
Selective ring-opening polymerization of glycidyl esters: a versatile synthetic platform for glycerol-based (co)polyethers†
Shan
Liu‡
a,
Lijun
Liu‡
 a,
Yubo
Zhou
a,
Ye
Chen
a and
Junpeng
Zhao
*ab
a,
Yubo
Zhou
a,
Ye
Chen
a and
Junpeng
Zhao
*ab
aFaculty of Materials Science and Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: msjpzhao@scut.edu.cn
bGuangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, South China University of Technology, Guangzhou 510640, China
First published on 20th May 2022
Abstract
Linear polyglycerols are highly valued for their excellent hydrophilicity and biocompatibility as well as their multihydroxy nature. We report here a convenient route for the controlled synthesis of polyglycerols through ring-opening polymerization (ROP) of commercialized glycidyl butyrates (GBs). Starting from enantiopure GBs, well-defined poly(glycidyl ester)s with controlled molar mass and stereoregularity are achieved thanks to the chemoselectivity of the bicomponent metal-free catalyst that prevents both transesterification and epimerization. The pendant butyrate groups are readily cleaved by organobase-catalyzed methanolysis, yielding linear polyglycerols that inherit isotacticity and regioregularity from the parent polymers. Copolymerization of SGB and propylene oxide occurs in a random manner resulting in a series of narrowly distributed copolyethers with a precisely regulated number of pendant hydroxyl groups to afford tunable aqueous thermosensitivity. The method is further extended to construct polyglycerol-based amphiphilic and double-hydrophilic copolyethers by block copolymerization of RGB with tert-butyl glycidyl ether and ethylene oxide, respectively.
Introduction
Poly(ethylene oxide) (PEO) is the most widely used synthetic polymer in bio-related technologies in virtue of its excellent hydrophilicity and biocompatibility,1,2 but is intrinsically limited by the low degree of functionality as it usually bears a maximum of two reactive groups at the chain ends for (bio)conjugation or further derivatization.3 Linear polyglycerol (PGC, also termed polyglycidol) has also attracted much attention as another epoxide-derived hydrophilic polyether, as well as a multifunctional alternative (with one pendant hydroxymethyl group per repeating unit) to PEO.4–6 It has been reported that PGC exhibits comparable or even superior water-solubility, biocompatibility,7,8 fouling-resistant ability, etc.,9–11 as compared with PEO, and is therefore considered an ideal complement or replacement in many PEO-dependent applications.2 For instance, oligoglycerols and their fatty acid esters are approved by the FDA to be used as food and pharmaceutical additives.12,13 Moreover, the inherent multiple hydroxyl groups of PGC can be chemically modified to incorporate a rich catalogue of reactive pendant groups such as carboxyl, azide, allyl, thiol, etc., thereby facilitating the application of PGC in biomedical areas such as encapsulation, and storage and transport of biomolecules where two or more functions of the polymer are desired.4,5,14Linear PGC, however, has hardly been involved in industrial manufacturing to date (only diglycerol and triglycerol are commercially available) because the direct synthesis of linear PGC from glycidol through ring-opening polymerization (ROP) is enormously challenging (most probably impossible). The inimer nature of glycidol leads only to branched or hyperbranched polyethers.7,8 Therefore, it becomes indispensable to use hydroxyl-protected glycidol derivatives as monomers. So far, the successful synthesis of well-defined linear PGC has been accomplished by the ROP of trimethylsilyl glycidyl ether, ethoxyethyl glycidyl ether (EEGE) and other acetal-based glycidyl ethers, tert-butyl glycidyl ether (tBGE), allyl glycidyl ether, and isopropylidene glyceryl glycidyl ether followed by acid hydrolysis and/or Pd/C hydrogenation of the protective groups to liberate free pendant hydroxyl groups.4,5,15–23
Carboxylic ester is another commonly employed protection for hydroxyl groups which can be facilely and, in most cases, quantitatively formed and cleaved. Typically, the radical polymerization of vinyl acetate (VAc) followed by the alkaline hydrolysis of the pendant ester groups of poly(vinyl acetate) is a long-established and industrialized technique to obtain poly(vinyl alcohol) (PVA), the most commercially important water-soluble plastic for large-scale uses (Scheme 1a).24 As an oxygenated heterochain analog of PVA, however, linear PGC has not been obtained from ester-protected glycidol derivatives, i.e. glycidyl (carboxylic) esters. The main reason lies in that carboxylic esters easily undergo side reactions, typically transesterification, under strongly basic or (Lewis) acidic conditions that are usually used for the ROP of epoxides,25,26 which would cause impaired pendant groups and disordered polyether structures. The only successful example of synthesizing poly(glycidyl esters) may be the selective ROP of glycidyl methacrylate, affording reactive polyethers with pendant methacrylate groups which provide multiple possibilities for post-polymerization modifications and curing.27–30 Considering that glycidyl esters, including pure enantiomers, may be readily accessible by e.g. esterification of glycidol, epoxidation of allyl esters, or substitution of epichlorohydrin by sodium carboxylate,31,32 and that some are already commercially available, we are interested to pave a new route to linear PGC by exploring the selective ROP of glycidyl esters (Scheme 1b).
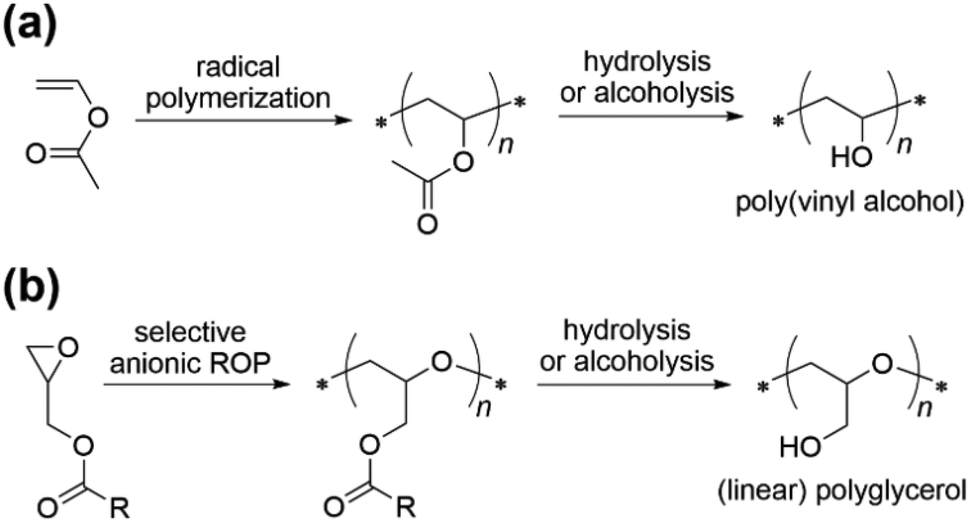 | ||
| Scheme 1 Comparative illustration of the well-established synthetic route to PVA through the radical polymerization of VAc and the route proposed in this study for linear PGC through the selective anionic ROP of glycidyl carboxylic esters. | ||
Since the initial attempts using alkylaluminum compounds for monomer-activated ROP of various epoxides by Carlotti and Deffieux,33–35 two-component Lewis pair initiators/catalysts have thrived in the last few decades. The triethylborane(Et3B)-organobase system, first reported by Feng et al.,36 has been among the most attractive ones in recent years for the ROP of epoxides and copolymerization of epoxides with non-epoxide monomers (e.g. cyclic anhydrides and compounds with cumulated double bonds).37–55 It is usually considered that Et3B, a metal-free analog to alkylaluminum, acts as both an epoxide activator and an (oxyanionic) chain-end coordinator to ensure greatly enhanced catalytic efficiency and diminish side reactions associated with high basicity and/or high nucleophilicity. It was found that such a Lewis pair with excess Et3B allows well-controlled and strictly chemoselective (transesterification-free) ROP of epoxide monomers, including ethylene oxide (EO), propylene oxide (PO), glycidyl ethers, etc., in the presence of small-molecule and/or macromolecular carboxylic esters (as the end groups, main-chain components, or solvents).56–58 We thus envisaged that such a unique character could be utilized for the chemoselective ROP of glycidyl esters with fully suppressed transesterification on the monomer and the polyether pendant groups, so as to facilitate the precise and versatile synthesis of linear PGC and other glycerol-based (hydroxymethyl-carrying) copolyethers.
Results and discussion
Enantiopure glycidyl butyrates (GBs), including (R)-(−)-glycidyl butyrate (RGB) and (S)-(+)-glycidyl butyrate (SGB), can be produced on a large scale by the enzymatic resolution of their racemic mixtures.31,59,60 Commercially available RGB and SGB are thus selected as representative glycidyl ester monomers in this study. The combination of a phosphazene base (tBuP2) and excess Et3B is employed as the bicomponent metal-free catalyst with benzyl alcohol (BA) as the initiator (Scheme 2a and Table 1).61 The ROP of RGB is conducted for the first time at room temperature (RT; 23–25 °C) in bulk with a target degree of polymerization (DP) of 70 and a low catalyst loading ([RGB]0/[BA]0/[tBuP2]/[Et3B] = 70/1/0.05/0.15) which was previously shown to lead to efficient and transesterification-free ROP of epoxides in the presence of carboxylic esters.56–58 However, the ROP proceeds much slower in this case as at least 9 days are needed to reach full monomer conversion as shown by the 1H NMR spectrum of the withdrawn crude products (entry 1 in Table 1, Fig. S1†), which is most probably due to the lower concentration of the epoxy group in bulk caused by the relatively higher molecular weight of GB and/or the higher steric hindrance of the propagating hydroxy species as compared with representative epoxide monomers (e.g. EO and PO).61 One benefit of using this bicomponent catalyst is that the catalytic activity can be finely regulated by varying the ratio and amounts of the Lewis acid and/or base.57 Indeed, adding more Et3B to increase the loaded ratio of [tBuP2]/[Et3B] from 1/3 to 1/5 leads to a markedly higher polymerization rate as the monomer is fully consumed in 56 h (entry 2 in Table 1). Full monomer conversions are also achieved in 36 and 72 h when the target DP is varied to 30 and 280, respectively (entries 3 and 4 in Table 1). Note that the overall catalyst loading is quadrupled for the highest DP to ensure an acceptable reaction time. | ||
| Scheme 2 (a) General scheme for the chemo- and regioselective ROP of GBs catalyzed by tBuP2-Et3B and initiated by BA followed by methanolysis to obtain (stereoregular) linear PGCs. (b) Synthesis of P(PO-co-RGC) with tunable aqueous thermosensitivity by the random copolymerization of PO and SGB followed by the methanolysis of the pendant butyrate groups. (c) Synthesis of amphiphilic/double-hydrophilic PSGC-b-PtBGE/PSGC-b-PEO by the block copolymerization of RGB and tBGE/EO via sequential monomer addition followed by the methanolysis of the pendant butyrate groups. | ||
| Entry | Mon. | [Mon.]0/[init.]0/[tBuP2]/[Et3B]b | Time (h) |
M
n,th![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (kg mol−1) c (kg mol−1) |
M
n,NMRd (kg mol−1) |
M
n,SECe (kg mol−1) |
Đ
Me |
|---|---|---|---|---|---|---|---|
| a Performed at RT in bulk with BA or BDM (for entries 8 and 9) as the initiator. Monomer conversion reaches 100% in all cases. b Molar feed ratio of the monomer, initiator, tBuP2, and Et3B. c Theoretical number-average molar mass calculated from the feed ratio of the monomer and initiator. d Calculated from the 1H NMR spectrum of the isolated product. e Apparent number-average molar mass and molar mass distribution determined by SEC in THF (35 °C, PS standards). f Random copolymerization of PO and SGB. g Block copolymerization of RGB and tBGE/EO by sequential monomer addition. For entry 16, SEC is performed in DMF (50 °C, PS standards). | |||||||
| 1 | R GB | 70/1/0.05/0.15 | 220 | 10.2 | 9.9 | 6.7 | 1.15 |
| 2 | R GB | 70/1/0.05/0.25 | 56 | 10.2 | 10.5 | 6.9 | 1.12 |
| 3 | R GB | 30/1/0.05/0.25 | 36 | 4.4 | 4.5 | 3.9 | 1.09 |
| 4 | R GB | 280/1/0.2/1.0 | 72 | 40.5 | 40.6 | 15.3 | 1.13 |
| 5 | S GB | 30/1/0.05/0.25 | 36 | 4.4 | 4.5 | 4.0 | 1.08 |
| 6 | S GB | 70/1/0.05/0.25 | 56 | 10.2 | 10.6 | 7.1 | 1.15 |
| 7 | S GB | 280/1/0.2/1.0 | 72 | 40.5 | 41.6 | 19.3 | 1.09 |
| 8a | R GB | 30/1/0.1/0.5 | 36 | 4.5 | 4.5 | 4.3 | 1.09 |
| 9a | R GB | 280/1/0.2/1.0 | 72 | 40.5 | 40.5 | 37.9 | 1.06 |
| 10 | racGB | 70/1/0.05/0.25 | 56 | 10.2 | 10.8 | 7.4 | 1.16 |
| 11f | PO + SGB | 95/5/1/0.1/0.4 | 18 | 6.3 | 6.4 | 9.2 | 1.06 |
| 12f | PO + SGB | 90/10/1/0.1/0.4 | 18 | 6.8 | 6.8 | 9.7 | 1.07 |
| 13f | PO + SGB | 80/20/1/0.1/0.4 | 18 | 7.6 | 7.5 | 9.8 | 1.06 |
| 14f | PO + SGB | 50/50/1/0.1/0.4 | 18 | 10.2 | 10.8 | 10.5 | 1.09 |
| 15g | R GB + tBGE | 70/70/1/0.1/0.4 | 48 + 48 | 19.3 | 18.9 | 13.0 | 1.09 |
| 16g | R GB + EO | 70/120/1/0.1/0.4 | 48 + 5 | 15.5 | 16.1 | 22.2 | 1.15 |
Size exclusion chromatography (SEC) conducted in tetrahydrofuran (THF) shows unimodal and narrow molar mass distribution (ĐM < 1.15) for lower target DPs (30 and 70), and a small shoulder towards the high-molar-mass side for the highest DP (280) most probably caused by adventitious water which acts as a difunctional initiator (Fig. 1a). The apparent number-average molar mass (Mn,SEC) continuously increases with an increase in the target DP, indicating that the molar mass of the ROP product is controlled by the feed ratio of the monomer and the initiator. The deviation of Mn,SEC from the theoretical molar mass (Mn,th) is in part due to the use of polystyrene (PS) standards for calibration. For the largest DP, the large deviation may also be caused by protic impurities such as glycidol and butyric acid, in addition to water, which add to the total amount of initiators. Similar SEC results are obtained by the ROP of SGB performed under the same conditions and using the same target DPs (entries 5–7 in Table 1; Fig. 1b).
 | ||
| Fig. 1 Left: SEC traces (RI signals, THF, 35 °C) of the crude products of the chemoselective ROP of RGB (a) and SGB (b) with different target DPs (entries 2–7 in Table 1). Right: 1H NMR spectra (in CDCl3) of the isolated PRGB (a) and PSGB (b) with a DP of 30. | ||
The well-defined structures of PRGB and PSGB are identified in 1H NMR and 13C NMR spectra which exhibit all the expected signals of the α-end benzyloxyl (derived from BA), hydroxyl-neighboring methine (at the ω-end), aliphatic ether methylenes and methines (in the polyether backbone), and pendant butyrate groups (Fig. 1 and S2, S3†). The number-average molar mass (Mn,NMR) calculated by comparison between the signal integrals of PGB main body and α-end methylene conforms well with Mn,th. The absence of the proton signals of carboxylic esters derived from secondary alcohols, e.g. –CH2CH(CH2OCOC3H7)OCOC3H7 which would have appeared at ca. 5.0 ppm,25 together with the low ĐM, rules out the possibility of transesterification between the propagating hydroxy chain end and the ester groups on the polymer pendants or the unreacted monomers even with the highest targeted DPs (Fig. S4 and S5†). Therefore, it is confirmed that the controlled and chemoselective ROP of GBs can be achieved with an acid-excess bicomponent metal-free catalyst. It is noted that the number of hydroxyl-neighboring methines at the ω-end is larger than that of BA-derived α-end methylenes according to the integrals of the corresponding proton signals, indicating the existence of impurities acting as extra initiators.
1,4-Benzenedimethanol (BDM) is used as a difunctional initiator for the ROP of RGB with a low target DP (entry 8 in Table 1). Controlled molar mass, low ĐM, and high end-group fidelity are achieved as evidenced by SEC and 1H NMR results (Fig. 2a). The matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) spectrum of the isolated product depicts a single mass population with a constant interval of 144.2 Da (the molecular weight of RGB), further verifying the achievement of the expected BDM-centered α,ω-dihydroxyl PRGB structure and that the butyrate groups remain intact during the ROP. In comparison with the SEC profile containing a shoulder peak shown by BA-initiated PGBs with the highest DP, a monomodal and narrow distribution is obtained with BDM as the initiator (entry 9 in Table 1), which is supportive of the presence of a trace amount of water acting as a difunctional initiator in the previous case (Fig. S6†). It needs to be noted that this experiment was done with thoroughly purified RGB by prolonged drying several times over CaH2 and distillation. Therefore, the distinctly increased molar mass of PRGB clearly shows that the large deviation between the measured and theoretical molar masses observed above for DP = 280 (entries 4 and 7) is due to the presence of impurities in the monomer acting as extra initiators. The racemic mixture of RGB and SGB (racGB) is also polymerized with a target DP of 70 which shows a similar reaction time and results in enantiopure monomers (entry 10 in Table 1).
 | ||
| Fig. 2 MALDI-TOF MS spectra (left), SEC traces (middle; RI signals, DMF, 50 °C, PS standards; with Mn,SEC and ĐM noted), and 1H NMR spectra (right; with Mn,NMR noted) taken in CDCl3 (upper) or DMSO-d6 (lower) of (a) BDM-initiated PRGB (entry 8 in Table 1; DP = 30) and (b) the PSGC obtained after methanolysis. Mass signals in the MALDI-TOF MS spectra correspond to polymers ionized by sodium cations. The calculated masses of (a) [BDM-RGB24]Na+ and (b) [BDM-SGC24]Na+ are 3621.2 and 1939.1 Da, respectively. | ||
Since there are two Lewis basic sites in one monomer, we are interested to understand the interactions between Et3B and the oxygen atoms in the epoxy ring and the pendant ester group by conducting DFT calculations (Fig. S7†). A minimum value is observed in a potential energy surface for the interaction between Et3B and the epoxy group, while the interaction energy of Et3B and the ester carbonyl group decreases monotonically as the atom distance increases. It is thus suggested that Et3B preferentially interacts with epoxides and the plausible conjugation between Et3B and carbonyl is unfavorable, probably due to the steric effect. Such results are consistent with our earlier assumption that the relatively slow polymerization rate with a low amount of Et3B is likely ascribed to the bulkiness and low bulk concentration of GBs, rather than a competitive interaction of Et3B by pendant ester groups.
Next, organobase-catalyzed methanolysis of PGBs is performed to liberate the pendant hydroxyl groups and afford linear PGCs (Scheme 2a). 1H NMR analysis shows that methanolysis is completed in 12 h at RT by the addition of an organobase, (1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), used in a small amount (5% of the ester groups). The SEC peak obtained in N,N-dimethylformamide (DMF), however, reveals a distinct shift to the low-elution-volume (high-molar-mass) side after methanolysis, which is probably because PGCs are more solvophilic than the parent PGBs in DMF. Narrow and unimodal distributions for all PGC samples are maintained (Fig. 2b and S8†), indicating the chemical inertness of the polyether backbone under the methanolysis conditions. Note that using DMF as the mobile phase and PS samples as the standards for PGB and PGC may have caused a considerable overestimation of their molar masses. However, DMF is the only one among commonly used SEC eluents that can well-dissolve both PGB and PGC for making comparisons before and after methanolysis. The disappearance of the signals of carbonyl-neighboring n-propyl and pendant methylene groups (–CH2OCOC3H7), the appearance of hydroxymethyl signals (–CH2OH, 3.2–3.5 ppm) generated in the 1H NMR spectrum of the isolated product, and the fitting measured molar masses (e.g., Mn,NMR = 2.4 kg mol−1, Mn,th = 2.4 kg mol−1 in Fig. 2b) further indicate the complete removal of the butyryl groups and the successful transformation of PGBs into PGCs with the same DP. The expected linear PGC structure is also confirmed by the MALDI-TOF MS spectrum which is the absence of the products caused by incomplete methanolysis or by other side reactions. Owing to the poor solubility of PGC in common organic solvents such as THF, 2-methyl THF, acetone, ethyl acetate, dichloromethane, and chloroform, the generated PGC is easily isolated by precipitation with the catalyst residuals and small-molecule byproducts (i.e. methyl butyrate), which are completely removed. Also note that similar results are obtained when 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) instead of TBD is used as the methanolysis catalyst with the same loaded amount and reaction time (Fig. S9†).
The microstructures of PGBs and the corresponding PGCs with a DP of 70 derived from RGB, SGB, and racGB (products in entries 2, 6, and 10 in Table 1) are analyzed by the use of their 13C NMR spectra. PRGB and PSGB present singlet signals for the backbone methylene and methine carbons and the pendant methylene carbon adjacent to the backbone, indicating that the epimerization of the enantiopure GBs is absent during the ROP so that highly isotactic structures are achieved. Also, such a result strongly suggests that the opening of the epoxy ring occurs selectively through sterically favored β-scission so that chiral configuration inversion is minimal.62 In this way, isotactic PGBs with stringent head-to-tail regioregularity are obtained. On the other hand, PracGB shows distinctly broader and multiple peaks for the same carbons (Fig. 3a), indicating that RGB and SGB copolymerize in a random manner leading to an atactic structure. The stereoregularity difference between PRGB/PSGB and PracGB is also implied by the forms of the isolated polymer samples. While PracGB remains as a viscous liquid under all storage conditions, PRGB and PSGB turn into white solids when stored in a fridge at 4 °C or −20 °C or even at RT with an adequate time. The (semi)crystalline feature of the isotactic PGBs is also revealed by the wide-angle X-ray diffraction profiles of the solidified samples which exhibit five strong diffraction peaks (Fig. S10†). Differential scanning calorimetry (DSC) shows glass transition temperatures (Tgs) of −57.3 °C, −55.7 °C and −63.9 °C for PRGB, PSGB and PracGB, and melting points (Tms) of 38.7 °C and 40.4 °C for PRGB and PSGB, respectively (Fig. S10†). Note that Tm is taken from the first heating run owing to the slow recrystallization of the melted PGBs. However, a stereocomplex may not be formed when PRGB and PSGB are mixed in a 1:1 mass ratio as suggested by DSC results. This is understood as a result of the bulky pendant butyrate group which hinders the ordered co-crystallization of PRGB and PSGB segments. Much effort is being made to better reveal and understand the crystallinity of stereoregular PGBs and other poly(glycidyl esters).
 | ||
| Fig. 3 (a) 13C NMR spectra (in CDCl3) of PRGB, PSGB, and PracGB with a DP of 70 (the isolated products in entries 2, 6, and 10 in Table 1, respectively). (b) 13C NMR spectra (in DMSO-d6) of the corresponding PSGC, PRGC, and PracGC obtained by TBD-catalyzed methanolysis and precipitation in THF. | ||
The stereoregularity of the parent PGBs is fully inherited by the PGCs obtained after methanolysis, as revealed by the singlet signals of PSGC/PRGC and the split signals of PracGC in their 13C NMR spectra, especially for the main-chain methine carbons (Fig. 3b). The obtained linear PSGC and PRGC, though highly stereo- and regio-regular, appear to be amorphous under all storage conditions (also indicated by DSC measurements, Fig. S11†), which is in accordance with previous reports.4,63,64 No stereocomplexation is suggested for the PRGC and PSGC blend either. In fact, the properties of stereoregular PGCs are highly under-investigated in the literature.63,64 Therefore, our method provides a useful tool to exploit this specific area, given that there are well-established synthetic routes to enantiopure GBs.31,59,60
The statistical copolymerization of PO and SGB is carried out aiming at P(PO-co-SGC)s with an adjustable number of pendant hydroxyl groups and properties. The molar feed ratio of the monomers (in total), BA, and the catalytic components ([M]0/[BA]0/[tBuP2]/[Et3B]) is fixed at 100/1/0.1/0.4 to target a constant DP of 100, while the fraction of SGB in the mixed monomer is varied to be 5%, 10%, 20%, and 50% (Scheme 2b, entries 11–14 in Table 1). Both monomers are fully consumed within 18 h in all cases. SEC presents narrow and unimodal molar mass distributions, indicating that the copolymerization is well-controlled. The expected P(PO-co-SGB) structures with molar masses and comonomer compositions agreeing well with the theoretical values are confirmed by 1H NMR data (Fig. 4 and S12–S14†). Plots of monomer conversion versus reaction time obtained from the experiment with equally loaded molar amounts of PO and SGB (entry 14 in Table 1) show that the two monomers react at comparable rates (with PO incorporated in the copolymer slightly faster) and that the molar mass of the copolyether increases steadily with a low ĐM maintained throughout the copolymerization (Fig. S15†). In contrast to PPO and PSGB homopolymers, the 13C NMR spectrum of P(PO50-co-SGB50) displays evidently broadened and split signals especially for the backbone methine and methylene carbons of the SGB units, indicating the altered chemical environment of these carbon atoms (Fig. S16†). An integral ratio of 0.26 is obtained for the SGBSGBSGB linkage, which is very close to that calculated for a perfect statistical binary copolymerization reaction ([SGBSGBSGB] = 0.25). These results support the achievement of well-defined P(PO-co-SGB) with a highly random sequence distribution of the two monomeric units.
 | ||
| Fig. 4 SEC traces (RI signals, THF, 35 °C, PS standards) and 1H NMR spectra (in CDCl3) of (a) P(PO50-co-SGB50), corresponding to entry 14 in Table 1, and (b) P(PO50-co-RGC50) obtained after methanolysis treatment. | ||
Methanolysis of the copolyethers is conducted in the same manner as described above for PGBs, with 5 mol% of TBD, relative to the ester groups, as the transesterification catalyst. A complete transformation of the pendant butyrates into hydroxymethyl groups and achievement of well-defined P(PO-co-RGC)s with total DPs and low ĐMs, and compositions of the two monomeric units inherited from the parent P(PO-co-SGB)s are confirmed by SEC and 1H NMR data (Fig. 4 and S12–S14†). While the P(PO-co-SGB) precursors do not dissolve in water regardless of temperature and SGB contents, the hydroxyl-liberated P(PO-co-RGC)s are all able to form homogeneous and transparent water solutions after being kept in a fridge (4 °C) for 2 h at a concentration of 1.0 mg mL−1. The aqueous thermosensitivity of the copolyethers is investigated by temperature-dependent light transmittance (turbidity) experiments. P(PO-co-RGC)s with the molar fractions of RGC units being 5%, 10%, and 20%, respectively, exhibit lower critical solution temperatures (LCSTs) of 15 °C, 21 °C, and 33 °C at 50% transmittance during the heating process (Fig. 5). Clearly, the hydrophilicity of the copolyether enhances as more RGC units are incorporated. The solution of P(PO-co-RGC) with 50 mol% of RGC units shows a transparent solution in the whole range of temperatures investigated with no LCST being observed. Therefore, our method fulfils the controlled synthesis of multifunctional copolyethers with randomly distributed pendant hydroxyl groups and readily tunable aqueous thermosensitivity, similar to the previously reported (cesium hydroxide)-catalyzed copolymerization of PO and EEGE followed by acid hydrolysis.65
 | ||
| Fig. 5 Temperature dependence of light (λ = 600 nm) transmittance given by the 1.0 mg mL−1 aqueous solutions of P(PO-co-RGC)s with varying molar fractions of RGC units (total DP = 100). | ||
The utility of glycidol esters for the construction of glycerol-based polyether structures is further addressed by the block copolymerization of RGB with tBGE or EO via sequential monomer addition, followed by methanolysis treatment of the PRGB block (Scheme 2c; entries 15 and 16 in Table 1). The target DP for PRGB is 70 in both cases and 70/120 for PtBGE/PEO. The livingness of the BA-initiated ROP of the epoxides, catalyzed by the acid-excess metal-free Lewis pair ([BA]0/[tBuP2]/[Et3B] = 1/0.1/0.4), and the formation of PRGB-b-PtBGE and PRGB-b-PEO diblock copolymers are demonstrated by the full conversion of the monomers and the increase of molar mass after the addition of the second monomer (Fig. 6). A protrusion to the low-molar-mass side is observed in the SEC peak of PRGB-b-PEO, which may indicate that the initiation of the ROP of EO by the secondary hydroxyl group at the PRGB end is slower than the propagation of the PEO chain with a primary hydroxy active species. This issue would be circumvented if the order of monomer addition was reversed as we observed previously in the block copolymerization of EO and PO.61 The methanolysis occurs selectively and completely on the PRGB blocks for both PRGB-b-PtBGE and PRGB-b-PEO as evidenced by SEC and 1H NMR results (Fig. S17 and S18†), leading to the expected PSGC-b-PtBGE and PSGC-b-PEO diblock copolymers with fully inherited block lengths of their precursors. The amphiphilic and double-hydrophilic natures of these PGC-based block copolymers, together with the multifunctional and biocompatible features of the PGC blocks, may forge them into useful building materials for e.g. nanosized carriers and controlled release systems.
 | ||
| Fig. 6 (a) SEC traces (THF, 35 °C, RI signals, PS standards) of the PRGB precursor and the PRGB-b-PtBGE diblock copolymer (crude products, entry 15 in Table 1). (b) SEC traces (DMF, 50 °C, RI signals, PS standards) of the PRGB precursor and PRGB-b-PEO diblock copolymer (crude products, entry 16 in Table 1). | ||
Conclusions
In summary, we have shown that commercially available and enantiopure glycidol carboxylic esters, i.e. RGB and SGB, and their racemic mixture (racGB), can undergo chemoselective ROP using an alcoholic initiator and a bicomponent Lewis pair type metal-free catalyst comprising a strong phosphazene base (tBuP2) and excess Et3B. Potential side reactions including transesterification and epimerization are fully suppressed so that well-defined and tacticity-controlled polyethers bearing one carboxylic ester pendant group per repeating unit are readily achieved. The pendant butyrate groups can be completely and selectively cleaved by methanolysis catalyzed by an organobase (TBD) to afford linear PGCs with a controlled molar mass, a low molar mass distribution, and high regio- and stereoregularity. GBs can also copolymerize with PO in a highly random manner and with tBGE/EO in a one-pot sequential manner to afford, respectively, random and block copolymers with controlled compositions. TBD-catalyzed methanolysis also occurs selectively on pendant butyrate groups for these copolymers, resulting in glycerol-based random copolyethers with tunable aqueous thermosensitivity and block copolyethers with amphiphilic and double-hydrophilic nature. Therefore, our study shows that metal-free selective ROP of GB followed by organobase-catalyzed alcoholysis can serve as a useful alternative to the previously established methods based on e.g. alkali alcoholate or (onium alkoxide)-aluminum catalyzed ROP of EEGE (an acetal-protected glycidol monomer) and acid hydrolysis of PEEGE for the synthesis of well-defined linear PGC. The commercial availability and well-established synthetic routes for GBs, especially the enantiopure ones, may facilitate the large-scale production of (stereoregular) linear PGCs. Also, the controlled and chemoselective synthesis of GB-based (co)polyethers may be extended to other glycidol esters to create a new subcatalog of aliphatic polyethers with multiple functionalities and new properties.Author contributions
S. L. and L. L. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (52022031, 21734004, 21971075) and the Fund of Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates (2019B030301003) is acknowledged.References
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- B. Obermeier, F. Wurm, C. Mangold and H. Frey, Angew. Chem., Int. Ed., 2011, 50, 7988–7997 CrossRef CAS PubMed.
- A. Thomas, S. S. Müller and H. Frey, Biomacromolecules, 2014, 15, 1935–1954 CrossRef CAS PubMed.
- H. Keul and M. Möller, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3209–3231 CrossRef CAS.
- H. Zhang and M. W. Grinstaff, Macromol. Rapid Commun., 2014, 35, 1906–1924 CrossRef CAS PubMed.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- M. Imran ul-haq, B. F. Lai, R. Chapanian and J. N. Kizhakkedathu, Biomaterials, 2012, 33, 9135–9147 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Langmuir, 2009, 25, 5703–5712 CrossRef CAS PubMed.
- M. Weinhart, I. Grunwald, M. Wyszogrodzka, L. Gaetjen, A. Hartwig and R. Haag, Chem. – Asian J., 2010, 5, 1992–2000 CrossRef CAS PubMed.
- M. Weinhart, T. Becherer, N. Schnurbusch, K. Schwibbert, H. J. Kunte and R. Haag, Adv. Eng. Mater., 2011, 13, B501–B510 CrossRef.
- H. Frey and R. Haag, Rev. Mol. Biotechnol., 2002, 90, 257–267 CrossRef CAS PubMed.
- M. Calderon, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef CAS PubMed.
- Z. Li and Y. Chau, Bioconjugate Chem., 2009, 20, 780–789 CrossRef CAS PubMed.
- M. Erberich, H. Keul and M. Möller, Macromolecules, 2007, 40, 3070–3079 CrossRef CAS.
- D. Taton, A. Le Borgne, M. Sepulchre and N. Spassky, Macromol. Chem. Phys., 1994, 195, 139–148 CrossRef CAS.
- J. Song, L. Palanikumar, Y. Choi, I. Kim, T.-Y. Heo, E. Ahn, S.-H. Choi, E. Lee, Y. Shibasaki, J.-H. Ryu and B.-S. Kim, Polym. Chem., 2017, 8, 7119–7132 RSC.
- E. Hwang, K. Kim, C. G. Lee, T.-H. Kwon, S.-H. Lee, S. K. Min and B.-S. Kim, Macromolecules, 2019, 52, 5884–5893 CrossRef CAS.
- J. Baek, M. Kim, Y. Park and B.-S. Kim, Macromol. Biosci., 2021, 21, 2100251 CrossRef CAS PubMed.
- A. Dworak, I. Panchev, B. Trzebicka and W. Walach, Macromol. Symp., 2000, 153, 233–242 CrossRef CAS.
- M. Gervais, A.-L. Brocas, G. Cendejas, A. Deffieux and S. Carlotti, Macromolecules, 2010, 43, 1778–1784 CrossRef CAS.
- M. Gervais, A.-L. Brocas, G. Cendejas, A. Deffieux and S. Carlotti, Macromol. Symp., 2011, 308, 101–111 CrossRef CAS.
- F. Wurm, J. Nieberle and H. Frey, Macromolecules, 2008, 41, 1909–1911 CrossRef CAS.
- V. Goodship and D. Jacobs, Polyvinyl alcohol: materials, processing and applications, Smithers rapra technology, 2009 Search PubMed.
- S. Hu, J. Zhao and G. Zhang, ACS Macro Lett., 2015, 5, 40–44 CrossRef.
- J. P. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 1693–1698 CrossRef CAS.
- A. L. Labbé, A.-L. Brocas, E. Ibarboure, T. Ishizone, A. Hirao, A. Deffieux and S. P. Carlotti, Macromolecules, 2011, 44, 6356–6364 CrossRef.
- M. T. Muñoz, M. Palenzuela, T. Cuenca and M. E. Mosquera, ChemCatChem, 2018, 10, 936–939 CrossRef.
- N. Bicak and B. Karagoz, Polym. Bull., 2006, 56, 87–93 CrossRef CAS.
- B. Karagoz and N. Bicak, Eur. Polym. J., 2008, 44, 106–112 CrossRef CAS.
- Y. J. K. Araujo, N. P. Avvari, D. R. Paiva, D. P. D. Lima and A. Beatriz, Tetrahedron Lett., 2015, 56, 1696–1698 CrossRef CAS.
- P. Muppa, R. Postma, C. Schoolderman, S. Rens and K. Stoitsas, U.S. Patent No9174955, 2015 Search PubMed.
- A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS.
- S. P. Carlotti, A. L. Labbé, V. Rejsek, S. P. Doutaz, M. Gervais and A. Deffieux, Macromolecules, 2008, 41, 7058–7062 CrossRef CAS.
- A. Labbé, S. Carlotti, C. Billouard, P. Desbois and A. Deffieux, Macromolecules, 2007, 40, 7842–7847 CrossRef.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- C.-J. Zhang, H.-Y. Duan, L.-F. Hu, C.-H. Zhang and X.-H. Zhang, ChemSusChem, 2018, 11, 4209–4213 CrossRef CAS PubMed.
- S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Nat. Commun., 2019, 10, 293–301 CrossRef PubMed.
- J.-L. Yang, H.-L. Wu, Y. Li, X.-H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- L.-F. Hu, C.-J. Zhang, H.-L. Wu, J.-L. Yang, B. Liu, H.-Y. Duan and X.-H. Zhang, Macromolecules, 2018, 51, 3126–3134 CrossRef CAS.
- N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2019, 52, 2431–2438 CrossRef CAS.
- M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 1593–1598 CrossRef CAS PubMed.
- M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, ACS Macro Lett., 2019, 8, 1594–1598 CrossRef CAS PubMed.
- Z. Chen, J.-L. Yang, X.-Y. Lu, L.-F. Hu, X.-H. Cao, G.-P. Wu and X.-H. Zhang, Polym. Chem., 2019, 10, 3621–3628 RSC.
- M. Jia, D. Zhang, G. W. de Kort, C. H. R. M. Wilsens, S. Rastogi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 5297–5307 CrossRef CAS PubMed.
- V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS.
- C.-J. Zhang, S.-Q. Wu, S. Boopathi, X.-H. Zhang, X. Hong, Y. Gnanou and X.-S. Feng, ACS Sustainable Chem. Eng., 2020, 8, 13056–13063 CrossRef CAS.
- L.-F. Hu, D.-J. Chen, J.-L. Yang and X.-H. Zhang, Molecules, 2020, 25, 253–263 CrossRef CAS PubMed.
- B. Zhang, H. Li, H. Luo and J. Zhao, Eur. Polym. J., 2020, 134, 109820 CrossRef CAS.
- H. Li, G. He, Y. Chen, J. Zhao and G. Zhang, ACS Macro Lett., 2019, 8, 973–978 CrossRef CAS PubMed.
- H.-Y. Ji, D.-P. Song, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2019, 21, 6123–6132 RSC.
- N. Patil, S. Bhoopathi, V. Chidara, N. Hadjichristidis, Y. Gnanou and X. Feng, ChemSusChem, 2020, 13, 5080–5087 CrossRef CAS PubMed.
- J. K. Varghese, N. Hadjichristidis, Y. Gnanou and X. Feng, Polym. Chem., 2019, 10, 3764–3771 RSC.
- G. W. Yang, Y. Y. Zhang, R. Xie and G. P. Wu, Angew. Chem., Int. Ed., 2020, 59, 16910–16917 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-W. Yang and G.-P. Wu, Synlett, 2022, 33, 8–15 CrossRef CAS.
- Y. Chen, S. Liu, J. Zhao, D. Pahovnik, E. Žagar and G. Zhang, ACS Macro Lett., 2019, 8, 1582–1587 CrossRef CAS PubMed.
- S. Liu, T. Bai, K. Ni, Y. Chen, J. Zhao, J. Ling, X. Ye and G. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478–15487 CrossRef CAS PubMed.
- S. Liu, L. Liu, Y. Chen and J. Zhao, Chem. Commun., 2020, 56, 12186–12189 RSC.
- W. E. Ladner and G. M. Whitesides, J. Am. Chem. Soc., 1984, 106, 7250–7251 CrossRef CAS.
- A. L. Margolin, Enzyme Microb. Technol., 1993, 15, 266–280 CrossRef CAS PubMed.
- Y. Chen, J. Shen, S. Liu, J. Zhao, Y. Wang and G. Zhang, Macromolecules, 2018, 51, 8286–8297 CrossRef CAS.
- W.-M. Ren, T.-J. Yue, X. Zhang, G.-G. Gu, Y. Liu and X.-B. Lu, Macromolecules, 2017, 50, 7062–7069 CrossRef CAS.
- A. Haouet, M. Sepulchre and N. Spassky, Eur. Polym. J., 1983, 19, 1089–1098 CrossRef CAS.
- C. C. Price and E. J. Vandenberg, Coordination polymerization, Plenum Press, New York, 1983 Search PubMed.
- M. Schömer and H. Frey, Macromolecules, 2012, 45, 3039–3046 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional SEC traces, and NMR spectra. See DOI: https://doi.org/10.1039/d2py00551d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |