
Applications of MAX phases and MXenes as catalysts
Iuliana M.
Chirica
 ab,
Anca G.
Mirea
a,
Ştefan
Neaţu
a,
Mihaela
Florea
*a,
Michel W.
Barsoum
*c and
Florentina
Neaţu
*a
ab,
Anca G.
Mirea
a,
Ştefan
Neaţu
a,
Mihaela
Florea
*a,
Michel W.
Barsoum
*c and
Florentina
Neaţu
*a
aNational Institute of Materials Physics, 405A Atomistilor Street, 077125 Magurele, Romania. E-mail: mihaela.florea@infim.ro; florentina.neatu@infim.ro
bUniversity of Bucharest, Faculty of Physics, 405 Atomistilor Street, 077125 Magurele, Romania
cDepartment of Materials Science & Engineering, Drexel University, Philadelphia, PA 19104, USA. E-mail: barsoumw@drexel.edu
First published on 28th July 2021
Abstract
MAX phases and MXenes are important materials that have recently gained great popularity due to their special properties, which render them particularly useful in many applications, including catalytic ones. This can be seen in the large number of publications that appear annually on these materials and their applications. This review aims to evaluate MAX phases and MXenes as materials for heterogeneous, non-electrocatalytic, catalytic applications. The review begins with a brief introduction to the MAX phase and MXene properties that recommend them as potential materials for heterogeneous catalytic applications, followed by four sections grouped according to the processes in which they have already proven effective. These include supports to activate the C–H or C–O bonds in applications such as dehydrogenation of light or aromatic alkanes, methanol formation from CH4, dry reforming, and CO oxidation or the water gas shift reaction (Section 2), and their use in fine chemical reactions (Section 3) and in chemical degradation (Section 4). The last section deals with photocatalytic applications (Section 5). The review ends by highlighting the huge potential of these materials for a wide range of heterogeneous catalytic applications as well as the challenges ahead.
 Iuliana M. Chirica | Research team, part of the group Catalytic Material and Catalysis, at the National Institute of Materials Physics, Romania (NIMP) (https://www.infim.ro). From left to right: front row- Dr Mihaela Florea (senior researcher), Iuliana M. Chirica (PhD student), and Dr Anca G. Mirea (junior researcher) and back row- Dr Stefan Neatu (senior researcher) and Dr Florentina Neatu (senior researcher). The research of the NIMP team is focused on the preparation, characterization and study of the physical properties of new materials connected to high technology products and devices. Their main research areas deal with heterogeneous catalysis, fuel cells and electrochemistry, materials surface science and energy conversion. |
 Michel W. Barsoum | Prof. Michel W. Barsoum is a Distinguished Professor in the Department of Materials Science and Engineering at Drexel University. He is an internationally recognized leader in the areas of MAX phases and their 2D derivatives, MXenes. He has published over 500 refereed publications. His h index is >110 and he was in the highly cited researchers list in 2018, 2019 and 2020. He is the author of MAX Phases: Properties of Machinable Carbides and Nitrides and the leading textbook in his field, Fundamentals of Ceramics. In 2020, he was awarded the International Ceramics Prize for basic science by the World Academy of Ceramics. |
1. Introduction
The MAX phases (Mn+1AXn) and their derivatives, MXenes (Mn+1XnTx), (with n = 1–4, M = early transition metal, A = A-group element, X = carbon or nitrogen and Tx = various surface terminations, such as –O, –OH and –F) are a relatively new class of materials that have been discovered in the last two decades.1–3 They have impressed us by their multitude of physical and chemical properties. These materials manage to bring together two completely different classes of materials (ceramics and metals) by combining important properties from both classes, such as high thermal and electrical conductivity, mechanical strength and low density. Moreover, MAX phases also possess resistance to high temperatures or oxidation.Presently, over 150 MAX phases4 and more than 30 MXenes with different compositions have been synthesized.5 The unique properties of MAX phases arise from their lamellar structure, metallic/covalent bonds, and the relatively weak bonds between the M and A layers.
The presence of oxygenated terminations preserves the high conductivity even in polar media.2 Thus, as a result of all these interesting properties, MAX phases and MXenes are highly desirable and attractive materials for catalysis, but despite all evidence that recommends them as catalysts, their catalytic properties were investigated only recently6 and fortunately the interest is increasing exponentially (see Fig. 1) and is expected to grow considerably in the future.
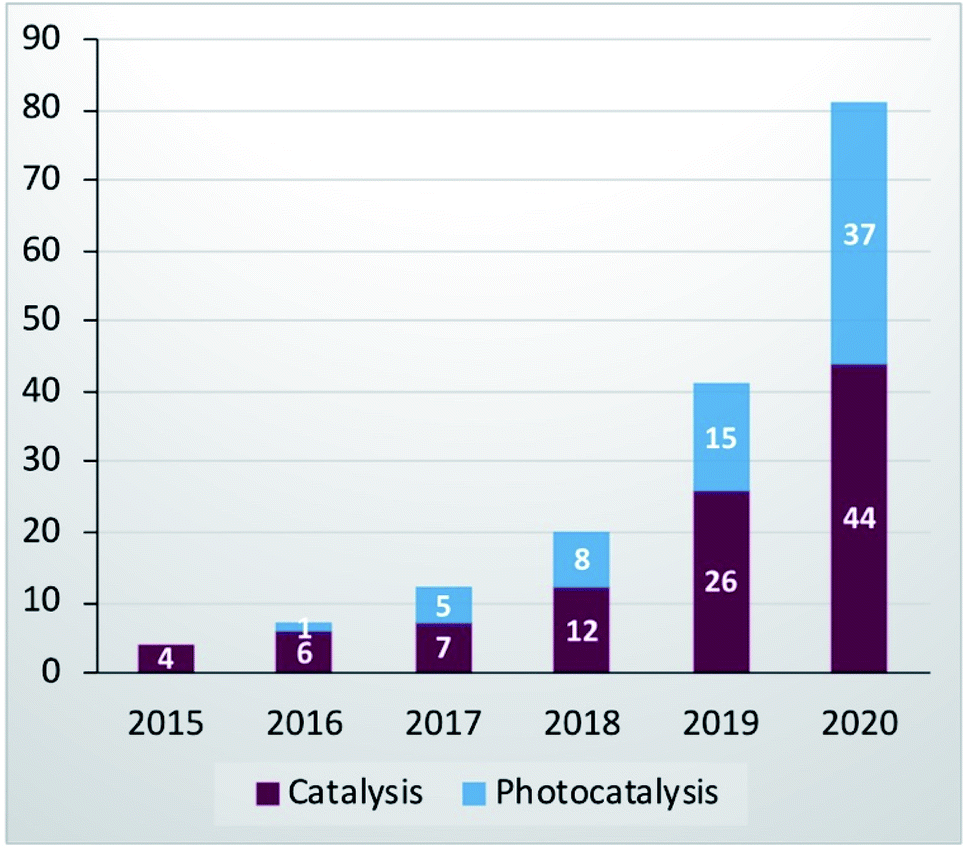 | ||
| Fig. 1 MAX phase and MXene publications in catalysis and photocatalysis fields (data collected from http://www.webofknowledge.com) (May 2021, electrocatalysis was excluded). | ||
Up to now, there are several excellent reviews about MXenes and MXene-based materials that focus on the preparation, properties, and potential applications.1–3,5,7–10 However, a comprehensive and systematic review on recent progress in the use of MAX phases and MXene-based materials in catalytic applications is lacking. In this review, we intend to present general aspects to be considered and to outline which are currently the most efficient catalytic systems based on these materials in: (i) catalytic processes centred on C–H and C–O activation, (ii) fine chemical reactions, (iii) catalytic degradation, and (iv) photocatalytic applications. An application roadmap is depicted in Scheme 1. Note that there are already several review papers up to 2020 that cover photocatalytic materials.11–14 Here we thus focus only on the last two years (2020 and 2021). Moreover, in this review we do not discuss MAX phases and MXene materials as catalysts for electrochemical applications. Readers can refer to other comprehensive reviews in that area.15–17
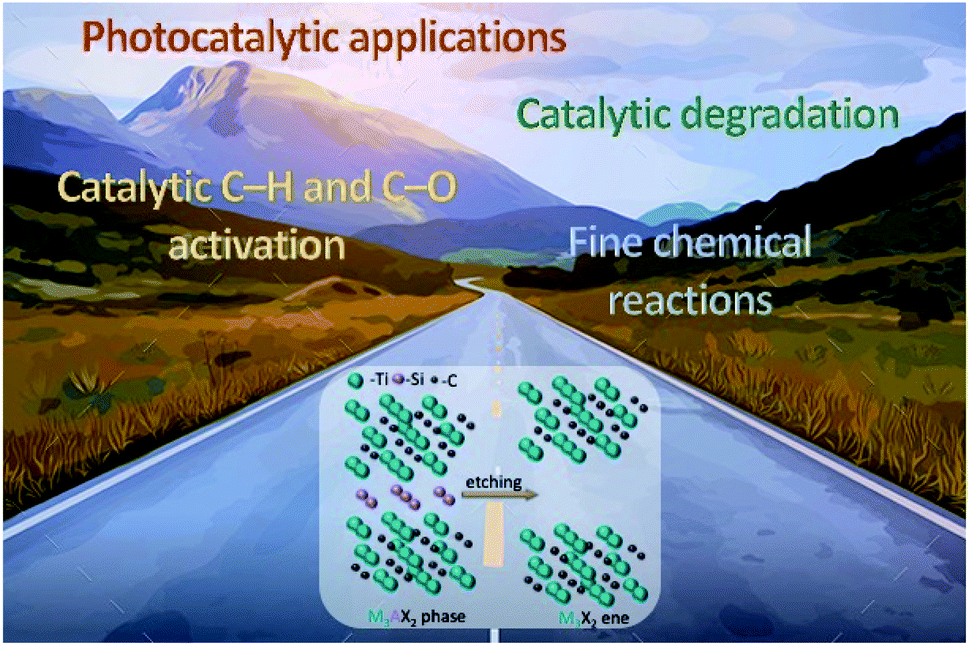 | ||
| Scheme 1 Application road map of the MAX phases and their MXene derivatives. | ||
2. Processes based on C–H and C–O activation (theoretical and experimental studies)
The mind map illustrated in Scheme 2 summarizes the materials used in C–H and C–O bond activation and their corresponding catalytic reactions.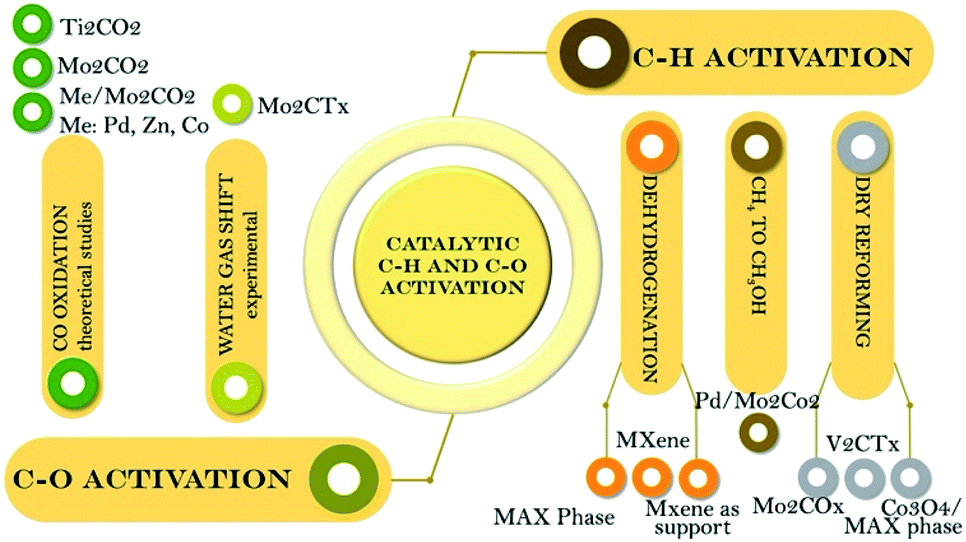 | ||
| Scheme 2 Types of MAX phases and MXenes used in processes based on C–H and C–O activation. | ||
2.1. C–H activation
Direct dehydrogenation (DDH) and oxidative dehydrogenation (ODH) of hydrocarbons are important reactions for obtaining alkene monomers, that are, in turn, key intermediates in the polymer industry. Some of the important monomers include n-butene or 1,3-butadiene starting from n-butane18 or styrene synthesized from ethylbenzene.19
Usually these processes require high temperatures (∼600 °C) and excess steam and suffer from catalyst deactivation due to the formation of coke. The MAX phases and MXenes show activity and long-term stability in these types of reactions.
Oxidative reactions in general, and ODH in particular, are recognised to take place through redox mechanisms, meaning that the catalyst in a higher oxidation state oxidizes the reactants while it is being reduced, and the oxygen re-oxidizes the catalyst surface to restart a new cycle.20,21 This mechanism – also known as the Mars–van-Krevelen mechanism – is associated with oxide materials and usually metallic oxides or metals supported on metallic oxides.22
The most intriguing part is that the MAX phases, while presenting combined metallic and ceramic properties, do not possess an oxygen lattice, which apparently is a prerequisite for oxidative reactions. However, Ti3AlC2 is not only active (24% conversion) in n-butane ODH, but it is also quite selective (a total selectivity of 50% for butenes and butadiene), which is a remarkable result.6 It is difficult to achieve active and selective catalysis simultaneously because conventional conditions to activate n-butane are too harsh and typically lead to total oxidation of n-butane (CO2 and H2O). The authors supposed that in the absence of an oxygen lattice, the adsorption of oxygen highly depends on the defect content of the surface oxide, which can also act as active sites. HRTEM data showed the presence of a large number of defects (point, domain or layer defects), while positron annihilation lifetime spectroscopic analyses confirmed the presence of Ti monovacancy defects and vacancy clusters and voids. The presence of a thin oxide layer on the surface (Al–Ti–O), with a high oxygen content (HRTEM, XPS, and EDX mapping) that is believed to have synergy with the electron reservoir below it, is believed to be responsible for the Ti3AlC2 activity in n-butane ODH. To confirm this assumption, computational evaluation of the possible mechanisms was considered, starting from the presumption that the stable ternary phase of Al2TiO5 is present on the surface and it is oxygen terminated. First, the DFT calculations showed that butane adsorbs relatively easily, through the formation of a C–O bond on the selected surface. Also, the DFT calculations indicated that the oxidative dehydrogenation of n-butane is energetically favourable on the MAX phase surface and moreover, the presence of defects and the existence of superficial non-stoichiometric oxide surface layers rich in oxygen vacancies promote this material as a potential catalyst, enabling the transformation of n-butane into butene or/and butadiene.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at the catalyst interface.24,25
O) at the catalyst interface.24,25
The results obtained by Diao et al.23 showed that Ti3C2Tx was not only suitable for this catalytic reaction, but also increased ethylbenzene's DDH activity to 92 μmol m−2 h−1, ∼8 times higher than that of graphene. Combining experimental and theoretical first-principles calculations, it was concluded that the surface C–Ti–O groups and the layered structure of MXenes, that facilitate the mass transfer and adsorption–desorption processes, are responsible for the catalytic performance of Ti3C2Tx in the ethylbenzene dehydrogenation reaction. Noteworthily, the selectivity of ethylbenzene DDH on Ti3C2Tx was 97.5%, indicating very few side products. Thus, experimentally, the layered structure of MXenes has been demonstrated to bring an important value to the DDH reaction. Interestingly, the MAX phase precursor – Ti3AlC2 – reactivity was close to zero.
Fig. 2 shows the mechanism details of ethylbenzene dehydrogenation on Ti3C2Tx. The functional groups, Tx, found on the surface were C–Ti–O (high percentage), C–Ti–OH and C–Ti–F. At higher temperatures, the last two functionalities are prone to transform into C–Ti–O groups, that are believed to be responsible for the impressive activity of Ti3C2Tx. Additionally, Ti3C2Tx has the advantage to remain active for a long time (40 h) without any deactivation. Transmission electron microscope, TEM, images evidenced the absence of coke on the surface, underscoring the stability of this material for ethylbenzene DDH. Besides the experimental evidence, the authors performed theoretical calculations to highlight both the MXene properties and the active sites responsible for this improved activity.23
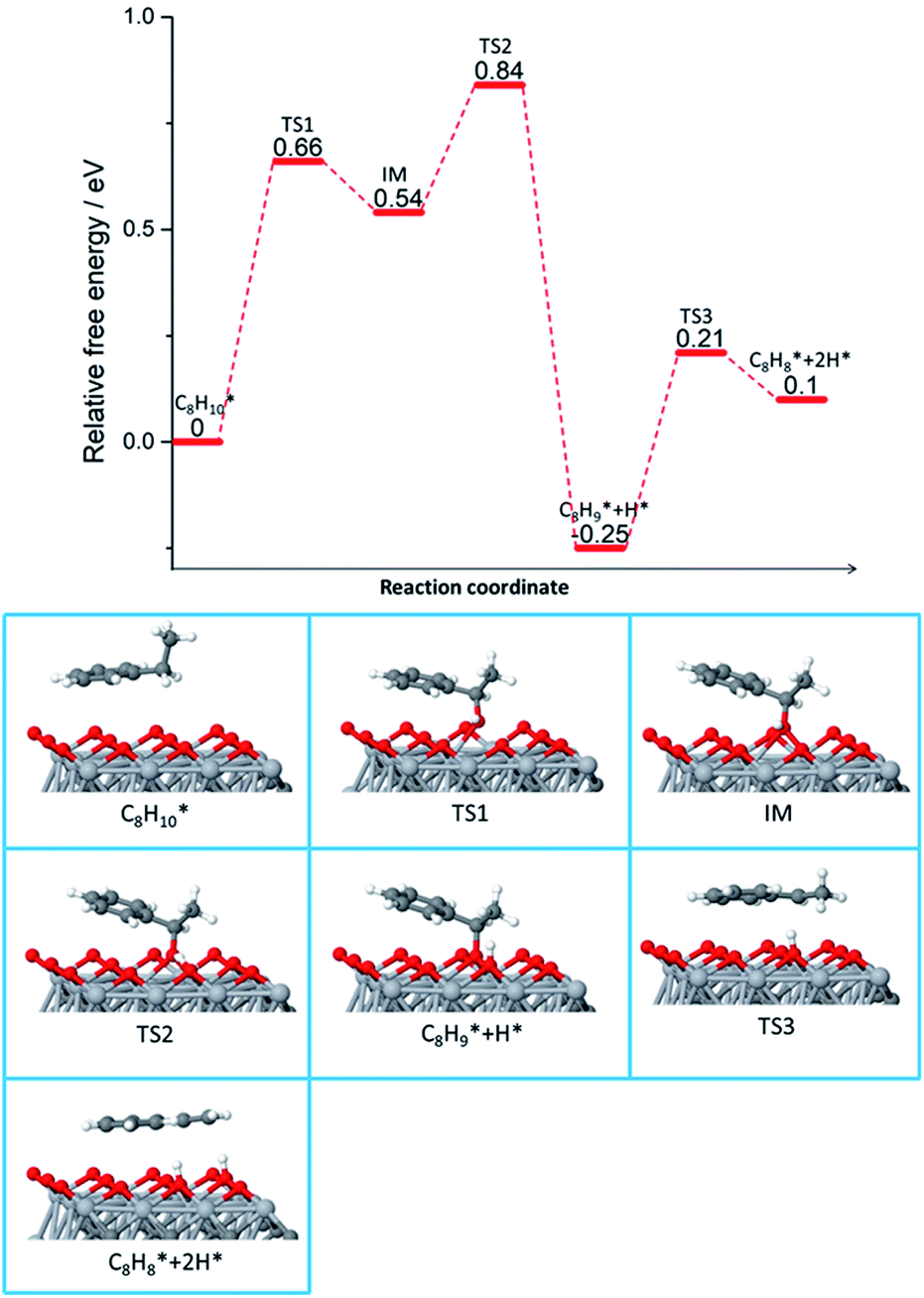 | ||
| Fig. 2 Reaction pathway and important structures of ethylbenzene dehydrogenation on Ti3C2O2. Ti is shown in light gray, C in dark gray, O in red, and H in white. Reprinted with permission from ref. 23, copyright 2018 American Chemical Society. | ||
Computational studies also demonstrated that C–Ti–O can facilitate ethylbenzene dehydrogenation by approaching ethylbenzene in a parallel way and activating the C–H bond (0.66 eV) with O found on the surface. The second dehydrogenation is possible due to the abundance of O on the MXene surface and diffusion of hydrogen to neighbouring O (Fig. 2).
Such an interaction drives the formation of bimetallic structures that form ordered intermetallic compounds (IMCs) that are quite thermodynamically stable. This type of interaction can occur at lower temperatures facilitating the control of the noble metal particle size. For example, Li et al.27 synthesized Pt3Ti and Pt3Nb IMCs with a Cu3Au type structure by supporting Pt on Ti3C2Tx and Nb2CTx MXenes, respectively. These compounds are extremely active in light alkane dehydrogenation. The reactivity is due to the Pt–M ordered intermetallic structures, which were evidenced by high resolution TEM microscopy (see Fig. 3), EXAFS and XANES.
 | ||
| Fig. 3 TEM characterization of 1 wt% Pt/MXene catalysts. (a) Representative HAADF-STEM image of the 1% Pt/Ti3C2Tx catalyst. (b) (111) surface of Pt3Ti NPs. The inset shows a simulated STEM image of the Pt3Ti(111) surface. The simulated image is in good agreement with the experimental results. (c) Schematic illustration of reactive metal-support interactions in Pt/MXene catalysts and the structure of L12-ordered intermetallic Pt3Ti. (d) Representative HAADF STEM image of the 1% Pt/Nb2CTx catalyst. (e) Pt–Nb NPs viewed along [111], the inset shows the FFT pattern of the NPs. (f) IFFT pattern of the NPs in (e). The inset shows the enlarged image showing the superlattice of the NPs. Scale bars: (a, d, e) 2 nm, and (b) 500 pm. Reprinted from ref. 27 (open access). | ||
These materials were tested in propane and isobutane dehydrogenation. In terms of activity, Pt/Ti3C2Tx and Pt/Nb2CTx were comparable with Pt/SiO2 (15%), but they showed higher selectivity (90–95%) than standard catalysts (60%) in propylene formation. The influence of particle size on selectivity (smaller particles, higher selectivity) was excluded because the amount of intermetallic compounds was higher than that on Pt/SiO2. The increase in selectivity was related to the formation of IMCs. Contrary to classical catalysts, these materials, as explained by density functional theory, DFT, preserve the selectivity even at high values of activity and are stable under long-term reaction conditions. DFT showed that in the first step of the reaction, Pt(111) found in Pt/SiO2 is more energetically favourable to adsorb and activate the C–H bond than Pt3Ti(111), but in the following steps, the formation of secondary products is not favoured on Pt3Ti(111). The experimental results and DFT calculations show that this is due to the lower barrier desorption of olefins compared with the deep dehydrogenation energy calculated on Pt3Ti(111), in contrast to the behaviour observed for Pt(111), thus explaining the high selectivity of Pt/MXene catalysts.
| CH4 + CO2 →2CO + 2H2, ΔH0298K = 247 kJ mol−1 | (1) |
Theoretical and experimental results indicated that CH4 activation is the rate-limiting step, in agreement with the results of Wang et al.29 The characterization data, such as XANES and XPS , suggested that 2D Mo2COx contains Mo, with an average oxidation state of +4, that is the active phase for dry reforming of CH4. This was also supported by DFT calculations, which showed that CH4 activation on oxygen sites to form *OCH3 and *OH is endergonic by 105 kJ mol−1, which is 45 kJ mol−1 less favourable than CH4 activation on the Mo sites of the same surface. Another key step in the dry reforming of CH4 is the oxidation of either CH* or C* species on the oxycarbide surface by adsorbed oxygen (O* or structural oxygen)31 to produce CO*. In the case of 2D-Mo2COx, and in contrast to metallic surfaces,32 DFT calculations showed that the preferred mechanism is the direct oxidation of adsorbed C* by adsorbed O* to form CO*.
The idea of depositing MXenes on silica was to prevent the thermal transformation into bulk Mo2C and MoO2 phases, in this way opening new avenues for the application of 2D Mo-carbides and oxycarbides for high temperature heterogeneous catalysis. Indeed, the nanosheet morphology of 2D-Mo2COx was maintained during catalysis and regeneration processes. Moreover, the 2D nature provides a very high utilization of Mo. For instance, when normalized by the weight of Mo, the initial CH4 consumption rate of 2D-Mo2COx/SiO2 was ∼10–200 times higher than the rates that have been reported for other Mo2C based catalysts under similar reaction conditions.
This was not the case when multilayer V2CTx MXene (m-V2CTx) was used as the catalyst in DRM. Carrero et al. revealed that V2CTx MXene transforms during the reaction into an oxycarbide with the following composition: V2O3–V8C7/m-V2CTx after exposure to CH4 and CO2.33 This oxy-carbide phase shows interesting activity for DRM, converting about 78% CH4 and 82% CO2 with CH4 and CO2 conversion rates of 153.4 mmolCH4 molV−1 min−1 and 178.2 mmolCO2 molV−1 min−1, respectively. The authors concluded that these results are comparable to those of Ni-based catalysts34 and are about four times higher than those of the parent bulk V2AlC MAX phase or VC.
The same V-based MXene was also reported as a good catalyst for dehydroaromatization of CH4.35 The multi-layered, ML, 2D V2CTx transformed CH4 in benzene at 700 °C, with similar results to those of the benchmark Mo/ZSM-5 catalyst (11.8% activity, 4.84% yield at a formation rate of 1.9 mmol gcat−1 h−1). Besides the great importance of CH4 dehydroaromatization (MDA) to produce liquid aromatics, this reaction is also extremely important from a fundamental point of view to establish the reaction mechanism for C–H activation. The correlation of experimental data and operational Raman spectroscopy evidenced the presence of 2D-VC and 2D-VO and the formation of C2H2, H2, and benzene. The authors propose the V–C bond as an active site for C–H activation, followed by the formation of dimers, between the V2CTx layers (∼7.0 Å) (see Fig. 4). Oligomers and benzene formation requires acidic sites, which are associated with the presence of terminal functional groups (e.g., OH and O−) and/or are due to the presence of the unreacted V2AlC MAX phase. Further studies are needed to conclusively better understand this reaction.
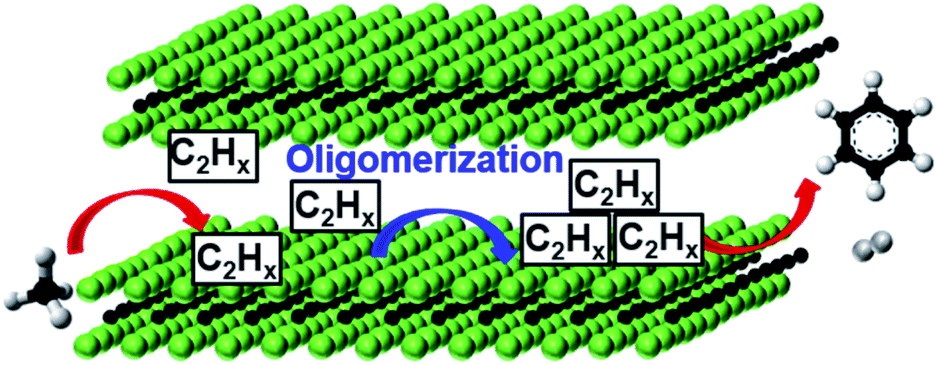 | ||
| Fig. 4 MDA reaction scheme of m-V2CTx MXene forming C6H6 and H2 as the main reaction products. Reprinted with permission from ref. 35, copyright 2020 Wiley Online Library. | ||
This material attracted interest due to the fact that it is stable at high temperatures.36 Unfortunately, like in the benchmark catalyst, deactivation due to the deposition of graphitized C occurs. The deactivation rate of the MXene, however, is lower. Comparing the catalytic activity in DRM for the two MXenes, Mo2CTx and V2CTx, it was observed that Mo2CTx is not active at all, while V2CTx is active but only after the in situ transformation into an oxycarbide with the following composition: V2O3–V8C7/m-V2CTx. By supporting Mo2CTx on silica and after CO2 treatment, a composite, Mo2COx/SiO2, that is as active as V2O3–V8C7/m-V2CTx was obtained.
Based on the results obtained for direct dehydrogenation of n-butane, Ronda-Lloret et al.37 surface modified Ti2AlC with Co3O4, to allow butane dry reforming. For butane dry reforming, the acidity of the material and also the good dispersion of the active sites are important considerations. Ti2AlC has 10 times lower acidity than Al2O3 and its use as a support should generate a catalyst which deactivates slower than the benchmark catalysts (Co3O4/Al2O3 and Co3O4/TiO2). Indeed, Co3O4/Ti2AlC has a lower activity (35%) than the Co3O4/Al2O3 (60%) catalyst, but has a much higher selectivity to CO and H2. The use of Ti2AlC as a support generates special agglomeration of Co3O4 particles in the form of hollow spheres, 90–500 nm in diameter, with small voids (6–30 nm), that are unlike the Co3O4 particles supported on alumina or titania. The appearance of hollow spheres and small voids is associated with the special features of the MAX phase containing Ti–C and Ti–Al bonds, which allows the Kirkendall effect (different diffusion rates of the cations and anions during oxidation) to take place on the surface. Contrary to TiC, which decomposes to graphite and TiO2, Ti2AlC is quite stable under dry reforming conditions. With time, the Co3O4/Ti2AlC surface becomes covered with carbon nanotubes, but the rate of coke deposition is much lower than that of the benchmark catalysts.
2.2. C–O activation
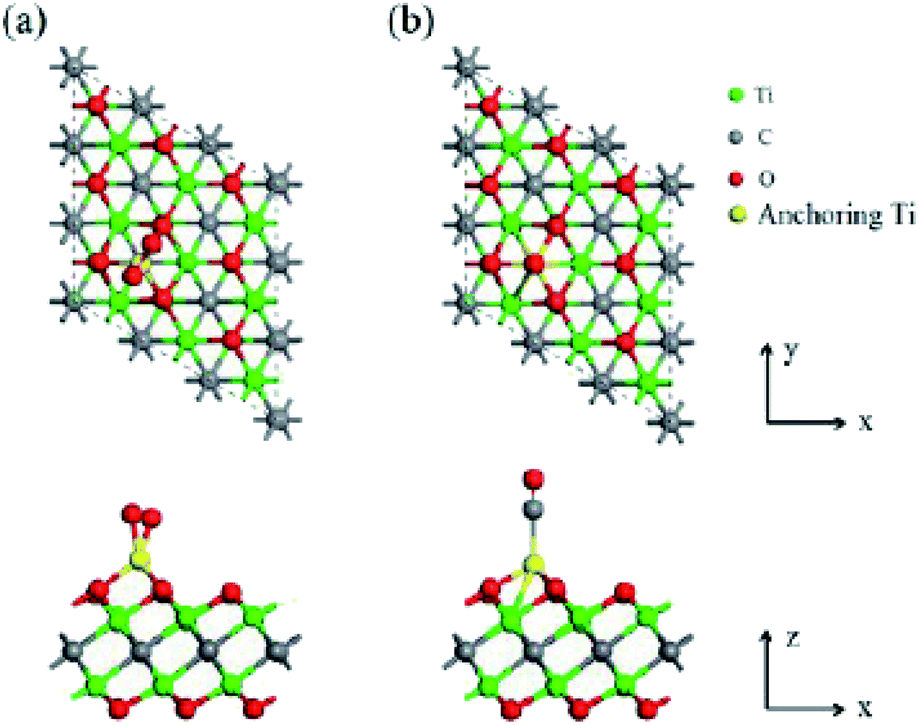 | ||
| Fig. 5 Energetically most favorable structures of (a) O2 and (b) CO adsorbed on Ti-anchored Ti2CO2 (top (upper) and side (lower) views). Reprinted with permission from ref. 38, copyright 2016 Royal Society of Chemistry. | ||
Data obtained using first-principles calculations indicated that the Eley–Rideal mechanism may be preferred under specific experimental conditions. Moreover, due to the fact that the energy barriers are comparable to many noble metal catalysts, the results demonstrated that Ti/Ti2CO2 does not need a noble metal to exhibit high activity for CO oxidation.
Another theoretical study of CO oxidation was reported using MXene as a support for single Pd atoms anchored on Mo2CO2 monolayers.39 The key point of this study was the comparison of pristine and defective Mo2CO2 monolayers with an oxygen vacancy (denoted as OV–Mo2CO2). In line with previous results,38 Pd/OV–Mo2CO2 was found to be highly active for CO oxidation and can achieve comparable catalytic activity to those containing noble metals. Moreover, the authors found that Pd/OV–Mo2CO2, due to the fact that single Pd atoms can modify the electronic structure, has higher catalytic activity than iron embedded graphene,40 copper embedded graphene,41 Pd-anchored graphene oxide and Pd-embedded vacancy graphene42 or Pd@TiO2(110)43 studied under the same reaction conditions. Contrary to the results obtained for Ti anchored on Mo2CO2 (ref. 38) for which the Eley–Rideal mechanism is preferred, for Pd supported on Mo2CO2, among the three mechanisms studied, Eley–Rideal, Langmuir–Hinshelwood and termolecular Eley–Rideal, the latter is more preferable because of a small reaction barrier (0.49 eV) and can occur at lower temperatures. Moreover, the adsorbed CO molecules themselves could promote O2 activation to easily form CO2, due to the fact that CO could transfer electrons to the O2–2π* orbitals and induce O–O bond scission, in a process similar to the one found in gold cluster catalysts. It is demonstrated that defective MXenes may be used as supports for stable SACs, in agreement with other reported results.38
Indeed, in 2019, also on the basis of first-principles calculations, different metals (M), such as Fe, Co, Ni, Cu, Zn, Ru, Rh, Ag, Ir, Pt, and Au were decorated on defective Mo2CO2−δ monolayers and investigated as SAC candidates for a low-temperature CO oxidation reaction.44 This study showed that the binding energy (Ebind) variations as the M atomic number increased (see Fig. 6) are similar for the three transition metal periods, but the M-surface binding becomes much weaker toward the end of the period, due to the fact that Cu, Zn, Ag, and Au have fully occupied d orbitals, so less free orbitals are available to participate in bonding with the MXene atoms.
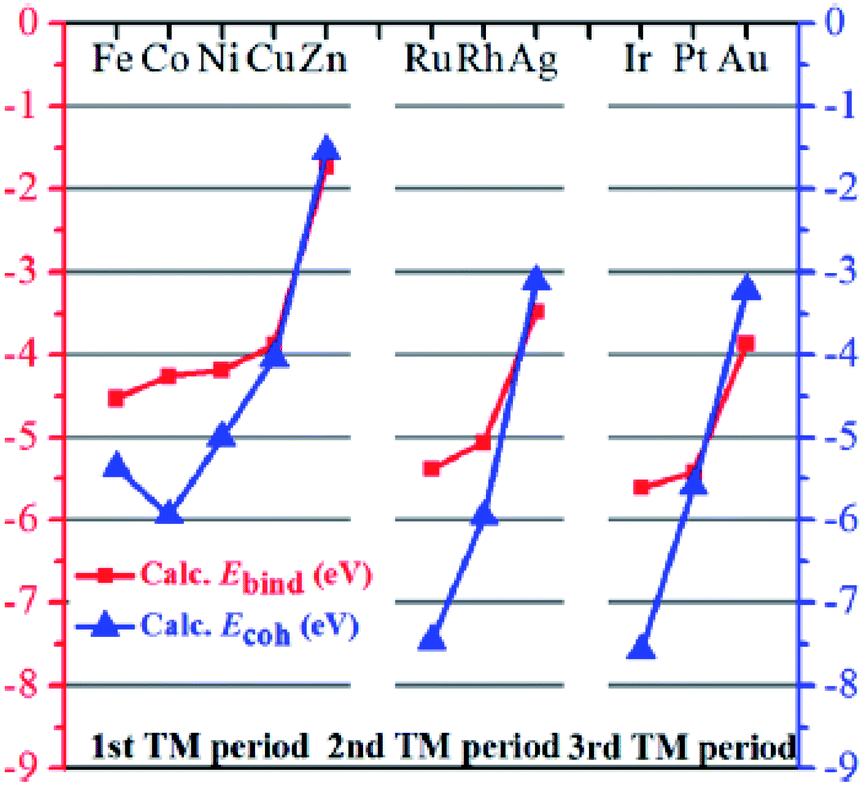 | ||
| Fig. 6 Calculated binding energy (Ebind, red squares, left y-axis) and the corresponding bulk cohesive energy (Ecoh, blue triangles, right y-axis) for M/Mo2CO2−δ (M = Fe, Co, Ni, Cu, Zn, Ru, Rh, Ag, Ir, Pt, and Au). Reprinted with permission from ref. 44, copyright 2019 John Wiley and Sons. | ||
From the calculated data it seems that Zn/Mo2CO2−δ has the weakest binding strength because of the fully filled 3d and 4s orbitals. Also, Zn is the only M that has a negative Ediff value (Ediff is the difference between the binding energy and cohesive energy), while the other metals (Fe, Co, Ni, Cu, Ru, Rh, Ir, and Pt) have positive Ediff values, indicating a strong tendency for clustering and they are, at least in this respect, not promising SAC candidates.
Therefore, based on well-established intuitive criteria concerning metal sintering, CO poisoning, and O2 adsorption strength, the Zn/Mo2CO2−δ system was selected for reactivity calculations as a function of CO concentrations. The authors suggested that at low CO concentrations the oxidation reaction will predominantly proceed via the Eley–Rideal mechanism as it has a small energy barrier of 0.15 eV. In this case also, the main conclusion was that Zn/Mo2CO2−δ represents a potential candidate for low-temperature SAC applications that have a lower energy barrier for CO oxidation than that of noble metals and other 2D SAC systems.44
Along the same lines, to design stable SACs, a systematic screening was performed using first principles calculations based on density functional theory.45 More precisely, this study concerns different metal atoms such as Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn, that are deposited on different MXene surfaces with a M2C stoichiometry (M = Ti, V, Cr, Zr, Nb, Mo, Hf, Ta, and W).45 As in the previous case,44 Zn atoms attached on MXene surfaces appear as the most stable SAC due to the fact that surface diffusion is hindered by moderate energy barriers. The calculation data also showed that all 3d metal studied atoms interact exothermically with MXene substrates and that the properties of the SAC can be tuned by suitably matching the two components of the resulting M@MXene. The results obtained are in good agreement with analogous SAC studies on graphene46 and graphynes.47
In 2020, using the same approach, Co deposited on Mo2CO2 was used to investigate the CO oxidation by O2.48 In agreement with previous results, it was shown that Co strongly bonds to a defective Mo2CO2 surface, and that CO adsorption is more favourable than O2 adsorption. The relatively small activation energy barriers indicate that Eley–Rideal, Langmuir–Hinshelwood and termolecular Eley–Rideal mechanisms are all possible at low temperatures.
Unfortunately, no experimental studies on the CO oxidation reaction using MXene as the support were found in the literature, but we hope that the theoretical data presented in this review will pave the way for experimentalists to develop practical experiments in the near future. We emphasize here that before starting such experiments, the oxygen partial pressure in the system must be estimated and compared to that of the oxidation of the metal used. If the partial pressure is higher, they will oxidize which may or may not be useful.
The first experimental example involving CO activation on MXene is the water gas shift, WGS, reaction. In 2019, Deeva et al. demonstrated the stability of layered MXene structures based on Mo, in reducing environments when heated up to 550–600 °C. At higher temperatures (>730 °C), bulk β-Mo2C was evidenced.49 To probe the catalytic properties, Mo2CTx was tested in the WGS reaction. In Fig. 7, its catalytic activity is compared with that of samples treated at 500 °C and reference β-Mo2C. As expected, due to the surface functional groups, Mo2CTx showed a higher activity than the other catalysts. The highest activity was recorded at 520 °C with a peak consumption rate of CO of ca. 100 μmol (CO) g (Mo)−1 s−1. Interestingly, this value is comparable to weight normalized rates of CO consumption obtained for the conventional Fe-based WGS catalyst, i.e., Fe/Ce/Co oxide,50 as well as for a 3.9 wt% Pt/Al2O3 catalyst.51 At temperatures >700 °C, the activity of Mo2CTx diminished; however, the conversion values were still higher than those of other tested samples. It is important to note here that this Mo2CTx catalyst showed high stability in O-rich environments at high temperatures, as depicted in Fig. 7b and confirmed by XRD analysis, which shows that the tested Mo2CTx contained only trace amounts of MoO2.
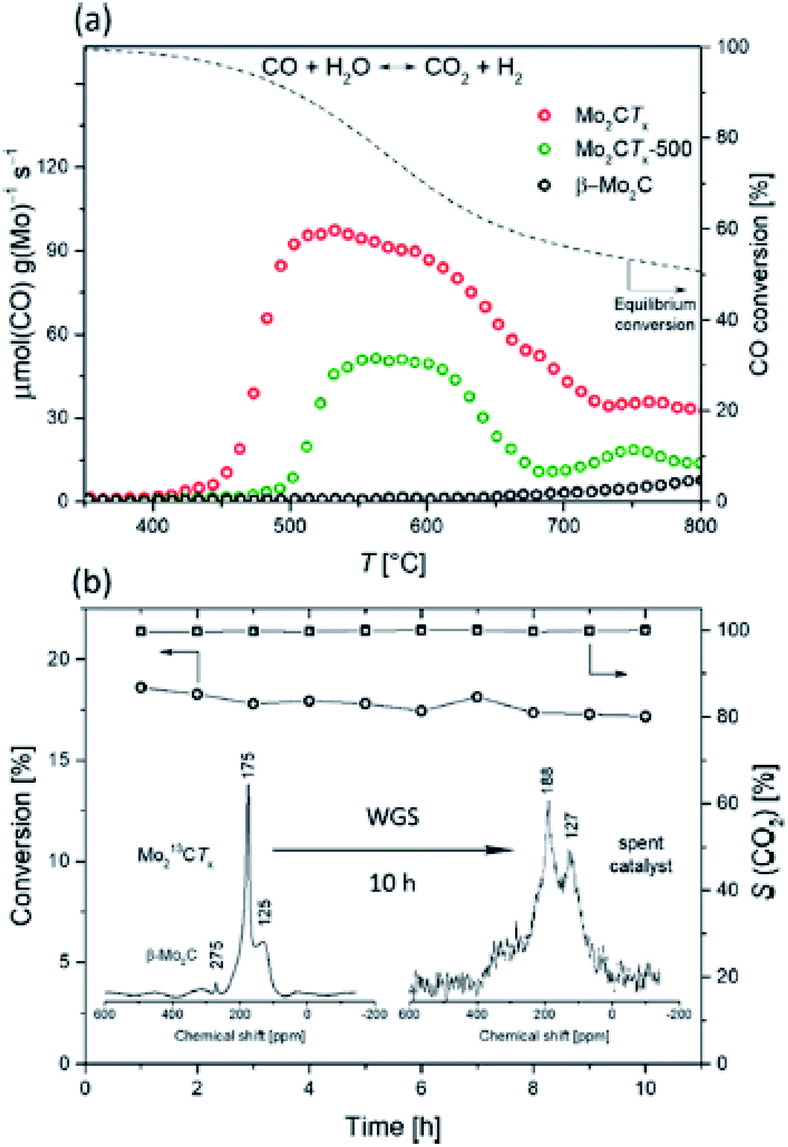 | ||
Fig. 7 (a) WGS catalytic activity of Mo2CTx, Mo2CTx-500, and β-Mo2C and (b) WGS stability test of Mo2CTx at 500 °C and 13C MAS NMR (400 MHz, spinning rate 10 kHz) of Mo213CTx before (1200 scans) and after (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans) the WGS catalytic test. Reprinted with permission from ref. 49 copyright 2019 American Chemical Society. 000 scans) the WGS catalytic test. Reprinted with permission from ref. 49 copyright 2019 American Chemical Society. | ||
These results represent the first proof demonstrating that Mo2CTx is a robust catalyst for high-temperature catalytic applications and that it is particularly stable against oxidation to MoO2.
3. Fine chemical reactions
Fine chemical reactions are an important class of chemical transformations, in which MXenes are starting to be explored52–55 as catalysts due to their interesting properties (Scheme 3).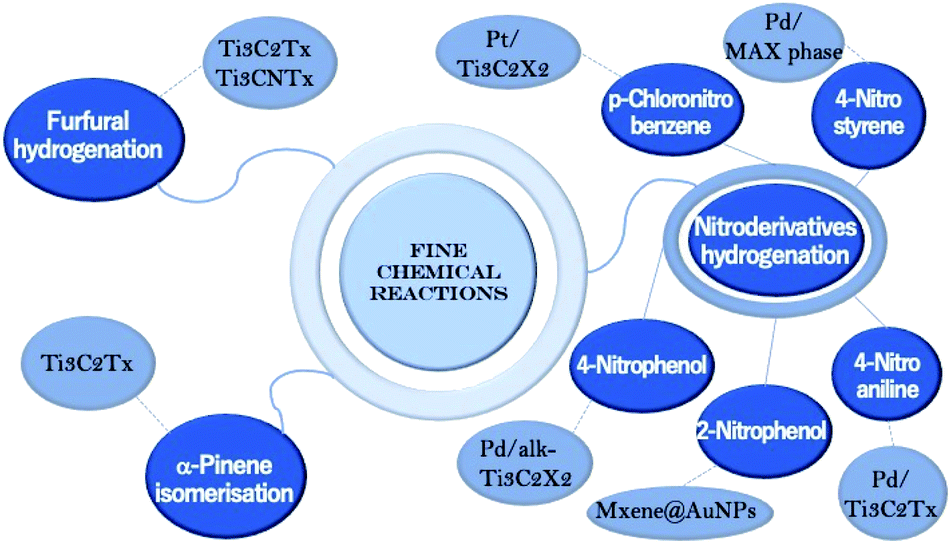 | ||
| Scheme 3 Types of MAX phases and MXenes used in fine chemical reactions. | ||
3.1. Isomerization of α-pinene
Recently, Zielińska et al.56 tested Ti3C2Tx as a catalyst for the α-pinene isomerization reaction to camphene, one of the most pervasive monoterpenes. The target point of this catalytic process is to produce camphene with high selectivity because it is of great importance in the perfumery industry,57 as an intermediate in organic synthesis. Lately, there have also been studies58 confirming its anticancer properties.The catalytic activity of Ti3C2 and exfoliated-Ti3C2 (ex-Ti3C2) was studied in α-pinene isomerisation and it was found that ex-Ti3C2 displays higher activity (60% conversion) than the unmodified Ti3C2 catalyst (48% conversion) and similar selectivity to camphene (∼59 mol%).56 There are many key factors that can explain the superior catalytic activity of ex-Ti3C2, particularly the higher specific surface area and the higher amount of acid sites of this catalyst.
Briefly, it was shown that ex-Ti3C2 shows much higher activity (100 mol% conversion of α-pinene in 7 h of process) compared with the multilayered Ti3C2. Moreover, the catalytic reactions were performed with smaller amounts of the catalyst and without solvent. The kinetic model was studied and the proposed pattern was well fitted to the experimental data for the isomerization of α-pinene; the reactions over both catalysts followed first-order kinetics. Additionally, the selectivity of the product of interest at 100% conversion is ∼2 times higher for Ti3C2 MXenes compared with other Ti-based catalysts such as Ti-SBA-15 and Ti-MCM-41. Moreover, undesired dimerization and polymerization reactions were not detected.56
3.2. Hydrogenation of nitroderivates
Catalytic hydrogenation reactions are among the most important reactions concerning the industrial production of chemicals.59,60Selective hydrogenation is difficult,61 especially when it is possible to hydrogenate more than one functional group per molecule of reactant.62 Heterogeneous metal-based catalysts possess chameleonic properties and are good materials to be developed for selective catalytic hydrogenation toward desired reaction products.63 So, in the case of nitroderivates, the selective reduction of the –NO2 groups while preserving intact the other reducible groups is highly desirable, but not easy. Typically, the nitrostyrene molecule, usually used as substrate, is considered to be the most demanding because the –CC bond is quite sensitive and, consequently, more susceptible to be reduced than –NO2.64
Li et al.65 reported the synthesis of functionalized core–shell MXene composites made by a layer-by-layer (LbL) self-assembly strategy via amine-containing polyethyleneimine (PEI) and carboxyl-containing polyacrylic acid (PAA), with the general formula MXene–COOH@(PEI/PAA)n@AuNPs (see Fig. 8). The catalytic potential of the following two selected nanocomposites with different shell layers (i.e., MXene–COOH@(PEI/PAA)2@AuNPs and MXene–COOH@(PEI/PAA)10@AuNPs) was investigated for catalytic reduction of 2-nitrophenol (2-NA) and 4-nitrophenol (4-NP). The composites revealed remarkable stability and repeatability (see Table 1, entries 1–4). Based on previous results reported a few years ago,66 the obtained nanocomposite suspension was added to 2-NA or 4-NP with a fresh NaBH4 mixture. After 8 consecutive cycles, the MXene–COOH@(PEI/PAA)10@AuNPs showed enhanced stability and catalytic properties compared to their MXene–COOH@(PEI/PAA)2@AuNP counterpart, with less LbL assembled shell structures, indicating the importance of LbL self-assembly for the improvement of porous structures and anchored sites on the surface.
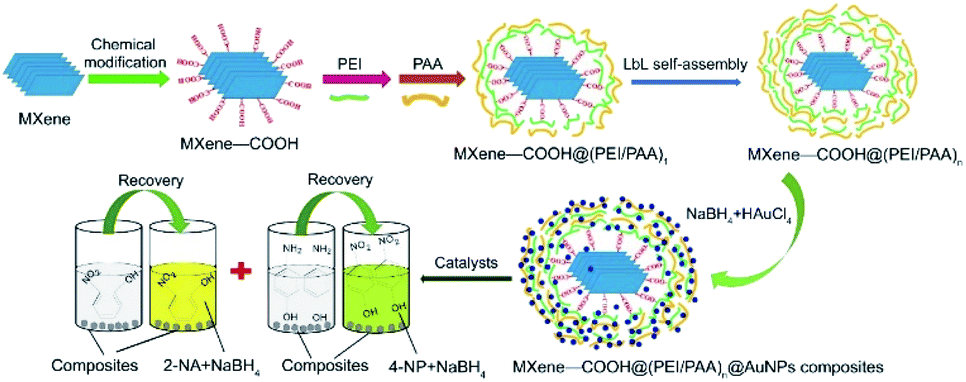 | ||
| Fig. 8 Graphic design of synthesis and catalysis of MXene–COOH@(PEI/PAA)n@AuNP nanocomposites. Reproduced with permission from ref. 65, copyright 2018 Elsevier. | ||
| Entry | Catalyst | Reactant | Conv. [%] | Selectivity [%] | Reaction time | Reducing agent | Ref. |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: ethanol/water mixture with a volume percent of 28% (total volume: 7.0 mL); 14%; 42%; 56%; 100%. b Methanol/water mixture with a volume percent of 28% (total volume: 7.0 mL). c Propanol/water mixture with a volume percent of 28% (total volume: 7.0 mL). d 1,4-Dioxane/water mixture with a volume percent of 28% (total volume: 7.0 mL). | |||||||
| 1 | MXene–COOH@(PEI/PAA)2@AuNPs | 2-Nitrophenol | 70 (8 cycles) | — | 60 min | — | 65 |
| 2 | MXene–COOH@(PEI/PAA)2@AuNPs with fresh NaBH4 | 4-Nitrophenol | 74 (8 cycles) | — | 57 min | ||
| 3 | MXene–COOH@(PEI/PAA)10@AuNPs with fresh NaBH4 | 2-Nitrophenol | ∼90 (8 cycles) | — | 36 min | ||
| 4 | MXene–COOH@(PEI/PAA)10@AuNPs with fresh NaBH4 | 4-Nitrophenol | ∼90 (8 cycles) | — | 30 min | ||
| 5 | Pd/alk–Ti3C2X2 in aqueous solution | 4-Nitrophenol | 99.9 | 100 | 70 min | H2 | 52 |
| 6 | Pd/Ti3C2Tx⊂graphene hydrogels | 4-Nitroaniline | 91 | — | 1 min | NaBH4 | 69 |
| 7 | Pd/Ti3C2Tx films on a poly(vinylidene fluoride) (PVDF) substrate | 4-Nitroaniline | 62.9 | — | 1 min | ||
| ∼80 | — | 2 min | |||||
| ∼98 | — | 5 min | |||||
| 8 | Pd/Ti3C2Tx⊂graphene hydrogels | 4-Chloronitrobenzene | ∼15 | — | 1 min | ||
| ∼25 | — | 2 min | |||||
| ∼45 | — | 5 min | |||||
| 9 | Pd/Ti3C2Tx⊂graphene hydrogels | 4-Bromonitrobenzene | ∼48 | — | 1 min | ||
| ∼55 | — | 2 min | |||||
| ∼60 | — | 5 min | |||||
| 10 | Pd/Ti3C2Tx⊂graphene hydrogels | 4-Nitrotoluene | ∼63 | — | 1 min | ||
| ∼72 | — | 2 min | |||||
| ∼78 | — | 5 min | |||||
| 11 | Pd/Ti3C2Tx⊂graphene hydrogels | 4-Nitrophenol | ∼70 | — | 1 min | ||
| ∼75 | — | 2 min | |||||
| ∼79 | — | 5 min | |||||
| 12 | Ti3SiC2 | 4-Nitrostyrene | <1 | 100 | 24 h | H2 1.1 MPa | 71 |
| 3 | 100 | H2 2.5 MPa | |||||
| 13 | Ti2AlC | 4-Nitrostyrene | <1 | 100 | H2 1.1 MPa | ||
| 1 | 100 | H2 2.5 MPa | |||||
| 14 | Ti3AlC2 | 4-Nitrostyrene | <1 | 100 | H2 1.1 MPa | ||
| 4 | 13 | H2 2.5 MPa | |||||
| 15 | 0.05 wt% Pd/Ti3SiC2 | 4-Nitrostyrene | 100 | 0 | 24 h | H2 | 71 |
| 16 | 0.0005 wt% Pd/Ti3SiC2 | 4-Nitrostyrene | 100 | 0 | H2 | ||
| 17 | 0.0005 wt% Pd/Ti3SiC2_DP | 4-Nitrostyrene | 100 | 25 | H2 | ||
| 18 | Pd/Ti3SiC2_mix1 | 4-Nitrostyrene | 4 | 100 | H2 | ||
| 19 | Pd/Ti3SiC2_mix2 | 4-Nitrostyrene | 59 (cycle 1) | 73 | H2 | ||
| 100 (cycle 2) | 93 | ||||||
| 21 | Pd/Ti3SiC2_mix3 | 4-Nitrostyrene | 100 | 58 | H2 | ||
| 22 | Pd/Ti3SiC2_mix4 | 4-Nitrostyrene | 100 | 10 | H2 | ||
| 23 | Pd/TiC_mix2 | 4-Nitrostyrene | 100 (cycle 1) | — | H2 | ||
| 18 (cycle 2) | — | ||||||
| 25 | Pd/Ti2AlC_mix2 | 4-Nitrostyrene | 52 | 27 | H2 | ||
| 26 | Pd/Ti3AlC2_mix2 | 4-Nitrostyrene | 100 | — | H2 | ||
| 27 | Pd/Ti3C2Tz_mix2 | 4-Nitrostyrene | 90 (cycle 1) | — | H2 | ||
| 25 (cycle 2) | — | ||||||
| 29 | Pd/Ti3SiC2_mix2 | 3-Nitrostyrene | 100 | 96 | 24 h | H2 | 71 |
| Cinnamaldehyde | 94 | 3 (cinnamyl alcohol) | |||||
| 80 (hydro-cinnamaldehyde) | |||||||
| Trans-β nitrostyrene | 9 | 13 | |||||
| 4-Nitrophenol | 18 | 83 | |||||
| 30 | Ni/Ti3SiC2 | 4-Nitrostyrene | 1 | 100 | 24 h | H2 | 71 |
| 31 | Pt/Ti3C2Tx-D-AB | 4-Chloronitrobenzene | 100a | 99.5 | 1 h | H2 | 72 |
| 94a | 92.4 | ||||||
| 99.8a | 95.2 | ||||||
| 96.9a | 92.9 | ||||||
| 2.6a | 67 | ||||||
| 98.2b | 95 | ||||||
| 78.1c | 90.8 | ||||||
| 39.3d | 83.3 | ||||||
| 32 | Pt/Ti3C2Tx-D-SB | 4-Chloronitrobenzene | 40.4a | 84.3 | |||
| 33 | Pt/Ti3C2Tx-D-AB | 2-Chloronitrobenzene | 100a | 96.5 | 1.5 h | ||
| 34 | Pt/Ti3C2Tx-D-AB | 3-Chloronitrobenzene | 100a | 95.8 | 2 h | ||
| 35 | Pt/Ti3C2Tx-D-AB | 4-Nitrotoluene | 100a | 98.2 | |||
| 36 | Pt/Ti3C2Tx-D-AB | 2-Nitrotoluene | 100a | 94.7 | |||
| 37 | Pt/Ti3C2Tx-D-AB | 2-Methoxi-1-nitrobenzene | 100a | 95.7 | 3 h | ||
| 38 | Pt/Ti3C2Tx-D-AB | 4-Nitrophenol | 100a | 97.4 | 1.5 h | ||
| 39 | Pt/Ti3C2Tx-D-AB | 2-Fluoronitrobenzene | 100a | 94.7 | |||
| 40 | Pt/Ti3C2Tx-D-AB | Nitrobenzene | 100a | 99.2 | 1 h | ||
Fan et al.52 used Pd/alk–Ti3C2X2 (alk = alkaline) as catalysts for the hydrogenation of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in an aqueous solution.
The Pd/alk–Ti3C2X2 synthesis is described in Fig. 9, and the method used provides well dispersed active sites of Pd on the alk–Ti3C2X2 surface. This good dispersion in turn is responsible for the complete conversion and selective hydrogenation of 4-NP to 4-AP in 70 min. Additionally, the catalyst is stable showing no significant loss of catalytic efficiency after seven cycles, making it a promising candidate for numerous catalytic applications.
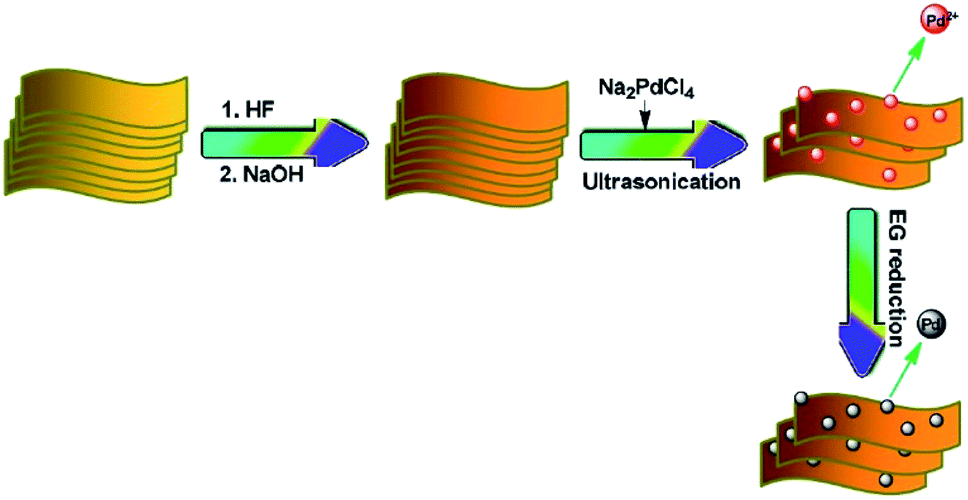 | ||
| Fig. 9 Schematic representation of the synthesis procedure of Pd/alk–Ti3C2X2. Reproduced from ref. 52 (open access). | ||
Noble metals such as Pd are intensively used in catalysis.67,68 Therefore, Xie et al.69 described the synthesis of Pd/Ti3C2Tx encapsulated in 3D graphene networks, henceforth referred to as Pd/Ti3C2Tx⊂graphene hydrogels, as potent and easily retrievable catalysts for efficient hydrogenation of 4-nitroaniline (4-NA) to p-phenylenediamine (PPD) in the presence of NaBH4 as the reducing agent. The reaction is quite fast – 91% conversion after 1 min of reaction (see Fig. 10a) – demonstrating that the porous structure of the 3D Pd/Ti3C2Tx⊂graphene hydrogel is key for the accessibility of active sites for catalytic applications (see Table 1, entries 6–11).
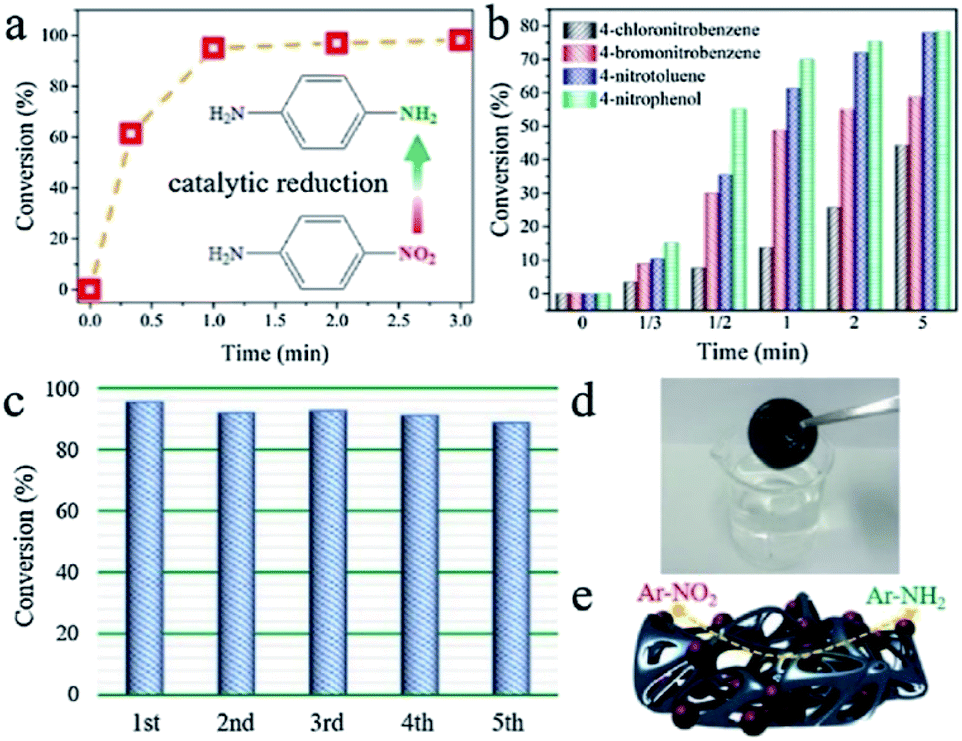 | ||
| Fig. 10 Catalytic performance of the Pd/Ti3C2Tx⊂graphene hydrogel for (a) hydrogenation of 4-NA; (b) hydrogenation of other nitroderivates; (c) cycling performance for 4-NA dehydrogenation; (d) picture of the catalyst after the hydrogenation reaction; (e) schematic illustration of the catalytic dehydrogenation reaction. Reproduced with permission from ref. 69, copyright 2020 Elsevier. | ||
Moreover, this hydrogel was tested for selective hydrogenation reactions with many other nitroaromatic molecules (e.g., 4-chloronitrobenzene, 4-bromonitrobenzene, 4-nitrotoluene, and 4-nitrophenol) in order to prove the generality of this catalyst. The results (Fig. 10b) show that this catalyst results in exceptional catalytic performance with high conversion values for all these different nitro-compounds, mostly for those with electron donating functional groups (such as –CH3 and –OH) compared to molecules with electron withdrawing substituents (i.e. –Cl and –Br) under the same reaction conditions.
Furthermore, it has been shown that the hydrogel is stable after successive recycle tests for the catalytic hydrogenation of 4-NA (see Fig. 10c).
To compare the results obtained with the Pd/Ti3C2Tx⊂graphene hydrogel, Xie et al.69 also prepared Pd/Ti3C2Tx films deposited on the poly-vinylidene fluoride (PVDF) substrates. In this case, the conversion of 4-NA over the Pd/Ti3C2Tx catalyst was 63% after 1 min of reaction, which was much lower than the 91% obtained for the Pd/Ti3C2Tx⊂graphene hydrogel. The integrity of Pd/Ti3C2Tx films was preserved during the catalytic processes due to sodium ions and water intercalation into the Ti3C2Tx layers, also reported in other studies.70
In a recent study, Trandafir et al.71 explored the use of MAX phases as supports for Pd nanoparticles (NPs), at remarkably low concentrations, for the chemoselective hydrogenation of a functionalized nitro-compounds.
The new generation of Pd NPs supported on the Ti3SiC2 catalyst used in the selective hydrogenation reaction of 4-NS achieved a high value of turnover frequency (TOF) of 4.7 × 103 h−1, that represents roughly a 100-fold increase over that of the most selective catalyst known to date (see Fig. 11a). 4-AS is the main product sought here, but other reaction products were identified (i.e., 4-ethylnitrobenzene (4-EN) and 4-ethylaniline (4-EA)) depending on the reaction pathway (see Fig. 11b).
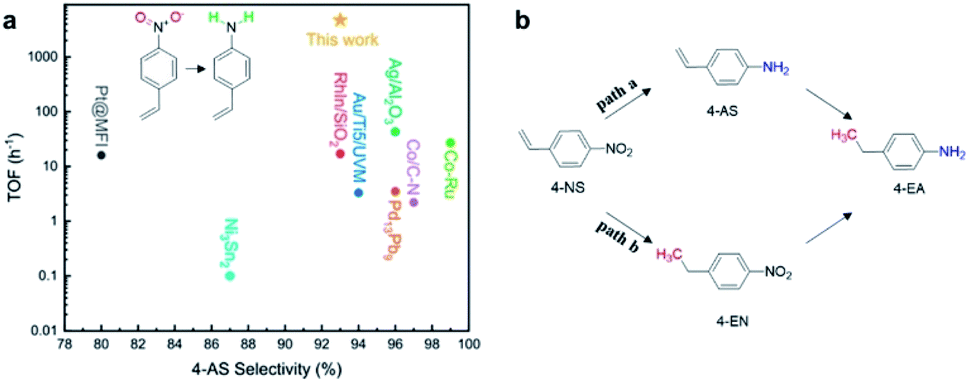 | ||
| Fig. 11 (a) Comparison of the highest TOF values reported in the literature for catalysts in 4-NS hydrogenation to 4-AS, and (b) reaction pathways for the hydrogenation reaction of 4-NS. Reproduced with permission from ref. 71, copyright 2020 American Chemical Society. | ||
Different Pd supporting methods were explored (wet impregnation, deposition–precipitation, and mechanical mixing) and the authors found that the number of accessible sites appears to depend on the preparation method.
Actually, a good dispersion is obtained using a deposition–precipitation method compared with the large clusters formed through the impregnation method that consequently limited the access to the active sites. Diluting the Pd content by mechanically mixing the impregnated Pd samples with Ti3SiC2 was a good strategy to better control the Pd amount. A total conversion of 4-NS to 4-AS with a selectivity of 93% was obtained with an optimal loading of very well dispersed 130 ppm Pd. The authors also showed that higher Pd loadings resulted in selectivity losses. The high chemoselectivity of the Pd/MAX phase catalyst can be assigned to the synergetic effect between the Pd nanoparticle size and dispersion and also to the non-Ti containing oxides (viz. presence of SiO2 as one of the native oxides formed in the Ti3SiC2 phase, the other being titania) that preferentially activate the nitro group. The proof of concept was realised by using the best material as the catalyst for different substrates (see Table 1 entries 12–30).
Additionally, Pt nanoparticles on Ti3C2Tx-based MXenes as efficient catalysts have also been investigated recently72 for selective hydrogenation of p-chloronitrobenzene (p-CNB) to p-chloroaniline (p-CAN) (see Fig. 12b). Using ammonia borane (AB) as a mild reducing agent compared with sodium borohydride (SB), highly dispersed and nano-sized metallic Pt crystallites were uniformly decorated on Ti3C2Tx nanosheets (see Fig. 12a). It was concluded that the reducing agent strongly influenced the particle size and distribution of the Pt NPs on the Ti3C2Tx-D surface. It is remarkable that this catalyst can catalyse the complete conversion of p-CNB to p-CAN with 99.5% selectivity, and exhibits a superior catalytic activity to that of Pt/Ti3C2Tx-D-SB synthesized with SB (see Table 1, entries 31–32). The excellent performance of Pt/Ti3C2Tx-D-AB can be assigned to the well-dispersed Pt nanoparticles, abundant surface electron-efficient Pt(0) and also to the synergistic catalysis between hydrophilic Pt/Ti3C2Tx-D-AB and water (used as a co-solvent).
 | ||
| Fig. 12 (a) Schematic representation of the preparation process of Pt/Ti3C2Tx-D samples with different reducing agents; (b) overview of the selective hydrogenation reaction of p-CNB to p-CNA with Pt–MXene based catalysts. Reproduced with permission from ref. 72, copyright 2020 Royal Society of Chemistry. | ||
The Pt/Ti3C2Tx-D-AB catalyst was relatively stable for six cycles, after which it began to deactivate due to Pt aggregation as confirmed by TEM.
Chen et al.72 also tested Pt/Ti3C2Tx-D-AB in the hydrogenation of a series of nitroaromatic compounds to their corresponding amines with high efficiency (see Table 1, entries 33–40), with conversions that reached 100% in 1–3 h. The target products' selectivity is higher than 94% in all tests which indicates that Pt/Ti3C2Tx-D-AB is a flexible catalyst that can be used to obtain a series of aromatic amines.
3.3. Furfural hydrogenation
During the last decade, increased attention has been paid to selective biomass transformation. One of the most promising molecular platforms derived from hemicelluloses73 is furfural, mainly considering the value added chemicals obtained by different catalytic reactions such as furfuryl alcohol.Recently, Naguib et al.74 reported the catalytic activity of Ti3C2Tx and Ti3CNTx MXenes for the hydrogenation of furfural to furfuryl alcohol with the scope to understand the role of the presence of nitrogen in the support for carbonyl hydrogenation. Both catalysts had similar activities (36–42%) and selectivities (49–52%). It is worth mentioning that these results are similar or superior when compared with those of TiO2, metal carbides or other noble metal-free catalysts under the same conditions. The main difference between Ti3C2Tx and Ti3CNTx was observed in terms of stability; Ti3CNTx was stable for six consecutive runs compared with Ti3C2Tx which deactivated faster. This behaviour was explained by the differences of surface terminations, which varied as a function of MXene composition. Ti3C2Tx is richer in surface terminations (especially OH), facilitating the intercalation of the reaction between the layers (leading to faster deactivation). The presence of nitrogen in Ti3CNTx significantly lowered the OH terminations and hindered the interaction of the surface with the reactants.
DFT calculations were performed to elucidate the furfural hydrogenation mechanism for Ti3CNTx. Apparently, TiO–H Brønsted acid sites present on the MXene surface bind to the furfural molecule by the CO functional group, which is further hydrogenated via the addition of hydride dissociated on the metallic active site.
The bifunctionality of MXenes given by the presence of both metal and OH terminations which act as acids render these materials quite interesting for a wide range of catalytic reactions. Also, this study revealed that the surface terminations can have an influence on material stability, and suggests that MXene surfaces can be tuned in order to target certain reactions.
4. Catalytic degradation
4.1. Short overview
Pharmaceuticals and nonsteroidal anti-inflammatory drugs are used intensively for the treatment of human and animal diseases. The vast majority of drugs used end up in wastewater or groundwater, and removing or destroying them becomes a very difficult task due to their low or non-biodegradability.75 Similar to pharmaceuticals, dyes also often end up polluting water systems and their removal is a subject of major interest. Dyes are used in many industries and the most common are methylene blue, rhodamine B, methyl orange or congo red.Like dyes, pesticides can have a negative impact on humans and animals due to their toxic characteristics. For example, 2,4-dichlorophenoxyacetic acid was frequently found in drinking water in high concentrations due to its high solubility. The development of methods to remove this material from the environment is of great importance because its presence can cause endocrinal disturbance and chromosomal mutations in human lymphocytes. It can also endanger aquatic life.76
MAX phases and MXene-based heterojunction materials have been shown to be effective catalysts in the degradation of organic compounds such as pharmaceuticals,75,77,78 dyes,79,80 pesticides76 and other compounds (see Scheme 4).81,82
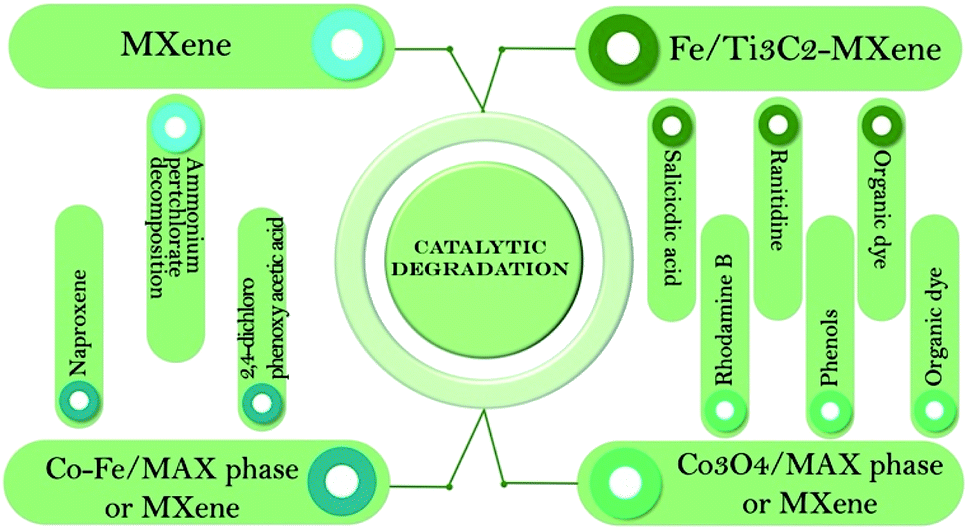 | ||
| Scheme 4 Types of MAX phases and MXenes used in catalytic degradation reactions. | ||
4.2. Ti3C2–MXenes modified with iron compounds
Ding et al.77 described the synthesis of a novel nanocomposite catalytic material obtained by supporting α-Fe2O3 NPs onto multi-layered Ti3C2 and its use as a heterogeneous activator of peroxymonosulfate (PMS) for the degradation of salicylic acid. Salicylic acid is a phenolic compound widely used in the pharmaceutical, cosmetics and food industries. Due to the fact that it has been detected constantly in environmental water systems and that it represents a potential threat for ecological systems and humans, various methods to eliminate it from the aqueous environment have been developed. The authors claim that MXenes were selected for supporting α-Fe2O3 nanoparticles because of the presence on their surface of different termination groups such as –O and –OH, which provide better aqueous dispersibility and strong anchoring of the free cations through electrostatic interactions.77Three types of nanocomposites were synthesized, FM-1, FM-2, and FM-3, using different concentrations of FeCl3·6H2O of 0.5 M, 1 M and 2 M, respectively. The α-Fe2O3/MXene nanocomposites present high efficiency and stability during the activation process of PMS to produce free radicals for the degradation of salicylic acid in aqueous solutions. Under neutral conditions, the removal of the salicylic acid was ≈97% using the FM-2 catalyst nanocomposite. The catalytic system FM-2/PMS displayed low dissolution of metal ions and reusability during five successive runs. A possible mechanism of PMS activation was proposed by the authors based on EPR spectra, quenching tests, XPS and in situ Raman characterization.
Fig. 13 shows the mechanism which involves 4 steps. Salicylic acid is degraded by the reactive radicals generated during the PMS activation process. Unfortunately, the known instability of MXenes in water was not taken into account.
 | ||
| Fig. 13 Schematic diagram of the possible mechanism of PMS activation by FM nanocomposites. Reprinted with permission from ref. 77, copyright 2020 Elsevier. | ||
Another example of drugs present in aquatic systems that require their removal from water is ranitidine, due to its carcinogenic effect. Ranitidine is frequently used as a histamine H2-receptor antagonist and is utilized for the treatment of ulcers and gastroesophageal reflux, due to the presence of furan rings in its structure.78
Zang et al.78 reported obtaining magnetic zero-valent iron (nZVI)@Ti3C2-based MXene nanosheets via an in situ reductive deposition, which efficiently succeed in degrading ranitidine in the presence of H2O2, a Fenton-like process (see Fig. 14). The proposed mechanism for catalytic degradation of ranitidine in the presence of H2O2 and (nZVI)@Ti3C2-based MXene nanosheets (see Fig. 14) could be divided into five steps: (i) molecules of ranitidine diffuse and are adsorbed on the surface of the (nZVI)@Ti3C2 nanosheets; (ii) the nZVIPs undergo oxidative corrosion; (iii) cyclic transformation of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe(III)/Fe(II) occurs; (iv) ferrous iron activates H2O2 molecules; (v) hydroxyl radicals react with ranitidine. In this scheme the Ti3C2-based MXene plays the role of a cathode, favouring a direct electron transfer reaction. The nZVI cores behave as an electron donor and the iron-based oxide outer shell promotes a series of Fe(III)/Fe(II) conversions.
Fe(III)/Fe(II) occurs; (iv) ferrous iron activates H2O2 molecules; (v) hydroxyl radicals react with ranitidine. In this scheme the Ti3C2-based MXene plays the role of a cathode, favouring a direct electron transfer reaction. The nZVI cores behave as an electron donor and the iron-based oxide outer shell promotes a series of Fe(III)/Fe(II) conversions.
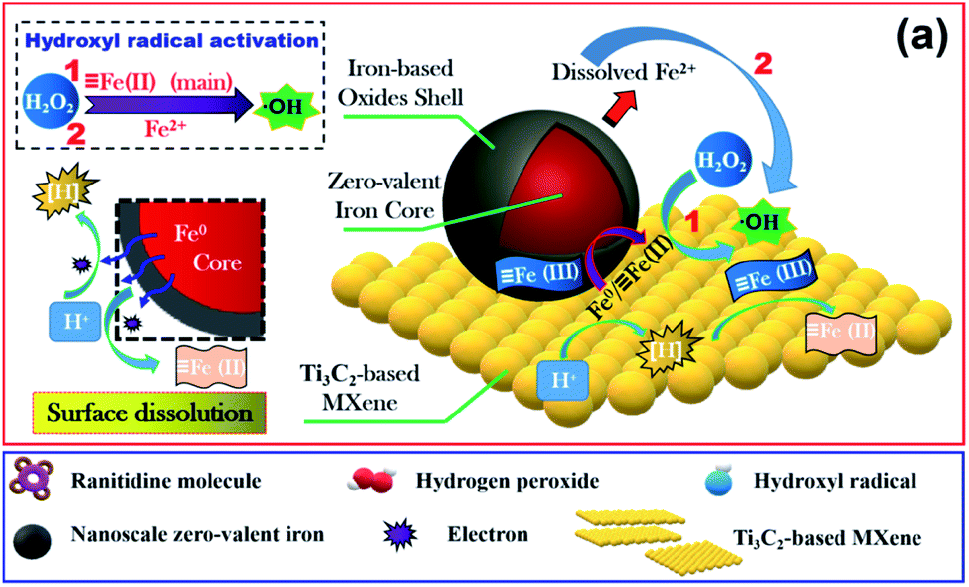 | ||
| Fig. 14 Schematic of the mechanism of ranitidine catalytic degradation by the (nZVI)@Ti3C2/H2O2 system. Reprinted with permission from ref. 78, copyright 2021 Elsevier. | ||
Apart from drug removal, a Fenton-like catalyst, Fe3O4/Ti3C2–MXene proved to be efficient also in organic dye degradation. Zang et al.80 developed, for the first time, a one-pot synthesis method for magnetic MXene composites (Ti3C2-MNPs) which incorporated Fe3O4 nanoparticles into Ti3C2 nanosheets via thermal treatment (see Fig. 15).
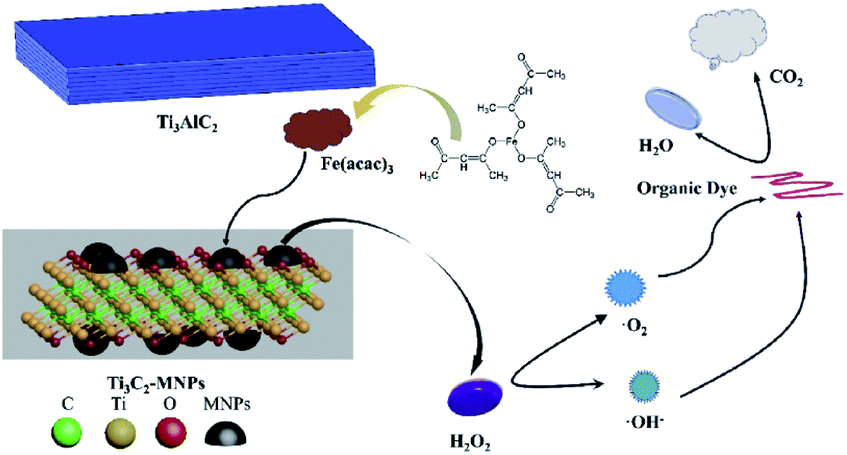 | ||
| Fig. 15 Schematic of the synthesis of the Ti3C2/Fe3O4 system and its utilisation in dye degradation (reproduced with permission from ref. 80, copyright 2020 Elsevier). | ||
The hybrid Ti3C2/Fe3O4 system presents high degradation efficiency of organic dyes. The optimal conditions occur at pH = 3, a catalyst concentration and H2O2 concentration of 0.5 g L−1 and 10 mmol L−1, respectively, and at a temperature = 40 °C. The degradation mechanism involves the presence of hydroperoxyl radicals (˙OOH) and superoxide radicals (O2˙−).80 Here again H2O2 is a very strong oxidizing agent and whether the MXene flakes would survive for times that are useful for an application has to be carefully considered.
4.3. MAX phases and MXenes modified with Co3O4
As in the case of Fe, Co also degraded organic dyes or phenols when it was deposited on MXenes and the MAX phases. Thus, nanocomposites of Co3O4 particle-modified MXene (MXene–Co3O4) synthesized by the solvothermal method showed good performance for catalytic degradation of methylene blue and rhodamine B (see Fig. 16).79 The capacity of MXene–Co3O4 to degrade methylene blue and rhodamine B can be attributed to the positive synergistic effect of MXene, dye and Co3O4 in the presence of H2O2. Cationic dye molecules were adsorbed on the MXene surfaces due to the electrostatic attraction and this process increased the concentration of dye molecules anchored on the surface of MXene–Co3O4, leading to a high catalytic degradation rate. Also, the hydrophilicity of MXenes facilitated the dispersion of MXene–Co3O4 in water and improved the contact between molecules, and H2O2 was effectively catalyzed to produce free OH radical species that promoted dye degradation.79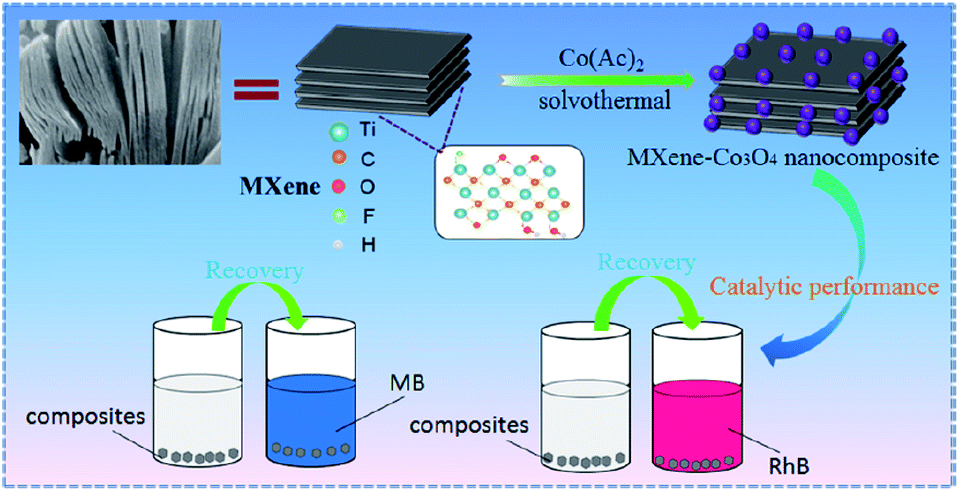 | ||
| Fig. 16 Schematic representation of the synthesis of the Ti3C2/Co3O4 system and catalytic process of dye degradation. Reprinted with permission from ref. 79, copyright 2021 American Chemical Society. | ||
Wang et al.81 reported the synthesis of sandwiched Co3O4/Ti3C2Tx composites as a heterogeneous catalyst to activate PMS for bisphenol A catalytic degradation, another toxic compound for humans and animals, released from polymers in the fabrication of water bottles, sport equipment, coatings of food containers, etc.83 The Co3O4/Ti3C2Tx composites were obtained by impregnation and the highest catalytic activity was obtained when the loading of Co3O4 was about 20% on Ti3C2Tx (see Fig. 17). In addition, the Co3O4/Ti3AlC2 composite activates PMS in a wide range of pH (4–10) and shows good catalytic performance with the existence of anions such as NO3− and Cl−. Oxidizing species detected in the Co3O4/Ti3C2Tx/PMS system were SO4˙− and ˙OH radicals.
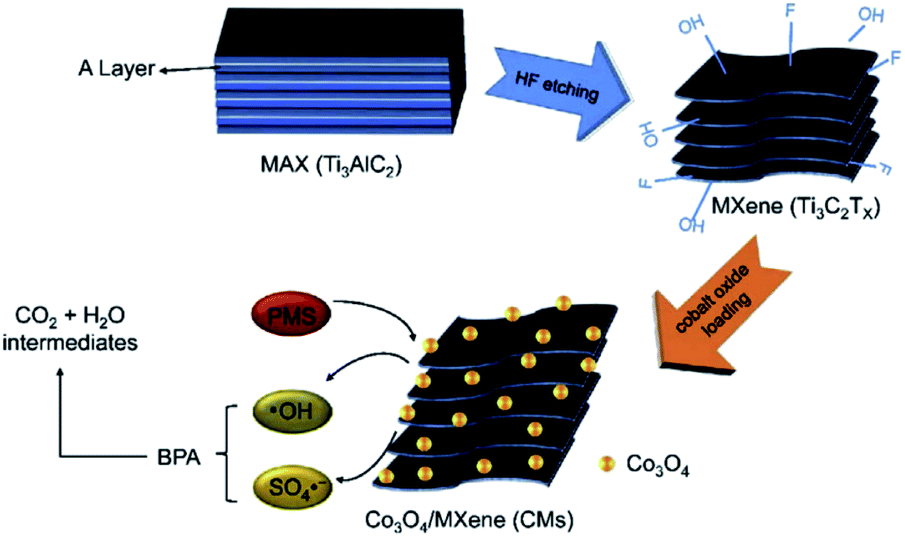 | ||
| Fig. 17 Schematic of the synthesis of the Ti3AlC2/Co3O4 system and its utilisation in BPA catalytic degradation. Reprinted with permission from ref. 81, copyright 2018 Elsevier. | ||
4.4. MAX phases and MXenes modified with iron and cobalt
The high activity of Fe and Co supported on the MAX phases and MXenes observed in catalytic degradation encouraged the researchers to also continue and explore the synergy between the two elements (Fe and Co). Fayyaz et al.75 described the synthesis of a heterogeneous nanocomposite catalyst of MXene nanosheets (synthesized from Ti3AlC2) functionalized with CoFe2O4 nanoparticles that can be used for the activation of persulfate (PS) to degrade naproxen in water. Naproxen is a nonsteroidal anti-inflammatory drug used in the treatment of rheumatoid arthritis and acute muscle pain75 and is similar to salicylic acid; it contaminates environmental water systems and its removal is a subject of major interest.The experiments described by the authors confirm that neither CoFe2O4 nanoparticles nor MXenes alone can activate PS, but the CoFe2O4@MXene system proved to be an efficient catalyst for PS activation. A proposed mechanism for naproxen degradation with persulfate activated by this nanohybrid system is described in Fig. 18.
 | ||
| Fig. 18 Proposed mechanism for naproxen degradation with PS activated by CoFe2O4@MXene. Reprinted with permission from ref. 75, copyright 2021 Elsevier. | ||
Coordination interactions of Co and Fe moieties with MXene functional groups modulate redox reactions of stable Co/Fe/Ti couples. The electrons are transferred from PS to form SO4˙−/S2O8˙− by thermodynamic oxidation of Co(II)/Fe(II)/Ti(II) and reduction of Co(III)/Fe(III)/Ti(IV). By activating PS with the CoFe2O4@MXene, radical reactive species such as ˙OH, SO4˙−, and O2˙− and the corresponding nonradical 1O2 are formed, which significantly promotes naproxen degradation.
Moreover, the naproxen adsorbed on the CoFe2O4@MXene acts as an electron donor and transfers two electrons to PS that is, in turn, reduced to 2SO42−. Naproxen was degraded approximately 99% in the presence of PS at 1 g L−1 of CoFe2O4@MXene dosage.
Composites such as Fe2CoTi3O10–MXene were described by Ding et al.76 as a heterogeneous catalyst for oxidative degradation of 2,4-dichlorophenoxyacetic acid based on PMS activation. These composites could efficiently activate PMS and, under neutral conditions, approximately 98% of 2,4-dichlorophenoxyacetic acid was removed. In the catalytic oxidation process, radicals such as ˙OH and SO4˙− are generated and the main activation mechanism for PMS can be represented by the electron transfer between PMS and ternary transition metal (Fe, Co and Ti) active sites on the catalyst surface.76
It is our duty to inform the readers that the catalytic degradations presented in Sections 4.1–4.4, that use Ti-based MXenes, are useful from the academic/fundamental point of view, showing some synergetic effects between the supported Fe and Co transition metals and the Ti-based MXenes. Unfortunately, Ti-based MXenes are not stable in water for a long time, and therefore any practical applications are not feasible.
In addition, when studying the Fenton degradation of drugs in aquatic systems, researchers should take into account that the use of H2O2 will also result in faster oxidation of MXenes and their transformation into metal oxides will occur.
4.5. MXenes for ammonium perchlorate decomposition
Recently, Jiao et al.82 showed that Ti3C2 has intrinsic catalytic activity for the thermal decomposition of ammonium perchlorate-based molecular perovskites such as [(H2dabco)[NH4(ClO4)3]. Experiments revealed that adding 10 wt% Ti3C2 to the reaction mixture reduced the decomposition temperature by 43.8 °C at a heating rate of 10 °C min−1.The catalytic mechanism (Fig. 19) proposed by the authors assumed that the thermal decomposition of ammonium perchlorate-based molecular perovskites is mainly determined by the stability of the cage-like framework structures of the substrate which were composed of NH4+ cations and ClO4− anions.82
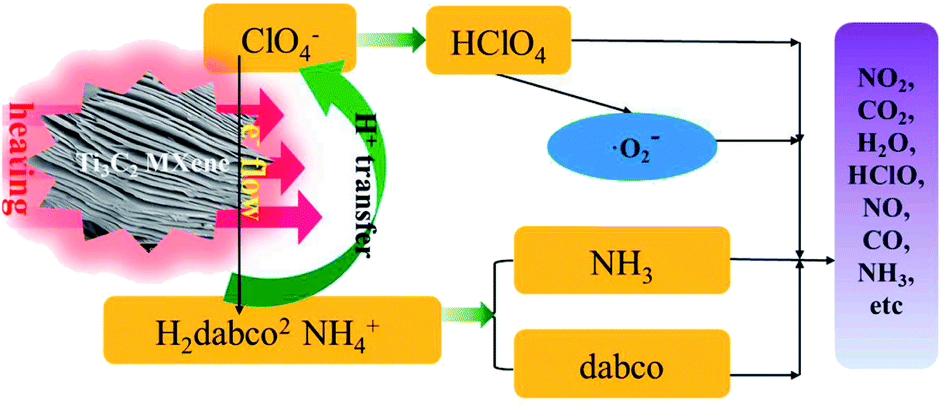 | ||
| Fig. 19 Schematic of the catalysis decomposition mechanism of [(H2dabco)[NH4(ClO4)3] with Ti3C2 MXene. Reprinted with permission from ref. 82, copyright 2020 Elsevier. | ||
The inherent catalytic properties of MXenes can realise the thermal decomposition of ammonium perchlorate-based molecular perovskites by lowering the decomposition temperature and activation energy, and increasing the heat release.82
Along the same lines, Cu2O supported on Ti3C2 MXenes was found to provide a higher catalytic activity compared with Ti3C2 MXenes or Cu2O alone, in the thermal decomposition of ammonium perchlorate (AP).84 Combining the advantages of Cu2O and MXenes, good thermal conductivity, improved heat transfer rate of the reaction system and also high surface area and lamellar structure of MXenes, the thermal decomposition temperature of ammonium perchlorate was significantly reduced by 121.4 °C compared to that of the pure AP.
In 2021, Li et al.85 demonstrated that Ti3C2 MXene-supported CuO nanocomposites also significantly decrease the decomposition temperature of AP from 425.0 °C to 324.9 °C and increase the exothermic heat from 295.2 J g−1 of AP to 1272.9 J g−1.
These results prove that the MXene used as a support for CuO or Cu2O could be a viable solution to enhance the catalytic performance in ammonium perchlorate decomposition, which is of major interest.
5. Applications of MXenes in photocatalysis
5.1. Short overview
Photocatalysis can be described as the acceleration of a photoreaction in the presence of a catalyst, called a photocatalyst, that is often stable to the photolysis conditions and attains a large number of oxidative conversions per active site without significant degradation of its redox catalytic capacity.86 In the case of heterogeneous photocatalysis, upon photoexcitation of several semiconductor photocatalysts nonhomogeneously suspended in either aqueous or nonaqueous solutions or in gaseous mixtures, the photogenerated electron–hole pairs migrate to the external surface and initiate simultaneous oxidation and reduction reactions.86,87 Depending on the reaction media, the photogenerated electrons and holes might interact directly with the adsorbate molecules (e.g., the photocatalytic overall water splitting reaction) or create reactive species (e.g., hydroxyl and superoxide radicals) that undergo non-selective oxidative processes to break down different substances including organic materials, pesticides, dyes or bacteria, among others.88The major hurdle that needs to be overcome in this area is the recombination of photogenerated electrons and holes. In this regard, the combination of MXenes with various semiconductor photocatalysts led to a marked increase in photoactivity. Such phenomena are caused by the structural merits of MXenes including (i) abundant hydrophilic groups (–OH and –O) present on the external surfaces that afford considerable interaction with various semiconductors to form composite heterostructured photocatalysts, (ii) strong redox activities of surface metal sites (e.g., Ti, Nb or V) existing on the MXene surfaces which are greater than the redox reactivity of carbon materials,89 and (iii) due to their metallic character, MXenes possess excellent electron conductivity that guarantees efficient charge transport.90
As a typical representative of MXene materials, Ti3C2Tx features high electrical conductivity, surface hydrophilicity, and tuneable electronic properties that makes it a suitable co-catalyst in various photocatalytic applications. Thus, benefitting from the Schottky effect (due to the high work functions of Ti3C2Tx 3.9–4.8 eV depending on the surface termination in comparison with those of most well-established semiconductor photocatalysts)91 and the accompanying built-in electric field, the photogenerated charges of several semiconductors interfaced with Ti3C2Tx can be effectively separated and, from a photocatalytic point of view, this process enhances the activity of the photocatalytic system.
Due to their excellent metallic conductivity (the measured conductivity of single-layer Ti3C2Tx was reported to be up to 6.76 × 105 S m−1, which is comparable to that of graphene films that is situated in the range of 5 × 106 to 6.4 × 106 S m−1),92,93 MXenes can boost the activity of semiconductor photocatalysts. In comparison with graphene, however, the use of MXenes as co-catalysts brings several advantages.94,95 For instance, in the construction of graphene-based semiconductor photocatalysts, the majority of the literature data report the use of graphene oxide (GO), followed by a reduction process to generate reduced graphene oxide (RGO).96 The adoption of this synthesis route causes a significant loss in electronic conductivity due to the presence of numerous point defects, and the reported electrical conductivity of RGO (<1 × 103 S m−1)97 is about two orders of magnitude below the conductivity of Ti3C2Tx. Another interesting discovery shows that MXenes can serve as hole acceptors during the photocatalytic reactions, a fact that was never reported in the case of graphene-based semiconductor photocatalysts.94
On the other hand, use of Ti3C2Tx as a co-catalyst comes with a big disadvantage: its low stability in water. However, within the formed photocatalytic composite, Ti3C2Tx should not be seen as the Trojan horse, since the oxidized product is TiO2, which itself possesses excellent photoactivity. Moreover, as will be shown later in Tables 2–4, the stability and the photoactivity of the photocatalyst composite obtained by the in situ approach (e.g., solvothermal procedure) are significantly greater than those obtained by the electrostatic self-assembly process.
| Photocatalyst | Preparation methods | Reaction conditions | Light source | Sacrificial reagent | H2 production [μmol g−1 h−1] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|---|
| a Results included in studies published starting from 2020 onwards only; AA: ascorbic acid; UCN: ultrathin carbon nitride; BQD: black phosphorus quantum dots; TEOA: triethanolamine; DI: deionized water; HCN: heptazine-based crystalline carbon nitride; np: information not provided by authors. | |||||||
| Pt/ZnIn2S4/Ti3C2Tx (3 wt% Pt) | Solvothermal procedure | 20 mg catalyst dispersed in 40 mL TEOA (10 v%) | 300 W Xe lamp (λ ≥ 400 or 420 nm) | TEOA | 6482 (≥400 nm) | 20.41 (400 nm) | 105 |
| 3475 (≥420 nm) | 11.14 (420 nm) | ||||||
| Chlorophyll–Ti3C2Tx | Electrostatic self-assembly process | 3 mg catalyst dispersed in 3 mL AA (55 mM) | 350 W Xe lamp (λ ≥ 400 nm) | AA | 52 | np | 106 |
| MXene@Au@CdS | Stepwise approach | 4 mg catalyst dispersed in 80 mL sacrificial agent aqueous solution | 300 W Xe lamp (λ ≥ 420 nm) | 0.25 M Na2SO3 and 0.35 M Na2S | 17070.43 |
np | 107 |
| TiO2–Ti3C2/Ru | Stepwise approach | 10 mg catalyst dispersed in 80 mL methanol (10 v%) | 300 W Xe lamp (350–780 nm) | Methanol | 235.3 | 14.33 (350 nm) | 108 |
| p-g-C3N4/Ti3C2Tx | Electrostatic self-assembly process | 40 mg catalyst dispersed in 60 mL TEOA (10 v%) | 300 W Xe lamp | TEOA | 982.8 | np | 109 |
| CdLa2S4/Ti3C2 | Hydrothermal procedure | 50 mg catalyst dispersed in 100 mL sacrificial agent aqueous solution | 300 W Xe lamp (λ ≥ 420 nm) | 0.25 M Na2SO3 and 0.35 M Na2S | 11182.4 |
15.6 (420 nm) | 110 |
| BiVO4@ZnIn2S4/Ti3C2 | Ultrasonic assisted self-assembly process | 60 mg catalyst | 300 W Xe lamp (λ ≥ 420 nm) | No scavenger | 102.67 | 2.9 (460 nm) | 111 |
| g-C3N4/Ti3C2 | Stepwise approach | 50 mg catalyst dispersed in 100 mL TEOA (10 v%) | 300 W Xe lamp (λ ≥ 420 nm) | TEOA | 116.2 | np | 112 |
| CdS@Ti3C2@CoO | Stepwise approach | 20 mg catalyst dispersed in 100 mL DI | 300 W Xe lamp (λ ≥ 420 nm) | No scavenger | 134.46 | np | 113 |
| 2 wt% Pt/UCN/Ti3C2Tx/BQD | Stepwise approach | 10 mg catalyst dispersed in 78 mL TEOA (10 v%) | 300 W Xe lamp (λ ≥ 420 nm) | TEOA | 18420 |
17.6 (420 nm) | 101 |
| Cd0.5Zn0.5S/Ti3C2 | Hydrothermal procedure | 50 mg catalyst dispersed in 100 mL DI or seawater | 300 W Xe lamp (λ ≥ 420 nm) | 0.25 M Na2SO3 and 0.35 M Na2S | 9.07 | 43.28 (420 nm, DI splitting and AM 1.5G) | 114 |
| 1 wt% Pt/HCN/Ti3C2 | Ionothermal method | 20 mg catalyst dispersed in 100 mL TEOA (10 v%) | 3 W LED lamp (420 nm) | TEOA | 4225 | 14.6 (420 nm) | 115 |
| CdS nanorods/Ti2C3Tx | Hydrothermal procedure | 20 mg catalyst dispersed in 100 mL TEOA (20 v%) | 300 W Xe lamp | TEOA | 63.53 | 2.28 | 116 |
| TiO2–C/CdS synthesized from Ti3C2/CdS | Calcination | 50 mg catalyst dispersed in 100 mL lactic acid (20 v%) | 300 W Xe lamp (λ ≥ 420 nm) | Lactic acid | 1480 | np | 102 |
| 0.5 wt% Pt/TiO2@C synthesized from Ti3C2 | Calcination | 35 mg catalyst dispersed in 100 mL TEOA (10 v%) | Solar simulator AM 1.5G | TEOA | 160.42 | np | 103 |
The application of Ti3C2Tx in the field of photocatalysis is summarized in this review by the following four aspects: (i) the photocatalytic hydrogen, H2, evolution reaction (HER) through water splitting, (ii) photocatalytic carbon dioxide, CO2, reduction reaction (CO2RR), (iii) photocatalytic degradation reactions of various pollutants, and (iv) nitrogen photofixation and other photocatalytic/photo-assisted processes (see Scheme 5). As many reviews were recently published on this topic,11–14 here we only report the studies published in 2020–2021. Moreover, the aim of this section is not to provide an exhaustive review of this area, but to present general aspects to be considered, and then to outline which are currently the most efficient photocatalytic systems comprising Ti3C2Tx.
 | ||
| Scheme 5 MXenes in photocatalysis. | ||
5.2. Applications in the hydrogen evolution reaction
As stated above, after photogeneration, the electron–hole pairs initiate simultaneous oxidation and reduction reactions on the outer surfaces of the photocatalyst. In order to maintain charge neutrality during the photocatalytic process, the consumption rate of electrons and holes must be equal. For this reason, most of the studies on the HER are carried out with an excess of electron donor reagents.98 Thus, while the holes are quenched and used by different hole scavengers, the electrons reduce the H+ species, followed by the formation of intermediate adsorbed H* states and finally the production of H2 molecules.99In the field of H2 generation through photocatalytic water splitting, the first synthesized MXene, Ti3C2Tx, is also the most widely used because it forms Schottky junctions that promote the separation of photogenerated charge carriers. Besides this, the DFT calculated Gibbs free energy for atomic H adsorption on the surface of O-terminated Ti3C2 (|ΔGH*| = 0.00283 eV)89 is much lower than that of the highly active and well-known HER catalyst, Pt (ΔGH* ≈ −0.09 eV)100 and thus much closer to the most desired |ΔGH*| value which should be zero.99
Table 2 summarizes several examples of Ti3C2Tx-based photocatalysts used in the most recent years for the HER. The synthetic approaches to combine different semiconductor photocatalysts with Ti3C2Tx and several reaction parameters are also presented. These reports demonstrated that Ti3C2Tx could greatly improve the photoactivity compared to solely the base photocatalyst. The highest H2 production activity and apparent quantum efficiency (AQE) of 18.42 mmol h−1 g−1 and 17.6% (at 420 nm), respectively, were obtained when Ti3C2 nanosheets were mixed with ultrathin 0D/2D black phosphorus/ultrathin carbon nitride heterojunctions that were loaded with 2 wt% Pt and tested in photocatalytic water splitting in the presence of triethanolamine as a hole scavenger.101
Besides its role as a co-catalyst, Ti3C2Tx can be used as a precursor for the generation of TiO2-based active photocatalysts through a simple thermal degradation process.102,103 Thus, flash oxidation of 2D Ti3C2Tx in air resulted in the formation of a hybrid structure of thin sheets of disordered graphitic carbon decorated with nanocrystalline anatase.104 In this regard, Table 2 presents two of the most recent experiments comprising the use of Ti3C2Tx as a precursor for obtaining photocatalysts with remarkable photoactivity toward the HER (see ref. 92 and 93).
5.3. Applications in the carbon dioxide reduction reaction, CO2RR
In comparison with photocatalytic water splitting, the photocatalytic reduction of CO2 is a much more complicated process, due to several aspects which include but are not limited to the following: (i) although water is the most suitable hydrogen donor for CO2 reduction, in aqueous solutions, the HER competes with the CO2RR; (ii) while basic pH increases CO2 solubility in water, this is not necessarily advantageous since, under these conditions, the real species that are present would be carbonates or bicarbonates, which are more difficult to reduce than CO2 itself and, in this way, the advantages of high solubility, meaning the high concentration of the substrate around the photocatalyst is lost; (iii) performing the CO2RR under gas phase conditions might lead to the formation of less volatile reaction products that can strongly adsorb or deposit on the photocatalyst surface and act as poisons; (iv) in cases in which the concentration of the CO2RR products is low, the impurities (e.g., adsorbed airborne organic compounds) present on the surface of the photocatalyst might lead to misleading results concerning the real activity of the photocatalysts.117Considering all these difficulties, it is not surprising that the number of studies on the CO2RR are much less than those on water splitting. More importantly, the instability of the MXene carbon atoms causing interference during the CO2RR limits the application of Ti3C2Tx-based photocatalysts in the CO2RR in comparison with its use for photocatalytic water splitting.
As shown in Table 3, among the limited reports in recent years, Ti3C2Tx, as a co-catalyst, promotes the separation of charge carriers, leading to the formation of solely C1 products (i.e., CO and CH4), while the lack of the formation of C2 products (e.g., formaldehyde, formic acid or oxalic acid) indicates that Ti3C2Tx cannot change the energy barrier of the base photocatalyst.
| Photocatalyst | Preparation methods | Reaction conditions | Light source | Products and yield [μmol g−1 h−1] | Ref. |
|---|---|---|---|---|---|
| a Results included in studies published starting from 2020 onwards only. | |||||
| FAPbBr3/Ti3C2 | Deposition | 3 mg catalyst, 0.5 mL deionized water and CO2 (1 bar) | 300 W Xe lamp (λ ≥ 420 nm) | 283.41 (CO) | 118 |
| 17.7 (CH4) | |||||
| Cu2O/Ti3C2Tx | Hydrothermal procedure | 30 mg catalyst coated onto a quartz plate and CO2 (<1 atm) | 300 W Xe lamp | 17.5 (CO) | 119 |
| 1.0 (CH4) | |||||
| g-C3N4/Ti3C2Tx | Calcination | 50 mg catalyst uniformly dispersed on a 28.26 cm2 glass sheet, 5 mL water and 1 mL CO2 gas | 300 W Xe lamp | 2.1 (CH4) | 120 |
| 4 (CO) | |||||
| g-C3N4/TiO2/C synthesized from Ti3C2Tx | Electrostatic self-assembly and calcination | Certain amounts of catalyst and water and CO2 (ca. 1 atm) | Arc Xe lamp | 8.65 (CO) | 121 |
| 1.23 (CH4) | |||||
A great enhancement of the photoactivity toward the CO2RR was obtained by Que et al.118 by anchoring inorganic halide perovskite FAPbBr3 (formamidinium lead bromide) quantum dots on Ti3C2 nanosheets to form a FAPbBr3/Ti3C2 composite with a Schottky heterojunction. Thus, under visible light irradiation, an optimal electron consumption rate of 717 μmol g−1 h−1 is obtained, which corresponds to a ≈ 2-fold improvement over that of pristine FAPbBr3 QDs (344 μmol g−1 h−1).
5.4. Applications in photocatalytic degradation reactions
Degradation of both atmospheric and aquatic organic contaminants can be efficiently performed through heterogeneous photocatalysis. Here the photocatalytic degradation process follows three main steps: (i) photogeneration of electron–hole pairs upon light irradiation, (ii) formation of highly active radicals (i.e., ˙OH, O2˙−, and HOO˙) on the surface as a result charge separation, and (iii) redox processes initiated by these radicals.While the photogenerated holes react with surface bound water molecules or hydroxyl groups (OH−) to produce the hydroxyl radical (˙OH), photogenerated electrons interact with oxygen and generate anionic superoxide radicals (O2˙−). These are the most common radicals formed on the surface of photocatalysts, with the hydroperoxyl radical (HOO˙) being produced through a O2˙− protonation process.
The ˙OH radicals are extremely powerful oxidizing agents that initiate a non-selective oxidative attack over the adsorbed organic molecules or those that are close to the photocatalyst surface, causing them to mineralize to an extent depending upon their structure and stability level.122 On the other hand, the O2˙− radicals not only initiate further oxidation processes but also prevent charge recombination.
It should be noted that during the photocatalytic degradation processes, the inherent presence of oxidative radicals in the reaction medium, the UV light irradiation and the photothermal effect induced by the near-infrared irradiation may oxidize the Ti3C2Tx surfaces.94,123 On the other hand, the target dye molecules (especially the cationic ones) can be adsorbed on the MXene surfaces and might change/affect their surface and electronic states.124,125 In this regard, it is clear that the reactions and interactions between Ti3C2Tx and the aqueous environment are complicated and more detailed investigations (preferably operando and in situ) are needed to reveal the true working mechanism of the photocatalytic degradation processes on the surface of MXene-based composites.
As shown in Table 4, the photocatalytic degradation reactions of various pollutants (e.g., methylene blue, methyl orange, bisphenol A, rhodamine B, diclofenac, tetracycline or tetracycline hydrochloride, among others) or the removal of toxic metals (i.e., reduction of Cr(VI) anions) from water are the most studied applications of Ti3C2Tx-based photocatalysts. Once again, the introduction of Ti3C2Tx within a photocatalytic composite improves the photoactivity mainly due to the promotion of the separation of charge carriers and the presence of abundant surface groups and active sites where the targeted molecules are easily adsorbed.
| Photocatalyst | Preparation methods | Reaction conditions | Light source | Substrate of degradation | Oxygenic species | Removal rate/rate constants [%]/[min−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a Results included in studies published starting from 2020 onwards only; RhB: rhodamine B; CIP: ciprofloxacin; BPA: bisphenol A; TC: tetracycline; PMS: peroxymonosulfate; DCF: diclofenac; MO: methyl orange; TCH: tetracycline hydrochloride; iN: isopropyl amine modified; MB: methylene blue; PFOA: perfluorooctanoic acid; MR: methyl red; np: information not provided by authors. | |||||||
| ZnO–Ti3C2 | Ultrasonic assisted self-assembly process | 35 mg catalyst dispersed in 50 mL RhB (10 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | RhB | np | 98/0.0077 | 133 |
| Bi2WO6/C3N4/Ti3C2 | Hydrothermal procedure | 30 mg catalyst dispersed in 200 mL CIP (10 mg L−1) | 300 W Xe lamp | CIP | h+/O2˙− | 0.058 | 134 |
| TiO2(TiO2−x)/Ti3C2 | Stepwise synthesis | 5 mg catalyst dispersed in 50 mL BPA (15 mg L−1) | 300 W Hg lamp (λ < 365 nm) | BPA | ˙OH/O2˙− | 90.5 | 135 |
| Ti3C2/Ti3+–TiO2 | Stepwise synthesis | 100 mg catalyst coated on a glass slide. Continuous flow (20 sccm) of acetaldehyde (500 ppm) | 500 W Xe lamp (λ ≥ 420 nm) | Acetaldehyde | ˙OH/O2˙− | 27 | 136 |
| α-Fe2O3/ZnFe2O4@Ti3C2 | Ultrasonic assisted self-assembly process | 20 mg catalyst dispersed in 100 mL RhB (10 mg L−1) or Cr(VI) (10 mg L−1) | 300 W Xe lamp (λ ≥ 400 nm) | RhB | O2˙−/h+/e− | ∼98/0.02686 | 137 |
| Cr(VI) | |||||||
| Ti3C2/TiO2 | Solvothermal procedure | 20 mg catalyst dispersed in 50 mL RhB (20 mg L−1) | 300 W Xe lamp | RhB | h+ | 93.7 | 138 |
| TiO2/g-C3N4 synthesized from Ti3C2 | Calcination | 60 mg catalyst dispersed in 100 mL of pollutants | 300 W Xe lamp (λ ≥ 400 nm) | TC | O2˙−/˙OH | 0.02442 | 139 |
| CIP | 0.01675 | ||||||
| TC (15 mg L−1); CIP (3 mg L−1); BPA (5 mg L−1); RhB (20 mg L−1) | BPA | 0.01935 | |||||
| RhB | 0.05586 | ||||||
| BiOBr/TiO2/Ti3C2Tx | Hydrothermal procedure | 20 mg catalyst dispersed in 50 mL RhB | 300 W Xe lamp (λ ≥ 420 nm) | RhB | ˙OH/O2˙− | 99.8 | 140 |
| Ti3C2/TiO2/BiOCl | Solvothermal procedure | 50 mg catalyst dispersed in 50 mL RhB (10 mg L−1) or TC (20 mg L−1) | 500 W Xe lamp AM 1.5G | RhB | O2˙−/˙OH | 84/0.0143 | 141 |
| TC | 0.0157 | ||||||
| TiO2/Ti3C2Tx | Solvothermal procedure | Certain amount of catalyst dispersed in 100 mL MO | 500 W Hg lamp | MO | Np | 92 | 142 |
| Ti3C2/BiPO4 | Hydrothermal procedure | 100 mg catalyst dispersed in 50 mL RhB (10 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | RhB | O2˙−/˙OH | 100/0.12469 | 143 |
| ZnO/Ti3C2 | Hydrothermal procedure | 50 mg catalyst dispersed in 200 mL MB (10 mg L−1) | 40 W Hg lamp (365 nm) | MB | O2˙−/˙OH | 83.97/0.03357 | 144 |
| Ti3C2/TiO2 | Hydrothermal oxidation method | Certain amount of catalyst (200 mg L−1) and PFOA (20 μM) | Hg lamp (254 nm) | PFOA | Np | 99.9 | 145 |
| CoAl-LDHs/Ti3C2Tx | Electrostatic self-assembly process | 100 mg catalyst dispersed in 100 mL TCH | 300 W Xe lamp (λ ≥ 420 nm) | TCH | O2˙− > h+ > ˙OH | 96.67 | 146 |
| Ag/Ag3PO4/Ti3C2 | Electrostatic self-assembly process | 50 mg catalyst dispersed in 100 mL MO (20 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | MO | h+, e− | 93/0.044 | 147 |
| 40 mg catalyst dispersed in 80 mL Cr(VI) (10 mg L−1) | Cr(VI) | h+, e− | 61/0.014 | ||||
| TiO2/Ti3C2 | Stepwise synthesis | 40 mg catalyst dispersed in 50 mL RhB (20 μM) | Solar simulator AM 1.5G | RhB | ˙OH > O2˙− | 95 | 148 |
| CaIn2S4/Ti3C2Tx | Hydrothermal procedure | 50 mg catalyst dispersed in 50 mL TCH (20 mg L−1) or Cr(VI) (20 mg L−1) | 400 W metal halogen lamp (λ ≥ 420 nm) | TCH | O2˙−/h+ | 96 | 149 |
| Cr(VI) | e− | 98 | |||||
| Sm-g-C3N4/Ti3C2 | Stepwise synthesis | 20 mg catalyst dispersed in 100 mL CIP (20 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | CIP | O2˙−/h+ | 99 | 150 |
| BiOBr/Ti3C2 | Solvothermal procedure | 10 mg catalyst dispersed in 50 mL RhB (20 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | RhB | O2˙− | 99.3/0.23043 | 151 |
| iN-Ti3C2/TiO2 | Stepwise synthesis | 10 mg catalyst dispersed in 50 mL MB (20 mg L−1) | 300 W Hg lamp (λ < 365 nm) | MB | ˙OH | 0.02642 | 152 |
| WO3/Ti3C2/In2S3 | Ultrasonic assisted self-assembly process | 5 mg catalyst dispersed in 10 mL BPA (10 mg L−1) | 30 W Xe lamp | BPA | O2˙− | 97.6 | 153 |
| 10 mg catalyst dispersed in 20 mL Cr(VI) (20 mg L−1) | Cr(VI) | e− | 99.8 | ||||
| NiCo2S4/Ti3C2 | Hydrothermal procedure | 5 mg catalyst dispersed in 50 mL RhB (50 mg L−1) | 250 W Xe lamp (400–800 nm) | RhB | O2˙−/˙OH | 100 | 154 |
| Ti3C2/SnNb2O6 | Ultrasonication-assisted hydrothermal method | 15 mg catalyst dispersed in 60 mL TCH (10 mg L−1) and RhB (10 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | TCH | ˙OH | 70/0.0145 | 155 |
| RhB | h+ | 98/0.0579 | |||||
| Ti3C2/Ag2S | Ultrasonic assisted self-assembly process | 50 mg catalyst dispersed in 100 mL MB (20 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | MB | O2˙− > e− > h+ > ˙OH | 98/0.08222 | 156 |
| MB | 99.2/0.99198 | ||||||
| RhB | 99.54/0.99538 | ||||||
| MO | 96.98/0.96985 | ||||||
| MR | 87.45/0.87451 | ||||||
| ZnS/Ti3C2 | Ultrasonic assisted self-assembly process | 35 mg catalyst dispersed in 50 mL RhB (10 mg L−1) | 300 W Xe lamp (λ ≥ 420 nm) | RhB | np | 0.02464 | 157 |
5.5. Applications in N2 photofixation and other photocatalytic/photo-assisted processes
In addition to photocatalytic applications presented above, there are many other cases in which Ti3C2Tx, is used as a co-catalyst in photocatalytic systems for different reduction and oxidation processes, such as the N2 reduction reaction,126–129 photocatalytic bireforming of methane,130 NO oxidation131 or photo-assisted organic transformations,132 among others.The N2 reduction reaction (NRR) under light irradiation, a process known as N2 photofixation, has been investigated over different Ti3C2Tx-based materials. Thus, Huo et al.127 recently developed a highly active photocatalyst toward near-infrared light-driven N2 photofixation by fabricating a plasmonic hybrid Ti3C2Tx/TiO2 structure. The photocatalytic system attained a NH3 production rate of 422 μmol g−1 h−1 under full-spectrum irradiation. The authors also demonstrate that oxygen vacancies found on the surface served as the active centers for the adsorption and the activation of N2 gas molecules.
By using Ti3C2Tx as a precursor, Qian et al.128 synthesized, through a one-step calcination approach, oxygen vacancy-rich C/TiO2 materials that showed remarkable activity for the NRR. Thus, by using H2O and CH3OH as proton sources, they were able to produce NH3 at rates of 41 μmol g−1 h−1 and 84 μmol g−1 h−1, respectively. Once again, the importance of oxygen vacancies in the efficiency of N2 adsorption and activation was revealed by electron spin-resonance spectroscopy, ESR, and temperature programmed desorption of nitrogen, N2-TPD, experiments which showed that the chemisorption of N2 is much more efficient on the surface of C/TiO2 possessing higher concentrations of oxygen vacancies than in the case of commercial TiO2.
In line with the above, Gao et al.129 reported the performance of the NRR over the Ti3C2Tx/TiO2/Co composite synthesized by introducing Co into the MXene@TiO2 catalysts in which the TiO2 nanoparticles were derived from the in situ growth on the surface of MXene nanosheets. The optimal photocatalyst (containing 0.5 wt% Co) shows a NH4+ production rate of 110 μmol g−1 h−1 without the addition of any hole scavengers and under UV-vis light irradiation conditions.
The photocatalytic bireforming of CH4 under visible light irradiation over Ti3C2 nanosheets coupled with a 2D g-C3N4/TiO2 heterojunction was investigated by Khan et al.130 The authors demonstrate that the as-prepared photocatalytic composite shows CO and H2 production rates of 48.4 and 83.2 μmol g−1, reaching apparent quantum yields as high as 0.41 and 0.7%, respectively.
Another example of the use of gas phase photocatalysis over Ti3C2Tx-based composites is the photocatalytic oxidation of nitric oxide (NO). In line with this research, Li et al.131 reported the use of a Ti3C2/g-C3N4 composite with an enhanced photocatalytic NO removal efficiency. Thus, the authors performed the photocatalytic oxidation of NO, and through monitoring by in situ diffuse reflectance infrared spectroscopy, they demonstrated that the final products were nitrite and nitrate instead of the undesired and toxic NO2.
Visible-light induced one-pot hydrogenation and amidation of nitroaromatics with carboxylic acids was recently investigated by Jiang et al.132 The authors prepared Pt/N–TiO2/Ti3C2 heterojunctions by in situ growth of TiO2 on Ti3C2 nanosheets and then N doped the TiO2 with melamine, followed by the deposition of Pt nanoparticles. Thus, by using the 3 wt% Pt/N–TiO2/Ti3C2 photocatalyst and in the presence of a K3PO4 additive, the hydrogenation and amidation reaction of nitrobenzene with acetic acid reached 100% conversion with 100% chemoselectivity to acetanilide.
6. Concluding remarks
In this review we have shown that the MAX phases and MXenes, with different chemical compositions, can be considered as potential catalysts, as well as supports, creating synergy with various active species. In some cases, their catalytic activity was similar, or even better, compared with that of noble metal containing catalysts, highlighting, once again, their catalytic capabilities. At the current stage, these materials are proving to be extremely promising in various catalytic applications; however their surface chemistry and thermal stability are not fully understood.Currently, MXenes cannot be applied in industrial processes as their large-scale production is not yet possible due to the HF involved in the synthesis that cannot be used in large quantities. Progress is expected in the synthesis method to avoid the use of HF, so that the remarkable properties observed for these materials in heterogeneous catalytic reactions can be improved further, to make their industrialization possible.
Depending on the application, these materials have been shown to be extremely resistant to coke, or if deactivation has been observed, it has been much slower than in the case of standard catalysts. One of the best ways to counteract this issue is by tuning the chemical composition and surface terminations.
Finally, we would like to draw attention to the two subclasses of these materials that have not yet been explored in heterogeneous catalytic reactions, other than electrochemical ones, namely the double M′M′′AX phase, obtained by alloying a MAX phase with another metal, and i-MXenes (in-plane ordered MXenes).158 Usually, alloying of a MAX phase with another metal leads to chemically disordered solid solutions that can be an advantage in catalytic applications which are dependent on surface defects. Likewise, in the case of i-MXenes, the etching process results in 2D MXene structures with ordered vacancies, ordered divacancies or disordered vacancies with huge potential in catalytic applications.158 The development of these materials is still in its infancy, but we predict a rapid evolution because the presence of vacancies and how they can influence the properties of the final catalytic material, is not only scientifically rewarding for any researchers, but also has major implications in the development of new materials for a variety of practical applications.
As compared with the oxide-based catalysts, MAX phases and MXenes have not been widely investigated as catalysts in traditional heterogeneous processes. However, further theoretical and experimental concerted studies will pave the way for new heterogeneous applications, such as selective oxidation of different hydrocarbons to value added compounds, and also in total oxidation reactions. Furthermore, appropriate modification of their surface or intercalation between the layers of different chemical compounds, will open new avenues for MAX phases and MXenes to be used as heterogeneous catalysts (e.g., oxidative coupling of amines to imines).
Lastly a word of caution: MXenes are unstable in aqueous environments in which there is dissolved oxygen. In some cases, the oxidation occurs by hydrolysis,123 and thus reducing the dissolved oxygen does not solve the problem. There are ways to mitigate oxidation159,160 but whether such approaches solve the long-term problem, especially in an open system, is an open question at this time. If this problem is not solved and/or shown not to be an issue, then all the work in that domain will be totally academic with no practical applications. Along the same lines and somewhat ironically, it follows that oxidizing the MXenes to their more stable oxides and possibly C104 as some have done, may be an advisable and fruitful way forward.
Summarizing, it is clear that there is room for improvement in the design and synthesis of MAX phases and MXene based catalysts and in this regard several strategies can be adopted, such as tuning the chemical composition, ratios between elements, or surface terminations. As shown by the studies presented in this review, the deposition of different chemical species (transition metal oxides and metals) on MAX phase or MXene materials can lead to new different compounds/structures that have never been reported as a “classical support”, which leads us to believe that these materials are not sufficiently explored, and represent a vast reservoir of both knowledge and potential applications.
Author contributions
I. M. C. contributed to Section 3, A. G. M. contributed to Section 4, S. N. contributed to Section 5, while M. F., M. W. B. and F. N. contributed to the remaining sections with scientific writing, illustrations and table drafting. M. F., M. W. B. and F. N. supervised the research work and contributed to funding acquisition, outline drafting, reviewing, and editing the overall text, illustrations and tables of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant of the Ministry of Research, Innovation and Digitization, CNCS/CCCDI – UEFISCDI, project number PN-III-P1-1.1-TE-2019-1969, within PNCDI III. MWB acknowledges the support of the Division of Materials Research of NSF (DMR 1740795).References
- M. W. Barsoum, Prog. Solid State Chem., 2000, 28, 201–281 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS.
- M. W. Barsoum, MAX Phases: Properties of Machinable Ternary Carbides and Nitrides, Wiley-VCH Verlag, 2013 Search PubMed.
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, Trends Chem., 2019, 1, 210–223 CrossRef CAS.
- L. Verger, V. Natu, M. Carey and M. W. Barsoum, Trends Chem., 2019, 1, 656–669 CrossRef CAS.
- W. H. K. Ng, E. S. Gnanakumar, E. Batyrev, S. K. Sharma, P. K. Pujari, H. F. Greer, W. Zhou, R. Sakidja, G. Rothenberg, M. W. Barsoum and N. R. Shiju, Angew. Chem., Int. Ed., 2018, 57, 1485–1490 CrossRef CAS PubMed.
- P. Eklund, M. Beckers, U. Jansson, H. Högberg and L. Hultman, Thin Solid Films, 2010, 518, 1851–1878 CrossRef CAS.
- Z. M. Sun, Int. Mater. Rev., 2011, 56, 143–166 CrossRef CAS.
- P. Eklund, J. Rosen and P. O. Å. Persson, J. Phys. D: Appl. Phys., 2017, 50, 113001 CrossRef.
- L. Verger, C. Xu, V. Natu, H.-M. Cheng, W. Ren and M. W. Barsoum, Curr. Opin. Solid State Mater. Sci., 2019, 23, 149–163 CrossRef CAS.
- J. K. Im, E. J. Sohn, S. Kim, M. Jang, A. Son, K.-D. Zoh and Y. Yoon, Chemosphere, 2021, 270, 129478 CrossRef CAS PubMed.
- L. Yu, B. Liu, Y. Wang, F. Yu and J. Ma, J. Power Sources, 2021, 490, 229250 CrossRef CAS.
- Q. Zhong, Y. Li and G. Zhang, Chem. Eng. J., 2021, 409, 128099 CrossRef CAS.
- X. Li, Y. Bai, X. Shi, N. Su, G. Nie, R. Zhang, H. Nie and L. Ye, Mater. Adv., 2021, 2, 1570–1594 RSC.
- Y. Zhang, X. Zhang, C. Cheng and Z. Yang, Chin. Chem. Lett., 2020, 31, 931–936 CrossRef CAS.
- C. Yang, H. Huang, H. He, L. Yang, Q. Jiang and W. Li, Coord. Chem. Rev., 2021, 435, 213806 CrossRef CAS.
- Z. Xia, Q. Huang and S. Guo, FlatChem, 2019, 17, 100129 CrossRef CAS.
- M. A. Gibson and J. W. Hightower, J. Catal., 1976, 41, 420–430 CrossRef CAS.
- E. H. Lee, Catal. Rev., 1974, 8, 285–305 CrossRef.
- G. Busca, in Heterogeneous Catalytic Materials, ed. G. Busca, Elsevier, Amsterdam, 2014, pp. 375–419 Search PubMed.
- C. Doornkamp and V. Ponec, J. Mol. Catal. A: Chem., 2000, 162, 19–32 CrossRef CAS.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- J. Diao, M. Hu, Z. Lian, Z. Li, H. Zhang, F. Huang, B. Li, X. Wang, D. S. Su and H. Liu, ACS Catal., 2018, 8, 10051–10057 CrossRef CAS.
- J. Diao, Z. Feng, R. Huang, H. Liu, S. B. A. Hamid and D. S. Su, ChemSusChem, 2016, 9, 662–666 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS.
- Z. Li, Y. Cui, Z. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E. Shi, J. T. Miller, F. H. Ribeiro and Y. Wu, Nat. Catal., 2018, 1, 349–355 CrossRef CAS.
- Z. Li, L. Yu, C. Milligan, T. Ma, L. Zhou, Y. Cui, Z. Qi, N. Libretto, B. Xu, J. Luo, E. Shi, Z. Wu, H. Xin, W. N. Delgass, J. T. Miller and Y. Wu, Nat. Commun., 2018, 9, 5258 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X. Sun, Y. Gao, C. Zhao, S. Deng, X. Zhong, G. Zhuang, Z. Wei and J. Wang, Adv. Theory Simul., 2019, 2, 1800158 CrossRef.
- A. Kurlov, E. B. Deeva, P. M. Abdala, D. Lebedev, A. Tsoukalou, A. Comas-Vives, A. Fedorov and C. R. Müller, Nat. Commun., 2020, 11, 4920 CrossRef CAS.
- C. M. Damaskinos, M. A. Vasiliades and A. M. Efstathiou, Appl. Catal., A, 2019, 579, 116–129 CrossRef CAS.
- L. Foppa, M.-C. Silaghi, K. Larmier and A. Comas-Vives, J. Catal., 2016, 343, 196–207 CrossRef CAS.
- R. Thakur, A. VahidMohammadi, J. Smith, M. Hoffman, J. Moncada, M. Beidaghi and C. A. Carrero, ACS Catal., 2020, 10, 5124–5134 CrossRef CAS.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, T. D. Nguyen-Phan, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola and J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7455 CrossRef CAS.
- R. Thakur, M. Hoffman, A. VahidMohammadi, J. Smith, M. Chi, B. Tatarchuk, M. Beidaghi and C. A. Carrero, ChemCatChem, 2020, 12, 3639–3643 CrossRef CAS.
- R. Thakur, A. VahidMohammadi, J. Moncada, W. R. Adams, M. Chi, B. Tatarchuk, M. Beidaghi and C. A. Carrero, Nanoscale, 2019, 11, 10716–10726 RSC.
- M. Ronda-Lloret, V. S. Marakatti, W. G. Sloof, J. J. Delgado, A. Sepúlveda-Escribano, E. V. Ramos-Fernandez, G. Rothenberg and N. R. Shiju, ChemSusChem, 2020, 13, 6401–6408 CAS.
- X. Zhang, J. Lei, D. Wu, X. Zhao, Y. Jing and Z. Zhou, J. Mater. Chem. A, 2016, 4, 4871–4876 RSC.
- C. Cheng, X. Zhang, M. Wang, S. Wang and Z. Yang, Phys. Chem. Chem. Phys., 2018, 20, 3504–3513 RSC.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CrossRef CAS.
- E. H. Song, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2011, 115, 3678–3683 CrossRef CAS.
- T.-T. Jia, C.-H. Lu, K.-N. Ding, Y.-F. Zhang and W.-K. Chen, Comput. Theor. Chem., 2013, 1020, 91–99 CrossRef CAS.
- J. L. Shi, J. H. Wu, X. J. Zhao, X. L. Xue, Y. F. Gao, Z. X. Guo and S. F. Li, Nanoscale, 2016, 8, 19256–19262 RSC.
- C. Cheng, X. Zhang, Z. Yang and K. Hermansson, Adv. Theory Simul., 2019, 2, 1900006 CrossRef.
- H. Oschinski, Á. Morales-García and F. Illas, J. Phys. Chem. C, 2021, 125, 2477–2484 CrossRef CAS.
- M. Manadé, F. Viñes and F. Illas, Carbon, 2015, 95, 525 CrossRef.
- S. Kim, A. Ruiz Puigdollers, P. Gamallo, F. Viñes and J. Y. Lee, Carbon, 2017, 120, 63 CrossRef CAS.
- S. H. Talib, S. Baskaran, X. Yu, Q. Yu, B. Bashir, S. Muhammad, S. Hussain, X. Chen and J. Li, Sci. China Mater., 2021, 64, 651–663 CrossRef CAS.
- E. B. Deeva, A. Kurlov, P. M. Abdala, D. Lebedev, S. M. Kim, C. P. Gordon, A. Tsoukalou, A. Fedorov and C. R. Müller, Chem. Mater., 2019, 31, 4505 CrossRef CAS.
- D. Damma, T. Boningari and P. G. Smirniotis, Mol. Catal., 2018, 451, 20 CrossRef CAS.
- G. Wang, J. A. Schaidle, M. B. Katz, Y. Li, X. Pan and L. T. Thompson, J. Catal., 2013, 304, 92 CrossRef CAS.
- G. Fan, X. Li, C. Xu, W. Jiang, Y. Zhang, D. Gao, J. Bi and Y. Wang, Nanomaterials, 2018, 8, 141 CrossRef.
- Z. Li and Y. Wu, Small, 2019, 15, 1–10 Search PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- B. Zielińska, A. Wróblewska, K. Maślana, P. Miądlicki, K. Kiełbasa, A. Rozmysłowska-Wojciechowska, M. Petrus, J. Woźniak, A. M. Jastrzębska, B. Michalkiewicz and E. Mijowska, Appl. Catal., A, 2020, 604, 117765 CrossRef.
- J. B. Sharmeen, F. M. Mahomoodally, G. Zengin and F. Maggi, Molecules, 2021, 26, 666 CrossRef CAS PubMed.
- N. Girola, C. R. Figueiredo, C. F. Farias, R. A. Azevedo, A. K. Ferreira, S. F. Teixeira, T. M. Capello, E. G. A. Martins, A. L. Matsuo, L. R. Travassos and J. H. G. Lago, Biochem. Biophys. Res. Commun., 2015, 467, 928–934 CrossRef CAS.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS.
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steinei and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- M. Crespo-Quesada, F. Cardenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- G. Vile, D. Albani, N. Almora-Barrios, N. López and J. Pørez-Ramirez, ChemCatChem, 2016, 8, 21–33 CrossRef CAS.
- H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- K. Li, T. Jiao, R. Xing, G. Zou, Q. Zhao, J. Zhou, L. Zhang and Q. Peng, Green Energy Environ., 2018, 3, 147–155 CrossRef.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- L. Zhang, Z. J. Shao, X. M. Cao and P. Hu, J. Phys. Chem. C, 2018, 122, 20337–20350 CrossRef CAS.
- X. Xie, Z. Wu and N. Zhang, Chin. Chem. Lett., 2020, 31, 1014–1017 CrossRef CAS.
- M. Ghidiu, J. Halim, S. Kota, D. Bish, Y. Gogotsi and M. W. Barsoum, Chem. Mater., 2016, 28, 3507–3514 CrossRef CAS.
- M. M. Trandafir, F. Neaţu, I. M. Chirica, Ş. Neaţu, A. C. Kuncser, E. I. Cucolea, V. Natu, M. W. Barsoum and M. Florea, ACS Catal., 2020, 10, 5899–5908 CrossRef CAS.
- Q. Chen, W. Jiang and G. Fan, Dalton Trans., 2020, 49, 14914–14920 RSC.
- J. P. Lange, E. Van Der Heide, J. Van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- M. Naguib, W. Tang, K. L. Browning, G. M. Veith, V. Maliekkal, M. Neurock and A. Villa, ChemCatChem, 2020, 12, 5733–5742 CrossRef CAS.
- A. Fayyaz, K. Saravanakumar, K. Talukdar, Y. Kim, Y. Yoon and C.M. Park, Chem. Eng. J., 2021, 407, 127842 CrossRef CAS.
- M. Ding, W. Chen, H. Xu, Z. Shen, T. Lin, K. Hu, Q. Kong, G. Yang and Z. Xie, Chem. Eng. J., 2019, 378, 122177 CrossRef CAS.
- M. Ding, W. Chen, H. Xu, Z. Shen, T. Lin and K. Hu, J. Hazard. Mater., 2020, 382, 121064 CrossRef CAS.
- Y. Ma, X. Lv, D. Xiong, X. Zhao and Z. Zhang, Appl. Catal., B, 2021, 284, 119720 CrossRef CAS.
- S. Luo, R. Wang, J. Yin, T. Jiao, K. Chen, G. Zou, L. Zhang, J. Zhou, L. Zhang and Q. Peng, ACS Omega, 2019, 4, 3946–3953 CrossRef CAS PubMed.
- Y. Cui, M. Liu, H. Huang, D. Zhang, J. Chen and L. Mao, Ceram. Int., 2020, 46, 11593–11601 CrossRef CAS.
- Y. Liu, R. Luo, Y. Li, J. Qi, C. Wang, J. Li, X. Sun and L. Wang, Chem. Eng. J., 2018, 347, 731–740 CrossRef CAS.
- K. Han, X. Zhang, P. Deng, Q. Jiao and E. Chu, Vacuum, 2020, 180, 109572 CrossRef CAS.
- L. Zhao, Y. Ji, D. Kong, J. Lu, Q. Zhou and X. Yin, Chem. Eng. J., 2016, 303, 458–466 CrossRef CAS.
- Y. Gao, L. Wang, Z. Li, A. Zhou, Q. Hu and X. Cao, Solid State Sci., 2014, 35, 62–65 CrossRef CAS.
- K. Li, Y. Lei, J. Liao and Y. Zhang, Inorg. Chem. Front., 2021, 8, 1747–1761 RSC.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS.
- F. Neaţu, L. E. Abramiuc, M. M. Trandafir, R. F. Negrea, M. Florea, C. M. Teodorescu and S. Neaţu, ChemCatChem, 2020, 12, 4642–4651 CrossRef.
- A. O. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189–218 CrossRef CAS.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS.
- H.-J. Lin, Q.-L. Mo, S. Xu, Z.-Q. Wei, X.-Y. Fu, X. Lin and F.-X. Xiao, J. Catal., 2020, 391, 485–496 CrossRef CAS.
- T. Schultz, N. C. Frey, K. Hantanasirisakul, S. Park, S. J. May, V. B. Shenoy, Y. Gogotsi and N. Koch, Chem. Mater., 2019, 31, 6590–6597 CrossRef.
- X. Sang, Y. Xie, M.-W. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. C. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS.
- J. Krupka and W. Strupinski, Appl. Phys. Lett., 2010, 96, 82101 CrossRef.
- X. Xie and N. Zhang, Adv. Funct. Mater., 2020, 30, 2002528 CrossRef CAS.
- C. Yu, L. Peng, Y. Zhu, G. Xie, Z. Wu, X. Xie and N. Zhang, J. Mater. Chem. A, 2021 10.1039/D1ta02736k.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 8240–8254 RSC.
- C. Gómez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef PubMed.
- Ş. Neaţu, M. Puche, V. Fornés and H. Garcia, Chem. Commun., 2014, 50, 14643–14646 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. Song, L. Hou, B. Long, A. Ali and G. J. Deng, J. Colloid Interface Sci., 2021, 584, 474–483 CrossRef CAS.
- N. Zhao, Y. Hu, J. Du, G. Liu, B. Dong, Y. Yang, J. Peng, J. Li and M. Zhai, Appl. Surf. Sci., 2020, 530, 147247 CrossRef CAS.
- J. Liu, X. Kong, Y. Li, S. Zhu, Y. Liang, Z. Li, S. Wu, C. Chang, Z. Cui and X. Yang, Appl. Surf. Sci., 2020, 530, 147283 CrossRef CAS.
- M. Naguib, O. Mashtalir, M. R. Lukatskaya, B. Dyatkin, C. Zhang, V. Presser, Y. Gogotsi and M. W. Barsoum, Chem. Commun., 2014, 50, 7420–7423 RSC.
- G. Zuo, Y. Wang, W. L. Teo, A. Xie, Y. Guo, Y. Dai, W. Zhou, D. Jana, Q. Xian, W. Dong and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 11287–11292 CrossRef CAS PubMed.
- Y. Li, X. Chen, Y. Sun, X. Meng, Y. Dall'Agnese, G. Chen, C. Dall'Agnese, H. Ren, S. Sasaki, H. Tamiaki and X. Wang, Adv. Mater. Interfaces, 2020, 7, 1902080 CrossRef CAS.
- J. Yin, F. Zhan, T. Jiao, W. Wang, G. Zhang, J. Jiao, G. Jiang, Q. Zhang, J. Gu and Q. Peng, Sci. China Mater., 2020, 63, 2228–2238 CrossRef CAS.
- Y. Liu, Y.-H. Li, X. Li, Q. Zhang, H. Yu, X. Peng and F. Peng, ACS Nano, 2020, 14, 14181–14189 CrossRef CAS PubMed.
- J. Kang, S. Byun, S. Kim, J. Lee, M. Jung, H. Hwang, T. W. Kim, S. H. Song and D. Lee, ACS Appl. Energy Mater., 2020, 3, 9226–9233 CrossRef CAS.
- L. Cheng, Q. Chen, J. Li and H. Liu, Appl. Catal., B, 2020, 267, 118379 CrossRef CAS.
- X. Du, T. Zhao, Z. Xiu, Z. Xing, Z. Li, K. Pan, S. Yang and W. Zhou, Appl. Mater. Today, 2020, 20, 100719 CrossRef.
- J. Li, L. Zhao, S. Wang, J. Li, G. Wang and J. Wang, Appl. Surf. Sci., 2020, 515, 145922 CrossRef CAS.
- Z. Ai, K. Zhang, B. Chang, Y. Shao, L. Zhang, Y. Wu and X. Hao, Chem. Eng. J., 2020, 383, 123130 CrossRef CAS.
- G. Zeng, Y. Cao, Y. Wu, H. Yuan, B. Zhang, Y. Wang, H. Zeng and S. Huang, Appl. Mater. Today, 2021, 22, 100926 CrossRef.
- J. Li, J. Li, C. Wu, Z. Li, L. Cai, H. Tang, Z. Zhou, G. Wang, J. Wang, L. Zhao and S. Wang, Carbon, 2021, 179, 387–399 CrossRef CAS.
- B. Sun, P. Qiu, Z. Liang, Y. Xue, X. Zhang, L. Yang, H. Cui and J. Tian, Chem. Eng. J., 2021, 406, 127177 CrossRef CAS.
- Ş. Neaţu, J. Maciá-Agulló and H. Garcia, Int. J. Mol. Sci., 2014, 15, 5246–5262 CrossRef.
- M. Que, Y. Zhao, Y. Yang, L. Pan, W. Lei, W. Cai, H. Yuan, J. Chen and G. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 6180–6187 CrossRef CAS PubMed.
- J. Zhang, J. Shi, S. Tao, L. Wu and J. Lu, Appl. Surf. Sci., 2021, 542, 148685 CrossRef CAS.
- X. Li, Y. Bai, X. Shi, J. Huang, K. Zhang, R. Wang and L. Ye, Appl. Surf. Sci., 2021, 546, 149111 CrossRef CAS.
- Y. Yang, D. Zhang, J. Fan, Y. Liao and Q. Xiang, Sol. RRL, 2021, 5, 2000351 CrossRef CAS.
- A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003–37026 RSC.
- T. Habib, X. Zhao, S. A. Shah, Y. Chen, W. Sun, H. An, J. L. Lutkenhaus, M. Radovic and M. J. Green, npj 2D Mater. Appl., 2019, 3, 8 CrossRef.
- O. Mashtalir, K. M. Cook, V. N. Mochalin, M. Crowe, M. W. Barsoum and Y. Gogotsi, J. Mater. Chem. A, 2014, 2, 14334–14338 RSC.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- C. Hao, Y. Liao, Y. Wu, Y. An, J. Lin, Z. Gu, M. Jiang, S. Hu and X. Wang, J. Phys. Chem. Solids, 2020, 136, 109141 CrossRef CAS.
- T. Hou, Q. Li, Y. Zhang, W. Zhu, K. Yu, S. Wang, Q. Xu, S. Liang and L. Wang, Appl. Catal., B, 2020, 273, 119072 CrossRef CAS.
- J. Qian, S. Zhao, W. Dang, Y. Liao, W. Zhang, H. Wang, L. Lv, L. Luo, H.-Y. Jiang and J. Tang, Adv. Sustainable Syst., 2021, 5, 2000282 CrossRef CAS.
- W. Gao, X. Li, S. Luo, Z. Luo, X. Zhang, R. Huang and M. Luo, J. Colloid Interface Sci., 2021, 585, 20–29 CrossRef CAS PubMed.
- A. A. Khan, M. Tahir and A. Bafaqeer, Energy Fuels, 2020, 34, 9810–9828 CrossRef CAS.
- J. Li, Q. Zhang, Y. Zou, Y. Cao, W. Cui, F. Dong and Y. Zhou, J. Colloid Interface Sci., 2020, 575, 443–451 CrossRef CAS PubMed.
- H. Jiang, Z. Hu, C. Gan, B. Sun, S. Kong and F. Bian, Mol. Catal., 2021, 504, 111490 CrossRef CAS.
- X. Liu and C. Chen, Mater. Lett., 2020, 261, 127127 CrossRef CAS.
- K. Wu, S. Song, H. Wu, J. Guo and L. Zhang, Appl. Catal., A, 2020, 608, 117869 CrossRef CAS.
- Z. Miao, G. Wang, X. Zhang and X. Dong, Appl. Surf. Sci., 2020, 528, 146929 CrossRef CAS.
- X. Wang, Y. Yang, G. Lu, G. Shi, Y. Wang, R. Wang, X. Xie and J. Sun, Appl. Surf. Sci., 2020, 531, 147101 CrossRef CAS.
- H. Zhang, M. Li, C. Zhu, Q. Tang, P. Kang and J. Cao, Ceram. Int., 2020, 46, 81–88 CrossRef CAS.
- L. Chen, X. Ye, S. Chen, L. Ma, Z. Wang, Q. Wang, N. Hua, X. Xiao, S. Cai and X. Liu, Ceram. Int., 2020, 46, 25895–25904 CrossRef CAS.
- Z. Wu, Y. Liang, X. Yuan, D. Zou, J. Fang, L. Jiang, J. Zhang, H. Yang and Z. Xiao, Chem. Eng. J., 2020, 394, 124921 CrossRef CAS.
- T. Xu, J. Wang, Y. Cong, S. Jiang, Q. Zhang, H. Zhu, Y. Li and X. Li, Chin. Chem. Lett., 2020, 31, 1022–1025 CrossRef CAS.
- H. Liu, C. Yang, X. Jin, J. Zhong and J. Li, Colloids Surf., A, 2020, 603, 125239 CrossRef CAS.
- J. Chen, H. Zheng, Y. Zhao, M. Que, X. Lei, K. Zhang and Y. Luo, J. Phys. Chem. Solids, 2020, 145, 109565 CrossRef CAS.
- L. Zhang, J. Yang, T. Xie, S. Feng and L. Xu, Mater. Des., 2020, 192, 108772 CrossRef CAS.
- Q. Luo, J. Yang, Y. Wu and Q. Cai, Micro Nano Lett., 2020, 15, 764–768 CrossRef CAS.
- H. Song, Y. Wang, Z. Ling, D. Zu, Z. Li, Y. Shen and C. Li, Sci. Total Environ., 2020, 746, 141009 CrossRef CAS PubMed.
- B. Shao, Z. Liu, G. Zeng, Y. Liu, Q. Liang, Q. He, T. Wu, Y. Pan, J. Huang, Z. Peng, S. Luo, C. Liang, X. Liu, S. Tong and J. Liang, Appl. Catal., B, 2021, 286, 119867 CrossRef CAS.
- B. Sun, F. Tao, Z. Huang, W. Yan, Y. Zhang, X. Dong, Y. Wu and G. Zhou, Appl. Surf. Sci., 2021, 535, 147354 CrossRef CAS.
- N. My Tran, Q. Thanh Hoai Ta and J.-S. Noh, Appl. Surf. Sci., 2021, 538, 148023 CrossRef CAS.
- Z. Zhuge, X. Liu, T. Chen, Y. Gong, C. Li, L. Niu, S. Xu, X. Xu, Z. A. Alothman, C. Q. Sun, J. G. Shapter and Y. Yamauchi, Chem. Eng. J., 2020, 127838 Search PubMed.
- M. Yu, H. Liang, R. Zhan, L. Xu and J. Niu, Chin. Chem. Lett., 2021, 32, 2155–2158 CrossRef CAS.
- X. Meng, J. Song, J. Ren and J. Zhu, CrystEngComm, 2021, 23, 1507–1516 RSC.
- T. Ke, S. Shen, K. Rajavel, K. Yang and D. Lin, J. Hazard. Mater., 2021, 402, 124066 CrossRef CAS.
- Z. Yuan, H. Huang, N. Li, D. Chen, Q. Xu, H. Li, J. He and J. Lu, J. Hazard. Mater., 2021, 409, 125027 CrossRef CAS.
- S. Vigneshwaran, C. M. Park and S. Meenakshi, Sep. Purif. Technol., 2021, 258, 118003 CrossRef CAS.
- H. Wang, L. Chen, Y. Sun, J. Yu, Y. Zhao, X. Zhan and H. Shi, Sep. Purif. Technol., 2021, 265, 118516 CrossRef CAS.
- X. Feng, Z. Yu, Y. Sun, M. Shan, R. Long and X. Li, Sep. Purif. Technol., 2021, 266, 118606 CrossRef CAS.
- X. Liu, Q. Liu and C. Chen, Vacuum, 2021, 183, 109834 CrossRef CAS.
- B. Ahmed, A. El Ghazaly and J. Rosen, Adv. Funct. Mater., 2020, 30, 2000894 CrossRef CAS.
- X. Zhao, D. E. Holta, Z. Tan, J.-H. Oh, I. J. Echols, M. Anas, H. Cao, J. L. Lutkenhaus, M. Radovic and M. J. Green, ACS Appl. Nano Mater., 2020, 3, 10578–10585 CrossRef CAS.
- V. Natu, J. L. Hart, M. Sokol, H. Chiang, M. L. Taheri and M. W. Barsoum, Angew. Chem., Int. Ed., 2019, 58, 12655–12660 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |