
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental evidence of ion migration in aged inorganic perovskite solar cells using non-destructive RBS depth profiling†
Taimoor
Hussain
abc,
Kalsoom
Fatima
a,
Arfa
Anjum
b,
Turab Ali
Abbas
c,
Ishaq
Ahmad
 c,
Azhar
Fakharuddin
*d and
Muhammad
Sultan
*ae
c,
Azhar
Fakharuddin
*d and
Muhammad
Sultan
*ae
aNational Centre for Physics, Quaid-i-Azam University Campus, 44000 Islamabad, Pakistan. E-mail: taimoorhussain380@gmail.com; kalsoomfatima94@yahoo.com; muhammad.sultan@kum.edu.pk
bDepartment of Physics, Quaid-i-Azam University, 45320 Islamabad, Pakistan. E-mail: arfaanjum1996@gmail.com
cExperimental Physics Department (EPD), National Centre for Physics, Quaid-i-Azam University Campus, 44000 Islamabad, Pakistan. E-mail: aliturabb@gmail.com; ishaq.ahmad@ncp.edu.pk
dDepartment of Physics, University of Konstanz, D-78457 Konstanz, Germany. E-mail: azhar-fakhar.uddin@uni-konstanz.de
eDepartment of Physics, Kohsar University Murree, 47150 Punjab, Pakistan. E-mail: sultan@ncp.edu.pk
First published on 6th August 2022
Abstract
Hybrid halide perovskites have made breakthroughs in a range of optoelectronic devices due to their favorable properties. Perovskites show mixed ionic electronic conductivity, which leads to current–voltage transients at timescales of milliseconds to hours and is responsible for hysteresis in the devices. Under the device-operating conditions, ions present in the bulk of the perovskites migrate towards the electrodes due to electrostatic effects, react with the interfacial layers and, thus, deteriorate the device performance and stability. Most experimental techniques used for probing ion migration in perovskites are indirect or destructive. In this work, we use Rutherford back scattering (RBS), which is a non-destructive technique, to investigate the elemental composition profile and interface diffusion between various layers to disentangle the extrinsic and intrinsic ion migration in inorganic halide perovskites (IHPs, e.g., CsPbI2Br). The experimental results suggest that I− ions are the most mobile and diffuse into adjacent layers in the CsPbI2Br-based inverted perovskite solar cells, followed by Cs+ ions. We also show that oxygen leads to intrinsic and extrinsic ion migration and that ion migration takes place in aged devices even in the absence of external stress.
Introduction
Hybrid halide perovskites have gained considerable attention globally due to their excellent optoelectronic properties, i.e., long carrier lifetimes, high defect tolerance and radiative efficiencies,1 large ambipolar free carrier mobilities,2 tunable bandgaps between 1.25 eV and ∼3 eV,3–5 high absorption coefficients and low-cost processing.6 These properties have enabled breakthroughs in a range of optoelectronic devices such as light-emitting diodes (LEDs),7 lasing devices,8 photodetectors,9 resistive memory devices,10 and solar cells.11 A certified power conversion efficiency (PCE) of 25.5% has been reported for perovskite solar cells (PSCs),12 which is already comparable to the market-dominating silicon solar cells. In spite of their in-device performance, concerns remain, however, regarding the operational stability of perovskites under the device-operating conditions, especially in perovskite materials that use a volatile organic component. Perovskites have been shown to be chemically reactive with adjacent charge-transport layers as well as with oxygen, moisture, light, applied bias and heat.13The compositional engineering of perovskite materials and the use of encapsulation techniques have greatly reduced degradation that is caused by extrinsic (or environmental) factors.14–16 Still, the intrinsic instability of perovskites that arises from their ionic nature remains a challenge. On the one hand, ion migration influences the various electronic processes in devices, such as the in-operando formation of p–i–n structures,17,18 phase segregation in mixed-halide perovskites,19 hysteresis in the current–voltage curves (for PSCs)20 and current–voltage–luminance curves (in LEDs), and an unstable device output with the time span ranging from a few seconds to several hours,21,22 among others. On the other hand, ion migration leads to irreversible changes in the devices, for example, due to diffusion between the perovskite layer and interfacial charge extraction/injection layers,23,24 which is a key reason for degradation in the devices.25 In the archetypical MAPbI3 perovskite, the phenomenon of ion migration arises mainly because of the presence of volatile organic ions, i.e., methylammonium (MA+), and halides, i.e., iodine (I−),25,26 when exposed to ambient air, humidity, or heat.27 Replacing MA+ with FA+ exhibits superior thermal and structural stability and results in a lower ion migration and current–voltage hysteresis in FAPbI3 devices.28 The volatile MA+ can also be replaced with an inorganic counterpart, such as cesium (Cs+), which shows improved operational stability,29–32 reduced ion migration and also higher structural stability.33–35 One must, however, note that if the amount of Cs+ is more than 15%, the α-CsPbI3 phase is disordered and changes to the δ-CsPbI3 phase, which leads to degradation of the CsPbI3 perovskite.36,37 In such a case, J–V hysteresis is still witnessed in these inorganic perovskites, suggesting that ion migration is still a detrimental process.38,39
Amongst the various plausible explanations on the origin of the hysteresis,40,41 ion migration is considered to be the most likely one.20,42–44 Theoretical studies, i.e., density-functional theory (DFT), have suggested that the small activation energy of the elements leads to the migration of vacancies and interstitials in the PSCs.42,43 Furthermore, some indirect techniques, i.e., Kelvin probe force microscopy (KPFM),45 photothermal-induced resonance (PTR)46 and impedance spectroscopy (IS)47,48 have also been utilized to study ion migration in conventional PSCs. Most of the aforementioned techniques are indirect and investigate ion migration without any elemental sensitivity. The direct elemental investigation of ion migration could validate the existing understanding of this process. Recently, time-of-flight secondary ion mass spectrometry (ToF-SIMS), which is a direct elemental technique, has been used.49 ToF-SIMS is a destructive technique, in which secondary ions are sputtered by primary ions, that does not investigate the sample in its real state in the multilayer architecture.49 These various techniques have been employed to understand the phenomena of ion migration in hybrid organic–inorganic perovskites, such as in MAPbI3 and triple-cation perovskites.50 For IHPs, it is generally anticipated that they are inherently more stable than their hybrid counterparts; however, photoinduced halide segregation has also been reported in Cs-based perovskites.51 Therefore, we emphasize that, contrary to a common perception that IHPs are inherently more stable than their organic counterparts, these perovskite materials also show signs of instability and ion migration.
In this work, we use Rutherford backscattering spectroscopy (RBS),52,53 which is a powerful, direct, and to some extent non-destructive technique, to investigate the stoichiometry,54 elemental composition,54 interfacial diffusion,55 and the intrinsic and extrinsic ion migration across the various layers of inorganic PSCs. The simplex codes SIMNRA and XRUMP are used to analyze the experimental data. To decouple the effect of various interfacial layers, we analyzed a perovskite thin film deposited on glass (glass/CsPbI2Br); a perovskite thin film deposited on a nickel oxide hole-transport layer and ITO (glass/ITO/NiOx/CsPbI2Br); and an inverted solar-cell stack with an architecture of glass/ITO/NiOx/CsPbI2Br/C60/Cu. To understand the phenomena of degradation and diffusion without any external stress, which leads to ion migration, the samples were prepared and sealed in a glove box under a nitrogen environment. The samples were stored at room temperature for a year and opened just before the RBS analysis. The devices were measured immediately after the fabrication (which led to initial biasing of the cells immediately after fabrication). The RBS measurements were performed without applying any potential and external stimulus.
Results
The deconvoluted RBS spectrum along with fitting of a single layer of CsPbI2Br on the glass substrate is shown in Fig. 1. The solid black diamonds represent the RBS experimental data, the red line shows the envelope of fitting, and the colored lines represent deconvolution of the spectra originating from the different elements present in the CsPbI2Br perovskite and glass substrate. A complementary analysis to identify the elements was carried out using particle induced X-ray emission (PIXE; see Fig. S4a, ESI†). | ||
| Fig. 1 RBS spectrum with fitting for the IHP (CsPbI2Br) deposited on glass, excited from the IHP side. The RBS spectrum is deconvoluted for various elements present in the film architecture, e.g., Pb, Cs, I, Br, Si, Ca, and O. | ||
The deconvoluted peaks that correspond to each element are shown in Fig. 1. The first right-hand edge at 1700 keV corresponds to Pb; the central highest peak at 1600 keV indicates the presence of Cs+ and I−, which are not differentiable due to the non-uniform concentrations of the elements with depth, which is observed in depth profile (Table S1, ESI†); and edge at around 1525 keV corresponds to Br−. The broad edges at 200–1200 keV represent the substrate elements Ca, Si and O, respectively.
The compositional results for the IHP are given in the depth-profile analysis (Table S1, ESI†), where the single IHP layer is split into multiple layers. The first two layers in the table belong to the IHP, which shows notable changes in the concentration of I−,27 and minor changes in the Cs+ and Br concentrations.27 The latter indicates a slight change in the uniformity of the IHP concentration along the film thickness (which we refer to as the depth in this work). The detection of oxygen in the third layer indicates its diffusion from the glass substrate to the IHP layer. Layer 4 indicates the interface and interfacial diffusion between the IHP and glass, and the fifth layer belongs to the glass substrate.
The precise overlap of the SIMNRA program-fitting envelope with the experimental results shows that the fitting nicely reproduces the experimental results shown in Fig. 1. The spectrum of Pb2+ is symmetric, whereas the rest of the elements, i.e., I−, Br−, and Cs+, show asymmetric spectra. This asymmetric shape of I−, Br−, and Cs+ indicates that these ions have a non-uniform concentration across the film thickness, as observed in the depth-profile analysis (Table S1, ESI†). This change in concentration with depth is significant for I−, affirming the previous observation by deQuilettes et al.56 of light-induced halide migration by I− in the neat film. The change in concentration with depth is modest for Br− and Cs+ and is negligible for Pb2+, affirming that Pb2+ is the least mobile ion.50 Beyond asymmetry, the elemental spectra also show a tail that extends to the lower channel side, indicating the diffusion of IHP towards the substrate due to mobile ions, i.e., I−, Br− and Cs+, which were also quantitatively analyzed as an interface (layers 4 and 5, as shown in the depth profile of Table S1, ESI†).
To validate the RBS experimental results of the IHP deposited on glass (Fig. 1), the SIMNRA program was used to simulate an ideal device considering the respective kinematic factors and cross-section of the IHP. Simulations were performed with a uniform (ideal) composition of the IHP throughout the depth along with an interface with glass as shown in the depth profile (Table S1, ESI†), considering the real experimental parameters to compensate the instrumental contribution. The simulated envelope of the IHP deposited on glass is shown in Fig. 2(a), and symmetric edges of Cs+, Pb2+, I− and Br− are observed in the simulated envelope. The various elemental peaks that correspond to the simulated envelope are symmetric (Fig. 2(a)), while they are asymmetric in the experimental spectrum and its spectral fit results (Fig. 1).
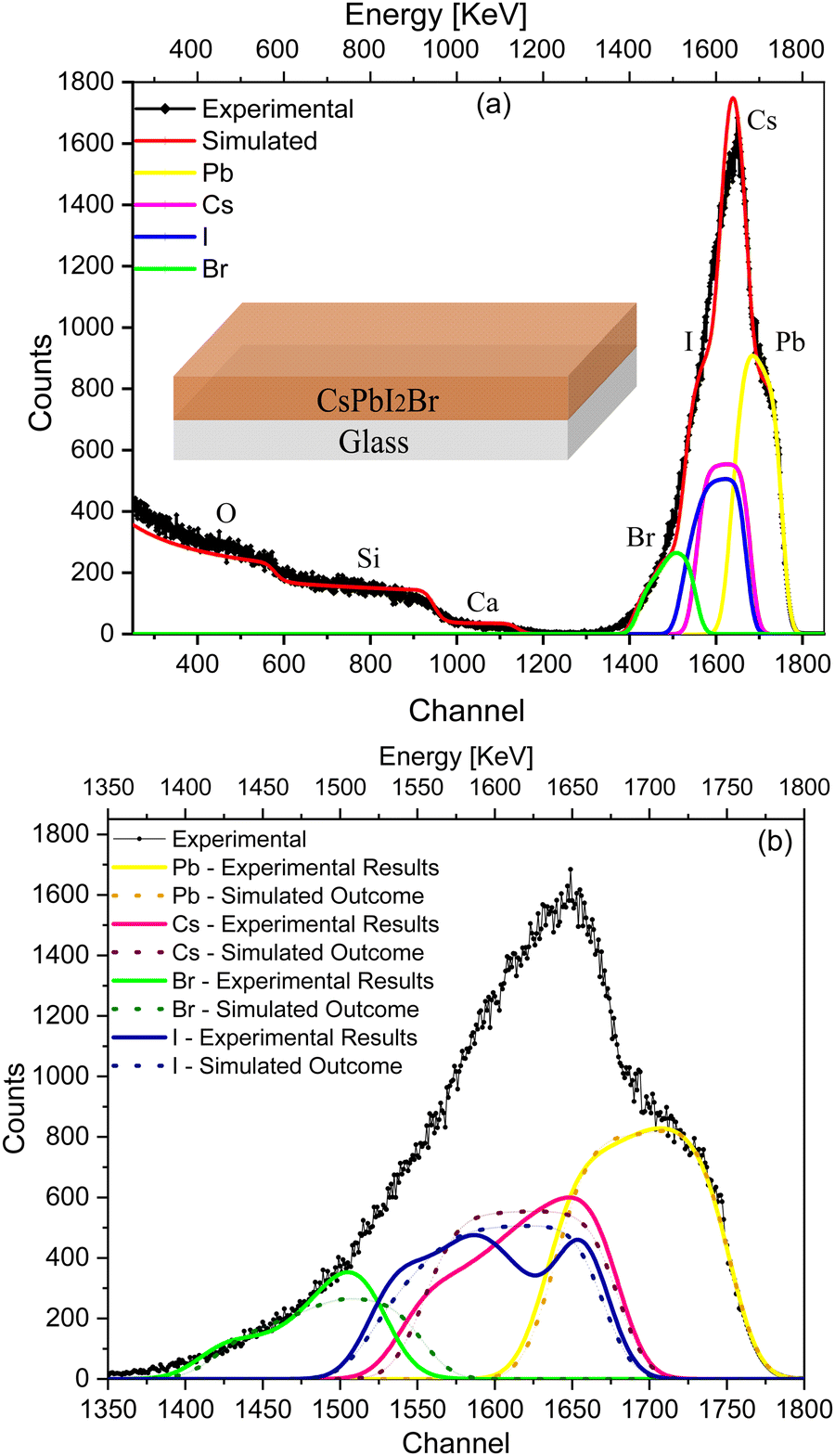 | ||
| Fig. 2 (a) Simulated envelope of RBS in comparison with the experimental data for IHP (CsPbI2Br) deposited on glass. (b) Comparison of the elemental (I, Br, Cs, and Pb) spectra of the simulated envelope (dotted lines) and the experimental fitting envelope (solid lines). | ||
Fig. 2(b) compares the simulated elemental spectra (dotted lines) with the experimental spectrum and the fitted elemental spectra (solid lines) of the IHP. A significant deviation in the simulated envelope from that of the experimental spectra is due to the non-uniform composition of I−, Br− and Cs+ along the depth (Table S1, ESI†), which affects the experimental results. The asymmetry in the case of I−, Br− and Cs+ (Fig. 2(b)), together with their extended tails, and the symmetry of their elemental peaks in Fig. 2(a) indicate the non-uniform stoichiometry of IHP across the depth.
As a complementary analysis, X-ray diffraction (XRD) and diffuse reflectance spectroscopic (DRS) measurements of the samples were recorded to examine the changes that arise after aging and re-crystallization (the aged samples were annealed again at 150 °C to check the recovery of the perovskite black phase) as shown in Fig. S2 (ESI†). The bandgap of the aged IHP film coated on glass is 2.79 eV, which upon re-crystallization changes to 1.90 eV, thereby confirming the recovery of the cubic phase after heating at 150 °C. The XRD of the freshly prepared samples showed the pure cubic phase, whereas the aged perovskite samples showed mixed. i.e., both yellow (orthorhombic) and black (cubic), phases with the yellow phase being dominant. Some additional peaks were also observed along with the yellow and black phases, such as CsI (cubic) and CsPb2Br5 (tetragonal). These peaks were not observed in freshly prepared samples, which confirms the occurrence of non-reversible degradation into IHP upon aging.
We extended our experiments to more complex multilayer architectures, e.g., half and full solar cell stacks, in order to evaluate the influence of the various interfaces in the devices and their correlation with the ion migration. We start by introducing a hole-transport layer, e.g., NiO, between the ITO-coated glass substrate and the perovskite (glass/ITO/NiOx/CsPbI2Br). The RBS spectrum with the fitting results of this multilayer sample along with deconvolution of the spectra is shown in Fig. 3. The conformational analysis for identification of the elements was carried out using Particle induced X-ray emission (PIXE; see Fig. S4b, ESI†).
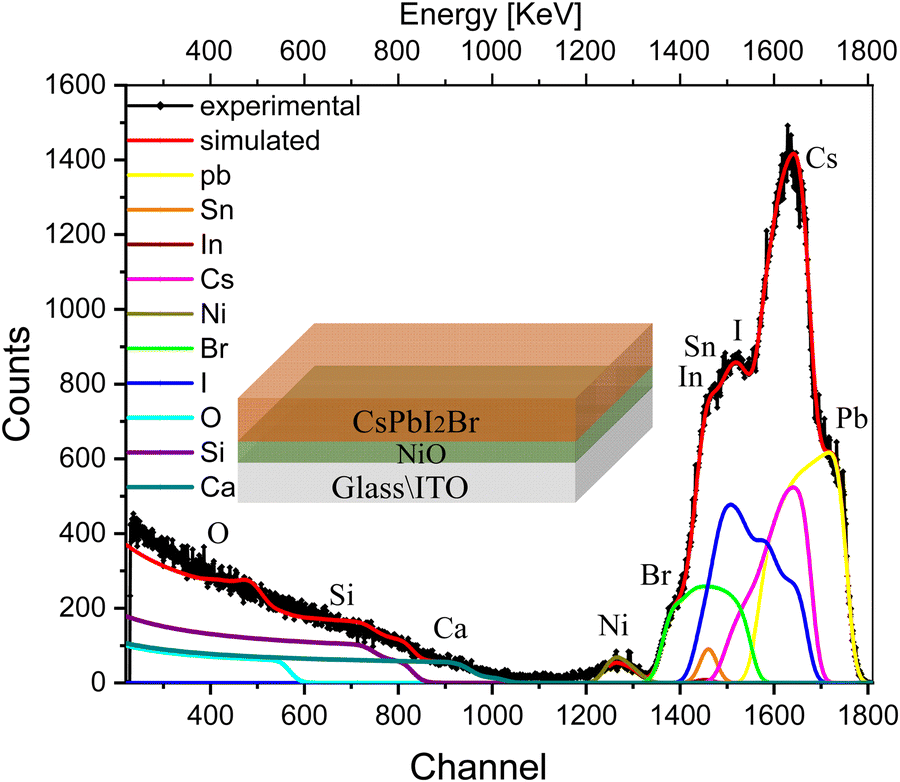 | ||
| Fig. 3 RBS spectrum with the fitting of multilayer samples, i.e., glass/ITO/NiOx/CsPbI2Br, excited from the IHP side, along with deconvolution of the elemental spectra. | ||
The indistinct shape of the peaks in the experimental spectra indicates non-uniformity in the multilayer sample with depth. The peaks corresponding to each element are diffused and cannot be distinguished separately in the spectrum. The experimental trend is well reproduced after splitting the samples into multilayers, and the compositional information of the sample is shown in the depth profile of Table S2 (ESI†). This suggests that the concentration of elements is non-uniform along the depth of the perovskite film (layers 1, and 2), which is substantially greater for I− and is smaller for Br− and Cs+. The next few layers (layers 3 and 4) illustrate the interface between IHP and HTL in which variations in the concentration of O and I− are more pronounced than Br− and Cs+. the subsequent few layers show the composition of the HTL. The interface layer in which the variation in concentration of O is observed and the last layer refer to the composition of glass elements.
We further recorded the RBS spectra of a complete device (glass/ITO/NiOx/CsPbI2Br/C60/Cu) that now includes both electron- and hole-transport layers as well as the top metal contact. The RBS spectrum of the complete device with fitting and deconvolution of the spectra for each element is shown in Fig. 4, while the ratios of various elements in the different layers are summarized in the depth profile of Table S3 (ESI†). As a complementary analysis for identification of the elements, particle induced X-ray emission (PIXE) was also carried out (see Fig. S4c, ESI†).
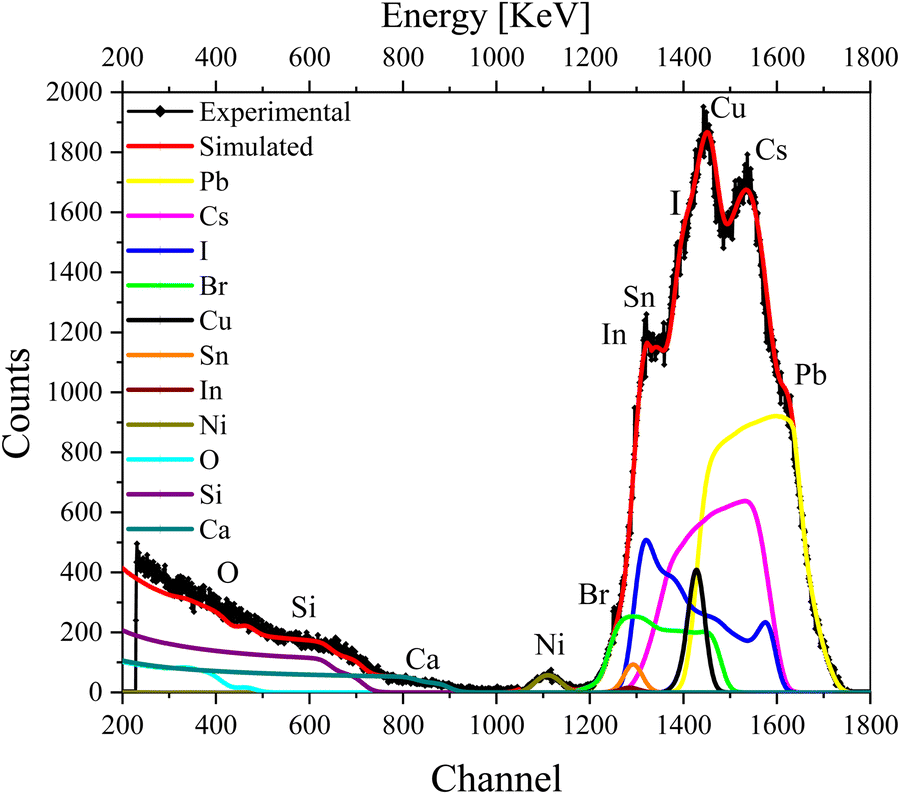 | ||
| Fig. 4 RBS spectrum of the complete device excited from the Cu contact side, with simulation and deconvolution of the elemental spectra for glass/ITO/NiOx/CsPbI2Br/C60/Cu. | ||
The first few layers in the depth profile of Table S3 (ESI†) correspond to the composition of the Cu contact, the electron-transport layer (ETL), and the interface between Cu and ETL, respectively. The IHP layer is split into further layers (layers 4–6) in the depth-profile analysis to reproduce the experimental data nicely. We note that the concentration of I−, Br− and Cs+ vary with the depth. The subsequent layers belong to the HTL and ITO (layers 7–9) in which the variation in the concentration of O is visible. As O is present in ITO/NiO and also seems to have diffused into the IHP, it gives rise to an interface that is mentioned in multiple layers in Table S3 (ESI†).
Discussions
A few observations can be drawn from RBS spectra of the various parts of the device stack and their subsequent conformal analysis. The elemental spectra of Sn, In, Pb, Cu and Ni are symmetric, whereas those of I−, Br− and Cs+ are asymmetric due to the non-uniform concentration with the depth (Fig. 1, 3 and 4). The variation in concentration is prominent for I−, while it is modest for Cs+ and Br−. This asymmetric spectrum of I−, Cs+ and Br− is due to mobile ions that have migrated from their original positions56 and have redistributed in the IHP. Slight changes in the spectra of Pb2+ (Fig. 3 and 4) with its symmetric shape suggest negligible migration of the Pb2+ ions, which confirms previous findings that Pb2+ is the least mobile ion as it has a large activation energy50,57 and only moves when complete degradation has occurred.58 The minor diffusion of I− ions can be witnessed from the simulated spectra that show a homogenous distribution (Fig. 2(a)) of ions across the entire film depth, which is symmetric as well.Another notable observation is the different distribution profiles of I−, Br− and Cs+ ions when deposited over different substrates. When deposited on glass, the elemental spectrum of I− shows a dual peak in the spectrum while the Cs+ ion shows a sharp spectrum (Fig. 1). This dual nature of I− shows that the stoichiometry of I− ions changes in the IHP. Its low activation energy (0.58 eV) and high diffusion coefficient of 10−12 cm2 s−1 suggest that I− acts as the majority ionic carriers that contribute to the mixed ionic–electronic conductivity of perovskites.43 The greater activation energy (0.83 eV)59 of Cs+ compared with I− makes it less mobile than I−, as suggested by first-principles calculations.60
When deposited on NiO (the HTL) or in the complete device, the RBS spectrum of I− is higher towards the lower channel side, suggesting a higher I− concentration near the HTL side (Fig. 3 and 4). An opposite trend is observed for Cs+, which shows a higher concentration towards the higher channel side, e.g., towards the surface of the perovskite film (the ETL side). This trend is similar to that observed in hybrid organic–inorganic perovskites, i.e., the accumulation of I− towards the HTL and MA+, FA+ towards the ETL, which was also observed in MA+- and FA+-based planar p–i–n device architectures after illumination.60–62 Similar trends were shown in freshly prepared samples without any bias and as well as under the application of an external bias.44,63 In triple-cation [(FA0.79MA0.16Cs0.05)0.97Pb(I0.84Br0.16)2.97] PSCs, a remarkable shift was also reported for Cs+ toward the HTL after 25 h of illumination, using TOF-SIMS.50 In the same study the FA+ and MA+ cations have also shown to be redistributed throughout the device.50 Similarly, the migration of I− towards the HTL was also observed by Vu et al.64 in the Cs-based inorganic perovskite (CsPbI3) upon illumination without an external potential, while iodide vacancies VI+ migrated towards the ETL side with different mobilities. In our work, the ion migration in the absence of an external stimulus is probably due to Schottky disorder, which is a dominant type of defect in IHPs that is associated with the formation of a stoichiometric amount of anion and cation vacancies.65,66 Quantum mechanical calculations also suggest that the origin of this phenomenon is the prevalence of ionic over electronic disorder in stoichiometric materials. Herein, Schottky defect formation provides a mechanism for self-regulating the concentration of charge carriers through the ionic compensation of charged point defects. The equilibrium charged vacancy concentration is predicted to exceed 0.4% at room temperature.66J–V hysteresis has also been measured under dark conditions at room temperature both in hybrid and inorganic perovskite solar cells, which suggests that ionic conduction takes place in the dark.67
Our experimental findings, which provide direct strong evidence of the diffusion of ions in aged IHP without any external stimulus at room temperature, i.e., 300 K, thus confirm the trends previously observed in the literature using indirect techniques, e.g., the accumulation of anions (I− and Br−) towards the HTL68,69 and redistribution of these ions together with Cs+.50 The migration of ions in our work (in the absence of biasing or photoexcitation) is linked to irreversible degradation.
Likewise, the distribution of Br− ions also seems to show a strong dependency on the underneath layers. On glass, Br− shows a tail extending towards the substrate while a more uniform spread is visible on the NiO HTL. In a complete device, the Br− ions70 redistribute and a slightly higher concentration is noted towards the HTL-side interface (Fig. 4 and Table S3, ESI†). This might be due to the fact that the migration of ions, e.g., the presence of the metallic cation Cs+ near the ETL and I− at the HTL side, generate a local electric field inside the perovskite films, as also proposed by Eames et al.,43 which pulls ions towards their opposing electrodes, e.g., cations toward the HTL and anions toward the ETL.61 This assumption is also confirmed by Zhang et al. in organic–inorganic-based planar p–i–n PSCs upon illumination.61 The redistribution of ions can alter the band structure at the interface and modulate the barrier height for charge collection/extraction. For example, this is extremely relevant for light-emitting diodes since, over time, as the ions redistribute, the barrier height for different charge injections will lead to a change in the operating voltage for the device.
Another notable observation is the mobility of Cs+ in the various device-architecture studies in this work (Tables S1–S3, ESI†). It has been suggested that if the amount of Cs+ is higher than 15%, the α-CsPbI3 phase converts to the δ-CsPbI3 phase, which leads to degradation36,37 and J–V hysteresis in the IHPs.67 When compared with a mobile organic counterpart, e.g., MA+ present in organic–inorganic hybrid halide perovskites, the elemental peak of Cs+ in our work is smooth and the slight changes in concentration confirm its less mobile nature compared with MA+ and FA+.46,61 We also note that the spectrum of Pb2+ is almost symmetric, showing negligible changes in its concentration in all the architectures investigated in this work, which was also reported earlier.50
To analyze the extrinsic ion migration in the complete device (glass/ITO/NiO/CsPbI2Br/C60/Cu), we also quantify the oxygen-sensitive part of the spectrum in Fig. 4. The spectrum of oxygen for the complete device (glass/ITO/NiO/CsPbI2Br/C60/Cu) is shown in Fig. 5.
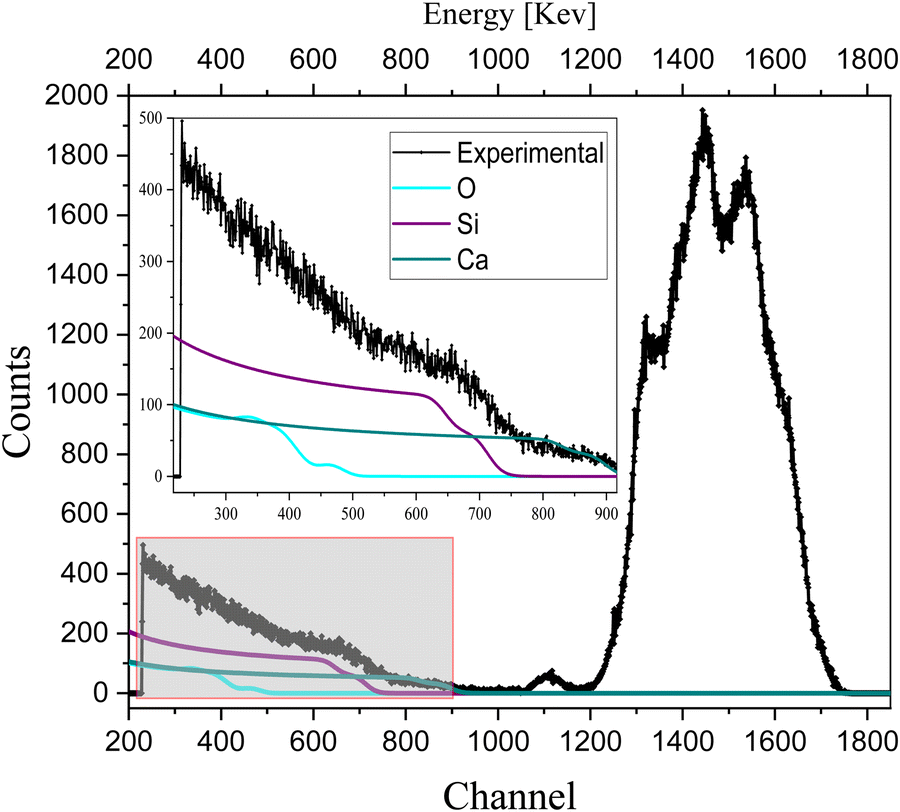 | ||
| Fig. 5 RBS spectrum with quantification of oxygen in a complete device (glass/ITO/NiO/CsPbI2Br/C60/Cu). | ||
The less sharp broad edge in the RBS spectrum of oxygen indicates its non-uniform concentration with depth, which is also observed in the depth profile of the complete device (as shown in the depth profile of Table S3, ESI†), confirms the extrinsic ion migration of oxygen from NiO to the IHP. Also, the detection of the interfaces in the depth profile of Table S2 (ESI†) indicated the migration of oxygen from NiO to the perovskite.
To understand the behavior of oxygen in the device, the RBS of the sample, i.e., glass/ITO/NiOx was analyzed (as shown in Fig. S1, ESI†) after 3 months and after 1 year (aged samples). Sharp peaks (as shown in Fig. S1a, ESI†) and broad peaks (as shown in Fig. S1b, ESI†) are observed, which illustrate that the concentration of Ni and O changes with the depth in NiO-based aged samples. The broad edge of O is of zigzag fashion (aged sample) due to the non-uniform concentration of oxygen with the depth, which supports the previous results for the complete device shown in Fig. 5, i.e., the mobile nature of oxygen and ion migration of oxygen from NiO to the IHP, and is referred as extrinsic ion migration into perovskites.
Conclusions
In summary, this work investigates the interfacial diffusion of ions across different layers of aged samples and suggests intrinsic ion migration in inorganic halide-based perovskites. SIMNRA fitting and simulation are used to quantify the experimental data. Our experimental results showed a non-uniform concentration of Cs+, I− and Br− in the device and perovskite films with different interfaces, which is due to the different diffusion rates of these ions. As these ions are displaced from their original position and diffuse into the adjacent layer, they trigger intrinsic ion migration in the perovskite film. Without applying any external stress, we observe the diffusion of ions, i.e., Br− and I− towards the HTL and Cs+ towards the ETL. The depth profiles of the interface between the charge-transport layers and the IHP verify the interfacial diffusion. The ion migration of low-atomic-number elements, particularly oxygen, was observed, which diffuses into different layers of the IHP devices and thus induces the extrinsic ion migration of oxygen atoms from the NiO layer into the IHP. This is one of the reasons for the degradation of the devices, contrary to the general understanding that perovskite material degrades with an electrical bias, light illumination or moisture. Overall, in this work, we provide experimental evidence of both extrinsic and intrinsic ion migration in PSCs using a non-destructive technique. We also experimentally verify that ion migration occurs in inorganic PSCs even without any external stress and that, remarkably, inorganic perovskites are no exception. Our findings extend the current understanding of the mobile nature of ions in PSCs, and suggest that more robust device architectures should include passivation and interfacial engineering approaches.Experimental methods
The device preparation was performed in an inert atmosphere using a glove box with a deposition system as described in detail in our earlier publication.71 Rutherford backscattering (RBS) was used in which high-energy ions (MeV), i.e., He++ with energies ranging from 0.5 to 2.0 MeV, are guided onto the sample, which are elastically backscattered due to the Coulomb repulsion force between the target and the incoming ion. Further details of the RBS technique are provided in the ESI.†The SIMNRA and XRUMP programs were used to simulate and validate the experimental RBS results, and a surface barrier detector was used to detect the backscattered He++ ion at an angle of 170° by target nuclei. Powder X-ray diffraction (pXRD) spectra were collected using a Brucker D-8 Advance instrument, employing Cu Kα1 radiation (λ = 1.5418 Å). The band-gap energies were obtained using a PerkinElmer Lambda 35 UV-Vis spectrophotometer.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
DAAD Project #57345608 and 57458981 financially supported this research project. The authors acknowledge Experimental Physics Department (EPD), National Centre for Physics (NCP), Islamabad, Pakistan for RBS measurements. AF acknowledges support from the Research Executive Agency (REA) under the framework of a Marie Skłodowska-Curie Individual Fellowship (project RADICEL; grant number 101030985). In addition, the authors would like to thank the Hybrid Nanostructures (HNS) group at the University of Konstanz, Germany for access to solar-cell fabrication and characterization facilities.References
- C. M. Wolff, P. Caprioglio, M. Stolterfoht and D. Neher, Adv. Mater., 2019, 31(52), 1902762 CrossRef CAS PubMed.
- B. Zhao, G. Niu, Q. Dong, J. Liu, N. Li, J. Li and L. Wang, J. Mater. Chem. A, 2018, 6(46), 23797–23804 RSC.
- J. W. Lee, D. J. Seol, A. N. Cho and N. G. Park, Adv. Mater., 2014, 26(29), 4991–4998 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13(4), 1764–1769 CrossRef CAS PubMed.
- Y. Ogomi, A. Morita, S. Tsukamoto, T. Saitho, N. Fujikawa, Q. Shen, T. Toyoda, K. Yoshino, S. S. Pandey, T. Ma and S. Hayase, J. Phys. Chem. Lett., 2014, 5(6), 1004–1011 CrossRef CAS PubMed.
- Y. Kuang, Y. Ma, D. Zhang, Q. Wei, S. Wang, X. Yang, X. Hong and Y. Liu, Nanoscale Res. Lett., 2020, 15(1), 213 CrossRef CAS PubMed.
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T.-W. Lee, Science, 2015, 350(6265), 1222–1225 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13(5), 476–480 CrossRef CAS PubMed.
- L. Dou, Y. Yang, J. You, Z. Hong, W.-H. Chang, G. Li and Y. Yang, Nat. Commun., 2014, 5(1), 5404 CrossRef CAS PubMed.
- J. Choi, S. Park, J. Lee, K. Hong, D.-H. Kim, C. W. Moon, G. D. Park, J. Suh, J. Hwang, S. Y. Kim, H. S. Jung, N.-G. Park, S. Han, K. T. Nam and H. W. Jang, Adv. Mater., 2016, 28(31), 6562–6567 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2(1), 591 CrossRef PubMed.
- NREL, Best Research-Cell Efficiencies: Emerging Photovoltaics. 2021.
- L. Schmidt-Mende, V. Dyakonov, S. Olthof, F. Ünlü, K. M. T. Lê, S. Mathur, A. D. Karabanov, D. C. Lupascu, L. M. Herz, A. Hinderhofer, F. Schreiber, A. Chernikov, D. A. Egger, O. Shargaieva, C. Cocchi, E. Unger, M. Saliba, M. M. Byranvand, M. Kroll, F. Nehm, K. Leo, A. Redinger, J. Höcker, T. Kirchartz, J. Warby, E. Gutierrez-Partida, D. Neher, M. Stolterfoht, U. Würfel, M. Unmüssig, J. Herterich, C. Baretzky, J. Mohanraj, M. Thelakkat, C. Maheu, W. Jaegermann, T. Mayer, J. Rieger, T. Fauster, D. Niesner, F. Yang, S. Albrecht, T. Riedl, A. Fakharuddin, M. Vasilopoulou, Y. Vaynzof, D. Moia, J. Maier, M. Franckevičius, V. Gulbinas, R. A. Kerner, L. Zhao, B. P. Rand, N. Glück, T. Bein, F. Matteocci, L. A. Castriotta, A. Di Carlo, M. Scheffler and C. Draxl, APL Mater., 2021, 9(10), 109202 CrossRef CAS.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5(20), 1501310 CrossRef.
- F. J. Ramos, T. Maindron, S. Béchu, A. Rebai, M. Frégnaux, M. Bouttemy, J. Rousset, P. Schulz and N. Schneider, Sustainable Energy Fuels, 2018, 2(11), 2468–2479 RSC.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53(42), 11232–11235 CrossRef CAS PubMed.
- M. Lenes, G. Garcia-Belmonte, D. Tordera, A. Pertegás, J. Bisquert and H. J. Bolink, Adv. Funct. Mater., 2011, 21(9), 1581–1586 CrossRef CAS.
- H. M. Zhang, H. Lin, C. J. Liang, H. Liu, J. J. Liang, Y. Zhao, W. G. Zhang, M. J. Sun, W. K. Xiao, H. Li, S. Polizzi, D. Li, F. J. Zhang, Z. Q. He and W. C. H. Choy, Adv. Funct. Mater., 2015, 25(46), 7226–7232 CrossRef CAS.
- P. Vashishtha, M. Ng, S. B. Shivarudraiah and J. E. Halpert, Chem. Mater., 2019, 31(1), 83–89 CrossRef CAS.
- A. Fakharuddin, M. K. Gangishetty, M. Abdi-Jalebi, S.-H. Chin, A. R. bin Mohd Yusoff, D. N. Congreve, W. Tress, F. Deschler, M. Vasilopoulou and H. J. Bolink, Nat. Electron., 2022, 5(4), 203–216 CrossRef CAS.
- A. Fakharuddin, W. Qiu, G. Croes, A. Devižis, R. Gegevičius, A. Vakhnin, C. Rolin, J. Genoe, R. Gehlhaar, A. Kadashchuk, V. Gulbinas and P. Heremans, Adv. Funct. Mater., 2019, 29(37), 1904101 CrossRef.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen, J. P. Correa Baena, A. Hagfeldt and M. Graetzel, Energy Environ. Sci., 2018, 11(1), 151–165 RSC.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5(1), 5784 CrossRef CAS PubMed.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5(9), 1511–1515 CrossRef CAS PubMed.
- X. Ren, L. Zhang, Y. Yuan and L. Ding, J. Semicond., 2021, 42(1), 010201 CrossRef CAS.
- M. H. Futscher, J. M. Lee, L. McGovern, L. A. Muscarella, T. Wang, M. I. Haider, A. Fakharuddin, L. Schmidt-Mende and B. Ehrler, Mater. Horiz., 2019, 6(7), 1497–1503 RSC.
- X. Yan, W. Fan, F. Cheng, H. Sun, C. Xu, L. Wang, Z. Kang and Y. Zhang, Nano Today, 2022, 44, 101503 CrossRef CAS.
- Y. Fan, H. Meng, L. Wang and S. Pang, Sol. RRL, 2019, 3(9), 1900215 CrossRef.
- J. Liang, P. Zhao, C. Wang, Y. Wang, Y. Hu, G. Zhu, L. Ma, J. Liu and Z. Jin, J. Am. Chem. Soc., 2017, 139(40), 14009–14012 CrossRef CAS PubMed.
- W. Xiang, S. Liu and W. Tress, Energy Environ. Sci., 2021, 14(4), 2090–2113 RSC.
- L. Zhang, T. Hu, J. Li, L. Zhang, H. Li, Z. Lu and G. Wang, Front. Mater., 2020, 6, 330 CrossRef.
- X. Jia, C. Zuo, S. Tao, K. Sun, Y. Zhao, S. Yang, M. Cheng, M. Wang, Y. Yuan, J. Yang, F. Gao, G. Xing, Z. Wei, L. Zhang, H.-L. Yip, M. Liu, Q. Shen, L. Yin, L. Han, S. Liu, L. Wang, J. Luo, H. Tan, Z. Jin and L. Ding, Sci. Bull., 2019, 64(20), 1532–1539 CrossRef CAS.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28(1), 284–292 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9(6), 1989–1997 RSC.
- S. Zhang, S. Wu, R. Chen, W. Chen, Y. Huang, H. Zhu, Z. Yang and W. Chen, J. Phys. Chem. Lett., 2019, 10(11), 2898–2903 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139(40), 14173–14180 CrossRef CAS PubMed.
- L. T. Schelhas, Z. Li, J. A. Christians, A. Goyal, P. Kairys, S. P. Harvey, D. H. Kim, K. H. Stone, J. M. Luther, K. Zhu, V. Stevanovic and J. J. Berry, Energy Environ. Sci., 2019, 12(4), 1341–1348 RSC.
- G. Eperon, G. Paterno, R. Hawke, A. Zampetti, A.-A. Haghighirad, F. Cacialli and H. Snaith, J. Mater. Chem. A, 2015, 3 Search PubMed.
- D. Y. Heo, S. M. Han, N. S. Woo, Y. J. Kim, T.-Y. Kim, Z. Luo and S. Y. Kim, J. Phys. Chem. C, 2018, 122(28), 15903–15910 CrossRef CAS.
- B. Chen, M. Yang, S. Priya and K. Zhu, J. Phys. Chem. Lett., 2016, 7(5), 905–917 CrossRef CAS PubMed.
- N. K. Elumalai and A. Uddin, Sol. Energy Mater. Sol. Cells, 2016, 157, 476–509 CrossRef CAS.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8(7), 2118–2127 RSC.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6(1), 7497 CrossRef CAS PubMed.
- M. T. Khan, P. Huang, A. Almohammedi, S. Kazim and S. Ahmad, iScience, 2021, 24(2), 102024 CrossRef CAS PubMed.
- Z. Xiao, Y. Yuan, Y. Shao, Q. Wang, Q. Dong, C. Bi, P. Sharma, A. Gruverman and J. Huang, Nat. Mater., 2015, 14(2), 193–198 CrossRef CAS PubMed.
- Y. Yuan, J. Chae, Y. Shao, Q. Wang, Z. Xiao, A. Centrone and J. Huang, Adv. Energy Mater., 2015, 5(15), 1500615 CrossRef.
- P. Srivastava, R. Kumar and M. Bag, Phys. Chem. Chem. Phys., 2021, 23(18), 10936–10945 RSC.
- M. Kim, H. Jun, H. Lee, H. Nahdi, D. Tondlier, Y. Bonnassieux, J.-É. Bourée and B. Geffroy, Eur. J. Inorg. Chem, 2021,(18), 4781–4789 CrossRef CAS.
- Z. Li, C. Xiao, Y. Yang, S. P. Harvey, D. H. Kim, J. A. Christians, M. Yang, P. Schulz, S. U. Nanayakkara, C.-S. Jiang, J. M. Luther, J. J. Berry, M. C. Beard, M. M. Al-Jassim and K. Zhu, Energy Environ. Sci., 2017, 10(5), 1234–1242 RSC.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119(5), 3418–3451 CrossRef CAS PubMed.
- W. Zhou, Y. Zhao, X. Zhou, R. Fu, Q. Li, Y. Zhao, K. Liu, D. Yu and Q. Zhao, J. Phys. Chem. Lett., 2017, 8(17), 4122–4128 CrossRef CAS PubMed.
- M. Aebli, N. Porenta, N. Aregger and M. V. Kovalenko, Chem. Mater., 2021, 33(17), 6965–6973 CrossRef CAS.
- N. Phung, M. Verheijen, A. Todinova, K. Datta, M. Verhage, A. Al-Ashouri, H. Köbler, X. Li, A. Abate, S. Albrecht and M. Creatore, ACS Appl. Mater. Interfaces, 2022, 14(1), 2166–2176 CrossRef CAS PubMed.
- M. A. Barreiros, L. C. Alves, M. J. Brites and V. Corregidor, Nucl. Instrum. Methods Phys. Res., Sect. B, 2017, 404, 211–218 CrossRef CAS.
- C. Liu, J. Yuan, R. Masse, X. Jia, W. Bi, Z. Neale, T. Shen, M. Xu, M. Tian, J. Zheng, J. Tian and G. Cao, Adv. Mater., 2021, 33(22), 1905245 CrossRef CAS PubMed.
- D. W. deQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulović, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7(1), 11683 CrossRef CAS PubMed.
- R. Xiaoxue, Z. Lixiu, Y. Yongbo and D. Liming, J. Semicond., 2021, 42(20120007), 010201 Search PubMed.
- Y. Yuan, Q. Wang, Y. Shao, H. Lu, T. Li, A. Gruverman and J. Huang, Adv. Energy Mater., 2016, 6(2), 1501803 CrossRef.
- A. P. Batra and L. M. Slifkin, J. Phys. Chem. Solids, 1976, 37(10), 967–969 CrossRef CAS.
- T. Zhang, X. Meng, Y. Bai, S. Xiao, C. Hu, Y. Yang, H. Chen and S. Yang, J. Mater. Chem. A, 2017, 5(3), 1103–1111 RSC.
- T. Zhang, S. H. Cheung, X. Meng, L. Zhu, Y. Bai, C. H. Y. Ho, S. Xiao, Q. Xue, S. K. So and S. Yang, J. Phys. Chem. Lett., 2017, 8(20), 5069–5076 CrossRef CAS PubMed.
- C. Das, M. Kot, T. Hellmann, C. Wittich, E. Mankel, I. Zimmermann, D. Schmeisser, M. Khaja Nazeeruddin and W. Jaegermann, Cell Rep. Phys. Sci., 2020, 1(7), 100112 CrossRef.
- J.-P. Correa-Baena, S.-H. Turren-Cruz, W. Tress, A. Hagfeldt, C. Aranda, L. Shooshtari, J. Bisquert and A. Guerrero, ACS Energy Lett., 2017, 2(3), 681–688 CrossRef CAS.
- T. H. Vu, W. Chen, X. Deng, C. F. J. Lau, S. Huang, A. Ho-Baillie, B. Jia and X. Wen, J. Phys. Chem. Lett., 2020, 11(1), 136–143 CrossRef CAS PubMed.
- H. Schmalzried, Ber. Bunsenges. Phys. Chem., 1964, 68(6), 608 CrossRef.
- A. Walsh, D. O. Scanlon, S. Chen, X. G. Gong and S.-H. Wei, Angew. Chem., Int. Ed., 2015, 54(6), 1791–1794 CrossRef CAS PubMed.
- M. Kim, H. Jun, H. Lee, H. Nahdi, D. Tondelier, Y. Bonnassieux, J.-É. Bourée and B. Geffroy, Eur. J. Inorg. Chem., 2021,(46), 4781–4789 CrossRef CAS.
- H. S. Choi and H. S. Kim, Materials, 2020, 13(17), 3868 CrossRef CAS PubMed.
- R. Guo, D. Han, W. Chen, L. Dai, K. Ji, Q. Xiong, S. Li, L. K. Reb, M. A. Scheel, S. Pratap, N. Li, S. Yin, T. Xiao, S. Liang, A. L. Oechsle, C. L. Weindl, M. Schwartzkopf, H. Ebert, P. Gao, K. Wang, M. Yuan, N. C. Greenham, S. D. Stranks, S. V. Roth, R. H. Friend and P. Müller-Buschbaum, Nat. Energy, 2021, 6(10), 977–986 CrossRef CAS.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6(1), 613–617 RSC.
- K. Fatima, M. Irfan Haider, A. Fakharuddin, Z. Akhter, M. Sultan and L. Schmidt-Mende, Sol. Energy, 2020, 211, 654–660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Includes depth profiles, experimental methods, i.e., RBS and PIXE results of IHP/glass, IHP/NiO/ITO/glass and Cu/C60/IHP/NiO/IO/glass. It also includes XRD and DRS results of freshly prepared and aged IHP/glass and RBS results of HTL/ITO/glass after 4 months and 1 year. See DOI: https://doi.org/10.1039/d2ma00199c |
| This journal is © The Royal Society of Chemistry 2022 |