
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis and applications of solid silylzinc reagents†
Revathi
Chandrasekaran
,
Feba Thomas
Pulikkottil
,
Krishna Suresh
Elama
and
Ramesh
Rasappan
 *
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Vithura, Thiruvananthapuram, Kerala 695551, India. E-mail: rr@iisertvm.ac.in
First published on 30th November 2021
Abstract
The increased synthetic utility of organosilanes has motivated researchers to develop milder and more practical synthetic methods. Silylzinc reagents, which are typically the most functional group tolerant, are notoriously difficult to synthesize because they are obtained by a pyrophoric reaction of silyllithium, particularly Me3SiLi which is itself prepared by the reaction of MeLi and disilane. Furthermore, the dissolved LiCl in silylzinc may have a detrimental effect. A synthetic method that can avoid silyllithium and involves a direct synthesis of silylzinc reagents from silyl halides is arguably the simplest and most economical strategy. We describe, for the first time, the direct synthesis of PhMe2SiZnI and Me3SiZnI reagents by employing a coordinating TMEDA ligand, as well as single crystal XRD structures. Importantly, they can be obtained as solids and stored for longer periods at 4 °C. We also demonstrate their significance in cross-coupling of various free alkyl/aryl/alkenyl carboxylic acids with broader functional group tolerance and API derivatives. The general applicability and efficiency of solid Me3SiZnI are shown in a wide variety of reactions including alkylation, arylation, allylation, 1,4-addition, acylation and more.
Introduction
Organosilicon compounds are widely utilized in various disciplines1 and employed in a broad range of organic transformations.2 Traditional silylating agents include silanes (R3SiH),2a,h disilanes,2f,l,3 silylboranes,2c,4 electrophilic silyl halides and nucleophilic silyl anions (Fig. 1a). While silanes are extensively utilized in hydrosilylations,2h,5 silyl halides are usually combined with organometallic reagents to forge C–Si bonds. Noteworthy examples are the recently developed silyl-Negishi and cross-electrophile coupling reactions by Watson6 and Shu7 and co-workers. The complementary silyl anions are also well established, and silylzinc reagents in general are one of the most appealing8 owing to their excellent functional group tolerance. Earlier in 1984, Oshima et al. described the synthesis of silylzinc reagents,9 and Oestreich and co-workers followed up with elegant applications.10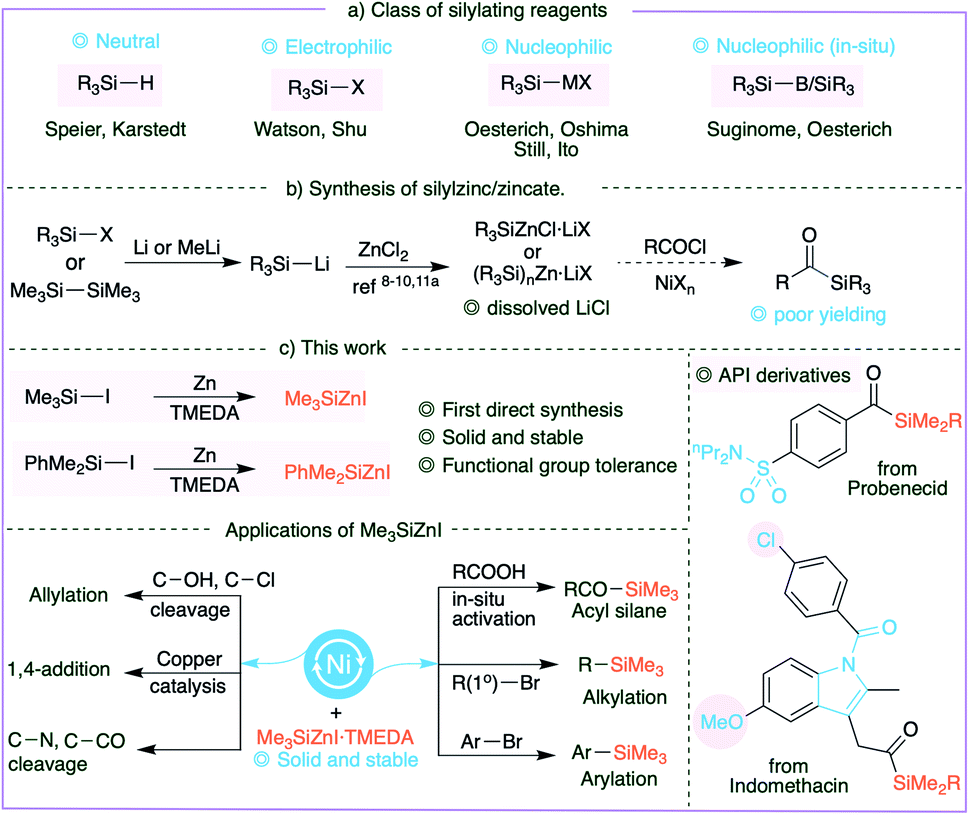 | ||
| Fig. 1 Synthesis of silylzinc reagents and acylsilanes. | ||
However, it is notoriously difficult to synthesize silylzinc reagents because it requires the preparation of silyllithium reagents.11 In particular, preparation of Me3SiLi necessitates treating MeLi with hexamethyldisilane in HMPA, evincing the remarkably high activation barrier required for effecting Si–X cleavage. Furthermore, the application of these reagents may be limited due to the dissolved lithium halides. The direct synthesis of silylzinc reagents is undoubtedly a desirable and economical route for the synthesis of silylzinc reagents, as it avoids the handling of pyrophoric silyllithium. Additional benefits may be obtained if the reagents are accessible as solids. Despite the fact that considerable advances have been made in the preparation of organozinc reagents, particularly by following Knochel's protocols,12 the existing methods are not amenable to the synthesis of silylzinc reagents through reductive metalation of zinc to silyl halides.
Acylsilanes, on the other hand, are versatile and unique building blocks in organic synthesis.13 They undergo a broad range of reactions including nucleophilic addition,14 transition-metal mediated reactions15 and intriguing transformations.16 Importantly they have been utilized as acyl anion synthetic equivalents via the Brook rearrangement.13d,f,14d,15a,17 It is ideal to employ carboxylic acid derivatives to synthesize acylsilanes; indeed, stoichiometric organosilicon reagents18 or disilanes19 or stannylsilanes20 in the presence of catalytic palladium at 110 °C have been employed to synthesize acylsilanes. Despite the initial achievements, the synthesis of acylsilanes still involves a tedious protocol, as seen by the contemporary literature that uses the classical multistep 1,3-dithiane protocol,21 or inconvenient protocols.16a Our attempts to couple silylzinc (prepared from silyllithium) and acid chlorides yielded poor results, perhaps owing to the dissolved LiCl, emphasizing the need for a new and facile synthetic strategy.
Herein we describe the first direct preparation of silylzinc reagents from silyl iodides and subsequent cross-coupling reactions with in situ prepared acid chlorides to generate a library of acylsilanes with broad functional group tolerance. In addition, the newly developed solid Me3SiZnI was successfully employed in a broad range of organic transformations including alkylation, arylation, allylation, 1,4-addition, acylation, cross-coupling via C–N bond cleavage, Brook rearrangement and decarboxylation.
Results and discussion
We began our studies with silyl iodide 1a which was prepared from the corresponding PhMe2SiH.6,22 It has been reported in the literature that the activation of zinc is crucial and there are multiple methods known for the activation of zinc23 including HCl wash,24 Me3SiCl,25 iodine,26 ultrasound27 and dibromoethane activation.28 We have also followed these protocols for the activation of zinc and subjected them to reductive metalation to silyl iodide 1a, and the results are summarized in Table 1. As expected, no silylation was observed in the absence of an activator or additive (entry 1). Initially, activation was carried out with Me3SiCl (entries 2 and 5); later iodine (entries 3 and 6–8) and DBE (entry 5) were employed to activate Zn. In general, THF was not suitable (entries 1, 2, and 5), and the C–O bond in THF was cleaved under reflux conditions (Fig. 1 and S5–S7 in the ESI†); a similar observation was reported during the synthesis of silylmagnesium reagents.29 The group of Knochel extensively studied the role of LiCl in the preparation of organometallic reagents including organozinc reagents.12 The same group reported the combination of LiCl, dibromoethane and Me3SiCl which is highly efficient for the preparation of alkyl zinc reagents.12e Unfortunately, the inclusion of LiCl as an additive or DBE as an activator was inconsequential (entries 4 and 5).|
|
|||
|---|---|---|---|
| Entry | Activator | Additive, solvent, temperature | 2a (M) |
| a Reaction conditions: additive/activator: Me3SiCl (0.12 eq.), iodine (0.03/0.05 eq.), DBE (0.03 eq.), LiCl (1.0 eq.), TMEDA (1.1 eq.), NMP/DMA (1.5 eq.). b Me3SiCl (1.5 mol%) was used. c An average of 5 isolated runs. M: molarity, DBE: 1,2-dibromoethane, NMP: N-methyl-2-pyrrolidone, DMA: dimethylacetamide, ND: not detected. | |||
| 1 | — | THF, 60 °C | ND |
| 2 | TMSCl | THF, 60 °C/toluene, 70 °C | ND |
| 3 | Iodine | DMA, 80 °C/toluene, 70 °C | ND |
| 4 | — | LiCl, toluene, 70 °C | ND |
| 5 | DBE | LiCl, toluene, 70 °C/THF, 60 °C | NDb |
| 6 | Iodine | TMEDA, toluene, 66 °C, 63 h | 0.21 |
| 7 | Iodine | TMEDA, toluene, 70–90 °C, 63 h | 0.68c |
| 8 | Iodine | NMP/DMA, toluene, 85 °C | ND |

In the backdrop of our recent findings on the preparation of Me3SiMgI from Me3SiI30 and the literature reports on polar solvents such as dimethylacetamide,26,31 hexamethylphosphoramide,32N,N-dimethylformamide,26,27,31 dimethylsulfoxide,32 acetonitrile,33 tetramethylurea,27a CH3CN–pyridine33 and N-methyl-2-pyrrolidone (NMP27a) favoring the generation of organozinc reagents, we envisioned that amine additives may assist the formation of silylzinc reagents. Pleasingly, we observed a successful reductive metalation of zinc when an industrially friendly solvent,34 toluene, along with 1.1 eq. of TMEDA was employed as an additive (entry 6). An increase in the reaction temperature also increased the concentration of PhMe2SiZnI (entry 6 vs. 7). As shown in entry 8, no reactivity was observed with either NMP or DMA as additives. The methodology is not limited to the synthesis of PhMe2SiZnI 2a; even the relatively less reactive Me3SiI was successfully employed to generate more productive Me3SiZnI 2b with 0.56 M (average of 4 runs) concentration. Attempts to synthesize Et3SiZnI from Et3SiI were made, and although a concentration of 0.16 M was obtained for Et3SiZnI, subsequent application utilizing Et3SiZnI was unsuccessful. Using Me3SiBr instead of Me3SiI was likewise unsuccessful, and a poor concentration of 0.04 M was observed. Moreover, these silylzinc reagents (in toluene) stored at 4 °C did not decompose even after six months. Importantly, we obtained the structures of PhMe2SiZnI·TMEDA and Me3SiZnI·TMEDA from single crystal XRD for the first time (Fig. 2). It is apparent from these structures that TMEDA stabilizes 2a and 2b, the driving force for the reductive metalation.
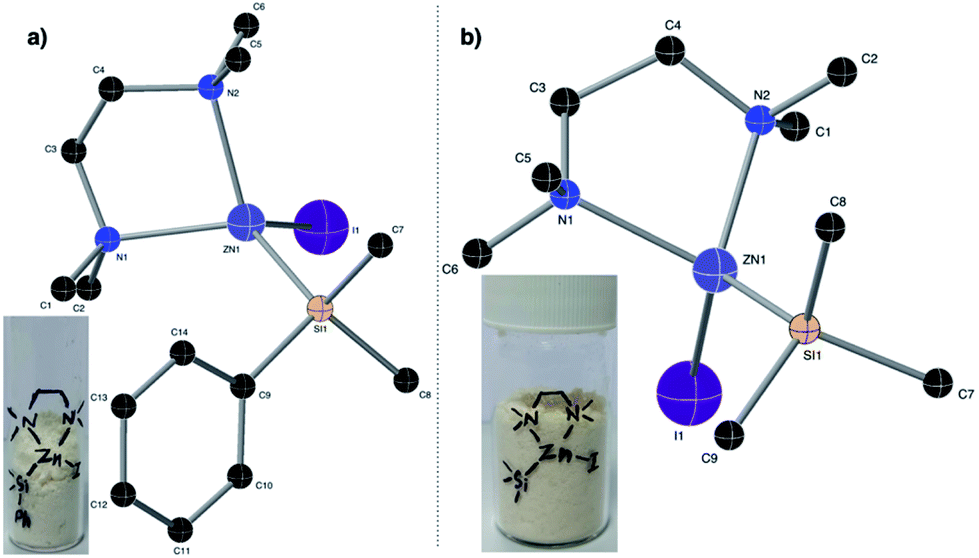 | ||
| Fig. 2 X-ray crystal structures of (a) PhMe2SiZnI·TMEDA and (b) Me3SiZnI·TMEDA complexes; the hydrogen atoms have been omitted for clarity. | ||
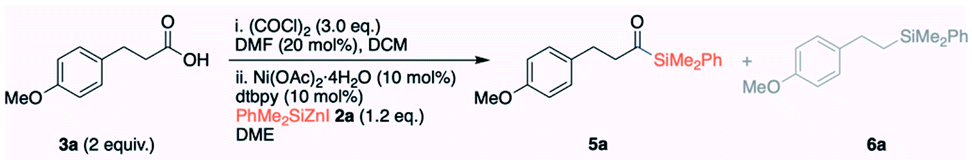
At the outset of our studies, we screened a range of nickel catalysts to obtain the acylsilane 5a (see ESI S11 and S12† for complete tables). The initial experiments were carried out with the isolated acid chloride 4a derived from 3-(4-methoxyphenyl)propanoic acid 3a; however, the later experiments (entries 8–16 and 20) were carried out with free carboxylic acid 3avia in situ generation of acid chloride 4a. When 10 mol% of NiBr2·diglyme was employed, we observed 60% of the cross-coupling product 5a (entry 1, Table 2). The yield of 5a was greatly improved by increasing the reaction temperature from 45 °C to 60 °C (entry 1 vs. 2). It is important to note that the activation of NiBr2·diglyme (10 mol%) requires 20 mol% of PhMe2SiZnI. The other nickel catalysts including NiCl2, Ni(acac)2 and NiBr2bpy afforded 5a in only 29%, 66% and 21% yields, with the hydrolyzed acid 3a being formed as the major byproduct (entries 3–5). To our surprise, the hydrated and air-stable Ni(OAc)2·4H2O afforded the cross-coupled product 5a in 86% yield (entry 6). A library of ligands was also screened: a simple bipyridine caused a significant reduction in the yield (entry 8), and TMEDA and dppe gave poor yields (entries 9 and 10). The non-polar toluene was not compatible (entry 11). A remarkable improvement in yield to 95% was obtained in DME (entry 12), although the dielectric constant (polarity) and coordinating ability of DME do not significantly vary from those of THF.
Pleasingly, the reaction can be performed at room temperature without a compromise in the yield (entry 13). Interestingly, 5 mol% of Ni(OAc)2·4H2O afforded the cross-coupled 5a in 86% isolated yield (entry 14). A further decrease in the loading of catalyst (2 mol%) decreased the yield of 5a (entry 15). When Ni(COD)2 was employed as a catalyst, 1.5 eq. of carboxylic acid 3a was sufficient to obtain excellent yield (entry 16). Virtually no cross-coupled product 5a was observed in the absence of Ni(OAc)2·4H2O (entry 17). Notably, (PhMe2Si)2Zn8a,h and PhMe2SiZnCl8j reagents prepared from silyllithium were found to be incompatible and product 5a was observed in only 23% yield (entry 18); the dissolved LiCl may be at play. The detrimental effect of LiCl was confirmed by the addition of 1 eq. of LiCl to the standard reaction that significantly reduced the yield (entry 19). To expand the synthetic utility, we also conducted the reaction on a 4.2 mmol scale using 5 mol% of Ni(OAc)2·4H2O and obtained 5a in 80% isolated yield (entry 20). We also found that the mode of addition is critical to obtain a consistent result; PhMe2SiZnI must be added dropwise at the end of sequential addition, and a change in the sequence resulted in a significant loss of yield.
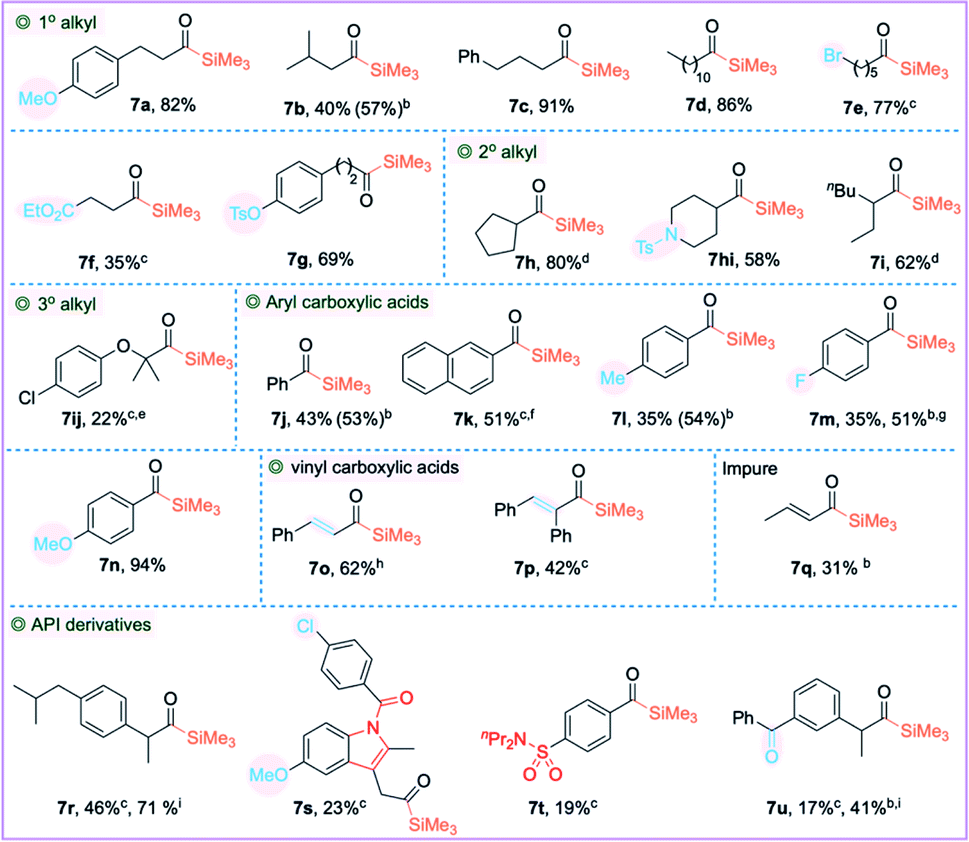
Having identified the optimal reaction conditions (Table 2, entry 14), we proceeded to examine the scope of carboxylic acids. The primary alkyl carboxylic acids 3a–3g including the long-chain alkyl carboxylic acid afforded the cross-coupled products 5a–5g in good to excellent yields. The α-branched secondary alkyl carboxylic acids 3h–3i had no impact on the efficiencies and afforded the corresponding acylsilanes 5h–5i in very good yields. Aryl carboxylic acids 3j–3no including π-extended naphthalene-2-carboxylic acid 3k and alkenyl carboxylic acids 3o–3q were also compatible, affording the corresponding acylsilanes in good yields. The moderate yield concerning 5bc, 5m, 5mn and 5r–5u can be attributed to the incomplete reaction, as we observed a significant amount of PhMe2SiH at the end of the reaction. We have also observed traces of the decarbonylated byproduct during the synthesis of acylsilanes 5mn, 5no, 5p, 5r and 5t–5u. Various functional groups such as ethers (3a, 3n, 3no, and 3s), alkyl/aryl bromides (3e and 3mn), chloride 3s and medicinally relevant fluoride 3m were tolerated, affording the corresponding cross-coupled products in good to high yields. Carbonyls did not impede the reactivity of silylzinc reagents; ester 3f, ketone 3u and amide 3s were compatible, despite their moderate yields. The O-tosyl protecting group 3g was also compatible. The structurally complex API substances did not impede the reaction; indomethacin 3s, probenecid 3t, anti-inflammatory drug ibuprofen 3r, and ketoprofen 3u afforded the corresponding silylated products 5r–5u. As we observed lower yields on a few substrates, we needed to design a complementary copper catalysis to obtain an increased yield. In place of Ni(OAc)2·4H2O, we employed catalytic CuI along with LiCl additive, which resulted in higher yields for substrates 5m and 5u. For substrate 5r, a dual system incorporating both Ni(OAc)2·4H2O and CuI resulted in a significant increase in yield (40% vs. 89%).
|
|
|||
|---|---|---|---|
| Entry | Deviation from the above | Temp., time | 5a (%) |
| a Reaction conditions: 0.39 mmol of 3a, 0.234 mmol of PhMe2SiZnI·TMEDA 2a (0.66 M in toluene), 0.0195 mmol of Ni(OAc)2·4H2O and dtbpy, 0.17 M (overall concentration). Isolated acid-chloride 4a was used in entries 1–7 and 17–19; in situ acid-chloride 4a was used in entries 8–16 and 20. b Yield determined by 1H NMR using 1,3,5-trimethoxy benzene as an internal standard. c 1.3 eq. of PhMe2SiZnI. d Incomplete reaction; unreacted starting material was observed in the crude NMR. e 1.0 eq. of PhMe2SiZnI was used. f Reactions were repeated at least 10 times throughout the project. DME: 1,2-dimethoxy ethane, yield in parentheses is isolated yield. | |||
| 1 | NiBr2·diglyme, THF | 45 °C, 4 h | 60c |
| 2 | NiBr2·diglyme, THF | 60 °C, 4 h | 92 |
| 3 | NiCl2, THF | 60 °C, 4 h | 29d |
| 4 | Ni(acac)2, THF | 60 °C, 4 h | 66 |
| 5 | NiBr2bpy, THF | 60 °C, 4 h | 21 |
| 6 | THF | 60 °C, 4 h | 86 |
| 7 | 20 mol% of CuI or CuCN | 60 °C, 4 h | 2 |
| 8 | bpy instead of dtbpy, THF | 60 °C, 4 h | 70d |
| 9 | TMEDA instead of dtbpy, THF | 60 °C, 4 h | 28d |
| 10 | dppe instead of dtbpy, THF | 60 °C, 4 h | 10 |
| 11 | Toluene | 60 °C, 4 h | 38 |
| 12 | None | 60 °C, 4 h | 95 |
| 13 | None | rt, 12 h | 90 |
| 14 | 5 mol% Ni(OAc)2·4H2O | rt, 12 h | 94 (86)f |
| 15 | 2 mol% Ni(OAc)2·4H2O | rt, 12 h | 52 |
| 16 | Ni(COD)2 (1.5 eq. 3a), THF | rt, 12 h | 95 |
| 17 | Without Ni(OAc)2·4H2O | rt, 12 h | 2 |
| 18 | Silylzinc from PhMe2SiLi | rt, 12 h | 23 |
| 19 | 1 eq. LiCl additive | rt, 12 h | 42 |
| 20 | 4.2 mmol of 2a | rt, 12 h | (80) |
Importantly, the reaction is also compatible with Me3SiZnI 2b, affording the widely used TMS derived acylsilanes 7 in good to excellent yields. It was necessary to increase the loading of the catalyst to 10 mol% to improve the yield. Similar to PhMe2SiZnI 2a, Me3SiZnI 2b was also an efficient reagent (Table 4): primary and secondary alkyl carboxylic acids 3a–3i with various functional groups afforded the cross-coupled products 7a–7i in good to excellent yields. Aryl and vinyl carboxylic acids 3j–3q were also compatible to yield the corresponding acylsilanes 7j–7q in good yields. The API derivatives 7r–7u were also silylated albeit in lower to moderate yields. In addition, the sterically hindered tertiary alkyl carboxylic acid 3ij afforded acylsilane 7ij in lower yield; an incomplete reaction and decarbonylative protonation may account for the lower yield. Both Ni(OAc)2·4H2O and Ni(COD)2 afforded the acylsilane 7ij in similar yields. N-Tosyl protected isonipecotic acid 3hi was compatible affording the acylsilane 7hi in 58% isolated yield. In general, aryl carboxylic acids gave traces of decarbonylated products and increasing the reaction temperature further increased the undesired decarbonylative silylation. It is worth noting that the decarbonylation can generate catalytically inactive LnNi(CO)x species.35 Additionally, copper catalysis was employed to improve the yield of product 7m. Importantly, the dual Ni(OAc)2/CuI catalytic system significantly improved the yields for substrates 7r and 7u. Gram-scale synthesis of acylsilanes 5e (3.5 mmol), 7a (5 mmol), and 7e (4 mmol) was promising with 82%, 70% and 76% isolated yields. Additional screening using Ni(COD)2 and anhydrous Ni(OAc)2 was done for the low yielding substrates, but no further improvement was observed.
A hypothetical mechanism is shown in Fig. 3. When Ni(OAc)2 was exposed to Me3SiZnI or PhMe2SiZnI, we observed the formation of disilane with the concomitant formation of a low valent nickel complex, I. It has been reported in the literature that the use of organozinc reagents in cross-coupling reactions can lead to either Ni(0)12b,36 or Ni(I)37 intermediate complexes. The low valent nickel complex I undergoes oxidative addition with the in situ generated acid chloride to generate the intermediate complex II. Subsequent transmetallation to intermediate III and reductive elimination affords the acylated product 5 or 7 while regenerating the active catalyst I.
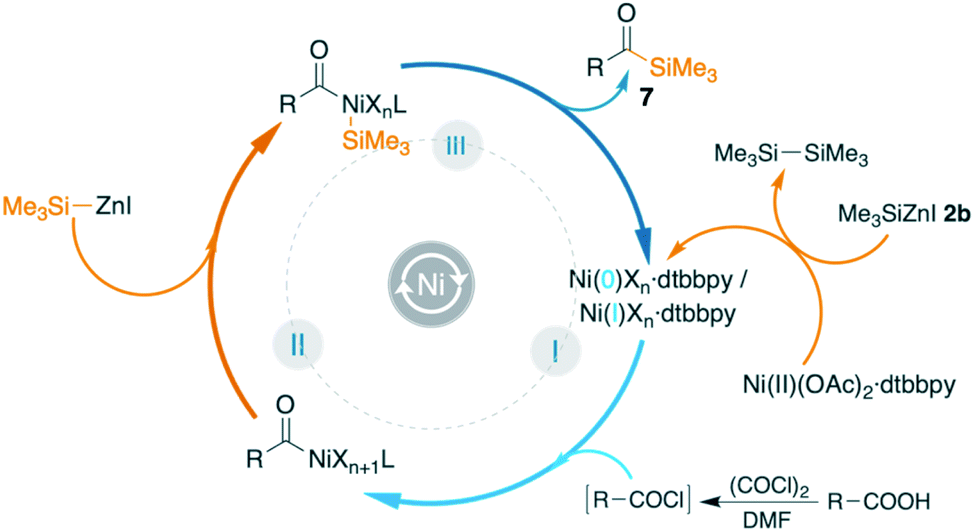 | ||
| Fig. 3 Mechanistic hypothesis. | ||
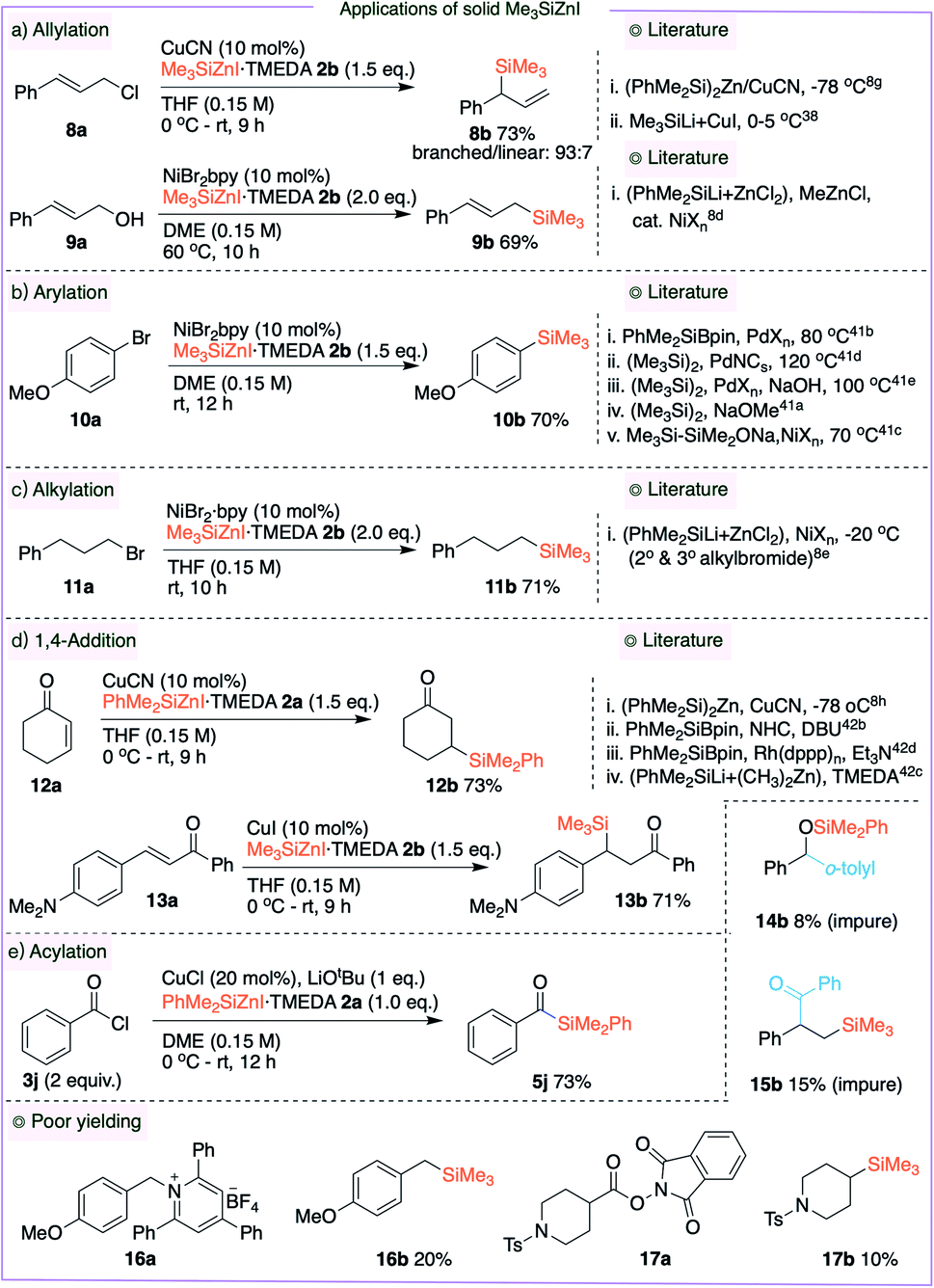
In Tables 2–4, the silylzinc reagents 2a and 2b were employed as 0.68 M and 0.56 M solutions, respectively. While studying the stability of these reagents, we discovered that they are quite stable in the form of a solid. To study the general applicability of this new solid Me3SiZnI 2b, we performed a wide range of organic transformations and the results are summarized in Fig. 4. Initially, both branched and linear allylsilanes 8b and 9b were synthesized through allylation. In the presence of catalytic CuCN, allyl chloride 8a was treated with 2b to generate the branched allylated product 8b in 73% isolated yield.8g,38 Using allylic alcohol 9a in the presence of a nickel catalyst, the linear silane 9b was likewise produced in 69% yield as a single isomer.8d,39 Because aryl silanes are commonly used in synthetic applications,40 we employed 2b in a nickel-mediated cross-coupling process of aryl bromide 10a, which resulted in the synthesis of aryl silane 10b in 70% isolated yield.41 Cross-coupling of a 2o-alkyl bromide and PhMe2SiZnCl was recently realized,8e and we were ecstatic to see that the new reagent 2b was effective in undergoing the cross-coupling reaction with 1o-alkyl bromide 11a, yielding the alkyl silane 11b in a 71% isolated yield. Fortunately, the copper-mediated 1,4-addition of 2a and 2b with enones 12a and 13a yielded the β-silyl ketones 12b and 13b in 73% and 71% isolated yields, respectively.8h,42 Acylsilane 5j was also synthesized by coupling of acid chloride 3j and 2a in the presence of CuCl. We also employed 2a in a novel Brook-rearrangement/cross-coupling of benzaldehyde and bromoarene;43 however, 14b was observed in poor yield. A similar result was obtained in a multi-component reaction between styrene, acid chloride and 2b to yield product 15b.44 Pyridinium salt 16a and redox ester 17a underwent cross-coupling reactions with 2b to afford alkyl silanes 16b and 17b in only 20% and 10% isolated yields respectively, indicating the need for dedicated studies. Samples of solid Me3SiZnI 2b were stored at three different temperatures (rt, 4 °C and −23 °C) for a period of two months and subjected in the cross-coupling of 10a to afford the aryl silane 10b. The silylzinc 2b samples stored at 4 °C and −23 °C were equally effective as the freshly prepared reagent; however, Me3SiZnI (two different batches) stored at room temperature gave poor yield (ESI S52†). It is worth noting that Apeloig and Zhivotovskii et al. described the synthesis of solid [(Me3Si)3Si]2Zn (stored in a glovebox),45 which was later used in silylzincation of alkynes.46
|
|
|---|
| a Reaction conditions: 2 eq. of 3, 1.1 eq. of PhMe2SiZnI·TMEDA 2a (0.66 M in toluene), 5 mol% of Ni(OAc)2·4H2O, 5 mol% of dtbpy, 1,2-dimethoxy ethane (0.17 M). b Unreactive PhMe2SiZnI·TMEDA was observed as PhMe2SiH. c NMR yield. d Conditions: 2 eq. of 3, 1 eq. of PhMe2SiZnI·TMEDA (0.71 M in toluene), 20 mol% of CuI, 1 eq. of LiCl, 1,2-dimethoxy ethane (0.15 M overall concentration), 0 °C – rt. e A decarbonylative byproduct was observed. f 10 mol% Ni(OAc)2·4H2O, 10 mol% 1,10-phen, and 20 mol% of CuI were added. g The Brook rearrangement was observed. |

|

|
|
|---|
| a 2 eq. of 3; 1.2 eq. of Me3SiZnI·TMEDA 2b (0.56 M in toluene, 1.19 mmol); 10 mol% Ni(OAc)2·4H2O; 10 mol% dtbpy; 1,2-dimethoxy ethane (0.17 M). b NMR yield. c A decarbonylative byproduct was observed. d Dimerization of 3 to a diketone was observed. e Ni(COD)2 (10 mol%) instead of Ni(OAc)2·4H2O. f Decomposition on silica was observed. g Conditions: 2 eq. of 3, 1eq. of PhMe2SiZnI·TMEDA (0.71 M in toluene), 20 mol% of CuI, 1 eq. of LiCl, 1,2-dimethoxy ethane (0.15 M overall concentration), 0 °C – rt. h 6 mmol of 2b was employed. i 10 mol% Ni(OAc)2·4H2O, 10 mol% 1,10-phen, 20 mol% of CuI was added. |
|
 | ||
| Fig. 4 Synthetic applications of solid Me3SiZnI. | ||
Conclusions
In summary, for the first time, we have developed a method for direct synthesis of silylzinc reagents from silyl iodides and their structures were confirmed by single-crystal XRD. Unlike the use of pyrophoric silyllithium in the synthesis of silylzinc reagents, the current method offers a simplified direct method to access them from silyl halides. The absence of dissolved lithium/magnesium salts in these reagents could be beneficial for various chemical processes. We have also demonstrated the practical synthesis of acylsilanes from unactivated alkyl acid chlorides by nickel, copper and dual catalysis. The methodology is compatible with various functional groups, and accommodates sterically hindered secondary and tertiary alkyl carboxylic acids. Structurally complex and API molecules are also silylated conveniently. The general synthetic utility of these reagents is shown in a broad range of reactions. Further study to expand the synthetic scope of these reagents is currently underway in our laboratory.Author contributions
The manuscript was written through the contributions of all authors. R. C., F. T. P. and K. S. performed the experiments. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge financial support from the Science and Engineering Research Board, Ramanujan Fellowship SB/S2/RJN059/2015, CSIR (Council of Scientific and Industrial Research, 02(0409)/21/EMR-II) and IISER Trivandrum. RC acknowledges CSIR, FTP acknowledges IISER Trivandrum and KS acknowledges DST-Inspire for fellowships.Notes and references
- (a) A. Franz and S. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed; (b) P. L. Fuchs, Handbook of Reagents For Organic Synthesis, Reagents For Silicon-Mediated Organic Synthesis, John Wiley & Sons, 2011 Search PubMed; (c) Y. Apeloig, The Chemistry of Organic Silicon Compounds, Wiley, 1989 Search PubMed.
- (a) Y. Minami and T. Hiyama, Chem.–Eur. J., 2019, 25, 391 CrossRef CAS PubMed; (b) S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16 CrossRef; (c) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402 CrossRef CAS PubMed; (d) L. Xu, L. Li, G. Lai and J. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC; (e) T. H. Chan and D. Wang, Chem. Rev., 1992, 92, 995 CrossRef CAS; (f) P. Xiao, L. Gao and Z. Song, Chem.–Eur. J., 2019, 25, 2407 CrossRef CAS PubMed; (g) A. Hameed, R. D. Alharthy, J. Iqbal and P. Langer, Tetrahedron, 2016, 72, 2763 CrossRef CAS; (h) M. Greenhalgh, Iron-catalysed hydrosilylation of alkenes and alkynes, Springer International Publishing, Cham, 2016, p. 33 Search PubMed; (i) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190 CrossRef CAS; (j) B. M. Trost and Z. T. Ball, Synthesis, 2005, 2005, 853 CrossRef; (k) M. G. Voronkov, V. M. Dyakov and S. V. Kirpichenko, J. Organomet. Chem., 1982, 233, 1 CrossRef CAS; (l) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed; (m) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000 Search PubMed.
- (a) J. P. Morken, in Comprehensive Organic Synthesis II, 2014, p. 939 Search PubMed; (b) I. Beletskaya and C. Moberg, Chem. Rev., 1999, 99, 3435 CrossRef CAS PubMed; (c) M. B. Ansell, O. Navarro and J. Spencer, Coord. Chem. Rev., 2017, 336, 54 CrossRef CAS; (d) M. Suginome and Y. Ito, J. Chem. Soc., Dalton Trans., 1998, 1925 RSC; (e) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221 CrossRef CAS PubMed.
- (a) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29 CrossRef CAS; (b) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010 RSC.
- (a) S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349 CrossRef PubMed; (b) B. Marciniec, Comprehensive Handbook on Hydrosilylation, Pergamon, 1992 Search PubMed; (c) B. Marciniec, Hydrosilylation, Springer, 2008 Search PubMed; (d) B. Marciniec and J. Guliński, J. Organomet. Chem., 1993, 446, 15 CrossRef CAS; (e) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603 RSC; (f) A. K. Roy, in Adv. Organomet. Chem., Elsevier, 2007, p. 1 Search PubMed; (g) J. L. Speier, in Catalysis and Organic Syntheses: Advances in Organometallic Chemistry, Elsevier, 1979, p. 407 Search PubMed.
- A. P. Cinderella, B. Vulovic and D. A. Watson, J. Am. Chem. Soc., 2017, 139, 7741 CrossRef CAS PubMed.
- J. Duan, K. Wang, G. L. Xu, S. Kang, L. Qi, X. Y. Liu and X. Z. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083 CrossRef CAS PubMed.
- (a) M. Oestreich, G. Auer and B. Weiner, Synthesis, 2006, 2006, 2113 CrossRef; (b) A. Hensel and M. Oestreich, Chem.–Eur. J., 2015, 21, 9062 CrossRef CAS PubMed; (c) Y. Y. Kong and Z. X. Wang, Adv. Synth. Catal., 2019, 361, 5440 CrossRef CAS; (d) B. Yang and Z.-X. Wang, Org. Lett., 2019, 21, 7965 CrossRef CAS PubMed; (e) C. Chu, Y. Liang and G. Fu, J. Am. Chem. Soc., 2016, 138, 6404 CrossRef CAS PubMed; (f) C. Fopp, E. Romain, K. Isaac, F. Chemla, F. Ferreira, O. Jackowski, M. Oestreich and A. Perez-Luna, Org. Lett., 2016, 18, 2054 CrossRef CAS PubMed; (g) D. Vyas and M. Oestreich, Chem. Commun., 2010, 46, 568 RSC; (h) M. Oestreich and B. Weiner, Synlett, 2004, 2139 CrossRef CAS; (i) L. Zhang and M. Oestreich, Org. Lett., 2018, 20, 8061 CrossRef CAS PubMed; (j) H. Azuma, K. Okano and H. Tokuyama, Chem. Lett., 2011, 40, 959 CrossRef CAS.
- Y. Morizawa, H. Oda, K. Oshima and H. Nozaki, Tetrahedron Lett., 1984, 25, 1163 CrossRef CAS.
- (a) C. K. Hazra and M. Oestreich, Org. Lett., 2012, 14, 4010 CrossRef CAS PubMed; (b) A. Perez-Luna, M. Oestreich, C. Fopp, K. Isaac, E. Romain, F. Chemla, F. Ferreira and O. Jackowski, Synthesis, 2017, 49, 724 Search PubMed.
- (a) W. C. Still, J. Org. Chem., 1976, 41, 3063 CrossRef CAS; (b) E. Yamamoto, S. Ukigai and H. Ito, Synlett, 2017, 28, 2460 CrossRef CAS.
- (a) P. Knochel, M. Schade, S. Bernhardt, G. Manolikakes, A. Metzger, F. Piller, C. Rohbogner and M. Mosrin, Beilstein J. Org. Chem., 2011, 7, 1261 CrossRef CAS PubMed; (b) X. Wu, X. Li, W. Huang, Y. Wang, H. Xu, L. Cai, J. Qu and Y. Chen, Org. Lett., 2019, 21, 2453 CrossRef CAS PubMed; (c) D. Wang, H. Niu, M. Xie, G. Qu, H. Wang and H. Guo, Org. Lett., 2014, 16, 262 CrossRef CAS PubMed; (d) S. J. Edeson, E. J. M. Maduli, S. Swanson, P. A. Procopiou and J. P. A. Harrity, Eur. J. Org. Chem., 2016, 2016, 83 CrossRef CAS; (e) A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040 CrossRef CAS PubMed; (f) C. Feng, D. Cunningham, Q. Easter and S. Blum, J. Am. Chem. Soc., 2016, 138, 11156 CrossRef CAS PubMed.
- (a) X. Wang, F. Liu, Y. Li, Z. Yan, Q. Qiang and Z. Q. Rong, ChemCatChem, 2020, 12, 5022 CrossRef CAS; (b) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540 RSC; (c) G. Boyce, S. Greszler, J. Johnson, X. Linghu, J. Malinowski, D. Nicewicz, A. Satterfield, D. Schmitt and K. Steward, J. Org. Chem., 2012, 77, 4503 CrossRef CAS PubMed; (d) W. H. Moser, Tetrahedron, 2001, 57, 2065 CrossRef CAS; (e) P. C. B. Page, S. S. Klair and S. Rosenthal, Chem. Soc. Rev., 1990, 19, 147 RSC; (f) A. G. Brook, Acc. Chem. Res., 1974, 7, 77 CrossRef CAS.
- (a) J. Feng and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 8211 CrossRef CAS PubMed; (b) J. Rong, R. Oost, A. Desmarchelier, A. Minnaard and S. Harutyunyan, Angew. Chem., Int. Ed., 2015, 54, 3038 CrossRef CAS PubMed; (c) J. González, J. Santamaría and A. Ballesteros, Angew. Chem., Int. Ed., 2015, 54, 13678 CrossRef PubMed; (d) R. Lettan, C. Galliford, C. Woodward and K. Scheidt, J. Am. Chem. Soc., 2009, 131, 8805 CrossRef CAS PubMed; (e) R. B. Lettan, T. E. Reynolds, C. V. Galliford and K. A. Scheidt, J. Am. Chem. Soc., 2006, 128, 15566 CrossRef CAS PubMed.
- (a) J. R. Schmink and S. W. Krska, J. Am. Chem. Soc., 2011, 133, 19574 CrossRef CAS PubMed; (b) S. Ramgren and N. Garg, Org. Lett., 2014, 16, 824 CrossRef CAS PubMed.
- (a) X. Lu, J. Zhang, L. Xu, W. Shen, F. Yu, L. Ding and G. Zhong, Org. Lett., 2020, 22, 5610 CrossRef CAS PubMed; (b) M. Sasaki, Y. Kondo, T. Moto-ishi, M. Kawahata, K. Yamaguchi and K. Takeda, Org. Lett., 2015, 17, 1280 CrossRef CAS PubMed; (c) Y. Matsuya, K. Wada, D. Minato and K. Sugimoto, Angew. Chem., Int. Ed., 2016, 55, 10079 CrossRef CAS PubMed; (d) M. Decostanzi, J. Godemert, S. Oudeyer, V. Levacher, J.-M. Campagne and E. Leclerc, Adv. Synth. Catal., 2016, 358, 526 CrossRef CAS; (e) J.-H. Ye, L. Quach, T. Paulisch and F. Glorius, J. Am. Chem. Soc., 2019, 141(41), 16227 CrossRef CAS PubMed; (f) B. Huang, M. Wei, E. Vargo, Y. Qian, T. Xu and F. D. Toste, J. Am. Chem. Soc., 2021, 143, 17920 CrossRef CAS PubMed; (g) C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18605 CrossRef CAS PubMed.
- (a) D. Priebbenow, J. Org. Chem., 2019, 84, 11813 CrossRef CAS PubMed; (b) F. Tang, P.-J. Ma, Y. Yao, Y.-J. Xu and C.-D. Lu, Chem. Commun., 2019, 55, 3777 RSC; (c) H. J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F. F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157 CrossRef CAS; (d) K. Ito, H. Tamashima, N. Iwasawa and H. Kusama, J. Am. Chem. Soc., 2011, 133, 3716 CrossRef CAS PubMed; (e) M. Sasaki, Y. Kondo, M. Kawahata, K. Yamaguchi and K. Takeda, Angew. Chem., Int. Ed., 2011, 50, 6375 CrossRef CAS PubMed; (f) J. Chen, C. Ding, W. Liu, X. Hou and L. Dai, J. Am. Chem. Soc., 2010, 132, 15493 CrossRef CAS PubMed; (g) R. Unger, F. Weisser, N. Chinkov, A. Stanger, T. Cohen and I. Marek, Org. Lett., 2009, 11, 1853 CrossRef CAS PubMed; (h) S. N. Greszler and J. S. Johnson, Org. Lett., 2009, 11, 827 CrossRef CAS PubMed; (i) R. Lettan, C. Woodward and K. Scheidt, Angew. Chem., Int. Ed., 2008, 47, 2294 CrossRef CAS PubMed; (j) Y. Obora, Y. Ogawa, Y. Imai, T. Kawamura and Y. Tsuji, J. Am. Chem. Soc., 2001, 123, 10489 CrossRef CAS PubMed; (k) K. Takeda, M. Fujisawa, T. Makino, E. Yoshii and K. Yamaguchi, J. Am. Chem. Soc., 1993, 115, 9351 CrossRef CAS; (l) H. J. Reich, J. J. Rusek and R. E. Olson, J. Am. Chem. Soc., 1979, 101, 2225 CrossRef CAS; (m) A. G. Brook, J. Am. Chem. Soc., 1958, 80, 1886 CrossRef CAS; (n) N. Lee, C.-H. Tan and D. Leow, Asian J. Org. Chem., 2019, 8, 25 CrossRef CAS.
- (a) K. Jahyo, L. Jae Hyoung, K. Koan Seong, J. Jae Uk and P. Chongsuh, Tetrahedron Lett., 1987, 28, 3261 CrossRef; (b) J. P. Picard, R. Calas, J. Dunogues, N. Duffaut, J. Gerval and P. Lapouyade, J. Org. Chem., 1979, 44, 420 CrossRef CAS; (c) I. Fleming and U. Ghosh, J. Chem. Soc., Perkin Trans. 1, 1994, 257 RSC; (d) A. Capperucci, A. Degl'Innocenti, C. Faggi, A. Ricci, P. Dembech and G. Seconi, J. Org. Chem., 1988, 53, 3612 CrossRef CAS; (e) B. F. Bonini, M. Comes-Franchini, G. Mazzanti, U. Passamonti, A. Ricchi and P. Zani, Synthesis, 1995, 1995, 92 CrossRef.
- (a) K. Yamamoto, S. Suzuki and J. Tsuji, Tetrahedron Lett., 1980, 21, 1653 CrossRef CAS; (b) K. Yamamoto, A. Hayashi, S. Suzuki and J. Tsuji, Organometallics, 1987, 6, 974 CrossRef CAS.
- F. Geng and R. E. Maleczka Jr, Tetrahedron Lett., 1999, 40, 3113 CrossRef CAS.
- (a) B. Wu and R. Zhu, ACS Catal., 2020, 10, 510 CrossRef CAS; (b) X. Deng, G. Zhou, J. Tian and R. Srinivasan, Angew. Chem., Int. Ed., 2020, 22, 7024 Search PubMed.
- A. Kunai, T. Sakurai, E. Toyoda, M. Ishikawa and Y. Yamamoto, Organometallics, 1994, 13, 3233 CrossRef CAS.
- (a) L. Zhu, R. M. Wehmeyer and R. D. Rieke, J. Org. Chem., 1991, 56, 1445 CrossRef CAS; (b) E. Erdik, Tetrahedron, 1987, 43, 2203 CrossRef CAS.
- M. S. Newman and F. J. Evans, J. Am. Chem. Soc., 1955, 77, 946 CrossRef CAS.
- (a) G. Picotin and P. Miginiac, J. Org. Chem., 1987, 52, 4796 CrossRef CAS; (b) J. K. Gawroński, Tetrahedron Lett., 1984, 25, 2605 CrossRef.
- S. Huo, Org. Lett., 2003, 5, 423 CrossRef CAS PubMed.
- (a) K. Takagi, Chem. Lett., 1993, 22, 469 CrossRef; (b) K. Takagi, Y. Shimoishi and K. Sasaki, Chem. Lett., 1994, 23, 2055 CrossRef; (c) R. F. Abdulla, Aldrichimica Acta, 1988, 21, 31 CAS.
- M. Bellassoued, M. Gaudemar, A. El Borgi and B. Baccar, J. Organomet. Chem., 1985, 280, 165 CrossRef CAS.
- R. Goddard, C. Krüger, N. A. Ramadan and A. Ritter, Angew. Chem., Int. Ed., 1995, 34, 1030 CrossRef CAS.
- V. Murugesan, V. Balakrishnan and R. Rasappan, J. Catal., 2019, 377, 293 CrossRef CAS.
- T. N. Majid and P. Knochel, Tetrahedron Lett., 1990, 31, 4413 CrossRef CAS.
- K. Takagi, N. Hayama and S. Inokawa, Bull. Chem. Soc. Jpn., 1980, 53, 3691 CrossRef CAS.
- (a) C. Gosmini, M. Amatore, S. Claudel and J. Périchon, Synlett, 2005, 2171 CrossRef CAS; (b) I. Kazmierski, C. Gosmini, J.-M. Paris and J. Périchon, Tetrahedron Lett., 2003, 44, 6417 CrossRef CAS.
- (a) D. S. Ziegler, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 6701 CrossRef CAS PubMed; (b) M. Balkenhohl, D. Ziegler, A. Desaintjean, L. Bole, A. Kennedy, E. Hevia and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 12898 CrossRef CAS PubMed.
- C. A. Malapit, J. R. Bour, S. R. Laursen and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 17322 CrossRef CAS PubMed.
- (a) J. Hu, H. Wu, C. Li, W. Sheng, Y. Jia and J. Gao, Chemistry, 2011, 17, 5234 CrossRef CAS PubMed; (b) R. Jana, T. Pathak and M. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- (a) G. Schwarzwalder, C. Matier and G. Fu, Angew. Chem., Int. Ed., 2019, 58, 3571 CrossRef CAS PubMed; (b) R. Soler-Yanes, I. Arribas-Álvarez, M. Guisán-Ceinos, E. Buñuel and D. J. Cárdenas, Chem.–Eur. J., 2017, 23, 1584 CrossRef CAS PubMed; (c) A. Oelke, J. Sun and G. Fu, J. Am. Chem. Soc., 2012, 134, 2966 CrossRef CAS PubMed; (d) V. Phapale, E. Buñuel, M. García-Iglesias and D. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790 CrossRef CAS PubMed; (e) G. Jones, J. Martin, C. McFarland, O. Allen, R. Hall, A. Haley, R. Brandon, T. Konovalova, P. Desrochers, P. Pulay and D. Vicic, J. Am. Chem. Soc., 2006, 128, 13175 CrossRef CAS PubMed.
- J. G. Smith, S. E. Drozda, S. P. Petraglia, N. R. Quinn, E. M. Rice, B. S. Taylor and M. Viswanathan, J. Org. Chem., 1984, 49, 4112 CrossRef CAS.
- R. Moser, T. Nishikata and B. H. Lipshutz, Org. Lett., 2009, 12, 28 CrossRef PubMed.
- (a) L. T. Ball, M. Green, G. C. Lloyd-Jones and C. A. Russell, Org. Lett., 2010, 12, 4724 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem.–Eur. J., 2012, 18, 2931 CrossRef CAS PubMed; (c) J. Xie, K. Sekine, S. Witzel, P. Krämer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16648 CrossRef CAS PubMed; (d) K. Gondo, J. Oyamada and T. Kitamura, Org. Lett., 2015, 17, 4778 CrossRef CAS PubMed; (e) M. J. Harper, E. J. Emmett, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2017, 139, 12386 CrossRef CAS PubMed; (f) W. E. Brenzovich, J.-F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728 CrossRef CAS PubMed; (g) M. Shibata, H. Ito and K. Itami, J. Am. Chem. Soc., 2018, 140, 2196 CrossRef CAS PubMed; (h) K. Funaki, T. Sato and S. Oi, Org. Lett., 2012, 14, 6186 CrossRef CAS PubMed.
- (a) A. Postigo and R. A. Rossi, Org. Lett., 2001, 3, 1197 CrossRef CAS PubMed; (b) H. Guo, X. Chen, C. Zhao and W. He, Chem. Commun., 2015, 51, 17410 RSC; (c) K. Hitoshio, H. Yamagishi, J. Shimokawa and H. Yorimitsu, Chem. Commun., 2021, 57, 6867 RSC; (d) T. Nagata, T. Inoue, X. Lin, S. Ishimoto, S. Nakamichi, H. Oka, R. Kondo, T. Suzuki and Y. Obora, RSC Adv., 2019, 9, 17425 RSC; (e) E. Shirakawa, T. Kurahashi, H. Yoshida and T. Hiyama, Chem. Commun., 2000, 1895 RSC.
- (a) G. Auer and M. Oestreich, Chem. Commun., 2006, 311 RSC; (b) J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712 CrossRef PubMed; (c) A. Vaughan and R. D. Singer, Tetrahedron Lett., 1995, 36, 5683 CAS; (d) C. Walter, G. Auer and M. Oestreich, Angew. Chem., Int. Ed., 2006, 45, 5675 CrossRef CAS PubMed; (e) T. Kitanosono, L. Zhu, C. Liu, P. Xu and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 15422 CrossRef CAS PubMed; (f) K.-s. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898 CrossRef CAS PubMed.
- M. Takeda, K. Yabushita, S. Yasuda and H. Ohmiya, Chem. Commun., 2018, 54, 6776 RSC.
- D. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858 CrossRef CAS.
- R. Dobrovetsky, Y. Kratish, B. Tumanskii, M. Botoshansky, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2012, 51, 4671 CrossRef CAS PubMed.
- E. Romain, K. Vega-Hernández, F. Guégan, J. Sanz García, C. Fopp, F. Chemla, F. Ferreira, H. Gerard, O. Jackowski, S. Halbert, M. Oestreich and A. Perez-Luna, Adv. Synth. Catal., 2021, 363, 2634 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2089058 and 2089062. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06038d |
| This journal is © The Royal Society of Chemistry 2021 |