
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA strained intramolecular P/Al-FLP and its reactivity toward allene†
Patrick
Federmann
,
Tamino
Bosse
,
Siad
Wolff
 ,
Beatrice
Cula
,
Christian
Herwig
and
Christian
Limberg
*
,
Beatrice
Cula
,
Christian
Herwig
and
Christian
Limberg
*
Institut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489, Berlin, Germany. E-mail: Christian.limberg@chemie.hu-berlin.de
First published on 2nd November 2022
Abstract
FLPs featuring aluminum–phosphane interactions, spring-loaded by a rigid biphenylene linker, have been accessed through a route where trimethyltin units at phosphane-functionalized organic backbones are exchanged by an AlCl2 moiety. Upon contact with substrates like CO2 these are readily bound by the Al/P site with release of strain. The system could also be utilized for a unique reactivity, namely the activation of allene.
Since the discovery of frustrated Lewis pairs (FLP) in 2006 a large number of small molecules have been activated and even catalytic applications have been reported by both inter- and intramolecular systems.1 Their reactivity is primarily influenced by the HOMO–LUMO gap between the Lewis acid and base (often a combination of group 13 and 15 elements).2 For intramolecular FLPs in which the two reactive centers are separated by a linker the spatial distance is also of decisive importance. It is generally assumed that the latter should be in a range of 3–5 Å, as with larger distances cooperative interaction of the Lewis acid and base with a substrate is impeded; shorter distances lead to an interaction between the HOMOLB and LUMOLA such that the potential is (partly) quenched.3 This can be exemplified considering four intramolecular FLPs from the literature with Lewis acidic B(C6F5)2 and Lewis basic phosphine moieties bound to different rigid linker scaffolds (Fig. 1): at 5.67 Å, the reactive centers fixed to a dibenzofuran scaffold (A)4 are too far apart, while the short distance of 2.06 Å mediated by an acenaphthyl scaffold (D)5 originates from a pronounced interaction. For both molecules no reactivity toward substrates was observed. In contrast, the xanthene- and biphenylene-based systems (B and C) readily react with dihydrogen, nitrous oxide, terminal alkynes, and dioxygen.4,6 Solid state structures of the active FLPs to determine the P⋯B distances could not be determined, yet spectroscopic data indicate no interaction between the reactive sites, which is in line with the widely accepted assumption that these backbones prevent an interaction.7
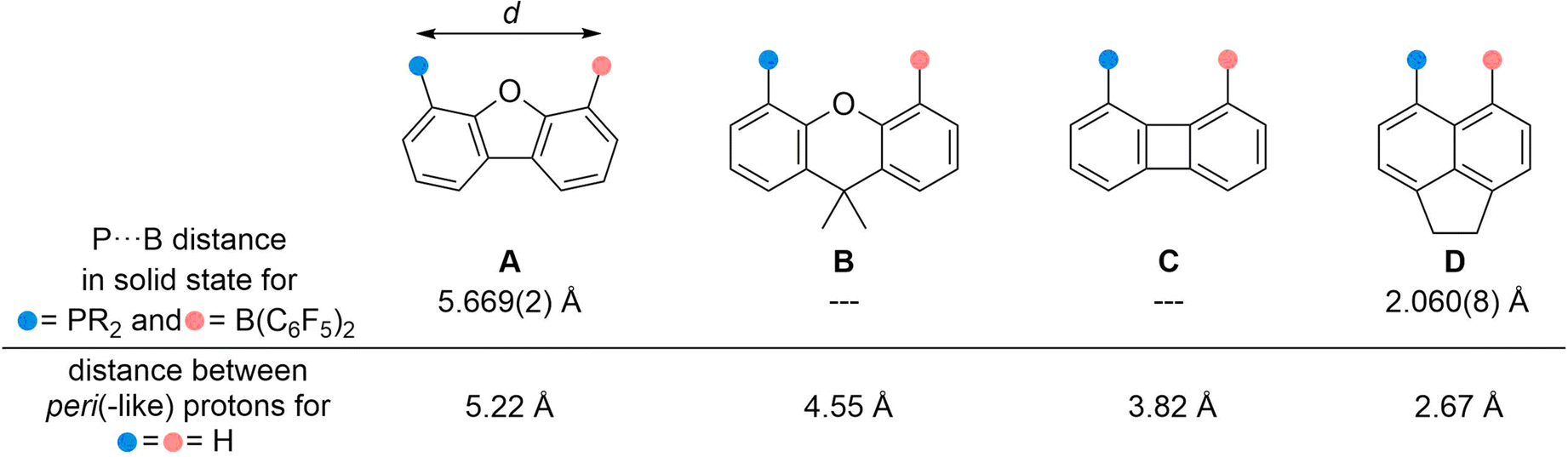 | ||
| Fig. 1 Intramolecular P/B-based FLPs on rigid carbon scaffolds to adjust the P⋯B distance. | ||
An analogous chemistry utilizing aluminum as acceptor has not been explored for such a variety of linker scaffolds because of the synthetic difficulties associated with its higher intrinsic Lewis acidity compared to boron.8
Reported examples feature close arrangements of Lewis acid and base and often an interaction is observed that stabilizes the Lewis acidic alane, thereby rendering the FLP more exothermic and synthetically easier to access.8,9
In previous work we reported the synthesis of xanthene-based P/Al-FLPs via an aluminum-tin exchange between a stannylated backbone and a methylaluminum precursor.10 However, two equivalents of the respective alane reagent were required, as the envisaged FLP upon formation immediately spans a further alane precursor molecule, which short-circuits the FLP. This was shown to not be detrimental for reactivity, as the bridging alane molecules are readily displaced by external substrates with O-donors such as THF or CO2, which get activated between the phosphine and the two alane functions, rendering the system an inter-/intramolecular hybrid.
Herein, we present the exploitation of the aluminum-tin exchange route for the synthesis of a P/Al-based FLP with a highly Lewis acidic acceptor site on the biphenylene backbone. This was envisaged to permit a small degree of interaction between the Lewis acidic and basic moieties thus leading to a reactive intramolecular 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 P/Al-FLP with a large spatial separation.
:1 P/Al-FLP with a large spatial separation.
Following the same synthetic strategy as for the xanthene-based P/Al-FLPs the precursor for an Al–Sn exchange [8-trimethylstannyl-1-biphenylenyl]diphenylphosphine 1-Ph, was synthesized from the parent 1,8-dibromobiphenylene11 in an overall yield of 79% over two steps (Scheme 1). Single crystal XRD analysis confirms the successful attachment of the diphenylphosphine and trimethylstannyl units in a distance (dP–Sn ∼ 4.02 Å) typical for the biphenylene backbone.
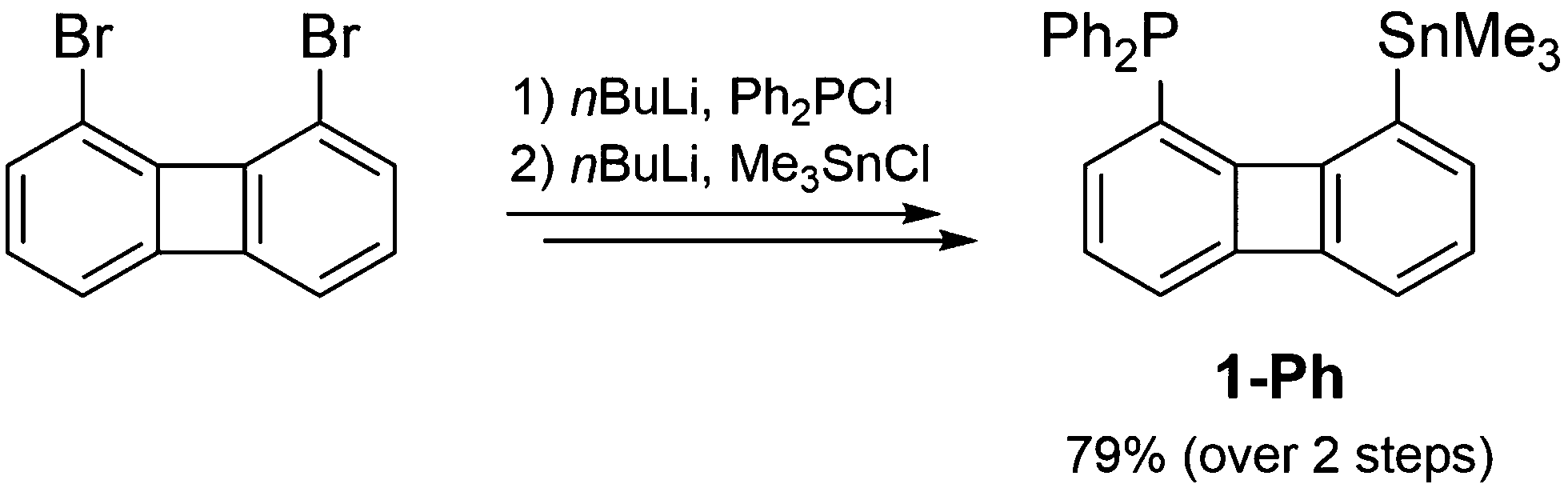 | ||
| Scheme 1 Synthesis of the precursor for an Al–Sn exchange 1-Ph. | ||
The reactivity of the precursor 1-Ph toward one equivalent of MeAlCl2 in C6D6 was monitored by NMR spectroscopy. In the 1H NMR spectrum recorded after 15 minutes at room temperature an intense signal with tin satellites at δ = 0.00 ppm was noted which could be assigned to Me4Sn. The absence of resonances of the starting material 1-Ph indicated a complete reaction. In the 31P{1H} NMR spectrum the formation of a new species resonating as a broadened singlet at δ = −19.1 ppm (ν½ ≈ 60 Hz) was observed. Due to the high selectivity of the reaction the new compound Biph(PPh2)(AlCl2), 2-Ph, could be isolated as a highly air and water sensitive solid with satisfying purity simply through removal of the solvent and Me4Sn in vacuo (Scheme 2). Our attempts to detect the 27Al{1H} NMR resonance of 2-Ph, which would provide information about the coordination number of the alane in solution remained unsuccessful.12
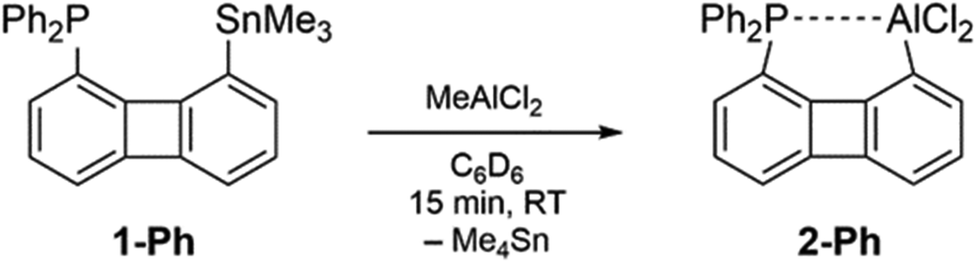 | ||
| Scheme 2 Al–Sn exchange starting from 1-Ph and MeAlCl2 to give 2-Ph. | ||
The solid-state structure determined via single crystal XRD analysis confirms the successful introduction of the AlCl2 moiety beside the Ph2P unit on the biphenylene scaffold in 1,8 positions (Fig. 2). Most conspicuous is the bending of both substituents towards each other, so that compared with 1-Ph a much shorter peri-like distance of dP–Al = 2.5456(6) Å results. The ratio between this distance and the sum of the covalent radii (228 pm) is r = 1.12 indicating a strong interaction.13r values in this magnitude are usually observed for element 13 and 15 combinations on the (ace)naphthalene scaffold,7e.g. the corresponding acenaphthalene compound Ace(PPh2)(AlCl2) exhibits a P–Al distance of 2.4305(6) Å (r = 1.07).14 The bay angles P1–C8–C7 113.6(1)° and Al1–C1–C6 111.6(1)° deviate significantly from 120° indicating a high strain within the molecule. The donation of the phosphine's lone pair into the empty pz orbital triggers a rehybridization at the aluminum center, and hence the sum of the angles around the aluminum center 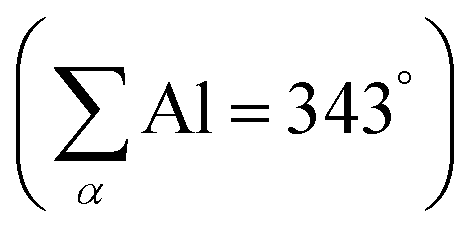 substantially differs from 360°. The tetrahedral character was determined to be 92%.15
substantially differs from 360°. The tetrahedral character was determined to be 92%.15
 | ||
| Fig. 2 Molecular structure of 2-Ph. H atoms omitted for clarity; thermal ellipsoids drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: P1–C8 1.807(1), Al1–C1 1.960(1), Al1-P1 2.5456(6), Cl1–Al1–Cl2 111.39(2), Cl1–Al1–C1 115.46(4), C1–Al1–Cl2 116.36(4). | ||
By calculation (B3PW91/6-311+G(2df,p)) of an isodesmic reaction (see ESI†) a peri-interaction energy16 of α-PIE(P → Al) = −84.9 kJ mol−1 was revealed (compare α-PIE = −106.5 kJ mol−1 for Ace(PPh2)(AlCl2)).
To investigate in how far this prominent P → Al interaction in 2-Ph diminishes the Lewis acidity of the alane function, which an external substrate experiences, the Fluoride Ion Affinity (FIA) was calculated and the Gutmann–Beckett acceptor number (AN) was ascertained experimentally (see ESI†). Notably, in spite of the repulsion between the phosphine and the negatively charged fluoridoaluminiate unit in the calculated product the FIA is 412 kJ mol−1. By reaction with the neutral compound Et3PO an AN of 82 was determined which is as high as reported for B(C6F5)3 and close to AlCl3 (AN = 87)17 despite the +I effect of the biphenylene substituent and the proximity to the Ph2P unit. These results exemplify the capability of the P–Al bond to open up when an external donor is presented. Consequently, 2-Ph reacts with O-donors like Et2O or THF to the corresponding adducts, and evaporation/heating did not lead back to 2-Ph. Interestingly, unlike in case of the xanthene-based analogue,10,18 no ring-opening reaction of THF was observed, which confirms the importance of the appropriate distance for a certain activation process.
Next, we investigated the capability of 2-Ph to activate CO2 – a benchmark substrate in FLP chemistry.1,8,9a,9b,19 Therefore, a solution of 2-Ph in DCM-d2 was treated with 2 bar CO2. 1H and 31P{1H} NMR spectra recorded subsequently confirmed the full conversion of 2-Ph and the selective formation of a new product (Scheme 3). In the ATR-IR spectrum recorded for the solid isolated after evaporation of all volatiles two bands in the characteristic carbonyl region were found at ν = 1720 cm−1 and 1643 cm−1 (ν = 1680 cm−1 and 1598 cm−1 when 13CO2 was used) underpinning the formation of the CO2 adduct 3-Ph.
 | ||
| Scheme 3 Activation of CO2 by 2-Ph. | ||
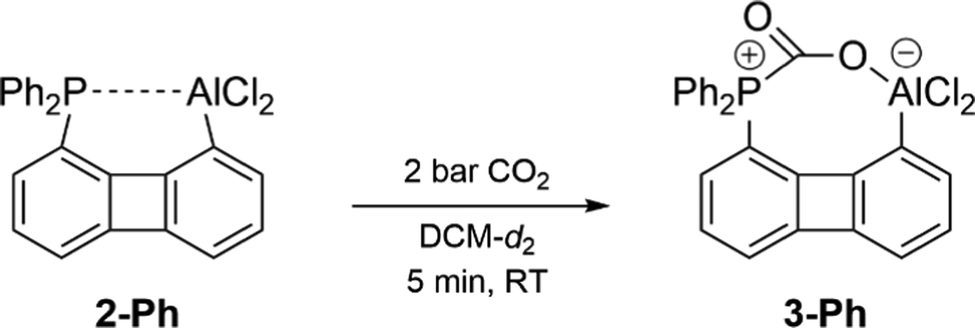
The solid-state structure of 3-Ph determined by single crystal XRD analysis revealed the successful fixation of CO2 within the reaction pocket generated in the course of breaking the P–Al bond (Fig. 3). As expected, a P–C and an Al–O bond have been formed but in contrast to the CO2 adduct of the previously reported xanthene-based P/Al-FLP10 the second oxygen atom doesn’t interact with another Al center. Hence, the CO2 molecule has been activated asymmetrically through breaking of one of the π-bonds, so that one C–O bond (1.28 Å) is longer than the other (1.20 Å). Even under high vacuum CO2 is not released again. Having proven the general capability of FLP 2-Ph to react with small molecules despite the strong P → Al interaction a more challenging substrate, lacking an O-donor was targeted. We considered allene to be an interesting candidate, since – in spite of a rich chemistry of allenes reacting with Lewis acidic boranes, particularly in hydroboration reactions with HB(C6F5)220 – the activation of simple allenes by an FLP remains unknown, which is particularly surprising as reports on the conversion of isolated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds are extensive.1c In a study of Melen and coworkers B(C6F5)3 and a bulky phosphine were added to different 2,3-butadienones, leading to transformations, in which the ketone group plays an active role.21 The reactivity of non-activated (and activated) allenes towards a P/B-FLP has only been investigated in a predictive computational study but has not been achieved experimentally.22
C bonds are extensive.1c In a study of Melen and coworkers B(C6F5)3 and a bulky phosphine were added to different 2,3-butadienones, leading to transformations, in which the ketone group plays an active role.21 The reactivity of non-activated (and activated) allenes towards a P/B-FLP has only been investigated in a predictive computational study but has not been achieved experimentally.22
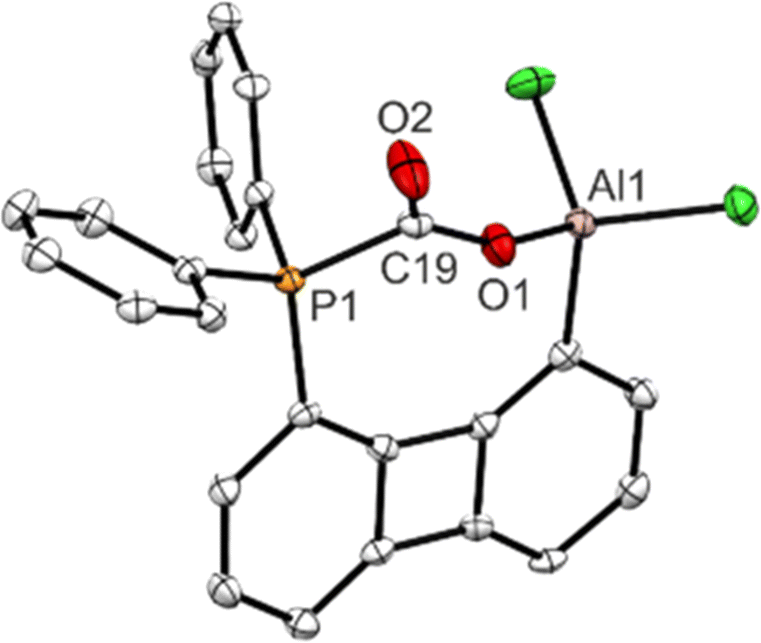 | ||
| Fig. 3 Molecular structure determined for one of the two independent molecules within the asymmetric unit of 3-Ph. H atoms omitted for clarity; thermal ellipsoids drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: P1–C19 1.883(3), C19–O1 1.281(3), C19–O2 1.202(3), O1–Al1 1.784(2), O1–C19–O2 128.2(3), Al1–O1–C19 150.5(2). | ||
When a sample of 2-Ph in C6D6 was exposed to a pressure of 1.6 bar propadiene a very slow reaction at room temperature could be observed as indicated by a rising resonance at δ = 23.0 ppm in the 31P{1H} NMR spectrum. Heating to 70 °C accelerates the reaction, leading to full consumption of 2-Ph within 3 days. In the 1H NMR spectrum, three doublets were found at chemical shifts of δ = 6.59, 5.00, and 1.65 ppm with 1H–31P coupling constants of J = 53.3, 24.4, and 19.8 Hz, respectively, and an integral ratio of 1:1:2. These spectroscopic data are in line with the regioselective formation of an allene-FLP-complex, 4-Ph (Scheme 4). Attempts to isolate the product failed, however, from the crude oil obtained after evaporation of the reaction mixture crystals suitable for XRD analysis formed after standing for several weeks.
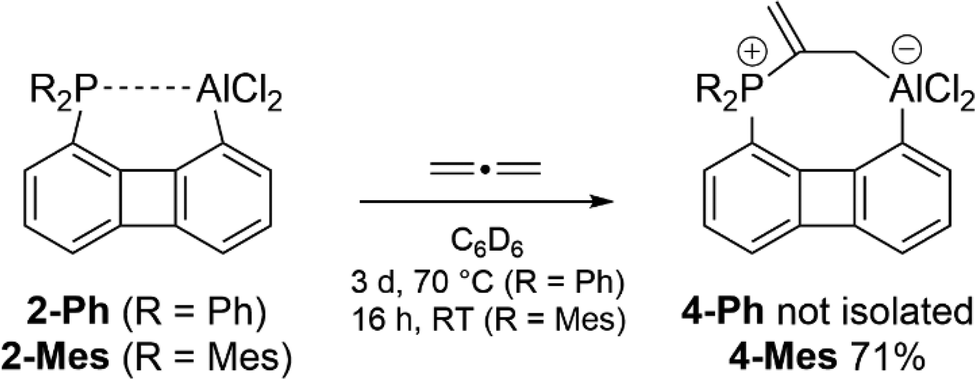 | ||
| Scheme 4 Activation of propadiene by 2-R. | ||
The solid-state structure (Fig. 4) reveals that the allene has indeed been captured in such a way that the phosphine binds to the central carbon atom of the allene and the aluminum center to a terminal one. As can be seen from the C–C bond length (1.500(2) Å) between these carbon atoms, the activation of the allene by the FLP cleaves the π-bond. Compared to free allene (d(CC) = 1.308 Å) also the C19–C27 bond (1.338(2) Å) is elongated. This can be rationalized by the increasing p-character of the central carbon atom which rehybridizes from sp to sp2, adapting a trigonal planar coordination sphere. The carbon atom bound to aluminum changes from a sp2 to a sp3 hybridization.
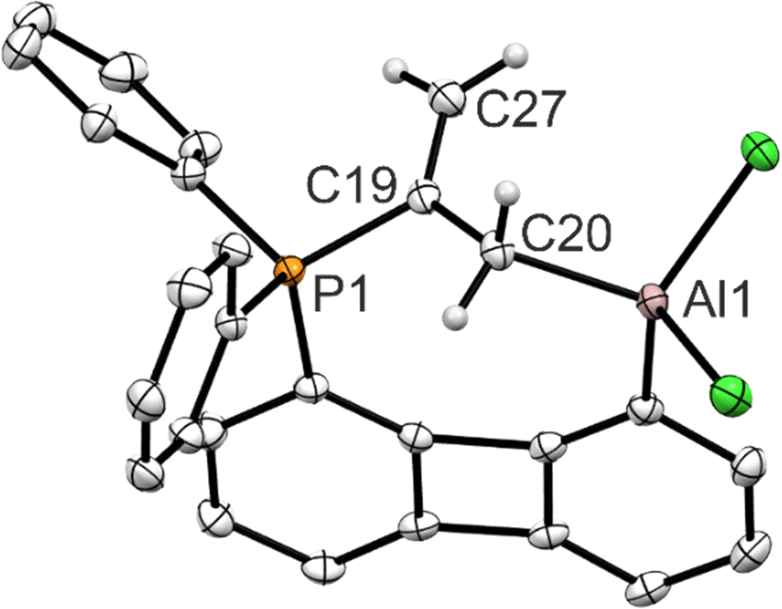 | ||
| Fig. 4 Molecular structure of 4-Ph. H atoms omitted for clarity except for the allene unit; thermal ellipsoids drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: P1–C19 1.811(2), C20–Al1 1.976(2), C20–C19–C27 125.8(2). | ||
We were able to isolate and fully characterize the product of allene activation when the substituents of the phosphine in 1-Ph were altered from phenyl to mesityl. Following the route described above, 1-Mes could be converted into 2-Mes by MeAlCl2 and the resulting solution was exposed to a slight excess of C3H4. Within 16 hours at room temperature the allene adduct 4-Mes precipitated from the reaction mixture and could be isolated in 71% yield. The far more rapid reaction found for 2-Mes toward allene in contrast to 2-Ph can be explained by the fact that the mesityl substituents, cause both a weaker interaction between the P and Al centers due to their increased steric demand and an improved nucleophilicity of the phosphorus atom due to the electron donating properties.
In conclusion, we report here the convenient synthesis of an intramolecular P/Al-based FLP on the biphenylene backbone via aluminum-tin exchange. It was shown that by steric strains the reactive centers can overcome the large distance spanned by the scaffold to coordinatively saturate the highly Lewis acidic AlCl2 moiety. Despite this interaction the FLP exhibits pronounced reactivity toward a common substrate like carbon dioxide and also binds allene which has not been activated by an FLP before. Future research will aim at exploiting the latter for organic transformations.
We thank Dr André Dallmann for the 27Al NMR spectroscopy measurements. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 387284271 – SFB 1349.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (d) D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 Search PubMed; (e) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 Search PubMed.
- Z. Mo, A. Rit, J. Campos, E. L. Kolychev and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 3306–3309 CrossRef CAS PubMed.
- L. L. Zeonjuk, N. Vankova, A. Mavrandonakis, T. Heine, G. V. Röschenthaler and J. Eicher, Chem. – Eur. J., 2013, 19, 17413–17424 Search PubMed.
- Z. Mo, E. L. Kolychev, A. Rit, J. Campos, H. Niu and S. Aldridge, J. Am. Chem. Soc., 2015, 137, 12227–12230 CrossRef CAS PubMed.
- S. Furan, E. Hupf, E. Lork, S. Mebs and J. Beckmann, Eur. J. Inorg. Chem., 2017, 3302–3311 Search PubMed.
- J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2018, 54, 6344–6347 Search PubMed.
- O. Sadek, G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2021, 50, 5777–5805 RSC.
- M.-A. Courtemanche, J. Larouche, M.-A. Légaré, W. Bi, L. Maron and F.-G. Fontaine, Organometallics, 2013, 32, 6804–6811 CrossRef CAS.
- (a) J. Boudreau, M.-A. Courtemanche and F.-G. Fontaine, Chem. Commun., 2011, 47, 11131–11133 Search PubMed; (b) C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 Search PubMed; (c) F. Bertini, F. Hoffmann, C. Appelt, W. Uhl, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Organometallics, 2013, 32, 6764–6769 Search PubMed; (d) H. S. Zijlstra, J. Pahl, J. Penafiel and S. Harder, Dalton Trans., 2017, 46, 3601–3610 RSC.
- P. Federmann, R. Müller, F. Beckmann, C. Lau, B. Cula, M. Kaupp and C. Limberg, Chem. – Eur. J., 2022, 28, e202200404 CrossRef CAS PubMed.
- F. Kutter, E. Lork and J. Beckmann, Z. Anorg. Allg. Chem., 2018, 644, 1234–1237 CrossRef CAS.
- R. Benn, E. Janssen, H. Lehmkuhl and A. Rufínska, J. Organomet. Chem., 1987, 333, 155–168 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- E. Hupf, E. Lork, S. Mebs, L. Chęcińska and J. Beckmann, Organometallics, 2014, 33, 7247–7259 CrossRef CAS.
- H. Höpfl, J. Organomet. Chem., 1999, 581, 129–149 CrossRef.
- E. Hupf, E. Lork, S. Mebs and J. Beckmann, Organometallics, 2015, 34, 3873–3887 Search PubMed.
- M. A. Beckett, D. S. Brassington, S. J. Coles and M. B. Hursthouse, Inorg. Chem. Commun., 2000, 3, 530–533 CrossRef CAS.
- P. Federmann, C. Herwig, F. Beckmann, B. Cula and C. Limberg, Organometallics, 2021, 40, 4143–4149 CrossRef CAS.
- (a) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed; (b) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796–1797 Search PubMed; (c) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2011, 50, 8396–8399 CrossRef PubMed; (d) E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 1651–1662 RSC; (e) B. Waerder, M. Pieper, L. A. Körte, T. A. Kinder, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015, 54, 13416–13419 Search PubMed.
- (a) A. Averdunk, M. Hasenbeck, T. Müller, J. Becker and U. Gellrich, Chem. – Eur. J., 2022, 28, e202200470 CrossRef CAS PubMed; (b) U. Gellrich, Eur. J. Org. Chem., 2021, 4707–4714 CrossRef CAS.
- R. L. Melen, L. C. Wilkins, B. M. Kariuki, H. Wadepohl, L. H. Gade, A. S. K. Hashmi, D. W. Stephan and M. M. Hansmann, Organometallics, 2015, 34, 4127–4137 CrossRef CAS.
- H. Mondal, M. Ghara and P. K. A. Chattaraj, Chem. Phys. Lett., 2021, 774, 138623 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, computational details, crystallographic data. CCDC 2179785 and 2179787–2179789. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05640b |
| This journal is © The Royal Society of Chemistry 2022 |