
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the ring-opening polymerization of sulfur-containing monomers
Vishal B.
Purohit
a,
Marlena
Pięta
a,
Joanna
Pietrasik
 b and
Christopher M.
Plummer
*a
b and
Christopher M.
Plummer
*a
aInternational Centre for Research on Innovative Biobased Materials (ICRI-BioM)—International Research Agenda, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland. E-mail: christopher.plummer@p.lodz.pl
bInstitute of Polymer and Dye Technology, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 9th August 2022
Abstract
Inspired by the uniqueness of ring-opening polymerization and the ubiquity of sulfur-containing heterocycles, much attention has been paid to the synthesis of sulfur-containing polymers as they can provide novel materials with a broad range of applications ranging from stimuli responsiveness to (bio)degradability. Within this review article an overview is presented regarding various ROP technologies (ROP/rROP/ROMP) which are further categorized by monomer class, detailing significant developments over the previous decades which cement the importance of sulfur-containing monomers in modern polymer chemistry. This article aims to explore the chemistry involved in the ring-opening polymerization of sulfur-containing cyclic monomers by summarizing the state-of-the-art, as well as providing an overview of the fundamental challenges involved in synthesizing sulfur-containing polymers, including potential future directions.
 Vishal B. Purohit | Vishal Purohit completed his Ph.D. in 2017 under the supervision of Dr K. H. Patel and Prof. D. K. Raval at S. P. University (India), where he studied NHC-transition metal catalyzed C–H activation reactions. Between 2017–2020 he was employed as an Assistant Professor at Shri A. N. Patel PG Institute of Science and Research (India). During 2020–2021, he worked as a Postdoc with Prof. Karol Grela at the University of Warsaw (Poland). In November 2021, he joined the group of Dr Christopher Plummer as a Postdoc at Lodz University of Technology (Poland), working on the synthesis of new monomers for ring-opening polymerization. |
 Marlena Pięta | Marlena Pięta completed her Ph.D. in 2018 at Lodz University of Technology (TUL), Poland in the area of organic synthesis, specifically the synthesis of chiral azaheterocycles with cytotoxic activity. In 2019 she worked as researcher in the process chemistry department in Ryvu Therapeutics, Poland. Since 2020 she has been employed as a post-doctorate at TUL. In 2020–2021 she worked on the synthesis of biologically active compounds and profluorescent probes for ROS in a medicinal chemistry group, and from 2022 onwards she is working on the synthesis of new monomers for radical ring-opening polymerization (rROP) in a polymer chemistry group. |
 Joanna Pietrasik | Joanna Pietrasik received her Ph.D. in 2003 at Lodz University of Technology (TUL), Poland; next she spent 2 years at Carnegie Mellon University (CMU), USA, in the group of Prof. Matyjaszewski as a postdoc. She obtained her habilitation degree in 2014. She now holds the position of professor at TUL. Her research activity is dedicated to polymer synthesis, inorganic particles, surface modifications, as well as hybrid materials and nanocomposite properties. |
 Christopher M. Plummer | Christopher Plummer completed his Ph.D. in 2016 at RMIT University, Australia in the area of organic synthesis, specifically the synthesis of oxygenated heterocycles. Between 2016–2019 he was employed as a post-doctorate at Sun Yat-sen University (SYSU), China working on the post-modification of polyolefins. In 2019–2021 he worked as researcher in an industry-funded position at Aix-Marseille University, France, working on the synthesis of new monomers for radical ring-opening polymerization (rROP). Following this, he took up a position as Junior Principal Investigator at Lodz University of Technology (TUL), Poland, and is a presently a group leader in polymer chemistry. |
1. Introduction
The introduction of sulfur atom(s) into chemical compounds is an active area of research, both in the synthesis of new active pharmaceutical ingredients (APIs),1–3 and in the synthesis of new polymers with enhanced mechanical, electrical, and physicochemical properties.4 As a consequence, the synthesis of sulfur-containing polymers has received considerable attention due to unique properties that sulfur can instil including optical features, chemical resistance, self-healing capabilities, and heavy-metal capturing abilities, among others.5,6 In comparison to a parent material, the inclusion of even small quantities of sulfur atoms can significantly alter the physicochemical properties. Even functional groups can be significantly altered by the inclusion of sulfur atoms. For example, a thioester functional group possesses a weak resonance in comparison to its (oxo)ester counterpart, this phenomenon making thioesters relatively electrophilic and therefore more susceptible to nucleophilic attack. Consequently, relative to polyesters, polythioesters can be degraded by additional mechanisms including aminolysis, trans-thioesterification and oxidative hydrolysis.7Ring-opening polymerization (ROP) is a powerful chain-growth polymerization technique that enables the facile incorporation of heteroatoms into a polymer, facilitating access to a broad range of materials. Over the past forty years, various ROP processes including anionic, cationic, coordination–insertion, organocatalytic and radical methods have been explored to target polymers with specific and controllable properties. Based on the polymerization mechanism, ROP can generally be classified into three primary categories: ring-opening polymerization (ROP), radical ring-opening polymerization (rROP), and ring-opening metathesis polymerization (ROMP).8,9 By the application of ROP and judicious choice of monomer, various sulfur-containing functionalities can be installed into a polymer backbone (Scheme 1).
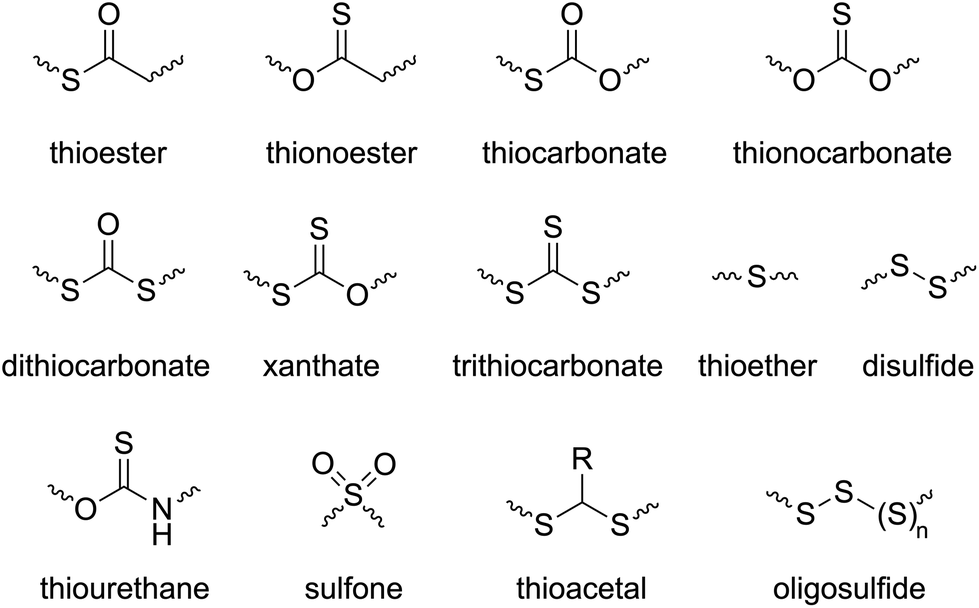 | ||
| Scheme 1 Sulfur-containing functional groups that can be incorporated into a polymer backbone by the proper choice of monomer and ROP method. | ||
For ring-opening polymerization (ROP), the mechanism typically involves either a cationic or anionic initiator species performing the ring-opening of a cyclic monomer to ultimately generate an ionic propagating species. In specific, anionic ROP (AROP) proceeds by nucleophilic attack of an initiator species on a polarized bond of a cyclic monomer to generate an anionic propagating species (Scheme 2a). Conversely, in cationic ROP (CROP), the most polarized functionality within a cyclic monomer attacks an electrophilic initiator species to form a cationic initiating species which then undergoes subsequent ring opening via either an SN1 or SN2 mechanism (Scheme 2b). These processes then repeat until all monomer is consumed or the reaction quenched, forming a polymer. The nature of the initiating and propagating species determines both the rate of polymerization and the control over the macromolecular structure; with carefully tailored conditions allowing for the synthesis of polymer architectures including star-shaped, branched, statistical and block copolymers. Depending on the initiator system and conditions applied, different degrees of activation of either the monomer or propagating chain-end are obtained.10,11 For example, the difference in ROP behaviour between ε-caprolactone (ε-CL) and L-lactide (LLA) towards cationic and anionic initiating species: cationic ROP of ε-CL with diphenyl phosphate (DPP) proceeds with good control at a moderate rate, while poor control is reported for LLA. Conversely, the reversed trend is observed with the anionic catalyst, 1,8-diazabicycloundec-7-ene (DBU).12–14
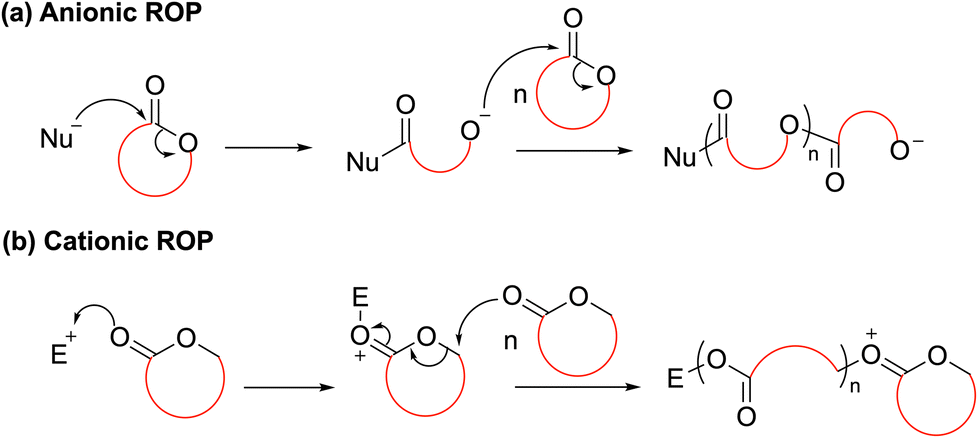 | ||
| Scheme 2 General mechanism of (a) anionic and (b) cationic (SN2) ROP. Note that cationic polymerization can also occur by an SN1 mechanism (not shown). | ||
A mechanistic variation named radical ring-opening polymerization (rROP) is a polymerization technique that involves the polymerization of cyclic monomers via a free-radical pathway.15,16 As a radical polymerization methodology, rROP is thereby compatible with free-radical polymerization (FRP), as well as reversible-deactivation radical polymerization (RDRP)17 methods which provide precise polymerization control. It has been stated that rROP can be considered to combine the advantages of both ROP and FRP in one polymerization system, i.e. it can produce polymers incorporating heteroatoms and/or functional groups in the polymer backbone, alongside the robustness and mild conditions of an FRP process.15,16 A primary challenge of employing rROP monomers is the mechanistic reactivity competition between the radical ring-opening process and the direct radical propagation of the vinylic bond (i.e., propagation without ring-opening). This mechanistic competition frequently gives rise to polymers containing both ring-opened and ring-retained units within the same polymer backbone (Scheme 3).
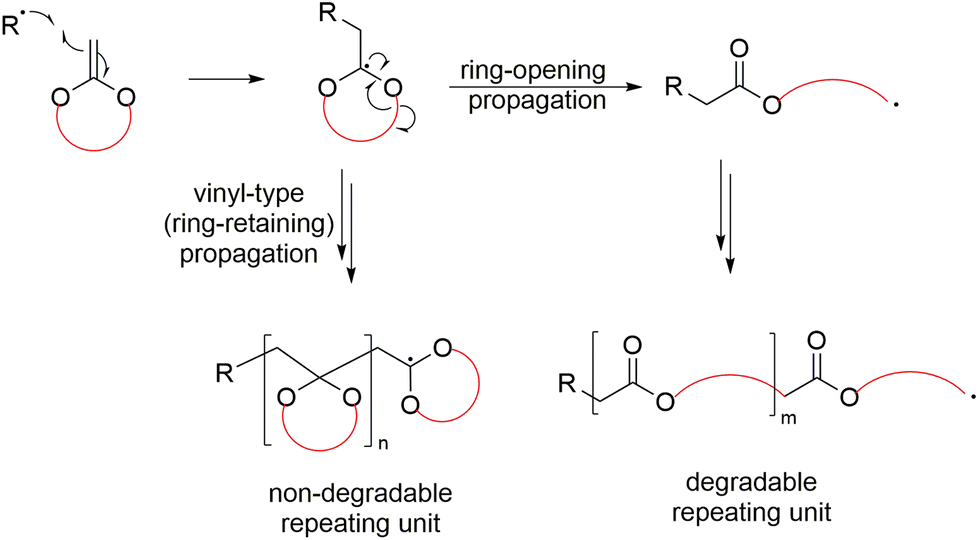 | ||
| Scheme 3 Competition between radical ring-opening and ring-retaining (vinyl type) polymerization. | ||
Another polymerization technique within this family, ring-opening metathesis polymerization (ROMP), is a transition metal alkylidene-triggered process in which cyclic olefins experience ring-opening and are concomitantly joined together to form a polymer chain (Scheme 4).18 Similar to Ziegler–Natta type polymerizations, ROMP is also a chain-growth polymerization that belongs to the family of polyinsertions. Initiation begins with the coordination of a transition metal alkylidene complex to a cyclic olefin to form a metallocyclobutane intermediate, followed by [2 + 2] cycloaddition, and finally cycloreversion to generate a linear propagating species. In principle, all the steps within ROMP are readily reversible and therefore exist as an equilibrium between polymerization and depolymerization.19 Recent developments in ROMP provide a versatile strategy for the construction of functional polymers with controlled molecular weights and architectures, including block copolymers.19–21 Moreover, ROMP displays living polymerization characteristics for many monomer types, particularly strained and bicyclic olefins.22
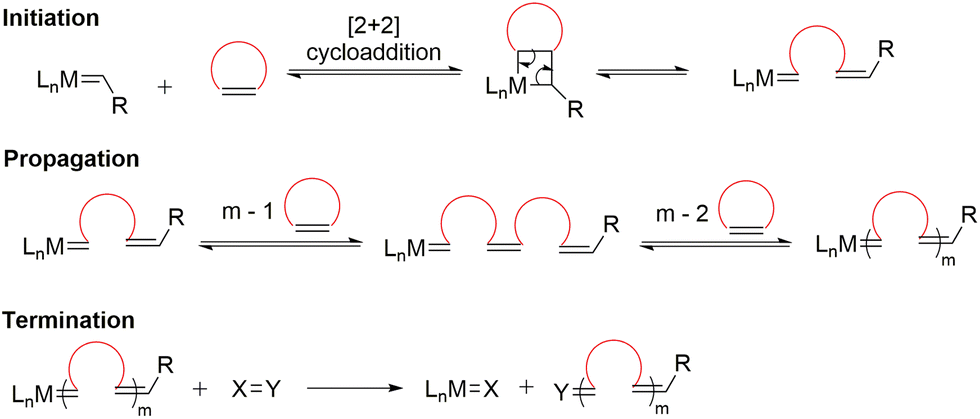 | ||
| Scheme 4 General mechanism of ROMP. | ||
The ROP of sulfur-containing monomers is a broad research area with several recent developments. Accordingly, there have been multiple excellent review articles published that overlap with the topics discussed herein, including polythioesters,23,24 thiocarbonyl monomers,25 sulfur chemistry in polymers,26 and degradable polymers by rROP.15 In consideration of the significance of both sulfur-containing polymers and ROP within the field of polymer science, we herein present an overview of the ROP of sulfur-containing monomers, principally those in which the sulfur–containing moiety is part of the heterocyclic unit. This article categorizes monomers by their primary functional group, and by the polymerization methodology applied i.e. ROP, rROP, ROMP, and additionally includes some miscellaneous polymerization techniques which are not readily classified. In principle, this review primarily considers the relevant literature from the past two decades, but also includes pioneering research to give a thorough overview of the subject.
2. Ring-opening polymerization (ROP)
2.1 Cyclic thioesters
In pioneering work on the ring-opening polymerization of thioesters, in 1968 Overberger and Weise reported the anionic ROP of 7- and 6-membered thiolactones, ε-thiocaprolactone (M1) and δ-thiovalerolactone (M2), respectively, using potassium tert-butoxide (KOtBu) at 155 °C. Interestingly, it was reported that the 5-membered analogue γ-thiobutyrolactone (M3a) was not polymerizable under identical conditions (Scheme 5).27 However, in 2020, Illy et al. reported the anionic ring-opening copolymerization (ROCOP) of M3a with functionalized epoxides using a tBu-P4/BnOH initiator system to obtain poly(ester-alt-thioether)s.28 Upon performing the polymerization at low temperatures (35–50 °C), the ROCOP featured controlled polymerization characteristics and narrow dispersities (Đ < 1.22). This methodology was later extended for the preparation of renewable poly(ester-alt-thioether)s using bio-based thiolactone monomer M3b and various functionalized epoxides.29,30 | ||
| Scheme 5 (a) ROP and (b) monomer structures of thiolactones. | ||
In 2015, Kiesewetter et al. described the thiol-initiated organocatalytic ROP of M1 to obtain polymers with broad molecular weight distributions (Đ = 1.4–1.7) but with unsatisfactory Mn values generally lower than 33.0 kg mol−1 due to trans-thioesterification side reactions.31 The suppression of such undesired trans-thioesterification was later reported achievable using fused or bridged-ring thiolactone monomers. In 2017, Avérous and coworkers reported the enzymatic ROP of M1 mediated by Novozyme 435, yielding the corresponding polythioester with low molar mass and narrow dispersity (Mn = 4.4 kg mol−1, Đ = 1.10).32 The methodology was extended to the ROCOP of M1 with ε-caprolactone by applying the lipase in toluene at 70 °C, yielding statistical copolymers with Mn values ranging from 2.9 to 10.8 kg mol−1. In 2018, Destarac and coworkers studied the alcohol initiated and tin(II) octoate (SnOct2) catalyzed ROCOP of substituted γ-thiolactones (M4a–c) with trimethylene carbonate to synthesize new functional poly(carbonate-co-thioester)s.33 Nevertheless, copolymers with very low molecular weights (Mn = 1.6–6.2 kg mol−1) were obtained, providing evidence to support the hypothesis of ring stability for γ-thiolactones towards ROP.
In an unusual report, Bowman et al. demonstrated the ROP of fused-ring thiolactone M5 to furnish DNA-mimicking polythioesters which displayed dynamic and responsive properties, depolymerization capability, and narrow dispersities in the range of 1.2–1.3.34 However, the highest Mn of the prepared polyM5 was merely 7.6 kg mol−1, with a poor monomer conversion of 27%. Bridged bicyclic thiolactone M6 was also reported to be active towards ROP, providing polymers with crystallinity values that could be readily tuned by polymerization conditions to provide samples with Tm values ranging from 166 to 213 °C, this value being directly related to tacticity which modulates the crystallinity.35PolyM6 was found to be chemically recyclable (i.e. depolymerizable) in both bulk and solution, and as a proof of concept a recycled M6 sample was successfully repolymerized. Interestingly, the topology of the resulting polymers was also found to be dependent on the catalyst system. For example, MALDI-TOF indicated that linear polymers were obtained using mixed catalyst systems such as tBu-P4/BnOH and IMes/BnOH, while cyclic polymers were exclusively obtained using IMes alone. Later, Lu et al. carried out the base-catalyzed controlled ROP of bridged-ring thiolactone monomer series M7a–c to afford chemically recyclable polymers with Mn values as high as 259 kg mol−1 with dispersities generally below 1.15.36 Upon treatment with DBU at 50 °C, polyM7 analogues showed quantitative depolymerization in <2 min.37
A literature survey revealed that only six reports are available describing the ROP of β-propiothiolactones (i.e., M8–10). Their ROPs were carried out with nucleophilic initiators such as amine, thiol, ammonium carboxylate,38–40 as well as transition metal catalysts.41 Initial attempts at the polymerization of β-thiolactone monomer M8 were reported to be complicated by the uncontrolled nature of the polymerization. Suzuki and coworkers reported the synthesis of polythioesters via the ROP of a cysteine-derived β-thiolactone M9,42 but the resulting polymers were of relatively low molar mass (Mn < 10.0 kg mol−1) with relatively broad dispersities (Đ = 1.6–2.4), and were also reported difficult to depolymerize. Xiong et al. hypothesized that the introduction of geminal dimethyl groups to the four-membered scaffold could accelerate the ring-closure step of the depolymerization, by the so-called “Thorpe–Ingold effect”.43 To this effect they synthesized a series of monomers from naturally occurring amino acid D-penicillamine, differing only in amine substitution (M10a–d). The corresponding polymers were found to readily depolymerize in the presence of DBU at 65 °C by a domino-like “zipper” process. Interestingly, PolyM10c was reported to depolymerize to a racemic monomer mixture at high temperature (>65 °C), but could also depolymerize to give enantiopure samples of M10c at reduced temperatures (i.e. 25 °C).
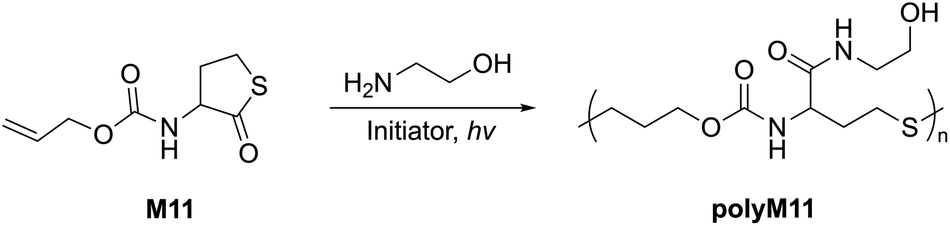
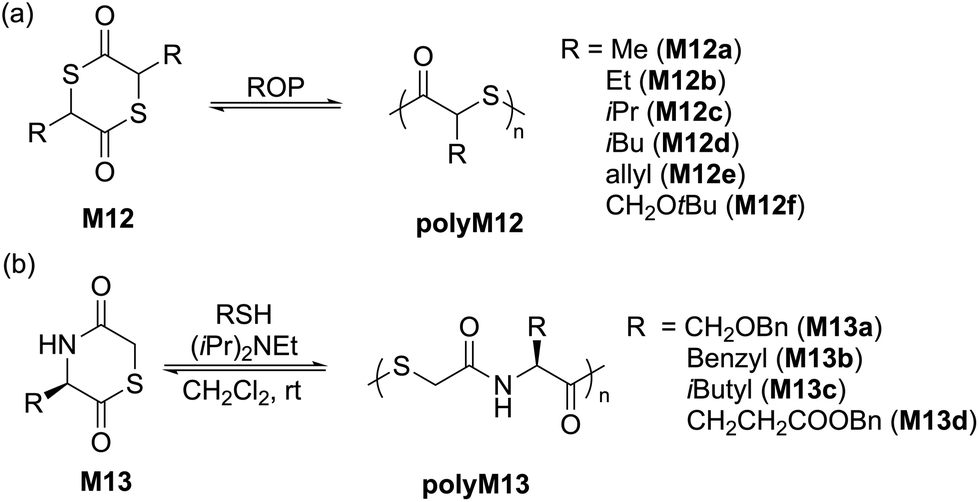
Another 5-membered thiolactone derivative M11 was polymerized by Du Prez et al. by the application of a one-pot amine-initiated ROP and thiol–ene conjugation polymerization. In this way, the polymerization is initiated when γ-thiolactone M11 is ring-opened by a primary amine, which thereby releases the thiol group to further react with the alkene functionality of another monomer unit, ultimately yielding polyM11 (Mn = 22.0 kg mol−1, Đ = 1.6) (Scheme 6).44 Suzuki et al. synthesised and polymerized thiolactide (M12a), the thioester analogue of lactide which is the precursor to commercially available polylactic acid (Scheme 7a).45 The polymerization was found to exist as an equilibrium, with monomer conversions reaching only 50% even under forcing bulk conditions. MALDI-TOF experiments revealed that the product contained cyclic impurities, and that thiol–thioester exchanges had also taken place during polymerization. Controlled polymerizations were then successfully performed using pyridine as a catalyst, however extended reaction times were reported to decrease the Mn which was ascribed to back-biting processes forming cyclic polymer. The authors also homopolymerized thioglycolide (M12b), as well as copolymerized thiolactide (M12a) and thioglycolide (M12b). In comparison to polylactide, polythiolactide (polyM12a) showed a lower glass transition temperature (48 °C versus −0.5 °C, respectively), and was additionally reported relatively more degradable by alkaline hydrolysis and UV photolysis.
 | ||
| Scheme 6 ROP of γ-thiolactone derivative via one-pot amine–thiol–ene conjugation. | ||
 | ||
| Scheme 7 ROP of (a) dithiolactones and (b) 1,4-thiazine-2,5-diones. | ||
Later, Tao and coworkers improved upon this protocol by performing the controlled ROP of various substituted dithiolactones M12a–f mediated by 4-dimethylaminopyridine (DMAP)/BnSH.46 Structurally diverse polythioesters with controlled molecular weights (Mn values up to 100.5 kg mol−1, Đ = 1.2–1.4) were obtained alongside desirable features such as high crystallinity, enhanced polymerizability and chemical recyclability. Noteworthy, the synthesized polythiolactide samples (polyM12a–f) were readily depolymerizable to their corresponding thiolactide monomers in quantitative yield. A variety of amide analogues i.e. 1,4-thiazine-2,5-diones (M13a–d) were also examined for their polymerizability, providing polymers with Mn values up to 15.5 kg mol−1 (Đ = 1.4–2.0) (Scheme 7b).47
2.2 Cyclic thionoesters
Endo and coworkers were the first researchers to investigate the anionic and cationic ROP of ε–thionocaprolactone (M14) (Scheme 8a). Interestingly, under cationic polymerization conditions (1 mol% BF3·Et2O in DCE), the ROP of M14 proceeded with quantitative monomer conversion via an inversion of substitution at the thionoester group to generate polythioester polyM14a exclusively with an Mn of 57.0 kg mol−1 (Đ = 1.95), with the Mn values rising alongside solvent polarity.48 Conversely, the anionic polymerization mechanism was reported to be dependent on the nature of the initiator.49 Polythionoesters (i.e.polyM14b) were exclusively obtained when organolithiums, lithium tert-butoxide and Grignard reagents were applied, whereas, KOtBu and DBU yielded mixed copolymers (i.e.polyM14c), which primarily consisted of thioester repeating groups. As an example, homopolymer polyM14b (Mn = 13.0 kg mol−1, Đ = 1.65) could be obtained using 2 mol% tert-BuLi at 100 °C, with almost quantitative monomer conversion. Mechanistic investigations suggested that the formation of the mixed copolymer backbone was due to S/O scrambling via an SN2 pathway owing to the considerably high polymerization temperatures (100 °C).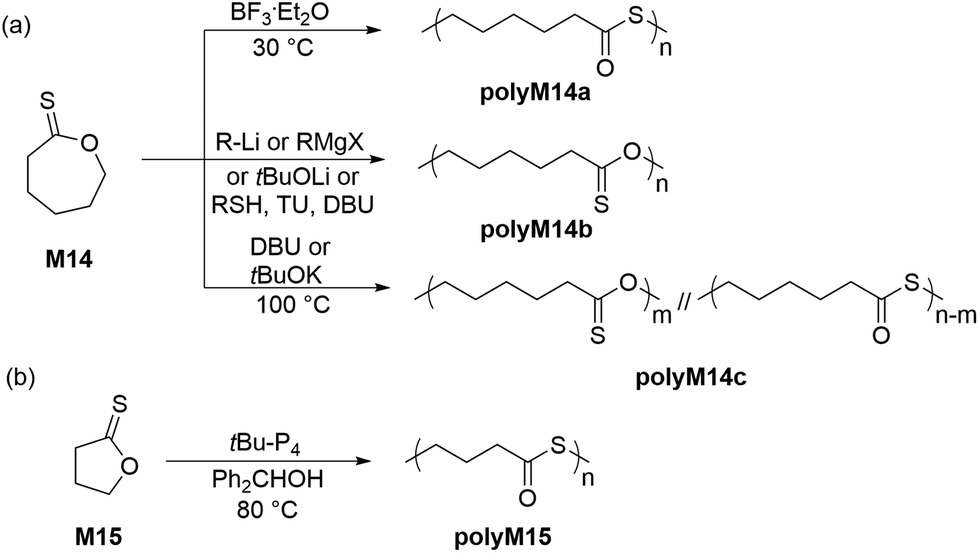 | ||
| Scheme 8 (a) ROPs of ε-thionocaprolactone (M14); (b) ROP of γ-thionobutyrolactone (M15). | ||
In 2016, Datta and Kiesewetter reported the alcohol (or thiol) initiated ROP of M14 at ambient temperature without carbonyl scrambling in the presence of DBU and thiourea (TU),50 leading to homopolymer poly(ε-thionocaprolactone) with Mn values between 12.7–22.5 kg mol−1 (Đ = 1.10–1.41). The ROP of M14 was hypothesized to be operative by dual activation i.e. H-bond activation of the thionoester group by thiourea and chain-end group activation by the base (DBU), thereby proceeding without carbonyl scrambling independent of the reaction conditions. More recently, Hong et al. demonstrated a tBu-P4 catalyzed ROP of γ-thionobutyrolactone (M15) to obtain the corresponding polyM15, the polymerization being driven by a thermodynamically irreversible S/O isomerization (Scheme 8b).51 This was the first report describing such irreversible ROP, coined “IROP”, which rendered the five-membered ring highly polymerizable at 80–100 °C. Quantitative monomer conversion was accomplished within only 4 h, providing polyM15 in 94% yield with Mn values up to 162 kg mol−1 (Đ = 1.47–1.66).
2.3 Sulfur-containing carbonates
Cyclic carbonates are a remarkable monomer class that display volume expansion during ROP,52 leading to biodegradable polycarbonates which are applied commercially for biomedical applications.53 Substitution of one or multiple oxygen atoms with sulfur atoms can potentially result in enhanced physical (e.g. increased crystallinity), chemical, thermal, mechanical, electrical and optical properties. In this regard, cyclic thiocarbonate monomers have thereby been designed for the synthesis of thiocarbonate-containing polymers. Depending on the nature of the covalent bonds and the number of sulfur atoms within the carbonate functionality, cyclic thiocarbonate monomers can be divided into three classes: (a) cyclic monothiocarbonates (–S–CO–O–), (b) cyclic xanthates (–S–CS–O–) and (c) cyclic thionocarbonates (–O–CS–O–). It should be noted that no reports are presently available within the literature regarding the ROP of monomers containing cyclic dithiocarbonate (–S–CO–S–) or trithiocarbonate (–S–CS–S–) functionality.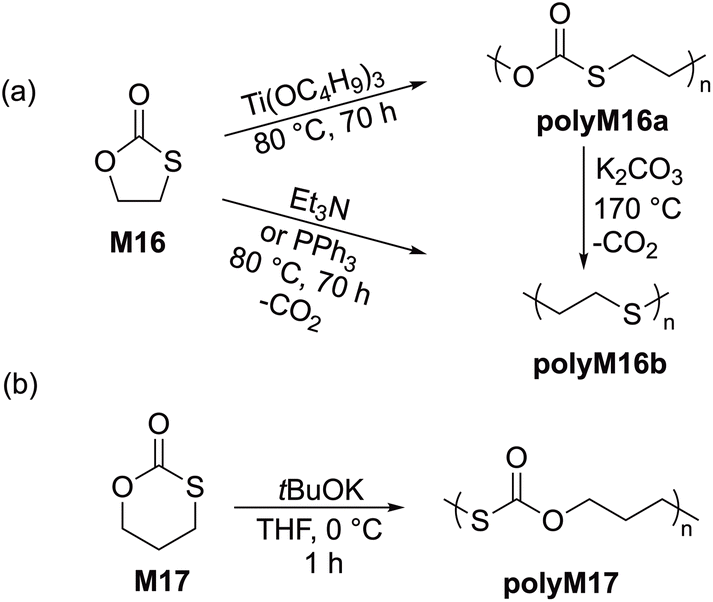 | ||
| Scheme 9 ROP of (a) five and (b) six-membered cyclic monothiocarbonate monomers. | ||
 | ||
| Scheme 10 (a) Cationic ROP of five-membered cyclic xanthate monomers (M18a–d) and the depolymerization of polyM18d; (b) catalytic isomerization of cyclic xanthates (M18a–d) when treated directly with TfOH. | ||
 | ||
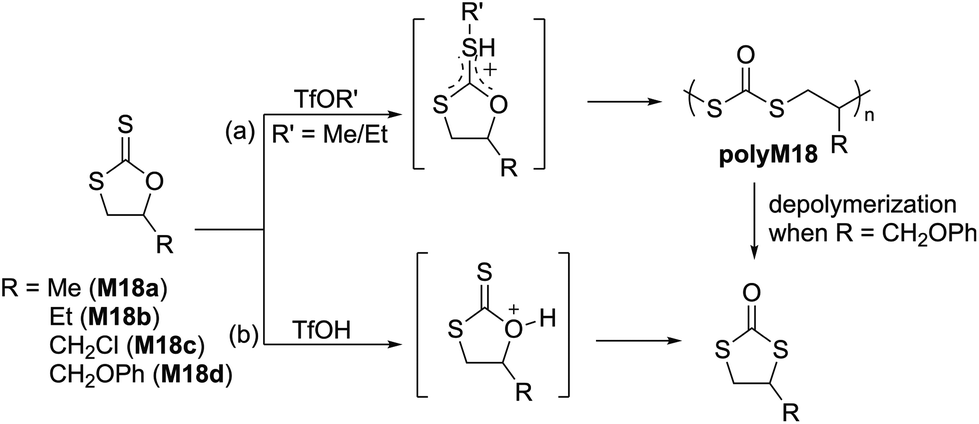
| Scheme 11 Cationic ROP of cyclic xanthates (a) involving neighboring group participation (NGP); (b) specific example showing the synthesis of mixed backbone polydithiocarbonate polyM20 obtained upon the polymerization of M20 initiated by TfOMe. | ||
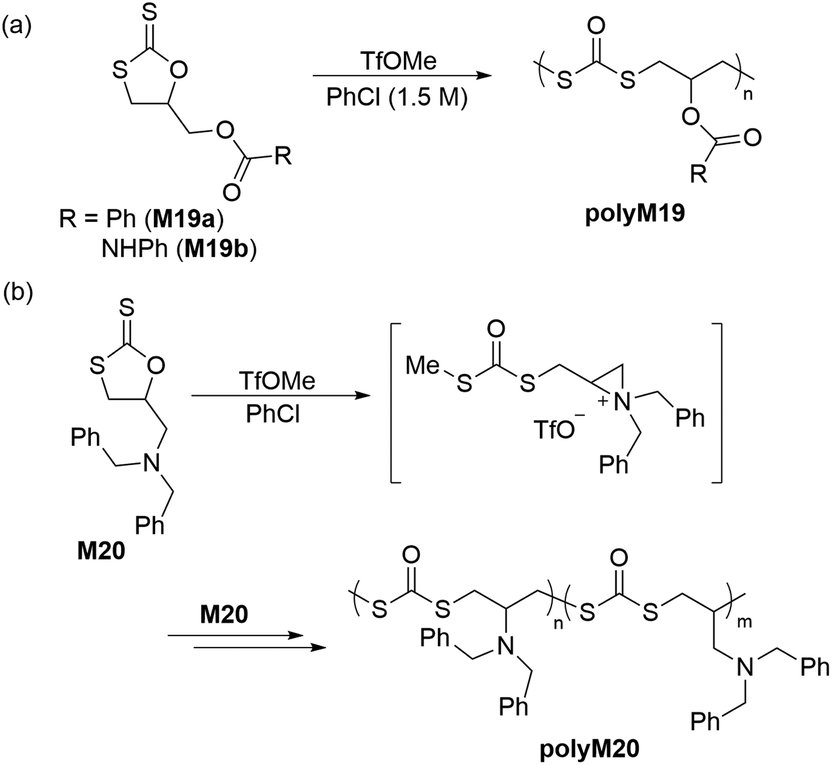
In 2018, Buchard et al. reported the ROP of cyclic xanthate monomers M21 and M22, both derived from the natural sugars 2-deoxy-D-ribose and D-xylose, respectively, using triazabicyclodecene (TBD) as an organocatalyst and 4–methylbenzyl alcohol as an initiator (Scheme 12).63 Polymers polyM21 and polyM22 with Mn values as high as 11.3 kg mol−1 (Đ = 2.2) and 15.7 kg mol−1 (Đ = 1.7) were obtained, respectively. MALDI-TOF analysis revealed the presence of both linear and cyclic topologies, in agreement with the limited polymerisation control. Moreover, NMR spectroscopic investigations on the ROP of xanthate monomer M22 suggested the occurrence of alternating ring-opening mechanisms, ultimately forming regioregular polymers with alternating C(S)S2 and C(S)O2 linkages (Scheme 12b).
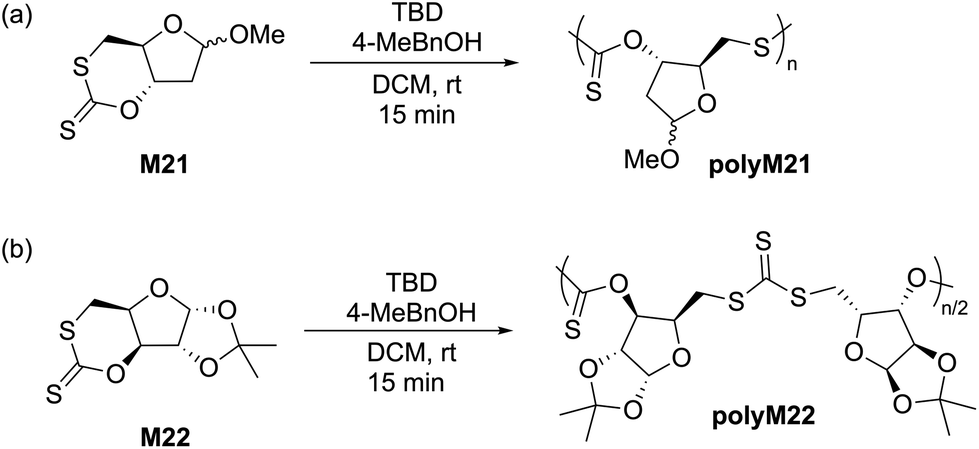 | ||
| Scheme 12 ROP of cyclic xanthate monomers (a) M21 and (b) M22 both derived from the natural sugars 2-deoxy-D-ribose and D-xylose, respectively, in the presence of triazabicyclodecene (TBD) as an organocatalyst and 4-methylbenzyl alcohol as an initiator. | ||
 | ||
| Scheme 13 (a) ROP and, (b) structures of cyclic thionocarbonate monomers. | ||
The cationic ROP of 5,5-dimethyl-1,3-dioxane-2-thione (M25) using various initiators such as TfOH, TfOMe, Et3OBF4 and BF3·Et2O all showed controlled polymerization behavior, giving the corresponding polythiocarbonates with Mn values ranging from 11.2 to 31.0 kg mol−1 with narrow molecular weight distributions (Đ = 1.04–1.15). It was reported that the cationic ROP of monomer M28 proceeded via NGP of the ester group to provide a homopolymer with an unexpected polymer backbone analogous to cyclic xanthate M19a/b (from Scheme 11a). Interestingly, the BF3·Et2O initiated ROP of M29 occurred without NGP to afford the expected corresponding polythiocarbonate with Mn = 11.0 kg mol−1 and Đ = 1.5.66
The anionic ROP of M32 in the presence of KOtBu provided the corresponding polythiocarbonate (Mn = 8.8 kg mol−1, Đ = 1.07) in quantitative yield, demonstrating the controlled nature of polymerization.67 The molecular weight of polyM32 was reported to increase linearly with monomer conversion, retaining a narrow molecular weight distribution. The cationic ROP of monomers M30 and M31 bearing fused- and spiro-adamantane moieties was also reported; for example the Mn and dispersity value obtained by the ROP of M30 with H2O/BF3·Et2O were 10.6 kg mol−1 and 1.44, respectively.68 However, M31 failed to undergo cationic ROP presumably because of the steric hindrance of the bulky adamantane moiety.69 Nevertheless, this monomer underwent cationic copolymerization with M23, M24 and M25 when initiated by BF3·Et2O or Et3OBF4 to afford the corresponding, albeit low molecular weight, copolymers. Buchard and coworkers also reported the ROP of cis-fused cyclic thionocarbonate M33 derived from 2-deoxy-D-xylose (Scheme 14).63PolyM33 samples with an Mn values up to 10.6 kg mol−1 (Đ = 1.5) were obtained, with close agreement between theoretical and experimental molecular weights. However, MALDI-TOF studies of polyM33 revealed the formation of a non-negligible quantity of cyclic polymer impurity, ascribed to backbiting of the polymer chain.
 | ||
| Scheme 14 ROP of a cis-fused cyclic thionocarbonate derived from 2-deoxy-D-xylose. | ||
2.4 Thiiranes
Since the 1920s it has been reported that thiiranes readily polymerize.70 Since then, there have been a multitude of methods reported for synthesis of polythioethers, with the most suitable being anionic in nature.71,72 However, high crystallinity and thereby insolubility in organic solvents have hampered any real-world applications of this polymer series, and thus further development to obtain polythioethers with improved properties remains ongoing. Polymerization experiments focused on the anionic ROP of thiirane derivatives revealed that simple and diverse nucleophiles can be utilized as initiators.73 In particular, thiolate species derived from thiols have been demonstrated as promising initiators for the ROP of propylene sulfide (M34) (Scheme 15a), resulting in pure regiochemical regulation (i.e., head-to-tail microstructure) within the polymer backbone.74–76 Interestingly, anionic ROP initiated by thiolates has been demonstrated to follow first order kinetics, hence possessing ‘living’ character.77 Similarly, the use of the naphthylsodium as an initiator has also demonstrated living character towards the ROP of propylene sulfide (M34) in THF, resulting in high molecular weight polymers (polyM34a) with Mn values up to 320 kg mol−1.78 Nishikubo reported the ROP of propylene sulfide (M34) initiated by quaternary tetraphenylphosphonium salts, with molecular weights of ca. 3000 g mol−1 (Đ = 1.18) being successfully obtained.79 Well-defined block copolymers of poly(styrene-b-M34) and poly(DMAA-b-M34) with Mn values of 3.9 and 3.8 kg mol−1, respectively, were also synthesized.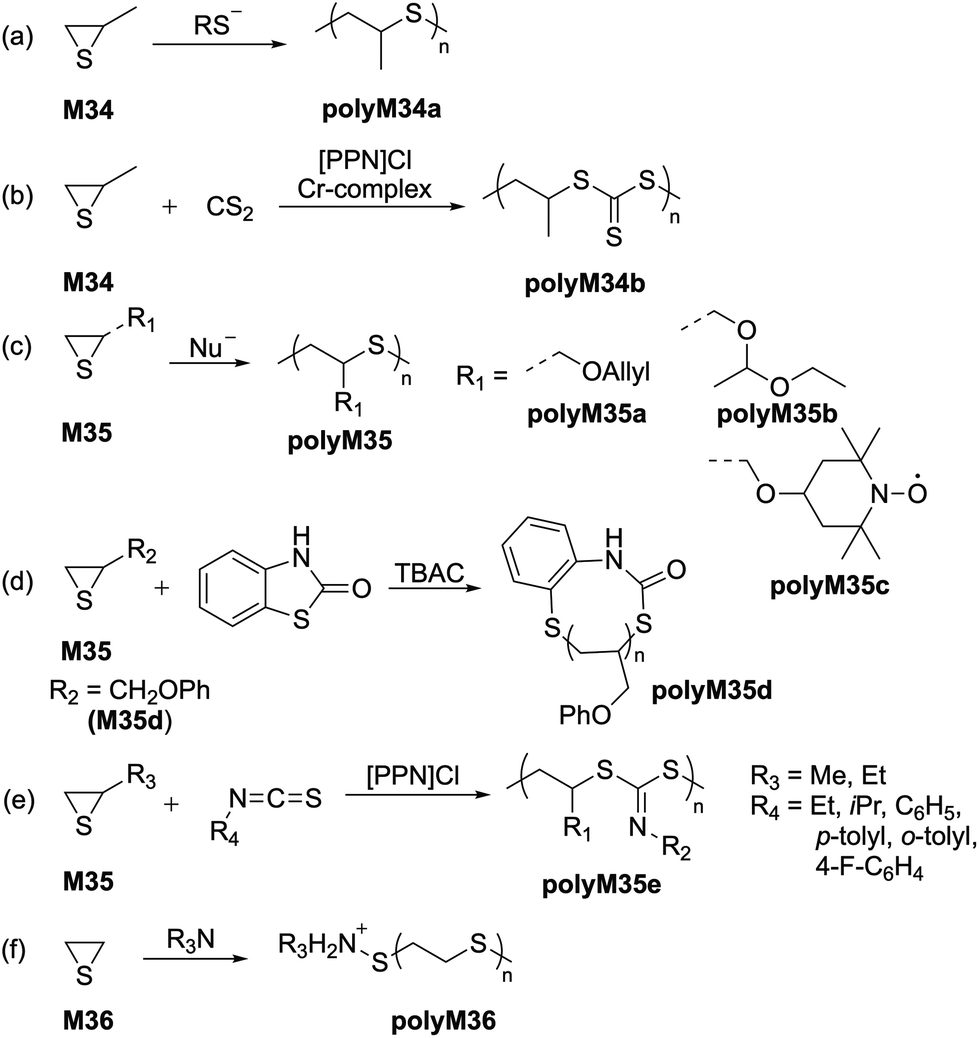 | ||
| Scheme 15 (a) Thiolate-initiated ROP of propylene sulfide; (b) ROCOP of propylene sulfide with CS2; (c) anionic ROPs of various thioglcidyl ether monomers; (d) REP of phenyl thioglcidyl ether (M35d); (e) ROCOP of episulfides with isothiocyanates and (f) tertiary amine-initiated ROP of ethylene sulfide. | ||
Nozaki and coworkers later reported the ROCOP of propylene sulfide (M34) with carbon disulfide using a bis(triphenylphosphine)iminium chloride ([PPN]Cl) and chromium initiator system to obtain alternating polytrithiocarbonates (polyM34b) (Scheme 15b).80 The anionic ROP of various thioglcidyl ether monomers (type M35) has also been reported to obtain polythioethers possessing distinct properties (Scheme 15c/d).81–84 For example, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)-substituted polythioether polyM35c has demonstrated potential applications in energy storage (Scheme 15c),81 while phenyl thioglcidyl ether (M35d) underwent ring-expansion polymerization using benzothiazol-2-one (BT) as a cyclic initiator in the presence of tetrabutylammonium chloride (TBAC) as a catalyst (Scheme 15d).82 The polymerization proceeded in a well-controlled manner to afford polyM35d (Mn = 6.15 kg mol−1, Đ = 1.18) with one BT moiety per macrocycle, as confirmed by MALDI-TOF analysis.
Recently, Zhu et al. reported similar ROCOPs of episulfides (M35) with isothiocyanates in the presence of [PPN]Cl (Scheme 15e).85 Using this methodology, a distinct class of sulfur-rich polymers (polyM35e) with varying substitutions were synthesized with Mn values between 27–78 kg mol−1. Spectroscopic investigations by 13C NMR confirmed the presence of S(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)S units in the polymer backbone, rather than S(CS)N units. Moreover, TGA and DSC analysis of these polymers revealed tuneable glass transition temperatures (Tg) over a range of 31–108 °C. Tertiary amines such as 1,4-diazabicyclo[2.2.2]octane (DABCO) were additionally reported to facilitate the ROP of ethylene sulfide (M36) to produce high molecular weight polymers through a zwitterionic mechanism (Scheme 15f), while polyamine-based initiators were reported to produce only oligomeric polyM36.86 Other nucleophiles were examined, however most of these resulted in the formation of oligomers in poor yield due to a slow initiation rate coupled with poor solubility in the reaction media.26,87–89
N)S units in the polymer backbone, rather than S(CS)N units. Moreover, TGA and DSC analysis of these polymers revealed tuneable glass transition temperatures (Tg) over a range of 31–108 °C. Tertiary amines such as 1,4-diazabicyclo[2.2.2]octane (DABCO) were additionally reported to facilitate the ROP of ethylene sulfide (M36) to produce high molecular weight polymers through a zwitterionic mechanism (Scheme 15f), while polyamine-based initiators were reported to produce only oligomeric polyM36.86 Other nucleophiles were examined, however most of these resulted in the formation of oligomers in poor yield due to a slow initiation rate coupled with poor solubility in the reaction media.26,87–89
2.5 Cyclic thiourethanes
The ROP of five-membered cyclic thiourethanes, in particular 1,3-oxazolidine-2-thiones, was first reported in 1966, however, these early studies did not report proper characterization for the obtained polymers.90 In 2003, the cationic ROP of cyclic thiourethane derivatives was re-revisited Endo and coworkers; they reported the living cationic ROP of an enantiopure cyclic thiourethane (M37) derived from L-serine using TfOMe in DCM at 30 °C (Scheme 16). The resulting chirally pure polythiourethane (Mn = 13.3 kg mol−1, Đ = 1.06) was obtained in quantitative yield with a head-to-tail structure.91,92 These results prompted the authors to examine the scope and limitation of this ROP system by exploring new monomer structures (M38–M43) and initiator systems (e.g. BF3·Et2O).93–95 For instance, the cationic polymerization of 1,3-oxazinane-2-thione (M42) in CH2Cl2 at 30 °C resulted in a polymer that was poorly soluble in organic media due to intermolecular H-bonding, while its N-benzyl analogue (M43) yielded a soluble polymer for which SEC analysis revealed a bimodal distribution. To eliminate this phenomenon, polymerization in nitrobenzene at 50 °C was performed, providing the corresponding polythiourethane in an excellent yield with a Mn of 20.4 kg mol−1 (Đ = 1.04).93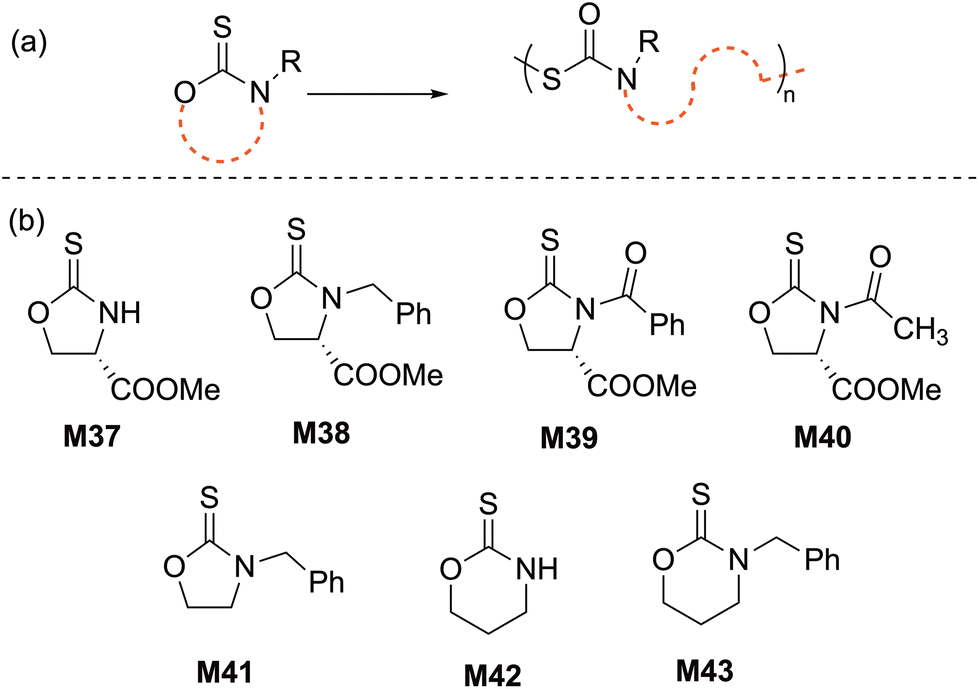 | ||
| Scheme 16 (a) ROP and (b) monomer structures of cyclic thiourethanes. | ||
2.6 Thioanhydrides
Ren and coworkers reported extensively the ROCOP of various substituted thioanhydrides with epoxides and episulfides to synthesize aliphatic and semiaromatic polythioester copolymers (Scheme 17). They initially reported the synthesis of well-defined aliphatic polythioesters by the controlled alternating copolymerization of a variety of cyclic thioanhydrides (M44a/b) and episulfides (M45), yielding copolymers of the type poly(M44-co-M45) with controlled molecular weights (Mn = 42.3–53.6 kg mol−1) and narrow dispersities (Đ = 1.25–1.35).96 Later, they explored the synthesis of semiaromatic polythioesters via the copolymerization of phthalic thioanhydrides (M44c/d) and epoxides (M46), catalyzed by a Cr(III)-complex to give polythioesters with Mn values up to 60.1 kg mol−1 (Đ = 1.29–1.94).97 However, NMR spectroscopic analysis revealed that the backbones of the resultant copolymers were composed of multiple repeating units including thioester, ester, and thioether–ester linkages owing to transesterification reactions. This difficulty was successfully overcome by the use of a bifunctional acid–base catalyst system composed of a Cr–salen complex with a covalently tethered TBD (i.e. a sterically hindered Lewis base), which significantly suppressed transesterification thereby yielding entirely alternating polythioesters.98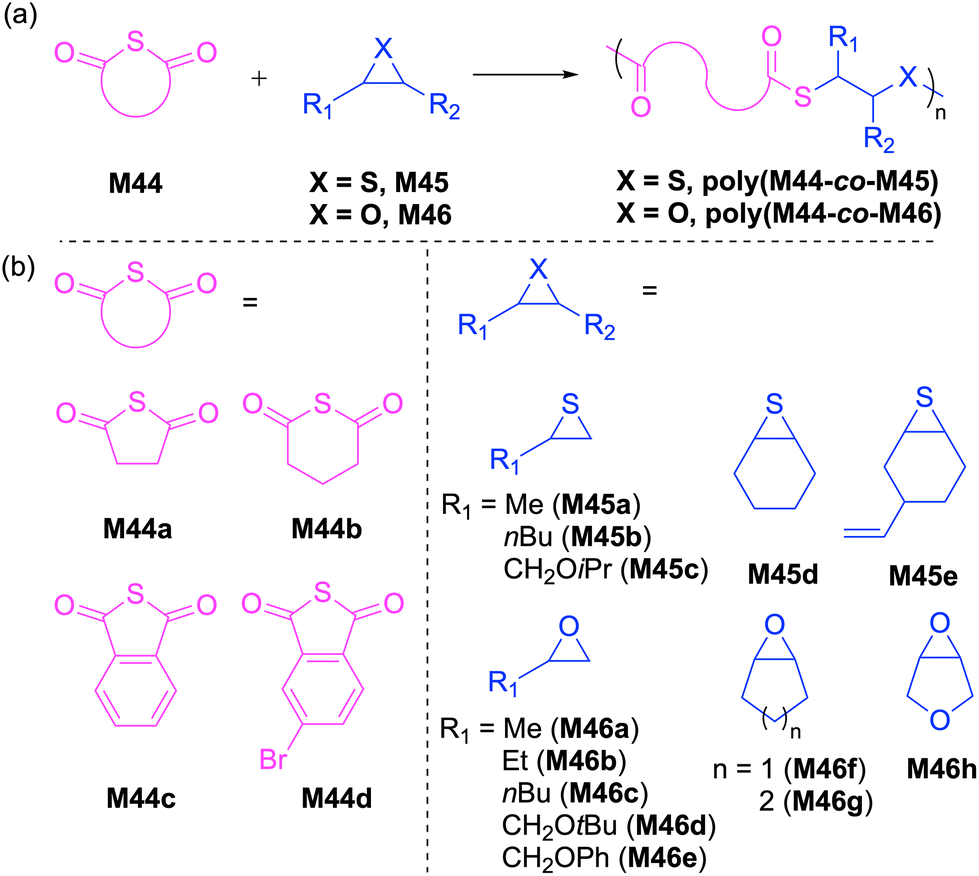 | ||
| Scheme 17 (a) ROCOP of cyclic thioanhydrides with epoxides/episulfides and (b) monomer structures. | ||
Recently, Chen et al. reported the synthesis of sequence-controlled polyester-b-poly(ester-alt-thioester)s via a one-pot copolymerization of phthalic thioanhydride (M44c), epoxides (M46) and anhydrides (M47) using phthalic acid as a bifunctional chain transfer agent (CTA) (Scheme 18).99 Spectroscopic investigations by 1H NMR indicated that the polymerization proceeded via two different polymerization mechanisms, namely, the ROCOP of anhydride/epoxide (high priority) and ROCOP of thioanhydride/epoxide (low priority). These observations suggest that the anhydride/epoxide copolymerization occurred initially, while the copolymerization of thioanhydride/epoxide did not proceed until after the full conversion of anhydride monomer. ABA type-triblock copolymers with Mn values ranging from 4.6 to 20.5 kg mol−1 (Đ = 1.27–1.50) were obtained with excellent chemoselectivity in presence of phthalic acid as a CTA, demonstrating the significant reactivity difference between anhydride and thioanhydride monomers.
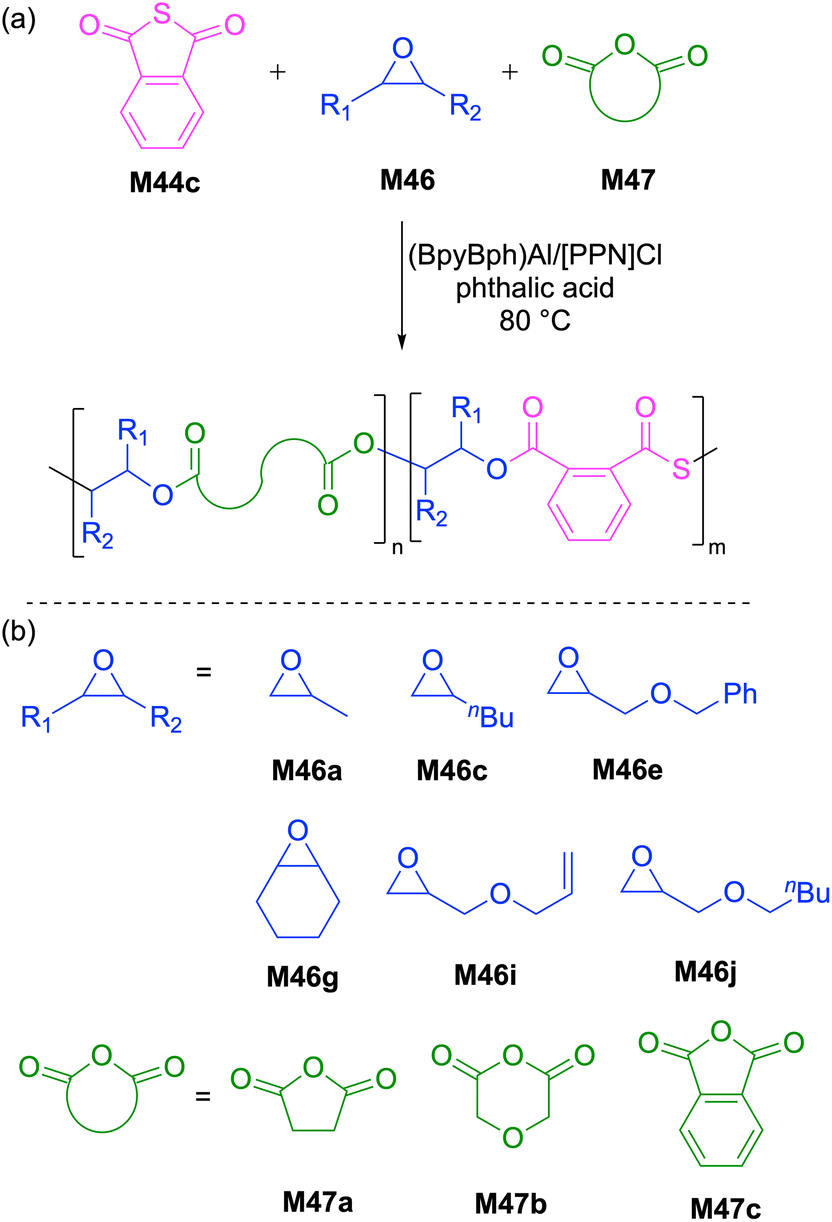 | ||
| Scheme 18 (a) One-step ring-opening copolymerization of various mixtures of heterocyclic monomers and (b) monomer structures. | ||
3. Radical ring-opening polymerization (rROP)
3.1 Cyclic vinylsulfones
In 1986 Cho and co-workers reported a new class of monomers, the cyclic vinylsulfones, that were able to polymerize by radical ring-opening polymerization (rROP) to give polyalkene sulfones (Scheme 19b).100–102 The AIBN/BPO-initiated polymerization of monomers M48–M51 was reported to be dependent upon the ring strain and steric bulk of the monomer (Scheme 19a). It was reported that complete ring-opening polymerization occurred for monomers M48 and M51, while monomers M49 and M50 underwent both ring-retaining and ring-opening propagation to give mixed copolymers. Among the six-membered monomers, M51 was reported to be comparatively more reactive due to the presence of additional functionalities (i.e. sulfonyl and methyl) which imparted additional ring-strain. Unfortunately, the resulting polymers were only soluble in concentrated H2SO4 and TFA (and DMSO in the case of polyM51), these properties preventing their full chemical analysis.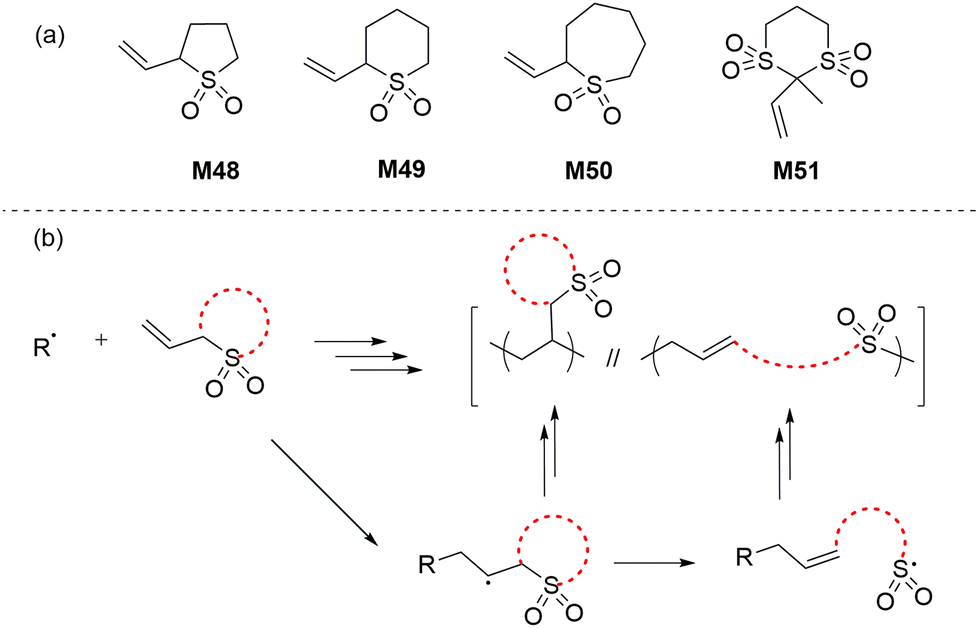 | ||
| Scheme 19 (a) Cyclic vinylsulfone monomers and (b) rROP mechanism of cyclic vinylsulfones. | ||
3.2 Cyclic allylic sulfides
In 1996, Evans and Rizzardo reported the rROP of cyclic allylic sulfide monomers to produce polysulfides containing 1,1-disubstituted alkene units along the backbone.103 The radical ring-opening process of these monomers produces an unusual thiyl radical which can propagate without significant competition from side reactions such as H-abstraction (Scheme 20). Bulk polymerizations of M53 and M55 were reported to proceed with complete ring-opening at 70 °C to give crystalline homopolymers of high molecular weight (Mn = ca. 6 × 105 kg mol−1).103 Complete monomer conversion was observed, although the polymers were reported insoluble in all organic solvents, with the exception of pyridine. It was reported that the introduction of methyl substituents (e.g., M56 and M57) provided mostly amorphous polymers which had improved solubility in organic media.104,105 The monomer M56 was also reported able to copolymerize with common acrylic and vinylic monomers such as methyl methacrylate and styrene.105 Yamada and Goto106 later investigated the photopolymerization of M62 in the presence of oxygen, demonstrating the robustness of this polymerization system towards oxygen inhibition.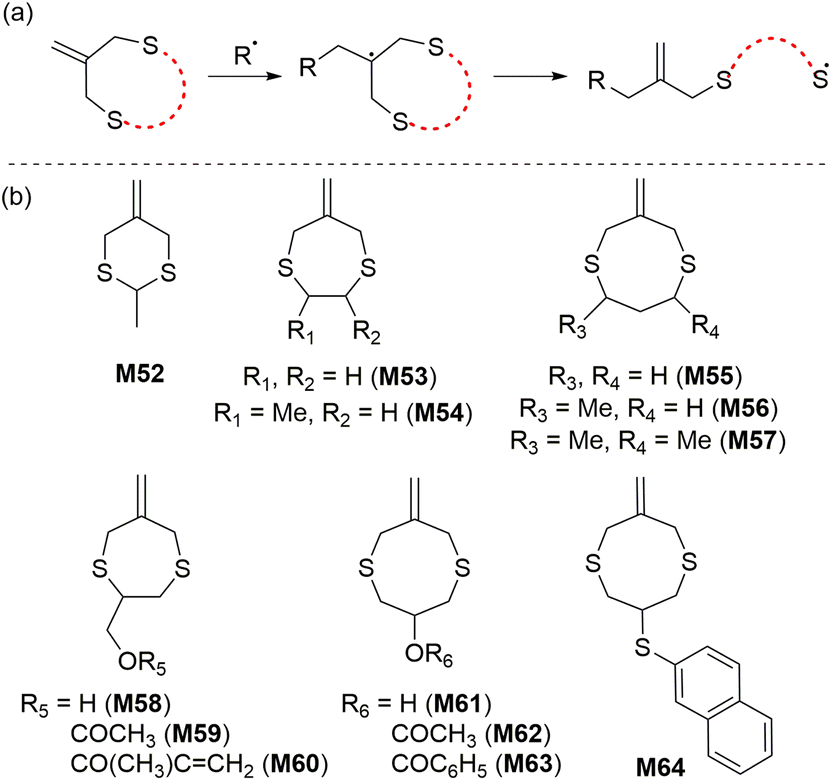 | ||
| Scheme 20 (a) Radical ring-opening and (b) structures of cyclic allylic sulfides. | ||
3.3 O,S/S,S-cyclic ketene acetal (CKA)
In 1984 Bailey and co-workers reported that the rROP of 5-membered O,S-CKA monomer M65 at 120 °C formed the corresponding thioester-containing polymer with 45% ring-opened units (Scheme 21).107 The copolymerization of styrene and M66 also provided a thioester-containing copolymer, which upon hydrolysis produced oligomeric fragments end-capped with a mercaptan and carboxylic acid groups as expected. The polymerization of 2-methylene-1,3-dithiane (M66) at 80 °C yielded a low molecular weight (700–1500 g mol−1) polymer possessing almost exclusively dithioacetal units by direct vinyl-type polymerization (i.e. without ring-opening).108 Similarly, its copolymerization with styrene and methyl methacrylate (MMA) proceeded without ring-opening.109 The homopolymerization of 2-methylene-1,3-dithiepane (M67) was reported not successful, for reasons unknown.108 | ||
| Scheme 21 Structures of O,S-/S,S-CKA monomers. | ||
3.4 Sulfide cyclic methacrylates
Reported by Rizzardo in 1994, a new class of sulfur-containing cyclic monomers named the sulfide cyclic methacrylates emerged as promising candidates for radical polymerization.110 As for the cyclic allylic sulfides, their propagation mechanism involves a thiyl radical (Scheme 22a). Exclusive ring-opening polymerization (i.e. with no ring-retaining propagation) and high reactivity were reported for this monomer class during both homopolymerization and copolymerization, although cross-linking was reported for the homopolymerization of M68 after 20% monomer conversion. However, this phenomenon was not observed for M69 due to the production of a trisubstituted alkene during the radical ring-opening process, thereby inhibiting radical addition to the alkene.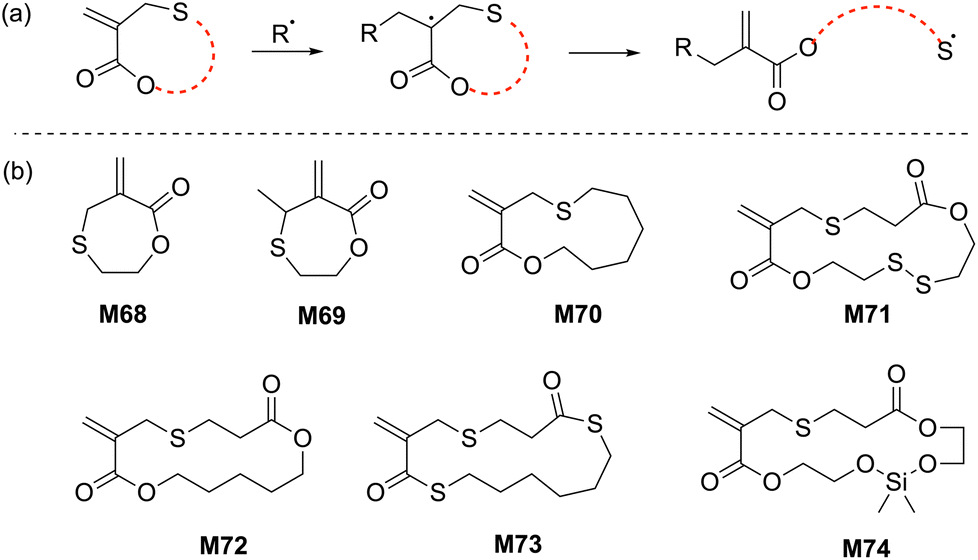 | ||
| Scheme 22 (a) General scheme for radical ring-opening of sulfide cyclic methacrylates and (b) structures of the cyclic/macrocyclic allylic sulfides used in rROP. | ||
Copolymerizations of M69 and M70 with styrene and MMA were reported to proceed with high monomer conversions (70–85%), producing their respective copolymers with Mn values between 1.7–19.2 kg mol−1 (Đ = 1.8–1.9). Later, Hawker extended this research by developing macrocyclic sulfide methacrylate monomers having ester, thioester, and disulfide functionalities within the monomer scaffold (Scheme 22b).111 RAFT copolymerizations of M70–M73 and MMA displayed good control over molecular masses (Mn up to 30 kg mol−1) and tuneable, but limited incorporation of macrocyclic allylic sulfide units into the copolymer (FM71 = 0.01–0.10). Different potential degradation modes of the copolymer backbone (such as basic/acidic, reductive, thiolate, and even enzymatic) can be envisioned by the proper choice of macrocyclic sulfide methacrylate monomer.
3.5 Macrocyclic allylic sulfones
To overcome the difficulties associated with the rROP of sulfide cyclic methacrylates, a new strategy named radical cascade-triggered ring-opening polymerization (RCT-ROP) was developed. Allylic sulfone groups were thus applied as “ring-opening triggers” via a radical cascade sequence that forms a sulfonyl radical which subsequently expels SO2 to generate a stabilized secondary alkyl radical (Scheme 23). The produced secondary alkyl radical is functionally analogous to that of polyacrylates and is therefore readily suited for controlled polymerization via RDRP techniques. In this way, Niu and co-workers112 synthesized a series of 12–16 membered macrocyclic allylic sulfones (M75–M78, Scheme 23) and successfully performed their RAFT polymerization to generate well-defined homopolymers and copolymers. For example, the AIBN initiated RAFT homopolymerization of M75 in toluene at 100 °C resulted in 62% monomer conversion within 2 h (Mn = 12.1 kg mol−1, Đ = 1.15) while its copolymerization with methyl acrylate (MA) (fM75 = 0.05) proceeded with 92% conversion to yield a copolymer with 12 mol% cyclic monomer incorporation (Mn = 10.2 kg mol−1, Đ = 1.17). However, unfavourable reactivity ratios were observed due to the faster incorporation of the macrocyclic allylic sulfone monomers in comparison to MA.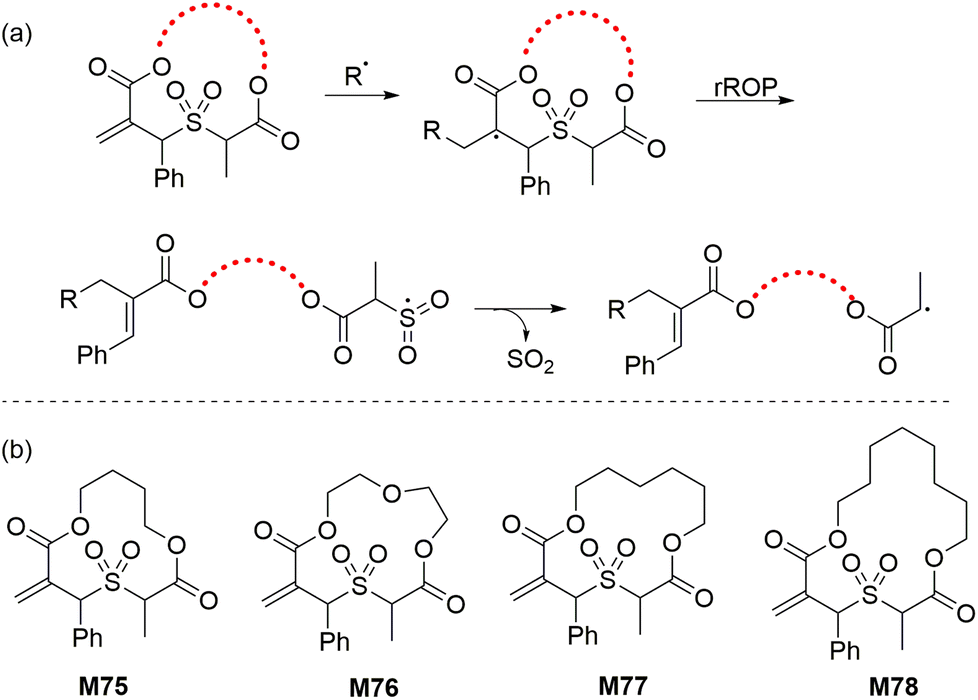 | ||
| Scheme 23 (a) RCT-ROP mechanism of a macrocyclic allylic sulfone and (b) structures of the various monomers polymerized by RCT-ROP. | ||
To overcome reactivity complications, the same group recently reported a photoinduced electron transfer-reversible addition fragmentation chain transfer (PET-RAFT) approach.113 For instance, the homopolymerization of M78 mediated by an iridium complex under 450 nm irradiation yielded polyM78 with an Mn of 9.8 kg mol−1 (Đ = 1.11), thereby showing excellent control over the polymerization. A diblock copolymer polyM75-b-M78 (Mn = 13.0 kg mol−1, Đ = 1.20) was also synthesized under similar conditions. It was demonstrated that propagation completely halted when the light source was removed, and then resumed efficiently after the light source was reintroduced. The photocontrolled copolymerization of M78 with MA at a feed composition of fM78 = 0.05 yielded a copolymer poly(M78-co-MA) with an Mn of 44.0 kg mol−1 (Đ = 1.28). Interestingly, fitting of the polymer compositional data into the Beckingham–Sanoja–Lynd integrated model114 determined the reactivity ratios of M78 and MA remained close to unity during the entire range of the monomer feed compositions (fM78 = 0–1). Furthermore, the copolymerization of M78 with other acrylic monomers at fM78 = 0.09 all exhibited excellent polymerization control and near-unity reactivity ratios, suggesting the general applicability of this method to a wide range of acrylates and acrylamides.
Following the success of RCT-ROP, Niu et al. developed a new series of monomers with even larger ring sizes (26- to 31-membered) which were also capable of undergoing radical-initiated cascade polymerization (M79–M81) (Scheme 24).115 It was proposed that a 1,6-diene-fused allylic alkylsulfone could undergo efficient SO2 extrusion during the cascade process to thereby form a stabilized alkyl radical to enable controlled chain growth. The synthesized macrocyclic structures (M79–M81) were successfully homopolymerized using RAFT at 85 °C for 40–45 h, displaying good control over molecular weights and dispersities (Mn = 4.1–20.1 kg mol−1, Đ = 1.21–1.39). The prepared polymers exhibited good thermal stability (∼5% mass loss at 310–340 °C), variable glass transition temperatures (Tg = 48–138 °C), and high susceptibility to degradation by sodium methoxide via cleavage of the ester and pyrrolidinone groups.
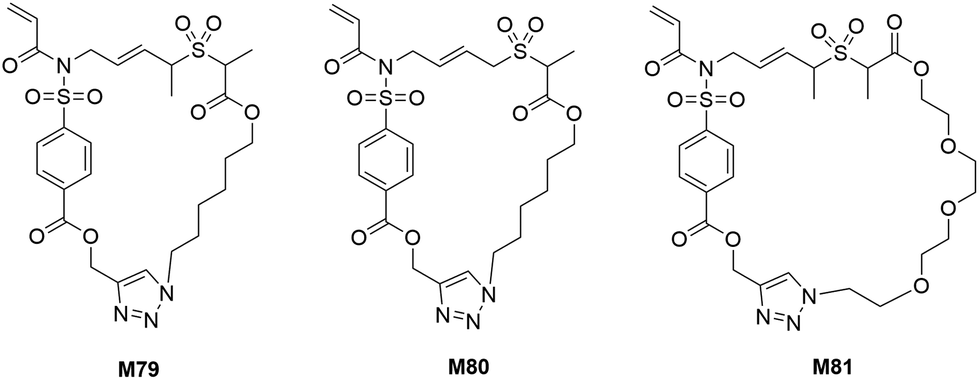 | ||
| Scheme 24 Structures of the macrocyclic allylic sulfone monomers. | ||
3.6 Thionolactones
Reported independently by Roth116 and Gutekunst117 in 2019, a new class of thionoester-containing rROP monomer recently emerged as good candidates to prepare degradable polymers. The polymerization of such cyclic thionolactone monomers leads to the insertion of thioester linkages into a polymer backbone via what was termed “thiocarbonyl addition-ring opening” (TARO) polymerization (Scheme 25).116 The introduction of thioester groups is advantageous when compared to their (oxo)ester counterpart as thioesters can be degraded by additional mechanisms including hydrolysis, aminolysis, trans–thioesterification and oxidative hydrolysis. Among the various thionolactone monomers reported, only dibenzo[c,e]oxepane-5-thione (M82), ε-thionocaprolactone (M14), and ε-thionodecalactone (M83) were successfully polymerizable by rROP.116–119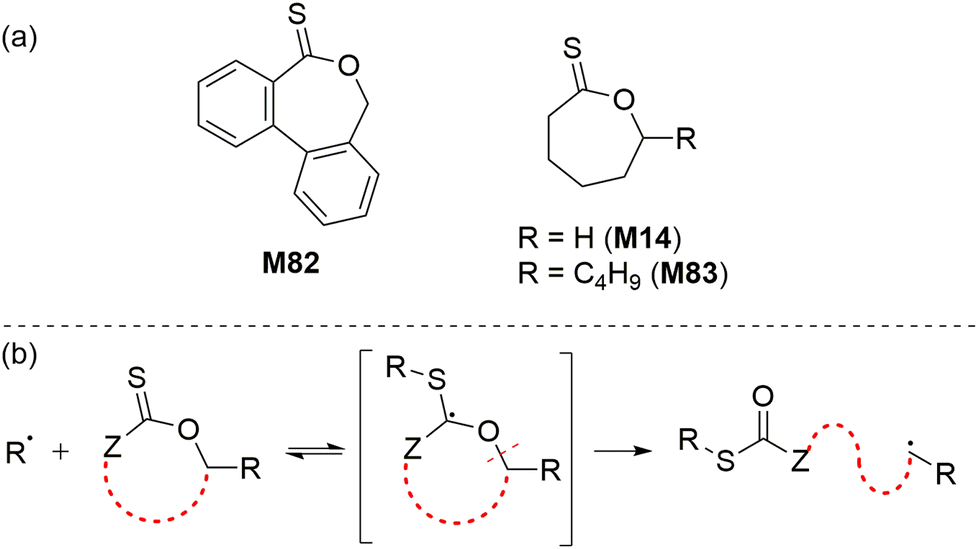 | ||
| Scheme 25 (a) Structures and (b) radical ring-opening of thionolactone monomers. | ||
Roth et al. performed the AIBN-initiated homopolymerization of M82 in acetonitrile, finding negligible conversion after heating at 80 °C overnight.116 Later, they improved the conversion up to 22% (Mn = 4.9 kg mol−1 and Đ = 2.0) by performing the polymerization in DMSO with the sequential addition of fresh AIBN over 27 days at 60 °C.120 However, it was noted that this method was impractically slow and failed to produce a well-defined homopolymer. When compared to CKA monomers, M82 is reported to be relatively stable under ambient conditions, however, heating M82 in DMSO at 150 °C was reported to lead its isomerization to the thioester (–C(O)S–) derivative.116 Attempted RAFT homopolymerizations of M82 also failed, having only <2% M82 conversion.116 Due to the stabilization of the propagating radical by the aromatic ring, M82 was reported to readily copolymerize with a variety of monomers including acrylates, acrylamides, acrylonitrile and maleimides (Scheme 26). Surprisingly, M82 was found to inhibit radical polymerizations of vinyl acetate and N-vinylcarbazole, and to retard the radical polymerization of styrene without being incorporated.116,117
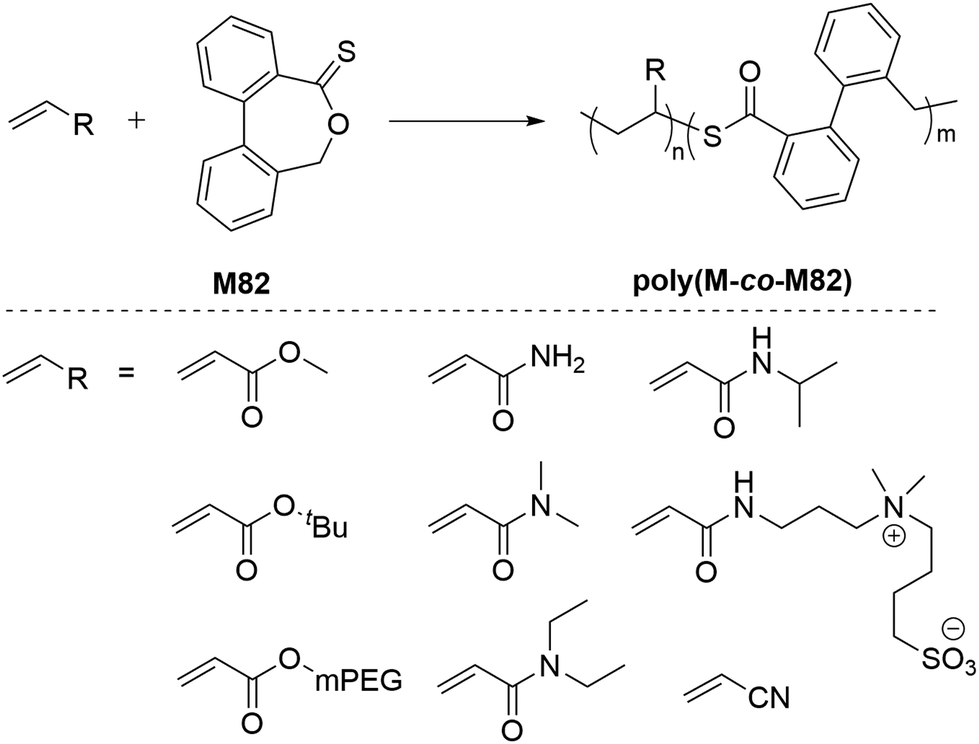 | ||
| Scheme 26 (a) Copolymerization of DOT (M82) with a vinyl monomer and (b) structures of various vinyl monomers. | ||
Roth and coworkers investigated the copolymerization of M82 and MA as a model monomer, which was reported to form alternating sequences of both monomers although it was noted that retardation (i.e. a lower-than-expected monomer consumption at a given time) was observed with an increased quantity of M82 within the feed.116 During the same period, Gutekunst117 published the RAFT copolymerizarion of M82 and tert-butyl acrylate (tBA) using a 5% DOT feed ratio. A variety of polymer molecular weights (Mn = 4.4–32.6 kg mol−1) with relatively low dispersities (Đ = 1.05–1.27) were obtained by varying the monomer-to-initiator ratio. It was reported that lower monomer conversions of M82 were obtained at 50% feed as a result of polymerization retardation. A well-defined diblock copolymer poly(tBA-co-M82)-b-poly(nBA-co-M82) was also prepared by RAFT polymerization from a poly(tBA-co-M82) macro-CTA to provide evidence of the livingness of the system. To verify the presence of thioester linkages within the copolymers, degradation experiments were performed by methanolysis (with NaOMe) and thiol–thioester exchange reaction (with cysteine methyl ester), giving broad low molecular weight oligomers (1.7–2.1 kg mol−1).117
Interesting, the RAFT copolymerization of M82 (50% feed ratio) with N-substituted maleimides produced well-defined alternating copolymers.120 The tendency to form alternating sequences was reported to be increased in the order of R = CH3 < Ph < C6F5. Interestingly, when N-phenylmaleimide and N-(pentafluorophenyl)maleimide were used as comonomers, defects in the alternating structure were more likely caused by faster incorporation of M82 leading to M82–M82 (degradable) sequences rather than MI–MI (nondegradable) sequences. M82 was also reported to copolymerize with a range of neutral and zwitterionic acrylamides to give copolymers that displayed temperature-responsive aqueous solubility behavior, tunable through the comonomer composition, as well via degradation.7 Roth also investigated the radical copolymerization of M82 with acrylonitrile but reported the copolymer was only soluble in DMSO and therefore difficult to analyse.116
After the reported success of thionolactone M82, other monomers in this series such as ε-thionocaprolactone (M14) were also investigated. Destarac and Harrisson reported well-defined copolymers of M14 and vinyl esters such as vinyl acetate (VAc) and vinyl pivalate (VPiv) using via both free-radical and RAFT polymerization.118 Using a 10% M14 feed, copolymerization with VAc was carried out to high conversion, producing poly(VAc-co-M14) copolymers having Mn values up to 22.8 kg mol−1 with good incorporation of M14. Spectroscopic investigations revealed that the majority of the incorporated M14 was present as the ring-opened thioester, while 36.5% of was incorporated in ring-retained form. The copolymerization M14 and N-vinyl pyrrolidinone was reported to result in polymerization retardation, low molar mass, high dispersity and little thionolactone incorporation, while attempts to homopolymerize M14 were reported completely unsuccessful.
Guillaneuf and co-workers also reported that two thionolactone monomers, M14 and ε-thionodecalactone (M83) were capable of performing rROP with VAc (Scheme 27).119 It was observed that at 10% feed of M14 the amount of thioester-linkages within the polymer became enriched, while above 20% feed the polymerization did not occur. In this way, the copolymerization of M83 with VAc (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 feed, respectively) provided a copolymer with 20% thioester repeating units (Mn = 16.6 kg mol−1). The authors later identified the initiation phase as the main hindrance to successful polymerization, hypothesizing that M14 was sequestering the AIBN-derived initiating radicals. In this way, copolymerizations of M14 with the CKA monomer 2-methylene-1,3-dioxepane (MDO) were performed, successfully providing homopolymer polyM14 (Mn = 16.8 kg mol−1 and Đ = 3.2), ascribed to its initiation by MDO-derived radicals. Interestingly, spectroscopic analysis of the copolymers revealed the presence of exclusively ring-opened units within the polymer backbone.118 To extend the scope of this protocol, block copolymers such as poly(DMMA)-b-(VAc-co-M14) and poly(DMMA-co-M82)-b-(VAc-co-M14) were synthesized via RAFT and demonstrated to be selectively degradable, thereby demonstrating the effectiveness of thionolactone monomers in preparing degradable polymers.
:90 feed, respectively) provided a copolymer with 20% thioester repeating units (Mn = 16.6 kg mol−1). The authors later identified the initiation phase as the main hindrance to successful polymerization, hypothesizing that M14 was sequestering the AIBN-derived initiating radicals. In this way, copolymerizations of M14 with the CKA monomer 2-methylene-1,3-dioxepane (MDO) were performed, successfully providing homopolymer polyM14 (Mn = 16.8 kg mol−1 and Đ = 3.2), ascribed to its initiation by MDO-derived radicals. Interestingly, spectroscopic analysis of the copolymers revealed the presence of exclusively ring-opened units within the polymer backbone.118 To extend the scope of this protocol, block copolymers such as poly(DMMA)-b-(VAc-co-M14) and poly(DMMA-co-M82)-b-(VAc-co-M14) were synthesized via RAFT and demonstrated to be selectively degradable, thereby demonstrating the effectiveness of thionolactone monomers in preparing degradable polymers.
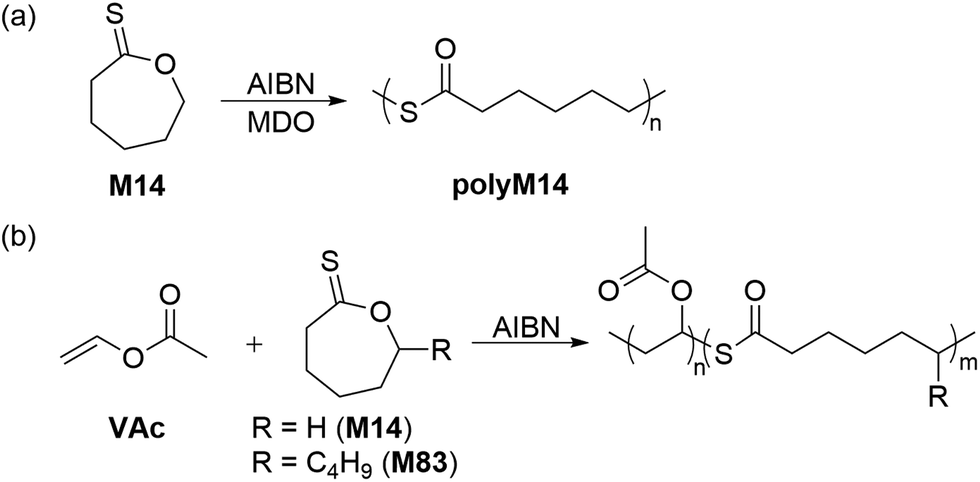 | ||
| Scheme 27 (a) Radical homopolymerization of M14 and (b) radical ring-opening copolymerizations of M14/M83 with VAc. | ||
4. Ring-opening metathesis polymerization (ROMP)
ROMP is a chain-growth strategy widely applied for the controlled synthesis of π-conjugated functional polymers. The mechanism of the ROMP process is based upon olefin metathesis which was initially proposed by Y. Chauvin and later significantly explored by R. H. Grubbs. A significant number of reports on the ROMP of a broad range of cyclic olefins are available in the literature, however, reports of the incorporation of sulfur-containing monomers are relatively scarce. Attempts to homopolymerize sulfur-containing cyclic olefins by ROMP failed due to the negative neighbouring group effect of sulfur.121 It was speculated that the coordination of sulfur to the transition metal within the catalyst (e.g. ruthenium) is more pronounced when the sulfur-containing linkage is present within the cyclic olefin due to the ease of formation of a five/six-membered ruthenocycle during the propagation step which may lead to catalyst decomposition. Nevertheless, disulfides and thioacetal groups have been successfully incorporated by ROMP into a polymer backbone.Hilf and Kilbinger reported the synthesis of thiol-functionalized ROMP polymers via ‘sacrificial synthesis’ using the unsaturated thioacetal monomer, 2-phenyl-1,3-dithiepine (M84).122 Diblock copolymers of exo-N-hexyl-2,3-norbornene dicarboximide (HNI) and exo-N-phenyl-2,3-norbornene dicarboximide (PNI) with unsaturated thioacetal monomer were synthesized by third-generation Grubbs catalyst (GIII)-catalyzed ROMP (Scheme 28). The thioacetal block of copolymer polyM84 was then cleaved by hydrogenation using RANEY® Ni to access the thiol functionalized polymer polyM84a (Mn = 5.9–33.0 kg mol−1, Đ = 1.09–1.28) which were then successfully employed for the coating of the gold nanoparticles.
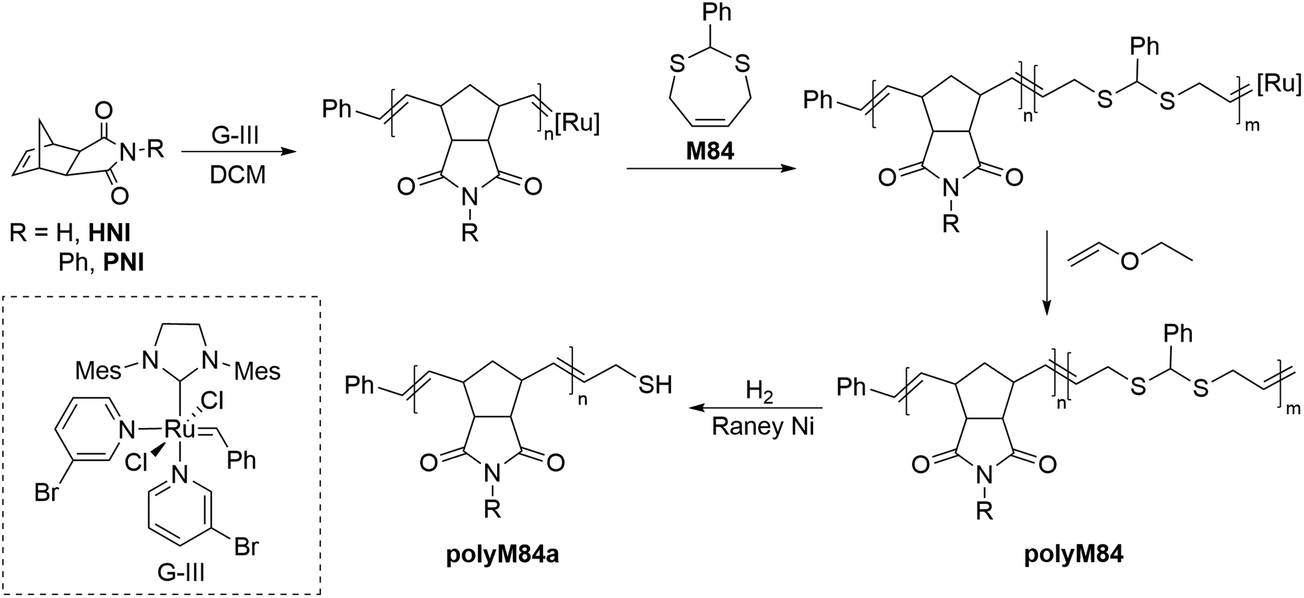 | ||
| Scheme 28 Sacrificial synthesis of thiol-functionalized polynorborneneimides. | ||
Chang and Emrick prepared disulfide-containing polyolefins by the ROMP of disulfide-containing cyclic olefin M85 with substituted cyclooctene monomers using G-III catalyst.121 This strategy allowed for the synthesis of a series of degradable block-copolymers (polyM85) with disulfide linkages within the polymer backbone, along with functional cyclooctene comonomers bearing pendent hydroxyl, amino, and pentafluorophenyl ester groups (Scheme 29). The disulfide monomer incorporation in the copolymer samples ranged from 5 to 27 mol%, with molecular weights between 12–34 kg mol−1. A significant decrease in the molecular weight of the copolymers was reported upon degradation with tri-n-butylphosphine (n-Bu3P) and 1-dodecanethiol, confirming the presence of the disulfide linkages. This ROMP strategy was further extended to terpolymerization with a phosphoester containing cyclic olefin monomer, providing polyM85a containing both disulfides and phosphoesters functionalities, which was also reported to exhibit degradability.
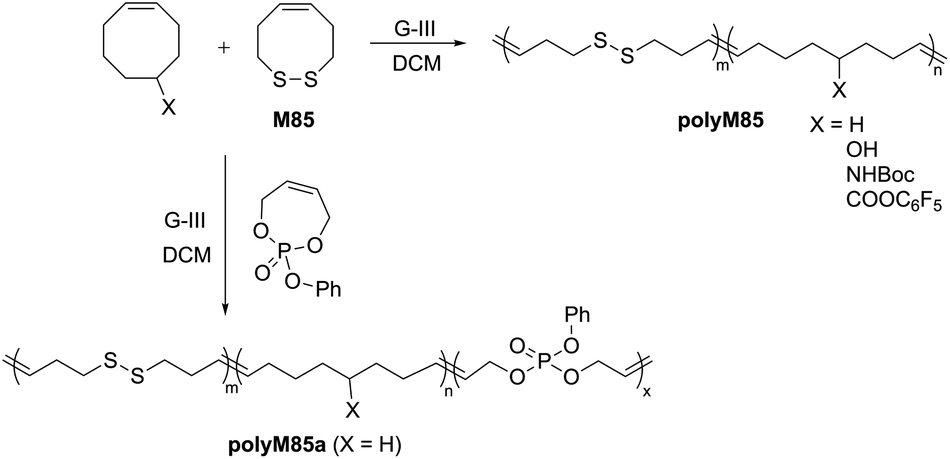 | ||
| Scheme 29 Synthesis of disulfide containing polyolefins by ROMP. | ||
Behrendt and Schlaad synthesized 16- and 30-membered macrocycles (M86a/b) based upon L-cystine which were polymerized by entropy-driven ROMP (ED-ROMP) to produce polyM86a/b containing disulfide and ester linkages within the polymer backbone (Scheme 30).123 Polymers with molar mass values of up to 80 kg mol−1 (Đ = 2) were obtained with ca. 80% monomer conversion using G-III as a catalyst. The synthesized polymers were successfully demonstrated to be degradable by reductive cleavage of the disulfide linkages. Subsequent kinetic studies on the synthesis of polyM86a revealed that the ruthenium catalyst (G-III) was deactivated during the polymerization, thereby limiting the achievable polymer molar mass. Such limitations were overcome by performing disulfide metathesis of the same macrocycle M86a to yield the polymer polyM86a′ (Mn = 180 kg mol−1) with an identical repeating unit structure, but with thiol chain ends.124
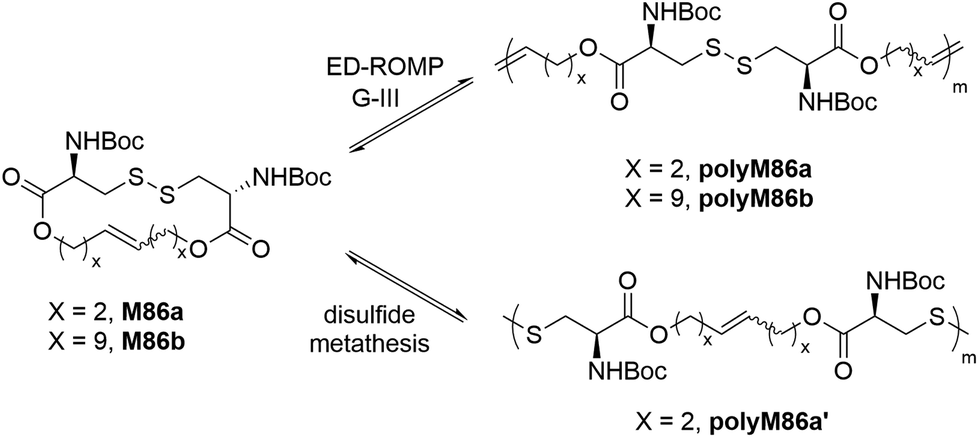 | ||
| Scheme 30 ED-ROMP and disulfide metathesis of macrocycles based on L-cystine. | ||
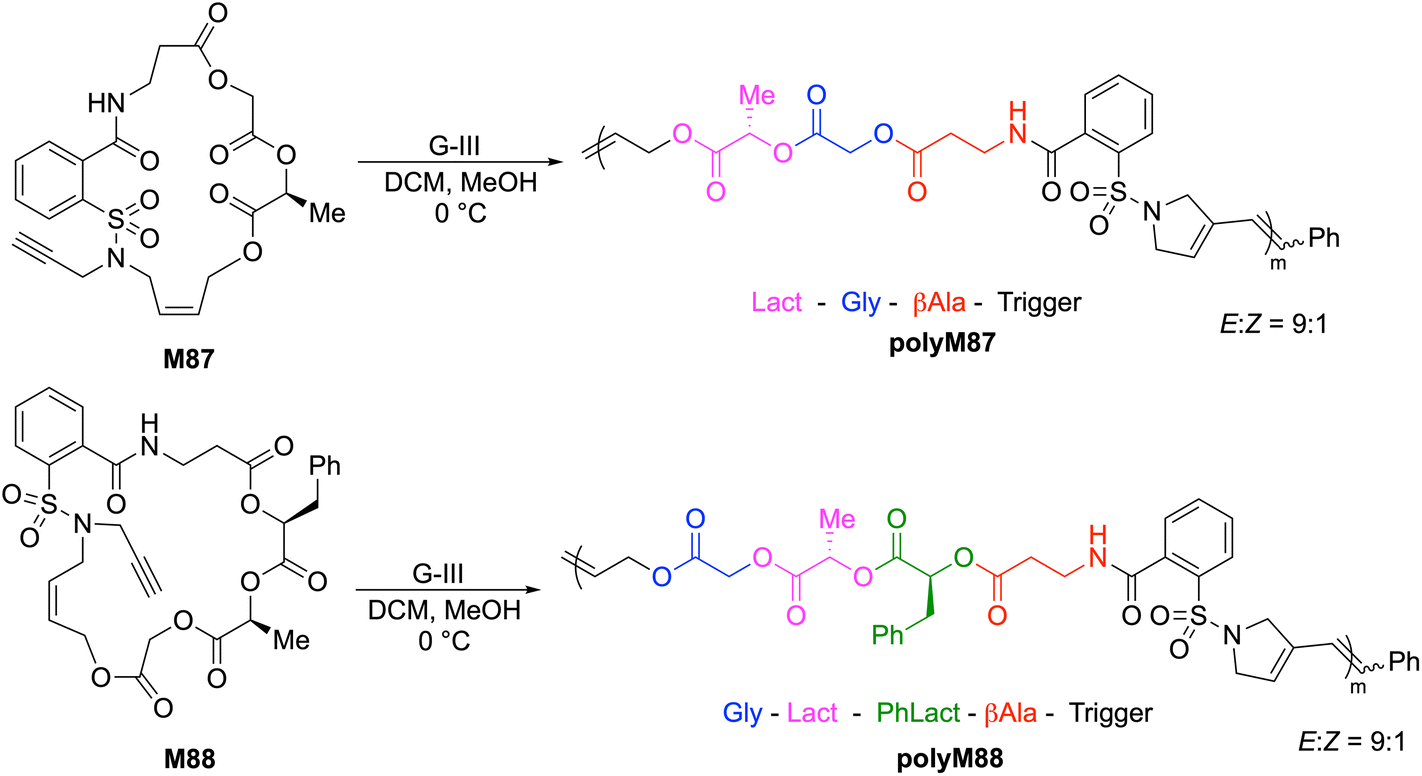
Gutekunst and Hawker synthesised two 20- and 23-membered sulfonamide-containing macrocycles (M87 and M88, respectively) designed in such a way that a polymerization trigger was attached to a series of glycolate (Gly), (S)-lactate (Lact), (S)-phenyllactate (PhLact), and β-alanine (β-Ala) units to give ABCD(E)-type sequences which are then displayed in the polymer backbone post-ROMP (Scheme 31).125 The ROMP of M87 and M88 thereby produced sequence-defined polymers with accurate control over molecular weights and low dispersities. For example, G-III catalyzed ROMP of M88 provided a polymer having an Mn value of 32.6 kg mol−1 (Đ = 1.26), with the polymer chain consisting of ca. 250 perfectly sequenced units. Upon degradation with TBD, SEC analysis of polyM88 revealed a dramatic decrease in the molecular weight after only 30 seconds, and complete conversion to oligomeric methyl esters with Mn values of ca. 200 g mol−1 after 2 h.
 | ||
| Scheme 31 ROMP of sequenced sulfonamide-containing macrocyclic monomers. | ||
5. Miscellaneous
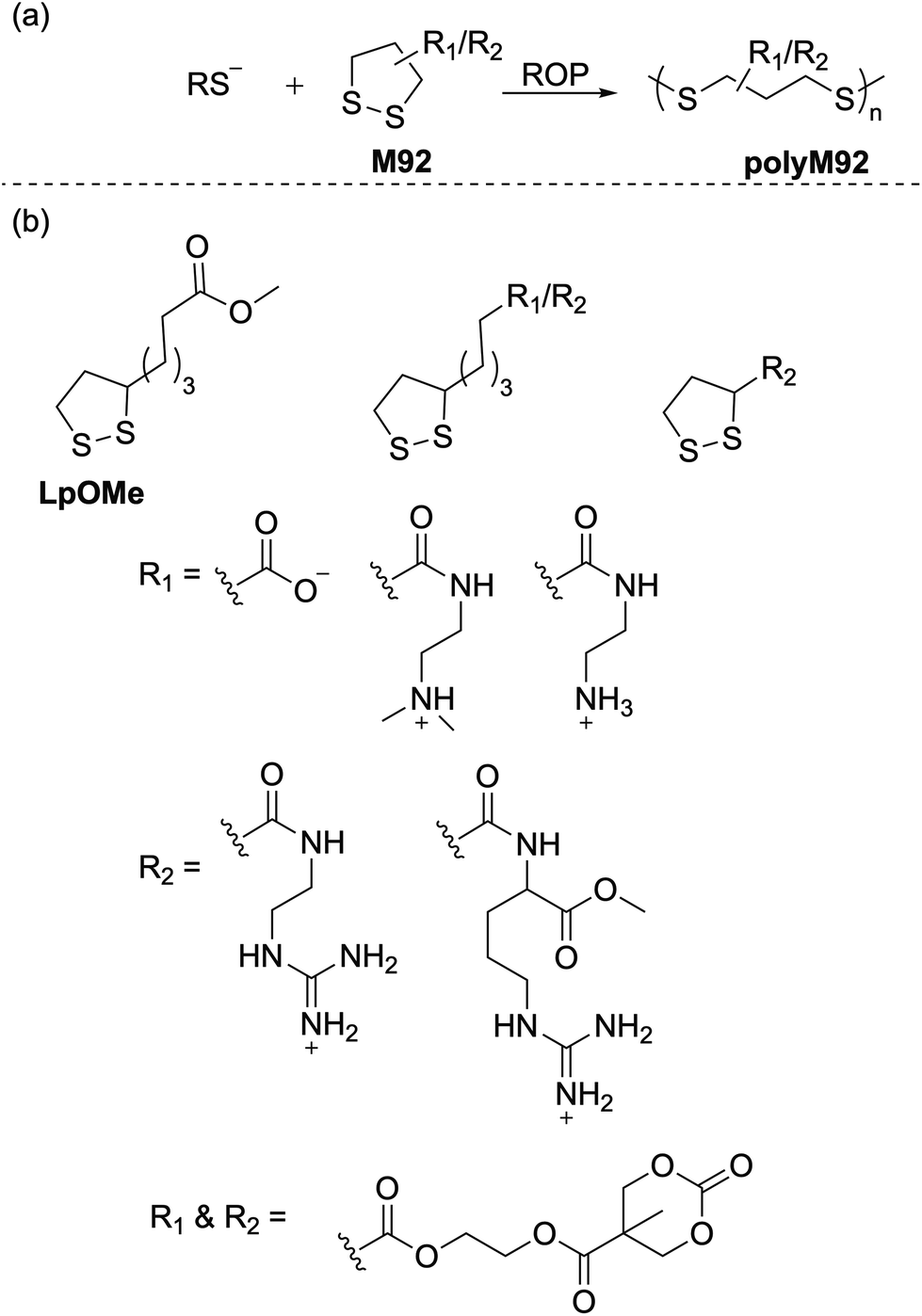
5.1 ROP of 1,2-dithiolanes
Calvin and coworkers were pioneers in the photoinduced ROP of 1,2-dithiolanes, demonstrating that the disulfide bond can be activated by the ring strain of the five-membered scaffold to obtain poly(1,2-dithiolane)s.126 More detailed studies to prepare poly(1,2-dithiolane)s with unambiguous molecular structures were later reported by Endo.127 They reported the synthesis of copolymers (Mn = ca. 5.0 kg mol−1) of lipoic acid (M89) and six-membered 1,2-dithiane. NMR spectroscopy of the resulting polymer confirmed a cyclic structure for the copolymer, and dynamic viscoelasticity measurements suggested mechanical entanglements of the cyclic polymer chains. Combined with other structural evidence as well as photo-decomposition studies, the authors concluded that the resulting polymers featured mechanically cross-linked polycatenane-type networks. Cyclic disulfide monomers are of particular interest in polymer chemistry as they can provide polymers with disulfide groups within the polymer backbone which can be readily degraded by reduction.:1 feed ratio) readily produced their respective copolymers poly(M89-co-EA) and poly(M90-co-EA) (Scheme 32).129 The radical polymerization of an LPA derivative, 2-acryloyloxyethyl lipoate (M91) having both cyclic disulfide and vinyl functionalities was also performed to prepare highly branched sulfur-containing degradable polymers.
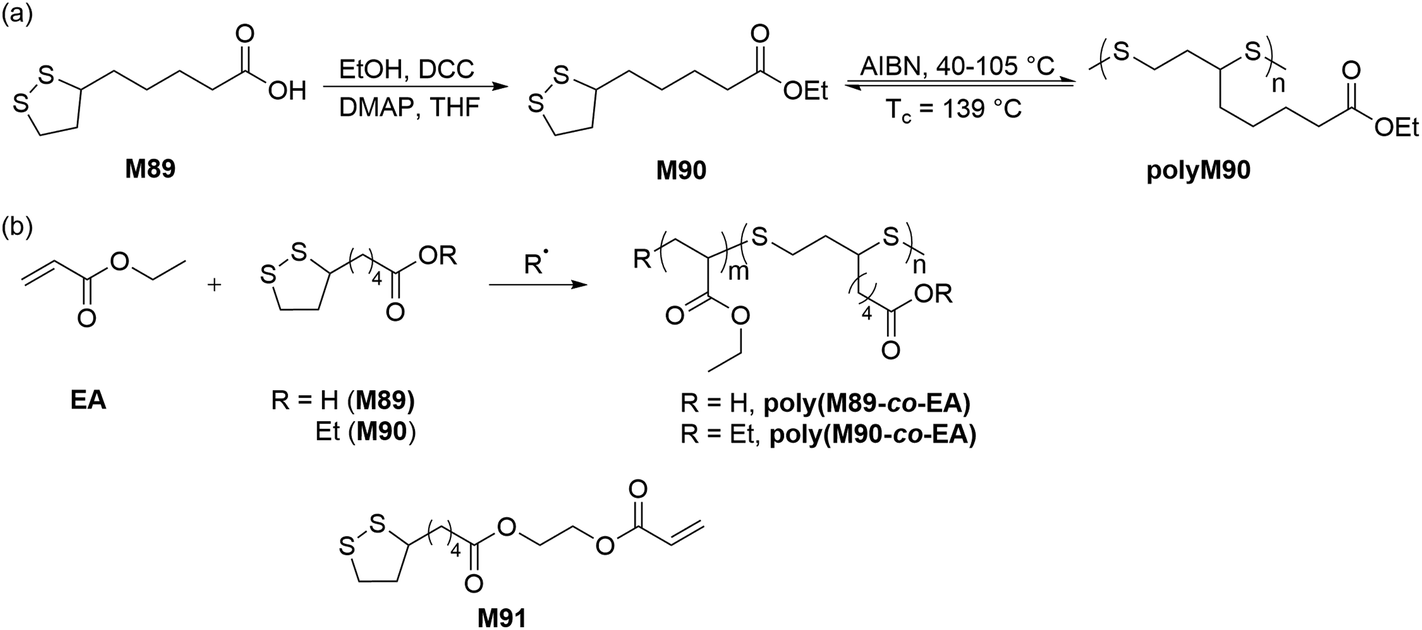 | ||
| Scheme 32 (a) Synthesis and polymerization of ethyl lipoate (M90) and degradation of polyM90; (b) radical copolymerizations of lipoic acid (M89) and ethyl lipoate (M90) with ethyl acrylate. | ||
 | ||
| Scheme 33 (a) Thiolate initiated ROP and (b) monomer structures of 1,2-dithiolanes. | ||
5.2 ROP of elemental sulfur (S8)
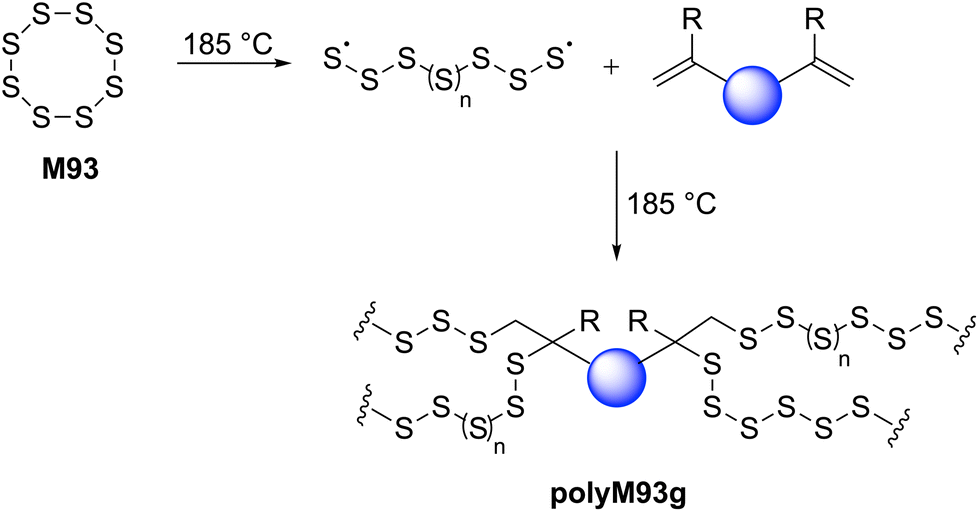
The incorporation of a high content of sulfur atoms into a polymer backbone can provide materials with several exotic and useful properties. For this reason, several research studies have been performed with the aim of utilizing elemental sulfur (S8, M93) for the preparation of new polymeric materials. Polymeric sulfur (polyM93a) has been synthesized by performing the ROP of orthorhombic α-S8 above its floor temperature (159 °C) (Scheme 34a).135 Under this temperature (i.e., under equilibrium polymerization conditions) the reaction becomes reversible, forming cyclic monomeric sulfur (S8) due the backbiting depolymerization by terminal sulfur radicals (Scheme 34a). Rapid quenching (using alkene or polyene derivatives) of the equilibrium melt polymerization of M93 has been applied to preserve the polymeric form of sulfur for prolonged durations. As displayed in Scheme 34, a variety of polymers have been synthesized by employing diverse polymerization strategies including anionic, cationic and free-radical copolymerizations of elemental sulfur (M93) with various comonomers. Four excellent reviews are available discussing such copolymerizations in detail,5,26,135,136 and thus this section merely intends to give a short overview of the research topic.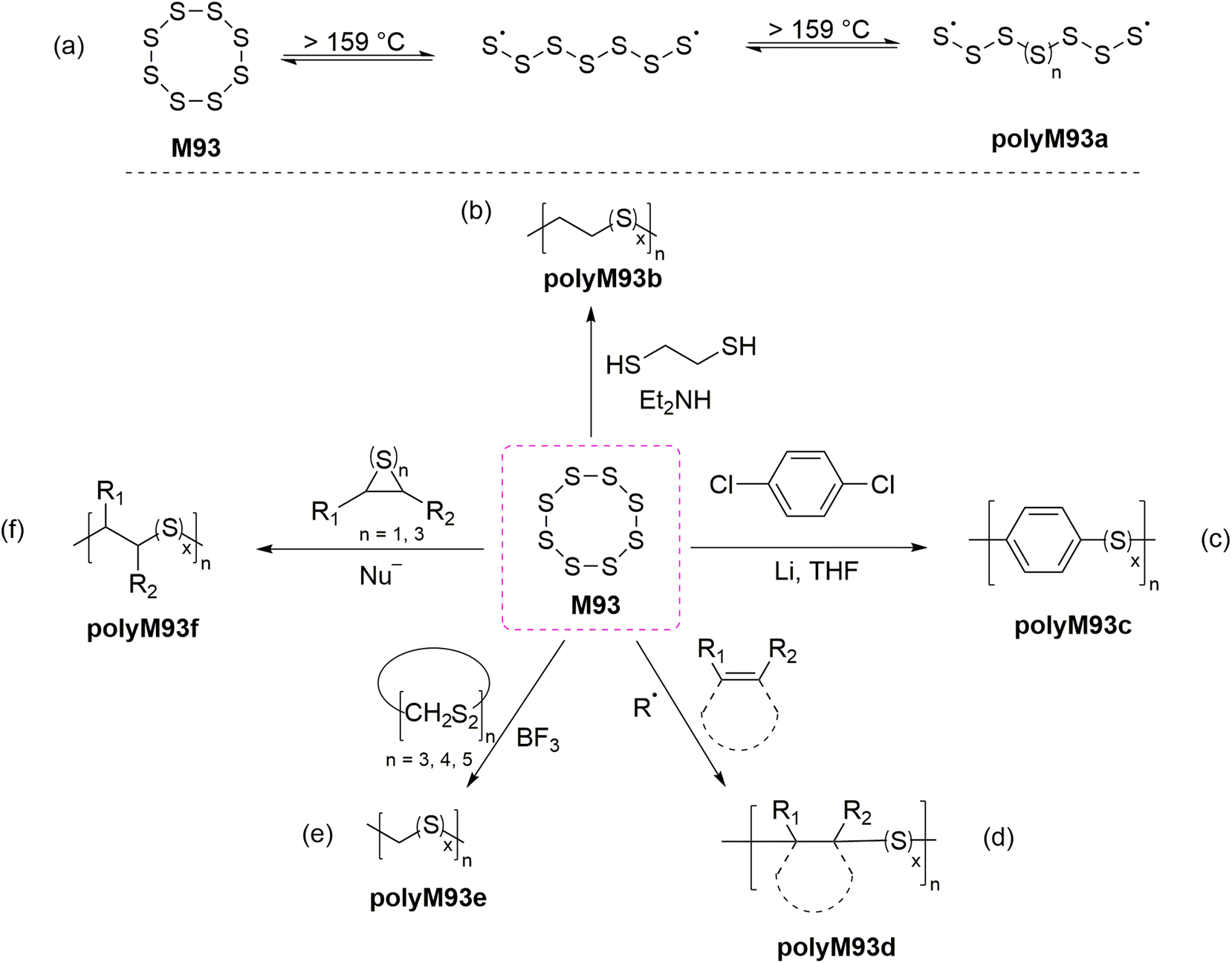 | ||
| Scheme 34 (a) Ring-opening homopolymerization of elemental sulfur (M93, S8) and ROCOP reactions of M93 (S8) with (b) ethane-1,2-dithiol, (c) p-dichlorobenzene, (d) olefins, (e) cyclic thioformaldehydes, and (f) cyclic sulfides. | ||
Bordoloi and coworkers reported the base-catalyzed ROCOP of M93 and ethane-1,2-dithiol to synthesize polysulfides (polyM93b) via a thiol–ene type reaction (Scheme 34b).137 Notably, as the molecular weight of the synthesized polymers increased during polymerization, the sulfur rank (a measure of flexibility and rigidity of inverse vulcanized polymers) was found to plateau. Later in 1981, Boscato et al. reported the synthesis of poly(phenylene polysulfane)s (polyM93c) through the alkali-mediated reaction of M93 and p-dichlorobenzene (Scheme 34c).138 High molecular weight linear polymers (Mn = 12.0 kg mol−1) with high sulfur content were obtained by this protocol. However, the post-polymerization removal of by-products (e.g., Li2Sn, LiCl) was reported problematic. The free-radical ROCOP of M93 and a variety of unsaturated monomers including vinylics, cyclic olefins and terpenoids was also reported (Scheme 34d).26,135 Spectroscopic characterization revealed that in-solution copolymerization produced polymers that had relatively low molecular weight and sulfur content, while bulk polymerization gave moderate molecular weight polymers (Mn ≤ 7000) and comparatively high sulfur content.139 Schmidt and Weissflog demonstrated the cationic ROCOP of M93 with cyclic thioformaldehydes using a Lewis acid (i.e., BF3) to obtain linear, semi-crystalline and colourless polysulfides (polyM93e) which were noted to partially decompose upon melting (Scheme 34e).140,141 The nucleophilic ring-opening of M93 has also been reported for the synthesis of linear polymers (polyM93f) by a ROCOP involving cyclic sulfides (thiiranes and trithioles) (Scheme 34f).142–144 These copolymers were found to have moderate sulfur rank and molar mass (Mn < 104 g mol−1).
During the past decade, the synthesis of polysulfide copolymers via inverse vulcanization between M93 and alkene cross-linkers has emerged as an important approach to valorize sulfur, which is generated as a by-product on a huge scale by the petrochemical industry.5,136,145 During inverse vulcanization, the S–S bonds within M93 first undergo homolytic cleavage to generate thiyl radicals which then initiate the ROP of other molecules, or undergo a thiol–ene reaction with the alkene additive (Scheme 35). Since its pioneering discovery by Pyun and co-workers in 2013,146 inverse vulcanization has been extended to a wide class of organic comonomers including styrenes, alkynes, naturally occurring olefins, allylics, maleimides, and nitriles.5,135 The polysulfide copolymers generated by this approach have found use in lithium–sulfur batteries,147 dye-sensitised solar cells,148 transparent IR lenses,149 controlled-release fertilizers,150 adhesives,151 antibacterial surfaces,152 and adsorbents for environmental remediation i.e. to remove heavy-metals from water.153 However, high reaction temperatures (typically 160–200 °C) are required for this transformation, generating polymeric products with irregular structures and poor solubility.
 | ||
| Scheme 35 Ring-opening copolymerization of elemental sulfur (M93) with vinyl monomer (i.e. inverse vulcanization). | ||
6. Conclusion
For next-generation materials, there is an urgent need to develop new polymers containing chemical diversity within the polymer backbone. The introduction of non-conventional functional groups into the polymer backbone offers the possibility to engineer materials that can respond to chemical stimuli and modulate their physicochemical properties. Recent developments in the ROP of sulfur-containing monomers have provided new routes to synthesize sulfur-containing polymers with specific targeted and controllable properties (e.g., refractive index). Polymers containing sulfur-based functionality within the backbone chain are also of great interest due their potential degradability, as well as wide usefulness in the fields of medicinal, biochemical, and material sciences.Despite excellent properties and immense applications, most conventional polymers suffer from limited degradability or chemical recyclability. Ring-opening polymerization is a powerful strategy to incorporate cleavable linkages into the backbone of polymers. Sulfur-containing functional groups can be selectively cleaved through multiple mechanisms and are therefore also promising candidates for nanotechnology applications such as drug delivery. In addition, polymers with disulfide linkages are also reported to show both degradable and self-healing behaviour. As an example of recent progress in the field, cyclic thionoester-containing rROP monomers have recently emerged as promising candidates to prepare (bio)degradable polymers. The rROP of these monomers has an advantage over conventional rROP systems in that it can selectively insert thioester linkages into a polymer backbone which can be selectively cleaved under mild and specific conditions.
The majority of polymers reported within this article have only been demonstrated on a bench scale and therefore remain at a proof-of-concept stage. For the widespread usage and commercialization of new sulfur-containing polymers, certain important factors will need to be considered including feedstock source, catalyst availability, and that prohibitive energy and material costs are avoided. Moreover, it could be considered advantageous if new monomer feedstock could be bio-sourced and therefore renewable. The development of purpose-driven catalysts that avoid the use of toxic and/or rare transition metals, are efficient, and readily removable from the final material is also paramount. Taking such factors into account, it can be predicated that further exploration of novel systems will lead the efficient preparation of new functional sulfur-containing polymers for use in next-generation materials.
To conclude, the ring-opening polymerization of sulfur-containing monomers is an incredibly productive field, and we hypothesize that the coming decade will be the theatre of important innovations in the design of sulfur-containing polymers with fascinating properties. Nonetheless, there remains much work to be performed in order to obtain this target of next-generation polymers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted as a part of the International Research Agendas PLUS programme of the Foundation for Polish Science, co-financed by the European Union under the European Regional Development Fund (MAB PLUS/2019/11).Notes and references
- M. Wang and X. Jiang, Sulfur–Sulfur Bond Construction in Sulfur Chemistry, Springer International Publishing, 2019, pp. 285–324 Search PubMed.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- H. Liu and X. Jiang, Chem. – Asian J., 2013, 8, 2546–2563 CrossRef CAS PubMed.
- A. Kultys, Sulfur-Containing Polymers in Encyclopedia of Polymer Science and Technology, Wiley, 4th edn, 2010 Search PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2014, 54, 185–267 CrossRef CAS.
- N. M. Bingham, Q. u. Nisa, S. H. L. Chua, L. Fontugne, M. P. Spick and P. J. Roth, ACS Appl. Polym. Mater., 2020, 2, 3440–3449 CrossRef CAS.
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- P. Dubois, O. Coulembier and J.-M. Raquez, Handbook of ring-opening polymerization, John Wiley & Sons, 2009 Search PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS PubMed.
- D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249–2256 RSC.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- K. Makiguchi, S. Kikuchi, K. Yanai, Y. Ogasawara, S.-i. Sato, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1047–1054 CrossRef CAS.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- A. G. W. Robert, H. Grubbs, D. J. O'Leary and E. Khosravi, Handbook of Metathesis, Wiley–VCH Verlag GmbH & Co. KGaA, 2015 Search PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- V. C. Gibson, Adv. Mater., 1994, 6, 37–42 CrossRef CAS.
- A. D. Benedicto, J. P. Claverie and R. H. Grubbs, Macromolecules, 1995, 28, 500–511 CrossRef CAS.
- S. Aksakal, R. Aksakal and C. R. Becer, Polym. Chem., 2018, 9, 4507–4516 RSC.
- H. Li, S. M. Guillaume and J.-F. Carpentier, Chem. – Asian J., 2022, e202200641 Search PubMed.
- N. M. Bingham, Z. Abousalman-Rezvani, K. Collins and P. J. Roth, Polym. Chem., 2022, 13, 2880–2901 RSC.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- C. G. Overberger and J. K. Weise, J. Am. Chem. Soc., 1968, 90, 3533–3537 CrossRef CAS.
- V. Puchelle, Y. Latreyte, M. Girardot, L. Garnotel, L. Levesque, O. Coutelier, M. Destarac, P. Guégan and N. Illy, Macromolecules, 2020, 53, 5188–5198 CrossRef CAS.
- S. Le Luyer, B. Quienne, M. Bouzaid, P. Guégan, S. Caillol and N. Illy, Green Chem., 2021, 23, 7743–7750 RSC.
- N. Illy, V. Puchelle, S. Le Luyer and P. Guégan, Eur. Polym. J., 2021, 153, 110490 CrossRef CAS.
- T. J. Bannin and M. K. Kiesewetter, Macromolecules, 2015, 48, 5481–5486 CrossRef CAS PubMed.
- S. W. Duchiron, E. Pollet, S. Givry and L. Avérous, Eur. Polym. J., 2017, 87, 147–158 CrossRef CAS.
- M. Langlais, O. Coutelier, S. Moins, J. De Winter, O. Coulembier and M. Destarac, Polym. Chem., 2018, 9, 2769–2774 RSC.
- S. Mavila, B. T. Worrell, H. R. Culver, T. M. Goldman, C. Wang, C.-H. Lim, D. W. Domaille, S. Pattanayak, M. K. McBride, C. B. Musgrave and C. N. Bowman, J. Am. Chem. Soc., 2018, 140, 13594–13598 CrossRef CAS PubMed.
- C. Shi, M. L. McGraw, Z.-C. Li, L. Cavallo, L. Falivene and E. Y.-X. Chen, Sci. Adv., 2020, 6, eabc0495 CrossRef CAS PubMed.
- J. Yuan, W. Xiong, X. Zhou, Y. Zhang, D. Shi, Z. Li and H. Lu, J. Am. Chem. Soc., 2019, 141, 4928–4935 CrossRef CAS PubMed.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS PubMed.
- D. Fleš and V. Tomašić, J. Polym. Sci., Part B: Polym. Lett., 1968, 6, 809–813 CrossRef.
- D. Fleš, V. Tomašić, M. Samsa, D. Ahmetović, B. Jerman and M. Fleš, J. Polym. Sci., Polym. Symp., 1973, 42, 321–329 CrossRef.
- B. Jerman and D. Fleš, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 1117–1125 CrossRef CAS.
- R. D. Adams, M. Huang and W. Huang, Organometallics, 1997, 16, 4479–4485 CrossRef CAS.
- M. Suzuki, K. Makimura and S.-i. Matsuoka, Biomacromolecules, 2016, 17, 1135–1141 CrossRef CAS PubMed.
- W. Xiong, W. Chang, D. Shi, L. Yang, Z. Tian, H. Wang, Z. Zhang, X. Zhou, E.-Q. Chen and H. Lu, Chem, 2020, 6, 1831–1843 CAS.
- P. Espeel, F. Goethals and F. E. Du Prez, J. Am. Chem. Soc., 2011, 133, 1678–1681 CrossRef CAS PubMed.
- M. Suzuki, A. Watanabe, R. Kawai, R. Sato, S.-i. Matsuoka and S. Kawauchi, Polymer, 2021, 215, 123386 CrossRef CAS.
- Y. Wang, M. Li, J. Chen, Y. Tao and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 22547–22553 CrossRef CAS PubMed.
- Y. Ura, M. Al-Sayah, J. Montenegro, J. M. Beierle, L. J. Leman and M. R. Ghadiri, Org. Biomol. Chem., 2009, 7, 2878–2884 RSC.
- F. Sanda, D. Jirakanjana, M. Hitomi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4057–4061 CrossRef CAS.
- F. Sanda, D. Jirakanjana, M. Hitomi and T. Endo, Macromolecules, 1999, 32, 8010–8014 CrossRef CAS.
- P. P. Datta and M. K. Kiesewetter, Macromolecules, 2016, 49, 774–780 CrossRef CAS PubMed.
- P. Yuan, Y. Sun, X. Xu, Y. Luo and M. Hong, Nat. Chem., 2022, 14, 294–303 CrossRef CAS PubMed.
- T. Takata, F. Sanda, T. Ariga, H. Nemoto and T. Endo, Macromol. Rapid Commun., 1997, 18, 461–469 CrossRef CAS.
- X. Chen, S. P. McCarthy and R. A. Gross, J. Appl. Polym. Sci., 1998, 67, 547–557 CrossRef CAS.
- K. Soga, H. Imamura and S. Ikeda, Makromol. Chem., 1975, 176, 807–811 CrossRef CAS.
- F. Sanda, J. Kamatani and T. Endo, Macromolecules, 1999, 32, 5715–5717 CrossRef CAS.
- W. Choi, F. Sanda, N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3853–3856 CrossRef CAS.
- W. Choi, F. Sanda and T. Endo, Macromolecules, 1998, 31, 2454–2460 CrossRef CAS.
- W. Choi, F. Sanda and T. Endo, Heterocycles, 2000, 52, 125–128 CrossRef CAS.
- F. Sanda, T. Shinjo, W. Choi and T. Endo, Macromol. Rapid Commun., 2001, 22, 363–366 CrossRef CAS.
- W. Choi, F. Sanda and T. Endo, Macromolecules, 1998, 31, 9093–9095 CrossRef CAS.
- S. Motokucho, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4459–4464 CrossRef CAS.
- S. Krishnamurthy, Y. Yoshida and T. Endo, Polym. Chem., 2022, 13, 267–274 RSC.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- F. N. Jones and S. Andreades, J. Org. Chem., 1969, 34, 3011–3014 CrossRef CAS.
- H. R. Kricheldorf and D.-O. Damrau, Macromol. Chem. Phys., 1998, 199, 2589–2596 CrossRef CAS.
- N. Nemoto, X. Xu, F. Sanda and T. Endo, Macromolecules, 2001, 34, 7642–7647 CrossRef CAS.
- B. Ochiai, K. Yoshii, D. Nagai and T. Endo, Macromolecules, 2004, 37, 2329–2331 CrossRef CAS.
- H. Kameshima, N. Nemoto, F. Sanda and T. Endo, Macromolecules, 2002, 35, 5769–5773 CrossRef CAS.
- N. Nemoto, Y. Ito and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 699–707 CrossRef CAS.
- M. Delepine, Bull. Soc. Chim. Fr., 1920, 27, 740 CAS.
- M. Sander, Chem. Rev., 1966, 66, 297–339 CrossRef.
- A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- S. Le Luyer, P. Guégan and N. Illy, Macromolecules, 2022, 55, 5430–5440 CrossRef CAS.
- S. Boileau, G. Champetier and P. Sigwalt, Makromol. Chem., 1963, 69, 180–192 CrossRef CAS.
- E. Nicol, C. Bonnans-Plaisance and G. Levesque, Macromolecules, 1999, 32, 4485–4487 CrossRef CAS.
- L. Wang, G. Kilcher and N. Tirelli, Macromol. Chem. Phys., 2009, 210, 447–456 CrossRef CAS.
- A. Rehor, N. Tirelli and J. A. Hubbell, Macromolecules, 2002, 35, 8688–8693 CrossRef CAS.
- J. C. Favier, S. Boileau and P. Sigwalt, Eur. Polym. J., 1968, 4, 3–12 CrossRef CAS.
- A. Nagai, N. Koike, H. Kudo and T. Nishikubo, Macromolecules, 2007, 40, 8129–8131 CrossRef CAS.
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116–15117 CrossRef CAS PubMed.
- K. Hatakeyama-Sato, H. Wakamatsu, S. Matsumoto, K. Sadakuni, K. Matsuoka, T. Nagatsuka and K. Oyaizu, Macromol. Rapid Commun., 2021, 42, 2000607 CrossRef CAS PubMed.
- A. Takahashi, R. Yuzaki, Y. Ishida and A. Kameyama, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2442–2449 CrossRef CAS.
- R. d'Arcy, A. Gennari, R. Donno and N. Tirelli, Macromol. Rapid Commun., 2016, 37, 1918–1925 CrossRef PubMed.
- M. Kuhlmann, S. Singh and J. Groll, Macromol. Rapid Commun., 2012, 33, 1482–1486 CrossRef CAS PubMed.
- X.-F. Zhu, R. Xie, G.-W. Yang, X.-Y. Lu and G.-P. Wu, ACS Macro Lett., 2021, 10, 135–140 CrossRef CAS PubMed.
- D. R. Morgan, G. T. Williams and R. T. Wragg, Eur. Polym. J., 1970, 6, 309–317 CrossRef CAS.
- C. Bonnans-Plaisance and G. Levesque, Macromolecules, 1989, 22, 2020–2023 CrossRef CAS.
- A. D. Aliev, I. P. Solomatina and B. A. Krentsel, Macromolecules, 1973, 6, 797–801 CrossRef CAS.
- E. J. Goethals, J. Macromol. Sci., Polym. Rev., 1968, 2, 73–144 CrossRef CAS.
- T. Mukaiyama, I. Kuwajima and K. Mizui, J. Org. Chem., 1966, 31, 32–34 CrossRef CAS.
- A. Nagai, T. Miyagawa, H. Kudo and T. Endo, Macromolecules, 2003, 36, 9335–9339 CrossRef CAS.
- A. Nagai, B. Ochiai and T. Endo, Chem. Commun., 2003, 3018–3019 RSC.
- D. Nagai, M. Sato, B. Ochiai and T. Endo, Macromolecules, 2004, 37, 3523–3525 CrossRef CAS.
- D. Nagai, M. Sato, B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4795–4803 CrossRef CAS.
- D. Nagai, M. Sato, B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4281–4289 CrossRef CAS.
- T.-J. Yue, M.-C. Zhang, G.-G. Gu, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 618–623 CrossRef CAS PubMed.
- L.-Y. Wang, G.-G. Gu, T.-J. Yue, W.-M. Ren and X.-B. Lu, Macromolecules, 2019, 52, 2439–2445 CrossRef CAS.
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS.
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS.
- I. Cho, S.-K. Kim and M.-H. Lee, J. Polym. Sci., Polym. Symp., 1986, 74, 219–226 CrossRef CAS.
- I. Cho and M.-H. Lee, J. Polym. Sci., Part C: Polym. Lett., 1987, 25, 309–311 CrossRef CAS.
- I. Cho and S. Y. Choi, Makromol. Chem., Rapid Commun., 1991, 12, 399–402 CrossRef CAS.
- R. A. Evans and E. Rizzardo, Macromolecules, 1996, 29, 6983–6989 CrossRef CAS.
- R. A. Evans and E. Rizzardo, Macromolecules, 2000, 33, 6722–6731 CrossRef CAS.
- S. Harrisson, T. P. Davis, R. A. Evans and E. Rizzardo, Macromolecules, 2001, 34, 3869–3876 CrossRef CAS.
- S. Yamada and Y. Goto, J. Photopolym. Sci. Technol., 2010, 23, 109–114 CrossRef CAS.
- W. J. Bailey, T. Endo, B. Gapud, Y.-N. Lin, Z. Ni, C.-Y. Pan, S. E. Shaffer, S.-R. Wu, N. Yamazaki and K. Yonezawa, J. Macromol. Sci., Part A: Pure Appl.Chem., 1984, 21, 979–995 CrossRef.
- S. Kobayashi, J. Kadokawa, S. Shoda and H. Uyama, J. Macromol. Sci., Part A: Pure Appl.Chem., 1991, 28, 1–5 CrossRef.
- S. Kobayashi, J. Kadokawa, Y. Matsumura, I. F. Yen and H. Uyama, J. Macromol. Sci., Part A: Pure Appl.Chem., 1992, 29, 243–250 CrossRef.
- R. A. Evans, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1994, 27, 7935–7937 CrossRef CAS.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- H. Huang, B. Sun, Y. Huang and J. Niu, J. Am. Chem. Soc., 2018, 140, 10402–10406 CrossRef CAS PubMed.
- W. Wang, Z. Zhou, D. Sathe, X. Tang, S. Moran, J. Jin, F. Haeffner, J. Wang and J. Niu, Angew. Chem., Int. Ed., 2022, 61, e202113302 CAS.
- N. A. Lynd, R. C. Ferrier and B. S. Beckingham, Macromolecules, 2019, 52, 2277–2285 CrossRef CAS.
- H. Huang, W. Wang, Z. Zhou, B. Sun, M. An, F. Haeffner and J. Niu, J. Am. Chem. Soc., 2019, 141, 12493–12497 CrossRef CAS PubMed.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2019, 55, 55–58 RSC.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- O. Ivanchenko, U. Authesserre, G. Coste, S. Mazières, M. Destarac and S. Harrisson, Polym. Chem., 2021, 12, 1931–1938 RSC.
- C. M. Plummer, N. Gil, P.-E. Dufils, D. J. Wilson, C. Lefay, D. Gigmes and Y. Guillaneuf, ACS Appl. Polym. Mater., 2021, 3, 3264–3271 CrossRef CAS.
- M. P. Spick, N. M. Bingham, Y. Li, J. de Jesus, C. Costa, M. J. Bailey and P. J. Roth, Macromolecules, 2020, 53, 539–547 CrossRef CAS.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- S. Hilf and A. F. M. Kilbinger, Macromolecules, 2009, 42, 4127–4133 CrossRef CAS.
- F. N. Behrendt and H. Schlaad, Polym. Chem., 2017, 8, 366–369 RSC.
- F. N. Behrendt, A. Hess, M. Lehmann, B. Schmidt and H. Schlaad, Polym. Chem., 2019, 10, 1636–1641 RSC.
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038–8041 CrossRef CAS PubMed.
- J. A. Barltrop, P. M. Hayes and M. Calvin, J. Am. Chem. Soc., 1954, 76, 4348–4367 CrossRef CAS.
- K. Endo and T. Yamanaka, Macromolecules, 2006, 39, 4038–4043 CrossRef CAS.
- M. Raeisi and N. V. Tsarevsky, J. Polym. Sci., 2021, 59, 675–684 CrossRef CAS.
- H. Tang and N. V. Tsarevsky, Polym. Chem., 2015, 6, 6936–6945 RSC.
- N. Sakai, M. Lista, O. Kel, S.-i. Sakurai, D. Emery, J. Mareda, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2011, 133, 15224–15227 CrossRef CAS PubMed.
- E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef CAS PubMed.
- J. Lu, H. Wang, Z. Tian, Y. Hou and H. Lu, J. Am. Chem. Soc., 2020, 142, 1217–1221 CrossRef CAS PubMed.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS.
- N. P. Tarasova, A. A. Zanin, E. G. Krivoborodov and Y. O. Mezhuev, RSC Adv., 2021, 11, 9008–9020 RSC.
- B. K. Bordoloi and E. M. Pearce, J. Appl. Polym. Sci., 1979, 23, 2757–2761 CrossRef CAS.
- J. F. Boscato, J. M. Catala, F. Clouet and J. Brossas, Polym. Bull., 1981, 4, 357–359 CrossRef CAS.
- B. R. Currell, A. J. Williams, A. J. Mooney and B. J. Nash, in New Uses of Sulfur, American Chemical Society, 1975, vol. 140, ch. 1, pp. 1–17 Search PubMed.
- M. Schmidt and E. Weissflog, Angew. Chem., Int. Ed. Engl., 1978, 17, 51–52 CrossRef.
- E. Weissflog and M. Schmidt, Z. Anorg. Allg. Chem., 1978, 445, 175–183 CrossRef CAS.
- S. Penczek, R. ŚLazak and A. Duda, Nature, 1978, 273, 738–739 CrossRef CAS.
- J. Wręczycki, D. M. Bieliński, M. Kozanecki, P. Maczugowska and G. Mlostoń, Materials, 2020, 13, 2597 CrossRef PubMed.
- J. Emsley and D. W. Griffiths, Phosphorus Sulfur Relat. Elem., 1980, 9, 227–230 CrossRef CAS.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- X. Liu, S. Wang, A. Wang, J. Chen, Z. Wang, Q. Zeng, W. Liu, Z. Li and L. Zhang, J. Phys. Chem. C, 2019, 123, 21327–21335 CrossRef CAS.
- P. Liu, J. M. Gardner and L. Kloo, Chem. Commun., 2015, 51, 14660–14662 RSC.
- J. Kuwabara, K. Oi, M. M. Watanabe, T. Fukuda and T. Kanbara, ACS Appl. Polym. Mater., 2020, 2, 5173–5178 CrossRef CAS.
- M. Mann, J. E. Kruger, F. Andari, J. McErlean, J. R. Gascooke, J. A. Smith, M. J. H. Worthington, C. C. C. McKinley, J. A. Campbell, D. A. Lewis, T. Hasell, M. V. Perkins and J. M. Chalker, Org. Biomol. Chem., 2019, 17, 1929–1936 RSC.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- J. A. Smith, R. Mulhall, S. Goodman, G. Fleming, H. Allison, R. Raval and T. Hasell, ACS Omega, 2020, 5, 5229–5234 CrossRef CAS PubMed.
- L.-A. Ko, Y.-S. Huang and Y. A. Lin, ACS Appl. Polym. Mater., 2021, 3, 3363–3372 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |