
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of protein–ligand interactions by SABRE†
Ratnamala Mandal,
Pierce Pham and
Christian Hilty*
and
Christian Hilty*
Department of Chemistry, Texas A&M University, 3255 TAMU, College Station, TX 77843, USA. E-mail: chilty@tamu.edu
First published on 31st August 2021
Abstract
Nuclear spin hyperpolarization through signal amplification by reversible exchange (SABRE), the non-hydrogenative version of para-hydrogen induced polarization, is demonstrated to enhance sensitivity for the detection of biomacromolecular interactions. A target ligand for the enzyme trypsin includes the binding motif for the protein, and at a distant location a heterocyclic nitrogen atom for interacting with a SABRE polarization transfer catalyst. This molecule, 4-amidinopyridine, is hyperpolarized with 50% para-hydrogen to yield enhancement values ranging from −87 and −34 in the ortho and meta positions of the heterocyclic nitrogen, to −230 and −110, for different solution conditions. Ligand binding is identified by flow-NMR, in a two-step process that separately optimizes the polarization transfer in methanol while detecting the interaction in a predominantly aqueous medium. A single scan Carr–Purcell–Meiboom–Gill (CPMG) experiment identifies binding by the change in R2 relaxation rate. The SABRE hyperpolarization technique provides a cost effective means to enhance NMR of biological systems, for the identification of protein–ligand interactions and other applications.
Introduction
Nuclear spin hyperpolarization has the potential to extend on biological applications of NMR by enhancing signals of molecules including ligands, enzyme substrates, proteins and others. Using hyperpolarized molecules, spectroscopy under conditions close to physiological concentration becomes possible. NMR detection is thus brought closer to the realm of widespread but less structurally specific detection methods for biological interactions, such as fluorescence spectroscopy.Hyperpolarization techniques are readily applicable for the detection of the binding of small molecules to proteins. Non-hyperpolarized NMR alone is a recognized technique for the screening of these interactions in drug discovery. The utility of hyperpolarization by dissolution dynamic nuclear polarization (D-DNP)1 has previously been demonstrated for ligand binding experiments. A reduction in the required target protein concentration can potentially enable NMR based ligand binding experiments with a broader range of proteins that are difficult to purify or that are unstable. D-DNP is capable of hyperpolarizing 13C, 1H, 19F and other nuclei in small molecules. One way of identifying binding of drug candidates is by observing the unique signals from 19F. Different methods of detection are applicable in the strong-, weak- and intermediate-binding regimes, reaching up to dissociation constants in the hundreds of micromolar.2 These include the direct detection of broadened signals from bound ligands, as well as of changes in other relaxation rates in the presence of fast exchange between free and bound forms. Broadening the range of possible applications, molecules containing 19F can be used as reporter ligands for the screening of non-fluorinated ligands in binding competition experiments.3
Spin hyperpolarization, such as of 1H, can transfer from a ligand to the macromolecule,4 transfer within two sites in a ligand,5 or transfer between two competitively binding ligands.6 Each of these cases offers a pathway to detecting the binding of the ligand. The polarization transfer provides added information on the proximity of the spins involved, which is useful to determine the structure of the binding epitope.7 Fast multi-dimensional and pseudo multi-dimensional NMR spectroscopy of the hyperpolarized ligand and macromolecule provide the necessary spin correlations.8 The structures are then determined with the assistance of simulations of the signals arising from a network of dipolar coupled spins in combination with computational optimization or scoring procedures.
The effectiveness of spin relaxation or polarization transfer parameters in identifying a small fraction of bound ligand increases with increasing molecular weight. For this reason, methods of hyperpolarized NMR for the detection of ligand binding are applicable to immobilized proteins, which can further reduce protein consumption in screening experiments.5 In addition to ligands, D-DNP can be used to directly hyperpolarize polypeptides, such as in the denatured form.9 The protein folding process and protein–protein interactions can be characterized in real-time on a time scale of several seconds. Similar information is available if proteins or nucleic acids receive polarization transfer from previously hyperpolarized water.10,11 Water protons exchange with labile protons on amide or hydroxy groups in macromolecules. Enhanced signals observed for the corresponding positions provide sequence specific information on solvent exposure and molecular dynamics.12
The above described modalities of applying hyperpolarization present significant advantages for specific, selective, and highly sensitive NMR spectroscopy of biological molecules. A barrier to entry into the use of these techniques is the added complexity in the instrumentation that is required to generate the hyperpolarized spin states. For example, a D-DNP instrument that is co-sited with an NMR spectrometer comprises an additional superconducting magnet with variable-temperature insert, a microwave source to saturate an electron spin transition, and a dissolution system.
Here, we demonstrate the application of an alternative hyperpolarization technique, signal amplification by reversible exchange (SABRE),13 to the detection of protein–ligand interactions. SABRE, based on para-hydrogen,14,15 is among the most inexpensive hyperpolarization methods. Molecular hydrogen spontaneously transfers to the anti-parallel para-spin state at cryogenic temperatures in the presence of a paramagnetic spin flip catalyst. The ordered spin state of para-hydrogen gas is then converted into hyperpolarization of nuclear spins on a target molecule. This conversion occurs either by catalytic hydrogenation in para-hydrogen induced polarization (PHIP), or by binding to a polarization transfer catalyst facilitating SABRE. Signal enhancements of at least thousands-fold for PHIP and hundreds-fold for SABRE are routinely obtained, whereby SABRE does not require a substrate undergoing a chemical change.
SABRE polarization transfer catalysts are organometallic complexes that bind an electron donating group in the target molecule, often an N-heterocycle, together with H2 in a co-planar arrangement. Ligands of the catalyst, like the N-heterocyclic carbenes, are chosen for an appropriate substrate exchange rate and to provide solubility. SABRE was successfully applied to hyperpolarize biological and bioactive molecules. Several drug molecules with nitrogen containing heterocycles that enable catalyst binding were hyperpolarized for 1H (ref. 16–18) and 15N.19–21 The structure of the target molecules, especially when containing substituents near the catalyst binding site necessitates the design of matching catalysts with requisite binding affinities that allow appropriate exchange rates for efficient polarization.22,23
Several methods have been developed for achieving SABRE in biocompatible solvents. These include developing water soluble catalysts,24,25 hyperpolarizing in D2O and ethanol mixtures,26,27 water addition to activated catalyst samples and subsequent methanol evaporation to achieve a methanol component as low as 10% (ref. 28) and aqueous reconstitution of the activated SABRE catalyst in pure D2O.29 Molecular probes for magnetic resonance imaging have been hyperpolarized using SABRE.30–32 These probes are designed to contain nuclei with long relaxation times, such as 15N, to retain polarization in biomedical imaging experiments.
A challenge in applications of several hyperpolarization techniques is the potential for interference of polarizing agents with the goal of the experiment. In D-DNP experiments, free radicals are needed to provide spin polarization. Radicals have been removed by chemical reaction, physical separation, or quenching in some in vitro, as well as in vivo applications. In SABRE, the analogous agent is the polarization transfer catalyst. This catalyst needs to be removed for in vivo experiments as the catalyst is identified as the main reason for toxicity of the SABRE approach.26 Catalyst removal by using chelating ligands33 and phase separation34 combined with metal scavengers35 has been developed to produce biocompatible hyperpolarized samples that are metal-free. Other experiments may have less strict requirements, however, when employing SABRE for monitoring of chemical reactions, the polarization transfer catalyst was reported to modulate the rates of organic reactions.36 Chelating agents, such as 1,10-phenanthroline and 2,2′-bipyridine, were introduced into the reaction mixture after completion of polarization transfer from para-hydrogen, to prevent or reduce effects from binding of other molecules to the catalyst.33
In this paper, we use SABRE to identify binding of a ligand to the trypsin protease. A ligand containing a well-known binding motif for trypsin, as well as a binding site for the polarization transfer catalyst in a different location, is hyperpolarized. The incompatibility of SABRE catalysts with proteins, as well as low polarization efficiency in water, is overcome by a two-step approach. The ligand is first hyperpolarized separately and subsequently injected with the protein for detection. We analyze the resulting changes in relaxation rates due to binding of the ligand, and discuss applications.
Results and discussion
Serine proteases including trypsin are inhibited by amidine containing ligands such as benzamidine, forming a salt bridge with an aspartate residue in the active site of the protein.37 Although the amidine group contains nitrogen atoms, its presence in the cationic form would prevent efficient catalyst binding. SABRE hyperpolarization of benzamidine was not observed using a typical catalyst [Ir(MeIMes)(COD)]Cl (COD = cyclooctadiene, MeIMes = 4,5-dimethyl-1,3-bis(2,4,6 trimethylphenyl)imidazol-2-ylidene). The putative ligand chosen for hyperpolarization was 4-amidinopyridine (Fig. 1). This molecule contains an N atom at the site most distant from the expected trypsin binding site. For the resulting molecule 4-amidinopyridine, SABRE hyperpolarization was observable using [Ir(MeIMes)(COD)]Cl, however, higher signal enhancements were obtained with the asymmetric catalyst [Ir(IMeMes)(COD)]Cl (IMeMes = 1-(2,4,6-trimethylphenyl)-3-methylimidazol-2-ylidene).38 The active form of this catalyst is in the following referred to as Ir(IMeMes). Signal enhancements ranging from −87 and −34 for 1.5 mM 4-amidinopyridine to −230 and −110 for 10 mM 4-amidinopyridine, for the H atoms in the ortho and meta positions with respect to the N was obtained. The nuclear spin polarization increases from 0.0032% for 1H at a magnetic field of 9.4 T at 298 K to 0.28% and 0.74% for the Ha enhancement values of −87 and −230, respectively. The spectrum for the 1.5 mM concentration is shown in Fig. 1. This condition was used for the protein–ligand interaction data at lower concentration (see below; Fig. 5). SABRE NMR spectra for the other ligand concentrations are included in ESI, Fig. S1.† This confirms that the heterocyclic ring promotes binding to the polarization transfer catalyst. The difference in polarization efficiency between the two catalysts is likely due to the reduced steric hindrance in the asymmetric catalyst, as the para-substituted substrates can also exhibit steric effects.39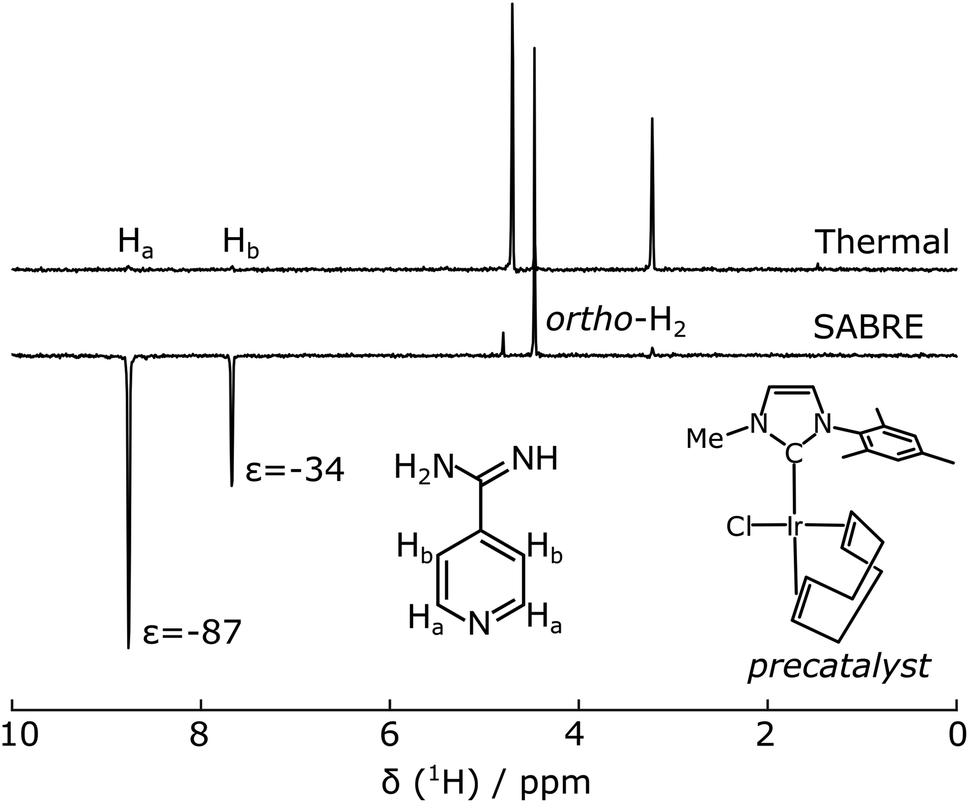 | ||
| Fig. 1 400 MHz NMR spectra of 1.5 mM 4-amidinopyridine with 0.3 mM Ir(IMeMes) polarization transfer catalyst in d4-methanol (a) non-hyperpolarized (“thermal”) and (b) SABRE after bubbling for 30 s in a 6.5 mT magnetic field at 294 K, followed by acquisition at 9.4 T. The structures of the 4-amidinopyridine, and of the precatalyst before activation, are inset. | ||
 | ||
| Fig. 2 Instrument for SABRE NMR measurements of ligand binding. The putative ligand interacts with para-hydrogen and polarization transfer catalyst at 6.5 mT. It is subsequently delivered to a sample loop. The ligand and protein samples are pushed by high-pressure syringe pumps through the Y-mixer, to a flow-cell in the NMR magnet, where the measurement takes place. | ||
 | ||
| Fig. 3 Hyperpolarized signals measured using a CPMG experiment. (a) Selected spectra from individual spin-echoes of 7.2 mM 4-amidinopyridine with polarization transfer catalyst in 36% methanol in final sample. (b) Signal decay and exponential fit of integrals are from (a). (c) Spectra from 6.8 mM 4-amidinopyridine, catalyst, and 3.9 mM chelating ligand 2,2′-bipyridine in 34% methanol in the final sample. (d) Signal decay and exponential fit of integrals are from (c). (e) Spectra from 5.9 mM 4-amidinopyridine, catalyst, 3.0 mM 2,2′-bipyridine and 0.33 mM trypsin in 30% methanol in final sample. (f) Signal decay and exponential fit of integrals are from (e). | ||
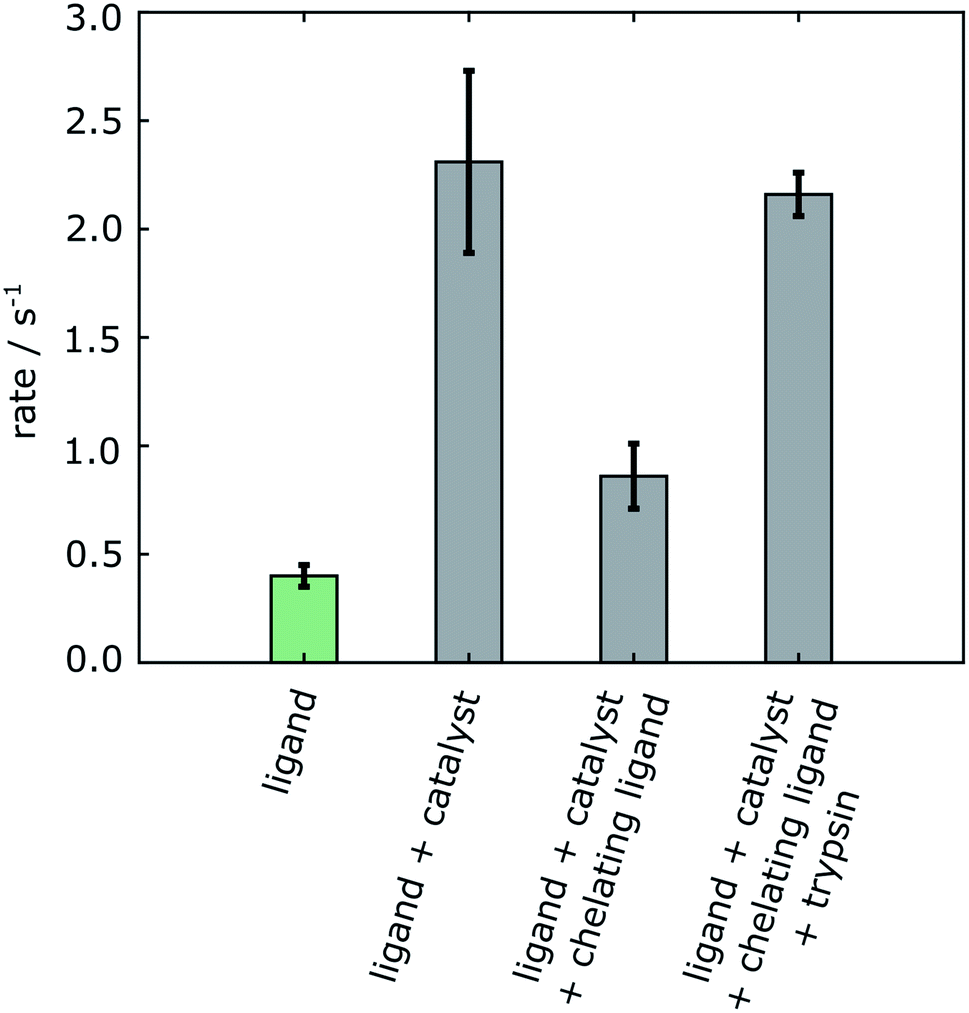 | ||
| Fig. 4 R 2 relaxation rates of 4-amidinopyridine. The green bar is from a non-hyperpolarized experiment in the absence of polarization transfer catalyst. The gray bars are from 4-amidinopyridine hyperpolarized by SABRE. Errors are shown as standard deviations from three separate measurements taken from different samples (Table S1†). | ||
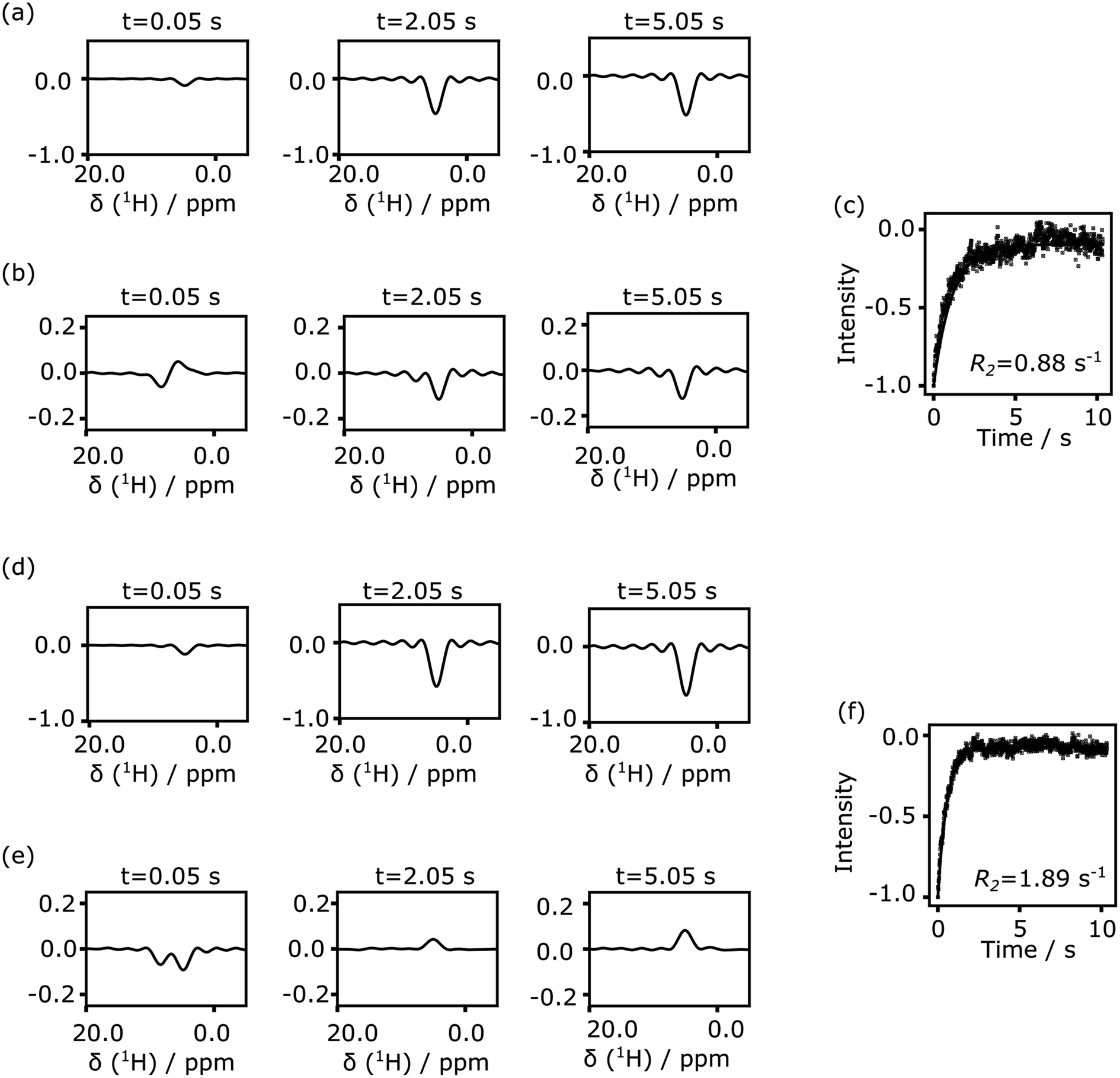 | ||
| Fig. 5 (a) Spectra from CPMG echoes of 134 μM 4-amidinopyridine in presence of polarization transfer catalyst and chelating ligand. (b) Spectra from CPMG echoes of 134 μM 4-amidinopyridine in presence of polarization transfer catalyst and chelating ligand after water signal subtraction. The final methanol fraction in the sample is 8.9%. (c) Integrated and fitted signals from (b). (d) Spectra of 125 μM 4-amidinopyridine and 7.2 μM trypsin in presence of catalyst and chelating ligand. (e) Spectra of 125 μM 4-amidinopyridine and 7.2 μM trypsin in presence of catalyst and chelating ligand after water signal subtraction. The final methanol fraction in the sample is 8.3%. (f) Integrated and fitted signals from (e). Where indicated, the reference water spectrum was subtracted after scaling to the maximum solvent signal intensity in each echo. All spectra are plotted at the same scale. | ||
Despite the ability to hyperpolarize 4-amidinopyridine, the methanol solvent used in Fig. 1 would not be conducive to biological applications such as the characterization of ligand binding. For this reason, SABRE polarization was attempted in a mixture of 50% v/v of methanol-d4 and D2O buffer. Under these conditions, the enhancement decreased from −93 to −3 for Ha and −46 to −1 for Hb, which would be insufficient for the experiment. The signals were further reduced if the protein was included in the mixture.
Given that the Ir(IMeMes) polarization transfer catalyst is incompatible with a one-pot reaction mixture that includes the protein, a two-step process was designed for characterizing the protein–ligand interactions using SABRE hyperpolarization. The molecule to be hyperpolarized separately underwent polarization transfer from para-hydrogen in methanol-d4, and was subsequently mixed with a protein solution. This two-step procedure is congruous with previous experiments employing D-DNP for the determination of ligand binding.40
For SABRE polarization, the solution of the putative ligand with polarization transfer catalyst in methanol-d4 underwent bubbling with para-hydrogen gas (Fig. 2). These conditions are optimal for polarization transfer. The sample was located in an electromagnet producing the required field of 6.5 mT (see also Experimental section). Following this polarization step, a discharge valve was opened. Under the pressure of the hydrogen gas, the solution was delivered to an injection valve with sample loop. Injection into a flow-cell installed in the 9.4 T NMR magnet was driven by water from high-pressure syringe pumps, simultaneously for the putative ligand and the protein sample. The two samples mixed in a Y-mixer prior to entering the NMR magnet. A stationary mixture in the flow-cell was obtained by switching the injection valve prior to NMR data acquisition.
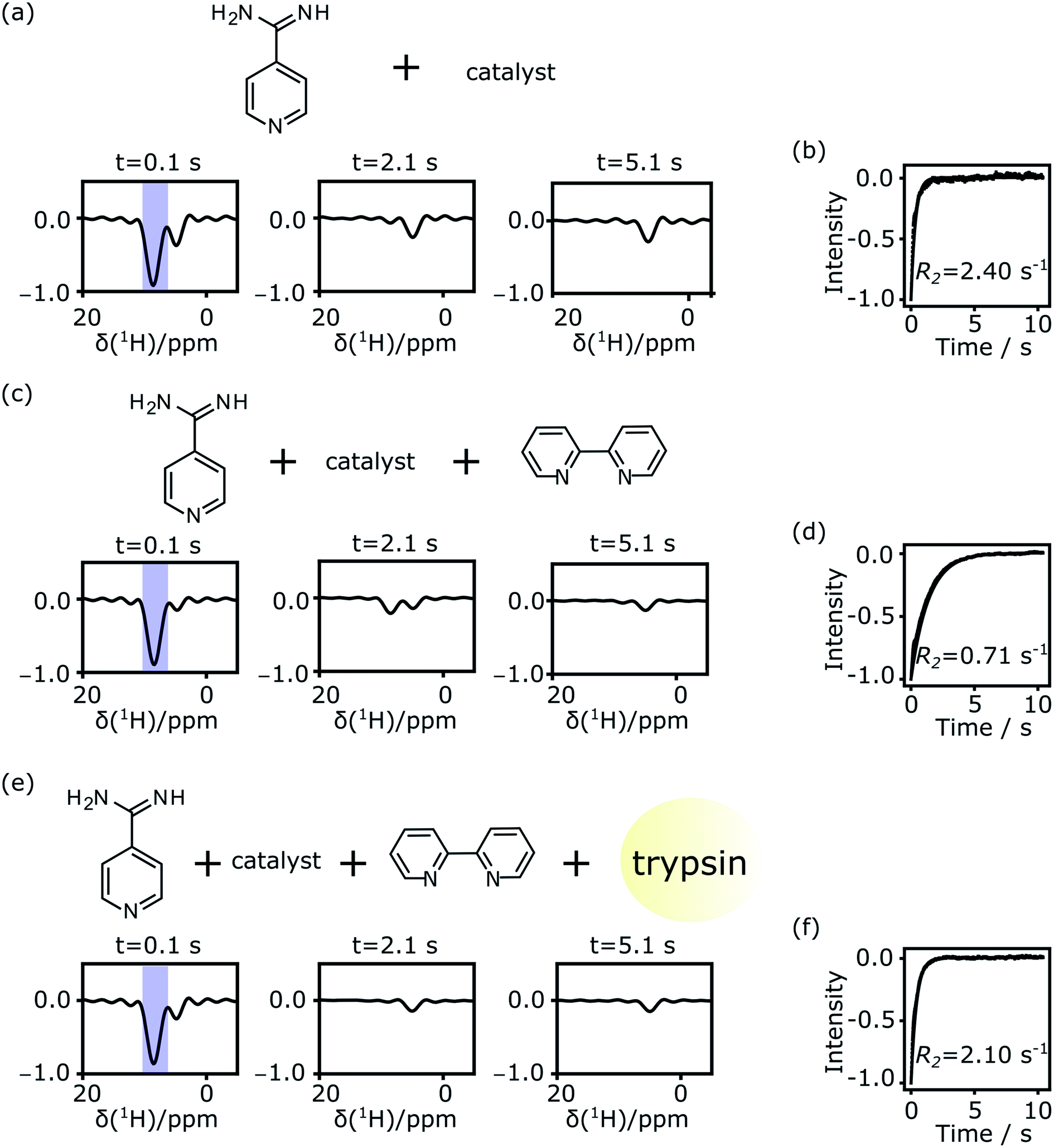
Single-scan Carr–Purcell–Meiboom–Gill (CPMG) NMR experiments were acquired to measure the transverse relaxation rate (R2) of the 1H spins of the putative ligand molecule. Spectra obtained from Fourier transforms of selected individual spin echoes are shown in Fig. 3. Fig. 3a contains signals from the ligand alone, where hyperpolarized ligand solution in methanol-d4 was mixed at a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (v/v) with a D2O buffer that did not contain any protein. The hyperpolarized signal from the putative ligand near 8 ppm is strong in the first echo, and decays during the experimental time. The water signal near 4.7 ppm was suppressed using selective pulses and pulsed field gradients. A residual water signal is visible in the spectra, as the syringe pumps used to drive the samples were filled with H2O. The spectral resolution is limited by the echo time in the CPMG experiment, which is 1.7 ms. The two peaks from the hyperpolarized 4-amidinopyridine molecule seen in Fig. 1 merge into one observed signal. Still, this signal of interest is well separated from the residual water signal and can be analyzed to result in an averaged relaxation rate for the putative ligand.
:7 (v/v) with a D2O buffer that did not contain any protein. The hyperpolarized signal from the putative ligand near 8 ppm is strong in the first echo, and decays during the experimental time. The water signal near 4.7 ppm was suppressed using selective pulses and pulsed field gradients. A residual water signal is visible in the spectra, as the syringe pumps used to drive the samples were filled with H2O. The spectral resolution is limited by the echo time in the CPMG experiment, which is 1.7 ms. The two peaks from the hyperpolarized 4-amidinopyridine molecule seen in Fig. 1 merge into one observed signal. Still, this signal of interest is well separated from the residual water signal and can be analyzed to result in an averaged relaxation rate for the putative ligand.
After integration of the signals from each echo, an exponential decay is observed (Fig. 3b). The R2 relaxation is obtained by fitting a single exponential curve, here resulting in a value of 2.40 s−1 for the 4-amidinopyridine without the presence of protein. This relaxation rate is much larger than R2 = 0.32 s−1 that was determined from a non-hyperpolarized NMR experiment for the same molecule (ESI, Fig. S2†). The difference in these relaxation rates was found to be due to the presence of the polarization transfer catalyst, as a direct consequence of binding of the molecule to the Ir center of the catalyst. Similar relaxation changes due to catalyst binding have previously been observed.33,39 After including a chelating ligand, 2,2′-bipyridine,33 with the buffer solution to trap the catalyst, the relaxation rate of the hyperpolarized signal was found to be slower, with R2 = 0.71 s−1 (Fig. 3c and d). Finally, upon the addition of the trypsin protein, the relaxation rate increased to R2 = 2.28 s−1 (Fig. 3e and f).
A summary of the measurements under the different experimental conditions, including several repetitions, is included in Table S1 (ESI†). The changes in the observed relaxation rates are represented in Fig. 4. The comparison of the non-hyperpolarized experiment with R2 relaxation rate 0.39 ± 0.06 s−1 (green bar) with the “ligand + catalyst” (first gray bar) indicates a significant relaxation effect due to the interaction of the hyperpolarized molecule with the polarization transfer catalyst with the R2 rate 2.30 ± 0.44 s−1. This effect is largely reversed by the addition of the 2,2′-bipyridine (second gray bar). The chelating agent therefore significantly improves the ability to measure the relaxation properties of the free ligand with values 0.86 ± 0.15 s−1. A smaller difference in the rates between the “ligand” (green bar) and the “ligand + catalyst + chelating ligand” (second gray bar), which narrowly exceeds the error limit, is likely due to a residual fraction of catalyst not trapped by the 2,2′-bipyridine chelating ligand. Finally, the inclusion of the protein leads to a significant increase in the relaxation rate with R2 values 2.16 ± 0.10 s−1 (third gray bar). This increase is due to the slower tumbling of the protein–ligand complex in solution, hence proving the binding of the ligand to the protein. Importantly, it can be seen that, firstly, the change in the relaxation rate that demonstrates the binding and therefore represents the result of the experiment is highly significant. Secondly, the change is only observable after removing the relaxation contribution that is introduced by the polarization transfer catalyst.
The result of this experiment is in agreement with competitive binding measurements of 4-amidinopyridine to trypsin measured by NMR (ESI, Fig. S3†). These measurements indicated a dissociation constant for 4-amidinopyridine that lies in-between the those of the related known ligands for trypsin, benzamidine and 4-(trifluoromethyl)benzene-1-carboximidamide.
The signals of the 2,2′-bipyridine chelating agent would appear in the same spectral region as the signals of interest from the ligand. An efficient transfer of hyperpolarization to 2,2′-bipyridine would not be expected because firstly, this molecule is not present in the sample during the SABRE hyperpolarization step in the experiment, and secondly its off-rate is slowed due to its ability to form a bidentate complex with Ir. Nevertheless, to ensure that the rates are determined from the ligand peaks of interest, a control experiment was performed, where the ligand was not included in the reaction mixture. The resulting spectra are shown in ESI, Fig. S4.† In this control experiment, no exponential decay is observed for the integrated spectral region, indicating an absence of signal contributions from the chelating ligand. Moreover, when the 2,2′-bipyridine is added to a sample of the activated catalyst and ligand 4-amidinopyridine for a one-pot experiment performed in the NMR tube, no SABRE hyperpolarized signals were observed (ESI, Fig. S5†).
Both the protein and the ligand concentration are lowered for the data of Fig. 5, to explore concentration limits under current experimental conditions. A smaller volume of ligand solution, 500 μl, with a stock concentration of 1.5 mM, was hyperpolarized. After mixing, the protein concentration reached to the single digit micromolar level, and the ligand was in the range of 100–150 μM. Under these conditions, the signal of the hyperpolarized ligand can be obscured by parts of the solvent line (Fig. 5a and d, top panels). In the bottom panels of these figures, the solvent signal was reduced by subtracting a reference spectrum that was scaled to the maximum solvent signal intensity. The binding of the ligand is identified by comparing the relaxation rates obtained from the fit in Fig. 5c and f.
Nuclear spin hyperpolarization offers significant advantages in the detection of protein–ligand interactions, by allowing a reduction in the ligand concentration. Under conditions of fast exchange between free and bound forms of the ligand, as is the case for 4-amidinopyridine and trypsin, the protein concentration can be reduced to a level several times below the ligand concentration. The reduction in concentration facilitates working with proteins that are unstable or difficult to purify.
Although the fluctuations in the echo signals seen in Fig. 5b and e are larger than those in Fig. 3, the experiment at these concentrations is not primarily limited by thermal noise in the spectra. The concentration of the hyperpolarized ligand in the final solution may be further reduced. Because SABRE hyperpolarization is typically most effective for a ligand concentration in the millimolar range,41 the concentration in the stock solution for hyperpolarization should not be arbitrarily reduced. Rather, the amount of stock solution that is used in the experiment could be lowered. This goal may preferably be combined with methods that introduce hyperpolarized gas into a smaller volume of liquid to minimize consumption of the ligand and the polarization transfer catalyst. For example, microfluidic techniques that introduce gases into liquids have previously been described.42 An experiment reducing the ligand concentration would further benefit from the addition of a technique that facilitates the rapid admixing of a small, microliter-range volume of ligand solution to the protein solution.
Several improvements would further increase achievable signals and lower the minimum ligand concentration. Additional water suppression or use of solvents with higher deuteration level would reduce fluctuations due to solvent signal overlap. The experiments could be performed using hydrogen gas with a higher para content. Here, 50% para-hydrogen was produced by cooling hydrogen gas to the temperature of 77 K using liquid nitrogen. Increasing the percentage by producing para-hydrogen at lower temperature can increase the signal enhancement by another factor of three.41 An additional improvement of at least a factor of two would be realized by changing the ligand concentration during the polarization step. As is known from the literature,41 optimal polarization efficiency is achieved in a range of catalyst and ligand concentrations, where a sufficient fraction of the ligand is bound to the catalyst. Based on the data in ESI, Fig. 1,† a ligand concentration of 10 mM during the polarization step would result in a higher signal enhancement. A lower concentration was used in this work in order to achieve a low ligand concentration after mixing with the protein at a moderate volume-to-volume ratio in the experimental setup as implemented. The mixing ratio could be further optimized to lower the volume of hyperpolarized ligand solution that is introduced, while increasing the ligand concentration in the hyperpolarization step.
In addition to the other reagents, the achievable signal enhancement depends on the hydrogen gas pressure. The pressure dependence of signal enhancement for this ligand and catalyst is shown in ESI, Fig. S6† over the range of 0.21 to 0.83 MPa. It is evident that the signal enhancement is close to reaching a plateau, indicating that the pressure used in the experiment is sufficient to achieve near saturation in the metal hydride formation and highest enhancements. The same pressure was also used to effectively drive the sample from the polarization vessel to the sample loop.
SABRE hyperpolarization using common polarization transfer catalysts is most readily achieved in polar organic solvents, here methanol. Apart from decreasing the protein concentration, a benefit of a large dilution factor upon mixing of the two solutions in this experiment is that the final concentration of the organic solvent component is reduced. The volume ratio of the experiments in Fig. 5 resulted in a methanol fraction after admixing the protein of <10%. Proteins are likely to retain their native structure in solutions with a low content of alcohol.43 Trypsin was previously found to retain the ability to bind a ligand in the presence 30% methanol.44 Measurements of trypsin catalytic activity confirmed similar initial reaction rate constants in 30% and 10% methanol compared to water, for the first 15 s or reaction time (ESI, Fig. S7†). Deactivation of the enzyme occurred after approximately 30 or 60 s, respectively, i.e. at a much longer time than the duration of the hyperpolarized NMR experiment. A further reduction of ligand solution volume as described above would entail the additional benefit of reducing the methanol concentration in the final sample.
The use of SABRE for the characterization of protein–ligand interactions can be expanded to other ligands containing appropriate functional groups. These may include the –NH2 (ref. 45) or –CN (ref. 46) groups, or the heterocyclic N as demonstrated here. In addition to protons, SABRE hyperpolarization can be achieved for other nuclei, including fluorine. 19F has been hyperpolarized by SABRE both directly and indirectly through the intermediary of a nearby proton.47,48 The method described here can be adapted for ligands containing this nucleus. Similar to previous D-DNP experiments,3 the observation of fluorine would avoid any interference from the solvent signal. Ligand derived SABRE hyperpolarization may in the future be used for studies of macromolecular structure at the binding site, by employing polarization transfer and using calculations similar to those demonstrated by other hyperpolarization methods.8 An additional generalization of the experiment includes the use of one molecule with weak affinity and fast exchange rate as a reporter ligand, which becomes displaced upon binding of another ligand.3,49 This approach would require the identification of only one SABRE hyperpolarizable ligand for screening of a library of other ligands.
Conclusion
In summary, we demonstrated the use of para-hydrogen derived hyperpolarization using the SABRE method for the determination of protein–ligand binding. The hyperpolarized small molecule contains a binding site for the protein, and at a distant location, for the polarization transfer catalyst. The use of flow-NMR allowed the experiment to be completed in predominantly an aqueous medium. The SABRE hyperpolarization method is cost-effective and can be added-on to standard NMR spectroscopy equipment. Hyperpolarization allows the reduction of protein concentration, enabling the screening of ligand binding in drug discovery and other applications.Experimental
Hydrogen gas enriched to a level of ∼50% para-content was prepared by passing room temperature hydrogen gas over iron(III) oxide spin-flip catalyst (Sigma-Aldrich, St Louis, MO) in a heat exchanger, which was immersed in liquid nitrogen (Caution: hydrogen gas is flammable and can form explosive mixtures with air. It should be exhausted through grounded metal piping. Eye protection is required for compressed gases). The ligand sample for hyperpolarization consisted of 20 mM 4-amidinopyridine hydrochloride (Alfa Aesar, Ward Hill, MA) in methanol-d4 (Cambridge Isotope Libraries, Andover, MA) (Caution: all chemicals require handling using gloves and eye protection. Methanol and 2,2′-bipyridine are toxic). The sample contained 3.5 mM of the precatalyst [Ir(IMeMes)(COD)]Cl, which was synthesized according to a previously established protocol.38 For the experiments at low concentration, this stock solution of ligand was diluted to 1.5 mM and 0.3 mM catalyst. For the SABRE experiments, the para-enriched hydrogen was bubbled through the sample solution at a pressure of 8.3 × 105 Pa and at 294 K. Bubbling was performed for 30 s at a field of 6.5 mT generated by a solenoid coil (diameter 22 cm and length 28 cm). After this polarization transfer step, the sample was pushed to a sample loop using the pressure of the H2 gas. The hyperpolarized sample was injected into a NMR flow-cell concomitantly with a sample of 50 mM sodium phosphate buffer in D2O (pH 7.5), or a sample of trypsin (Alfa Aesar) at 1 mM or 18 μM dissolved in the same buffer. Where indicated, 2,2′-bipyridine (Sigma-Aldrich) at 10 mM or 2.5 mM concentration was included with the protein solution. The two solutions mixed in a Y-mixer before entering the magnet. The sample injector that was used for this purpose is described elsewhere.50 Briefly, both samples were pushed from an injection loop made of poly ether ether ketone (PEEK) tubing of 0.5 mm inner diameter. Two high pressure syringe pumps (Models 500D and 1000D, Teledyne Isco, Lincoln, NE) were filled with water and used to transfer the sample from the injection loop to the Y-mixer and subsequently into the flow-cell. Flow rates were set to 110 ml min−1 and 150 ml min−1, respectively. The injection time was 128 ms, during which the pump was active before sample mixing. The time after mixing but before sample reaching flow cell was 1070 ms, and the stabilization time before triggering the NMR experiment was 500 ms. All measurements were performed with a TXI-probe (Bruker Biospin, Billerica, MA). A single scan CPMG experiment was performed to find the R2 relaxation rates of the 1H spins of the ligand 4-amidinopyridine hydrochloride. A water suppression sequence was used prior to collecting the echoes, where EBURP pulses of 20 ms were applied to selectively excite the solvent signal, followed by dephasing using pulsed field gradients (Gx,y,z = 70 G cm−1; 1 ms). For the CPMG block, a pulsing delay of 1696.2 μs was used, and 64 points were collected per echo. The total experiment time was 10.4 s.Author contributions
CH designed the project. RM performed experiments and analyzed data. PP prepared catalyst samples and performed experiments. All authors interpreted results and wrote manuscript.Conflicts of interest
Texas A&M University is seeking patent protection covering parts of this work.Acknowledgements
Financial support from the National Science Foundation (Grant 1900406) and the Welch Foundation (Grant A-1658) is gratefully acknowledged. We thank Quy Son Luu for assistance with sample characterization.References
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS.
- Y. Lee, H. Zeng, S. Ruedisser, A. D. Gossert and C. Hilty, J. Am. Chem. Soc., 2012, 134, 17448–17451 CrossRef CAS PubMed.
- Y. Kim and C. Hilty, Angew. Chem., Int. Ed., 2015, 54, 4941–4944 CrossRef CAS PubMed.
- H. Min, G. Sekar and C. Hilty, ChemMedChem, 2015, 10, 1559–1563 CrossRef CAS PubMed.
- Y. Wang and C. Hilty, Anal. Chem., 2020, 92, 13718–13723 CrossRef CAS PubMed.
- Y. Lee, H. Zeng, A. Mazur, M. Wegstroth, T. Carlomagno, M. Reese, D. Lee, S. Becker, C. Griesinger and C. Hilty, Angew. Chem., Int. Ed., 2012, 51, 5179–5182 CrossRef CAS.
- Y. Wang and C. Hilty, J. Med. Chem., 2019, 62, 2419–2427 CrossRef CAS PubMed.
- Y. Wang, J. Kim and C. Hilty, Chem. Sci., 2020, 11, 5935–5943 RSC.
- M. Ragavan, H.-Y. Chen, G. Sekar and C. Hilty, Anal. Chem., 2011, 83, 6054–6059 CrossRef CAS.
- M. Novakovic, G. L. Olsen, G. Pintér, D. Hymon, B. Fürtig, H. Schwalbe and L. Frydman, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2449–2455 CrossRef CAS PubMed.
- O. Szekely, G. L. Olsen, M. Novakovic, R. Rosenzweig and L. Frydman, J. Am. Chem. Soc., 2020, 142, 9267–9284 CrossRef CAS.
- J. Kim, R. Mandal and C. Hilty, J. Phys. Chem. Lett., 2019, 10, 5463–5467 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- H. Zeng, J. Xu, J. Gillen, M. T. McMahon, D. Artemov, J.-M. Tyburn, J. A. B. Lohman, R. E. Mewis, K. D. Atkinson, G. G. R. Green, S. B. Duckett and P. C. M. van Zijl, J. Magn. Reson., 2013, 237, 73–78 CrossRef CAS.
- R. Mandal, P. Pham and C. Hilty, ChemPhysChem, 2020, 21, 2166–2172 CrossRef CAS PubMed.
- H. J. Jeong, S. Min, H. Chae, S. Kim, G. Lee, S. K. Namgoong and K. Jeong, Sci. Rep., 2020, 10, 14290 CrossRef CAS PubMed.
- I. V. Skovpin, A. Svyatova, N. Chukanov, E. Y. Chekmenev, K. V. Kovtunov and I. V. Koptyug, Chem.–Eur. J., 2019, 25, 12694–12697 CrossRef CAS.
- O. G. Salnikov, N. V. Chukanov, A. Svyatova, I. A. Trofimov, M. S. H. Kabir, J. G. Gelovani, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2021, 60, 2406–2413 CrossRef CAS.
- D. A. Barskiy, R. V. Shchepin, A. M. Coffey, T. Theis, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, J. Am. Chem. Soc., 2016, 138, 8080–8083 CrossRef CAS.
- P. Pham and C. Hilty, Chem. Commun., 2020, 56, 15466–15469 RSC.
- J. F. P. Colell, A. W. J. Logan, Z. Zhou, J. R. Lindale, R. Laasner, R. V. Shchepin, E. Y. Chekmenev, V. Blum, W. S. Warren, S. J. Malcolmson and T. Theis, Chem. Commun., 2020, 56, 9336–9339 RSC.
- F. Shi, P. He, Q. A. Best, K. Groome, M. L. Truong, A. M. Coffey, G. Zimay, R. V. Shchepin, K. W. Waddell, E. Y. Chekmenev and B. M. Goodson, J. Phys. Chem. C, 2016, 120, 12149–12156 CrossRef CAS.
- J. F. P. Colell, M. Emondts, A. W. J. Logan, K. Shen, J. Bae, R. V. Shchepin, G. X. Ortiz, P. Spannring, Q. Wang, S. J. Malcolmson, E. Y. Chekmenev, M. C. Feiters, F. P. J. T. Rutjes, B. Blümich, T. Theis and W. S. Warren, J. Am. Chem. Soc., 2017, 139, 7761–7767 CrossRef CAS PubMed.
- A. Manoharan, P. J. Rayner, W. Iali, M. J. Burns, V. H. Perry and S. B. Duckett, ChemMedChem, 2018, 13, 352–359 CrossRef CAS.
- J.-B. Hövener, N. Schwaderlapp, R. Borowiak, T. Lickert, S. B. Duckett, R. E. Mewis, R. W. Adams, M. J. Burns, L. A. R. Highton, G. G. R. Green, A. Olaru, J. Hennig and D. von Elverfeldt, Anal. Chem., 2014, 86, 1767–1774 CrossRef.
- H. Zeng, J. Xu, M. T. McMahon, J. A. B. Lohman and P. C. M. van Zijl, J. Magn. Reson., 2014, 246, 119–121 CrossRef CAS.
- M. L. Truong, F. Shi, P. He, B. Yuan, K. N. Plunkett, A. M. Coffey, R. V. Shchepin, D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, K. W. Waddell, B. M. Goodson and E. Y. Chekmenev, J. Phys. Chem. B, 2014, 118, 13882–13889 CrossRef CAS PubMed.
- K. V. Kovtunov, B. E. Kidd, O. G. Salnikov, L. B. Bales, M. E. Gemeinhardt, J. Gesiorski, R. V. Shchepin, E. Y. Chekmenev, B. M. Goodson and I. V. Koptyug, J. Phys. Chem. C, 2017, 121, 25994–25999 CrossRef CAS.
- A. Svyatova, I. V. Skovpin, N. V. Chukanov, K. V. Kovtunov, E. Y. Chekmenev, A. N. Pravdivtsev, J.-B. Hövener and I. V. Koptyug, Chem.–Eur. J., 2019, 25, 8465–8470 CrossRef CAS.
- K. Shen, A. W. J. Logan, J. F. P. Colell, J. Bae, G. X. Ortiz, T. Theis, W. S. Warren, S. J. Malcolmson and Q. Wang, Angew. Chem., Int. Ed., 2017, 56, 12112–12116 CrossRef CAS.
- R. E. Mewis, M. Fekete, G. G. R. Green, A. C. Whitwood and S. B. Duckett, Chem. Commun., 2015, 51, 9857–9859 RSC.
- W. Iali, A. M. Olaru, G. G. R. Green and S. B. Duckett, Chem.–Eur. J., 2017, 23, 10491–10495 CrossRef CAS.
- D. A. Barskiy, L. A. Ke, X. Li, V. Stevenson, N. Widarman, H. Zhang, A. Truxal and A. Pines, J. Phys. Chem. Lett., 2018, 9, 2721–2724 CrossRef CAS.
- B. J. Tickner, P. J. Rayner and S. B. Duckett, Anal. Chem., 2020, 92, 9095–9103 CrossRef CAS.
- M. Renatus, W. Bode, R. Huber, J. Stürzebecher and M. T. Stubbs, J. Med. Chem., 1998, 41, 5445–5456 CrossRef CAS.
- C. M. Wong, M. Fekete, R. Nelson-Forde, M. R. D. Gatus, P. J. Rayner, A. C. Whitwood, S. B. Duckett and B. A. Messerle, Catal. Sci. Technol., 2018, 8, 4925–4933 RSC.
- E. V. Stanbury, P. M. Richardson and S. B. Duckett, Catal. Sci. Technol., 2019, 9, 3914–3922 RSC.
- Y. Kim, M. Liu and C. Hilty, Anal. Chem., 2016, 88, 11178–11183 CrossRef CAS.
- N. Eshuis, N. Hermkens, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, J. Am. Chem. Soc., 2014, 136, 2695–2698 CrossRef CAS PubMed.
- M. Malankowska, C. F. Martins, H. S. Rho, L. A. Neves, R. M. Tiggelaar, J. G. Crespo, M. P. Pina, R. Mallada, H. Gardeniers and I. M. Coelhoso, J. Membr. Sci., 2018, 545, 107–115 CrossRef CAS.
- K. Yamazaki, T. Iwura, R. Ishikawa and Y. Ozaki, J. Biochem., 2006, 140, 49–56 CrossRef CAS.
- S. M. Gopal, F. Klumpers, C. Herrmann and L. V. Schäfer, Phys. Chem. Chem. Phys., 2017, 19, 10753–10766 RSC.
- W. Iali, P. J. Rayner, A. Alshehri, A. J. Holmes, A. J. Ruddlesden and S. B. Duckett, Chem. Sci., 2018, 9, 3677–3684 RSC.
- R. E. Mewis, R. A. Green, M. C. R. Cockett, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. O. John, P. J. Rayner and D. C. Williamson, J. Phys. Chem. B, 2015, 119, 1416–1424 CrossRef CAS PubMed.
- A. M. Olaru, T. B. R. Robertson, J. S. Lewis, A. Antony, W. Iali, R. E. Mewis and S. B. Duckett, ChemistryOpen, 2017, 7, 97–105 CrossRef.
- N. V. Chukanov, O. G. Salnikov, R. V. Shchepin, A. Svyatova, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2018, 122, 23002–23010 CrossRef CAS.
- C. Dalvit, M. Flocco, S. Knapp, M. Mostardini, R. Perego, B. J. Stockman, M. Veronesi and M. Varasi, J. Am. Chem. Soc., 2002, 124, 7702–7709 CrossRef CAS.
- H.-Y. Chen and C. Hilty, ChemPhysChem, 2015, 16, 2646–2652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03404a |
| This journal is © The Royal Society of Chemistry 2021 |