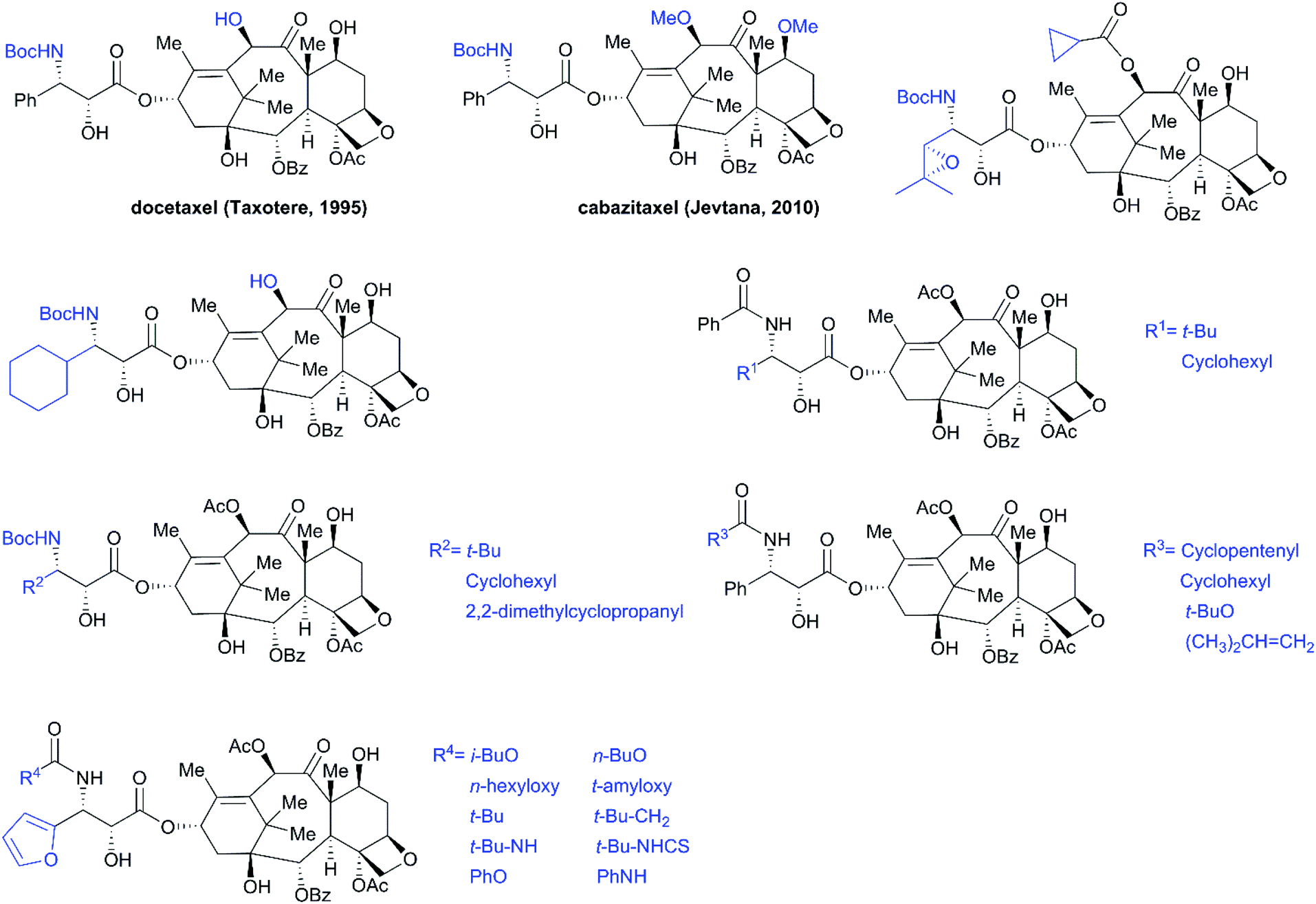
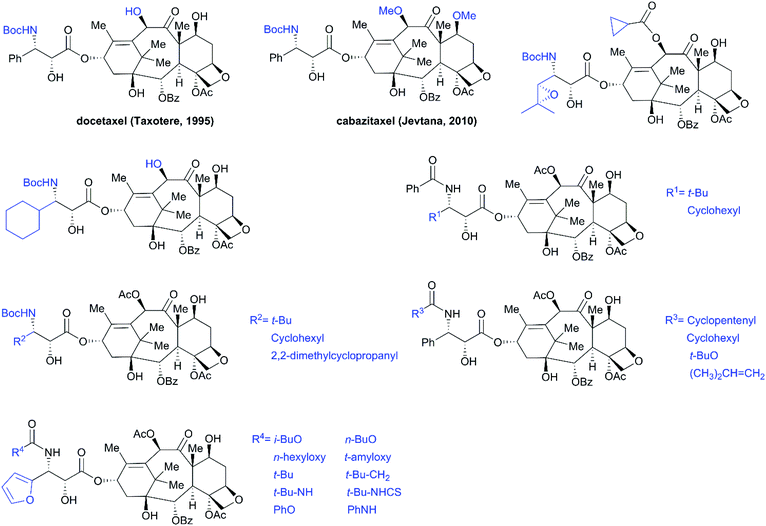
SAR studies revealed that the N-benzoyl-β-phenylisoserine side chain is essential for the antitumor activity of taxol, and this chain has attracted more extensive investigation than any other functional group of taxol.194–198 A number of analogs with a modified side chain exhibit stronger activity than taxol, some of which are listed in Fig. 12, including two marketed drugs, docetaxel (Taxotere®, 1995) and cabazitaxel (Jevtana®, 2010).
 |
| | Fig. 12 Examples of potent taxol analogs. | |
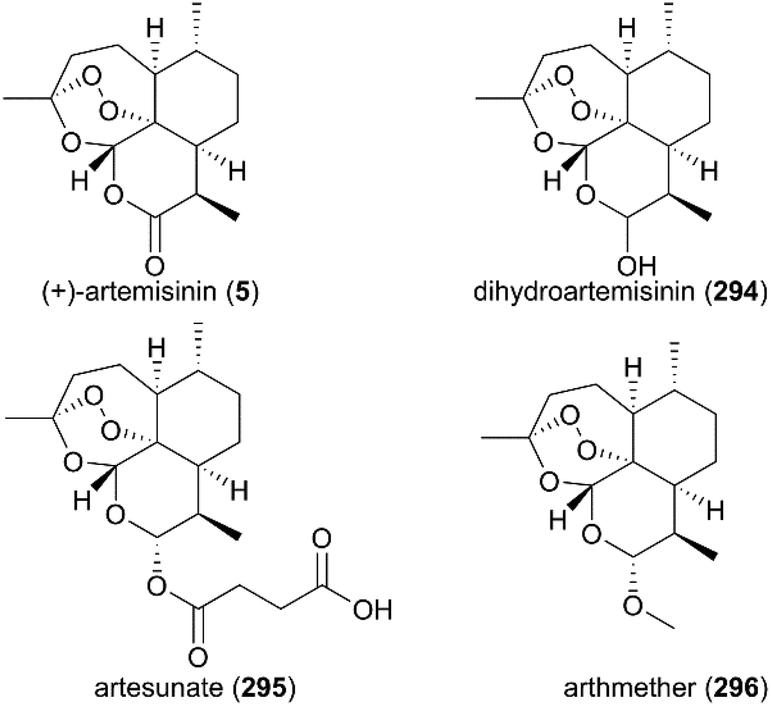
7. Artemisinin
(+)-Artemisinin (QingHaoSu) (Fig. 13, 5), the most used drug against malaria worldwide, was isolated from the traditional Chinese medicine plant Artemisia annua L. in 1972 by Youyou Tu, who shared the 2015 Noble Prize for this discovery,199 and (+)-artemisinin-based combination therapy is advocated by WHO. Artemisinin is a sesquiterpene lactone, and its structure is characterized by a unique 1,2,4-trioxane moiety, a pharmacophore that is a necessary endoperoxide bridge for the development of novel antimalaria drugs through semisynthesis of (+)-artemisinin. So far, pharmacological studies have not fully elucidated the exact mechanism of action of (+)-artemisinin, which is believed to be a pro-drug of carbon-centered free radical that plays a substantial role in antimalarial produced by cleavage of its endoperoxide ring inside erythrocytes. Upon bio-activation, the binding of (+)-artemisinin with low valent ions in hemin leads to reduction of the endoperoxide ring to produce an O radical, which subsequently generates a carbon free radical. This free radical is suggested to alkylate heme in the late stage of the parasite life cycle and react with reduced glutathione (GSH) in the early life-ring stage, after which cellular targets are selectively damaged.200–205 Due to its different mechanism of action, (+)-artemisinin shows higher efficacy against multi-drug-resistant malaria compared with the old antimalarial agent quinine and synthetic drug chloroquine. However, along with the widespread use of (+)-artemisinin-based combination therapy in the world have come reports of resistance from different regions. The mechanism of artemisinin-resistance is unclear enough and mainly attributed to the C580Y mutation in Kelch13 (K13) of parasites, and associated elevation of the lipid product phosphatidylinositol-3-phosphate (PI3P). It may influence host remodeling, functions of the apicoplast and food vacuole, all of which protect the parasites against artemisinin challenge.206,207 Therefore, there is an urgent need to develop novel drugs with new chemical scaffolds and, of course, different mechanisms of action would be excellent. But (+)-artemisinin is still sourced from plants with low yield, which limits the development of more efficient drugs. Therefore, exploring chemical synthesis methods, especially for the total synthesis of (+)-artemisinin, is important for academia and the pharmacy industry.
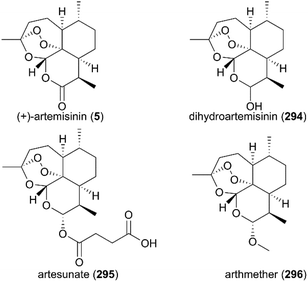 |
| | Fig. 13 (+)-Artemisinin and its derivatives. | |
7.1 Total synthesis of artemisinin
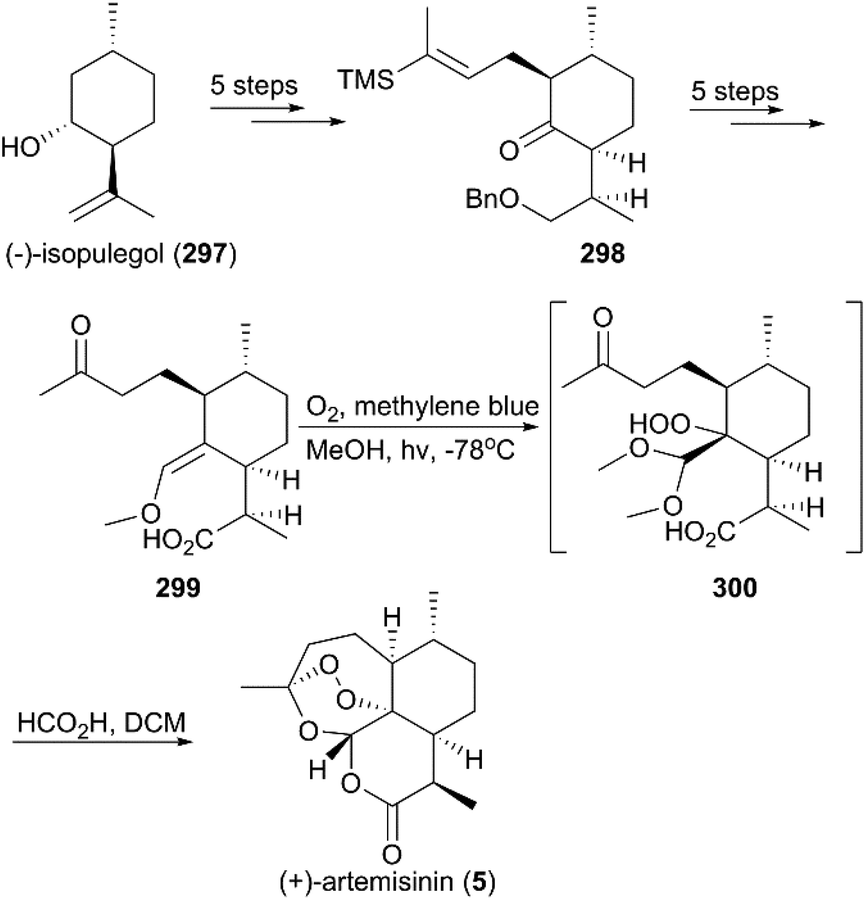
7.1.1 Schmid and Hofheinz's total synthesis of (+)-artemisinin.
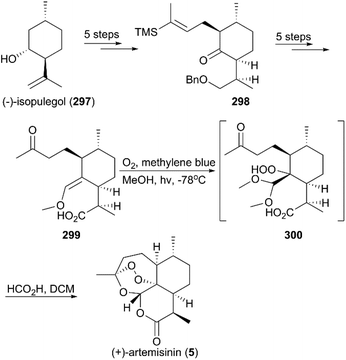
In the half-century since the discovery of (+)-artemisinin as an antimalarial drug with an unprecedented endoperoxide bridge structure, more than ten teams have completed its total synthesis.208,209 In 1983, scientists from Roche, Schmid and Hofheinz, first reported a 13-step route to synthesize artemisinin using (−)-isopulegol (297) as a starting material (Scheme 28).210 The alkylated menthone 298 was obtained from 297 with a 6:1 diastereomeric ratio in five steps of protection, oxidation, and alkylation reactions. Following the nucleophilic addition of a substituted trimethylsilane reagent, accompanied by deprotection and oxidation, enol ether 299 was generated after desilylation. With the key intermediate 299 in hand, photooxygenation was carried out at −78 °C, and the crude oxidation mixture was treated with acid; the target (+)-artemisinin (5) was achieved in 30% yield after crystallization.
 |
| | Scheme 28 Total synthesis of (+)-artemisinin by Schmid and Hofheinz. | |
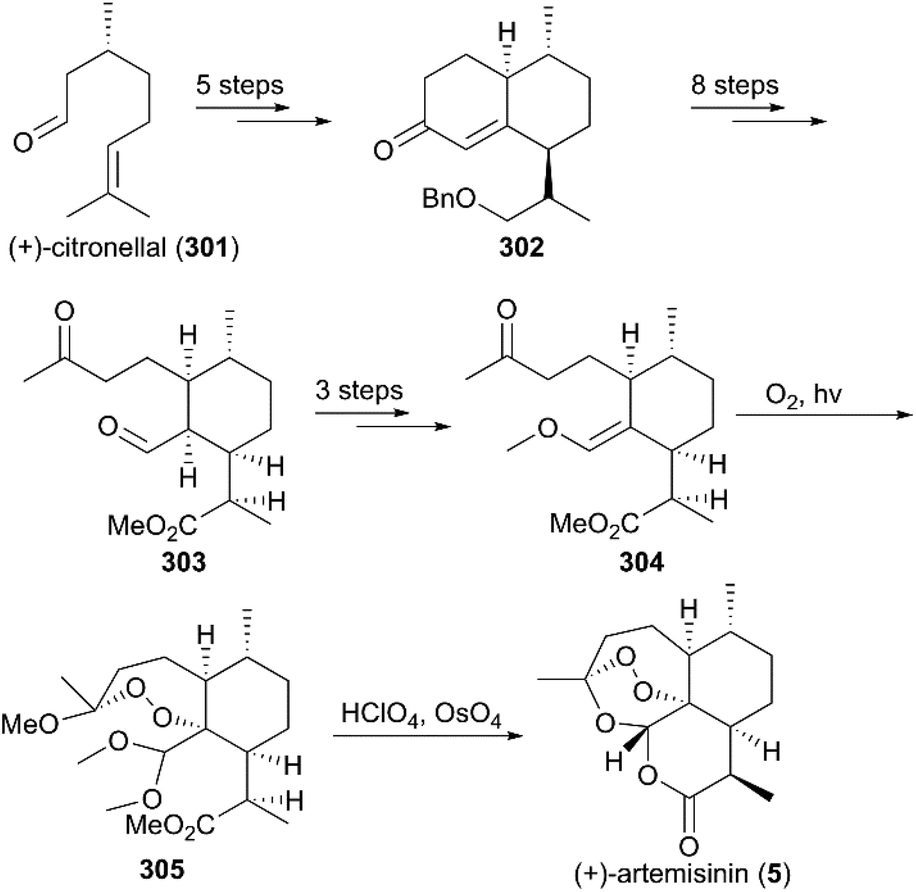
7.1.2 Zhou's total synthesis of (+)-artemisinin.
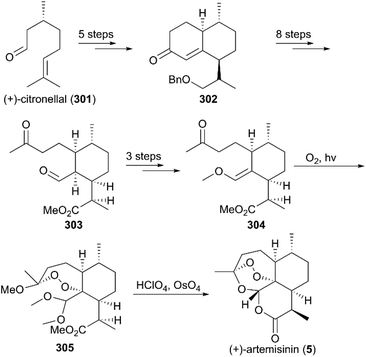
Three years later, Zhou and co-workers finished the total synthesis of (+)-artemisinin using R-(+)-citronellal (301) as raw material (Scheme 29).211R-(+)-citronellal (301) was subjected to two intramolecular cyclizations to produce α,β-unsaturated ketone 302, which was converted to aldehyde-ketone 303via eight consecutive steps. Subsequently, enol ether 304 was afforded by protecting group manipulations and the condensation of aldehyde carbonyl with trimethyl orthoformate. Photooxidation of the methanolic solution of 304 in the presence of oxygen and Rose Bengal at −78 °C provided intermediate 305, which was hydrolyzed in 70% HClO4 to give (+)-artemisinin (5) in 28% yield.
 |
| | Scheme 29 Total synthesis of (+)-artemisinin by Zhou. | |
7.1.3 Avery's total synthesis of (+)-artemisinin.
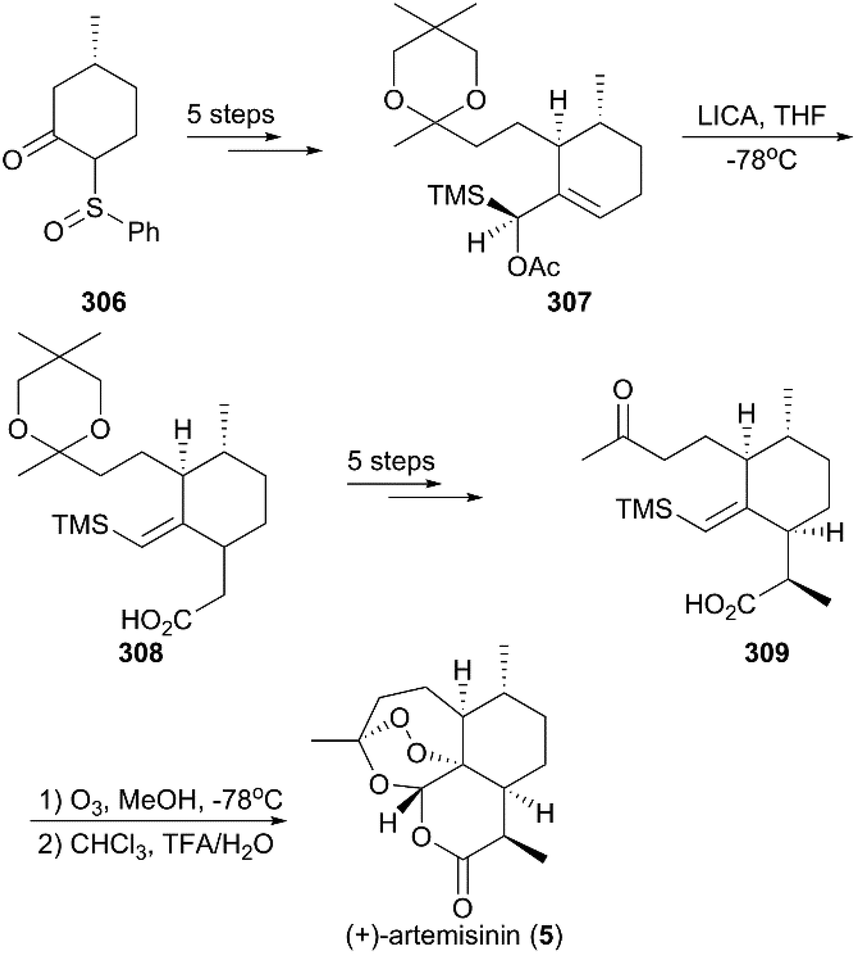
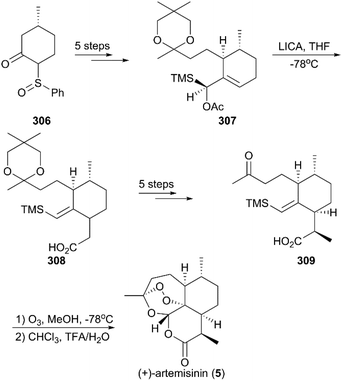
Soon after Zhou's work, Avery and co-workers reported their method for the total synthesis of (+)-artemisinin (Scheme 30).212 The known chiral sulfoxide 306 was used as the starting material, and five conversion steps gave rise to the diastereomerically pure silyl-acetate 307, which was converted to the cyclohexylacetate 308 by the exclusive Ireland–Claisen ester enolate rearrangement in the presence of lithium N-isopropylcyclohexylamide (LICA) as the base. Successive methylation, hydrolysis, and deketalization reactions generated the desired keto-acid 309. After exposure of the keto-acid 309 to ozone at −78 °C, the crude product was acidified with trifluoroacetic acid (TFA), and the synthesis of artemisinin was finished in a total of 12 steps. In 1992, Avery and co-workers completed the stereoselective total synthesis of 5 again, utilizing an optimized strategy with 10 steps.213
 |
| | Scheme 30 Total synthesis of (+)-artemisinin by Avery. | |
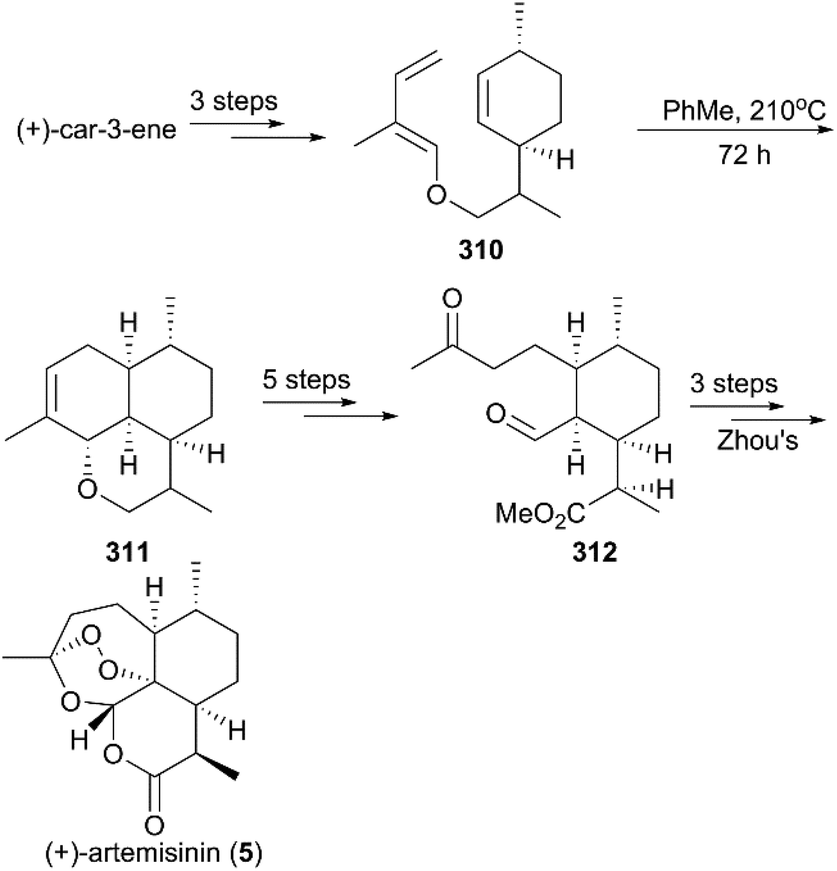
7.1.4 Ravindranathan's total synthesis of (+)-artemisinin.
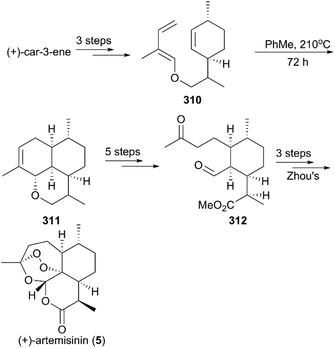
In 1990, Ravindranathan and co-workers utilized commercially available monoterpene (+)-car-3-ene as raw material to prepare the precursor enol ether 310, which was subjected to high temperature for 3 days to undergo an intramolecular Diels–Alder reaction, generating an epimeric mixture of ether 311 in a ratio of 3:2 (Scheme 31).214 After sequential oxidations, hydrolysis and methylation, the ketone-aldehyde 312 Zhou reported previously was obtained, and, followed by Zhou's protocol, the synthesis of (+)-artemisinin (5) was completed.
 |
| | Scheme 31 Total synthesis of (+)-artemisinin by Ravindranathan. | |
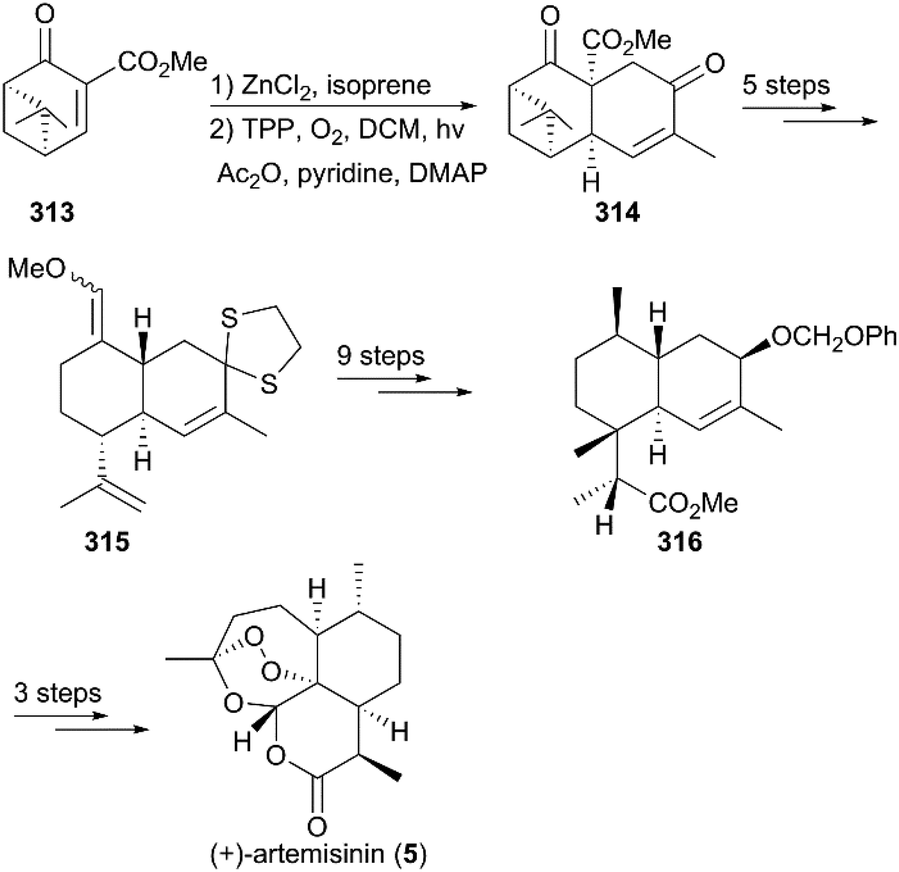
7.1.5 Liu's total synthesis of (+)-artemisinin.
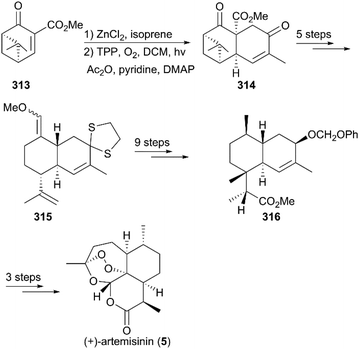
As shown in Scheme 32,215 the total synthesis of (+)-artemisinin by Liu and co-workers began with a ZnCl2-catalyzed intermolecular Diels–Alder addition of (+)-enone ester 313, followed by photooxygenation in the presence of 5,10,15,20-tetraphenyl-21H,23H-porphine and oxygen to give enedione 314, which was subjected to protection, fragmentation of the cyclobutane ring, and a Wittig reaction to generate a mixture of enone ethers (315). After nine conversion steps, including deprotection, reduction, a Mitsunobu reaction, and selective hydroboration–oxidation reaction, the ester 316 was obtained as a single stereoisomer. Finally, Schmid and Hofheinz's protocol for photooxygenation was applied to finish the preparation of 5.
 |
| | Scheme 32 Total synthesis of (+)-artemisinin by Liu. | |
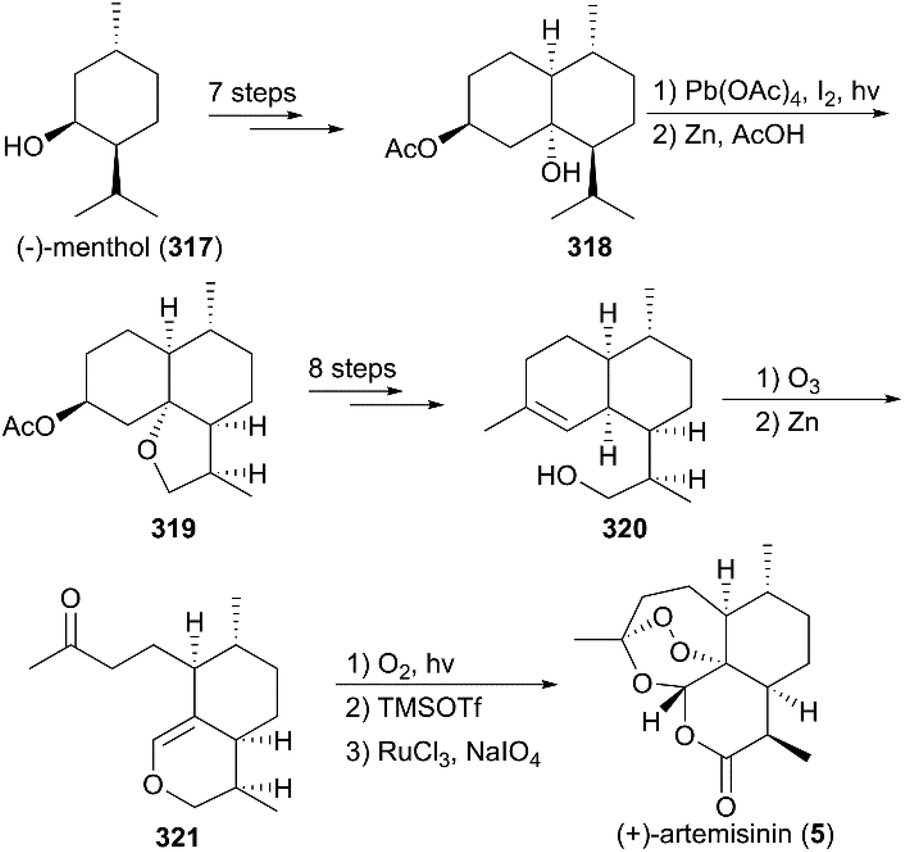
7.1.6 Bhonsle's total synthesis of (+)-artemisinin.
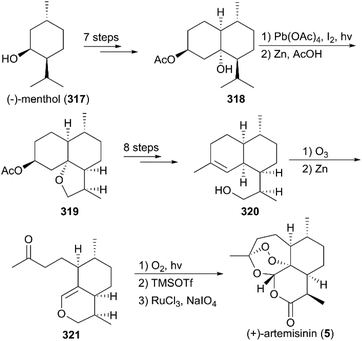
In Bhonsle and co-workers’ synthesis strategy (Scheme 33),216 (−)-menthol (317) was used as the starting material to prepare 318 by Jones oxidation, α-formylation, condensation with ethyl acetoacetate, epoxidation, and regioselective reductions, along with chemoselective acetylation. With intermediate 318 in hand, photooxygenation in the presence of lead tetraacetate and I2 was used to generate 319 as a major isomer. Next, alcohol 320 was obtained by consecutive saponification, oxidation, nucleophilic addition, and dehydration. Then, the natural product 5 was achieved using previously reported methods.
 |
| | Scheme 33 Total synthesis of (+)-artemisinin by Bhonsle. | |
7.1.7 Constantino's total synthesis of (+)-artemisinin.
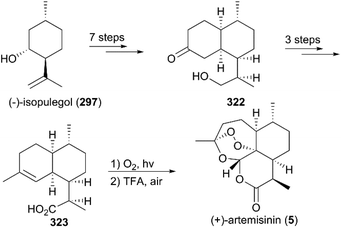
In 1996, Constantino and co-workers presented the total synthesis of 5, also using (−)-isopulegol (297) as the starting material (Scheme 34).217 In their strategy, diketone was obtained through oxidations and alkylation, and after condensation, dehydration, and some of the procedures from Bhonsle's work, acid 323 was generated and synthesis of (+)-artemisinin was achieved using a photooxygenation and additional oxidation by oxygen in the presence of trifluoroacetic acid.
 |
| | Scheme 34 Total synthesis of (+)-artemisinin by Constantino. | |
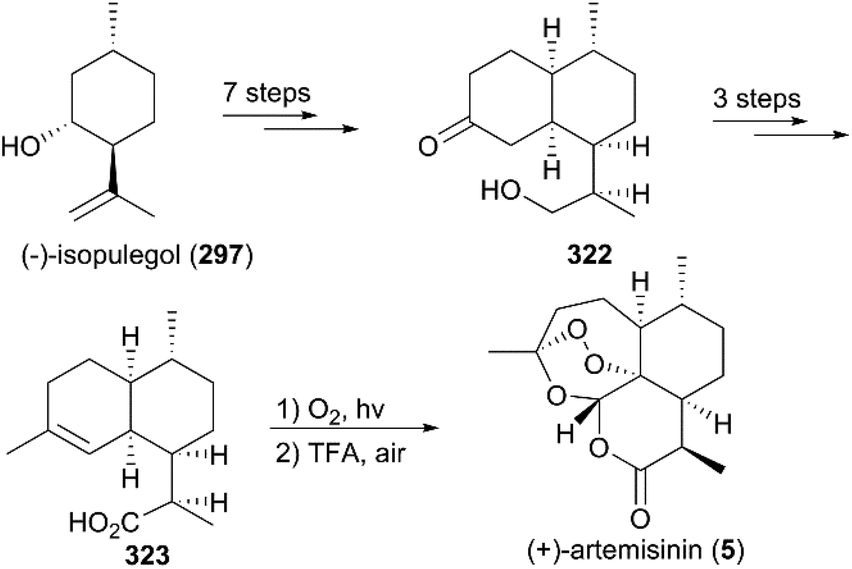
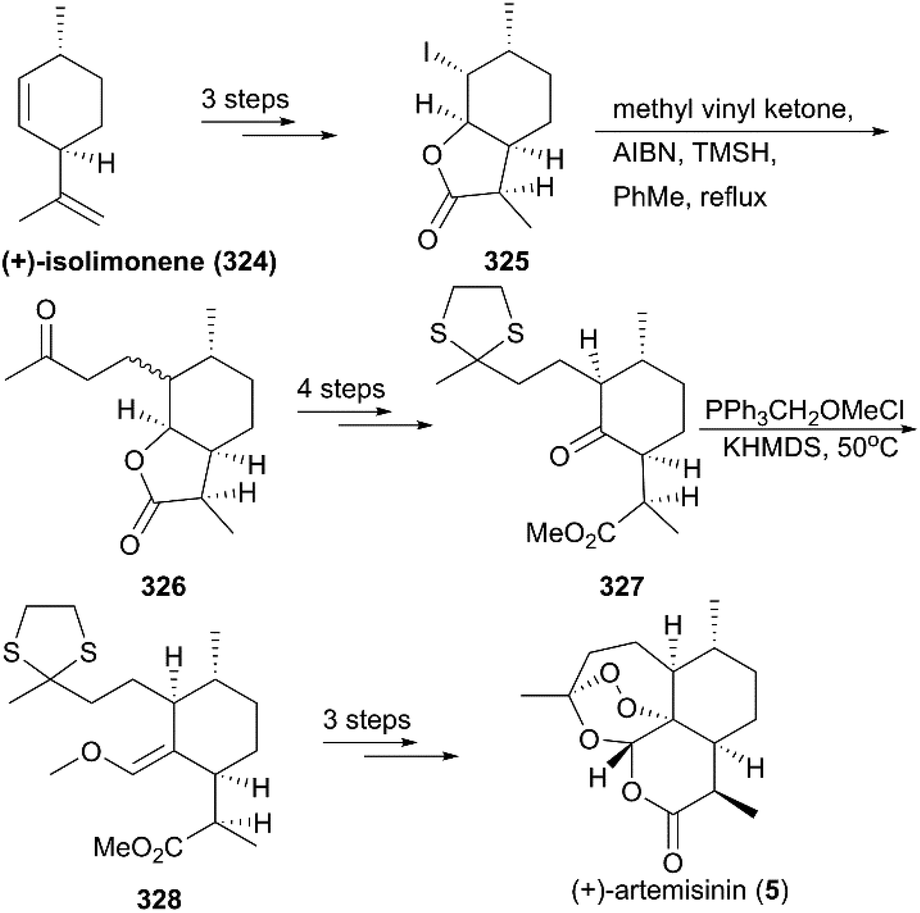
7.1.8 Yadav's total synthesis of (+)-artemisinin.
In 2003, Yadav and co-workers reported the total synthesis of natural product 5 in 11 steps using (+)-isolimonene 324 as a raw material (Scheme 35).218 In their strategy, an intermolecular radical reaction on the intermediate iodolactone 325 and a Wittig reaction on a ketone 327 were critical transformations. After deprotection and photooxygenation, synthesis of (+)-artemisinin was completed using previously reported procedures.
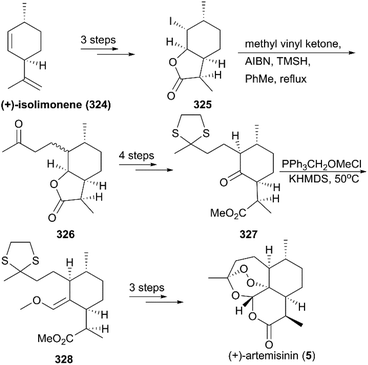 |
| | Scheme 35 Total synthesis of (+)-artemisinin by Yadav. | |
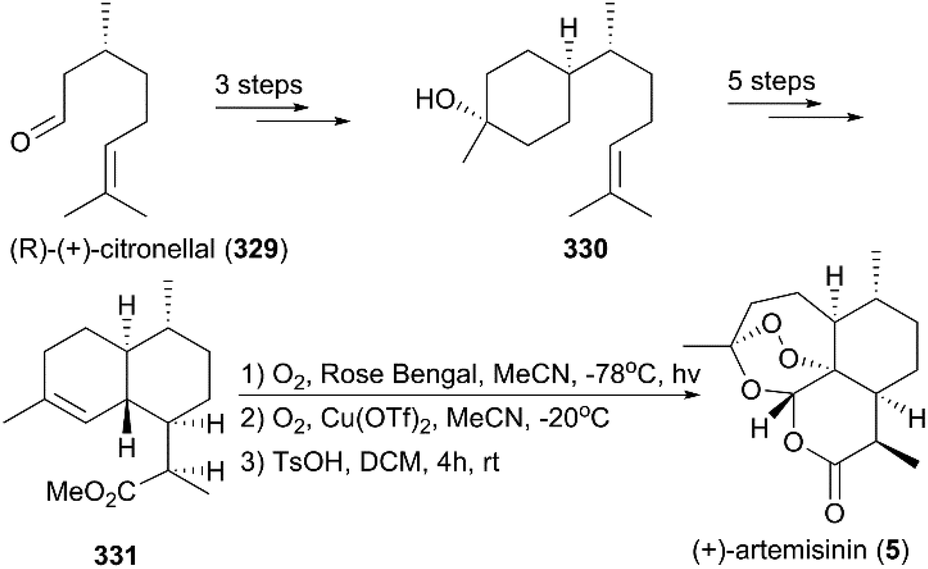
Seven years later, Yadav and co-workers altered the synthetic strategy by using (R)-(+)-citronellal 329 as a starting material instead of (+)-isolimonene 324 (Scheme 36).219 Their new route involved asymmetric 1,4-addition, aldol condensation, ene reaction, and regioselective hydroboration as key steps; synthesis of target molecule 5 was finished in 11 steps.
 |
| | Scheme 36 Alternative total synthesis of (+)-artemisinin by Yadav. | |
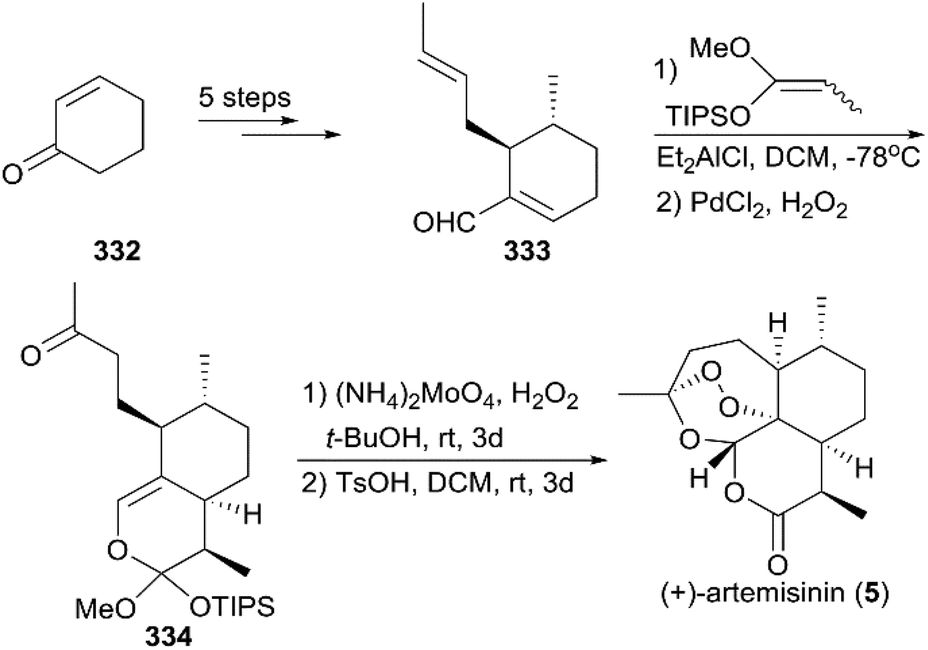
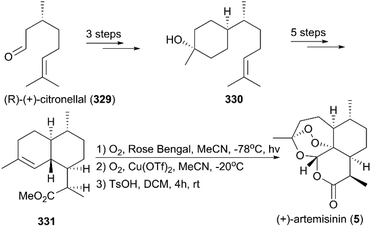
7.1.9 Cook's total synthesis of (+)-artemisinin.
In 2012, Zhu and Cook reported a highly efficient route for (+)-artemisinin synthesis in just five pots and a total of nine steps (Scheme 37).220 Their strategy began with the conversion of the simple and inexpensive starting material cyclohexenone 332 to α,β-unsaturated aldehyde 333 in five steps, including a one-pot conjugate addition/alkylation sequence and formylation of the sulfonylhydrazide in the presence of n-BuLi and N,N-dimethylformamide (DMF). A [4 + 2] annulation was explored for the installation of the six-membered lactone to generate enol ether 334. Different from the procedure that was previously reported, they applied a singlet oxygen generated from the decomposition of H2O2 by ammonium molybdate to oxidize the enol ether 334 in the final step. The conditions used for the final oxidative rearrangement to 5 were suitable for large-scale production.
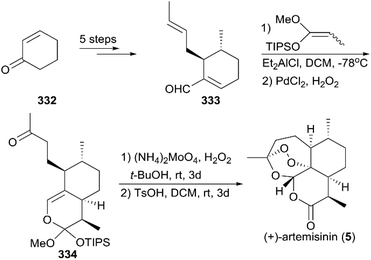 |
| | Scheme 37 Total synthesis of (+)-artemisinin by Cook. | |
7.1.10 Giannis’ total synthesis of (+)-artemisinin and antipode (−)-artemisinin.
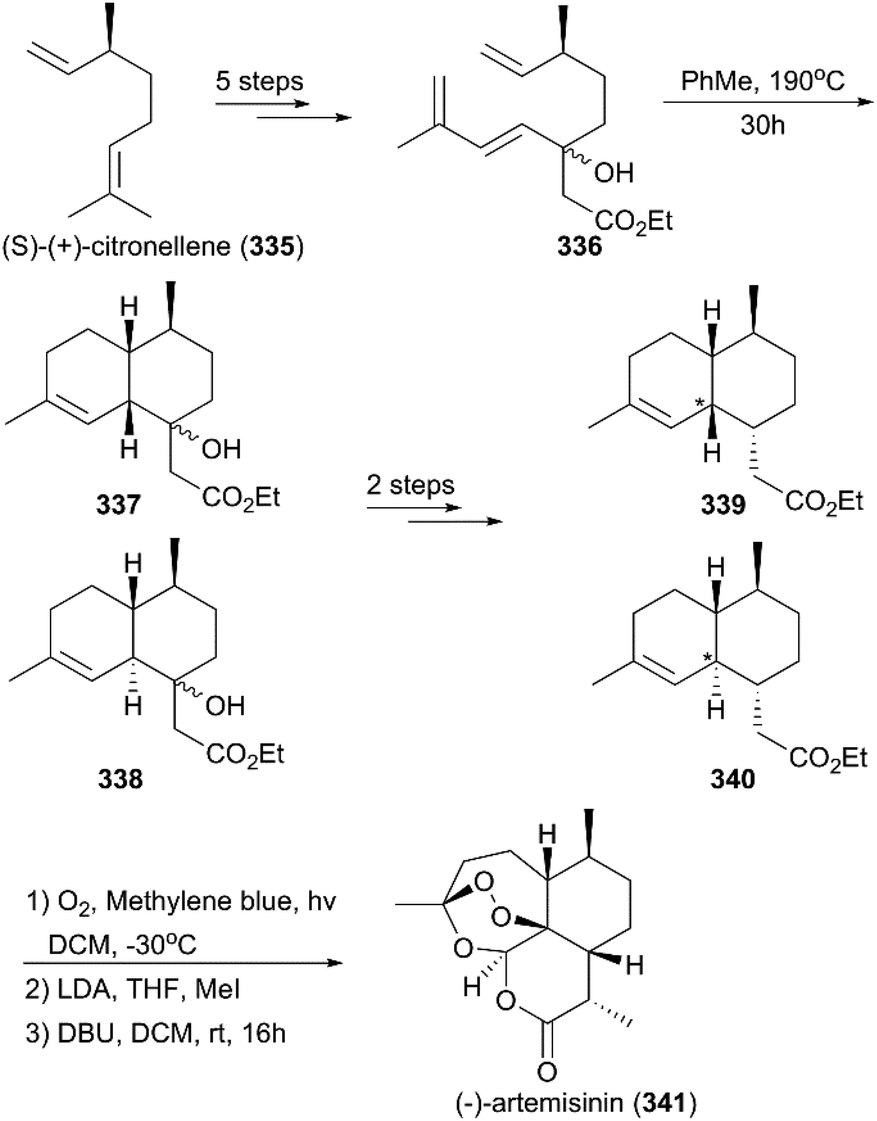
In 2018, Giannis and co-workers reported the total synthesis of (−)-artemisinin and (+)-artemisinin utilizing S-(+)- and R-(−)-citronellene (335) as starting materials, respectively (Scheme 38).221 After five steps, conversion of S-(+)-citronellene to a triene mixture (336) was achieved; this mixture was subjected to a thermal Diels–Alder reaction to generate artemisinic acid derivatives 337 and 338. Then, the alcohols of 337 and 338 were treated with Martin sulfurane to generate α,β-unsaturated esters, which were reduced by NiCl2/NaBH4 and Li/NH3 to produce esters 339 and 340, respectively. Subsequently, a previously reported procedure was applied to convert 339 and 340 to 9-desmethyl-(−)-artemisinin, which underwent methylation to produce (−)-artemisinin (341).
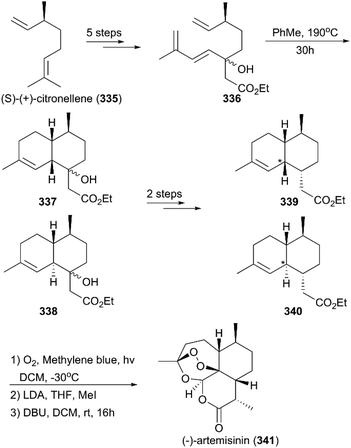 |
| | Scheme 38 Total synthesis of (−)-artemisinin by Giannis. | |
Although great advances in the total synthesis of artemisinin have been made by academic researchers over the past five decades, because of the complex structure of artemisinin and its derivatives and the drawbacks of the expensive starting material, long reaction routes, and excessive protecting group operations, no synthetic strategy is economically efficient enough for pharmaceutical industry-scale production to date. Therefore, more efforts are needed to design a total synthesis strategy that can be used by industry.
7.2 Structure–activity relationships of artemisinin derivatives
Since the discovery of artemisinin (5) as an excellent antimalaria agent, the structural analogs such as dihydroartemisinin (294), artemether (296), arteether (342), artesunic acid (295), and sodium artesunate (343) have been investigated (Fig. 14); this is because the poor physicochemical properties of artemisinin (6), including its low solubility in both oil and water, and short half-life (t1/2), limits its first-line clinical use. A large number of artemisinin derivatives and analogs have been explored in systematic SAR studies as part of an effort to seek more potential drug candidates.222–229
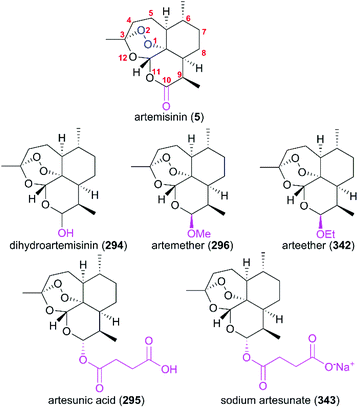 |
| | Fig. 14 The first-generation artemisinin derivatives. | |
In 2008, the first resistance to artemisinin was reported in Africa. Because of its worldwide use for a long time, WHO recommends artemisinin-based combination therapy (ACT) with different mechanisms and longer half-life instead of monotherapy using artemisinin to avoid the development and spread of resistance to artemisinin. The first-generation artemisinin-derived analogs dihydroartemisinin (294), artemether (296), arteether (342), artesunic acid (295), and sodium artesunate (343) were prepared by semisynthetic modifications at C10 with an acetal linkage; these analogs have improved physicochemical properties compared with their parent molecule, such as higher water-solubility. They are recommended by WHO to be used in combination with therapies such as ASAQ Winthrop (sodium artesunate-amodiaquine), Eurartesim (dihydroartemisinin-piperaquine), and ASMQ (sodium artesunate-mefloquine) for the treatment of malaria.
However, the drawbacks of analogs with the acid-sensitive acetal linkage include a short half-life and neurotoxicity. To overcome these drawbacks, the second-generation analogs without acetal groups were designed to improve the drug-like properties; in particular compounds 347 and 348 showed better activity (Fig. 15).230
 |
| | Fig. 15 The second-generation artemisinin derivatives. | |
The essential role of the peroxy bridge of artemisinin, which is the pharmacophore, in the development of antimalarials was established early after its discovery, and it was confirmed by the inactive analog deoxoartemisinin 349.231 The orientation of the peroxide bond in the chemical scaffold of analogs is also important. For example, the tricyclic analog 350, which has a flexible peroxide ring displayed weaker antimalarial activity compared with the parent molecule artemisinin (Fig. 16).232
 |
| | Fig. 16 Examples of artemisinin analogs. | |
A number of semisynthetic analogs with tricyclic 1,2,4-trixoanes in a rigid scaffold lacking the D ring of artemisinin were developed, and many of them exhibited potential antimalarial activities in lower nanomolar ranges, such as analogs 351, 352, and 353 (Fig. 17).
 |
| | Fig. 17 Examples of peroxy bridge-containing artemisinin analogs. | |
Most semisynthetic drug-like analogs with complex chemical scaffolds are probably not suitable for pharmaceutical industry-scale production. The natural product yingzhausu A (354) has an accessible endoperoxide ring and displays good antimalarial activity. Spurred by this finding, a large number of simple analogs with endoperoxides have been designed and some of them display good antimalarial activity and offer great potential as drug candidates (Fig. 18).233,234
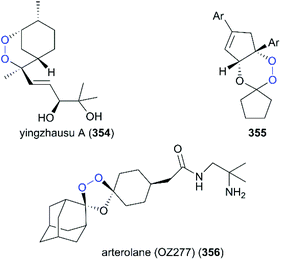 |
| | Fig. 18 Yingzhausu A and examples of analogs. | |
8. Ecteinascidins
Ecteinascidins are a family of marine natural products containing two or three tetrahydroisoquinoline subunits (Fig. 19). Many ecteinascidin members show antiproliferative activity against several tumor cell lines.235,236 Among these bioactive ecteinascidins, ecteinascidin 743 (Et-743, trabectedin, 357) was approved in 2007 for the treatment of advanced soft-tissue sarcoma and ovarian cancer, and lurbinectedin (363), one synthetic ecteinascidin derivative, was approved in 2020 for the treatment of metastatic small cell lung cancer.237,238 Et-743 was first isolated in 1990 from a Caribbean tunicate Ecteinascidia turbinate and its absolute stereochemistry was determined in 1992 with the help of the X-ray crystal structure of its N12-oxide derivative.239,240 Extensive studies of Et-743 show that this molecule functions by forming covalent adducts with DNA after binding to the minor groove of DNA and alkylating DNA at the N2 position of guanine.241–243 The adducts can bend the DNA structure toward the major groove,244,245 disrupt the interaction between DNA and DNA-binding proteins,246 and induce double-strand breaks (DBSs),247 all of which contribute to Et-743's antiproliferative activity on various cancer cell lines.
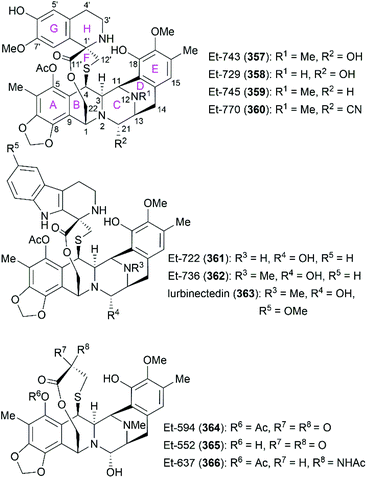 |
| | Fig. 19 Representative ecteinascidins and ecteinascidin derivatives and their ring lettering and carbon numbering system. | |
However, the natural source of Et-743 is limited: only 1.0 g of Et-743 can be isolated from about 1.0 ton of tunicate, which hampered its preliminary clinical studies.235,236 In 1996, Corey and co-workers reported the first synthetic route for Et-743, which was initially used for its industrial production.248 A semisynthetic route relying on Corey's synthetic route was developed in 2000 for the commercial manufacture of Et-743.249 Apart from its biological function, Et-743 also has several structural features that make it a quite challenging but attractive synthetic target: (1) three tetrahydroisoquinoline moieties with different substitution patterns; (2) eight rings including one bridged 10-membered lactone ring containing a sulfur atom; (3) seven chiral centers including one spiro center; and (4) a labile carbinolamine functional group. Continuous efforts from the synthetic community have led to outstanding achievements in the total synthesis of Et-743 and we discuss them in the following section.
8.1 Total synthesis of Et-743
8.1.1 Corey's total synthesis of Et-743.
Corey and co-workers achieved the first total synthesis of Et-743 and later (in 2000) they reported a more efficient synthetic route to the key intermediate 373 (Scheme 39).248,250 In their synthesis strategy, the chiral C13 (Et-743 numbering) stereocenter was established through enantioselective hydrogenation of the α,β-unsaturated ester 367 to generate the tyrosine derivative 368 with 96% ee. A similar approach was applied to the synthesis of another tyrosine derivative 370, which was transformed into the bridged lactone 371 through an intramolecular Mannich reaction. Condensation between the two tyrosine derivatives 369 and 371 and subsequent manipulation of the lactone led to lactol 372. Methanesulfonic acid promoted Mannich bisannulation of 372, leading to the successful construction of a bridged C/D ring system with establishment of the C11 stereocenter. Then, a Strecker reaction was used to construct amino nitrile 373, and subsequent manipulations led to pentacyclic compound 374 with the required methyl group on ring E.
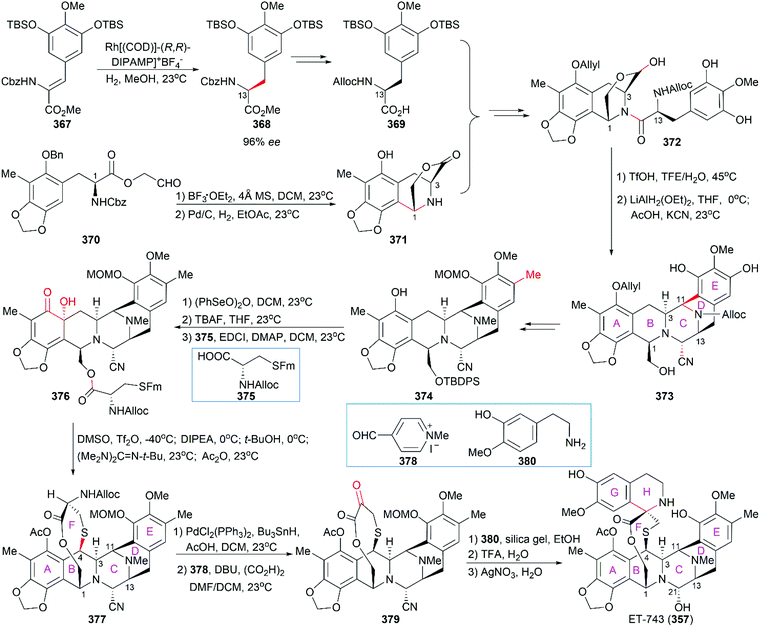 |
| | Scheme 39 Corey's synthetic route. | |
Position-selective hydroxylation with benzeneseleninic anhydride followed by the introduction of the cysteine derivative 375 generated 376, which was ready for the construction of the challenging 10-membered bridged lactone ring F. The construction of 377 from 376 involved a series of transformations: (1) activation of the tertial hydroxy group with the in situ-generated Swern reagent from Tf2O and DMSO, followed by addition of i-Pr2NEt, which led to o-quinone methide; (2) Barton's base-mediated removal of the Fm protecting group, and the subsequent nucleophilic addition of a sulfur ion to the quinone methide generating ring F; and (3) acetylation of the resulting phenoxide group with Ac2O. Removal of the Alloc protecting group and the subsequent transamination with 378 generated the keto lactone 379, which was utilized for the introduction of the third tetrahydroisoquinoline moiety through a Pictet–Spengler reaction with 380. Removal of the MOM protecting group from the C18 phenolic hydroxy group and stereoselective introduction of the C21 hydroxy group completed the total synthesis of Et-743.
8.1.2 Fukuyama's total synthesis of Et-743.
Fukuyama and co-workers reported their first-generation synthetic route to Et-743 in 2002 and continuous efforts in their lab led to a separate synthetic strategy in 2013.251,252 In their first-generation synthetic design,251 the C13 chiral stereocenter in the tyrosine derivative 382 was established through enantioselective hydrogenation, while a Mannich-type reaction between 383 and chiral template 384 stereoselectively constructed the C1 chiral center (Scheme 40). A four-component Ugi reaction including 382, 386, p-methoxyphenyl isocyanide (387) and acetaldehyde led to 388, which was transformed into the enamide compound 389 containing ring C. An intramolecular Heck reaction was applied for the formation of the bridged C/D ring system in tetracyclic compound 390, which was converted to aldehyde 391 after several manipulations. Subsequent hydrogenolytic removal of the benzyl group led to spontaneous cyclization to form the pentacyclic compound 392 with the desired stereoconfiguration at C4. Protecting group manipulations followed by the introduction of the cysteine derivative provided 393, which was utilized for the installation of the crucial ring F. Selective removal of the acetyl group gave thiol, which cyclized to form the ring F upon exposure to TFA under high dilution conditions in TFE, presumably via the o-quinone methide intermediate. Subsequent acetylation of the phenolic hydroxy group gave rise to 394, which could be converted to Et-743 in a manner similar to that utilized in Corey's total synthesis.
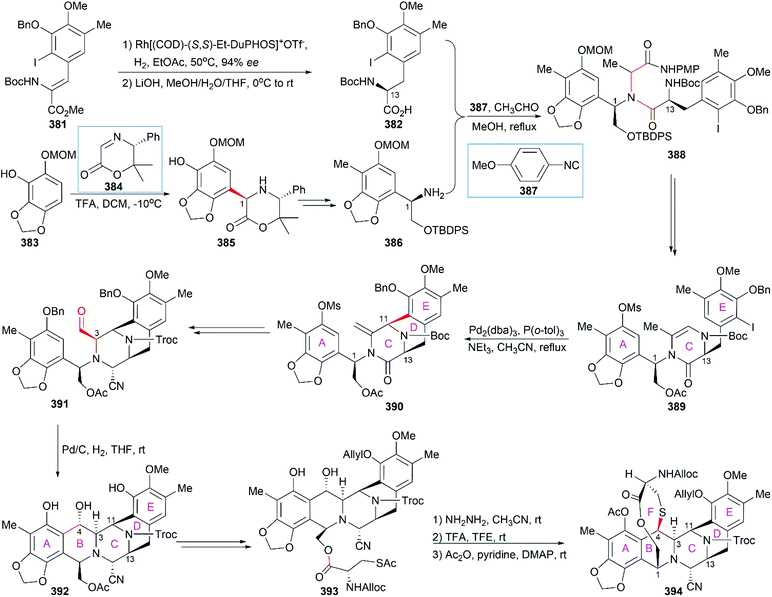 |
| | Scheme 40 Fukuyama's first-generation synthetic route. | |
In their optimized second-generation synthetic route, the diacetylated diketopiperazine 395, which was derived from L-glutamic acid, was the only chiral source (Scheme 41).252 Perkin condensation of 395 with aldehyde 396 followed by protection of the amide and hydrogenation stereoselectively constructed the C13 chiral center. Selective reduction of the imide carbonyl group after hydrazinolysis of the acetyl group converted 397 to a suitable intermediate for construction of the bridged C/D ring system through Mannich-type reaction, which was performed in the presence of TFA. Tetracyclic enamide 399, which was derived from 398 with the installation of the methyl group on ring E and several other manipulations, was used as a platform for the introduction of ring A. A regio- and stereo-selective Heck reaction of 399 with a diazonium salt derived from amine 400 successfully led to 401. Manipulation of 401 led to the dialdehyde equivalent 402, which cyclized to form the pentacyclic compound 403 upon heating in m-xylene. Reduction of 403 with Red-Al gave oxazolidine 404, a suitable precursor for the introduction of the cysteine derivative to give 405, an analog of 393, which could be similarly converted to Et-743.
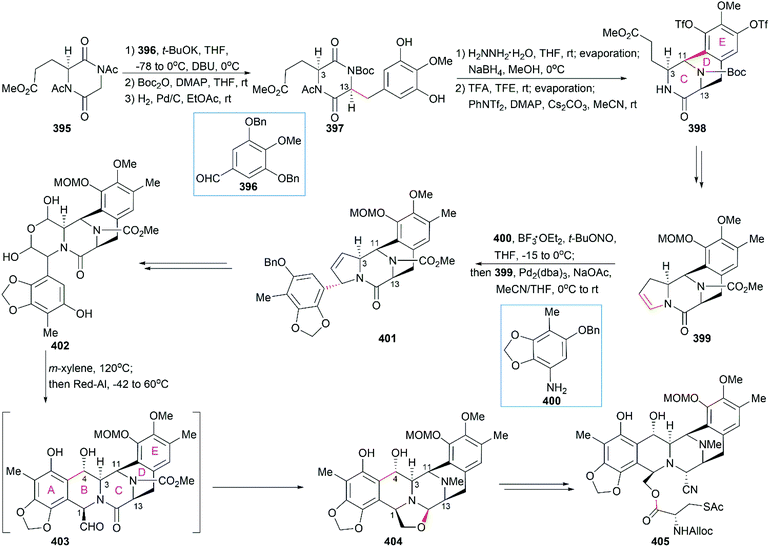 |
| | Scheme 41 Fukuyama's second-generation synthetic route. | |
8.1.3 Zhu's total synthesis of Et-743.
Zhu's synthesis of Et-743 started with preparing the right tetrahydroisoquinoline moiety (D/E ring system) by condensation of chiral amino alcohol 406 with Garner's aldehyde (S)-407 (Scheme 42).253 It is worth noting that the stereochemistry of the newly generated chiral center C11 was solely controlled by the absolute configuration of 406. Assembly of 409 with racemic 410 gave coupled product 411 in 68% yield with its C1 epimer in 23% yield, the diastereoselectivity of which may be explained by an SN1 mechanism. Construction of the piperazine ring C was achieved through oxidation of the hydroxy group of 412 followed by ZnCl2-catalyzed Strecker reaction to give 413. TFA-promoted cyclization of 414 concomitant with MOM protecting group removal successfully installed ring B in 415 with the desired stereoconfiguration at C4, which was similar to the ring B formation strategy utilized in Fukuyama's first-generation synthetic route. Removal of the acetate group followed by the introduction of the cysteine derivative led to 416, from which ring F was installed through a one-pot deprotection/cyclization mediated by TFA. Compound 417 could be converted into Et-743 following Corey's protocol.
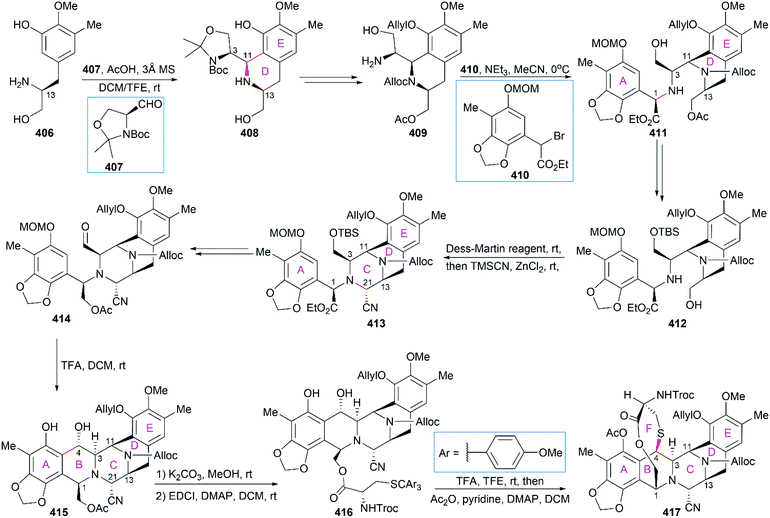 |
| | Scheme 42 Zhu's synthetic route. | |
8.1.4 Danishefsky's formal total synthesis of Et-743.
Starting with asymmetric reduction of the ketone moiety in 418 using Noyori transfer-hydrogenation conditions followed by a Mitsunobu reaction with diphenylphosphorylazide securely fashioned the C1 stereoconfiguration in 421 (Scheme 43).254 The left tetrahydroisoquinoline moiety (A/B ring) was formed from 421 through an acid-promoted Pomerantz–Fritsch reaction. Condensation of 422 with the chiral tyrosine derivative 423 afforded 424, which was converted into the cyclization precursor 425. Aldehyde 425 successfully cyclized to give rise to the pentacyclic compound 426 through a CHF2CO2H-promoted vinylogous Pictet–Spengler reaction. DMDO-mediated epoxidation of the double bond in the advanced intermediate 427 followed by amide nitrogen atom-promoted epoxide ring-opening and subsequent reduction of the iminiumion produced compound 428 with the desired stereoconfiguration at C3 and C4. It is worth noting that a large excess of sodium cyanoborohydride was required for the high yield of 428. Further manipulations on 428 led to 429, which was an intermediate in Fukuyama's total synthesis of Et-743.251
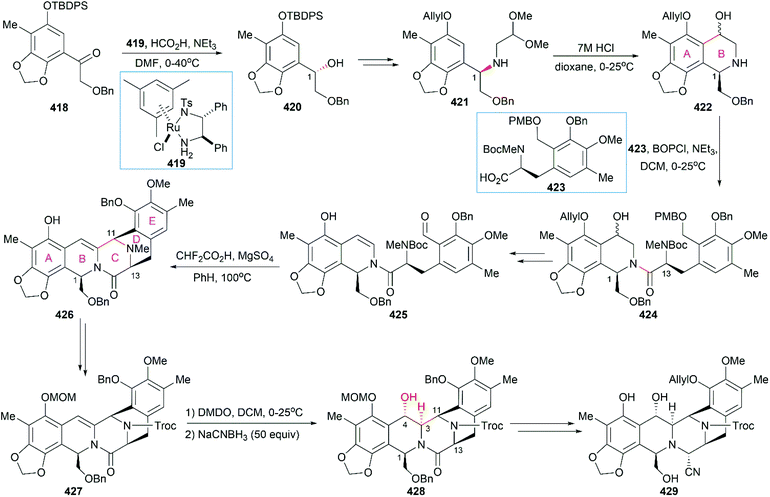 |
| | Scheme 43 Danishefsky's synthetic route. | |
8.1.5 Williams’ formal total synthesis of Et-743.
In Williams’ synthetic route,255 stereoselective condensation of the phenol 430 with (R)-Garner's aldehyde 431 in the presence of Ti(OiPr)4 produced the anti-product 432 (Scheme 44). Condensation of the chiral amine 433 with ethyl glyoxalate led to an imine intermediate, which proceeded to form the left tetrahydroisoquinoline 434 with the desired C1 stereocenter established through an AIBN/Bu3SnH-mediated radical cyclization. Removal of the acetonide from 436 with Dowex 50W-X8 cationic resin in methanol followed by desilylation and subsequent Swern oxidation produced 437. When treated with TFA, 437 successfully underwent a Pictet–Spengler reaction to give the pentacyclic compound 438, although with low regioselectivity on the E ring. Since several protecting group manipulations could transform 438 into 427, which was an advanced intermediate in Danishefsky's formal total synthesis, Williams and co-workers completed their formal total synthesis of Et-743.
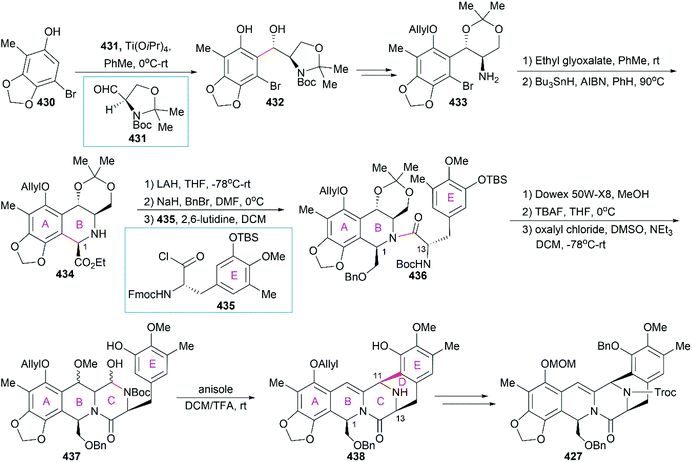 |
| | Scheme 44 Williams’ synthetic route. | |
8.1.6 Ma's total synthesis of Et-743.
In 2019, Ma and co-workers reported an efficient total synthetic route to Et-743 and lurbinectedin, in which the left and right tetrahydroisoquinoline moieties were constructed from the common intermediate 439 (Scheme 45).256 Starting from the chiral amino alcohol 439, which was prepared from Cbz-protected (S)-tyrosine, a precursor for the left tetrahydroisoquinoline moiety 440 was obtained through a Pictet–Spengler reaction. After oxidation of the phenol 440 to quinone 441, the authors carried out detailed screenings for optimized conditions to form benzo[1,3]dioxole through a light-mediated remote C–H bond activation. They finally found that blue-LED irradiation of a THF solution of 441 could smoothly transform quinone 441 into phenol 442. It should be pointed out that this transformation was an essential step in their synthesis because the attempts to synthesize 442 from 443 through a direct Pictet–Spengler reaction under various conditions failed. Condensation of aldehyde 444 with amine 439 successfully provided 445, which contained the desired two tetrahydroisoquinoline moieties. TFA-promoted deprotection of 446 followed by Strecker reaction and BCl3-mediated removal of the two benzyl groups produced amino nitrile 447, which could be transformed into Et-743 in a manner similar to that in Corey's synthetic route.
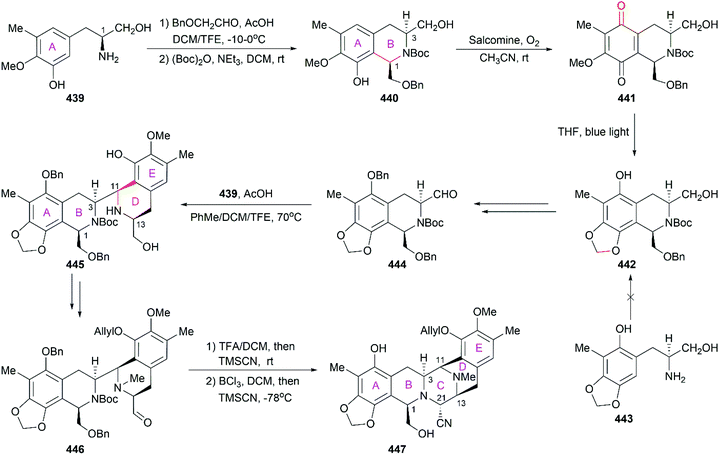 |
| | Scheme 45 Ma's synthetic route. | |
8.2 Structure–activity relationships of ecteinascidin derivatives
Although the reported total synthetic routes are not amenable to scaling-up for manufacturing ET-743 on an industrial scale, transformations developed in these studies provide access to diverse ecteinascidin derivatives, some of which are listed in Fig. 20. These synthesized ecteinascidin derivatives make SAR studies of Et-743 possible and have led to the development of more anti-tumor molecules including the approved drug lurbinectedin (363) and PM00104 (468), which is in phase II clinical trials. We give a short discussion about the SAR studies on ecteinascidin derivatives in the following section (Fig. 20 and Tables 2, 3).
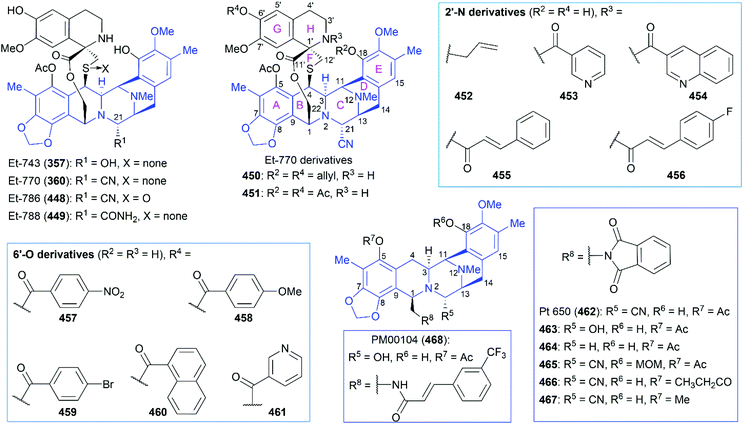 |
| | Fig. 20 Representative ecteinascidin derivatives for SAR study. | |
Table 2 Cytotoxicities of Et-770 and related derivatives to three carcinoma cell linesa
| Entry |
Compound |
IC50 (nM) |
| HCT116b |
QG56b |
DU145b |
|
Values given are IC50 in nM and original activity data can be found in ref. 259–262.
HCT116: human colon cancer cell line; QG56: human lung cancer cell line; DU145: human prostate cancer cell line.
|
| 1 |
Et-770 (360) |
0.71 |
1.6 |
1.6 |
| 2 |
Et-788 (449) |
3.5 × 102 |
1.3 × 103 |
9.9 × 102 |
| 3 |
450
|
54 |
1.6 × 102 |
1.0 × 102 |
| 4 |
451
|
1.3 |
9.5 |
4.4 |
| 5 |
452
|
0.50 |
1.8 |
1.2 |
| 6 |
453
|
0.13 |
0.40 |
0.72 |
| 7 |
454
|
0.014 |
0.071 |
0.087 |
| 8 |
455
|
0.053 |
0.33 |
0.24 |
| 9 |
456
|
0.010 |
0.12 |
0.041 |
| 10 |
457
|
0.26 |
0.96 |
0.37 |
| 11 |
458
|
0.31 |
1.7 |
0.62 |
| 12 |
459
|
15 |
36 |
16 |
| 13 |
460
|
13 |
36 |
24 |
| 14 |
461
|
0.20 |
1.0 |
0.51 |
Table 3 Cytotoxicities of Pt 650 and related derivatives to four carcinoma cell linesa
| Entry |
Compound |
IC50 (nM) |
| A-549b |
HCT116b |
A375b |
PC-3b |
|
Values given are IC50 in nM and original activity data can be found in ref. 257.
A-549: human lung cancer cell line; HCT116: human colon cancer cell line; A375: human melanoma cell line; PC-3: human prostate cancer cell line.
|
| 1 |
Et-743 (357) |
1.0 |
0.50 |
0.15 |
0.70 |
| 2 |
Pt 650 (462) |
0.95 |
0.38 |
0.17 |
0.55 |
| 3 |
463
|
1.0 |
0.61 |
0.20 |
0.53 |
| 4 |
464
|
2.2 × 103 |
1.1 × 103 |
6.1 × 102 |
8.5 × 102 |
| 5 |
465
|
2.3 × 102 |
74 |
66 |
98 |
| 6 |
466
|
2.1 |
1.2 |
0.51 |
2.9 |
| 7 |
467
|
3.1 |
1.4 |
0.55 |
3.1 |
After achieving the first total synthesis of Et-743, Corey and co-workers synthesized a series of Et-743 analogs from a common intermediate, which led to the discovery of one simpler and more stable anti-tumor molecule named Pt 650 (phthalascidin, 462).257 In 2002, Saito and co-workers isolated Et-770 (360), an analog of Et-743, from the pretreated Thai tunicate Ecteinascidia thurstoni with potassium cyanide.258 Both Pt 650 and Et-770 showed antiproliferative activities on various cancer cell lines. Synthesis and evaluation of more synthesized analogs of Pt 650 and Et-770 revealed the structural features for high potent biological activities.257–263
A cyano or a hydroxy group at the C21 position is essential and is responsible for alkylating DNA. Et-788 (449) with an amide group at C21 and 464 with hydrogen at C21 showed a significant decrease in activity compared with their corresponding parent compounds.257,261 Oxidation of the sulfur atom in Et-770 led to Et-786, which resulted in dramatically diminished cytotoxicity.259 Protection of the C18 and C6′ phenolic hydroxy groups with allyl (450) or acetate (451) decreased cytotoxicity.259,260 Protection of the C18 phenolic hydroxy group with MOM (465) also resulted in diminished antitumor activity.257
Modifications at the 2′-N position revealed that the 2′-N-allylated compound 452 showed activity similar to that of Et-770 (360), while several 2′-N amide derivatives including nitrogen-containing heterocycles (453 and 454) and cinnamoyl derivatives (455 and 456) exhibited improved cytotoxicity.260–262 Notably, 454 and 456, respectively, exhibited approximately 50- and 70-fold higher cytotoxicity to the HCT116 cancer cell line than Et-770 (360).261
Evaluation of the cytotoxicity of a series of C6′ aromatic ester derivatives showed that 457 and 458 with a nitro group or a methoxy group on the benzene ring exhibited activities similar to Et-770 (360).262 However, derivatives with a bromo group on the benzene ring (459) or a naphthyl group (460) showed significantly decreased cytotoxicity. The incorporation of a nitrogen-containing heterocyclic ester group (461) resulted in a moderate increase in activity. For Pt 650 analogs, derivatives possessing a propionyl (466) or a methyl (467) protecting group on the C5 phenolic hydroxy group were slightly less effective than the parent compound Pt 650 (462).257 These results indicate the promising role of total synthesis in the future development of ecteinascidin-type and related natural products into drugs.
9. Calicheamicin γI1
Calicheamicin γI1 (474) is a potent antitumor antibiotic that was isolated from Micromonospora echinospora ssp in 1986. One year later, its structure was defined as an enediyne with a bicyclo[7.3.1]tridec-9-ene-2,6-diyne system, a unique N–O glycosidic linkage, and a methyl trisulfide moiety; other members of the calicheamicin family were also discovered (Fig. 21).264–266 Several natural products containing the enediyne were also found in the 1980s, such as Neocarzinostatin chromophore (476),267 Esperamicin A1 (477),268 and Dynemicin A (478)269 (Fig. 21). Calicheamicin γI1 displays excellent activity against Gram-negative and -positive bacteria at concentrations lower than 1 pg mL−1. It demonstrated extreme toxicity to all cells, indicating that it has extraordinary potency against cancer cells. Investigation of its mechanism showed that calicheamicin γI1 targets and binds to the DNA minor groove, and then undergoes the Bergman cyclization after bioreductive activation to produce a biradical, 1,4-didehydrobenzene, which subsequently abstracts hydrogen atoms from the deoxyribose backbone of DNA to generate sugar radicals. The latter undergo oxygen-dependent reactions and finally induce double-strand DNA breaks.270–272 Because of its excellent activity and site-specific targeting, gemtuzumab ozogamicin (Mylotag®), a calicheamicin γI1 derivative, was approved by the FDA in 2000 as an antibody–drug conjugate (ADC) to treat CD-33-positive myeloid leukemia. Unfortunately, 10 years later it was withdrawn from the market because of its serious side effects. In 2017, inotuzumab ozogamicin (Besponsa®), also derived from calicheamicin γI1, was approved by the FDA as an ADC medication to treat relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia. All in all, the fascinating structure and highly potent drug-likeness characteristics of calicheamicin γI1 have spurred chemists to meet the huge challenge of developing strategies for the total synthesis of calicheamicin γI1.
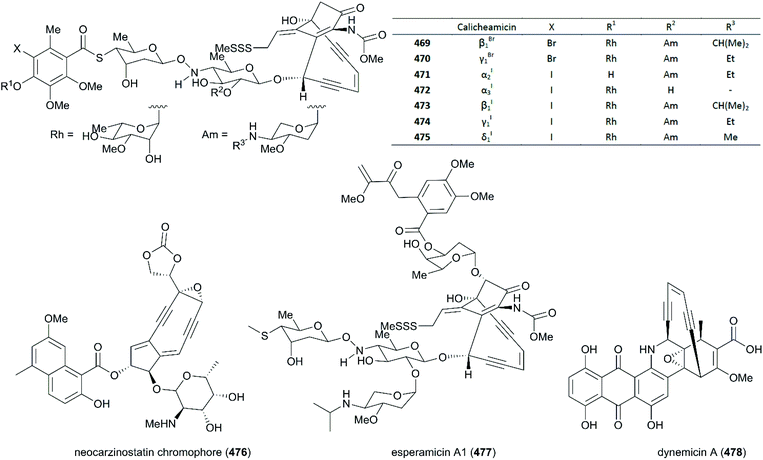 |
| | Fig. 21 Examples of natural products containing enediyne. | |
9.1 Total synthesis of calicheamicin γI1
9.1.1 Nicolaou's total synthesis of calicheamicin γI1.
In 1992, Nicolaou and co-workers reported the first approach for the synthesis of calicheamicin γI1 using the strategy of splitting the parent molecule into two advanced intermediates, an oligosaccharide fragment and an aglycon precursor.273–276 First, the building blocks 479 and 480 were glycosylated in the presence of AgClO4 and SnCl2 to produce α-disaccharide as the major product, which underwent selective deprotection followed by regioselective oxidation of C4-OH to generate hydroxy ketone 481. The active ketone was successfully condensated with hydroxylamine 482 using PPTS as a catalyst to produce trisaccharide 483. The introduction of thionoimidazolide instead of benzoyl generated 484, which proceeded the [3,3]-sigmatropic rearrangement and removal of the imidazoformyl to forge trisaccharide 485 for coupling with aryl saccharide 489. The latter was prepared by the glycosidation of saccharide unit 486 with fully substituted phenol 487 and subsequent interconversion of protecting groups. Photodeprotection of an o-nitrobenzyl glycoside of trisaccharide 490 generated lactol, and Schmidt's protocol was used to generate the trichloroacetimidate 491 from 490 (Scheme 46).
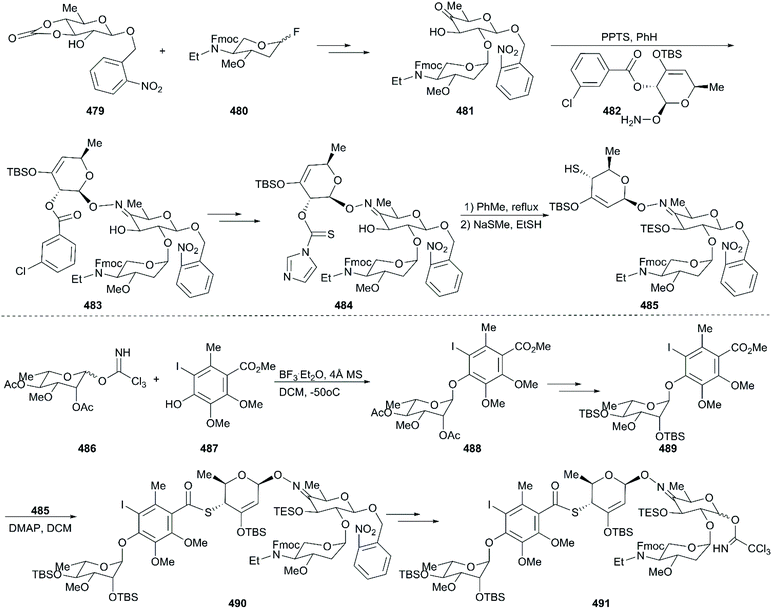 |
| | Scheme 46 Nicolaou's preparation of tetrasaccharide building block. | |
The critical synthesis of aglycon portion is to construct the cyclohexenone precursor of the bicyclic enediyne scaffold using the [3 + 2] nitrile oxide cycloaddition reaction (Scheme 47). The commercially available tetronic acid 492 was used to prepare the aldoxime precursor 493, which underwent an intramolecular 1,3-dipolar cycloaddition reaction using aqueous sodium hypochlorite solution in DCM as an oxidation system to generate isoxazoline 494. After removal of the benzoate group and the Jones oxidation of the resultant secondary hydroxy group, the ketone intermediate was produced. Subsequent addition of lithium(trimethylsilyl)acetylide and acetylation generated intermediate 495 with high stereoselectivity. After careful removal of MEM ether, the resultant secondary alcohol was subjected to Swern oxidation, generating keto isoxazole which was treated with methyl (triphenylphosphoranylidene)acetate followed by protecting group interconversions to generate the olefination product 496 as a single geometrical isomer. This product was subsequently coupled with (Z)-chloroenyne using a Pd(PPh3)4-CuI catalytic system to produce the protected enediyne 498. Cleavage of the isoxazole with molybdenum hexacarbonyl (Mo(CO)6), removal of the TMS group, and phthaloylation of amine gave rise to the desired intermediate 499. The new secondary hydroxy group produced through exposure of 499 to potassium hexamethyldisilazide in toluene was mesylated and subsequently inverted by a neighboring ester group to furnish the lactone 500 in high yield. After exchanging protecting groups and double reduction using DIBAL/NaBH4, the required aglycon intermediate 501 was produced by benzoylation. With the tetrasaccharide donor 491 and aglycon 501 in hand, Schmidt glycosidation was applied to form the coupled product 502 as a single isomer. Then, the target molecule calicheamicin γI1 (474) was generated after multi-step transformations.
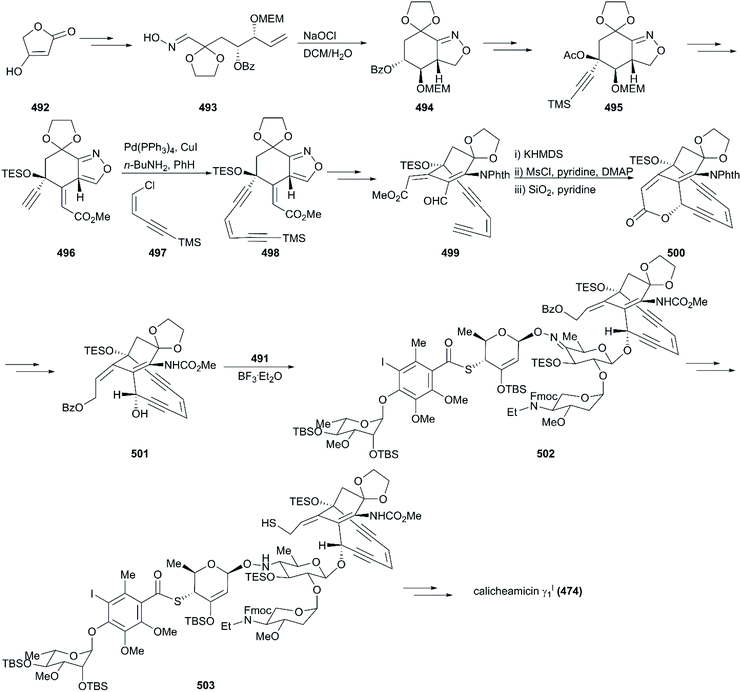 |
| | Scheme 47 Total synthesis of calicheamicin γI1 by Nicolaou. | |
9.1.2 Danishefsky's total synthesis of calicheamicin γI1.
Three years after Nicolaou's work, Danishefsky and co-workers also finished the total synthesis of calicheamicin γI1 (474).277–281 As shown in Scheme 48 tetrasaccharide 516 was constructed from the coupling of two disaccharide building blocks 508 and 514. After deiodination and triflation, the first disaccharide building block 508 was prepared from disaccharide fragment 507, which was forged by the coupling of glycal 506 and 505 in the presence of collidine-complexed iodonium perchlorate. For the second disaccharide, glycosyl donor 509 and acceptor 510 were subjected to the Schmidt procedure to build the aryl glycoside 511 with excellent yield. Subsequently, 511 was converted to acyl chloride 512, which was then successfully coupled with thiol 513 to provide disaccharide 514. Deprotonation of the carbamate nitrogen of disaccharide 514 using NaH as the base and subsequent alkylation with triflate 508 afforded tetrasaccharide 515, which was converted to glycosyl donor 516.
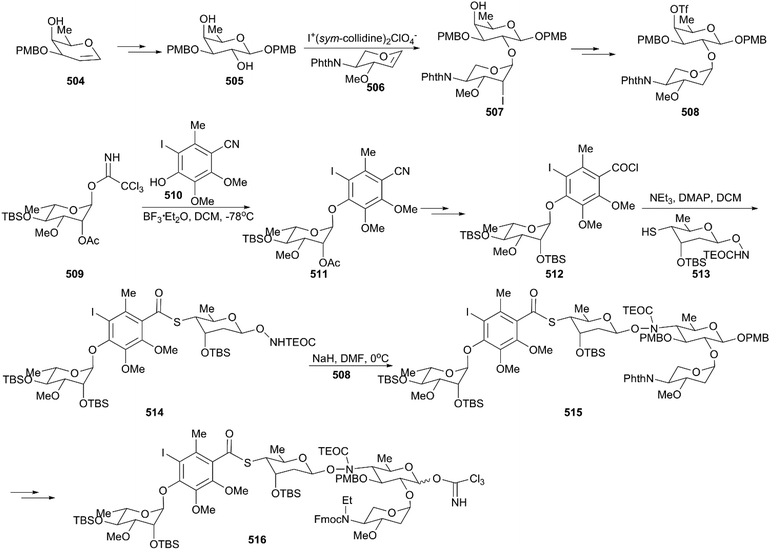 |
| | Scheme 48 Danishefsky's preparation of tetrasaccharide building block. | |
The synthesis of the aglycon unit started with the synthesis of unstable triol 518 from the commercially available benzoate 517; 518 was subjected to Becker oxidation to generate an epoxide and, following Dess–Martin oxidation, aldehyde 519 was obtained (Scheme 49). The aldehyde 519 was protected by treating with lithium N-methylanilide, and the keto group was attacked by the lithium acetylide generated from the parent enediyne to give adduct 520 after silylation of the newly formed alcohol by utilizing trimethylsilyl triflate. Then, 520 was subjected to potassium 3-ethyl-3-pentoxide, and the core skeleton 521 was obtained. Protecting group interconversions, acetolysis, and oxidative cleavage were employed to convert 521 to 522. Exposure of 522 to a solution of sodium azide afforded the desired product, azidoenone 523, which underwent subsequent key transformations including reduction of azide, an intramolecular Horner–Wadsworth–Emmons reaction, reduction using NaBH4, and a Mitsunobu reaction, generating trisulfide 524. Remarkably, the Schmidt protocol using silver triflate and molecular sieves was employed to couple trisulfide 524 with the tetrasaccharide donor 516 to forge 525 as a single isomer with moderate yield.
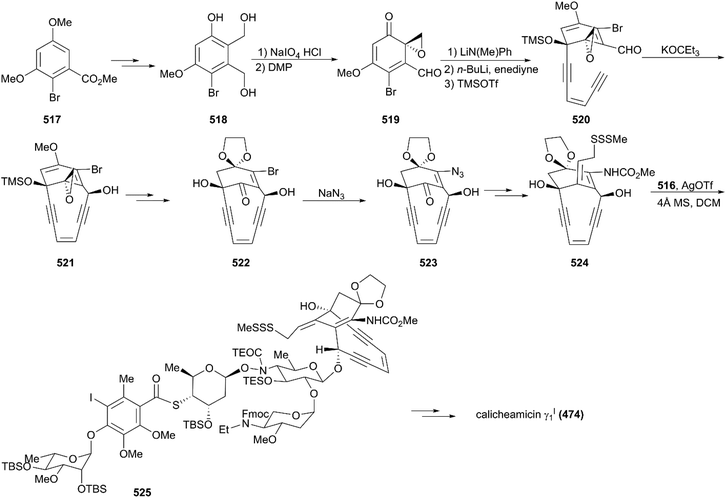 |
| | Scheme 49 Total synthesis of calicheamicin γI1 by Danishefsky. | |
Not long after the discovery of calicheamicin γI1, the structural analogs of calicheamicin, shishijimicin A (526)282 and namenamicin (527),283 were isolated from marine organisms (Fig. 22). Biological assays showed that they both displayed highly potent antitumor activity. Very recently, Nicolaou and co-workers finished the total synthesis of shishijimicin A (526)284,285 and namenamicin (527)286 using a protocol they developed previously, with some modifications. The details of these approaches are not discussed here. The derivatives and analogs of shishijimicin A were also constructed using the total synthesis strategy to explore the ADC payloads for clinical trials against tumors.287
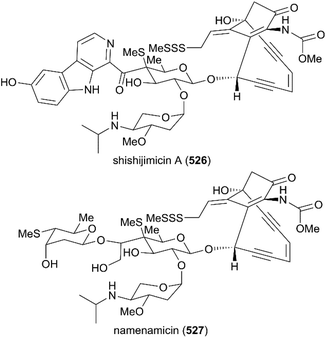 |
| | Fig. 22 The structures of shishijimicin A and namenamicin. | |
9.2 Structure–activity relationships of calicheamicin γI1 derivatives
As mentioned above, since calicheamicin γI1 was discovered as a 10-membered enediyne with highly potent cytotoxicity against tumors, a number of synthetic analogs have been designed and used as ADC payloads in preclinical evaluations and clinical tests against tumors. According to the mechanism of action described, the enediyne subunit of calicheamicin γI1 that plays an irreplaceable role in the generation of DNA lesions through the Bergman rearrangement is unchangeable, and the allylic trisulfide serves as a trigger to induce cycloaromatization. Studies of the semisynthetic aglycon analogs revealed that the role of carbohydrate fragments is to facilitate DNA binding and confer target specificity.270,288 To date, SAR research has mainly focused on the modification and simplification of oligosaccharide fragments or trisulfide groups.
The four calicheamicin γI1 analogs (Fig. 23), the natural products 471 and 472 and synthetic compounds 528 and 529, were all conjugated to CT-M-01 and the resultant derivatives were tested for therapeutic efficacy against MX-1 xenograft tumors. The results showed that compounds 472 and 528 with the DNA binding unit and the lack or modifications of amino sugar fragments showed stronger therapeutic efficacy than the parent molecule 474 and the derivatives 471 and 529, which missed the rhamnose at the DNA binding region. This result suggested that a rhamnose at the end of the binding region is required for ADC medications.289 CMB-401 (530) was further developed using a more stable amide-disulfide-based linker to conjugate it with hCTMO1 based on the structure of compound 528, and this compound was tested in phase I and phase II studies against epithelial ovarian cancer. However, no patients had objective responses in the phase II investigation. Scientists speculated that the sterically hindered disulfide failed to release the drug from the ADC payload.288
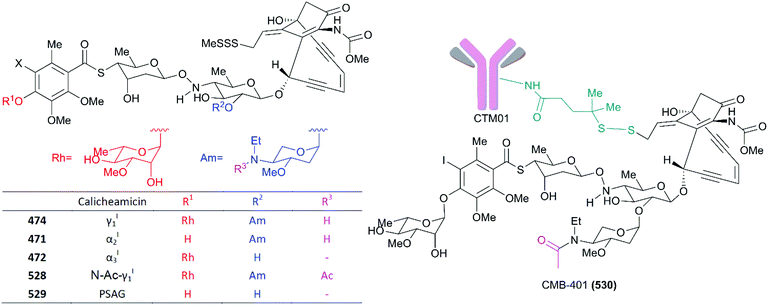 |
| | Fig. 23 Examples of calicheamicin γI1 derivatives(I). | |
Calicheamicin θI1 (Fig. 24, 531), a novel derivative of calicheamicin γI1, was developed using thioester to replace the methyl trisulfide and prepared through total synthesis by the Nicolaou group. After the test, calicheamicin θI1 was hypothesized to be a powerful biological tool for generating specific DNA lesions because of its highly potent and selective DNA cleaving, apoptosis-inducing, and cytotoxic properties.290 It was also discovered that calicheamicin θI1 (531) effectively suppressed liver metastases in a novel syngeneic model of murine neuroblastoma and CD19-positive malignancies.291,292 In some analogs, the allylic trisulfide was replaced by isopropyl disulfide or functionalized to improve the stability and solubility (Fig. 24, 532 and 533).293,294 Gemtuzumab ozogamicin (Mylotag®, 534) and inotuzumab ozogamicin (Besponsa®, 535) are ADC drugs approved to treat myeloid leukemia and B-cell malignancies by the FDA in 2000 and 2017, respectively. Because of the important role that the linker plays in the effective release of the calicheamicin derivative from the conjugation product, allowing it to access the DNA target, a linker, 4-(4-acetylphenoxy)butanoic acid, was added to condensate drug and antibody. This conjugate showed stability in physiological buffer (pH 7.4) and the calicheamicin derivative was efficiently released inside lysosomes (pH 4.0) (Fig. 25).295
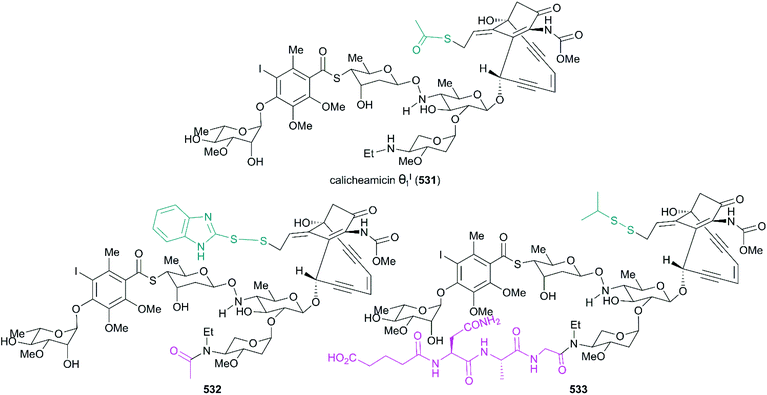 |
| | Fig. 24 Examples of calicheamicin γI1 derivatives(II). | |
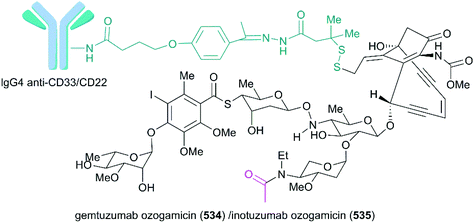 |
| | Fig. 25 Structures of the marketed ADC drugs gemtuzumab ozogamicin and inotuzumab ozogamicin. | |
10. Conclusions
In this review, we discussed the total synthesis and SAR studies of eight representative families of natural products applied in the treatment of various human diseases. Although just the tip of the iceberg, these examples demonstrate the essential role of total synthesis in natural product drugs’ discovery: development of efficient transformations for natural products synthesis, especially the deep-seated structural modifications that are otherwise tough to achieve; and providing adequate and sustainable supplies of natural products and their analogs for clinical studies and use. From another standpoint, total syntheses of natural products, which often contain complex structural features, also promote the development of synthetic chemistry by inspiring the discovery of new reactions and reagents for rapid molecular assembly and structural diversifications. In combination with the development of other disciplines, such as synthetic biology and chemical biology, chemical synthesis will continue to play a profound role in functional mechanism elucidation, new biological target identification, and natural product medicinal chemistry.
Abbreviations
| AcOH | Acetic acid |
| AIBN | 2,2′-Azobisisobutyronitrile |
| BHT | Butylated hydroxytoluene |
| Boc2O | Di-tert-butyl dicarbonate |
| BOPCl | Bis(2-oxo-3-oxazolidinyl)phosphinic chloride |
| BPO | Benzoyl peroxide |
| CSA | Camphorsulfonic acid |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DEAD | Diethyl azodicarboxylate |
| 1,2-DFB | 1,2-Difluorobenzene |
| DIPAMP | (Ethane-1,2-diyl)bis[(2-methoxyphenyl)(phenyl)phosphine] |
| DIPEA |
N,N-Diisopropylethylamine |
| DMAP | 4-(N,N-Dimethylamino)pyridine |
| DMDO | Dimethyldioxirane |
| DME | 1,2-Dimethoxyethane |
| DMF |
N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| Dppp | 1,3-Bis(diphenylphosphino)propane |
| DTBMP | 2,6-Di-tert-butyl-4-methylpyridine |
| EDCI |
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride |
| HFIP | Hexafluoroisopropanol |
| HMPA | Hexamethylphosphoramide |
| IBX | 2-Iodoxybenzoic acid |
| KHMDS | Potassium bis(trimethylsilyl)amide |
| LDA | Lithium diisopropylamide |
| LICA | Lithium N-isopropylcyclohexylamide |
| LiDBB | Lithium di-tert-butylbiphenyl |
| LiHMDS | Lithium bis(trimethylsilyl)amide |
| LTMP | Lithium tetramethylpiperidide |
|
m-CPBA |
meta-Chloroperoxybenzoic acid |
| MesLi | Mesityllithium |
| MS | Molecular sieve |
| MsCl | Methanesulfonyl chloride |
| NaHMDS | Sodium bis(trimethylsilyl)amide |
| NBS |
N-Bromosuccinimide |
| NMO | 4-Methylmorpholine N-oxide |
| NTf2 | Bis(trifluoromethylsulfonyl)imide |
| Pd2(dba)2 | Bis(dibenzylideneacetone)palladium |
| PhNTf2 | Bis(trifluoromethanesulfonyl)aniline |
| PMP | 1,2,2,6,6-Pentamethylpiperidine |
| PPTS | Pyridinium p-toluenesulfonate |
| Red-Al | Sodium bis(2-methoxyethoxy)aluminiumhydride |
| [Rh(COD)Cl]2 | Chloro(1,5-cyclooctadiene)rhodium(I) dimer |
| Rt | Room temperature |
| SARs | Structure–activity relationships |
| TBAF |
tert-Butylammonium fluoride |
| TBAI |
tert-Butylammonium iodide |
| TBSOTf |
tert-Butyldimethylsilyl trifluoromethanesulfonate |
| TESOH | Triethylsilanol |
| Tf2O | Trifluoromethanesulfonic anhydride |
| TFA | Trifluoroacetic acid |
| TFAA | Trifluoroacetic anhydride |
| TFE | 2,2,2-Trifluoroethanol |
| TfOH | Trifluoromethanesulfonic acid |
| THF | Tetrahydrofuran |
| TMEDA | Tetramethylethylenediamine |
| TMSCl | Chlorotrimethylsilane |
| TMSCN | Trimethylsilyl cyanide |
| TMSH | Trimethylsulfonium hydroxide |
| TMSimid | 1-(Trimethylsilyl)imidazole |
| TMSOH | Trimethylsilanol |
| TPAP | Tetrapropylammonium perruthenate |
| TPP | Triphenylphosphine |
| TsOH |
p-Toluenesulfonic acid |
Author contributions
X. Q. conceived the study; G. L., M. L. and X. Q. prepared the manuscript; all authors discussed the contents and commented on the manuscript.
Conflicts of interest
The authors declare no conflict of interests.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 21971018). We gratefully acknowledge the Beijing Municipal Government and Tsinghua University for financial support.
References
- G. M. Cragg and D. J. Newman, Natural products: a continuing source of novel drug leads, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- N. R. Farnsworth, O. Akerele, A. S. Bingel, D. D. Soejarto and Z. Guo, Medicinal plants in therapy, Bull. W. H. O., 1985, 63, 965–981 CAS.
-
D. T. Courtwright, Forces of habit: drugs and the making of the modern world, Harvard University Press, 2002, P36 Search PubMed.
-
V. Cechinel-Filho, Plant Bioactives and drug discovery John Wiley & Sons, Hoboken, New Jersey, 2011 Search PubMed.
- G. M. Cragg, D. J. Newman and K. M. Snader, Natural products in drug discovery and development, J. Nat. Prod., 1997, 60, 52–60 CrossRef CAS PubMed.
- K. S. Lam, New aspects of natural products in drug discovery, Trends Microbiol., 2007, 15, 279–289 CrossRef CAS PubMed.
- B. L. DeCorte, Underexplored opportunities for natural products in drug discovery, J. Med. Chem., 2016, 59, 9295–9304 CrossRef CAS PubMed.
- Z.-C. Wu and D. L. Boger, The quest for supernatural products: the impact of total synthesis in complex natural products medicinal chemistry, Nat. Prod. Rep., 2020, 37, 1511–1531 RSC.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, the International Natural Product Sciences Taskforce and C. T. Supuran, Natural products in drug discovery: advances and opportunities, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- M.-J. U. Ferreira, Natural products in drug discovery and human health, Phytochem. Rev., 2021, 20, 1–4 CrossRef CAS.
- C. A. Dehelean, I. Marcovici, C. Soica, M. Mioc, D. Coricovac, S. Iurciuc, O. M. Cretu and I. Pinzaru, Plant-derived anticancer compounds as new perspectives in drug discovery and alternative therapy, Molecules, 2021, 26, 1109–1138 CrossRef CAS PubMed.
- A. Nelson and G. Karageorgis, Natural product-informed exploration of chemical space to enable bioactive molecular discovery, RSC Med. Chem., 2021, 12, 353–362 RSC.
- J. Skubnik, V. Pavlickova, T. Ruml and S. Rimpelova, Current Perspectives on Taxanes: Focus on Their Bioactivity, Delivery and Combination Therapy, Plants, 2021, 10, 569–603 CrossRef CAS PubMed.
- C. Hobson, A. N. Chan and G. D. Wright, The antibiotic resistome: a guide for the discovery of natural products as antimicrobial agents, Chem. Rev., 2021, 121, 3464–3494 CrossRef CAS PubMed.
- P. Fernandes and E. Martens, Antibiotics in late clinical development, Biochem. Pharmacol., 2017, 133, 152–163 CrossRef CAS PubMed.
- R. I. Aminov, A brief history of the antibiotic era: lessons learned and challenges for the future, Front. Microbiol., 2010, 1, 1–7 Search PubMed.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, Impact of natural products on developing new anti-cancer agents, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS PubMed.
-
P. Kittakoop, Anticancer drugs and potential anticancer leads inspired by natural products, Elsevier B.V., 2015 Search PubMed.
- A. Mazumder, C. Cerell and M. Diederich, Natural scaffolds in anticancer therapy and precision medicine, Biotechnol. Adv., 2018, 36, 1563–1585 CrossRef CAS PubMed.
- M. Huang, J.-J. Lu and J. Ding, Natural Products in Cancer Therapy: Past, Present and Future, Nat. Prod. Bioprospect., 2021, 11, 5–13 CrossRef PubMed.
- A. J. Goodman, B. L. Bourdonnec and R. E. Dolle, Mu opioid receptor antagonists: recent developments, ChemMedChem, 2007, 2, 1552–1570 CrossRef CAS PubMed.
-
H. Buschmann, T. Christoph, E. Friderichs, C. Maul and B. Sundermann, Analgesics-from chemistry and pharmacology to clinical application, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed.
- L. H. Miller and X. Su, Artemisinin: discovery from the Chinese herbal garden, Cell, 2011, 146, 855–858 CrossRef CAS PubMed.
- N. S. Tibon, C. H. Ng and S. L. Cheong, Current progress in antimalarial pharmacotherapy and multi-target drug discovery, Eur. J. Med. Chem., 2020, 188, 111983–112007 CrossRef CAS PubMed.
- H. Madhav and N. Hoda, An insight into the recent development of the clinical candidates for the treatment of malaria and their target proteins, Eur. J. Med. Chem., 2021, 210, 112955–112995 CrossRef CAS PubMed.
- D. J. Newman, G. M. Cragg and K. M. Snader, Natural products as sources of new drugs over the period 1981–2002, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the last 25 Years, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs from 1981 to 2014, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- F. W. Serturner, Darstellung der reinen Mohnsäure (Opiumsaure) nebst einer Chemischen Untersuchung des Opiums mit vorzüglicher Hinsicht auf einen darin neuentdeckten Stoff und die dahin gehörigen Bemerkungen, J. Pharm., 1806, 14, 47–93 Search PubMed.
- B. H. Novak, T. Hudlicky, J. W. Reed, J. Mulzer and D. Trauner, Morphine synthesis and biosynthesis—an update, Curr. Org. Chem., 2000, 4, 343–362 CrossRef CAS.
- P. R. Blakemorea and J. D. White, Morphine, the proteus of organic molecules, Chem. Commun., 2002, 1159–1168 RSC.
- J. Zezula and T. Hudlicky, Recent progress in the synthesis of morphine alkaloids, Synlett, 2005, 388–405 CAS.
-
U. Rinner and T. Hudlicky, in Alkaloid synthesis, ed. H.-J. Knolker, Springer Science & Business Media, 2012, vol. 309, pp. 33–66 Search PubMed.
- A. S. Yekkirala, A. E. Kalyuzhny and P. S. Portoghese, Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective μ-δ agonists, ACS Chem. Neurosci., 2010, 1, 146–154 CrossRef CAS PubMed.
- A. S. Yekkirala, M. L. Banks, M. M. Lunzer, S. S. Negus, K. C. Rice and P. S. Portoghese, Clinically employed opioid analgesics produce antinociception via μ-δ opioid receptor heteromers in Rhesus monkeys, ACS Chem. Neurosci., 2012, 3, 720–727 CrossRef CAS PubMed.
- J. W. Reed and T. Hudlicky, The quest for a practical synthesis of morphine alkaloids and their derivatives by chemoenzymatic methods, Acc. Chem. Res., 2015, 48, 674–687 CrossRef CAS PubMed.
-
T. M. Kutchan, in The alkaloids: chemistry and biology, ed. G. A. Cordell, Academic Press, London, 1998, vol. 50, pp. 257–316 Search PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. F. Interrante and C. D. Smolke, Complete biosynthesis of opioids in yeast, Science, 2015, 349, 6252–6257 CrossRef PubMed.
- J. M. Gulland and R. Robinson, Mem. Proc. – Manchester Lit. Philos. Soc., 1925, 69, 79 CAS.
- M. Gates and G. Tschudi, The synthesis of morphine, J. Am. Chem. Soc., 1952, 74, 1109–1110 CrossRef CAS.
- M. Gates and G. Tschudi, The synthesis of morphine, J. Am. Chem. Soc., 1956, 78, 1380–1393 CrossRef CAS.
- M. F. Mackay and D. C. Hodgki, A crystallographic examination of the structure of morphine, J. Chem. Soc., 1955, 3261–3267 RSC.
- Q. Li and H. Zhang, Research progress on the synthesis of morphine alkaloids, Chin. J. Org. Chem., 2017, 37, 1629–1652 CrossRef CAS.
- S. A. Chambers, J. M. DeSousa, E. D. Huseman and S. D. Townsend, The DARK side of total synthesis: strategies and tactics in psychoactive drug production, ACS Chem. Neurosci., 2018, 9, 2307–2330 CrossRef CAS PubMed.
- C. Y. Hong, N. Kado and L. E. Overman, Asymmetric synthesis of either enantiomer of opium alkaloids and morphinans. Total synthesis of (−)- and (+)-dihydrocodeinone and (−)- and (+)-morphine, J. Am. Chem. Soc., 1993, 115, 11028–11029 CrossRef CAS.
- I. Iijima and K. C. Rice, Studies in the (+)-morphinan series I. An alternative conversion of (+)-dihydrocodeinone into (+)-codeine, Heterocycles, 1977, 6, 1157–1165 CrossRef CAS.
- J. D. White, P. Hrnciar and F. Stappenbeck, Asymmetric synthesis of (+)-morphine. The phenanthrene route revisited, J. Org. Chem., 1997, 62, 5250–5251 CrossRef CAS.
- B. M. Trost and W. Tang, An efficient enantioselective synthesis of (-)-galanthamine, Angew. Chem., Int. Ed., 2002, 41, 2795–2797 CrossRef CAS PubMed.
- B. M. Trost and W. Tang, Enantioselective synthesis of (-)-codeine and (-)-morphine, J. Am. Chem. Soc., 2002, 124, 14542–14543 CrossRef CAS PubMed.
- B. M. Trost, W. Tang and F. D. Toste, Divergent enantioselective synthesis of (-)-galanthamine and (-)-morphine, J. Am. Chem. Soc., 2005, 127, 14785–14803 CrossRef CAS PubMed.
- H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bissett, D. Ilceski and T. Hudlicky, Chemoenzymatic enantiodivergent total syntheses of (+)- and (-)-codeine, Tetrahedron, 2009, 65, 9862–9875 CrossRef CAS.
- M. Makarova, M. A. A. Endoma-Arias, H. E. D. Paz, R. Simionescu and T. Hudlicky, Chemoenzymatic total synthesis of ent-oxycodone: second-, third-, and fourth-generation strategies, J. Am. Chem. Soc., 2019, 141, 10883–10904 CrossRef CAS PubMed.
- Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Enantioselective synthesis of cis-hydrobenzofurans bearing all-carbon quaternary stereocenters and application to total synthesis of (-)-morphine, Nat. Commun., 2019, 10, 2507–2513 CrossRef PubMed.
- Y. S.-H. Hou, A. Prichina and G. Dong, Deconstructive asymmetric total synthesis of morphine-family alkaloid (-)-thebainone A, Angew. Chem., Int. Ed., 2021, 60, 13057–13064 CrossRef PubMed.
-
D. A. Williams, V. F. Roche and E. B. Roche, in Foye's principles of medicinal chemistry, ed. T. L. Lemke, D. A. Williams, V. F. Roche and S. W. Zito, Lippincott Williams & Wilkins, China, 7 edn, 2012, ch. 20, pp. 675–695 Search PubMed.
- K. Raynor, H. Kong, Y. Chen, K. Yasuda, L. Yu, G. I. Bell and T. Reisine, Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors, Mol. Pharmacol., 1994, 45, 330–334 CAS.
- I. D. Pogozheva, A. L. Lomize and H. I. Mosberg, Opioid receptor three-dimensional structures from distance geometry calculations with hydrogen bonding constraints, Biophys. J., 1998, 75, 612–632 CrossRef CAS PubMed.
- H. E. Hassan, S. L. Mercer, C. W. Cunningham, A. Coop and N. D. Eddington, Evaluation of the P-glycoprotein (Abcb1) affinity status of a series of morphine analogs: Comparative study with meperidine analogs to identify opioids with minimal P-glycoprotein interactions, Int. J. Pharm., 2009, 375, 48–54 CrossRef CAS PubMed.
- T. H. Jukes, Some historical notes on chlortetracycline, Rev. Infect. Dis., 1985, 7(5), 702–707 CrossRef CAS PubMed.
- A. C. Finlay, G. L. Hobby, S. Y. P′an, P. P. Regna, J. B. Routien, D. B. Seeley, G. M. Shull, B. A. Sobin, I. A. Solomons, J. W. Vinson and J. H. Kane, Terramycin, a new antibiotic, Science, 1950, 111, 85–87 CrossRef CAS PubMed.
- M. A. Darken, H. Berenson, R. J. Shirk and N. O. Sjolander, Production of tetracycline by Streptomyces aureofaciens in synthetic media, Appl. Microbiol., 1960, 8, 46–51 CrossRef CAS PubMed.
- J. R. D. McCormick, N. O. Sjolander, U. Hirsch, E. R. Jensen and A. P. Doerschuk, A new family of antibiotics: the demethyltetracyclines, J. Am. Chem. Soc., 1957, 79, 4561–4563 CrossRef CAS.
- I. Chopra and M. Roberts, Tetracycline
antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance, Microbiol. Mol. Biol. Rev., 2001, 65, 232–260 CrossRef CAS PubMed.
- D. E. Brodersen, J. William, M. Clemons, A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit, Cell, 2000, 103, 1143–1154 CrossRef CAS PubMed.
- M. C. Roberts, Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution, FEMS Microbiol. Rev., 1996, 19, 1–24 CrossRef CAS PubMed.
- S. R. Connell, D. M. Tracz, K. H. Nierhaus and D. E. Taylor, Ribosomal protection proteins and their mechanism of tetracycline resistance, Antimicrob. Agents Chemother., 2003, 47, 3675–3681 CrossRef CAS PubMed.
- T. H. Grossman, Tetracycline antibiotics and resistance, Cold Spring Harbor Perspect. Med., 2016, 6, a025387–a025387 CrossRef PubMed.
- C. R. Stephens, L. H. Conover, F. A. Hochstein, P. P. Regna, F. J. Pilgrim, K. J. Brunings and R. B. Woodward, Terramycin. VIII. Structure of aureomycin and terramycin, J. Am. Chem. Soc., 1952, 74, 4976–4977 CrossRef CAS.
- F. A. Hochstein, C. R. Stephens, L. H. Conover, P. P. Regna, R. Pasternack, P. N. Gordon, F. J. Pilgrim, K. J. Brunings and R. B. Woodward, The structure of terramycin, J. Am. Chem. Soc., 1953, 75, 5455–5475 CrossRef CAS.
- C. R. Stephens, L. H. Conover, R. Pasternack, F. A. Hochstein, W. T. Moreland, P. P. Regna, F. J. Pilgrim, K. J. Brunings and R. B. Woodward, Structure of aureomycin, J. Am. Chem. Soc., 1954, 76, 3568–3575 CrossRef CAS.
- D. L. J. Clive, Chemistry of the tetracyclines, Q. Rev., Chem. Soc., 1968, 22, 435–456 RSC.
- T. K. Allred, F. Manoni and P. G. Harran, Exploring the boundaries of “practical”: De novo syntheses of complex natural product-based drug candidates, Chem. Rev., 2017, 117, 11994–12051 CrossRef CAS PubMed.
- L. H. Conover, K. Butler, R. B. Johnston, R. B. Korst and R. B. Woodward, The total synthesis of 6-demethyl-6 deoxytetracycline, J. Am. Chem. Soc., 1962, 84, 3222–3224 CrossRef CAS.
- R. B. Woodward, The total synthesis of a tetracycline, Pure Appl. Chem., 1963, 6, 561–573 CAS.
- J. J. Korst, J. D. Johnston, K. Butler, E. J. Bianco, L. H. Conover and R. B. Woodward, The total synthesis of dl-6-demethyl-6-deoxytetracycline, J. Am. Chem. Soc., 1968, 90, 439–457 CrossRef CAS.
- A. I. Gurevich, M. G. Karapetyan, M. N. Kolosov, V. G. Korobko, V. V. Onoprienko, S. A. Popravko and M. M. Shemyakin, Synthesis of 12a-deoxy-5a,6-anhydrotetracycline. The first total synthesis of the naturally occuring tetracycline, Tetrahedron Lett., 1967, 8, 131–134 CrossRef.
- M. S. V. Wittenau, Preparation of tetracyclines by photooxidation of anhydrotetracyclines, J. Org. Chem., 1964, 29, 2746–2748 CrossRef.
- H. Muxfeldt and W. Rogalski, Tetracyclines. V. A total synthesis of (±)-6-deoxy-6-demethyltetracycline, J. Am. Chem. Soc., 1965, 87, 933–934 CrossRef CAS PubMed.
- H. Muxfeldt, G. Haas, G. Hardtmann, F. Kathawala, J. B. Mooberry and E. Vedejs, Tetracyclines. 9. Total synthesis of dl-terramycin, J. Am. Chem. Soc., 1979, 101, 689–701 CrossRef CAS.
- G. Stork, J. J. L. Clair, P. Spargo, R. P. Nargund and N. Totah, Stereocontrolled synthesis of (±)-12a -deoxytetracycline, J. Am. Chem. Soc., 1996, 118, 5304–5305 CrossRef CAS.
- K. Tatsuta, T. Yoshimoto, H. Gunji, Y. Okado and M. Takahashi, The first total synthesis of natural (−)-tetracycline, Chem. Lett., 2000, 29, 646–647 CrossRef.
- M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics, Science, 2005, 308, 395–398 CrossRef CAS PubMed.
- M. G. Charest, D. R. Siegel and A. G. Myers, Synthesis of (-)-tetracycline, J. Am. Chem. Soc., 2005, 127, 8292–8293 CrossRef CAS PubMed.
- J. D. Brubaker and A. G. Myers, A practical, enantioselective synthetic route to a key precursor to the tetracycline antibiotics, Org. Lett., 2007, 9, 3523–3525 CrossRef CAS PubMed.
- C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y.-M. Wang and A. G. Myers, A robust platform for the synthesis of new tetracycline antibiotics, J. Am. Chem. Soc., 2008, 130, 17913–17927 CrossRef CAS PubMed.
- D. A. Kummer, D. Li, A. Dion and A. G. Myers, A practical, convergent route to the key precursor to the tetracycline antibiotics, Chem. Sci., 2011, 2, 1710–1718 RSC.
- M. Ronn, Z. Zhu, P. C. Hogan, W.-Y. Zhang, J. Niu, C. E. Katz, N. Dunwoody, O. Gilicky, Y. Deng, D. K. Hunt, M. He, C.-L. Chen, C. Sun, R. B. Clark and X.-Y. Xiao, Process R&D of eravacycline: The first fully synthetic fluorocycline in clinical development, Org. Process Res. Dev., 2013, 17, 838–845 CrossRef CAS.
- A. Mullard, 2018 FDA drug approvals, Nat. Rev. Drug Discovery, 2019, 18, 85–89 CrossRef PubMed.
- W. Y. Zhang, C. Sun, D. Hunt, M. He, Y. Deng, Z. Zhu, C. L. Chen, C. E. Katz, J. Niu, P. C. Hogan, X. Y. Xiao, N. Dunwoody and M. Ronn, Process development and scale-up of fully synthetic tetracycline TP-2758: a potent antibacterial agent with excellent oral bioavailability, Org. Process Res. Dev., 2016, 20, 284–296 CrossRef CAS.
- G. G. Zhanel, K. Homenuik, K. Nichol, A. Noreddin, L. Vercaigne, J. Embil, A. Gin, J. A. Karlowsky and D. J. Hoban, The glycylcyclines, Drugs, 2004, 64, 63–88 CrossRef CAS PubMed.
-
A. R. Martin, in Wilson and Gisvold's textbook of organic medicinal and pharmaceutical chemistry, ed. J. N. Delgado and W. A. Remers, Lippincott-Raven Publishers, Philadelphia, 10th edn, 1998, pp. 210–215 Search PubMed.
- K. F. Benitz and H. F. Diermeier, Renal toxicity of tetracycline degradation products, Exp. Biol. Med., 1964, 115, 930–935 CrossRef CAS PubMed.
- M. J. Martell and J. H. Boothe, 6-deoxytetracyclines. VII. Alkylated aminotetracyclines possessing unique antibacterial activity, J. Med. Chem., 1967, 10, 44–46 CrossRef CAS PubMed.
- A. E. Yñigez-Gutierrez and B. O. Bachmann, Fixing the unfixable: the art of optimizing natural products for human medicine, J. Med. Chem., 2019, 62, 8412–8428 CrossRef PubMed.
- P. E. Sum and P. Petersen, Synthesis and structure-activity relationship of novel glycylcycline derivatives leading to the discovery of GAR-936, Bioorg. Med. Chem. Lett., 1999, 9, 1459–1462 CrossRef CAS PubMed.
- P. J. Petersen, N. V. Jacobus, W. J. Weiss, P. E. Sum and R. T. Testa, In vitro and in vivo antibacterial activities of a novel glycylcycline, the 9-t-butylglycylamido derivative of minocycline (GAR-936), Antimicrob. Agents Chemother., 1999, 43, 738–744 CrossRef CAS PubMed.
- L. Honeyman, M. Ismail, M. L. Nelson, B. Bhatia, T. E. Bowser, J. Chen, R. Mechiche, K. Ohemeng, A. K. Verma, E. P. Cannon, A. Macone, S. K. Tanaka and S. Levy, Structure-activity relationship of the aminomethylcyclines and the discovery of omadacycline, Antimicrob. Agents Chemother., 2015, 59, 7044–7053 CrossRef CAS PubMed.
- R. B. Clark, D. K. Hunt, M. He, C. Achorn, C.-L. Chen, Y. Deng, C. Fyfe, T. H. Grossman, P. C. Hogan, W. J. O'Brien, L. Plamondon, M. Rönn, J. A. Sutcliffe, Z. Zhu and X.-Y. Xiao, Fluorocyclines. 2. Optimization of the C-9 side-chain for antibacterial activity and oral efficacy, J. Med. Chem., 2012, 55, 606–622 CrossRef CAS PubMed.
- P. F. Wiley, K. Gerzon, E. H. Flynn, M. V. S. Jr., O. Weaver, U. C. Quarck, R. R. Chauvette and R. Monahan, Erythromycin. X.1 Structure of erythromycin, J. Am. Chem. Soc., 1957, 79, 6062–6070 CrossRef CAS.
- R. Place, F. O. R. The, C. Growikg, T. H. Office and H. Office, Erythromycin: another antibiotic, Lancet, 1952, 260, 232–233 Search PubMed.
- D. R. Harris, S. G. McGeachin and H. H. Mills, The structure and stereochemistry of erythromycin A, Tetrahedron Lett., 1965, 6, 679–685 CrossRef.
- A. Contreras and D. Vazquez, Cooperative and antagonistic interactions of peptidyl-tRNA and antibiotics with bacterial ribosomes, Eur. J. Biochem., 1977, 74, 539–547 CrossRef CAS PubMed.
- J. A. Dunkle, L. Xiong, A. S. Mankin and J. H. D. Cate, Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17152–17157 CrossRef CAS PubMed.
- J. Sutcliffe, A. Tait-Kamradt and L. W. Ondarck, Streptococcus pneumoniae and Streptococcus pyogenes resistantto macrolides but sensitive to clindamycin: a common resistance pattern mediated by an efflux system, Antimicrob. Agents Chemother., 1996, 40, 1817–1824 CrossRef CAS PubMed.
- D. Felminghama, R. Cantónb and S. G. Jenkins, Regional trends in β-lactam, macrolide, fluoroquinolone and telithromycin resistance among Streptococcus pneumoniae isolates 2001–2004, J. Infect., 2007, 55, 111–118 CrossRef PubMed.
- A. Canu, B. Malbruny, M. L. Coquemont, T. A. Davies, P. C. Appelbaum and R. Leclercq, Diversity of ribosomal mutations conferring resistance to macrolides, clindamycin, streptogramin, and telithromycin in streptococcus pneumoniae, Antimicrob. Agents Chemother., 2002, 46, 125–131 CrossRef CAS PubMed.
- S. Pestka, R. Vince, R. LeMahieu, F. Weiss, L. Fern and J. Unowsky, Induction of erythromycin resistance in staphylococcus aureus by erythromycin derivatives, Antimicrob. Agents Chemother., 1976, 9, 128–130 CrossRef CAS PubMed.
- N. E. Allen, Macrolide resistance in staphylococcus aureus: induction of macrolide-resistant protein synthesis, Antimicrob. Agents Chemother., 1977, 11, 661–668 CrossRef CAS PubMed.
- H. Ounissi and P. Courvalin, Nucleotide sequence of the gene ereA encoding the erythromycin esterase in Escherichia coli, Gene, 1985, 35, 271–278 CrossRef CAS PubMed.
- M. Arthur, D. Autissier and P. Courvalin, Analysis of the nucleotide sequence of the ereB gene encoding the erythromydn esterase type II, Nucleic Acids Res., 1986, 14, 4987–4999 CrossRef CAS PubMed.
- P. Kurath, P. H. Jones, R. S. Egan and T. J. Perun, Acid degradation of erythromycin A and erythromycin B, Experientia, 1971, 27, 362 CrossRef CAS PubMed.
- R. B. Woodward, E. Logusch, K. P. Nambiar, K. Sakan, D. E. Ward, B. W. Au-Yeung, P. Balaram, L. J. Browne, P. J. Card and C. H. Chen, Asymmetric total synthesis of erythromcin. 1. Synthesis of an erythronolide A secoacid derivative via asymmetric induction, J. Am. Chem. Soc., 1981, 103, 3210–3213 CrossRef.
- R. B. Woodward, B. W. Au-Yeung, P. Balaram, L. J. Browne, D. E. Ward, B. W. Au-Yeung, P. Balaram, L. J. Browne, P. J. Card and C. H. Chen, Asymmetric total synthesis of erythromycin. 2. Synthesis of an erythronolide A lactone system, J. Am. Chem. Soc., 1981, 103, 3213–3215 CrossRef CAS.
- R. B. Woodward, E. Logusch, K. P. Nambiar, K. Sakan, D. E. Ward, B. W. Au-Yeung, P. Balaram, L. J. Browne, P. J. Card and C. H. Chen, Asymmetric total synthesis of erythromycin. 3. Total synthesis of erythromycin, J. Am. Chem. Soc., 1981, 103, 3215–3217 CrossRef CAS.
- P. Breton, P. J. Hergenrother, T. Hida, A. Hodgson, A. S. Judd, E. Kraynack, P. R. Kym, W. C. Lee, M. S. Loft, M. Yamashita and S. F. Martin, Total synthesis of erythromycin B, Tetrahedron, 2007, 63, 5709–5729 CrossRef CAS.
- H. C. Kim and S. H. Kang, Total synthesis of azithromycin, Angew. Chem., Int. Ed., 2009, 48, 1827–1829 CrossRef CAS PubMed.
- S. Morimoto, Y. Takahashi, Y. Watanabe and S. Ōmura, Chemical modification of eryhromycins, J. Antibiot., 1984, 37, 187–189 CrossRef CAS PubMed.
- S. Djokić, G. Kobrehel, G. Lazarevski, N. Lopotar, Z. Tamburašev, B. Kamenar, A. Nagl and I. Vicković, Erythromycin series. Part 11. Ring expansion of erythromycin A oxime by the Beckmann rearrangement, J. Chem. Soc., Perkin Trans. 1, 1986, 1881–1890 RSC.
- J. W. McFarland, C. M. Berger, S. A. Froshauer, S. F. Hayashi, S. J. Hecker, B. H. Jaynes, M. R. Jefson, B. J. Kamicker, C. A. Lipinski, K. M. Lundy, C. P. Reese and C. B. Vu, Quantitative structure−activity relationships among macrolide antibacterial agents: in vitro and in vivo potency against pasteurella multocida, J. Med. Chem., 1997, 40, 1340–1346 CrossRef CAS PubMed.
- W. R. Baker, J. D. Clark, R. L. Stephens and K. H. Kim, Modification of macrolide antibiotics. Synthesis of 11-deoxy-11-(carboxyamino)-6-O-methy¡erythromycin A ll,12-(Cyclic esters) via an intramolecular Michael reaction of O-carbamates with an,/9-unsaturated ketone, J. Org. Chem., 1988, 53, 2340–2345 CrossRef CAS.
- D. Bertrand, S. Bertrand, E. Neveu and P. Fernandes, Molecular characterization of off-target activities of telithromycin: a potential role for nicotinic acetylcholine receptors, Antimicrob. Agents Chemother., 2010, 54, 5399–5402 CrossRef CAS PubMed.
- R. J. Owellen, C. A. Hartke, R. M. Dickerson and F. O. Hams, Inhibition of tubulin-microtubule polymerization by drugs of the Vinca alkaloid class, Cancer Res., 1976, 36, 1499–1502 CAS.
- R. H. Himes, Interactions of the catharanthus (Vinca) alkaloids with tubulin and microtubules, Pharmacol. Ther., 1991, 51, 257–267 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Microtubules as a target for anticancer drugs, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
-
V. F. Roche, in Foye's principles of Medicinal Chemistry, ed. T. L. Lemke, D. A. Williams, V. F. Roche and S. W. Zito, Lippincott Williams & Wilkins, China, 7 edn, 2012, ch. 37, pp. 1244–1250 Search PubMed.
- B. Gigant, C. Wang, R. B. Ravelli, F. Roussi, M. O. Steinmetz, P. A. Curmi, A. Sobel and M. Knossow, Structural basis for the regulation of tubulin by vinblastine, Nature, 2005, 435, 519–522 CrossRef CAS PubMed.
- R. L. Noble, C. T. Beer and J. H. Cutts, Role of chance observations in chemotherapy: vinca rosea, Ann. N. Y. Acad. Sci., 1958, 76, 882–894 CrossRef CAS PubMed.
- G. H. Svoboda, N. Neuss and M. Gorman, Alkaloids of vinca rosea Linn. (Catharanthus roseus G. Don.) V.: Preparation and characterization of alkaloids, J. Am. Pharm. Assoc., Sci. Ed., 1959, 48, 659–666 CrossRef CAS PubMed.
- G. H. Svoboda, Alkaloids of Vinca rosea. IX. Extraction and characterization of leurosidine and leurocristine, Lloydia, 1961, 24, 173–178 CAS.
- J. W. Moncrief and W. N. Lipscomb, Structures of leurocristine (vincristine) and vincaleukoblastine. X-ray analysis of leurocristine methiodide, J. Am. Chem. Soc., 1965, 87, 4963–4964 CrossRef CAS PubMed.
- N. Langlois, F. Gueritte, Y. Langlois and P. Potier, Application of a modification of the Polonovski reaction to the synthesis of vinblastine-type alkaloids, J. Am. Chem. Soc., 1976, 98, 7017–7024 CrossRef CAS PubMed.
- P. Mangeney, R. Z. Andriamialisoa, N. Langlois, Y. Langlois and P. Potier, Preparation of vinblastine, vincristine, and leurosidine, antitumor alkaloids from Catharanthus species (Apocynaceae), J. Am. Chem. Soc., 1979, 101, 2243–2245 CrossRef CAS.
- J. P. Kutney, L. S. L. Choi, J. Nakano, H. Tsukamoto, M. McHugh and C. A. Boulet, A highly efficient and commercially important synthesis of the antimuor Catharanthus alkaloids vinblastine and leurosidine from catharanthine and vindoline, Heterocycles, 1988, 27, 1845–1853 CrossRef CAS.
- M. E. Kuehne, P. A. Matson and W. G. Bornmann, Enantioselective syntheses of vinblastine, leurosidine, vincovaline, and 20′-epi-vincovaline, J. Org. Chem., 1991, 56, 513–528 CrossRef CAS.
- W. G. Bornmann and M. E. Kuehne, A common intermediate providing syntheses of ψ-tabersonine, coronaridine, iboxyphylline, ibophyllidine, vinamidine, and vinblastine, J. Org. Chem., 1992, 57, 1752–1760 CrossRef CAS.
- P. Magnus, J. S. Mendoza, A. Stamford, M. Ladlow and P. Willis, Nonoxidative coupling methodology for the synthesis of the antitumor bisindole alkaloid vinblastine and a lower-half analogue: solvent effect on the stereochemistry of the crucial C-15/C-18′ bond, J. Am. Chem. Soc., 1992, 114, 10232–10245 CrossRef CAS.
- S. Kobayashi, T. Ueda and T. Fukuyama, An efficient total synthesis of (-)-vindoline, Synlett, 2000, 883–886 Search PubMed.
- S. Yokoshima, T. Ueda, S. Kobayashi, A. Sato, T. Kuboyama, H. Tokuyama and T. Fukuyama, Stereocontrolled total synthesis of (+)-vinblastine, J. Am. Chem. Soc., 2002, 124, 2137–2139 CrossRef CAS PubMed.
- T. Kuboyama, S. Yokoshima, H. Tokuyama and T. Fukuyama, Stereocontrolled total synthesis of (+)-vincristine, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11966–11970 CrossRef CAS PubMed.
- T. Miyazaki, S. Yokoshima, S. Simizu, H. Osada, H. Tokuyama and T. Fukuyama, Synthesis of (+)-vinblastine and its analogues, Org. Lett., 2007, 9, 4737–4740 CrossRef CAS PubMed.
- H. Ishikawa, G. I. Elliott, J. Velcicky, Y. Choi and D. L. Boger, Total synthesis of (-)- and ent-(+)-vindoline and related alkaloids, J. Am. Chem. Soc., 2006, 128, 10596–10612 CrossRef CAS PubMed.
- H. Ishikawa, D. A. Colby and D. L. Boger, Direct coupling of catharanthine and vindoline to provide vinblastine: total synthesis of (+)- and ent-(-)-vinblastine, J. Am. Chem. Soc., 2008, 130, 420–421 CrossRef CAS PubMed.
- H. Gotoh, J. E. Sears, A. Eschenmoser and D. L. Boger, New insights into the mechanism and an expanded scope of the Fe(III)-mediated vinblastine coupling reaction, J. Am. Chem. Soc., 2012, 134, 13240–13243 CrossRef CAS PubMed.
- H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues, J. Am. Chem. Soc., 2009, 131, 4904–4916 CrossRef CAS PubMed.
- N. Wang, J. Liu, C. Wang, L. Bai and X. Jiang, Asymmetric total syntheses of (-)-jerantinines A, C, and E, (-)-16-methoxytabersonine, (-)-vindoline, and (+)-vinblastine, Org. Lett., 2018, 20, 292–295 CrossRef CAS PubMed.
-
L. S. Borman and M. E. Kuehne, in The Alkaloids, ed. A. Brossi and M. Suffness, Academic, San Diego, 1990, vol. 37, pp. 133–144 Search PubMed.
-
H. L. Pearce, in The Alkaloids, ed. A. Brossi and M. Suffness, Academic, San Diego, 1990, vol. 37, pp. 145–204 Search PubMed.
- J. Fahy, Modifications in the “upper” or velbenamine part of the vinca alkaloids have major implications for tubulin interacting activities, Curr. Pharm. Des., 2001, 7, 1181–1197 CrossRef CAS PubMed.
- J. E. Sears and D. L. Boger, Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure-function properties, Acc. Chem. Res., 2015, 48, 653–662 CrossRef CAS PubMed.
- H. Gotoh, K. K. Duncan, W. M. Robertson and D. L. Boger, 10′-Fluorovinblastine and 10′-fluorovincristine: synthesis of a key series of modified vinca alkaloids, ACS Med. Chem. Lett., 2011, 2, 948–952 CrossRef CAS PubMed.
- T. J. Barker, K. K. Duncan, K. Otrubova and D. L. Boger, Potent vinblastine C20′ ureas displaying additionally improved activity against a vinblastine-resistant cancer cell line, ACS Med. Chem. Lett., 2013, 4, 985–988 CrossRef CAS PubMed.
- E. K. Leggans, K. K. Duncan, T. J. Barker, K. D. Schleicher and D. L. Boger, A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedented tubulin binding steric tolerance: C20′ urea derivatives, J. Med. Chem., 2013, 56, 628–639 CrossRef CAS PubMed.
- A. Tam, H. Gotoh, W. M. Robertson and D. L. Boger, Catharanthine C16 substituent effects on the biomimetic coupling with vindoline: preparation and evaluation of a key series of vinblastine analogues, Bioorg. Med. Chem. Lett., 2010, 20, 6408–6410 CrossRef CAS PubMed.
- K. D. Schleicher, Y. Sasaki, A. Tam, D. Kato, K. K. Duncan and D. L. Boger, Total synthesis and evaluation of vinblastine analogues containing systematic deep-seated modifications in the vindoline subunit ring system: core redesign, J. Med. Chem., 2013, 56, 483–495 CrossRef CAS PubMed.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- M. A. Jordan, Mechanism of action of antitumor drugs that interact with microtubules and tubulin, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2, 1–17 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Microtubules as a target for anticancer drugs, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
-
A. Todd, P. W. Groundwater and J. H. Gill, in Anticancer Therapeutics: From Drug Discovery to Clinical Applications, John Wiley & Sons Ltd, 2018, pp. 211–232 Search PubMed.
- R. A. Holton, C. Somoza, H. B. Kim, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith and S. Kim, First total synthesis of taxol. 1. Functionalization of the B ring, J. Am. Chem. Soc., 1994, 116, 1597–1598 CrossRef CAS.
- R. A. Holton, H. B. Kim, C. Somoza, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith and S. Kim, First total synthesis of taxol. 2. Completion of the C and D rings, J. Am. Chem. Soc., 1994, 116, 1599–1600 CrossRef CAS.
- K. C. Nicolaou, Z. Yang, J. J. Liu, H. Ueno, P. G. Nantermet, R. K. Guy, C. F. Claiborne, J. Renaud, E. A. Couladouros, K. Paulvannan and E. J. Sorensen, Total synthesis of taxol, Nature, 1994, 367, 630–634 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Nantermet and H. Ueno, Novel chemistry of taxol. Retrosynthetic and synthetic studies, J. Chem. Soc., Chem. Commun., 1994, 295–296 RSC.
- S. J. Danishefsky, J. J. Masters, W. B. Young, J. T. Link, L. B. Snyder, T. V. Magee, D. K. Jung, R. C.
A. Isaacs, W. G. Bornmann, C. A. Alaimo, C. A. Coburn and M. J. D. Grandi, Total synthesis of baccatin III and taxol, J. Am. Chem. Soc., 1996, 118, 2843–2859 CrossRef CAS.
- P. A. Wender, N. F. Badham, S. P. Conway, P. E. Floreancig, T. E. Glass, C. Gränicher, J. B. Houze, J. Jänichen, D. Lee, D. G. Marquess, P. L. McGrane, W. Meng, T. P. Mucciaro, M. Mühlebach, M. G. Natchus, H. Paulsen, D. B. Rawlins, J. Satkofsky, A. J. Shuker, J. C. Sutton, R. E. Taylor and K. Tomooka, The pinene path to taxanes. 5. Stereocontrolled synthesis of a versatile taxane precursor, J. Am. Chem. Soc., 1997, 119, 2755–2756 CrossRef CAS.
- P. A. Wender, N. F. Badham, S. P. Conway, P. E. Floreancig, T. E. Glass, J. B. Houze, N. E. Krauss, D. Lee, D. G. Marquess, P. L. McGrane, W. Meng, M. G. Natchus, A. J. Shuker, J. C. Sutton and R. E. Taylor, The pinene path to taxanes. 6. A concise stereocontrolled synthesis of taxol, J. Am. Chem. Soc., 1997, 119, 2757–2758 CrossRef CAS.
- K. Morihira, R. Hara, S. Kawahara, T. Nishimori, N. Nakamura, H. Kusama and I. Kuwajima, Enantioselective total synthesis of taxol, J. Am. Chem. Soc., 1998, 120, 12980–12981 CrossRef CAS.
- H. Kusama, R. Hara, S. Kawahara, T. Nishimori, H. Kashima, N. Nakamura, K. Morihira and I. Kuwajima, Enantioselective total synthesis of (−)-taxol, J. Am. Chem. Soc., 2000, 122, 3811–3820 CrossRef CAS.
- T. Mukaiyama, I. Shiina, H. Iwadare, H. Sakoh, Y. Tani, M. Hasegawa and K. Satoh, Asymmetric total synthesis of Taxol, Chem. – Eur. J., 1999, 5, 121–161 CrossRef CAS.
- T. Doi, S. Fuse, S. Miyamoto, K. Nakai, D. Sasuga and T. Takahashi, A formal total synthesis of taxol aided by an automated synthesizer, Chem. – Asian J., 2006, 1, 370–383 CrossRef CAS PubMed.
- S. Hirai, M. Utsugi, M. Iwamoto and M. Nakada, Formal total synthesis of (−)-taxol through Pd-catalyzed eight-membered carbocyclic ring formation, Chem. – Eur. J., 2015, 21, 355–359 CrossRef CAS PubMed.
- K. Fukaya, Y. Tanaka, A. C. Sato, K. Kodama, H. Yamazaki, T. Ishimoto, Y. Nozaki, Y. M. Iwaki, Y. Yuki, K. Umei, T. Sugai, Y. Yamaguchi, A. Watanabe, T. Oishi, T. Sato and N. Chida, Synthesis of Paclitaxel. 1. Synthesis of the ABC ring of Paclitaxel by SmI2-mediated cyclization, Org. Lett., 2015, 17, 2570–2573 CrossRef CAS PubMed.
- K. Fukaya, K. Kodama, Y. Tanaka, H. Yamazaki, T. Sugai, Y. Yamaguchi, A. Watanabe, T. Oishi, T. Sato and N. Chida, Synthesis of paclitaxel. 2. Construction of the ABCD ring and formal synthesis, Org. Lett., 2015, 17, 2574–2577 CrossRef CAS PubMed.
- S. Jennewein, M. R. Wildung, M. Chau, K. Walker and R. Croteau, Random sequencing of an induced Taxus cell cDNA library for identification of clones involved in Taxol biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9149–9154 CrossRef CAS PubMed.
- R. Croteau, R. E. Ketchum, R. M. Long, R. Kaspera and M. R. Wildung, Taxol biosynthesis and molecular genetics, Phytochem.
Rev., 2006, 5, 75–97 CrossRef CAS PubMed.
- Y. Kanda, H. Nakamura, S. Umemiya, R. K. Puthukanoori, V. R. M. Appala, G. K. Gaddamanugu, B. R. Paraselli and P. S. Baran, Two-phase synthesis of taxol, J. Am. Chem. Soc., 2020, 142, 10526–10533 CrossRef CAS PubMed.
- Y. Kanda, Y. Ishihara, N. C. Wilde and P. S. Baran, Two-phase total synthesis of taxanes: tactics and strategies, J. Org. Chem., 2020, 85, 10293–10320 CrossRef CAS PubMed.
- A. Mendoza, Y. Ishihara and P. S. Baran, Scalable enantioselective total synthesis of taxanes, Nat. Chem., 2012, 4, 21–25 CrossRef CAS PubMed.
- D. G. I. Kingston, Taxol: The chemistry and structure-activity relationships of a novel anticancer agent, Trends Biotechnol., 1994, 12, 222–227 CrossRef CAS PubMed.
- W.-S. Fang and X.-T. Liang, Recent progress in structure activity relationship and mechanistic studies of taxol analogues, Mini-Rev. Med. Chem., 2005, 5, 1–12 CrossRef CAS PubMed.
- D. G. I. Kingston, The shape of things to come: structural and synthetic studies of taxol and related compounds, Phytochemistry, 2007, 68, 1844–1854 CrossRef CAS PubMed.
- Y. Fu, S. Li, Y. Zu, G. Yang, Z. Yang, M. Luo, S. Jiang, M. Wink and T. Efferth, Medicinal chemistry of paclitaxel and its analogues, Curr. Med. Chem., 2009, 16, 3966–3985 CrossRef CAS PubMed.
- J. Żwawiak and L. Zaprutko, A brief history of taxol, J. Med. Sci., 2014, 83, 47–52 CrossRef.
- D. G. I. Kingston, M. D. Chordia, P. G. Jagtap, J. Liang, Y.-C. Shen, B. H. Long, C. R. Fairchild and K. A. Johnston, Synthesis and biological evaluation of 1-deoxypaclitaxel analogues, J. Org. Chem., 1999, 64, 1814–1822 CrossRef CAS PubMed.
- H. Yuan, C. R. Fairchild, X. Liang and D. G. I. Kingston, Synthesis and biological activity of C-6 and C-7 modified paclitaxels, Tetrahedron, 2000, 56, 6407–6414 CrossRef CAS.
- Q. Cheng, T. Oritani, T. Horiguchi, T. Yamada and Y. Mong, Synthesis and biological evaluation of novel 9-functional heterocyclic coupled 7-deoxy-9-Dihydropaclitaxel analogue, Bioorg. Med. Chem. Lett., 2000, 10, 517–521 CrossRef CAS PubMed.
- L. L. Klein, Synthesis of 9-dihydrotaxol: a novel bioactive taxane, Tetrahedron Lett., 1993, 34, 2047–2050 CrossRef CAS.
- H. Ge, V. Vasandani, J. K. Huff, K. L. Audus, R. H. Himes, A. Seelig and G. I. Georg, Synthesis and interactions of 7-deoxy-, 10-deacetoxy, and 10-deacetoxy-7-deoxypaclitaxel with NCI/ADR-RES cancer cells and bovine brain microvessel endothelial cells, Bioorg. Med. Chem. Lett., 2006, 16, 433–436 CrossRef CAS PubMed.
- S. H. Chen, J. M. Wei and V. Farina, Taxol structure-activity relationships: synthesis and biological evaluation of 2-deoxytaxol, Tetrahedron Lett., 1993, 34, 3205–3206 CrossRef CAS.
- A. Datta, L. R. Jayasinghe and G. I. Georg, 4-Deacetyltaxol and 10-acetyl-4-deacetyltaxotere: synthesis and biological evaluation, J. Med. Chem., 1994, 37, 4258–4260 CrossRef CAS PubMed.
- K. A. Neidigh, M. M. Gharpure, J. M. Rimoldi, D. G. I. Kingston, Y. Q. Jiang and E. Hamel, Synthesis and biological evaluation of 4-deacetylpaclitaxel, Tetrahedron Lett., 1994, 35, 6839–6842 CrossRef CAS.
- R. Marder-Karsenti, J. Dubois, L. Bricard, D. Guénard and F. Guéritte-Voegelein, Synthesis and biological evaluation of D-ring-modified taxanes: 15(20)-azadocetaxel analogs, J. Org. Chem., 1997, 62, 6631–6637 CrossRef CAS.
- A. A. L. Gunatilaka, F. D. Ramdayal, M. H. Sarragiotto, D. G. I. Kingston, D. L. Sackett and E. Hamel, Synthesis and biological evaluation of novel paclitaxel (taxol) D-ring modified analogues, J. Org. Chem., 1999, 64, 2694–2703 CrossRef CAS PubMed.
- I. Ojima, J. C. Slater, E. Michaud, S. D. Kuduk, P.-Y. Bounaud, P. Vrignaud, M.-C. Bissery, J. M. Veith, P. Pera and R. J. Bernacki, Syntheses and structure−activity relationships of the second-generation antitumor taxoids: exceptional activity against drug-resistant cancer cells, J. Med. Chem., 1996, 39, 3889–3896 CrossRef CAS PubMed.
- S. M. Ali, M. Z. Hoemann, J. Aube, L. A. Mitscher, G. I. Georg, R. McCall and L. R. Jayasinghe, Novel cytotoxic 3′-(tert-Butyl) 3′-diphenyl analogs of paclitaxel and docetaxel, J. Med. Chem., 1995, 38, 3821–3828 CrossRef CAS PubMed.
- T. C. Boge, R. H. Himes, D. G. V. Velde and G. I. Georg, The effect of the aromatic rings of taxol on biological activity and solution conformation: synthesis and evaluation of saturated taxol and taxotere analogs, J. Med. Chem., 1994, 37, 3337–3343 CrossRef CAS PubMed.
- I. Ojima and S. Lin, Efficient asymmetric syntheses of β-lactams bearing a cyclopropane or an epoxide moiety and their application to the syntheses of novel isoserines and taxoids, J. Org. Chem., 1998, 63, 224–225 CrossRef CAS.
- I. Ojima, T. Inoue and S. Chakravarty, Enantiopure fluorine-containing taxoids: potent anticancer agents and versatile probes for biomedical problems, J. Fluor. Chem., 1999, 97, 3–10 CrossRef CAS.
- J. Wang, C. Xu, Y. K. Wong, Y. Li, F. Liao, T. Jiang and Y. Tu, Artemisinin, the magic drug discovered from traditional Chinese medicine, Engineering, 2019, 5, 32–39 CrossRef CAS.
- A. Robert, F. Benoit-Vical, C. Claparols and B. Meunier, The antimalarial drug artemisinin alkylates heme in infected mice, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13676–13680 CrossRef CAS PubMed.
- F. T. Aweeka and P. I. German, Clinical pharmacology of artemisinin-based combination therapies, Clin. Pharmacokinet., 2008, 47, 91–102 CrossRef CAS PubMed.
- L. Cui and X. Z. Su, Discovery, mechanisms of action and combination therapy of artemisinin, Expert Rev. Anti-Infect. Ther., 2009, 7, 999–1013 CrossRef CAS PubMed.
- J. Wang, C.-J. Zhang, W. N. Chia, C. C. Y. Loh, Z. Li, Y. M. Lee, Y. He, L.-X. Yuan, T. K. Lim, M. Liu, C. X. Liew, Y. Q. Lee, J. Zhang, N. Lu, C. T. Lim, Z.-C. Hua, B. Liu, H.-M. Shen, K. S. W. Tan and Q. Lin, Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum, Nat. Commun., 2015, 6, 10111–10122 CrossRef CAS PubMed.
- L. Tilley, J. Straimer, N. F. Gnädig, S. A. Ralph and D. A. Fidock, Artemisinin action and resistance in plasmodium falciparum, Trends Parasitol., 2016, 32, 282–296 CrossRef PubMed.
- M. Ouji, J.-M. Augereau, L. Paloque and F. Benoit-Vical, Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination, Parasite, 2018, 25, 1–12 CrossRef PubMed.
- C. H. Sibley, Understanding artemisinin resistance, Science, 2015, 347, 373–374 CrossRef CAS PubMed.
- A. A. Mbengue, S. S. Bhattacharjee, T. T. Pandharkar, H. Liu, G. Estiu, R. V. Stahelin, S. S. Rizk, D. L. Njimoh, Y. Ryan, K. Chotivanich, C. Nguon, M. Ghorbal, J. J. Lopez-Rubio, M. Pfrender, S. Emrich, N. Mohandas, A. M. Dondorp, O. Wiest and K. Haldar, A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria, Nature, 2015, 520, 683–687 CrossRef CAS PubMed.
- Z. Wang, L. Yang, X. Yang and X. Zhang, Advances in the chemical synthesis of artemisinin, Synth. Commun., 2014, 44, 1987–2003 CrossRef CAS.
- S. K. Sahu, P. K. Behera, P. Choudhury, S. Panda and L. Rout, Strategy and problems for synthesis of antimalaria artemisinin (qinghaosu), ChemistrySelect, 2020, 5, 12333–12344 CrossRef CAS.
- G. Schmid and W. Hofheinz, Total synthesis of qinghaosu, J. Am. Chem. Soc., 1983, 105, 624–625 CrossRef CAS.
- X.-X. Xu, J. Zhu, D.-Z. Huang and W.-S. Zhou, Total synthesis of arteannuin and deoxyarteannuin, Tetrahedron, 1986, 42, 819–828 CrossRef CAS.
- M. A. Avery, C. Jennings-White and W. K. M. Chong, The total synthesis of (+)-artemisinin and (+)-9-desmethyltemesinin, Tetrahedron Lett., 1987, 20, 4629–4632 CrossRef.
- M. A. Avery, W. K. M. Chong and C. Jennings-White, Stereoselective total synthesis of (+)-artemisinin, the antimalarial constituent of Artemisia annua L, J. Am. Chem. Soc., 1992, 114, 974–979 CrossRef CAS.
- T. Ravindranathan, M. A. Kumar, R. B. Menon and S. V. Hiremath, Stereoselective synthesis of artemisinin, Tetrahedron Lett., 1990, 31, 755–758 CrossRef CAS.
- H. J. Liu, W. L. Yeh and S. Yeu Chew, A total synthesis of the antimarial natural product (+)-qinghaosu, Tetrahedron Lett., 1993, 34, 4435–4438 CrossRef CAS.
- J. B. Bhonsle, B. Pandey, V. H. Deshpande and T. Ravindranathan, New synthetic strategies towards (+)-artemisinin, Tetrahedron Lett., 1994, 35, 5489–5492 CrossRef CAS.
- M. G. Constantino, M. B. Jr., G. V. J. Silva and J. Zukerman-Schpector, A novel asymmetric total synthesis of (+)-artemisinin, Synth. Commun., 1196, 26, 321–329 CrossRef.
- J. S. Yadav, R. S. Babu and G. Sabitha, Stereoselective total synthesis of (+)-artemisinin, Tetrahedron Lett., 2003, 44, 387–389 CrossRef CAS.
- J. S. Yadav, B. Thirupathaiah and P. Srihari, A concise stereoselective total synthesis of (+)-artemisinin, Tetrahedron, 2010, 66, 2005–2009 CrossRef CAS.
- C. Zhu and S. P. Cook, A concise synthesis of (+)-artemisinin, J. Am. Chem. Soc., 2012, 134, 13577–13579 CrossRef CAS PubMed.
- J. Krieger, T. Smeilus, M. Kaiser, E.-J. Seo, T. Efferth and A. Giannis, Total synthesis and biological investigation of (−)-artemisinin: the antimalarial activity of artemisinin is not stereospecific, Angew. Chem., Int. Ed., 2018, 57, 8293–8296 CrossRef CAS PubMed.
- J. N. Cumming, D. Wang, S. B. Park, T. A. Shapiro and G. H. Posner, Design, synthesis, derivatization, and structure−activity relationships of simplified, tricyclic, 1,2,4-trioxane alcohol analogues of the antimalarial artemisinin, J. Med. Chem., 1998, 41, 952–964 CrossRef CAS PubMed.
- M. A. Avery, M. Alvim-Gaston, C. R. Rodrigues, E. J. Barreiro, F. E. Cohen, Y. A. Sabnis and J. R. Woolfrey, Structure−activity relationships of the antimalarial agent artemisinin. 6. The development of predictive in vitro potency models using CoMFA and HQSAR methodologies, J. Med. Chem., 2002, 45, 292–303 CrossRef CAS PubMed.
- M. A. Avery, S. Mehrotra, T. L. Johnson, J. D. Bonk, J. A. Vroman and R. Miller, Structure−activity relationships of the antimalarial agent artemisinin. 5. Analogs of 10-deoxoartemisinin substituted at C-3 and C-9, J. Med. Chem., 1996, 39, 4149–4155 CrossRef CAS PubMed.
- M. A. Avery, P. Fan, J. M. Karle, J. D. Bonk, R. Miller and D. K. Goins, Structure−activity relationships of the antimalarial agent artemisinin. 3. Total synthesis of (+)-13-carbaartemisinin and related tetra- and tricyclic structures, J. Med. Chem., 1996, 39, 1885–1897 CrossRef CAS PubMed.
- M. A. Avery, S. Mehrotra, J. D. Bonk, J. A. Vroman, D. K. Goins and R. Miller, Structure−activity relationships of the antimalarial agent artemisinin. 4. Effect of substitution at C-3, J. Med. Chem., 1996, 39, 2900–2906 CrossRef CAS PubMed.
- M. A. Avery, J. D. Bonk, W. K. M. Chong, S. Mehrotra, R. Miller, W. Milhous, D. K. Goins, S. Venkatesan and C. Wyandt, Structure-activity relationships of the antimalarial agent artemisinin. 2. Effect of heteroatom substitution at O-11: synthesis and bioassay of N-alkyl-11-aza-9-desmethylartemisinins, J. Med. Chem., 1995, 38, 5038–5044 CrossRef CAS PubMed.
- M. A. Avery, F. Gao, W. K. M. Chong, S. Mehrotra and W. K. Milhous, Structure-activity relationships of the antimalarial agent artemisinin. 1. Synthesis and comparative molecular field analysis of C-9 analogs of artemisinin and 10-deoxoartemisinin, J. Med. Chem., 1993, 36, 4264–4275 CrossRef CAS PubMed.
- M. K. Tiwari and S. Chaudhary, Artemisinin-derived antimalarial endoperoxides from bench-side to bed-side: Chronological advancements and future challenges, Med. Res. Rev., 2020, 40, 1220–1275 CrossRef CAS PubMed.
- R. K. Haynes, B. Fugmann, J. Stetter, K. Rieckmann, H.-D. Heilmann, H.-W. Chan, M.-K. Cheung, W.-L. Lam, H.-N. Wong, S. L. Croft, L. Vivas, L. Rattray, L. Stewart, W. Peters, B. L. Robinson, M. D. Edstein, B. Kotecka, D. E. Kyle, B. Beckermann, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke, U. Wollborn, K. Schmeer and A. Römer, Artemisone—a highly active antimalarial drug of the artemisinin class, Angew. Chem., Int. Ed., 2006, 45, 2082–2088 CrossRef CAS PubMed.
-
Y. Tu, From Artemisia annua L. to artemisinins: the discovery and development of artemisinins and antimalarial agents, Academic Press, 2017 Search PubMed.
- M. A. Avery, F. Gao, W. K. M. Chong, T. F. Hendrickson, W. D. Inman and P. Crews, Synthesis, conformational analysis, and antimalarial activity of tricyclic analogs of artemisinin, Tetrahedron, 1994, 50, 957–972 CrossRef CAS.
- T. N. C. Wells, R. H. V. Huijsduijnen and W. C. V. Voorhis, Malaria medicines: a glass half full?, Nat. Rev. Drug Discovery, 2015, 14, 424–442 CrossRef CAS PubMed.
-
S. L. Rawe, in Antimalarial Agents, Elsevier Ltd, 2020, pp. 99–132 Search PubMed.
- C. Cuevas and A. Francesch, Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem, Nat. Prod. Rep., 2009, 26, 322–337 RSC.
- V. H. Le, M. Inai, R. M. Williams and T. Kan, Ecteinascidins. A review of the chemistry, biology and clinical utility of potent tetrahydroisoquinoline antitumor antibiotics, Nat. Prod. Rep., 2015, 32, 328–347 RSC.
- S. Yuan, Y.-Q. Luo, J.-H. Zuo, H. Liu, F. Li and B. Yu, New drug approvals for 2020: Synthesis and clinical applications, Eur. J. Med. Chem., 2021, 215, 113284–113317 CrossRef CAS PubMed.
- K. K. C. Liu, S. M. Sakya, C. J. O'Donnell and J. Li, Synthetic approaches to the 2007 new drugs, Mini-Rev. Med. Chem., 2008, 8, 1526–1548 CrossRef CAS PubMed.
- K. L. Rinehart, T. G. Holt, N. L. Fregeau, J. G. Stroh, P. A. Keifer, F. Sun, L. H. Li and D. G. Martin, Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata, J. Org. Chem., 1990, 55, 4512–4515 CrossRef CAS.
- R. Sakai, K. L. Rinehart, Y. Guan and A. H.-J. Wang, Additional antitumor ecteinascidins from a Caribbean tunicate: crystal structures and activities in vivo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 11456–11460 CrossRef CAS PubMed.
- Y. Pommier, G. Kohlhagen, C. Bailly, M. Waring, A. Mazumder and K. W. Kohn, DNA sequence- and structure-selective alkylation
of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the caribbean tunicate Ecteinascidia turbinata, Biochemistry, 1996, 35, 13303–13309 CrossRef CAS PubMed.
- B. M. Moore, F. C. Seaman and L. H. Hurley, NMR-based model of an ecteinascidin 743-DNA adduct, J. Am. Chem. Soc., 1997, 119, 5475–5476 CrossRef CAS.
- B. M. Moore, F. C. Seaman, R. T. Wheelhouse and L. H. Hurley, Mechanism for the catalytic activation of ecteinascidin 743 and its subsequent alkylation of guanine N2, J. Am. Chem. Soc., 1998, 120, 2490–2491 CrossRef CAS.
- R. Garcia-Nieto, I. Manzanares, C. Cuevas and F. Gago, Bending of DNA upon binding of ecteinascidin 743 and phthalascidin 650 studied by unrestrained molecular dynamics simulations, J. Am. Chem. Soc., 2000, 122, 7172–7182 CrossRef CAS.
- M. Zewail-Foote and L. H. Hurley, Ecteinascidin 743: a minor groove alkylator that bends DNA toward the major groove, J. Med. Chem., 1999, 42, 2493–2497 CrossRef CAS PubMed.
- A. B. Herrero, C. Martin-Castellanos, E. Marco, F. Gago and S. Moreno, Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin, Cancer Res., 2006, 66, 8155–8162 CrossRef CAS PubMed.
- D. G. Soares, A. E. Escargueil, V. Poindessous, A. Sarasin, A. D. Gramont, D. Bonatto, J. A. P. Henriques and A. K. Larsen, Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13062–13067 CrossRef PubMed.
- E. J. Corey, D. Y. Gin and R. S. Kania, Enantioselective total synthesis of ecteinascidin 743, J. Am. Chem. Soc., 1996, 118, 9202–9203 CrossRef CAS.
- C. Cuevas, M. Perez, M. J. Martin, J. L. Chicharro, C. Fernandez-Rivas, M. Flores, A. Francesch, P. Gallego, M. Zarzuelo, F. D. L. Calle, J. Garcia, C. Polanco, I. Rodriguez and I. Manzanares, Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B, Org. Lett., 2000, 2, 2545–2548 CrossRef CAS PubMed.
- E. J. Martinez and E. J. Corey, A new, more efficient, and effective process for the synthesis of a key pentacyclic intermediate for production of ecteinascidin and phthalascidin antitumor agents, Org. Lett., 2000, 2, 993–996 CrossRef CAS PubMed.
- A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan and T. Fukuyama, Total synthesis of ecteinascidin 743, J. Am. Chem. Soc., 2002, 124, 6552–6554 CrossRef CAS PubMed.
- F. Kawagishi, T. Toma, T. Inui, S. Yokoshima and T. Fukuyama, Total synthesis of ecteinascidin 743, J. Am. Chem. Soc., 2013, 135, 13684–13687 CrossRef CAS PubMed.
- J. Chen, X. Chen, M. Bois-Choussy and J. Zhu, Total synthesis of ecteinascidin 743, J. Am. Chem. Soc., 2006, 128, 87–89 CrossRef CAS PubMed.
- S. Zheng, C. Chan, T. Furuuchi, B. J. D. Wright, B. Zhou, J. Guo and S. J. Danishefsky, Stereospecific formal total synthesis of ecteinascidin 743, Angew. Chem., Int. Ed., 2006, 45, 1754–1759 CrossRef CAS PubMed.
- D. Fishlock and R. M. Williams, Synthetic studies on Et-743. Assembly of the pentacyclic core and a formal total synthesis, J. Org. Chem., 2008, 73, 9594–9600 CrossRef CAS PubMed.
- W. He, Z. Zhang and D. Ma, A scalable total synthesis of the antitumor agents Et-743 and lurbinectedin, Angew. Chem., Int. Ed., 2019, 58, 3972–3975 CrossRef CAS PubMed.
- E. J. Martinez, T. Owa, S. L. Schreiber and E. J. Corey, Phthalascidin, a synthetic antitumor agent with potency and mode of action comparable to ecteinascidin 743, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3496–3501 CrossRef CAS PubMed.
- K. Suwanborirux, K. Charupant, S. Amnuoypol, S. Pummangura, A. Kubo and N. Saito, Ecteinascidins 770 and 786 from the Thai Tunicate Ecteinascidia thurstoni, J. Nat. Prod., 2002, 65, 935–937 CrossRef CAS PubMed.
- P. Puthongking, C. Patarapanich, S. Amnuoypol, K. Suwanborirux, A. Kubo and N. Saito, Chemistry of ecteinascidins. Part 2. Preparation of 6′-O-acyl derivatives of stable ecteinascidin and evaluation of cytotoxicity, Chem. Pharm. Bull., 2006, 54, 1010–1016 CrossRef CAS PubMed.
- P. Saktrakulkla, S. Toriumi, M. Tsujimoto, C. Patarapanich, K. Suwanborirux and N. Saito, Chemistry of ecteinascidins. Part 3: preparation of 2′-N-acyl derivatives of ecteinascidin 770 and evaluation of cytotoxicity, Bioorg. Med. Chem., 2011, 19, 4421–4436 CrossRef CAS PubMed.
- M. Tsujimoto, W. Lowtangkitcharoen, N. Mori, W. Pangkruang, P. Putongking, K. Suwanborirux and N. Saito, Chemistry of ecteinascidins. Part 4: preparation of 2′-N-acyl ecteinascidin 770 derivatives with improved cytotoxicity profiles, Chem. Pharm. Bull., 2013, 61, 1052–1064 CrossRef CAS PubMed.
- R. Toyoshima, N. Mori, T. Suzuki, W. Lowtangkitcharoen, K. Suwanborirux and N. Saito, Chemistry of ecteinascidins. Part 5: an additional proof of cytotoxicity evaluation of ecteinascidin 770 derivatives, Chem. Pharm. Bull., 2016, 64, 966–969 CrossRef CAS PubMed.
- N. Saito, Chemical research on antitumor isoquinoline marine natural products and related compounds, Chem. Pharm. Bull., 2021, 69, 155–177 CrossRef CAS PubMed.
- M. D. Lee, T. S. Dunne, M. M. Siegel, C. C. Chang, G. O. Morton and D. B. Borders, Calichemicins, a novel family of antitumor antibiotics. 1. Chemistry and partial structure of calichemicin .gamma.1I, J. Am. Chem. Soc., 1987, 109, 3464–3466 CrossRef CAS.
- M. D. Lee, T. S. Dunne, C. C. Chang, G. A. Ellestad, M. M. Siegel, G. O. Morton, W. J. McGahren and D. B. Borders, Calichemicins, a novel family of antitumor antibiotics. 2. Chemistry and structure of calichemicin .gamma.1I, J. Am. Chem. Soc., 1987, 109, 3466–3468 CrossRef CAS.
- M. D. Lee, J. K. Manning, D. R. Williams, N. A. Kuck, R. T. Testa and D. B. Borders, Calicheamicins, a novel family of antitumor antibiotics, J. Antibiot., 1989, 42, 1070–1087 CrossRef CAS PubMed.
- K. Edo, M. Mizugaki, Y. Koide, H. Seto, K. Furihata, N. Ōtake and N. Ishida, The structure of neocarzinostatin chromophore possessing a novel bicyclo-[7,3,0]dodecadiyne system, Tetrahedron Lett., 1985, 26, 331–334 CrossRef CAS.
- J. Golik, G. Dubay, G. Groenewold, H. Kawaguchi, M. Konishi, B. Krishnan, H. Ohkuma, K. Saitoh and T. W. Doyle, Esperamicins, a novel class of potent antitumor antibiotics. 3. Structures of esperamicins A1, A2, and A1b, J. Am. Chem. Soc., 1987, 109, 3462–3464 CrossRef CAS.
- M. Konishi, H. Ohkuma, K. Matsumoto, T. Tsuno, H. Kamei, T. Miyaki, T. Oki, H. Kawaguchi, G. D. Vanduyne and J. Clardy, Dynemicin A, a novel antibiotic with the anthraquinone and 1,5-diyn-3-ene subunit, J. Antibiot., 1989, 42, 1449–1452 CrossRef CAS PubMed.
- S. Walker, R. Landovitz, W. D. Ding, G. A. Ellestad and D. Kahne, Cleavage behavior of calicheamicin gamma 1 and calicheamicin T, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4608–4612 CrossRef CAS PubMed.
- R. Gébleux and G. Casi, Antibody-drug conjugates: Current status and future perspectives, Pharmacol. Ther., 2016, 167, 48–59 CrossRef PubMed.
- S. Yaghoubi, M. H. Karimi, M. Lotfinia, T. Gharibi, M. Mahi-Birjand, E. Kavi, F. Hosseini, K. S. Sepehr, M. Khatami, N. Bagheri and M. Abdollahpour-Alitappeh, Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy, J. Cell. Physiol., 2020, 235, 31–64 CrossRef CAS PubMed.
- K. C. Nicolaou, C. W. Hummel, E. N. Pitsinos, M. Nakada, A. L. Smith, K. Shibayama and H. Saimoto, Total synthesis of calicheamicin .gamma.1I, J. Am. Chem. Soc., 1992, 114, 10082–10084 CrossRef CAS.
- R. D. Groneberg, T. Miyazaki, N. A. Stylianides, T. J. Schulze, W. Stahl, E. P. Schreiner, T. Suzuki, Y. Iwabuchi, A. L. Smith and K. C. Nicolaou, Total synthesis of calicheamicin .gamma.1I. 1. Synthesis of the oligosaccharide fragment, J. Am. Chem. Soc., 1993, 115, 7593–7611 CrossRef CAS.
- A. L. Smith, E. N. Pitsinos, C. K. Hwang, Y. Mizuno, H. Saimoto, G. R. Scarlato, T. Suzuki and K. C. Nicolaou, Total synthesis of calicheamicin .gamma.1I. 2. Development of an enantioselective route to (-)-calicheamicinone, J. Am. Chem. Soc., 1993, 115, 7612–7624 CrossRef CAS.
- K. C. Nicolaou, C. W. Hummel, M. Nakada, K. Shibayama, E. N. Pitsinos, H. Saimoto, Y. Mizuno, K. U. Baldenius and A. L. Smith, Total synthesis of calicheamicin .gamma.1I. 3. The final stages, J. Am. Chem. Soc., 1993, 115, 7625–7635 CrossRef CAS.
- M. P. Cabal, R. S. Coleman and S. J. Danishefsky, Total synthesis of calicheamicinone: a solution to the problem of the elusive urethane, J. Am. Chem. Soc., 1990, 112, 3253–3255 CrossRef CAS.
- J. N. Haseltine, M. P. Cabal, N. B. Mantlo, N. Iwasawa, D. S. Yamashita, R. S. Coleman, S. J. Danishefsky and G. K. Schulte, Total synthesis of calicheamicinone: new arrangements for actuation of the reductive cycloaromatization of aglycon congeners, J. Am. Chem. Soc., 1991, 113, 3850–3866 CrossRef CAS.
- S. A. Hitchcock, S. H. Boyer, M. Y. Chu-Moyer, S. H. Olson and S. J. Danishefsky, A convergent total synthesis of calicheamicin γ1′, Angew. Chem., Int. Ed., 1994, 33, 858–862 CrossRef.
- R. L. Halcomb, S. H. Boyer, M. D. Wittman, S. H. Olson, D. J. Denhart, K. K. C. Liu and S. J. Danishefsky, Studies related to the carbohydrate sectors of esperamicin and calicheamicin: definition of the stability limits of the esperamicin domain and fashioning of a glycosyl donor from the calicheamicin domain, J. Am. Chem. Soc., 1995, 117, 5720–5749 CrossRef CAS.
- S. A. Hitchcock, M. Y. Chu-Moyer, S. H. Boyer, S. H. Olson and S. J. Danishefsky, A remarkable glycosidation reaction: the total synthesis of calicheamicin .gamma.1I, J. Am. Chem. Soc., 1995, 117, 5750–5756 CrossRef CAS.
- N. Oku, S. Matsunaga and N. Fusetani, Shishijimicins A−C, novel enediyne antitumor antibiotics from the ascidian didemnum proliferum1, J. Am. Chem. Soc., 2003, 125, 2044–2045 CrossRef CAS PubMed.
- L. A. McDonald, T. L. Capson, G. Krishnamurthy, W.-D. Ding, G. A. Ellestad, V. S. Bernan, W. M. Maiese, P. Lassota, R. A. Kramer and C. M. Ireland, Namenamicin, a new enediyne antitumor antibiotic from the marine ascidian polysyncraton lithostrotum, J. Am. Chem. Soc., 1996, 118, 10898–10899 CrossRef CAS.
- K. C. Nicolaou, Z. Lu, R. Li, J. R. Woods and T.-I. Sohn, Total synthesis of shishijimicin A, J. Am. Chem. Soc., 2015, 137, 8716–8719 CrossRef CAS PubMed.
- K. C. Nicolaou, R. Li, Z. Lu, E. N. Pitsinos, L. B. Alemany, M. Aujay, C. Lee, J. Sandoval and J. Gavrilyuk, Streamlined total synthesis of shishijimicin A and its application to the design, synthesis, and biological evaluation of analogues thereof and practical syntheses of PhthNSSMe and related sulfenylating reagents, J. Am. Chem. Soc., 2018, 140, 12120–12136 CrossRef CAS PubMed.
- K. C. Nicolaou, R. Li, Z. Lu, E. N. Pitsinos and L. B. Alemany, Total synthesis and full structural assignment of namenamicin, J. Am. Chem. Soc., 2018, 140, 8091–8095 CrossRef CAS PubMed.
- K. C. Nicolaou, R. Li, Q. Chen, Z. Lu, E. N. Pitsinos, A. Schammel, B. Lin, C. Gu, H. Sarvaiya, R. Tchelepi, A. Valdiosera, J. Clubb, N. Barbour, V. Sisodiya, J. Sandoval, C. Lee, M. Aujay and J. Gavrilyuk, Synthesis and biological evaluation of shishijimicin A-type linker-drugs and antibody–drug conjugates, J. Am. Chem. Soc., 2020, 142, 12890–12899 CrossRef CAS PubMed.
- A. M. Gillespie, T. J. Broadhead, S. Y. Chan, J. Owen, A. P. Farnsworth, M. Sopwith and R. E. Coleman, Phase I open study of the effects of ascending doses of the cytotoxic immunoconjugate CMB-401 (hCTMO1-calicheamicin) in patients with epithelial ovarian cancer, Ann. Oncol., 2000, 11, 735–741 CrossRef CAS PubMed.
- L. M. Hinman, P. R. Hamann, R. Wallace, A. T. Menendez, F. E. Durr and J. Upeslacis, Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics, Cancer Res., 1993, 53, 3336–3342 CAS.
- K. C. Nicolaou, T. Li, M. Nakada, C. W. Hummel, A. Hiatt and W. Wrasidlo, Calicheamicin θ1′: A rationally designed molecule with extremely potent and selective DNA cleaving properties and apoptosis inducing activity, Angew. Chem., Int. Ed., 1994, 33, 183–186 CrossRef.
- H. N. Lode, R. A. Reisfeld, R. Handgretinger, K. C. Nicolaou, G. Gaedicke and W. Wrasidlo, Targeted therapy with a novel enediyene antibiotic calicheamicin ϑI1 effectively suppresses growth and dissemination of liver metastases in a syngeneic model of murine neuroblastoma, Cancer Res., 1998, 58, 2925–2928 CAS.
- K. M. Bernt, A. Prokop, N. Huebener, G. Gaedicke, W. Wrasidlo and H. N. Lode, Eradication of CD19+ leukemia by targeted calicheamicin θ, Bioconjugate Chem., 2009, 20, 1587–1594 CrossRef CAS PubMed.
-
G. Chen, B. K. Alisher, H. Zhang, T. Zhu and Z. Miao, Calicheamicin antibody drug conjugates linking and amidoacetyl group to a sugar moiety on calicheamicin, WO/2017/172907, 2017.
-
T. Pillow, Calicheamicin-antibody-drug conjugates and methods of use, WO/2017/068511, 2017.
- A. D. Ricart, Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin, Clin. Cancer Res., 2011, 17, 6417–6427 CrossRef CAS PubMed.
Footnote |
| † These authors made equal contribution. |
|
| This journal is © the Partner Organisations 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab