
Stereochemistry and stoichiometry in aliphatic polyester photopolymers for 3D printing tailored biomaterial scaffolds†
S. L.
Brooks
a,
E. J.
Constant
b,
O. M.
King
c and
A. C.
Weems
 *abcd
*abcd
aDepartment of Mechanical Engineering, Ohio University, Athens, OH 45701. E-mail: weemsac@ohio.edu
bBiomedical Engineering Program, Ohio University, Athens, OH 45701
cDepartment of Molecular and Chemical Biology, Ohio University, Athens, OH 45701
dDepartment of Orthopedic Musculoskeletal and Neurological Institute, Translational Biosciences, Ohio University, Athens, OH 45701
First published on 12th March 2022
Abstract
Stereoselective aliphatic polyesters were synthesized through the ring opening copolymerization of cyclic anhydrides and epoxides using a tin catalyst to yield Mn ∼ 10–13 kDa macromolecules (Đ < 1.6). Isomerization of the cis poly(maleate-co-phenyl glycidol ether) (PMPGE) to the trans isomer poly(fumarate-co-phenyl glycidol ether) (PFPGE) may be used to tune physical properties such as viscosity, which displays an order of magnitude increase as PMPFE is isomerized to PFPGE. However, the formulated photopolymer resins consisting of a 4-arm thiol and the polyester photopolymer display Newtonian fluid behavior and viscosities of ∼0.5 Pa s, ideal for digital light processing (DLP) 3D printing. Enhancement of the thermal, thermomechanical, mechanical, and gravimetric properties was achieved through off-stoichiometric imbalances in the photosets. For example, with a 20% excess of alkene both photopolymers display gelation times comparable with commercial resins (∼5 s) and were 3D printed via DLP into complex porous tissue scaffolds. Finally, cytocompatibility with murine macrophages over 7 days demonstrated superior material surfaces compared to control tissue culture polystyrene, as determined by statistically increased aspect ratios (cell spreading) and proliferation, indicating the biomedical application potential of these materials.
Introduction
Aliphatic polyesters have been utilized as biomaterials due to their tunable physical properties suitable for a range of applications including orthopedic implants, wound repairing scaffolds, and regenerative healing guides, with additional added benefits including non-toxic degradable byproducts and hydrolytic degradability.1–5 The advent of ring opening polymerization (ROP) provided advantages over other synthetic routes for polyester production, including superior molecular weight and distribution control, mild reaction conditions, no by-product formation, and industrial scalability as demonstrated with poly(lactic acid), although these materials display limited functionality and post-processing flexibility.6 More recently, an important subset of ROP has been developed, leveraging the ring opening copolymerization (ROCOP) of cyclic anhydrides and epoxides which has broadened the range of applicable monomers as well as the composition of the resultant polymeric backbones. ROCOP is especially important as it provides the opportunity for installation of various functional groups, including allyl ethers and alkenes, while simultaneously enhancing degradation rate tailoring and maintaining cyto- and biocompatibility.7An additional benefit of ROCOP is the opportunity to leverage stereochemistry in certain resultant polyesters, which is of interest as different isomers may display different mechanical behaviors or reactivities.2,8,9 For example, maleate-containing polyesters may be isomerized to the fumarate (trans) isomer state, which has been demonstrated to increase the elastic modulus from 25 MPa to 66 MPa, the ultimate strength from 23 MPa to 39 MPa, and even alter crystallinity and thermal transitions.10,11 Other studies have focused on the processability of such formulations, where poly(fumarate) photopolymers are used preferentially over poly(maleate) polyesters in free radical crosslinking due to reactivity restraints, as noted by significant works from the Becker and Mikos groups.12–19
While poly(fumarate) copolymers, as well acrylate-, methacrylate- and epoxide-containing polymers, are commonly exploited photoset materials for free radical crosslinking, alternative routes may be more suitable for biomaterials applications. For example, acrylates have been linked with toxicity concerns, particularly concerning residual functional groups post-crosslinking.20,21 Thiol–ene “click” photochemistry is an advantageous alternative due to: (1) its bio-orthogonality; (2) the selective, efficient, mild, and robust nature of the reactions; (3) the broad library of compatible materials. However, the utilization of thiol–ene photoclick has been predominantly limited to post-fabrication functionalization or solution polymerizations.22–25 In spite of this, thiol–ene-type photopolymerizations display considerable promise in 3D printing of photopolymer resins, particularly when considering the flexibility of the chemistry which allows for leveraging off-stoichiometric ratios of resultant photosets in a controlled manner, unlike free radical photocrosslinking.26–28 Creating off-stoichiometric ratios in thiol–ene photopolymer resins would provide avenues towards tailoring surface chemistry post-3D printing as a means of controlling cellular response, and could additionally tailor bulk physical and advanced properties including shape memory (4D printable) materials, a field of rapidly growing interest for biomedicine.23,29–31 This off-stoichiometric approach has even been utilized for incorporating spatial components, allowing for a delayed final material cure after fabrication.32
In this study, we demonstrate the bulk synthesis and isomerization of poly(maleate-co-phenyl glycidol ether) (PMPGE) to poly(fumarate-co-phenyl glycidol ether) (PFPGE) for thiol–ene photopolymerization-type digital light processing (DLP) 3D printing which, unlike previous work with free radical photocrosslinking limited to PFPGE, may be used to produce both PMPGE and PFPGE photosets under similar conditions (Fig. 1). The stereochemistry may be controlled as a means of tuning physical properties of both the photopolymers and the resultant photoset 3D scaffolds. This is demonstrated using rheological, thermomechanical, and degradation studies, as well as photoreactivity and 4D behaviors (shape memory responsiveness) of the DLP-printed scaffolds. With the multiple layers of property tailoring available in this system, it is anticipated that these polyesters will be of significant interest in biomedicine, additive manufacturing, and performance polymers.
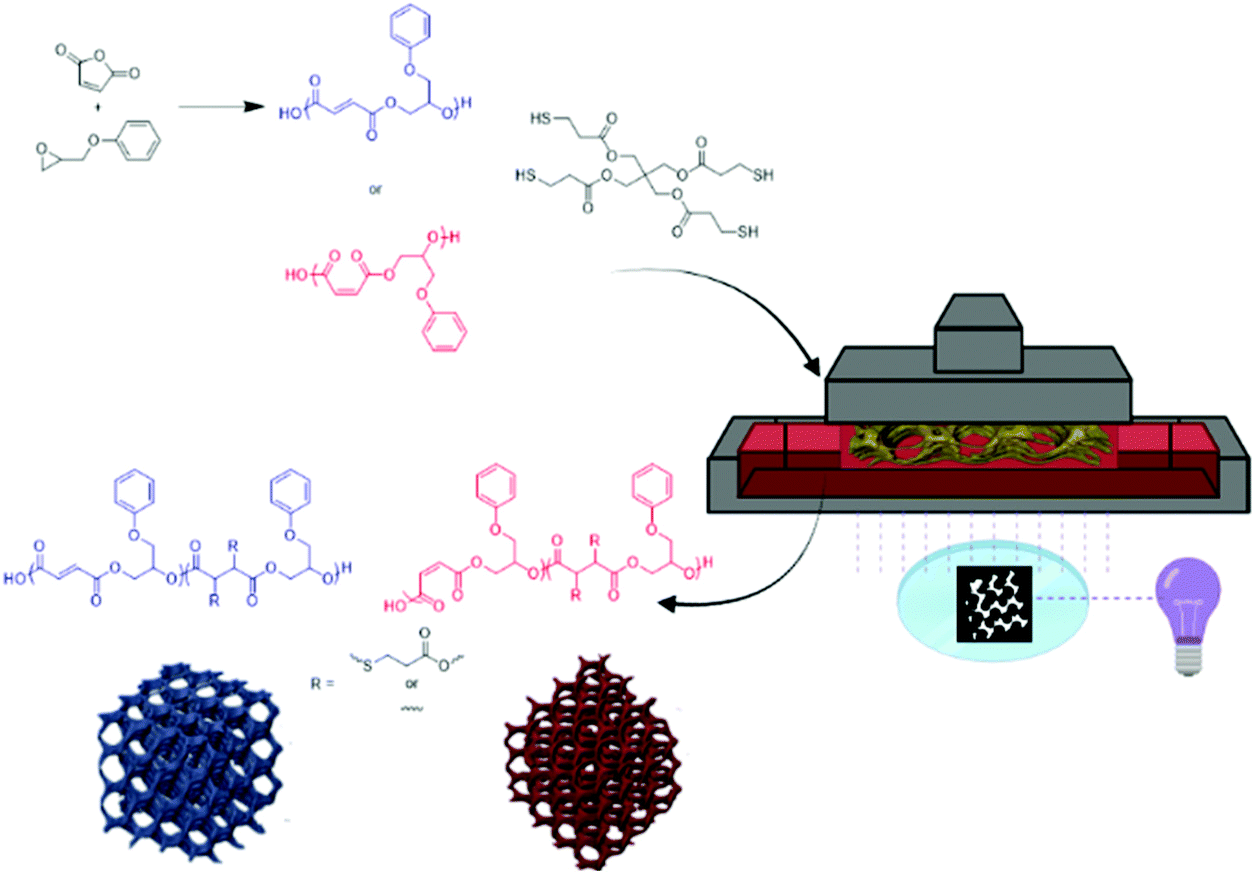 | ||
| Fig. 1 General schematic for the ROCOP for polyester photopolymers, which are subsequently used in DLP-type 3D printing of porous tissue scaffolds. | ||
Results
The ROCOP of maleic anhydride and phenyl glycidol ether (120 °C, 48 h), catalyzed by stannous octoate, yielded the cis isomer polyester PMPGE (Fig. 2A) which was then isomerized in the presence of diethylamine (HNEt2) to the trans isomer, PFPGE. The resultant polyesters display molecular weights between 9.2 kDa and 12.8 kDa, with dispersity ranging from 1.5–1.6 (Fig. 2B, and Table S1†). Polymerization and isomerization were confirmed by NMR and FT-IR (Fig. S1–S3†) using the characteristic shift in the alkene protons from 6.6 ppm to 6.3 ppm indicated in previous studies.12,33 Importantly, UV-vis spectroscopy of the polymers, which appears a deep red color, displayed a shift in the absorbance peak and full width half max (FWHM) with the PMPGE peak centered at 303 nm (FWHM = 20 nm) and the PFPGE peak centered at 309 nm (FWHM = 41 nm) (Fig. S4, and Table S2†). This behavior is promising for enhancing 3D printing spatial resolution and photopolymerization control using in situ inhibition, without requiring additional additives as demonstrated previously.26,27 | ||
| Fig. 2 Synthetic schematic of PMPGE through the ring opening copolymerization of maleic anhydride and phenyl glycidol ether and the subsequent isomerization into PFPGE (A). Respective SEC traces of PMPGE and PFPGE (B). Rheological characterization of PMPGE and PFPGE polymer viscoelasticity via an oscillatory temperature sweep (C, angular frequency of 10.0 rad s−1, range 5 °C to 195 °C at a rate of 2 °C min−1). | ||
The polyester thermal and viscoelastic properties, including viscosity and photo-crosslinking gelation kinetics, were characterized via rheology (Fig. 2C, Fig. S5, and Table S3†). Generally, the PFPGE photopolymer displays more robust thermal and viscoelastic behaviors relative to the PMPGE, as noted by significantly higher viscosity (η = 2.7 × 106 pa s) and complex moduli values (E* = 205 ± 6.23 Pa) compared to the PMPGE (η = 9.7 × 104 pa s, E* = 120 ± 5.56 Pa) at their respective flow temperatures (TF) (66.9 °C and 92.2 °C for PMPGE and PFPGE, respectively) when examined using uniaxial rotational shear, similar to previously reported results.10,15 The glassy nature of the PFPGE compared with the less viscous PMPGE is demonstrated by the difference in complex viscosity (η*), which is distinct until after the flow temperature at 157.5 °C, where both materials display similar storage moduli values.
The polyesters were then used to produce photopolymer resins for thiol–ene photochemistry, achieved by mixing different stoichiometric amounts of the polyester with an allyl-containing crosslinker, 1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (trione), and the 4-armed thiol pentaerythritol tetrakis(mercaptopropionic acid) (PETMP) in the presence of a commercial photoinitiator Irgacure 819. The stoichiometric balance was varied from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (alkene:thiol) up to 30:1 (the contribution of alkenes from the reactive diluent trione is accounted for in this ratio), as well as testing the free radical crosslinking in the absence of thiol (W/O Thiol). Rheologically, the 1:1 and 30:1 photopolymer resins of both isomers display primarily Newtonian fluid behavior in the examined range of 0.1–1000 s−1 yielding resins with consistently low viscosity (η = 0.5 Pa s) suitable for DLP-type 3D printing regardless of PETMP concentration (Fig. S6†) or stoichiometry ratio. Furthermore, the off-stoichiometric ratios could be used to tune photo-crosslinking gelation kinetics, where the photoresin storage moduli (G′) (Fig. 3B) dramatically increased (0.5 Pa to 1 × 106 Pa), as did other viscoelastic properties, as a function of irradiation time (Fig. S6, and Table S4†). Increasing the stoichiometric excess of alkene decreased the time to G′/G′′ crossover from 132.4 ± 3.6 s (1:1) to 8.7 ± 0.5 s (20:1) for PMPGE and 15.0 ± 8.8 s (1:1) to 5.6 ± 4.5 s (20:1) for PFPGE. At ratios exceeding the 20:1, the time to gelation increased by 36–107% for PMPGE and 55–166% for PFPGE.
:1 ratio (alkene:thiol) up to 30:1 (the contribution of alkenes from the reactive diluent trione is accounted for in this ratio), as well as testing the free radical crosslinking in the absence of thiol (W/O Thiol). Rheologically, the 1:1 and 30:1 photopolymer resins of both isomers display primarily Newtonian fluid behavior in the examined range of 0.1–1000 s−1 yielding resins with consistently low viscosity (η = 0.5 Pa s) suitable for DLP-type 3D printing regardless of PETMP concentration (Fig. S6†) or stoichiometry ratio. Furthermore, the off-stoichiometric ratios could be used to tune photo-crosslinking gelation kinetics, where the photoresin storage moduli (G′) (Fig. 3B) dramatically increased (0.5 Pa to 1 × 106 Pa), as did other viscoelastic properties, as a function of irradiation time (Fig. S6, and Table S4†). Increasing the stoichiometric excess of alkene decreased the time to G′/G′′ crossover from 132.4 ± 3.6 s (1:1) to 8.7 ± 0.5 s (20:1) for PMPGE and 15.0 ± 8.8 s (1:1) to 5.6 ± 4.5 s (20:1) for PFPGE. At ratios exceeding the 20:1, the time to gelation increased by 36–107% for PMPGE and 55–166% for PFPGE.
 | ||
| Fig. 3 Representative PFPMGE network structure after thiol–ene “click” crosslinking (A), photorheological analysis of PMPGE photopolymer resins storage moduli a function of irradiation time (E, angular frequency of 10.0 rad s−1, 25 °C, 405 nm wavelength, 20 W) (B). MAS multi-CP solid state 13C NMR spectra of photoset networks (C), comparing photocured and photothermally cured 30:1 PFPGE materials with photothermally cured 1:1 materials and the residual alkenes in the photoset materials during photoclick crosslinking as a function of irradiation time (determined from solution state NMR) (D). | ||
Spectroscopic analysis, conducted using both solution 1H NMR of 1:1 stoichiometrically balanced photoresins (1% Irgacure, irradiated at 405 nm, ambient conditions) and solid state (13C) NMR of solidified polymer films thermally or photothermally treated (comparing 1:1 and 30:1 stoichiometries), was used to probe residual alkenes and stereochemistry after the crosslinking and thermal treatment. Examination of the alkene regions of the photoresins during crosslinking (Fig. 2C, Fig. S7, and Table S5†) confirmed that the PMPGE displayed almost no free radical crosslinking, indicating that all gelation would be occurring as a result of thiol–ene crosslinking with ∼50% of alkenes consumed with seconds of exposure. Similarly, the PFPGE displayed a preference for thiol–ene crosslinking, with more than ∼80% of the alkenes consumed in the same time. The PFPGE films therefore could display free radical crosslinking in higher off-stoichiometries, as indicated by the new carbonyl peak at ∼175 ppm and carbonyl shoulder (extending to ∼178 ppm) in 13C NMR (Fig. 3D, and Fig. S8†). The 1:1 materials display two peaks associated with the ester carbonyl in the polyester backbone (173 ppm) and the ester linkages in the PETMP monomer (161 ppm). Photocuring the 30:1 sample produced a third carbonyl peak (167 ppm), likely associated with the unreacted backbone-ester linkages. Importantly, significant amounts of the free radically-crosslinked polyester species was found only after photothermally curing the samples, indicating that the photocrosslinking will be dominated by the thiol–ene reactions and the off-stoichiometry ratios will remain mostly fixed, likely as a result of the network formation preventing additional chain diffusion and crosslinking even with prolonged irradiation.
Thermal analysis of the photosets was performed using dynamic mechanical analysis (DMA) and differential scanning calorimetery (DSC). The differences in physical properties are likely linked with chain flexibility, where the thermoplastic PFPGE displayed a higher Tg (17.4 °C) while PMPGE displayed a Tg of 27.0 °C (Tgs determined by DSC).12 The PMPGE's Tg range from of 47.7 ± 2.7 °C (1:1) to a Tg of 81.0 ± 1.4 °C (30:1) (Fig. 4A) was broader compared to the PFPGE thermosets’ Tg, from 49.6 ± 1.0 °C (1:1) 59.7 ± 0.6 °C (30:1) (Fig. S9†). Close to the 1:1 stoichiometries, the thiol–ene crosslinking will result in similar networks between the two stereoisomer polyesters, hence the similar Tgs. At higher off-stoichiometry ratios, the additional free radical crosslinking between the fumarate moieties will alter network rigidity. DMA revealed a smaller Tg range for the PFPGE materials, with the 1:1 species displaying a Tg of 65.6 °C (tanδ peak, Fig. 4B) compared to PMPGE's Tg of 70.8 °C, while the PFPGE 30:1 thermosets displayed Tgs ∼85 °C. The range of the PMPGE Tgs extended to ∼115 °C, reflecting the broader range displayed during DSC analysis.
 | ||
| Fig. 4 Representative DSC thermograms (3rd cycle) of PMPGE materials with Tgs labeled (A) and DMA of tanδ as a function of temperature, comparing select PMGPE And PFPGE samples. DSC samples were heated at 10 °C min−1, DMA samples were heated at 2 °C min−1 (B). (n = 3). | ||
The off-stoichiometric ratios were generally found to increase in the Tgs more significantly compared with stereoisomer species, while viscoelasticity was seemingly more impacted by isomer species. The stoichiometric shift from 1:1 to 2:1 increased the Tg by 7–10 °C (Fig. S9 and Table S6†), and nearly a 60 °C increase for the shift from 1:1 and 30:1 (PMPGE) as measured by DSC. Viscoelastic properties were found to increase ∼2–3× with the isomerization of PMPGE to PFPGE (compared at respective Tgs). There was a significant jump in the storage and loss moduli of the 1:1 and 2:1 PFPGE photosets, but further increasing the off-stoichiometric ratios did not demonstrate the same significant enhancement as found with the Tgs.
Mechanical properties were probed for the photosets using uniaxial, monotonic tensile testing to failure at ambient conditions with 3D printed dogbone samples (Fig. 5A, and Fig. S10†). PMPGE samples displayed lower average elastic moduli (Fig. 5B), ultimate tensile strengths (UTS), and toughness compared with PFPGE, with no significant differences in strain at break (Table S7†). Importantly, increasing the off-stoichiometric ratios had a significant impact in the PMPGE materials while only marginally increasing mechanical properties in the PFPGE compositions. This is likely due to the presence of carbon–carbon bond formation in the PFPGE formulations, while the PMPGE materials will display behaviors more indicative of the role of residual alkenes. As a function of the resultant Tgs, a noticeable trend appears for the elastic moduli and the ultimate tensile strengths that is not found for the toughness and strain at break (Fig. S10†). With increasing off-stoichiometry, the elastic moduli is approximately doubled with the UTS is nearly tripled for the PMPGE. PFPGE materials, whose range is narrower as a result of the narrower resultant Tgs, display tunability only marginal tunability for these behaviors, as well. These results demonstrate an opportunity to directly tune the elastic modulus and UTS of the material system without statistically altering the toughness or strain at break simply through stereochemistry and stoichiometry control.
 | ||
| Fig. 5 Representative uniaxial tensile curves for PMPGE thermoset formulations (A). Box-and-whisker plots of PMPGE and PFPGE photoset elastic moduli (B) as well as elastic moduli as a function of Tg (C) and ultimate tensile strength as a function of Tg (D). Modified ASTM Type IV dogbones were tested at 5 mm min−1 at ambient atmosphere and temperature (n = 7). | ||
Both PMPGE and PFPGE photopolyester formulations, in stoichiometric and off-stoichiometric ratios, have the capability to be accurately and efficiently 3D printed via DLP with high resolution into complex porous tissue scaffolds (Fig. 6A and B) similar to those reported previously.26 The 4D behavior of the polyester scaffolds was tunable using either isomer species or off-stoichiometric ratios (Fig. S11†). In general, the increase in off stoichiometry resulted in decreased shape recovery rates, as did the isomerization from PMPGE to PFPGE (Fig. 6C and D). This is likely due to an interplay of the Tg and polymer backbone interactions. Additionally, all of the photosets displayed excellent strain fixation properties having strain recovery values less than 10% over the course of twelve hours at room temperature. This combination of tunable 4D behavior without a loss of strain fixation indicates that shape recovery times and conditions could be tailored across biomedical applications without a loss of performance.
 | ||
| Fig. 6 CAD rendering of the porous tissue scaffold (A), with the representative microCT image of 3D printed scaffolds of PMPGE (B) and PFPGE (C) 1:1, 3D printed via DLP. (Scale bar = 1 mm) (n = 5). (C) Representative PMPGE 1:1 4D printed scaffold during shape recovery at 50 °C; the kinetic response of strain recovery across the temperature range (D) (n = 3). | ||
Thermogravimetric analysis revealed that there is no variation in decomposition temperature for the off-stoichiometric ratios of the thermoset films, and the only significant decrease in the thermal decomposition temperature was found in the free-radical thermosets that did not contain PETMP (Fig. S12†). As the off-stoichiometric ratios increase, there is a relationship with the hydrolytic degradation rates consistent across the entire examined stoichiometry range (Fig. 7). Altering the stereoisomer state of the thermosets along with the off-stoichiometric ratios provides hydrolytic mass loss rate tunability. The PFPGE thermosets have a slower degradation rate than the PMPGE films, confirmed by gravimetric studies using accelerated and real time in vitro conditions for biomaterials (Fig. S13†). The PMPGE formulations, increasing the ratio from 1:1 to 1.1:1 and 1.5:1 speeds the degradation rate but when the ratio is increased with the 10:1 and W/O thiol formulations, the degradation is slowed significantly. This is then reversed again as the PMPGE 30:1 degraded along the rates of the lower ratios. With the PFPGE formulations, this trend is not repeated. As the off-stoichiometric ratio increases with the PFPGE formulations, so does the degradation rates as the 1:1, 1.1:1, and 1.5:1 ratios are fully degraded after 111 days while the 10:1, and W/O thiol formulations still have no significant mass loss. The PFPGE 30:1 formulation begins a downward trend in mass loss before the PFPGE 10:1, further solidifying what has been shown that increasing the off-stoichiometric rates of the thermosets is only beneficial to an extent, there is a point of diminishing returns.
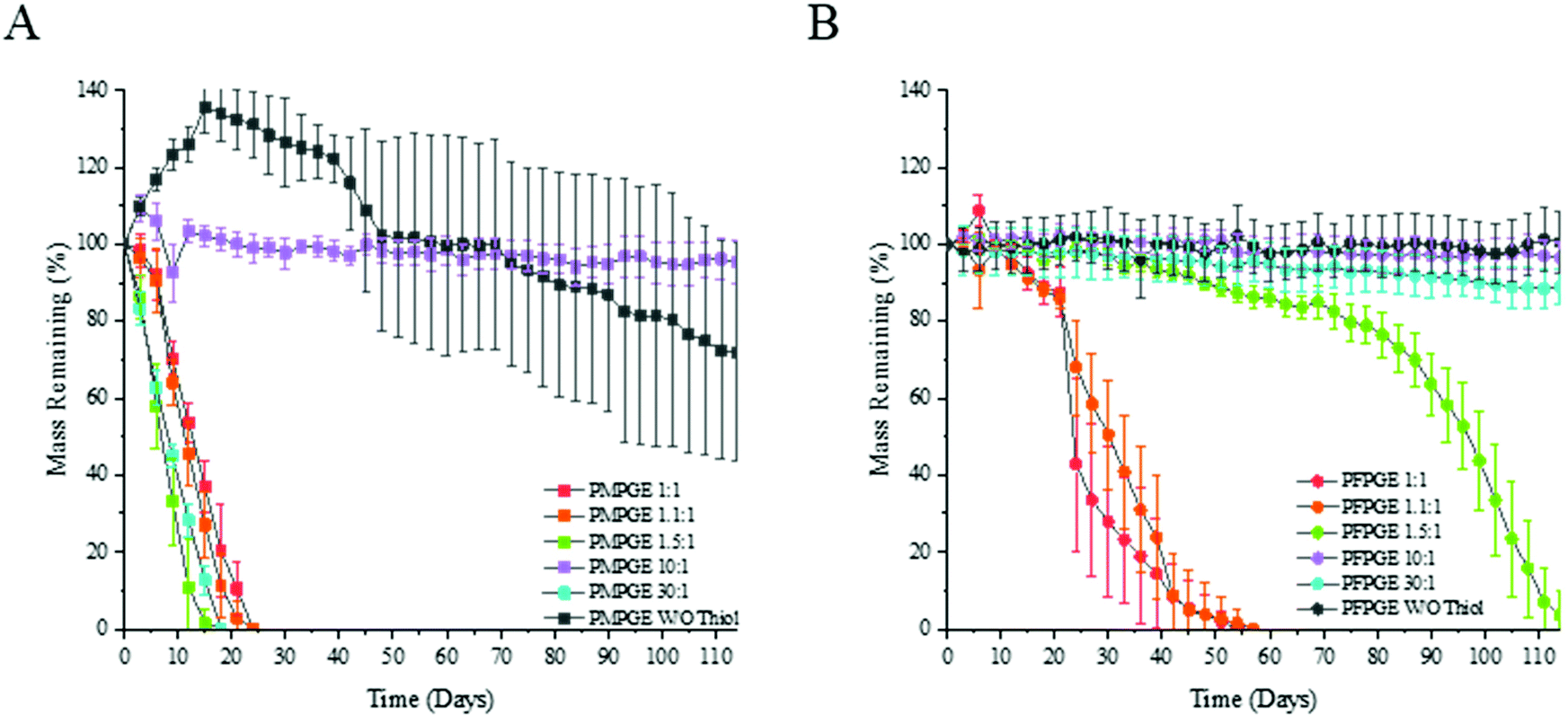 | ||
| Fig. 7 Accelerated gravimetric analysis of the 1:1, 1.1:1, 1.5:1, 10:1, 30:1, and without thiol crosslinker off stoichiometric ratios of the PMPGE (A) and PFPGE (B) photosets, with testing conducted at 37 °C, 1 Hz shaking, solution concentration 0.1 M NaOH (n = 7). | ||
To determine the cytocompatibility of the photoset PFPGE and PMPGE materials, both the 1:1 and 30:1 stoichiometric ratio were examined over 7 days using spin coated 2D films seeded with adherent RAW 264.7 murine macrophages. Macrophage proliferation was quantified using a MTS assay supplemented with cell counting, demonstrating cell multiplication on both isomer surfaces (Fig. 8A). Morphologically, macrophages cultured on glass had a mean aspect ratio of 1.92 on Day 2 and 1.39 on Day 7, indicating the cells did not spread on the surface but became more rounded (Fig. 8B–F, and Fig. S14†). By comparison, the mean aspect ratio of PMPGE 1:1 was 2.26 on Day 2 and 2.03 on Day 7, PMPGE 30:1 was 2.45 on Day 2 and 2.26 on Day 7. The mean aspect ratio of PFPGE 1:1 was 1.34 on Day 2 and 2.38 on Day 7, PFPGE 30:1 was 2.42 on Day 2 and 1.92 on Day 7, and all formulations display a statistically significant difference compared to the controls. Of note, the PMPGE materials display no statistical difference in aspect ratio over a 5 day period, indicating a superior cytocompatibility over the control and other polyester formulation, and a balanced stoichiometry resulted in greater filopodial extensions, indicating two methods for enhancing cytocompatibility in the photoset surfaces.34
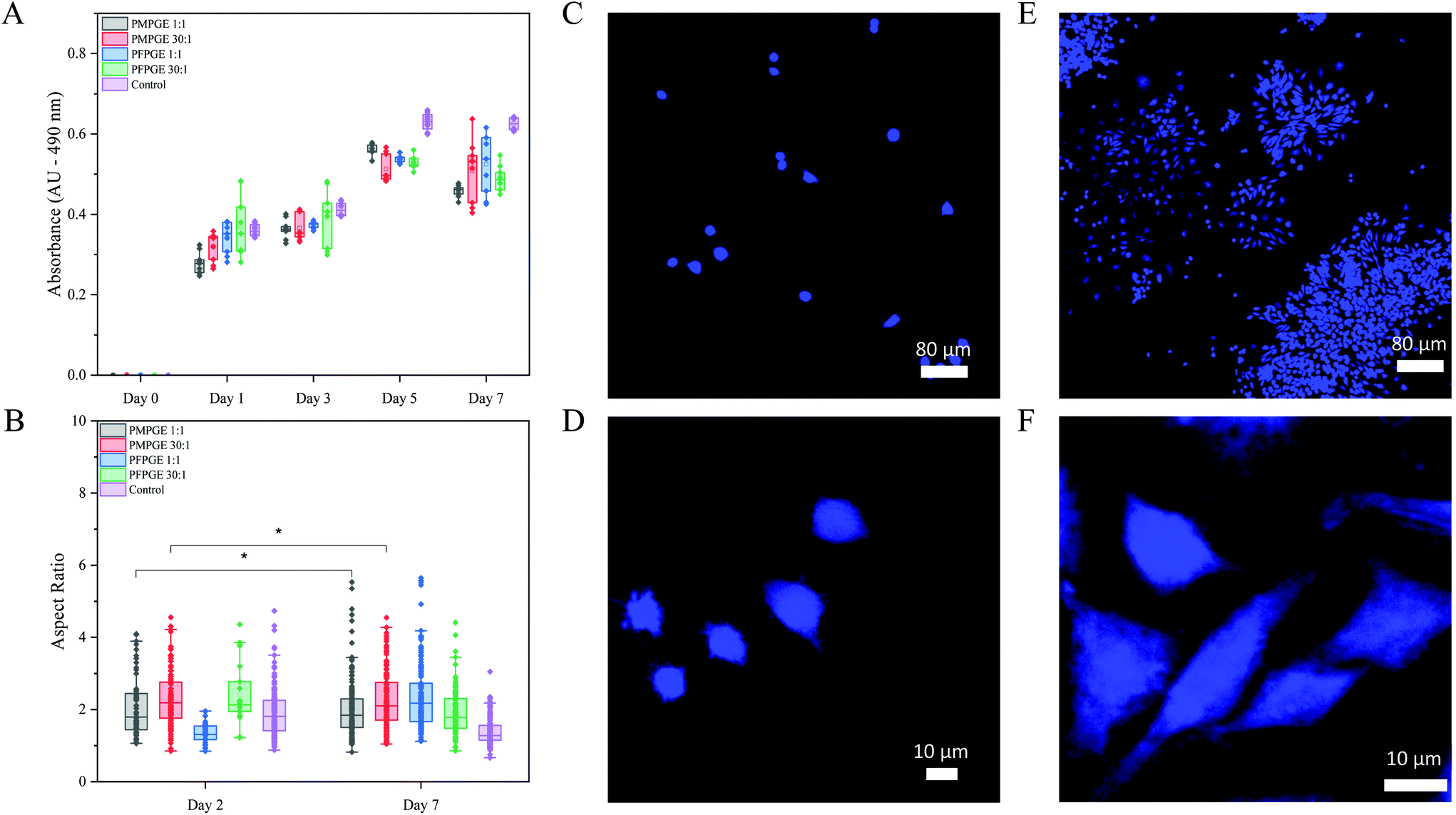 | ||
| Fig. 8 Cytocompatibility of PMPGE and PFPGE 1:1 and 30:1 photoset films using adherent murine macrophages (A) and the corresponding cell aspect ratios (B) over a 7-day period; representative fluorescent cell imaging comparing day 0 (C and D) with day 7 (E and F). *No significant difference (p = 0.05) (N = 2, n = 3). | ||
Conclusion
An aliphatic polyester system containing one of two stereoisomer polyesters (fumarate or maleate polyester copolymers) was synthesized by bulk ROCOP using stannous octoate in open air conditions. Material properties were demonstrated to be tunable through the alteration of polymer stereochemistry by leveraging thiol–ene click photochemistry stoichiometric crosslinking ratios. Unlike previous attempts leveraging similar polyesters, the use of thiol–ene chemistry provided the opportunity to probe both PMPGE and PFPGE thermosets, demonstrating the opportunity to dramatically control thermal and mechanical behaviors solely through stereochemistry and stoichiometry. Furthermore, the materials display tunable hydrolytic degradation rates as well as 4D behavior in the form of shape memory responsiveness. Both the PMPGE and PFPGE 1:1 and 30:1 materials demonstrated increasing macrophage proliferation and aspect ratio over a 7-day period. Leveraging excess alkenes in the resultant thermosets in conjunction with 3D printed complex architectures, makes these materials promising candidates for minimally invasive biomedical scaffolds and devices across multiple tissue systems.
Conflicts of interest
There are no known conflicts of interest.References
- Y. Z. Liping Ouyang, G. Jin, T. Lu, J. Li, Y. Qiao, C. Ning, X. Zhang, P. K. Chu and X. Liu, Biomaterials, 2016, 83, 115–126 CrossRef.
- D. Bandelli, J. Alex, C. Weber and U. S. Schubert, Macromol. Rapid Commun., 2020, 41, 1900560 CrossRef CAS PubMed.
- N. Olov, S. Bagheri-Khoulenjani and H. Mirzadeh, J. Biomed. Mater. Res., Part A, 2018, 106, 2272–2283 CrossRef CAS.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- M. Martínez de Sarasa Buchaca, F. de la Cruz-Martínez, J. Martínez, C. Alonso-Moreno, J. Fernández-Baeza, J. Tejeda, E. Niza, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, ACS Omega, 2018, 3, 17581–17589 CrossRef PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- J. M. Longo, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 15897–15900 CrossRef CAS PubMed.
- J. Slager and A. J. Domb, Adv. Drug Delivery Rev., 2003, 55, 549–583 CrossRef CAS PubMed.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS PubMed.
- C. J. Stubbs, J. C. Worch, H. Prydderch, M. L. Becker and A. P. Dove, Macromolecules, 2020, 53, 174–181 CrossRef CAS.
- J. Wang, X. Liu, Z. Jia, L. Sun, Y. Zhang and J. Zhu, Polymer, 2018, 137, 173–185 CrossRef CAS.
- Z.-Q. Wan, W.-M. Ren, S. Yang, M.-R. Li, G.-G. Gu and X.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 17636–17640 CrossRef CAS PubMed.
- L. G. Bracaglia, B. T. Smith, E. Watson, N. Arumugasaamy, A. G. Mikos and J. P. Fisher, Acta Biomater., 2017, 56, 3–13 CrossRef CAS PubMed.
- M. N. Cooke, J. P. Fisher, D. Dean, C. Rimnac and A. G. Mikos, J. Biomed. Mater. Res., Part B, 2003, 64, 65–69 CrossRef PubMed.
- Y. Luo, G. l. Le Fer, D. Dean and M. L. Becker, Biomacromolecules, 2019, 20, 1699–1708 CrossRef CAS PubMed.
- M. B. Wandel, C. A. Bell, J. Yu, M. C. Arno, N. Z. Dreger, Y. H. Hsu, A. Pitto-Barry, J. C. Worch, A. P. Dove and M. L. Becker, Nat. Commun., 2021, 12, 446 CrossRef CAS PubMed.
- J. A. Wilson, D. Luong, A. P. Kleinfehn, S. Sallam, C. Wesdemiotis and M. L. Becker, J. Am. Chem. Soc., 2018, 140, 277–284 CrossRef CAS PubMed.
- A. P. Kleinfehn, J. A. Lammel Lindemann, A. Razvi, P. Philip, K. Richardson, K. Nettleton, M. L. Becker and D. Dean, Biomacromolecules, 2019, 20, 4345–4352 CrossRef CAS PubMed.
- Y. Luo, C. K. Dolder, J. M. Walker, R. Mishra, D. Dean and M. L. Becker, Biomacromolecules, 2016, 17, 690–697 CrossRef CAS PubMed.
- B. J. Black, M. Ecker, A. Stiller, R. Rihani, V. R. Danda, I. Reed, W. E. Voit and J. J. Pancrazio, J. Biomed. Mater. Res., Part A, 2018, 106, 2891–2898 CrossRef CAS PubMed.
- M. A. Pemberton and B. S. Lohmann, Regul. Toxicol. Pharmacol., 2014, 69, 467–475 CrossRef CAS PubMed.
- E. A. Dhulst, W. H. Heath and J. M. Torkelson, Polymer, 2016, 96, 198–204 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- A. C. Weems, M. C. A. Arno, R. T. R. Huckstepp and A. P. Dove, Nat. Commun., 2021, 12, 1–14 CrossRef PubMed.
- D. Merckle, E. Constant, Z. Cartwright and A. Weems, Macromolecules, 2021, 54, 2681–2690 CrossRef CAS.
- A. C. Weems, K. R. D. Chiaie, J. C. Worch, C. J. Stubbs and A. P. Dove, Polym. Chem., 2019, 10, 5959–5966 RSC.
- C. F. Carlborg, T. Haraldsson, K. Öberg, M. Malkoch and W. van der Wijngaart, Lab Chip, 2011, 11, 3136–3147 RSC.
- O. Konuray, N. Areny, J. M. Morancho, X. Fernández-Francos, À. Serra and X. Ramis, Polymer, 2018, 146, 42–52 CrossRef CAS.
- F. Ejserholm, J. Stegmayr, P. Bauer, F. Johansson, L. Wallman, M. Bengtsson and S. Oredsson, Biomater. Res., 2015, 19, 19–19 CrossRef PubMed.
- C. F. Carlborg, A. Vastesson, Y. Liu, W. van der Wijngaart, M. Johansson and T. Haraldsson, J. Polym. Sci., Part A: Polym. Chem., 2014, 52(18), 2604–2615 CrossRef CAS.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- J. Y. Hsieh, M. T. Keating, T. D. Smith, V. S. Meli, E. L. Botvinick and W. F. Liu, APL Bioeng., 2019, 3, 016103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental data, including 1H and 13C NMR, FT-IR spectra; rheological, thermal, thermomechanical, and gravimetric analysis; quantification of material properties; cytocompatibility analysis. See https://doi.org/10.1039/d1py01405f |
| This journal is © The Royal Society of Chemistry 2022 |