
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect photo-induced reductive Heck cyclization of indoles for the efficient preparation of polycyclic indolinyl compounds†
Daohong
Yu
 abc,
Wai-Pong
To
b,
Yungen
Liu
a,
Liang-Liang
Wu
b,
Tingjie
You
b,
Jesse
Ling
e and
Chi-Ming
Che
*abde
abc,
Wai-Pong
To
b,
Yungen
Liu
a,
Liang-Liang
Wu
b,
Tingjie
You
b,
Jesse
Ling
e and
Chi-Ming
Che
*abde
aDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China. E-mail: cmche@hku.hk
bState Key Laboratory of Synthetic Chemistry, HKU-CAS Joint Laboratory on New Materials, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
cKey Laboratory of Organo-Pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou, 341000, China
dHKU Shenzhen Institute of Research and Innovation, Shenzhen, Guangdong 518057, China
eLaboratory for Synthetic Chemistry and Chemical Biology Limited, Units 1503-1511, 15/F, Building 17W, Hong Kong Science Park, New Territories, Hong Kong, China
First published on 16th September 2021
Abstract
The photo-induced cleavage of C(sp2)–Cl bonds is an appealing synthetic tool in organic synthesis, but usually requires the use of high UV light, photocatalysts and/or photosensitizers. Herein is described a direct photo-induced chloroarene activation with UVA/blue LEDs that can be used in the reductive Heck cyclization of indoles and without the use of a photocatalyst or photosensitizer. The indole compounds examined display room-temperature phosphorescence. The photochemical reaction tolerates a panel of functional groups including esters, alcohols, amides, cyano and alkenes (27 examples, 50–88% yields), and can be used to prepare polycyclic compounds and perform the functionalization of natural product analogues in moderate to good yields. Mechanistic experiments, including time-resolved absorption spectroscopy, are supportive of photo-induced electron transfer between the indole substrate and DIPEA, with the formation of radical intermediates in the photo-induced dearomatization reaction.
Introduction
Aryl halides are commonly used as building blocks in organic synthesis.1 Due to the high negative reduction potential and bond energy of C(sp2)–Cl bonds [e.g., for PhCl, Ered(PhCl/PhCl˙−) = −2.78 V vs. SCE;2 BDE (C–Cl) = 97.1 kcal mol−1],3 the reduction of aryl chloride to give an aryl radical usually requires harsh conditions, such as a strong base under thermal conditions4 or ultraviolet (UV) light irradiation5 is usually used. Highly reactive organic super-electron donors,6 consecutive visible light-induced electron transfer reactions7 and electrophotocatalysis8 are also employed to activate aryl chlorides, via one-electron reduction. Recent studies on photochemical C–Cl bond activation often entailed the use of a photocatalyst that initiates the reaction by electron transfer upon light irradiation.9For decades, UV light-excitation of aryl halides has been known to induce homolytic carbon–halide bond cleavage and lead to cyclization if an arene/alkene is in close proximity.10 Nonetheless, application studies of this strategy in the synthesis of indoles with complexity are sparse, as the decomposition of indole, photo-Fries rearrangement and photo-dehalogenation were usually observed, leading to low product yields and undesirable regioselectivity.10b,c,g Tin and radical initiators or transition metal catalysts are usually required for free radical cyclization10 or reductive Heck cyclization,11 in which the C–Br or C–I bond is involved (Scheme 1a). Reactions involving the C–Cl bond are comparatively less developed and they often entail an electron-rich phosphine ligand and high temperature.11c,f–i Reductive Heck cyclization without a radical initiator and transition metal catalyst has not been reported so far.
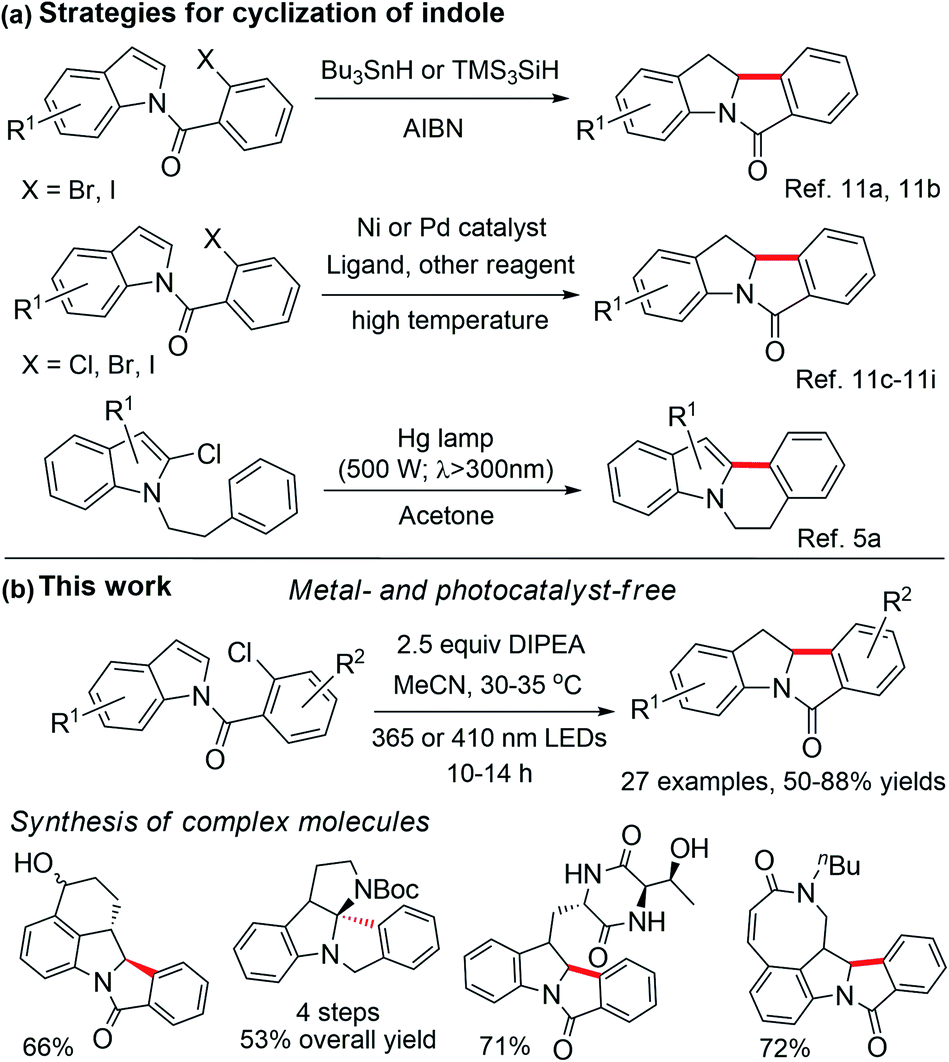 | ||
| Scheme 1 Reported strategies for (a) the cyclization of indole. (b) Our metal- and photocatalyst-free strategy for chloroarene activation and dearomatization of indole, and synthesis of complex molecules. | ||
We conjecture that organic compounds which could attain a reactive, triplet excited state in solution can be harnessed for achieving photoredox reactions without the need for a photocatalyst/photosensitizer. During our study on organic amide compounds, we found that N-(2-chlorobenzoyl)indoles display long-lived phosphorescence at room temperature, suggesting that this class of compounds may attain long-lived excited states in solution.12 Since carbonyl compounds exhibit rich photochemistry,13–16 we examined the photochemical reactivity of these N-benzoylindoles and discovered that they can undergo efficient photocyclization to give indolinyl compounds in the presence of a base. Herein, we describe a general light-induced Heck cyclization of aryl chlorides with broad substrate scope and its synthetic applications (Scheme 1b). During the preparation of the present manuscript, photo-induced [2 + 2] cycloaddition and reductive arylcarboxylation of indole derivatives which require the use of a photosensitizer were reported by several groups.17
Results and discussion
Considering both the halogen atom effect and n → π* transition of carbonyl groups in facilitating singlet-to-triplet intersystem crossing,18 we synthesized a class of indole compounds containing a 2-chlorobenzoyl group (e.g., N-(2-chlorobenzoyl)indole; S1). Upon excitation at 355 nm, S1 exhibits a broad, structureless emission band with λmax at ∼435 nm and decay time constants (τ) of 4.7 and 22 μs in the solid state at room temperature (Fig. 1). The pulse excitation (355 nm) of S1 at 77 K results in a short (∼0.2 s) blue afterglow visible to the naked eye. In 2-methyltetrahydrofuran at 77 K, the decay of the blue afterglow of S1 is lengthened to a few seconds (Fig. S3†). Nanosecond time-resolved absorption difference (ns-TA) spectroscopic measurement on S1 in MeCN revealed a positive ΔO.D. signal from 380 to ∼550 nm, which decays to the baseline in 1 μs (Fig. 1). Similarly, N-(2-chlorobenzoyl)-5-chloroindole (S3) in the solid form exhibits fluorescence with a peak maximum at 453 nm (τ = 58 ns; Fig. S4 and S5†) and a structured emission profile (500–700 nm), which slowly fades with a lifetime of 111 ms (Fig. S5†). The ns-TA spectra of S3 in degassed MeCN revealed a long-lived signal, which persists for at least 10 μs (Fig. S6†). The above two examples suggest that this class of amide compounds could attain a long-lived triplet excited state in solution upon light excitation.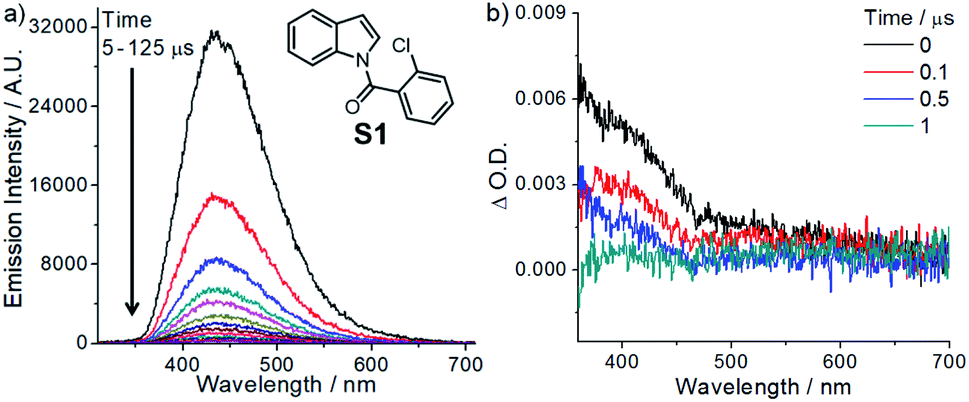 | ||
| Fig. 1 (a) Time-resolved emission spectra of S1 in the solid state (excitation, 355 nm; the arrow indicates the change in emission intensity). (b) ns-TA spectra of S1 (0.1 M) in degassed MeCN (excitation, 355 nm). | ||
We are intrigued to utilize the long-lived excited state of N-benzoylindoles for C(sp2)–Cl bond activation as proposed in Scheme 2. A long-lived excited state of N-(2-chlorobenzoyl)indole, A, undergoes a bimolecular redox reaction with the reductant (amine) to form the radical anion, B (reductive quenching by amine).19 This radical anion would undergo intramolecular C(sp2)–Cl bond activation, fragmenting into a phenyl radical C and chloride ion.20 The ensuing intramolecular radical cyclization of C generates benzylic radical D. Finally, the hydrogen atom abstraction21 with DIPEA˙+ or further reduction of D to carbanion E followed by protonation generates the reductive Heck cyclization product 1.
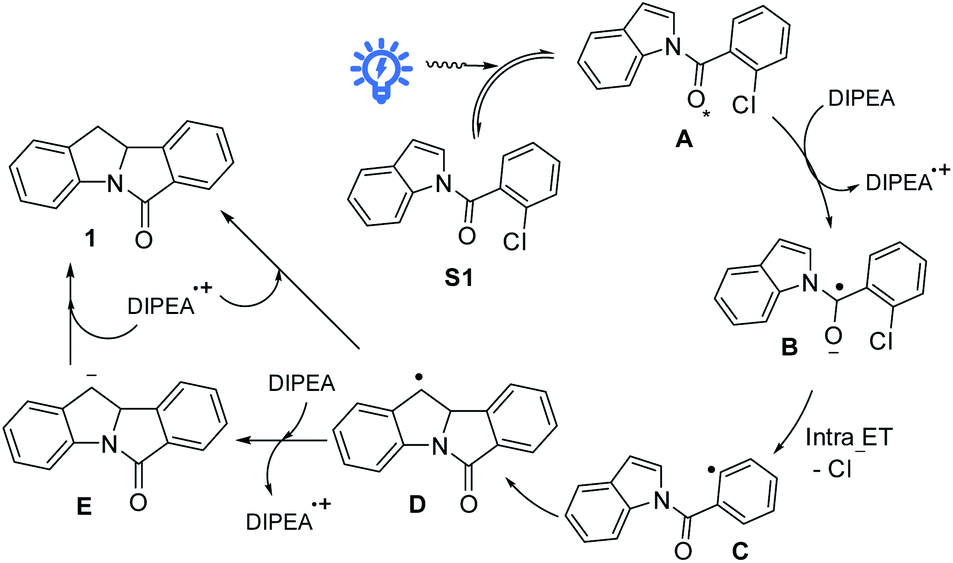 | ||
| Scheme 2 Proposed reaction pathway. | ||
We selected the reductive Heck cyclization of S1 under LED irradiation to examine the feasibility of photocyclization. Several sacrificial electron donors were screened. In the presence of 2.5 equivalents of N,N-diisopropylethylamine (DIPEA), the reaction proceeded smoothly with a 76% product yield under 410 nm LED irradiation (12 W; Fig. S1†), with an N-benzoylindole byproduct in 5% yield (Table 1, entry 1). The use of N,N,N′,N′-tetramethylethane-1,2-diamine (TMEDA) and Et3N also gave the product in 5–10% yields (Table 1, entries 2 and 3). Electron donors such as 1,4-diazabicyclo[2.2.2]octane (DABCO) or bases such as K2CO3 and pyridine did not afford the corresponding product, with S1 recovered quantitatively (Table 1, entries 4–7). As S1 was also fully recovered in the reaction without an additive (Table 1, entry 8), the reaction is unlikely to proceed via direct light-induced homolysis of S1. The product yield decreased to 21–33% and low substrate conversion was observed when DIPEA was decreased to 1 or fewer equivalents (Table S1, entries 12 and 13†). Shortening the reaction time was deleterious to the reaction (Table S1, entries 1–3†). The solvent effect was evaluated, with MeCN yielding the best results (Table S1, entries 4–11†). The reaction did not proceed when performed in the dark (Table 1, entry 9), or under 450 nm LEDs (Table 1, entry 10). Upon 365 nm LED irradiation for 12 h, the cyclized product was obtained in 80% yield (Table 1, entry 11), which is higher than that obtained with Ir(III) photocatalysts (Table S1, entries 18 and 19,† 50–59%). When the chlorine atom in substrate S1 was replaced by a bromo or iodo group, the reaction also proceeded to give 1 in 70% or 72% yield, respectively, under 410 nm LED irradiation (Table 2).
|
|
||||
|---|---|---|---|---|
| Entrya | Additive | X | Time (h) | Yield (%) |
| a S1 (0.2 mmol), DIPEA (0.5 mmol) and acetonitrile (2.0 mL), under argon and 410 LED irradiation at 30–35 °C for 8–12 hours. Yields were determined by 1H NMR using dibromomethane or 1,3,5-trimethoxybenzene as an internal standard. b The substrate was recovered completely. c Reaction conducted in the dark. d 450 nm LEDs were used. e 365 nm LEDs were used. f Under air. g Averaged isolated yield of three trials. | ||||
| 1 | DIPEA | 2.5 | 8 | 76 |
| 2 | TMEDA | 2.5 | 8 | 10 |
| 3 | Et3N | 2.5 | 8 | 5 |
| 4 | DABCO | 2.5 | 8 | 0b |
| 5 | K2CO3 | 2.5 | 8 | 0b |
| 6 | Me2S | 2.5 | 8 | 0b |
| 7 | Pyridine | 2.5 | 8 | 0b |
| 8 | — | — | 12 | 0b |
| 9c | DIPEA | 2.5 | 8 | 0b |
| 10d | DIPEA | 2.5 | 8 | 0b |
| 11e | DIPEA | 2.5 | 12 | 80 |
| 12e,f | DIPEA | 2.5 | 12 | 65 (52g) |
| a Substrate (0.5 mmol), DIPEA (1.25 mmol), MeCN (5 mL) under 410 nm LED irradiation at 30–35 °C for 10–14 h; isolated yield. b The reaction was irradiated with 365 nm LEDs. c Averaged yield of two trials in two independent laboratories. d N-(2,4,6-Trichlorobenzoyl)indole as the substrate. e X = Cl unless otherwise noted. |
|---|
|
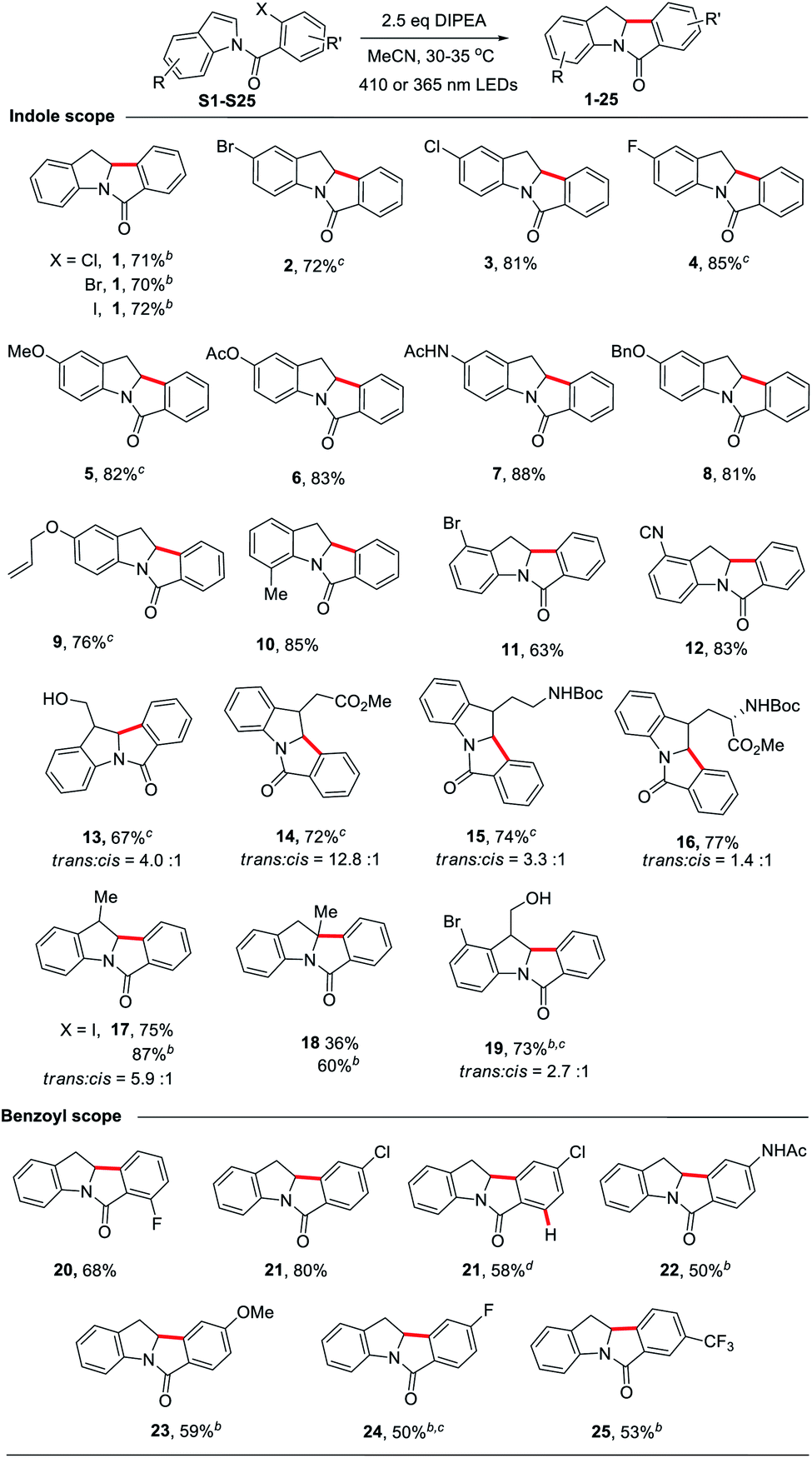
Under the optimized conditions, we examined the scope of the reductive Heck cyclization reaction (Table 2; see Scheme S1† for a list of substrates). A wide variety of functional groups, including both electron-donating and -withdrawing ones, were well tolerated and the corresponding products were obtained in good yields (1–16). The bromo or chloro substituent on the indole moiety remained intact (2–3, 11); this can benefit further transformation, such as the cross-coupling reaction. Sterically hindered N-(2-chlorobenzoyl)-2-methylindole (S18) afforded the product with one quaternary carbon centre in 60% yield with the use of 365 nm LEDs (18). The substrate with both bromo and hydroxyl groups on indole afforded the product in 73% yield (19).
We found that when both ortho C–Cl and C–F bonds are present, only C–Cl bond cleavage occurred, with a product yield of 68% (20). When N-(2,4-dichlorobenzoyl)indole was subjected to the reaction, only the ortho C–Cl bond reacted to afford 21 in 80% yield. Interestingly, for N-(2,4,6-trichlorobenzoyl)indole, which has two ortho C–Cl bonds, the cyclized product 21 with both ortho C–Cl bonds cleaved was obtained in 58% yield. We speculated that the reaction first gave the 2,4-dichloro-substituted cyclized product, followed by photoreduction with amine to give 21. Other electron-donating (AcNH and OMe) or electron-withdrawing (F and CF3) functional groups on the benzoyl moiety were tolerated, affording the corresponding products in moderate yields (22–25). However, no reaction occurred for substrates having phenol, aldehyde, pyridine or azide groups (Scheme S1†). When N-(2-chlorobenzoyl)-5-nitroindole (S28) was subjected to the reaction, the cyclization reaction did not occur; the nitro group was reduced to an amino group to give the amino product (S36) in 80% yield.22 Increasing the amount of DIPEA to 5 molar equivalents and extending the reaction time to 24 hours still resulted in S36 being the major product (81% yield).
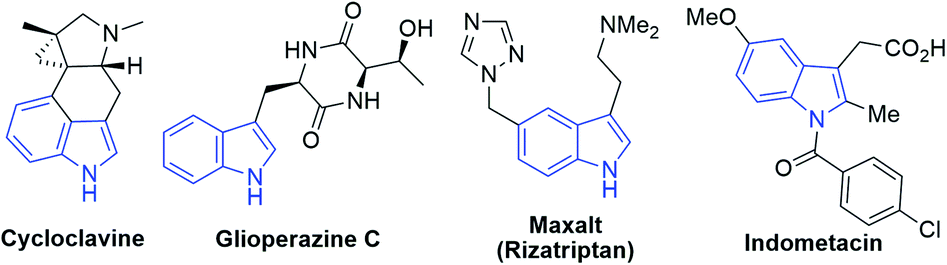
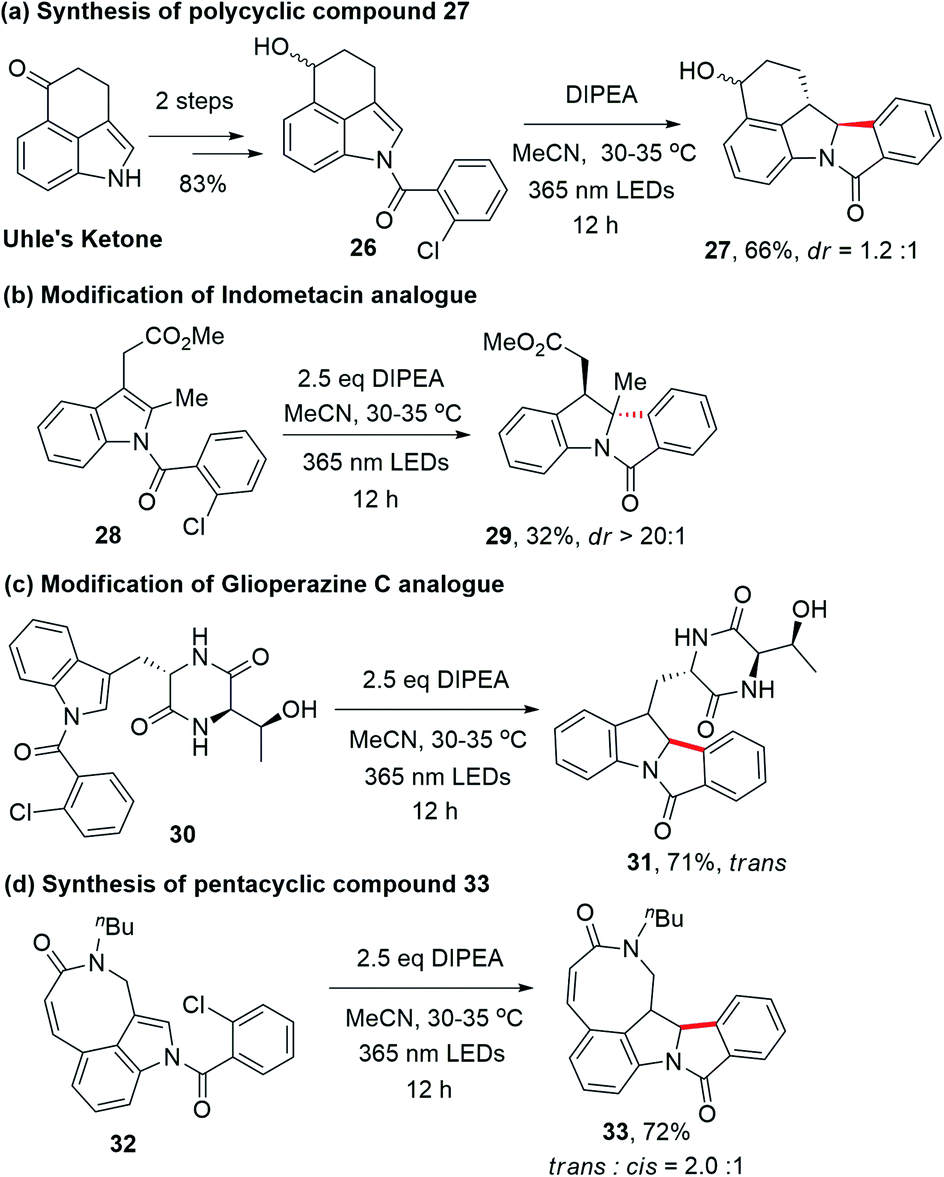
Since indole alkaloids are prevalent in natural products and pharmaceuticals (examples are shown in Scheme 3),23–25 this photocyclization strategy was applied to the modifications of indole-containing polycycles (Scheme 4). As shown in Scheme 4a, the treatment of Uhle's ketone,26 a key intermediate in the synthesis of lysergic acid,27 α-ergocryptine,27N-methylwelwistatin28 and cycloclavine,23,29 with 2-chlorobenzoyl chloride followed by reduction afforded 26 (Scheme S4†). By subjecting 26 to our photochemical reaction conditions, compound 27 was obtained in 66% yield (overall yield of 55% from Uhle's ketone). We also explored the modification of selected medicinal compounds and drug-like molecules. Indometacin analogue 28 cyclized into product 29 in 32% yield upon 365 nm LED excitation with excellent diastereoselectivity (Scheme 4b). Glioperazine C is known to display appealing bioactivity and anti-inflammatory effects. Similarly, when we subject compound 30, a structural analogue of anti-inflammatory alkaloid glioperazine C,25a to our photochemical reaction conditions, diketopiperazine 31 was produced in 71% yield as a single diastereomer without affecting the pre-existing stereogenic centres (Scheme 4c). Finally, tricyclic lactam 32 underwent photochemical cyclization to afford pentacyclic compound 33 in 72% yield (Scheme 4d).
 | ||
| Scheme 3 Natural products containing an indole moiety. | ||
 | ||
| Scheme 4 Modification of polycyclic compounds. | ||
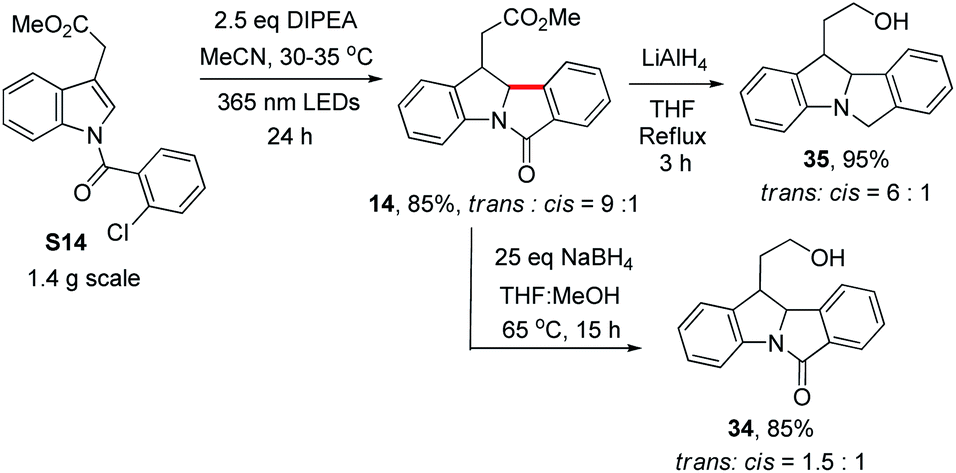
To demonstrate the synthetic utility of our method, we chose to derivatize 14, 15 and 16 (see Table 2), with their functionalized side chains serving as handles for further transformations. The reaction of S14 was scaled up to 1.4 grams. Product 14 (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis = 9:1) was obtained in 85% yield by prolonging the reaction time to 24 h. Further reduction of 14 was effected with LiAlH4 as the reductant, generating indoline 35 in 95% yield with a slight decrease in the trans:cis ratio. However, when 14 was reduced with NaBH4 in THF/MeOH, alcohol 34 was obtained in 85% yield but with a significant drop in the trans:cis ratio to 1.5:1 (Scheme 5). We noted that when 1 was treated with NaBH4 in a THF/CD3OD mixture at 65 °C, the incorporation of deuterium at C2 was observed, suggesting that the H at C2 could undergo exchange in the presence of a base and protic solvent (Fig. S7†), thus accounting for the drop in the trans:cis ratio.
:cis = 9:1) was obtained in 85% yield by prolonging the reaction time to 24 h. Further reduction of 14 was effected with LiAlH4 as the reductant, generating indoline 35 in 95% yield with a slight decrease in the trans:cis ratio. However, when 14 was reduced with NaBH4 in THF/MeOH, alcohol 34 was obtained in 85% yield but with a significant drop in the trans:cis ratio to 1.5:1 (Scheme 5). We noted that when 1 was treated with NaBH4 in a THF/CD3OD mixture at 65 °C, the incorporation of deuterium at C2 was observed, suggesting that the H at C2 could undergo exchange in the presence of a base and protic solvent (Fig. S7†), thus accounting for the drop in the trans:cis ratio.
 | ||
| Scheme 5 Further transformation of 14. | ||
Recently, we have developed an iron-catalyzed intramolecular C(sp3)–H insertion reaction of alkyl azides.30 To prepare complex indoline-containing compounds with this synthetic strategy, we converted trans-34 into azide 36. Using iron(II) phthalocyanine as the catalyst, pentacyclic indoline 37 was successfully prepared from 36 in 89% yield (27% from S14, 4 steps) (Scheme 6a). This indoline, incorporated with a quaternary aminal carbon centre, was characterized by X-ray crystallography (Fig. S14†). Compound 39 was synthesized using the same strategy in four steps, with an overall yield of 53% from S14 (Scheme 6b). A panobinostat31 derivative, 41, was obtained from 15 and methyl (E)-3-(4-(bromomethyl)-phenyl)acrylate 40 in moderate yield (Scheme 6c). Compound 31 was also synthesized in an overall 74% yield from 16 (Scheme 6d).
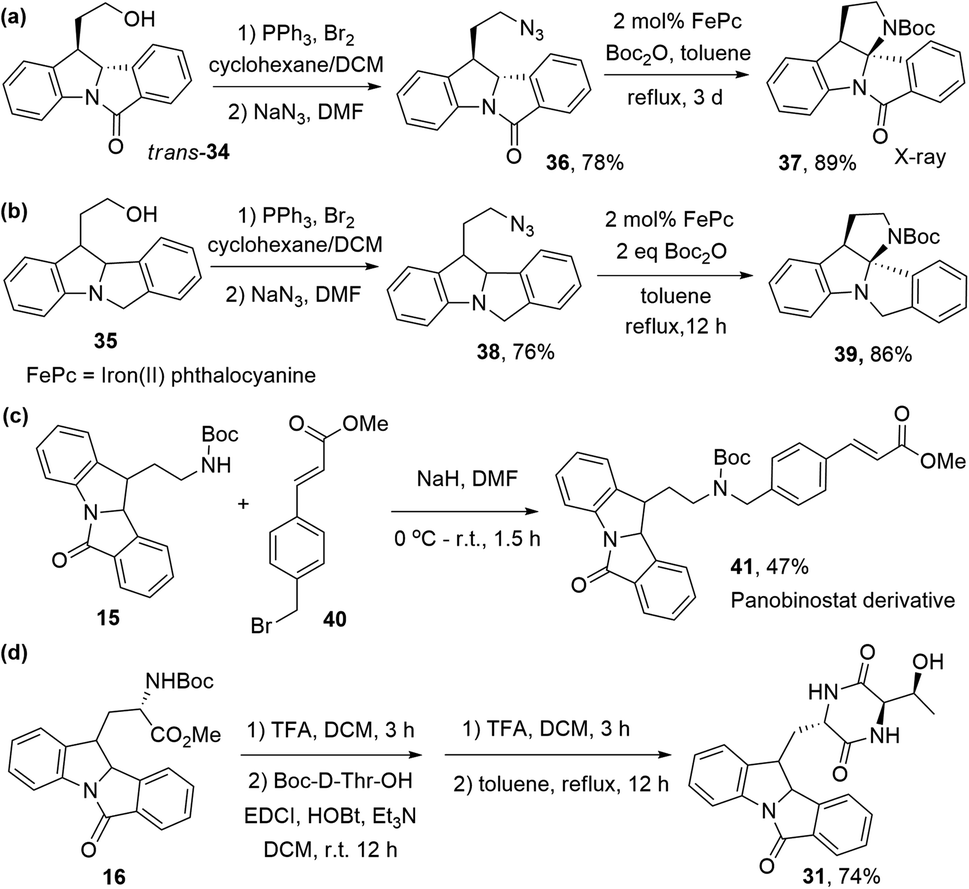 | ||
| Scheme 6 Synthesis of complex compounds. | ||
We conducted several experiments to examine the photochemical reaction and found the following: (1) when TEMPO was added, no product was formed and the substrate was recovered quantitatively, suggesting the involvement of radical(s) in the photochemical reaction (Table 3, entry 1). However, no TEMPO-trapped intermediate was detected by mass spectrometry. When 0.5 equivalent of butylated hydroxytoluene (BHT) was added, the product was obtained in only 50% yield (Table 3, entry 2). Hydroquinone and 1,4-dinitrobenzene are commonly used as single electron transfer (SET) inhibitors.32 With hydroquinone present, the product yield plummeted to 30% (Table 3, entry 3). No reaction was observed when 1-chloro-2,4-dinitrobenzene was added (Table 3, entry 4). These results indicate that a SET process is involved in the reaction.
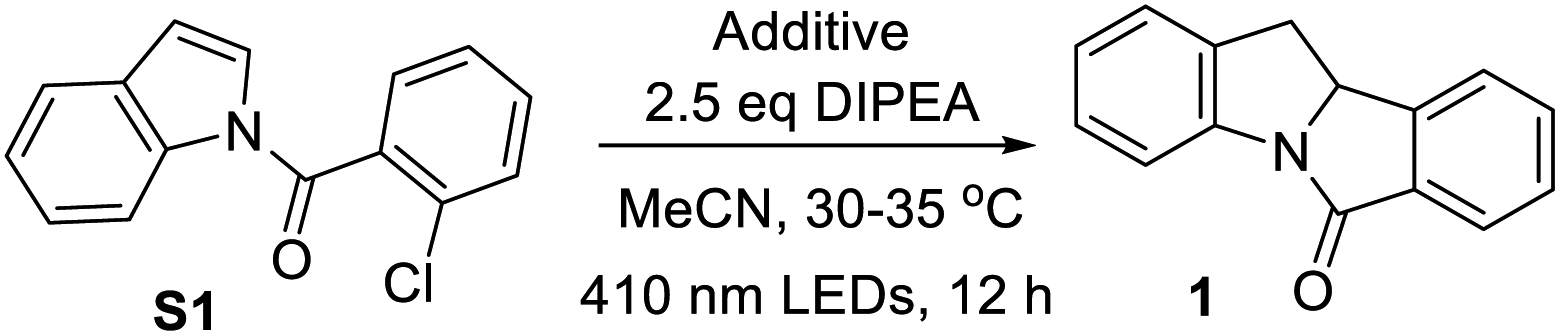
(2) The use of N-(2-chlorobenzyl)indole 42 (no carbonyl group) did not give the desired cyclized product, 43; the substrate remained intact (Scheme 7a). Hence, the presence of the carbonyl group is critical for the photochemical cyclization reaction. (3) When using CD3CN as the solvent, product 1 was obtained without deuteration (Scheme 7b). A solvent mixture with D2O, however, produced deuterated 1 with deuterium incorporated at the C3 position and C2 position to a lesser extent, (Scheme 7c). As D2O served as an external proton source in competition with DIPEA˙+, these results suggest the formation of the product as a consequence of protonation of anion intermediate E instead of hydrogen atom abstraction.10g,33 The deuteration at the C2 position could be a result of proton exchange after the product was formed (similar to the treatment of 1 with NaBH4 as aforementioned) because deuteration at the C2 position was also observed when the non-deuterated product 1 was subjected to the same reaction conditions shown in Scheme 7c (Fig. S7†). (4) When N-(2-chlorobenzoyl)-3-(2-bromoethyl)indole (S27) was subjected to the reaction, 44 was obtained in 30% yield and a spiro cyclopropane, 45, was formed in 43% yield. We surmised that the radical intermediate D′ is further reduced by DIPEA to afford the anion intermediate E′, giving product 45via nucleophilic substitution (Scheme 7d). The results depicted in Scheme 7b–d support the mechanism of protonation of cyclized anion intermediates E with water in solvent or an aminium radical cation (DIPEA˙+).17d,21,34 (5) The quantum yield for the photochemical conversion of S1 to 1 with 365 nm LEDs was estimated using potassium ferrioxalate as a chemical actinometer (see the ESI†); the photon flux of the LEDs was 3.91 × 10−6 einstein s−1. By subjecting S1 to the reaction conditions b depicted in Table 2, 89.2 μmol of 1 was formed in 40 minutes (18% yield). Thus, the quantum yield of the reaction is 2.1%, suggesting that a light-induced catalytic pathway is favored over a radical chain process.35
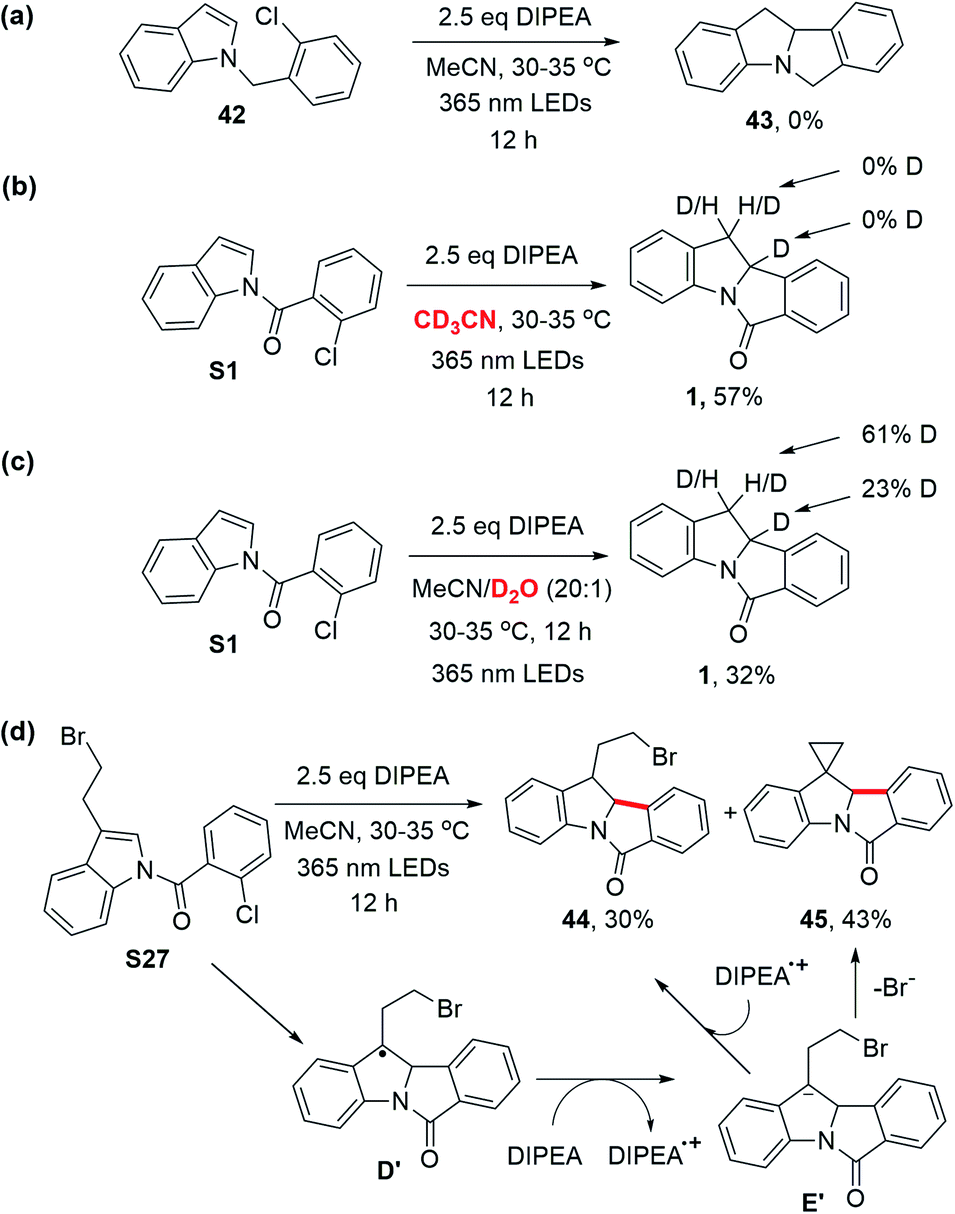 | ||
| Scheme 7 Mechanistic experiments. | ||
(6) ns-TA spectral measurements in degassed MeCN revealed that, at 4 μs after pulse excitation (355 nm), S1 or DIPEA alone showed no discernible signal. However, the solution containing both S1 and DIPEA displayed a broad ns-TA band (maximum at ∼500 nm) at the same time point (Fig. 2a). The signal with the ΔO.D. at 500 nm assigned to the ketyl radical anion36 was relatively long-lived showing a decay time constant of 28 μs. The excited state potential E(S1*/S1−) was estimated to be 1.15 V vs. Ag/AgCl (S1's E0–0 = 2.9 eV; Epc(S1/S1−) = −1.75 V), which is higher than the reported potential of E(DIPEA˙+/DIPEA) (0.9 V vs. Ag/AgCl).37 Therefore, upon excitation, S1 attains a triplet excited state, which accepts an electron from DIPEA to give one-electron reduced S1 (Scheme 2, species B) and DIPEA˙+. (7) In situ formation of electron donor–acceptor complexes is another strategy for light-induced C–Cl bond activation.38 In MeCN, DIPEA (0.25 M) shows an intense absorption at 250–300 nm and weak absorption beyond 325 nm. For indole substrate S1 (0.1 M), there is an intense absorption at 250–330 nm, with absorption tail ending at ∼390 nm (Fig. S11†). As the mixture of S1 and DIPEA used in the photochemical experiment showed no new absorption band, the possibility of in situ formation of an electron donor–acceptor complex is disfavored.38a,39
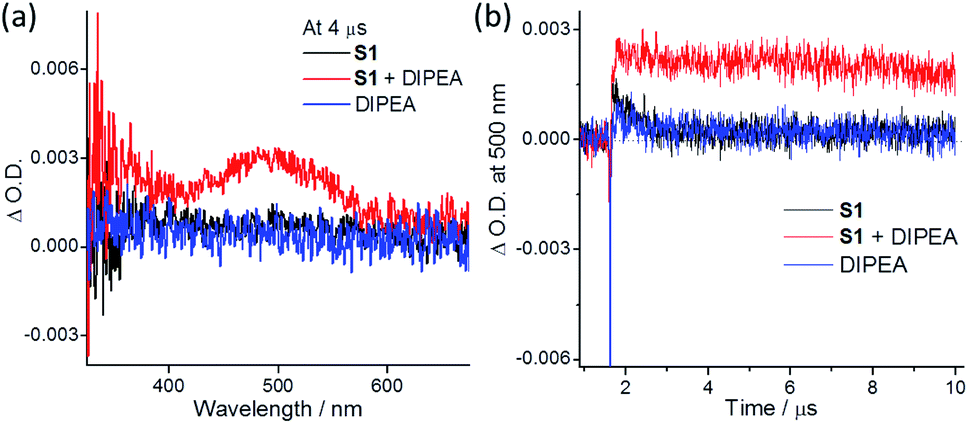 | ||
| Fig. 2 (a) ns-TA spectra recorded at 4 μs after pulse excitation at 355 nm, and (b) ΔO.D. intensity at 500 nm for degassed MeCN solutions containing S1 (0.1 M; black line), S1 (0.1 M) and DIPEA (0.25 M; red line) or DIPEA (0.25 M; blue line). | ||
DFT calculations (Fig. 3) revealed that the calculated transition state (TS) energy for carbon–halogen bond cleavage of radical-anion S1˙− is 8.9 kcal mol−1. On the other hand, the direct homolytic cleavage of the aryl–halogen bond in the triplet state (TS1′, 14.7 kcal mol−1) and concerted bond cleavage and C–C formation (TS1′′, 17.1 kcal mol−1) have higher TS energy. Therefore, the photoexcited S1 (triplet state) is more likely to undergo electron transfer with DIPEA to form the radical-anion species, which then undergoes C–Cl cleavage to form intermediate C. The subsequent cyclization of C is facile with a barrier of 9.5 kcal mol−1 (Fig. S16†). The computational results further lend support to the proposed radical-anion decomposition pathway as depicted in Scheme 2.
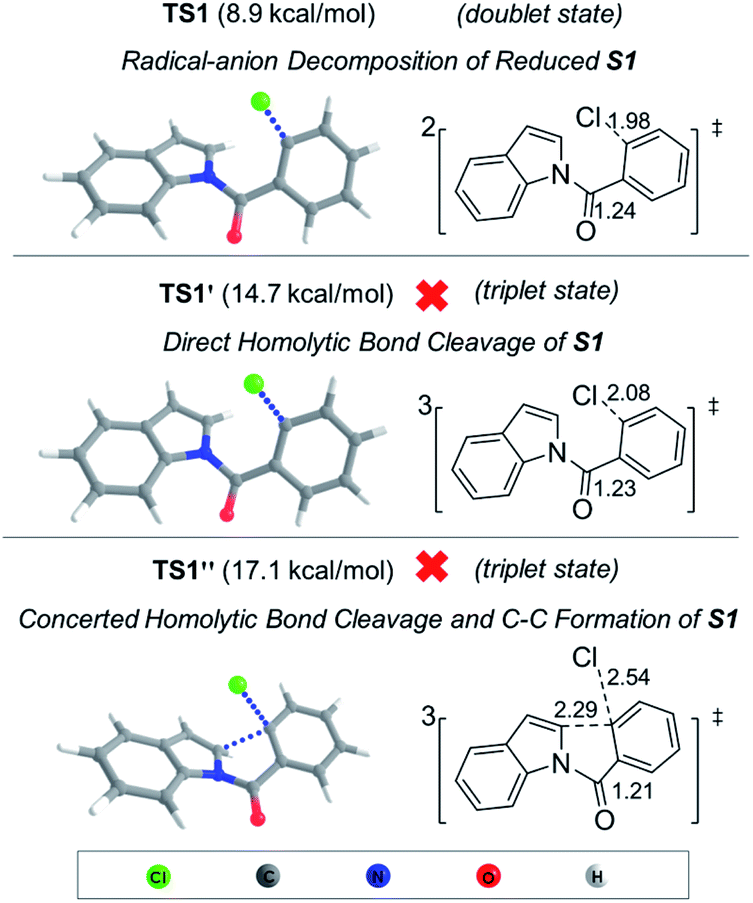 | ||
| Fig. 3 DFT calculated transition states at the M06-2X-D3/6-311++G**/PCM//B3LYP-D3/6-311G*/PCM level of theory. Critical bond lengths are given in Å. | ||
Conclusions
Herein, we have described a general and operationally simple direct photo-induced reductive Heck cyclization reaction of indole derivatives. This reaction has a high tolerance to functional groups, such as esters, amides, alcohols, allylics and alkenes. More importantly, this method can be applied to the functionalization of natural product derivatives and preparation of complex polycyclic indolinyl compounds.Data availability
CCDC 2101130, and 2101131 containing the supplementary crystallographic data for this paper. Experimental and computational data associated with this work are provided in the article. Additional data supporting the findings reported in this paper are available as ESI.† All other data are available from the authors upon reasonable request.Author contributions
D. Yu performed the majority of the reactions and wrote the first draft of the manuscript. W.-P. To conducted the time-resolved spectroscopic studies and photochemical reaction quantum yield measurement. L.-L. Wu performed the DFT calculations. T. You contributed to the iron-catalyzed reactions. W.-P. To, J. Ling, Y. Liu and C.-M. Che revised writing of the manuscript. Y. Liu helped C.-M. Che to supervise the research. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Guangdong Major Project of Basic and Applied Basic Research (2019B030302009), the Science, Technology and Innovation Commission of Shenzhen Municipality (JCYJ20180508162429786), the China Postdoctoral Science Foundation (2018M633043), the Laboratory for Synthetic Chemistry and Chemical Biology under the Health@InnoHK Program launched by the Innovation and Technology Commission and the CAS-Croucher Funding Scheme for Joint Laboratories. We also thank the Southern University of Science and Technology for financial support. We thank Dr Qingyun Wan for the help with DFT calculations. Dr Zhou Tang and Dr Hua-Hua Wang were thanked for their help with EPR analysis and optical rotation measurement, respectively.Notes and references
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed.
- (a) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS; (b) E. Shirakawa, K.-I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537 CrossRef CAS PubMed; (c) C. Adouama, M. E. Budén, W. D. Guerra, M. Puiatti, B. Joseph, S. M. Barolo, R. A. Rossi and M. Médebielle, Org. Lett., 2019, 21, 320 CrossRef CAS PubMed.
- (a) S.-C. Lu, X.-X. Zhang, Z.-J. Shi, Y.-W. Ren, B. Li and W. Zhang, Adv. Synth. Catal., 2009, 351, 2839 CrossRef CAS; (b) C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548 CrossRef CAS; (c) K. Chen, M. S. Cheung, Z. Lin and P. Li, Org. Chem. Front., 2016, 3, 875 RSC; (d) A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985 CrossRef CAS; (e) A. M. Mfuh, V. T. Nguyen, B. Chhetri, J. E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408 CrossRef CAS; (f) K. Chen, P. He, S. Zhang and P. Li, Chem. Commun., 2016, 52, 9125 RSC; (g) T. Fukuyama, Y. Fujita, H. Miyoshi, I. Ryu, S.-C. Kao and Y.-K. Wu, Chem. Commun., 2018, 54, 5582 RSC; (h) A. Steiner, J. D. Williams, J. A. Rincón, O. de Frutos, C. Mateos and C. O. Kappe, Eur. J. Org. Chem., 2019, 5807 CrossRef CAS.
- (a) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673 CrossRef CAS; (b) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed; (c) G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 9751 CrossRef CAS PubMed; (d) A. J. Smith, D. L. Poole and J. A. Murphy, Sci. China: Chem., 2019, 62, 1425 CrossRef CAS; (e) S. Rohrbach, R. S. Shah, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 11454 CrossRef CAS PubMed; (f) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124 CrossRef PubMed; (g) K. Liang, T. Li, N. Li, Y. Zhang, L. Shen, Z. Ma and C. Xia, Chem. Sci., 2020, 11, 2130 RSC; (h) K. Liang, Q. Liu, L. e. Shen, X. Li, D. Wei, L. Zheng and C. Xia, Chem. Sci., 2020, 11, 6996 RSC.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed; (b) A. Graml, I. Ghosh and B. König, J. Org. Chem., 2017, 82, 3552 CrossRef CAS PubMed; (c) C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023 RSC.
- (a) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087 CrossRef CAS PubMed; (b) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093 CrossRef CAS PubMed; (c) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732 CrossRef CAS PubMed.
- (a) H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266 CrossRef CAS PubMed; (b) M. Jiang, H. Li, H. Yang and H. Fu, Angew. Chem., Int. Ed., 2017, 56, 874 CrossRef CAS PubMed; (c) A. U. Meyer, T. Slanina, A. Heckel and B. König, Chem. Eur. J., 2017, 23, 7900 CrossRef CAS PubMed; (d) I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544 CrossRef CAS PubMed; (e) R. Matsubara, T. Yabuta, U. Md Idros, M. Hayashi, F. Ema, Y. Kobori and K. Sakata, J. Org. Chem., 2018, 83, 9381 CrossRef CAS PubMed; (f) T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646 CrossRef CAS PubMed; (g) D. Yu, W.-P. To, G. S. M. Tong, L.-L. Wu, K.-T. Chan, L. Du, D. L. Phillips, Y. Liu and C.-M. Che, Chem. Sci., 2020, 11, 6370 RSC; (h) F. Glaser, C. B. Larsen, C. Kerzig and O. S. Wenger, Photochem. Photobiol. Sci., 2020, 19, 1035 CrossRef CAS PubMed; (i) H. Cheng, T.-L. Lam, Y. Liu, Z. Tang and C.-M. Che, Angew. Chem., Int. Ed., 2020, 60, 1383 CrossRef PubMed; (j) M. Cybularczyk-Cecotka, J. Szczepanik and M. Giedyk, Nat. Catal., 2020, 3, 872 CrossRef CAS.
- Selected literature on the photochemistry of aryl halides: (a) M. Ohashi, K. Tsujimoto and K. Seki, J. Chem. Soc., Chem. Commun., 1973, 384a RSC; (b) M. Somei and M. Natsume, Tetrahedron Lett., 1973, 14, 2451 CrossRef; (c) W. Carruthers and N. Evans, J. Chem. Soc., Perkin Trans. 1, 1974, 1523 RSC; (d) G. R. Lenz, J. Org. Chem., 1974, 39, 2839 CrossRef CAS; (e) T. Kametani, T. Sugai, Y. Shoji, T. Honda, F. Satoh and K. Fukumoto, J. Chem. Soc., Perkin Trans. 1, 1977, 1151 RSC; (f) O. Tsuge, T. Hatta and H. Tsuchiyama, Chem. Lett., 1998, 155 CrossRef CAS; (g) J. Grimshaw and A. P. de Silva, Chem. Soc. Rev., 1981, 10, 181 RSC.
- Selected examples of thermal dearomatization of indoles: (a) W. Zhang and G. Pugh, Tetrahedron Lett., 1999, 40, 7591 CrossRef CAS; (b) W. Zhang and G. Pugh, Tetrahedron, 2003, 59, 3009 CrossRef CAS; (c) D. A. Petrone, A. Yen, N. Zeidan and M. Lautens, Org. Lett., 2015, 17, 4838 CrossRef CAS PubMed; (d) C. Shen, R.-R. Liu, R.-J. Fan, Y.-L. Li, T.-F. Xu, J.-R. Gao and Y.-X. Jia, J. Am. Chem. Soc., 2015, 137, 4936 CrossRef CAS PubMed; (e) R.-R. Liu, Y.-G. Wang, Y.-L. Li, B.-B. Huang, R.-X. Liang and Y.-X. Jia, Angew. Chem., Int. Ed., 2017, 56, 7475 CrossRef CAS PubMed; (f) M. Markovič, P. Koóš, T. Čarný and T. Gracza, Tetrahedron Lett., 2020, 61, 152370 CrossRef; (g) X. Qin, M. W. Y. Lee and J. S. Zhou, Angew. Chem., Int. Ed., 2017, 56, 12723 CrossRef CAS PubMed; (h) N. Zeidan, T. Beisel, R. Ross and M. Lautens, Org. Lett., 2018, 20, 7332 CrossRef CAS PubMed; (i) C. Shen, N. Zeidan, Q. Wu, C. B. J. Breuers, R.-R. Liu, Y.-X. Jia and M. Lautens, Chem. Sci., 2019, 10, 3118 RSC.
- (a) S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920 CrossRef CAS PubMed; (b) J. Chavez, J. Kimball, L. Ceresa, E. Kitchner, T. Shtoyko, R. Fudala, J. Borejdo, Z. Gryczynski and I. Gryczynski, J. Lumin., 2021, 230, 117724 CrossRef CAS; (c) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090 CrossRef CAS; (d) C. Haustein, W. D. Savage, C. F. Ishak and R. T. Pflaum, Talanta, 1989, 36, 1065 CrossRef CAS PubMed; (e) J. Chavez, L. Ceresa, E. Kitchner, J. Kimball, T. Shtoyko, R. Fudala, J. Borejdo, Z. Gryczynski and I. Gryczynski, J. Photochem. Photobiol., B, 2020, 208, 111897 CrossRef CAS PubMed; (f) S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W.-W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- (a) D. O. Cowan and R. L. Drisko, Photochemical Techniques and the Photochemistry of Ketones, in Elementsof organic photochemistry, Springer, Boston, 1976 Search PubMed; (b) P. Klán and J. Wirz, in Photochemistry of Organic Compounds, John Wiley & Sons, Hoboken, 2009, p. 227 CrossRef; (c) J. A. Dantas, J. T. M. Correia, M. W. Paixão and A. G. Corrêa, ChemPhotoChem, 2019, 3, 506 CrossRef CAS.
- (a) Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016 CrossRef CAS; (b) Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200 CrossRef CAS PubMed; (c) A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566 CrossRef CAS PubMed; (d) J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed.
- Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349 RSC.
- (a) C. Kerzig and M. Goez, Phys. Chem. Chem. Phys., 2015, 17, 13829 RSC; (b) M. Brautzsch, C. Kerzig and M. Goez, Green Chem., 2016, 18, 4761 RSC; (c) M. Schmalzbauer, I. Ghosh and B. König, Faraday Discuss., 2019, 215, 364 RSC; (d) M. Schmalzbauer, M. Marcon and B. König, Angew. Chem., Int. Ed., 2021, 60, 6270 CrossRef CAS PubMed.
- Recent examples of light-induced cyclization of indoles: (a) M. V. Popescu, A. Mekereeya, J. V. Alegre-Requena, R. S. Paton and M. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 23020 CrossRef CAS PubMed; (b) Z. Zhang, D. Yi, M. Zhang, J. Wei, J. Lu, L. Yang, J. Wang, N. Hao, X. Pan, S. Zhang, S. Wei and Q. Fu, ACS Catal., 2020, 10, 10149 CrossRef CAS; (c) M. Zhu, X. Zhang, C. Zheng and S.-L. You, ACS Catal., 2020, 10, 12618 CrossRef CAS; (d) W.-J. Zhou, Z.-H. Wang, L.-L. Liao, Y.-X. Jiang, K.-G. Cao, T. Ju, Y. Li, G.-M. Cao and D.-G. Yu, Nat. Commun., 2020, 11, 3263 CrossRef CAS PubMed; (e) M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. K. Gupta, N. A. Meanwell, A. Mathur and T. G. M. Dhar, J. Am. Chem. Soc., 2020, 142, 3094 CrossRef CAS PubMed; (f) M. S. Oderinde, A. Ramirez, T. G. M. Dhar, L. A. M. Cornelius, C. Jorge, D. Aulakh, B. Sandhu, J. Pawluczyk, A. A. Sarjeant, N. A. Meanwell, A. Mathur and J. Kempson, J. Org. Chem., 2021, 86, 1730 CrossRef CAS PubMed; (g) K. A. McDaniel and N. T. Jui, Org. Lett., 2021, 23, 5576 CrossRef CAS PubMed.
- (a) O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed; (b) W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592 CrossRef CAS; (c) T. Liu, G. Zhang, R. E. Evans, C. O. Trindle, Z. Altun, C. A. DeRosa, F. Wang, M. Zhuang and C. L. Fraser, Chem. Eur. J., 2018, 24, 1859 CrossRef CAS PubMed; (d) S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mat., 2018, 28, 1705045 CrossRef; (e) Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451 CrossRef CAS PubMed.
- (a) S. G. Cohen, G. A. Davis and W. D. K. Clark, J. Am. Chem. Soc., 1972, 94, 869 CrossRef CAS; (b) S. G. Cohen, A. Parola and G. H. Parsons, Chem. Rev., 1973, 73, 141 CrossRef CAS.
- (a) A. N. Santiago, K. Takeuchi, Y. Ohga, M. Nishida and R. A. Rossi, J. Org. Chem., 1991, 56, 1581 CrossRef CAS; (b) A. E. Lukach, D. G. Morris, A. N. Santiago and R. A. Rossi, J. Org. Chem., 1995, 60, 1000 CrossRef CAS; (c) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71 CrossRef CAS PubMed.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed.
- (a) T. Hirao, J. Shiori and N. Okahata, Bull. Chem. Soc. Jpn., 2004, 77, 1763 CrossRef CAS; (b) S. Gazi and R. Ananthakrishnan, Appl. Catal., B, 2011, 105, 317 CrossRef CAS.
- (a) F. R. Petronijevic and P. Wipf, J. Am. Chem. Soc., 2011, 133, 7704 CrossRef CAS PubMed; (b) W. Wang, J.-T. Lu, H.-L. Zhang, Z.-F. Shi, J. Wen and X.-P. Cao, J. Org. Chem., 2014, 79, 122 CrossRef CAS PubMed; (c) N. Netz and T. Opatz, J. Org. Chem., 2016, 81, 1723 CrossRef CAS PubMed.
- K. Douki, H. Ono, T. Taniguchi, J. Shimokawa, M. Kitamura and T. Fukuyama, J. Am. Chem. Soc., 2016, 138, 14578 CrossRef CAS PubMed.
- (a) P. Wang, L. Miao, H. Zhao, D. Wu, L. Wang, Y. Teng, H. Sun and P. Yu, J. Chem. Pharm. Res., 2015, 7, 85 CAS; (b) Y. Wang, P. Wang, H. Ma and W. Zhu, Expert Opin. Ther. Pat., 2013, 23, 1415 CrossRef CAS PubMed.
- (a) F. C. Uhle, J. Am. Chem. Soc., 1949, 71, 761 CrossRef CAS PubMed; (b) K. Teranishi, S. Hayashi, S.-i. Nakatsuka and T. Goto, Tetrahedron Lett., 1994, 35, 8173 CrossRef CAS.
- I. Moldvai, E. Temesvári-Major, M. Incze, É. Szentirmay, E. Gács-Baitz and C. Szántay, J. Org. Chem., 2004, 69, 5993 CrossRef CAS PubMed.
- M. Ruiz, P. López-Alvarado and J. C. Menéndez, Org. Biomol. Chem., 2010, 8, 4521 RSC.
- M. Incze, G. Dörnyei, I. Moldvai, E. Temesvári-Major, O. Egyed and C. Szántay, Tetrahedron, 2008, 64, 2924 CrossRef CAS.
- (a) K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947 CrossRef CAS PubMed; (b) T. You, S.-H. Zeng, J. Fan, L. Wu, F. Kang, Y. Liu and C.-M. Che, Chem. Commun., 2021 DOI:10.1029/d1cc04573c.
- P. G. Richardson, R. L. Schlossman, M. Alsina, D. M. Weber, S. E. Coutre, C. Gasparetto, S. Mukhopadhyay, M. S. Ondovik, M. Khan, C. S. Paley and S. Lonial, Blood, 2013, 122, 2331 CrossRef CAS PubMed.
- F. Wu, B. Huang and W. Huang, Chin. J. Org. Chem., 1993, 13, 449 CAS.
- (a) W. K. Smothers, K. S. Schanze and J. Satltiel, J. Am. Chem. Soc., 1979, 101, 1895 CrossRef CAS; (b) N. L. Holy, Chem. Rev., 1974, 74, 243 CrossRef CAS; (c) L. M. Dorfman, Acc. Chem. Res., 1970, 3, 224 CrossRef CAS.
- J. Du, L. R. Espelt, I. A. Guzei and T. P. Yoon, Chem. Sci., 2011, 2, 2115 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- Selected examples of the ketyl radical or radical anion of benzophenone: (a) C. Pac, K. Mizuno, T. Tosa and H. Sakurai, J. Chem. Soc., Perkin Trans. 1, 1974, 561 RSC; (b) T. S. Godfrey, J. W. Hilpern and G. Porter, Chem. Phys. Lett., 1967, 1, 490 CrossRef CAS; (c) M. von Raumer and P. Suppan, Chem. Phys. Lett., 1996, 250, 91 CrossRef CAS; (d) J. Pérez-Prieto, F. Boscá, R. E. Galian, A. Lahoz, L. R. Domingo and M. A. Miranda, J. Org. Chem., 2003, 68, 5104 CrossRef PubMed; (e) E. Haselbach, E. Vauthey and P. Suppan, Tetrahedron, 1988, 44, 7335 CrossRef CAS.
- Y. Imada, Y. Okaka and K. Chiba, ChemElectroChem, 2020, 7, 1619 CrossRef CAS.
- (a) J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017 CrossRef CAS PubMed; (b) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS PubMed; (c) J. Yuan, W.-P. To, Z.-Y. Zhang, C.-D. Yue, S. Meng, J. Chen, Y. Liu, G.-A. Yu and C.-M. Che, Org. Lett., 2018, 20, 7816 CrossRef CAS PubMed; (d) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed.
- L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photophysical data, DFT calculations and NMR spectral data. CCDC 2101130 and 2101131. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04258k |
| This journal is © The Royal Society of Chemistry 2021 |