
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMg(II) heterodinuclear catalysts delivering carbon dioxide derived multi-block polymers†
Gloria
Rosetto
,
Arron C.
Deacy
and
Charlotte K.
Williams
 *
*
Department of Chemistry, Chemistry Research Laboratory, 12 Mansfield Rd, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
First published on 26th August 2021
Abstract
Carbon dioxide derived polymers are emerging as useful materials for applications spanning packaging, construction, house-hold goods and automotive components. To accelerate and broaden their uptake requires both more active and selective catalysts and greater structural diversity for the carbon dioxide derived polymers. Here, highly active catalysts show controllable selectivity for the enchainment of mixtures of epoxide, anhydride, carbon dioxide and lactone. Firstly, metal dependent selectivity differences are uncovered using a series of dinuclear catalysts, Mg(II)Mg(II), Zn(II)Zn(II), Mg(II)Zn(II), and Mg(II)Co(II), each exposed to mixtures of bio-derived tricyclic anhydride, cyclohexene oxide and carbon dioxide (1 bar). Depending upon the metal combinations, different block structures are possible with Zn(II)Zn(II) yielding poly(ester-b-carbonate); Mg(II)Mg(II) or Mg(II)Co(II) catalysts delivering poly(carbonate-b-ester); and Mg(II)Zn(II) furnishing a random copolymer. These results indicate that carbon dioxide insertion reactions follow the order Co(II) > Mg(II) > Zn(II). Using the most active and selective catalyst, Mg(II)Co(II), and exploiting reversible on/off switches between carbon dioxide/nitrogen at 1 bar delivers precision triblock (ABA), pentablock (BABAB) and heptablock (ABABABA) polymers (where A = poly(cyclohexylene oxide-alt-tricyclic anhydride), PE; B = poly(cyclohexene carbonate), PCHC). The Mg(II)Co(II) catalyst also selectively polymerizes a mixture of anhydride, carbon dioxide, cyclohexene oxide and ε-caprolactone to deliver a CBABC pentablock copolymer (A = PE, B = PCHC C = poly(caprolactone), PCL). The catalysts combine high activity and selectivity to deliver new polymers featuring regularly placed carbon dioxide and biomass derived linkages.
Introduction
Carbon dioxide/epoxide copolymerization is a front-runner carbon dioxide utilization technology as it is truly catalytic, results in 20–50 wt% carbon dioxide uptake into the polymer backbone and significantly reduces the greenhouse gas emissions associated with polymer production.1–3 Further attractions include the widespread commercial availability and low-cost of epoxides, processes compatible with existing manufacturing infrastructure and polymers with properties suitable for applications spanning packaging, agricultural films, home-insulation materials, adhesives, coatings, flex-foams for furniture, clothing and automotive components, amongst others.4 To accelerate and broaden applications requires a greater diversity of carbon dioxide-containing materials and delivering these necessitates advances in the polymerization catalysis.5,6 One possibility is to copolymerize mixtures of carbon dioxide alongside other monomers so as to ‘place’ carbonate linkages selectively within more complex copolymer structures.7,8 More generally, devising atom economical and high yielding methods to control the sequences of both individual monomers and blocks in the polymer backbone is a major challenge.9,10In terms of controlling sequences within alternating polyesters, Coates and co-workers pioneered high activity Al(III) catalysts for epoxide/anhydride ring-opening copolymerization (ROCOP) and applied elegant post-functionalization reactions to install alternating sequences of imine and alkyl group substituents.11 Subsequently, we also exploited the high alternation of epoxide/anhydride ROCOP to install alternating but orthogonal functional groups producing amphiphilic polymers which self-assembled in aqueous solution.12 Wu and co-workers prepared self-healable thermoplastic elastomers from CO2/epoxide derived polycarbonates, with precision placement of dynamic cross-linking moieties to the side-chains.13 These materials showed a Young's modulus of 10 MPa and excellent elastic recovery. Meng and co-workers polymerized mixtures of propylene oxide (PO), cyclohexene oxide (CHO), phthalic anhydride (PA) and CO2 to form polymers with variable ester![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carbonate linkages.14 The materials showed high tensile strength (54 MPa), optical transparency and molar masses up to 70 kg mol−1: properties were competitive with polystyrene. Herein, efficient and one-pot polymerization catalyses afford both highly alternating monomer sequences within the blocks and highly selective block sequences within the polymer chain. Catalysts that selectively enchain monomer mixtures to provide specific and single block polymer structures are important targets.15–17
:carbonate linkages.14 The materials showed high tensile strength (54 MPa), optical transparency and molar masses up to 70 kg mol−1: properties were competitive with polystyrene. Herein, efficient and one-pot polymerization catalyses afford both highly alternating monomer sequences within the blocks and highly selective block sequences within the polymer chain. Catalysts that selectively enchain monomer mixtures to provide specific and single block polymer structures are important targets.15–17
In particular, being able to switch catalysts between different mechanisms is useful to diversify the block chemistries.16,18,19 In 2008, Coates and co-workers described the first example with a Zn(II) catalyst polymerizing a mixture of diglycholic anhydride (DGA), CHO and CO2 to produce a poly(ester-b-carbonate).20 The order of block enchainment was rationalized by >2000-fold faster rate for DGA vs. CO2 insertion into the propagating zinc alkoxide intermediate. Subsequently, many other equivalently selective and controlled homogenous mono-,21–24 dinuclear metal catalysts25,26 and organocatalysts were reported.27,28 A few catalysts are exceptions to the selectivity preference, including poorly defined heterogeneous catalysts that result in ill-controlled random or tapered copolymers.29–31
In 2014, we reported a new type of self-switchable polymerization catalysis, whereby a Zn(II)Zn(II) catalyst was directed between epoxide/carbon dioxide ROCOP and lactone ring-opening polymerization (ROP) to deliver well controlled poly(ester-b-carbonates).32,33 A year later, the same selectivity and block structures were observed when mixtures of epoxide, anhydride and lactone were polymerized by the di-Zn(II) catalyst.34 In 2016, a combined experimental and theoretical investigation revealed the ‘rules’ of switchable catalysis and allowed for the prediction of polymer structures from mixtures of epoxide, carbon dioxide, lactone and anhydride (Fig. 1).35 It was found that the energy barrier to CO2 insertion into the zinc-alkoxide intermediate was slightly lower than that for anhydride insertion (12.8 kcal mol−1 and 16 kcal mol−1 respectively), yet experimentally the polyester block formed before the polycarbonate (Fig. 1b). The zinc-carboxylate intermediate is significantly more stable than the corresponding zinc-carbonate and CO2 insertion would be expected to be reversible under the polymerization conditions. Thus, the carboxylate linkage provides a thermodynamic ‘sink’ driving the selective formation of poly(ester). Subsequently, a range of other metallic, organometallic and organo-catalysts were established to follow the same monomer sequence selectivity ‘rules’ and the switchable catalysis was shown to apply to different epoxides, anhydrides, lactones and heterocycles.15,28,36–42
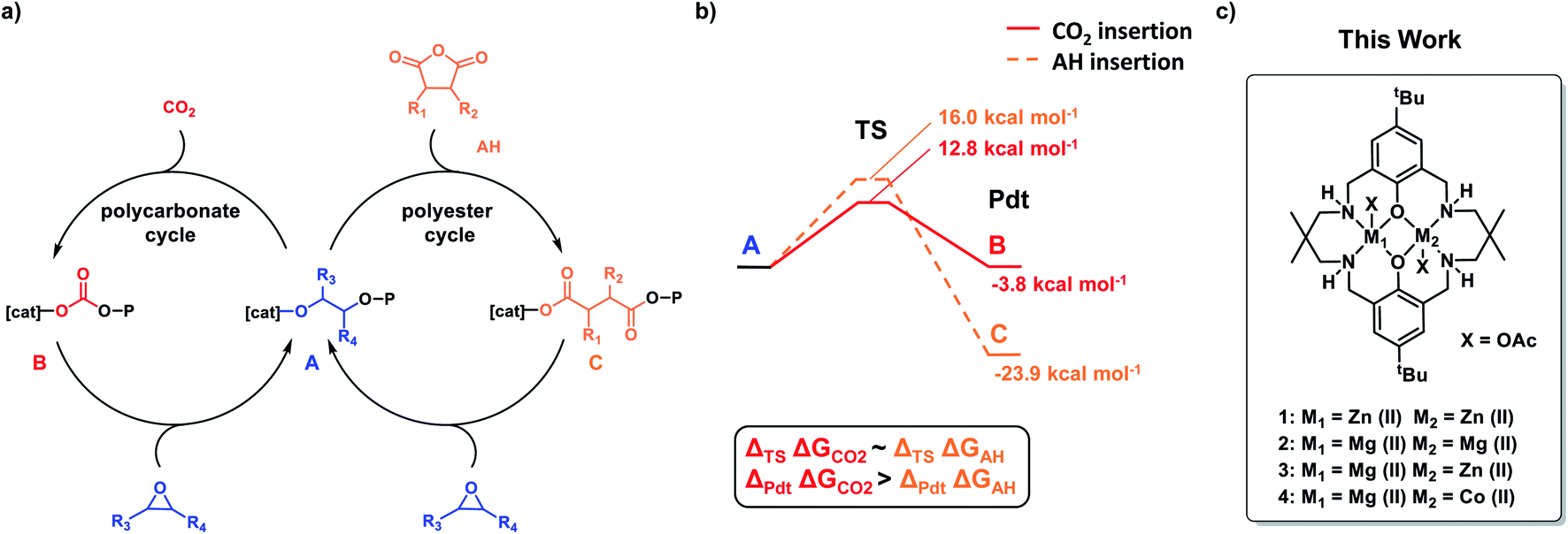 | ||
| Fig. 1 (a) Catalytic cycles for CO2/epoxide and anhydride (AH)/epoxide ROCOP, connected via the catalyst-alkoxide intermediate, A. (b) DFT calculated barriers to the selectivity limiting step to form products (Pdt), comparing carbon dioxide or anhydride insertion into the zinc alkoxide intermediate (A) (calculations were conducted using di-Zn(II) catalyst 1, cyclohexene oxide CHO, and phthalic anhydride PA). (c) Structures of the dinuclear catalysts 1–4 featured in this work. | ||
Despite the successes of switchable catalysis, almost all prior research has focussed on polyesters and -ethers, with a paucity of investigations of carbon dioxide linkage placement.26,43–47 This likely arises from the limited range of carbon dioxide/epoxide ROCOP catalysts and technical complexities in controlling the gas atmosphere since many catalysts require high pressures and use of stainless-steel reactors. Thus, there remain several notable absences from the emerging field of multi-block polymers produced by switchable catalysis. These include a lack of reports of multiple switching between epoxide/anhydride and carbon dioxide copolymerization to build up multi-block poly(ester-b-carbonate) structures and only one prior report of a CBABC pentablock structure combining blocks derived by lactone ROP and epoxide/anhydride/carbon dioxide ROCOP.48 It's also important to improve the activity of these switchable polymerization catalysts since current best-in-class catalysts fail to perform across multiple polymerization cycles, with turn-over-frequency values limited to 100 h−1 at 1 bar of CO2.25–27
Results
It was previously noted that the Zn(II)Zn(II) catalyst 1 exposed to a mixture of cyclohexene oxide, sterically hindered tricyclic anhydride (a bio-based monomer49,50) and carbon dioxide resulted in first anhydride and then carbon dioxide insertion forming a poly(ester-b-carbonate).26 Curiously, the analogous Mg(II)Mg(II) catalyst 2 showed the reverse order of insertion resulting in the formation of poly(carbonate-b-ester) – a unique selectivity (Fig. 1c).26 Overall, catalyst 2 is 17 times faster than 1, however, its activity remains low with a TOF = 77 h−1 ([2]:[TCA]:[CHO] = 1:100:1000, 1 bar CO2, 100 °C). The unexpected selectivity difference between Zn(II) and Mg(II) based catalysts motivated further investigation. Two recently reported heterodinuclear catalysts, Mg(II)Zn(II) (3) and Mg(II)Co(II) (4), show higher rates in anhydride/epoxide and CO2/epoxide ROCOP compared with their homodinuclear analogues (Fig. 1c).51,52 Both catalysts are synergic and in both cases the rate limiting step involves metal coordinated epoxide being attacked by a carbonate/carboxylate nucleophile. This means that the selectivity limiting step, i.e. carbon dioxide/anhydride insertion, is pre-rate limiting and as such there's no obvious way to predict selectivity from mixtures of carbon dioxide and anhydride but terpolymerization investigations are expected to reveal it.
Each catalyst was exposed to a mixture of 100 equiv. tricyclic anhydride, TCA, neat cyclohexene oxide (∼2000 equiv.), 10 equiv. 1,2-cyclohexenediol (CHD), and 1 bar carbon dioxide pressure, at 100 °C (Scheme 1a). The diol, CHD, functions as a chain transfer agent and results in the formation of α,ω-hydroxyl-telechelic polymers (Fig. S1†).3 The reactions were monitored using in situ IR spectroscopy by measuring the change in absorption intensity of characteristic anhydride (1780 cm−1) and polycarbonate (1230 cm−1) resonances against time. Catalyst 1 showed selective anhydride consumption and formation of poly(TCA-alt-CHO) (Fig. S5 and S6†), hereafter shortened to the abbreviation PE (Table 1). In contrast, catalyst 2 showed selective poly(cyclohexene carbonate), PCHC, formation without any PE (Fig. S7 and S8†) (Table 1, entries 1 and 2). Catalyst 3 (Zn(II)Mg(II)) showed the simultaneous formation of both PE and PCHC linkages in an approximate ratio 1:3 (PE:PCHC) by aliquot analysis using 1H NMR spectroscopy (Fig. S9†), indicative of faster carbon dioxide than anhydride consumption (Table 1, entry 3). The composition of the crude and purified polymer remained unchanged, as determined by 1H NMR spectroscopy, supporting the formation of a random copolymer, i.e. poly(carbonate-r-ester), rather than mixtures of different polymers (Fig. S10†). The polymer showed a single diffusion coefficient for all signals by DOSY NMR spectroscopy which, again, is indicative of copolymer formation rather than mixtures of PE and PCHC (Fig. S11†). New signals for the carbonyl resonances were observed in the 13C{1H} NMR spectrum (Fig. S12†). Catalyst 4 (Mg(II)Co(II)) showed equivalent selectivity to 2 and formed only polycarbonate without any polyester (Fig. S13 and S14†). Its activity was ∼6 times greater than 2, consistent with the previously observed synergy between Co(II) and Mg(II) centres (TOFMgMg = 112 h−1vs. TOFMgCo = 640 h−1, Table 1). All the catalysts showed well controlled polymerizations (Đ <1.2) and produced polyols.
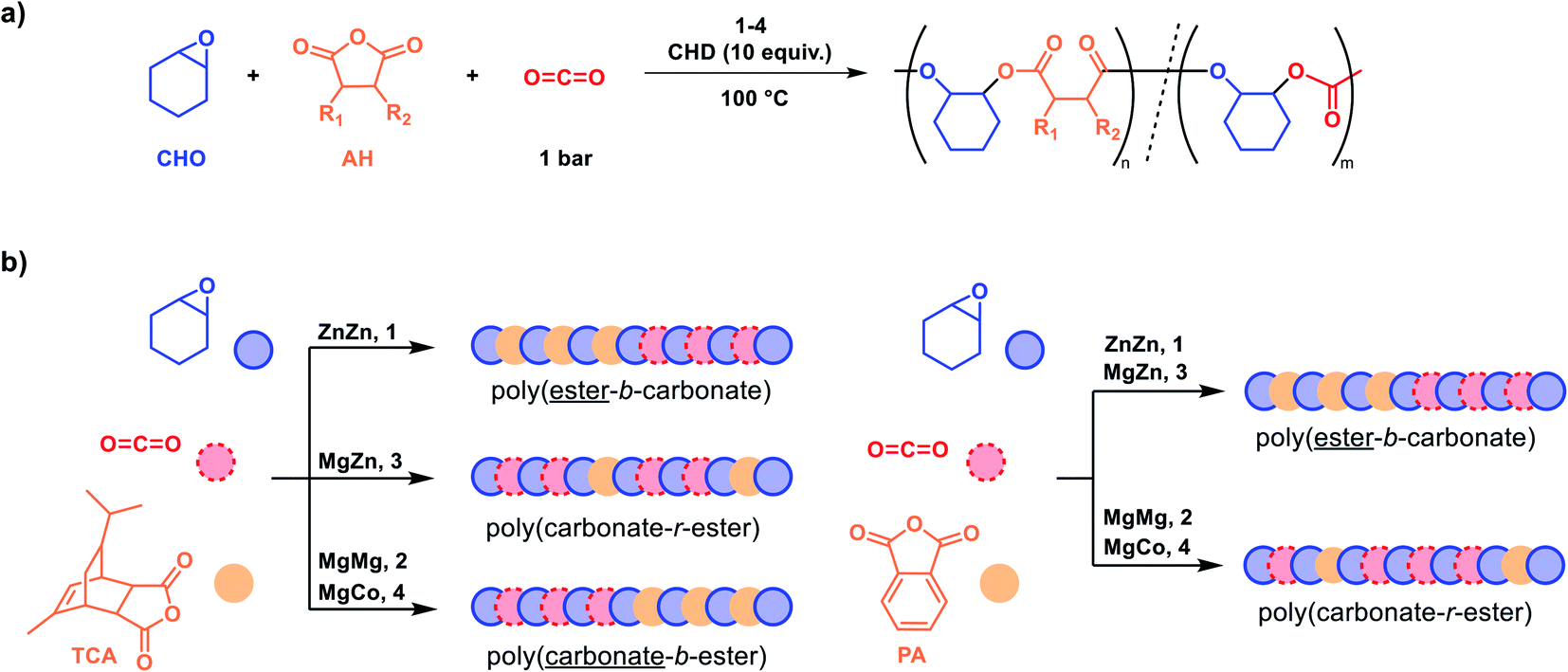 | ||
| Scheme 1 (a) Schematic showing the synthesis of different copolymers from mixtures of cyclohexene oxide (CHO), anhydride (AH), and CO2, with catalysts 1–4. (b) Summary of catalyst selectivity and the resulting polymer architectures. Note that changing from CO2 to N2 atmosphere is required when using catalysts 2 and 4 to ensure ester formation. | ||
| Entry | AH | Catalyst | Time (h) | TONb | TOF (h−1)c | Composition (%) | M n (kg mol−1) [Đ]d | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AH | CHO | PE | PCHC | PE | PCHC | |||||
|
a Polymerization conditions: [cat]:[CHD]:[AH]:[CHO] = 1:10:100:2000, 100 °C, 1 bar CO2.
b Turnover number (TON), number of moles of AH or CHO consumed/number of moles of catalyst. Moles consumed determined from the 1H NMR spectrum by comparing normalized integrals for polycarbonate (4.6 ppm), CHO (3.12 ppm), TCA (5.78 ppm), PA (7.90 ppm), PE (5.70–5.80 and 4.6 ppm) and PE′ (7.50 and 5.14 ppm) resonances. (Fig. S26 and S27).
c Determined by GPC, in THF, calibrated with narrow molar mass polystyrene standards. (Fig. S28–S36).
d Turnover frequency (TOF), TON/time (h).
e [cat]:[CHD]:[AH]:[CHO] = 1:10:100:6000 20 bar CO2.
|
||||||||||
| 1 | TCA | 1 | 7.7 | 82 | 82 | 11 | 0 | 100 | 0 | 1.7 [1.16] |
| 2 | 2 | 1.5 | 0 | 168 | 0 | 112 | 0 | 100 | 2.3 [1.15] | |
| 3 | 3 | 1.5 | 100 | 407 | 67 | 205 | 25 | 75 | 4.8 [1.14] | |
| 4 | 4 | 1.0 | 0 | 640 | 0 | 640 | 0 | 100 | 7.2 [1.13] | |
| 5 | PA | 1 | 5.7 | 70 | 70 | 13 | 0 | 100 | 0 | 1.3 [1.19] |
| 6 | 2 | 1.0 | 62 | 180 | 62 | 118 | 34 | 64 | 2.3 [1.14] | |
| 7 | 3 | 0.5 | 52 | 52 | 104 | 0 | 100 | 0 | 3.2 [1.11] | |
| 8 | 4 | 1.0 | 95 | 530 | 95 | 435 | 18 | 82 | 5.3 [1.09] | |
| 9e | 4 | 0.6 | 30 | 1440 | 50 | 2350 | 2 | 98 | 5.4 [1.25] | |
To understand better the differences in catalyst selectivity, the sterically hindered tricyclic anhydride was replaced with lower steric hindrance phthalic anhydride (PA, a commercial monomer used at scale in polymer production). Using either catalyst 1 or 3 resulted in faster anhydride insertion and formation of polyester, i.e. PA > CO2 (Table 1, entries 5 and 7) (Fig. S15–S17†). However, using 1 bar pressure of CO2 with either 2 or 4, formed a random copolymer with ∼1:4 (PE′:PC) ester:carbonate linkages implying CO2 > PA (Table 1, entry 6 and 8). In these cases, the aliquots taken throughout the polymerization showed consistent ratios indicating the random ester linkages were distributed within the polymer backbone (Fig. S18†). Characteristic peaks for poly(PA-alt-CHO) or PE′ (5.13 ppm), PCHC (4.61 ppm) and junction units (4.98 and 4.47 ppm) were observed in the 1H NMR spectrum (Fig. S18 and S19†). In situ IR spectroscopic analysis also showed the continual evolution of the carbonate (1230 cm−1) resonance and PA consumption (1779 cm−1) (Fig. S20†). Further, consistent uptake of carbon dioxide was observed throughout the reaction (Fig. S21†). Thus, both monomers, i.e. PA and CO2, are consumed throughout the polymerization but carbon dioxide uptake is faster. The resulting random copolymer shows a 13C{1H} NMR spectrum with carbonyl resonances (∼154 ppm) at signals distinct from either PE′ or PCHC, once again indicative of random ester enchainment (Fig. S22†). DOSY NMR analysis, of the purified polymer, displays a single diffusion coefficient consistent with all monomers being enchained in a single copolymer structure (rather than mixtures of different polymers) (Fig. S23†).
The most active catalyst, Mg(II)Co(II), shows a significant but slower rate for PA vs. carbon dioxide insertion at 1 bar pressure. It was envisaged that at a higher CO2 pressure (20 bar) the rate of carbon dioxide insertion might be biased to favour polycarbonate formation. The polymerization of CHO and PA under 20 bar CO2 resulted in nearly quantitative selectivity for PCHC, as confirmed using in situ IR spectroscopy (Fig. S24†). The resulting polymer comprises 98% carbonate and just 2% ester linkages (Fig. S25†). Releasing the carbon dioxide pressure (to <1 bar) after 35 min of reaction, resulted in anhydride consumption and formed a second random copolymer block, as confirmed by NMR spectroscopy (Fig. S25†). Apparently residual carbon dioxide, likely dissolved in solution, competes with anhydride in these insertion reactions. This finding is consistent with the insertion chemistry being kinetically controlled and with these catalysts being highly active for CO2/epoxide ROCOP, even under <1 bar CO2 pressure.53
The benefits of the heterodinuclear catalysts, especially 4, in terms of rate are also apparent compared to the homodinuclear catalysts. For example, at 1 bar pressure catalyst 4 is 20 times faster for CO2/CHO (TOF ∼32 h−1 for [1]:[CHO] = 1:1000, 1 bar CO2, 100 °C)26 and 7 times more active for PA/CHO ROCOP than the di-zinc catalyst 1 (Table 1, entries 5, 8). Compared with the equivalently selective, di-Mg(II) catalyst 2, its activity is 6 times higher for CO2/CHO ROCOP (Table 1, entries 2, 4). When applied under optimized conditions, catalyst 4 achieves an impressive activity of 2400 h−1 for CHO/CO2 ROCOP and maintains high PA/CHO activity even at higher catalyst dilution (Table 1, entry 9).
Multiblock carbon dioxide derived polymers
The most active catalyst 4, Mg(II)Co(II), selects for carbon dioxide insertion even under 1 bar pressure. We reasoned that it might be a useful candidate to make multi-block polymers if the gas atmosphere could be reversibly altered (Scheme 2). Such switching would be an appealing means to control carbonate linkage placement within the polymer but it remains unexplored in this field. A triple stainless-steel manifold was constructed allowing for reactions to be subjected to carbon dioxide, nitrogen or vacuum atmospheres (Fig. S37 and S38† for an illustration). Using this apparatus, reactions were conducted under 1 bar pressure of carbon dioxide or nitrogen and switching between these gases is achieved through vacuum–refill cycles; experiments showed that ∼6 such cycles are sufficient to reduce carbon dioxide levels in the reaction flask. It is also noted that provided such cycling occurs at temperatures <100 °C, there is no loss of any monomers (as evidenced through external ‘traps’ and mass balance experiments). The apparatus was used to polymerize mixtures of TCA, CHO and carbon dioxide, exposed to 12 μmol solutions of 4, in toluene; toluene was used to balance a rate law dependent upon epoxide concentration (i.e. maximise epoxide concentration for highest rates) and the increasing solution viscosity, leading to diffusion limitation to rate, for reactions conducted in neat epoxide at higher polymer molar mass. A ten-fold excess vs. catalyst of cyclohexane diol was also added to select for α,ω-hydroxyl-telechelic polymers (Scheme S1†). The reaction was monitored by regular removal of aliquots, always prior to changing the reaction gas. The gas switches were timed to ensure that each block showed a degree of polymerization (![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif)
![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) ) >6. This quantity was determined previously as the lower bound of confidence in complete block enchainment for multi-block polymer structures, given narrow dispersity molar mass distributions.54 Overall, in all cases, block polymer formation was confirmed by NMR spectroscopy, gel permeation chromatography (GPC) analysis and in situ IR spectroscopy (Fig. S40–S51†). Firstly, the reaction was conducted with just one switch – starting under carbon dioxide and, after 1 h, switching the gas with nitrogen. It was predicted that the catalyst should form an ABA triblock polymer based on the faster carbon dioxide than anhydride insertion chemistry, as mentioned previously (B = PCHC, A = PE; Table 2, entry 1). The reaction was conducted under carbon dioxide for 1.3 hours, at which point aliquot analysis revealed the selective formation of PCHC, showing a molar mass of 6.7 kg mol−1 (Ð = 1.15) (Fig. S40†). The reaction atmosphere gas was changed to N2 and the formation of PE was observed by in situ IR spectroscopy, as the intensity of the anhydride band at 1780 cm−1 decreased (Fig. S41†). Aliquots analysed by NMR spectroscopy confirmed the finding, as the anhydride's sharp doublet peak at 5.77 ppm turned into a broader signal (5.5–5.9 ppm) (Fig. S42†). The formation of an ABA-block polymer was indicated by NMR spectroscopy since block compositions match theoretical predictions based on monomer consumption, by an increase in the molar mass to 9.1 kg mol−1 and by DOSY NMR spectroscopy where a single diffusion coefficient was observed for all signals (Fig. S44†). Next, a BABAB pentablock was targeted by two changes in gas atmosphere – starting under carbon dioxide, switching to nitrogen and then switching back to carbon dioxide (Table 2, entry 2). The reaction was initiated under a CO2 atmosphere and PCHC formation was indicated by in situ IR spectroscopy (Fig. S45†) and aliquot analysis (NMR spectroscopy) (Fig. S46†); the polycarbonate had a molar mass of 7.5 kg mol−1 (Fig. S47†). The atmosphere was switched to N2 and after 2.3 h the complete consumption of the anhydride was observed as a plateau in the conversion vs. time data (IR spectroscopy) (Fig. S45†). The intermediary ABA block polymer showed a molar mass of 9.4 kg mol−1 (Fig. S47†). Finally, the atmosphere was switched back to CO2 allowing for polycarbonate block growth and the formation of a BABAB pentablock polymer with molar mass of 14.0 kg mol−1 (Fig. S47†).
) >6. This quantity was determined previously as the lower bound of confidence in complete block enchainment for multi-block polymer structures, given narrow dispersity molar mass distributions.54 Overall, in all cases, block polymer formation was confirmed by NMR spectroscopy, gel permeation chromatography (GPC) analysis and in situ IR spectroscopy (Fig. S40–S51†). Firstly, the reaction was conducted with just one switch – starting under carbon dioxide and, after 1 h, switching the gas with nitrogen. It was predicted that the catalyst should form an ABA triblock polymer based on the faster carbon dioxide than anhydride insertion chemistry, as mentioned previously (B = PCHC, A = PE; Table 2, entry 1). The reaction was conducted under carbon dioxide for 1.3 hours, at which point aliquot analysis revealed the selective formation of PCHC, showing a molar mass of 6.7 kg mol−1 (Ð = 1.15) (Fig. S40†). The reaction atmosphere gas was changed to N2 and the formation of PE was observed by in situ IR spectroscopy, as the intensity of the anhydride band at 1780 cm−1 decreased (Fig. S41†). Aliquots analysed by NMR spectroscopy confirmed the finding, as the anhydride's sharp doublet peak at 5.77 ppm turned into a broader signal (5.5–5.9 ppm) (Fig. S42†). The formation of an ABA-block polymer was indicated by NMR spectroscopy since block compositions match theoretical predictions based on monomer consumption, by an increase in the molar mass to 9.1 kg mol−1 and by DOSY NMR spectroscopy where a single diffusion coefficient was observed for all signals (Fig. S44†). Next, a BABAB pentablock was targeted by two changes in gas atmosphere – starting under carbon dioxide, switching to nitrogen and then switching back to carbon dioxide (Table 2, entry 2). The reaction was initiated under a CO2 atmosphere and PCHC formation was indicated by in situ IR spectroscopy (Fig. S45†) and aliquot analysis (NMR spectroscopy) (Fig. S46†); the polycarbonate had a molar mass of 7.5 kg mol−1 (Fig. S47†). The atmosphere was switched to N2 and after 2.3 h the complete consumption of the anhydride was observed as a plateau in the conversion vs. time data (IR spectroscopy) (Fig. S45†). The intermediary ABA block polymer showed a molar mass of 9.4 kg mol−1 (Fig. S47†). Finally, the atmosphere was switched back to CO2 allowing for polycarbonate block growth and the formation of a BABAB pentablock polymer with molar mass of 14.0 kg mol−1 (Fig. S47†).
| Entry | Block polymer structure | Reaction gas | Time (h) | TONe | Block polymer composition (%) | M n (kg mol−1) [Đ] |
|
T g (°C) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| TCA | CHO | PE | PCHC | |||||||
|
a All entries run at 100 °C at 1 bar pressure of CO2, 4 M CHO in Toluene.
b [cat]:[CHD]:[TCA]:[CHO] = 1:10:200:2000.
c [cat]:[CHD]:[TCA]:[CHO] = 1:10:300:3000.
d [cat]:[CHD]:[TCA]:[CHO] = 1:10:600:6000.
e Turnover number (TON), number of moles of TCA or CHO consumed/number of moles of catalyst. Moles consumed determined from the 1H NMR spectrum by comparing normalized integrals for PCHC (4.6 ppm), CHO (3.12 ppm), TCA (5.75 ppm) and PE (5.68 ppm) resonances.
f Determined by GPC, in THF, calibrated with narrow molar mass polystyrene standards.
g Determined from TON/number of growing chains (initiated by CHD only).
h Value for multi-block polymer determined by DSC, at 20 °C min−1 heating rate, and taken from the second heating/cooling cycle.
|
||||||||||
| 1b | ABA | CO2 | 1.3 | 0 | 740 | 0 | 100 | 6.7 [1.15] | 74 | 121 |
| N2 | 1.2 | 200 | 940 | 21 | 79 | 9.1 [1.17] | 10 | |||
| 2c | BABAB | CO2 | 1.0 | 0 | 750 | 0 | 100 | 5.7 [1.13] | 75 | 122 |
| N2 | 2.3 | 300 | 1050 | 29 | 71 | 9.4 [1.17] | 15 | |||
| CO2 | 1.7 | 300 | 1777 | 17 | 83 | 14.0 [1.12] | 37 | |||
| 3d | ABABABA | CO2 | 1.6 | 0 | 1020 | 0 | 100 | 6.0 [1.14] | 102 | 130 |
| N2 | 2.2 | 360 | 1380 | 26 | 74 | 11.1 [1.11] | 18 | |||
| CO2 | 1.7 | 360 | 2520 | 14 | 82 | 17.4 [1.09] | 57 | |||
| N2 | 4.5 | 600 | 2700 | 20 | 80 | 19.4 [1.07] | 12 | |||
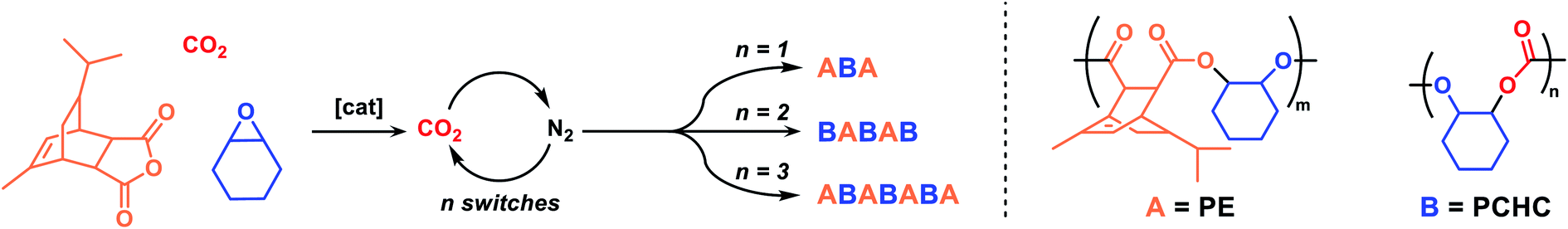 | ||
| Scheme 2 Multi-block polymer production using catalyst 4 with mixtures of TCA, CHO, and CO2. | ||
Lastly, an ABABABA heptablock polymer was prepared by three switches of gas atmosphere and following a similar protocol to that described for the pentablock polymer (Table 2, entry 3). In situ ATR-IR spectroscopy confirmed the monomer selectivity at each stage with formation of polycarbonate (1230 cm−1) or polyester (1780 cm−1), as the atmosphere was changed from carbon dioxide to nitrogen (Fig. 2a). Block polymer formation was also confirmed through NMR spectroscopy of aliquots (Fig. S49†) and GPC, with systematic increases to the polymer molar mass and narrow distributions in all cases (Fig. 2c).
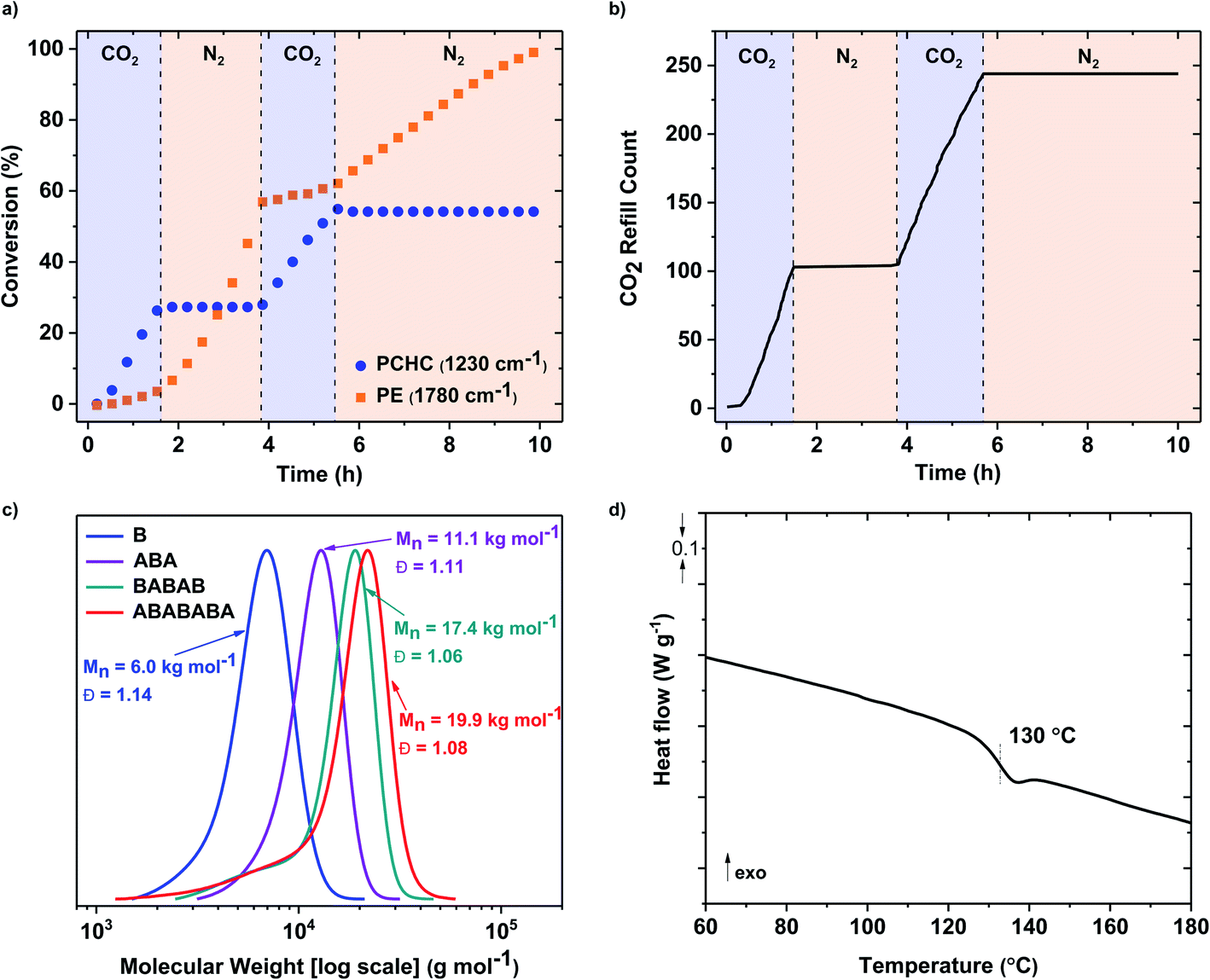 | ||
| Fig. 2 Data supporting the selective catalysis to form an ABABABA heptablock polymer. Polymerisation conditions 4: CHD:TCA:CHO 1:10:600:6000, 1 bar CO2, 100 °C, 4 M CHO in toluene. (a) In situ IR spectroscopy data are used to construct a plot of conversion vs. time showing selective formation of polycarbonate and polyester blocks. (b) The mass-flow controller output is used to determine the frequency of CO2 refill into the reaction vessel, corresponding to the consumption of CO2, as in (a). (c) Analysis of reaction aliquots using GPC, shows a continual evolution of molar mass after each block is formed with narrow dispersity distributions (note that at higher block configurations there is some low molar mass ‘tailing’ to data which may arise either from contamination by acetate initiated chains and/or from slower initiation from polymeric cyclohexanol end-capped chains). (d) DSC data for the heptablock polymer, determined at 20 °C min−1 heating rate, and taken from the second heating/cooling cycle. | ||
The three new multi-block polymers were also analysed using DSC, with all materials being amorphous and showing only a single glass transition temperature consistent with block miscibility (Fig. 2d, S50 and S51†). The Tg values of the multi-blocks increase with the number of blocks, although this may also correlate with the overall increase to molar mass. The Tg values are also all >100 °C and are consistent with previously reported values for PCHC (115 °C)55 and PE (118–126 °C).26,56,57 In general, polymers showing high glass transition temperatures are useful since a limitation of current aliphatic polyesters, produced by cyclic ester ring-opening polymerization, has been the moderate/low Tg which results in undesirable softening within useable temperature ranges.58
Pentablocks from CO2/anhydride/epoxide/lactone
In 2016, a DFT investigation of catalyst 1 predicted that a mixture of 4 monomers, namely phthalic anhydride (PA), carbon dioxide, cyclohexene oxide (CHO), and ε-caprolactone (CL), should form only CBABC-pentablock polymer structures (where A = poly(PA-alt-CHO), PE′; B = PCHC and C = polycaprolactone PCL).35 According to the analysis of both barriers and linkage stabilities, the insertion order, into the zinc alkoxide intermediate, should be: PA > carbon dioxide > caprolactone.35 In 2018, we reported difficulties in experimentally realising this prediction using a di-zinc catalyst because the PCHC chain end underwent backbiting to form cyclic carbonate faster than it initiated lactone ROP.48 Using catalyst 1, it was possible to prepare a pentablock BCACB-pentablock (where A = PE′, B = PCHC, C = poly(ε-decalactone), PDL) by adding carbon dioxide after selective catalysis using anhydride, epoxide and lactone to form a CAC triblock polymer.48 So far, no other catalysts have been shown to selectively enchain from the mixture of these four monomers. We posited that using catalyst 4, which has distinctive carbon dioxide/anhydride selectivity, should access other pentablock polymers and, in particular, allow production of CABAC patterns. Such an enchainment is predicted since PCHC formation occurs before PE from mixtures and by installing PE end-groups the undesirable PCHC back-biting reactions might be obviated allowing for initiation of lactone ROP, once the carbon dioxide is removed.Catalyst 4 was added to a solution of TCA (100 equiv.), ε-decalactone (DL, 100 equiv.), CHO (∼2000 equiv.), CHD (10 equiv.) and 1 bar pressure of CO2, at 100 °C. The catalysis proceeded with first formation of PCHC (polycarbonate) followed, after switching carbon dioxide for nitrogen gas, by formation of the PE block (TCA/CHO ROCOP). Once the anhydride was consumed, the reaction stopped and no polymerization of DL was observed, even after 8 hours with heating. Aliquot analysis at this point, by 1H NMR spectroscopy, confirmed formation of a BAB polymer (B = PE, A = PCHC) but also showed new low intensity signals at 4.93 ppm attributed to cyclohexene carbonate (∼1%) (Fig. S52†). The formation of cyclic carbonate is curious since it was absent from aliquots removed prior to complete anhydride consumption. Thus, it seems that the formation of cyclic carbonate is not a result of PCHC block degradation and consistent with this notion, there was no change to the polymer molar mass or dispersity values after failure to initiate DL ROP, as indicated by GPC. Rather, it is proposed that the cyclic carbonate forms from low levels of dissolved carbon dioxide, perhaps in equilibrium with vapour phases, which preferentially inserts into the PE block zinc alkoxide chain end-group. Such a finding would be consistent with the decomposition of PCHC end-groups to cyclic carbonate and failure to initiate DL ROP.48 To investigate this phenomenon, a mixture of 4, TCA, DL and CHO, heated to 100 °C, resulted in the formation of a triblock polymer of the form CAC (C = PDL, A = PE); aliquot analysis confirmed the formation of PE, followed by PDL blocks (Fig. S53 and S54†). This experiment confirms the feasibility of PE end-group initiation of DL ROP. It seems that the polymer end-group sterics/rigidity are not limiting pentablock formation. Accordingly, even low levels of contaminating carbon dioxide appear to result in preferential insertion into metal alkoxide intermediates and trigger a side-reaction in which the carbonate decomposes slowly over time to form cyclic carbonate. To investigate the importance of the relative rate of lactone initiation on pentablock formation, polymerizations using CL in place of DL were undertaken since CL propagates by a primary alkoxide (whereas DL propagates with a secondary alkoxide) (Fig. S55†). Further, there is already precedent for switch catalysis using mixtures of CHO, CO2 and CL to form block polymers.59 A mixture of 4, TCA (100 equiv.), CL (200 equiv.), CHO (∼2000 equiv.) and 1 bar pressure CO2, heated to 100 °C was polymerized, with the carbon dioxide being exchanged for nitrogen after 0.5 h (Fig. 3a). The catalyst selectively formed the target CABAC-pentablock polymer (where A = PE; B = PCHC and C = PCL) (Table 3). First, the polycarbonate PCHC forms consistent with the catalyst's fast carbon dioxide insertion and was confirmed by NMR spectroscopy where only resonances due to polycarbonate and the unreacted other monomers were observed (Fig. 3c). At this point, the carbon dioxide was switched for nitrogen and TCA/CHO ROCOP occurred. An aliquot removed after 2 hours, shows the growth of the polyester block and GPC analysis shows an increase in molar mass to 5.2 kg mol−1 (Fig. 3b). Once all the anhydride was converted, CL ROP occurred. GPC analysis of the aliquots demonstrate an increase in molar mass consistent with block polymer formation (Fig. 3b). The final aliquot, after PCL growth, shows a broader dispersity attributed to slower rates of initiation (from a secondary cyclohexanol group) vs. propagation (from a primary hydroxyl group) during CL ROP.60 The final pentablock polymer was analysed by DSC and showed a single Tg value of 86 °C, indicative of block miscibility and demonstrating the utility in incorporating ‘softer’ aliphatic polyesters (Fig. S59†).
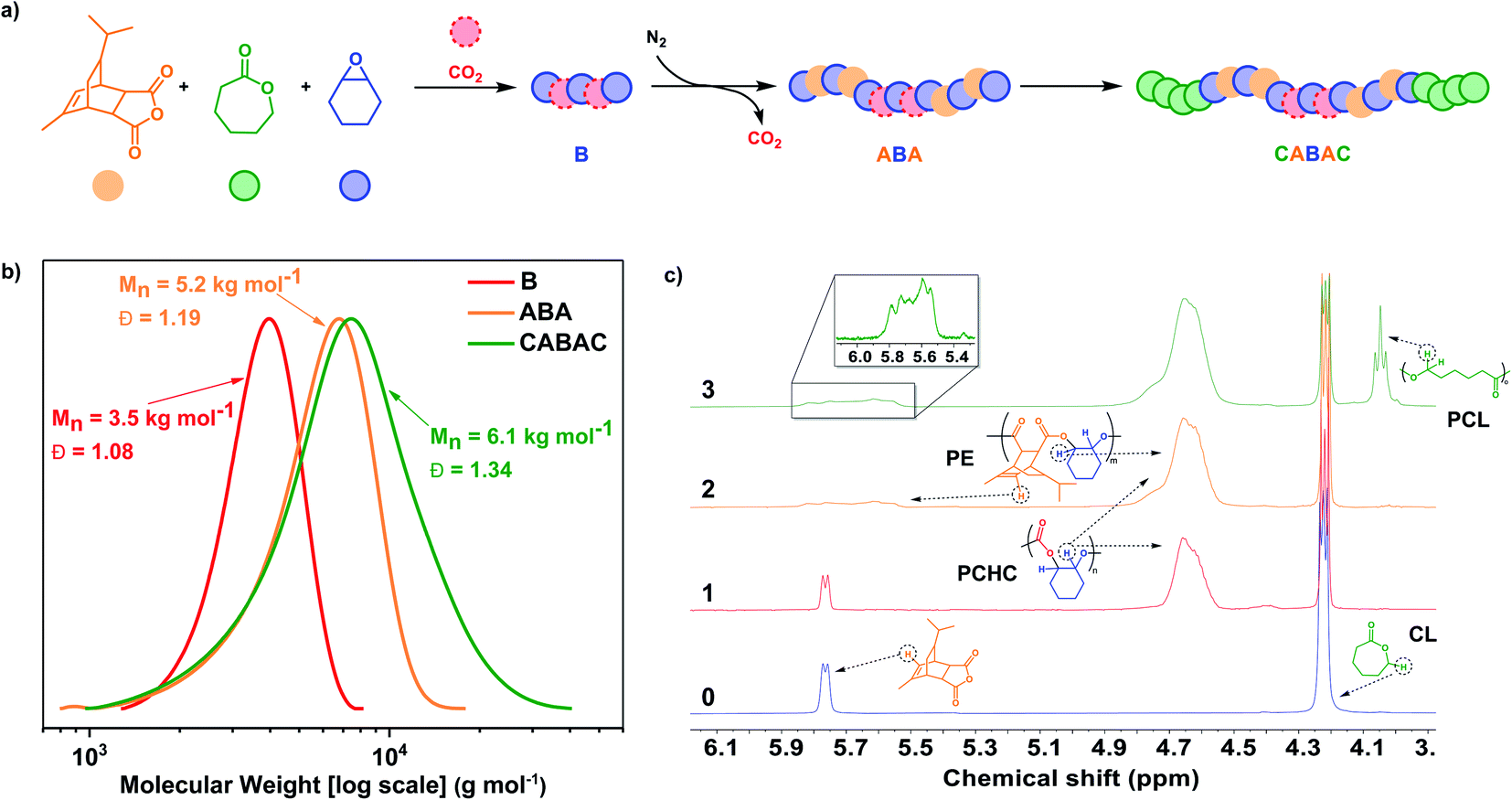 | ||
| Fig. 3 Selective polymerization catalysis using 4 and mixtures of TCA (orange), CL (green), CHO (blue), and carbon dioxide (red). Polymerization conditions: [4]:[CHD]:[TCA]:[CL]:[CHO] = 1:10:100:200:2000, 1 bar CO2, 100 °C (a) illustration of the block polymer structure and simplified representation using coloured ‘balls’ to represent different monomers. (b) GPC analysis of aliquots removed at different times (corresponding to entries 1, 2, and 3 in Table 3) during the reaction and showing the formation of PCHC, PE-PCHC-PE and PCL-PE-PCHC-PE-PCL. (c) Stack plot showing a selected region of the 1H NMR spectra of aliquots removed during formation of the polymer blocks. Spectrum 0 is the mixture before polymerization. The first block that formed is PCHC, shown by the resonance at 4.6 ppm, the second is PE, shown by the loss of the sharp doublet into a broad polymer signal at 5.8 ppm, and the final PCL block in 3 from the new signal at 4.0 ppm. For complete spectra see Fig. S56.† | ||
| Entry | Reaction gas | Time (h) | TONb | TOF (h−1)c | M n (kg mol−1) [Đ]d | ||
|---|---|---|---|---|---|---|---|
| TCA | CL | CHO | |||||
|
a [cat]:[CHD]:[TCA]:[CL]:[CHO] = 1:10:100:200:2000, 100 °C, entry 1 under 1 bar of CO2 for 0.5 h, Entry 2 under 1 bar of N2 for 1.5 h and 3 for 3.0 h.
b Turnover number (TON), number of moles of TCA or CHO consumed/number of moles of catalyst. Moles consumed determined by 1H NMR, by comparison of the relative integrals of the resonances due to monomer (TCA 5.73 ppm, CHO 3.08 ppm, CL 4.18 ppm) and polymer (PE 4.63 ppm, PCHC 4.63 ppm, PCL 4.02 ppm) (Fig. S56).
c Turnover frequency (TOF), TON/time (h).
d Measured by GPC, in THF, using narrow MW polystyrene standards to calibrate the instrument.
|
|||||||
| 1 | CO2 | 0.5 | 0 | 0 | 325 | 650 | 3.5 [1.08] |
| 2 | N2 | 1.5 | 100 | 0 | 425 | 67 | 5.2 [1.19] |
| 3 | N2 | 3.0 | 100 | 86 | 460 | 29 | 6.1 [1.34] |
Discussion
These polymerizations show that carbon dioxide insertion rates, are strongly metal dependent. Even though such rates are difficult to directly measure as they occur in the pre-rate limiting step in catalysis (k2 and k3 in Fig. 4a), they can be inferred from the monomer selectivity from mixtures. Given that other CO2/epoxide ROCOP catalysts also show zeroth order rate dependencies on carbon dioxide pressure, the findings are likely more broadly significant.19,61–64 Accordingly, for this series of catalysts the carbon dioxide insertion rates follow the trend Co(II) > Mg(II) > Zn(II). In these polymerizations, where selectivity occurs through competition between anhydride and carbon dioxide, catalysts featuring Mg(II) are selective for CO2 insertion, whereas the di-Zn(II) analogues are selective for anhydride insertion. Most other catalysts in this field are also highly selective for anhydride insertion.6,14,22,23,27,65,66 There are very few other examples of catalysts selective for carbon dioxide over anhydride.20,25,46 This year, Feng and co-workers reported specific conditions under which a triethyl borane and bis(triphenylphosphine)iminium chloride (PPNCl) catalyst system (I) formed random copolymers from mixtures of PA/CHO/CO2 (Fig. 4b).46 These reactions required 10 bar CO2 pressure and only yielded the randomized enchainment at ratios of PPNCl:Et3B:PA:CHO in the range 1:3:50–80:200, i.e. at very high loading of catalyst relative to PA.46 At lower catalyst loadings/higher PA proportion, the catalysts were deactivated. A further issue is that the polymers all show bimodal molar mass distributions which would preclude any selective block or multi-block formation. Finally, it should be noted that the organo-catalytic system must be very finely balanced since Lu and co-workers showed that the same catalyst system exposed to the same monomer ratios but at 1 bar CO2 pressure formed the expected poly(ester-b-carbonate) – i.e. anhydride insertion was selected.27 Another limitation of this organocatalyst system is its modest activity (TOFCHO/PA = 5 h−1, TOFCHO/CO2 = 3.5 h−1, PPNCl:Et3B:PA:CHO:CO2, 1:3:80:200, 16 h, 10 bar, 80 °C). In comparison catalyst 4 shows higher activity (TOFCHO/PA = 95 h−1, TOFCHO/CO2 = 460 h−1, 4:[CHD]:[AH]:[CHO] = 1:10:100:2000, 100 °C, 1 bar CO2, Table 1, entry 8). Coates and co-workers found that at CO2 pressures >27 bar, the Zn(II) catalyst (II) randomly inserted CO2 into the DGA/CHO polyester.20
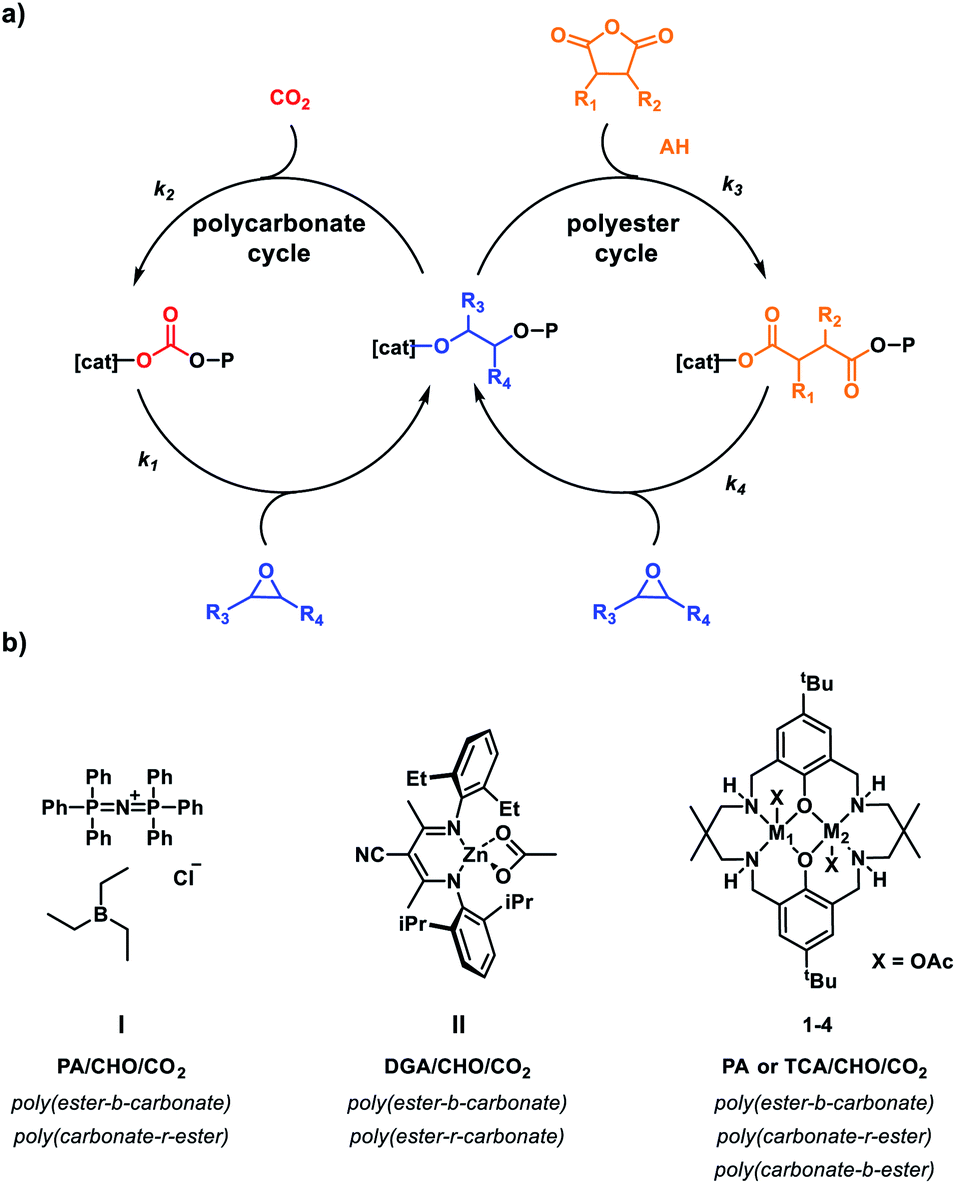 | ||
| Fig. 4 (a) Catalytic cycles for carbon dioxide/epoxide and anhydride/epoxide ROCOP which are accessed by the catalysts to build up block polymers. (b) Catalysts which exhibit unexpected selectivity for carbon dioxide vs. anhydride (k2 > k3) depending on reaction conditions (pressure, concentration, monomer, metal centre). | ||
In terms of future directions for these dinuclear catalysts, other catalysts containing Group 1 or 2 metals should be explored to understand whether they also show beneficial influences over carbon dioxide insertion chemistry. Recently highly active heterodinuclear Co(III)M(I) (M = Na, K) catalysts for carbon dioxide/epoxide ROCOP were reported and these species should also be explored in switchable catalysis.63,67 The findings also underscore the central importance of the metal selection in dictating both the polymerization activity (rate determining step) and selectivity (pre-rate limiting step). In particular, inexpensive and light Mg(II)-based catalysts appear to be beneficial in selecting for carbon dioxide placement over anhydride in more complex polymer structures.
This work demonstrates a straightforward method to make multi-block polymers. Until now, such structures were not investigated and any preparations would have required several sequential monomer additions; such processes can be difficult to control, time correctly and may also introduce impurities which compromise molar mass distributions. In contrast, here the best Mg(II)Co(II) catalyst yields, by simple gas environment switches, tri-, penta- and heptablock structures. This work also demonstrates its potential to sequentially access three different polymerization pathways: epoxide/anhydride ROCOP, carbon dioxide/epoxide ROCOP and lactone ROP. There is just one prior report of a di-Zn(II) catalyst able to enchain selectively from these four monomers.48 In comparison, 4 yields both a new multi-block structure and accelerates the rate by 10 fold for CHO/CO2 ROCOP and 3 fold for the AH/CHO ROCOP (TOFPCHC = 650 h−1 for 4vs. 6 h−1 for 1; TOFPE = 67 h−1 for 4vs. 27 h−1 for 1).48
Here, the focus has been on improving the catalytic selectivity, activity and in making new multi-block structures. These catalytic discoveries should be generalizable to a wide range of new structures and property investigation is warranted. Controlling the block sequence is obviously a means to moderate the polymer's glass transition temperature and end-group chemistry. Here, such selectivity relies upon the dinuclear catalysts being applied with excess (>10 equiv. vs. catalyst) of a chain transfer agent, such as cyclohexane diol, and the resulting polymers are low molar mass hydroxyl-telechelic, i.e. polyols.3 Such polyols could be useful to make higher polymers like polyurethanes, cross-linked resins or as surfactants.13,68,69 The new structures accessible using these catalysts should prove valuable in furthering understanding of how polymer composition and sequence influence the polyol rheology, viscosity, reactivity through end-group chemistry, thermal stability and degradation chemistry.4 It is now well-established that alkene-functionalized monomers, including vinyl-cyclohexene oxide, can be efficiently and quantitatively post-functionalized to install carboxylic acid, hydroxyl, amine and sulfonate functional groups to the polymer backbone.36,70–76 In future, combining these selective catalyses with post-functionalizations could deliver new surfactants, self-assembled nanostructures, colloids and electrolytes without needing to increase polymer molar mass.72,73 An important target for future study are catalysts featuring organometallic initiators since these species allow for reduced loading of chain transfer agent and access higher molar mass polymers.38 Such materials would be expected to undergo phase separation and there is emerging evidence that multi-block polymers may afford new nanostructures and improve upon thermal mechanical properties compared with equivalent di- or triblock structures.47,77,78
Conclusions
Highly active and selective switchable polymerization catalysts form multi-block polymers featuring ester and carbonate linkages, with predictable and controlled placement of both the monomers and the blocks in the overall polymer structure. The catalysts, coordinated by a macrocyclic ancillary ligand, feature both homo- and heterodinuclear combinations of metals [Mg(II)Mg(II), Zn(II)Zn(II), Zn(II)Mg(II) and Mg(II)Co(II)]. Catalysts featuring Mg(II) show a strong preference for carbon dioxide vs. anhydride insertion – an unusual finding since almost all other catalysts in this field show the inverse selectivity. Further, the Mg(II)Co(II) catalyst combines very high carbon dioxide selectivity with outstanding rates across both polymerization cycles, allowing for up to 20 fold improvement in activity compared to di-Mg(II) or di-Zn(II) catalysts. The Mg(II)Co(II) catalyst was used to make tri-, penta- and heptablock sequences from mixtures of carbon dioxide, anhydride (tricyclic anhydride, a bio-based monomer) and cyclohexene oxide. It also enchains selectively a four-component mixture (cyclohexene oxide, tricyclic anhydride, carbon dioxide and caprolactone) to make a single sequenced pentablock polymer. These results demonstrate the importance of continued focus on both catalytic activity and selectivity. They may also be significant in polymer materials contexts since by controlling the monomer mixture composition, it is feasible to control the end-group chemistry and glass transition temperature for the resulting polymers. With routes to control sequence in these polymers, in future their polymer chemistry, rheology, degradation chemistry and functional substituents should be investigated. Finally, it is noted that by using carbon dioxide and bio-derived anhydrides it is possible to increase the renewable content in these polymers which may offer benefits in terms of reduced greenhouse gas emissions and sustainability.Data availability
All experimental procedures, charactarisation, and NMR spectra for polymerizations can be found in the ESI.†Author contributions
GR conducted all experimental work, ACD and CKW supervised the research and all authors wrote the manuscript. CKW secured all research funding and leads the research group.Conflicts of interest
CKW is a director of econic technologies.Acknowledgements
The EPSRC (EP/S018603/1; EP/R027129/1), Oxford Martin School (Future of Plastics Programme) and SCG Chemicals (Studentship to GR) are acknowledged for research funding.References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-P. Wu and D. J. Darensbourg, Trends Chem., 2020, 2, 750–763 CrossRef CAS.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- C. Diaz and P. Mehrkhodavandi, Polym. Chem., 2021, 12, 783–806 RSC.
- G. X. De Hoe, M. T. Zumstein, B. J. Tiegs, J. P. Brutman, K. McNeill, M. Sander, G. W. Coates and M. A. Hillmyer, J. Am. Chem. Soc., 2018, 140, 963–973 CrossRef CAS PubMed.
- M. J. Sanford, N. J. Van Zee and G. W. Coates, Chem. Sci., 2018, 9, 134–142 RSC.
- N. Yi, T. T. D. Chen, J. Unruangsri, Y. Zhu and C. K. Williams, Chem. Sci., 2019, 10, 9974–9980 RSC.
- G.-W. Yang and G.-P. Wu, ACS Sustainable Chem. Eng., 2019, 7, 1372–1380 CrossRef CAS.
- S. Ye, W. Wang, J. Liang, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS.
- N. Zhu, X. Hu, Z. Fang and K. Guo, Prog. Polym. Sci., 2021, 117, 101397 CrossRef CAS.
- D. J. Darensbourg, Inorg. Chem. Front., 2017, 4, 412–419 RSC.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- J. Liang, S. Ye, S. Wang, M. Xiao and Y. Meng, Polym. J., 2021, 53, 3–27 CrossRef.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS.
- C. E. Koning, R. J. Sablong, E. H. Nejad, R. Duchateau and P. Buijsen, Prog. Org. Coat., 2013, 76, 1704–1711 CrossRef CAS.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- J. Y. Jeon, S. C. Eo, J. K. Varghese and B. Y. Lee, Beilstein J. Org. Chem., 2014, 10, 1787–1795 CrossRef PubMed.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- P. K. Saini, G. Fiorani, R. T. Mathers and C. K. Williams, Chem. - Eur. J., 2017, 23, 4260–4265 CrossRef CAS.
- J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202104495.
- Y. Liu, K. Deng, S. Wang, M. Xiao, D. Han and Y. Meng, Polym. Chem., 2015, 6, 2076–2083 RSC.
- Y. Liu, K. Huang, D. Peng and H. Wu, Polymer, 2006, 47, 8453–8461 CrossRef CAS.
- Y. Liu, M. Xiao, S. Wang, L. Xia, D. Hang, G. Cui and Y. Meng, RSC Adv., 2014, 4, 9503–9508 RSC.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- T. Stößer, T. T. D. Chen, Y. Zhu and C. K. Williams, Philos. Trans. R. Soc., A, 2018, 376, 20170066 CrossRef.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS PubMed.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668 CrossRef.
- S. Kernbichl, M. Reiter, J. Mock and B. Rieger, Macromolecules, 2019, 52, 8476–8483 CrossRef CAS.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- S. Zhu, Y. Zhao, M. Ni, J. Xu, X. Zhou, Y. Liao, Y. Wang and X. Xie, ACS Macro Lett., 2020, 9, 204–209 CrossRef CAS.
- W. T. Diment, T. Stößer, R. W. F. Kerr, A. Phanopoulos, C. B. Durr and C. K. Williams, Catal. Sci. Technol., 2021, 11, 1737–1745 RSC.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef.
- T. Stößer, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 CrossRef PubMed.
- A. Virachotikul, N. Laiwattanapaisarn, P. Wongmahasirikun, P. Piromjitpong, K. Chainok and K. Phomphrai, Inorg. Chem., 2020, 59, 8983–8994 CrossRef CAS.
- W.-B. Li, Y. Liu and X.-B. Lu, Organometallics, 2020, 39, 1628–1633 CrossRef CAS.
- F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Tejeda, J. Fernández-Baeza, C. Alonso-Moreno, A. M. Rodríguez, J. A. Castro-Osma and A. Lara-Sánchez, Inorg. Chem., 2020, 59, 8412–8423 CrossRef.
- V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS.
- Y. Li, Y.-Y. Zhang, L.-F. Hu, X.-H. Zhang, B.-Y. Du and J.-T. Xu, Prog. Polym. Sci., 2018, 82, 120–157 CrossRef CAS.
- T. T. D. Chen, Y. Zhu and C. K. Williams, Macromolecules, 2018, 51, 5346–5351 CrossRef CAS.
- D. Dakshinamoorthy, A. K. Weinstock, K. Damodaran, D. F. Iwig and R. T. Mathers, ChemSusChem, 2014, 7, 2923–2929 CrossRef CAS.
- N. J. Van Zee, M. J. Sanford and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 2755–2761 CrossRef CAS.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS.
- A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed.
- S. K. Raman, R. Raja, P. L. Arnold, M. G. Davidson and C. K. Williams, Chem. Commun., 2019, 55, 7315–7318 RSC.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- C. Koning, J. Wildeson, R. Parton, B. Plum, P. Steeman and D. J. Darensbourg, Polymer, 2001, 42, 3995–4004 CrossRef CAS.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- Y. Zhu, C. Romain, V. Poirier and C. K. Williams, Macromolecules, 2015, 48, 2407–2416 CrossRef CAS.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- J. Deng, M. Ratanasak, Y. Sako, H. Tokuda, C. Maeda, J.-y. Hasegawa, K. Nozaki and T. Ema, Chem. Sci., 2020, 11, 5669–5675 RSC.
- W. Lindeboom, D. A. X. Fraser, C. B. Durr and C. K. Williams, Chem.–Eur. J., 2021, 27(47), 12224–12231 CrossRef CAS PubMed.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- C.-H. Chang, C.-Y. Tsai, W.-J. Lin, Y.-C. Su, H.-J. Chuang, W.-L. Liu, C.-T. Chen, C.-K. Chen and B.-T. Ko, Polymer, 2018, 141, 1–11 CrossRef CAS.
- S. Pappuru and D. Chakraborty, Eur. Polym. J., 2019, 121, 109276 CrossRef CAS.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- D. J. Darensbourg and F.-T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS.
- G.-W. Yang, Y.-Y. Zhang, Y. Wang, G.-P. Wu, Z.-K. Xu and D. J. Darensbourg, Macromolecules, 2018, 51, 1308–1313 CrossRef CAS.
- B. Han, B. Liu, H. Ding, Z. Duan, X. Wang and P. Theato, Macromolecules, 2017, 50, 9207–9215 CrossRef CAS.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC.
- K. Deng, S. Wang, S. Ren, D. Han, M. Xiao and Y. Meng, ACS Appl. Mater. Interfaces, 2016, 8, 33642–33648 CrossRef CAS PubMed.
- D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC.
- D. J. Darensbourg, W.-C. Chung, C. J. Arp, F.-T. Tsai and S. J. Kyran, Macromolecules, 2014, 47, 7347–7353 CrossRef CAS.
- J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS.
- I. Lee, T. R. Panthani and F. S. Bates, Macromolecules, 2013, 46, 7387–7398 CrossRef CAS.
- E. Grune, M. Appold, A. H. E. Müller, M. Gallei and H. Frey, ACS Macro Lett., 2018, 7, 807–810 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03856g |
| This journal is © The Royal Society of Chemistry 2021 |