
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo metal-catalyzed SeCT therapy by a proapoptotic peptide†
Peni
Ahmadi‡
a,
Kyohei
Muguruma‡
b,
Tsung-Che
Chang
a,
Satoru
Tamura
c,
Kazuki
Tsubokura
a,
Yasuko
Egawa
a,
Takehiro
Suzuki
d,
Naoshi
Dohmae
d,
Yoichi
Nakao
e and
Katsunori
Tanaka
 *abf
*abf
aBiofunctional Synthetic Chemistry, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: kotzenori@riken.jp
bDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro, Tokyo 152-8552, Japan
cDepartment of Medicinal and Organic Chemistry, School of Pharmacy, Iwate Medical University, Yahaba, Iwate 028-3694, Japan
dBiomolecular Characterization Unit, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
eSchool of Advanced Science and Engineering, Department of Chemistry and Biochemistry, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo 169-8555, Japan
fBiofunctional Chemistry Laboratory, A. Butlerov Institute of Chemistry, Kazan Federal University, 18 Kremlyovskaya Street, Kazan 420008, Russia
First published on 2nd September 2021
Abstract
Selective cell tagging (SeCT) therapy is a strategy for labeling a targeted cell with certain chemical moieties via a catalytic chemical transformation in order to elicit a therapeutic effect. Herein, we report a cancer therapy based on targeted cell surface tagging with proapoptotic peptides (Ac-GGKLFG-X; X = reactive group) that induce apoptosis when attached to the cell surface. Using either Au-catalyzed amidation or Ru-catalyzed alkylation, these proapoptotic peptides showed excellent therapeutic effects both in vitro and in vivo. In particular, co-treatment with proapoptotic peptide and the carrier–Ru complex significantly and synergistically inhibited tumor growth and prolonged survival rate of tumor-bearing mice after only a single injection. This is the first report of Ru catalyst application in vivo, and this approach could be used in SeCT for cancer therapy.
Introduction
Cell surface engineering has been widely utilized to prepare cell-based drugs, referred to as a living drug, to treat a variety of diseases.1 Cellular function can be regulated by tagging certain functional molecules to the cell surface, such as the transport control of the living drug in vivo.2Multiple reports have described methods for cell surface modification, including chemical tagging of protein amines or thiols,3 enzymatic4 or metabolic tagging,5 hydrophobic insertion,6 and genetic techniques.7 Most of these approaches were developed for cell engineering under in vitro or ex vivo conditions for the purpose of preparing living drugs. However, only a few examples have highlighted direct applications of cell surface modification in vivo, e.g., genetic modification,8 protein-targeted approaches,9 metabolic glycoengineering,10 or glycan-targeted metal-catalyzed labeling.11,12 The main limitation on this research has been the difficulty of implementing a specific tagging reaction on targeted cells or organs in vivo.
We previously applied a selective chemical tagging method for cancer treatment in vivo, referred to as selective cell tagging (SeCT) therapy (Fig. 1),12 which consists of three components: a catalyst carrier, a catalyst, and a therapeutic molecule linked to the catalyst's substrate. In the first step of SeCT, the biocompatible catalyst is delivered to the target cell by the catalyst carrier (Step 1). The catalyst then performs a tagging reaction on its substrate, thus attaching the therapeutic molecule onto the targeted cell (Step 2). Finally, the attached molecule exerts its therapeutic effects (Step 3). SeCT in vivo does not utilize genetic or metabolic pathways, and should therefore be able to exert a therapeutic effect such as disruption of cancer cell function without damaging surrounding tissues. Very recently, we succeeded in a kind of cancer therapy in which a cyclic RGD (cRGD) peptide13 or a doxorubicin prodrug is used to tag targeted cancer cells, thereby treating the cells via the enhanced function of these therapeutic moieties relative to the un-tagged form.12 In addition, various combinations of the three components could provide therapeutic effects against various diseases based on regulation of cellular function by the SeCT. The combination of components could be changed according to the targeted cell or therapeutic purpose. Moreover, the success of the strategy depends on a number of factors such as the target specificity of the carrier, the biorthogonality of every component, the efficiency of the catalytic reaction, and the function of the therapeutic molecule.
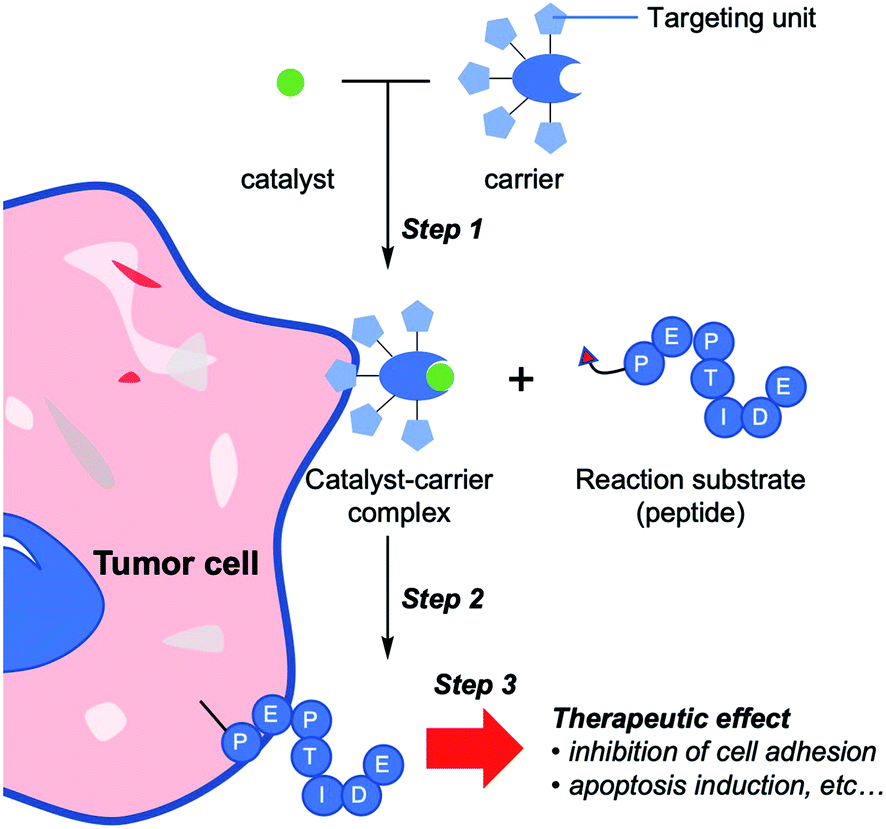 | ||
| Fig. 1 Schematic illustration of selective cell tagging (SeCT) strategy using therapeutic peptides. Step 1: selective delivery of catalyst to target cell by catalyst carrier. Step 2: conjugation of bioactive reaction substrates to the cell surface. Step 3: attached molecules exert therapeutic effects. | ||
Despite the benefits of SeCT therapy, the components available for this strategy are currently very limited. In particular, there is no example of a therapeutic substrate that can kill the tumor cells simply by covalent attachment to the cell surface. An ideal therapeutic molecule for SeCT therapy of cancer would minimize the unwanted side effects toward untargeted tissues and exert no therapeutic effect before attachment to the targeted cells. Transition-metal catalysts are an attractive candidate as a catalyst for application in the SeCT therapy because they can efficiently catalyze diverse organic reactions.14 However, many transition-metal catalysts are difficult to use in vivo because of their instability in aqueous or biological environments, and in previous work we have only succeeded in the in vivo application of gold-catalyzed amide bond formation using a propargyl ester (PE) compound as a substrate.11,12
In this study, we discovered a proapoptotic peptide that induces apoptosis only when it is covalently attached to the cell surface. In addition, we developed cell surface modification chemistry using a Ru catalyst and a benzyl fluoride (BnF) substrate. This approach yielded a more efficient modification than the previous Au-catalyzed amide bond formation. The Ru-catalyst system in combination with the proapoptotic peptide exerted an outstanding therapeutic effect on tumor growth inhibition in vivo following only a single administration. Based on these results, we propose that SeCT therapy represents an attractive alternative choice for cancer therapy.
Results and discussion
Cyclic RGD-coated human serum albumin [HSA(cRGD)] as a catalyst carrier
Human serum albumin (HSA) is a potential catalyst carrier because the hydrophobic pocket of HSA can protect the susceptible transition metal catalyst from quenchers present in body, such as glutathione.15 Furthermore, by coating the HSA surface with peptide or glycan as a targeting unit, the coated HSA can effectively target specific cells or organs based on the interaction between the coated unit and the distinctive proteins on the target cell surface.16In this study, cRGD peptide, which recognizes integrin and is frequently used as a drug delivery system,13 was used to target HSA to tumor cells. The cRGD-coated HSA [HSA(cRGD)] was prepared by mixing HSA and cRGD possessing succinimidyl ester. Mass analysis revealed that an average of five molecules of cRGD were conjugated onto the surface of HSA. To evaluate the targeting behavior of HSA(cRGD) in vivo, we intravenously injected Cy7.5-labeled HSA(cRGD) into mice bearing tumors derived from SW620 colon cancer cells. The imaging revealed rapid accumulation of fluorescence in tumors after only 4 h (Fig. 2b), indicating the excellent targeting ability of HSA(cRGD) in comparison with antibody17 or other cRGD conjugates.13 The fluorescence was retained after 24 h (Fig. S2†). This accumulation was caused by the dual effects of passive targeting by the enhanced permeability and retention (EPR) effect18 and active targeting by the cRGD unit. In addition, the complex of HSA(cRGD) with Au-Cou catalyst (HSA(cRGD)-Au) can be prepared via the 7-diethylaminocoumarin (DEAC) ligand, which binds to the hydrophobic pocket of HSA, as in our previous experiments11,12,15 (Fig. 2a). When DEAC binds to the pocket, the fluorescence intensity increases because its surrounding environment has become hydrophobic.19 Indeed, the fluorescence intensity of HSA(cRGD)-Au was 7-fold higher than that of the gold catalyst alone (Fig. 2c), indicating that the metal catalyst complex was successfully prepared for use of HSA(cRGD). The binding affinity of Au-Cou was determined as 39.2 μM of Kd by the measurement of fluorescence quenching induced by the ligand binding15 (Fig. S19†). The fluorescence quenching was inhibited by warfarin (ligand for the site I) not by ibuprofen (ligand for site II), so that Au-Cou mainly binds to site I20 (Fig. S20†). Based on observations regarding cancer targeting and complex formation with metal catalyst, we chose HSA(cRGD) as the cancer-targeting carrier for SeCT therapy in this study.
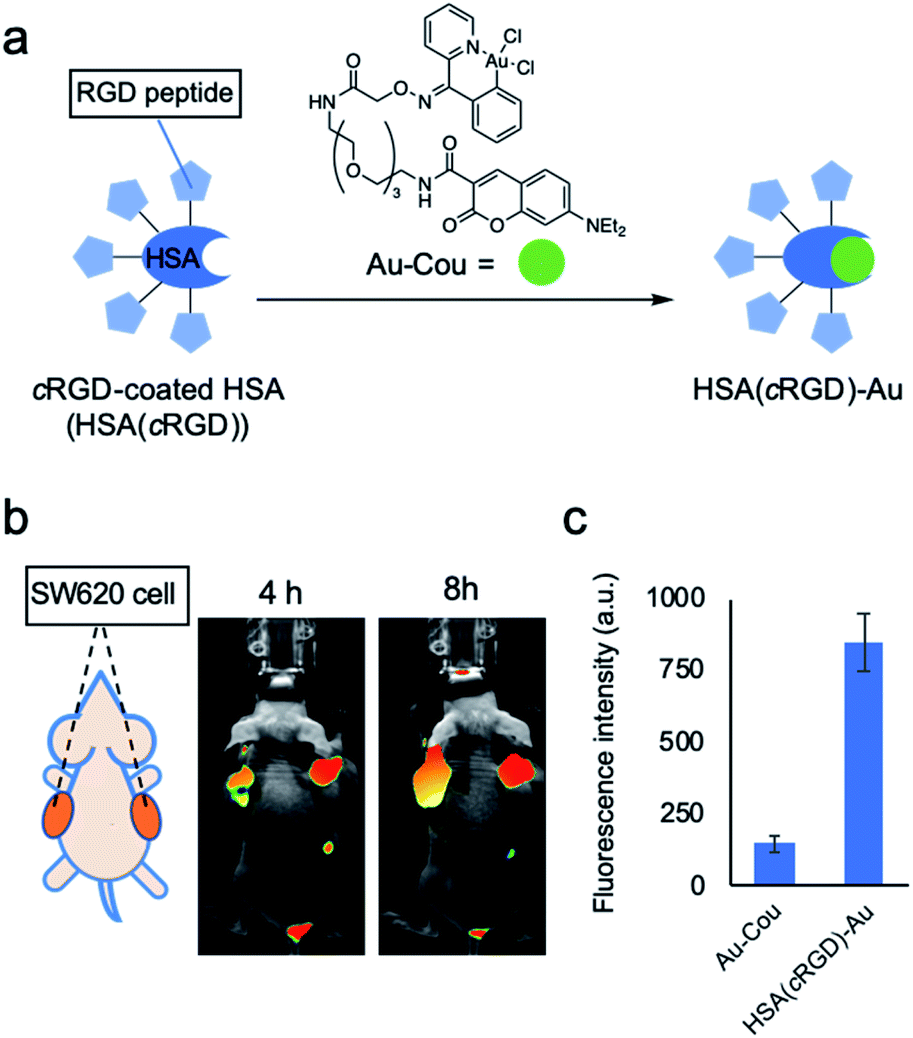 | ||
| Fig. 2 Properties of cRGD-coated HSA [HSA(cRGD)]. (a) Preparation of HSA(cRGD)-Au. (b) Fluorescence imaging of a mouse bearing a tumor of SW620 cells treated with Cy7.5-labeled HSA (10 nmol) after 4 or 8 h. (c) The change in fluorescence due to complex formation between HSA(cRGD) and Au-Cou (ex: 425 nm/em: 465 nm). Data are presented as means ± s.d. n = 3. | ||
Proapoptotic peptide with gold-catalyzed amide bond formation
Several studies have reported proapoptotic peptides,21 but the ideal substrate for SeCT therapy would only exert cytotoxicity when it is covalently attached to the cell surface. However, no such substrate has ever been reported. During our different project, we attempted in situ preparation of a bioactive cyclic peptide, such as a cyclin inhibitor,22 by gold-catalyzed amide bond formation using peptides possessing propargyl ester (PE) at the C-terminus (see ESI for more details; Fig. S1†). The cyclization reaction did not proceed, but we unexpectedly identified peptide 1 (Ac-GGKLFG-PE), which exerted cytotoxicity against SW620 cells in the presence of HSA(cRGD)-Au (20 μM) (Fig. 3a and b), from our peptide library. Because peptide 1 and HSA(cRGD)-Au were not cytotoxic on their own, and combination with PE compound and Au can cause tagging on the cellular surface (Fig. S4†), the observed cytotoxicity is based on the synergistic effect of covalent attachment of peptide 1 to the cell surface. In addition, other peptides with similar sequences, e.g., peptide 2 (Ac-GYKLFG-PE) generated by the Gly2Tyr substitution in peptide 1, had no cytotoxicity regardless of the presence or absence of the catalyst (Fig. 3b and S1†). Therefore, the cells were killed not only by covalent tagging of the cell surface, but also by a sequence-specific property of peptide 1.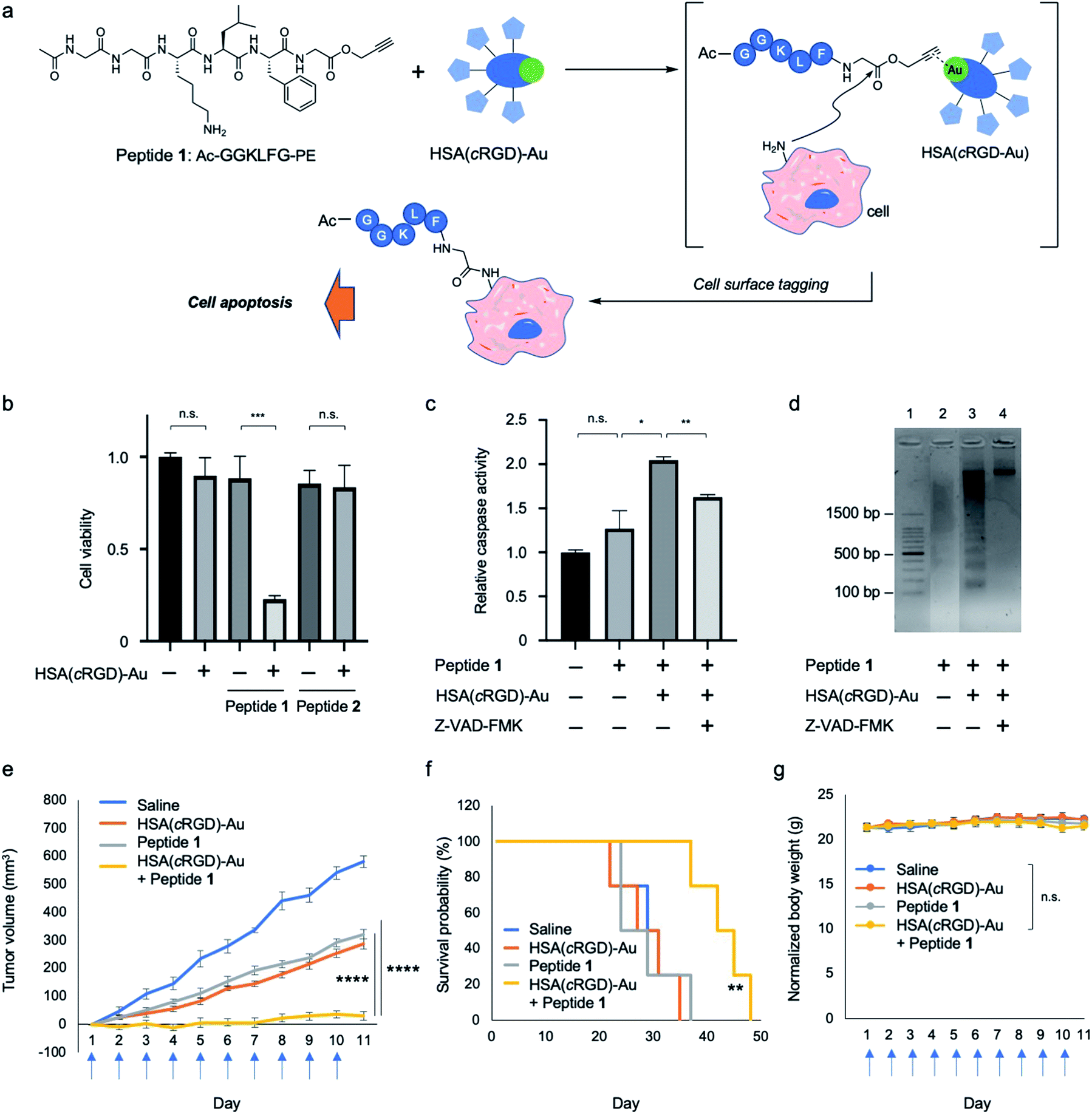 | ||
| Fig. 3 Cell surface modification with proapoptotic peptide 1 with propargyl ester attached at the C-terminus by Au-catalyzed amide bond formation (Au/PE). (a) Cell surface modification with proapoptotic peptide 1 and HSA(cRGD)-Au. (b) Cytotoxicity of peptide 1 or peptide 2 (600 μM) with or without HSA(cRGD)-Au (20 μM) in SW620 cells (n = 3, mean ± SE). (c) Caspase activities (n = 3, mean ± SE) in the presence of caspase inhibitor (Z-VAD-FMK). (d) Electrophoresis image of DNA fragmentation in cells treated with peptide 1 cells with or without HSA(cRGD)-Au. Effects of tumor therapy on (e) tumor volume, (f) survival probability, and (g) body weight changes of SW620-xenografted mice subjected to the following treatments: saline (n = 4, blue), HSA(cRGD)-Au (35.0 mg kg−1, n = 7, orange), peptide 1 (4.0 mg kg−1, n = 7, gray), and co-treatment of peptide 1 and HSA (cRGD)-Au (4.0 mg kg−1 and 35.0 mg kg−1, n = 7, yellow). Blue arrows under the horizontal axis indicate the day of treatment with compounds. Data are shown as means ± SE. n.s.: not significant, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001. | ||
Next, we evaluated the cell death pathway induced by peptide 1, in particular caspase activity, which is important for induction of apoptosis. A cell-based assay revealed that caspase activity was activated by peptide 1 in the presence of HSA(cRGD)-Au (Fig. 3c). In addition, cell viability was recovered by addition of caspase inhibitor Z-VAD-FMK (0.70 ± 0.04) to co-treatment with peptide 1 and HSA(cRGD)-Au (0.23 ± 0.02) (Fig. S3†). In addition, we examined DNA damage using a DNA ladder assay (Fig. 3d). The combination of peptide 1 and HSA(cRGD)-Au promoted DNA fragmentation, indicating DNA damage (Fig. 3d, lane 3), whereas the lane with peptide 1 alone (Fig. 3d, lane 2) showed slightly DNA fragmentation, which was caused by DMSO including in medium.23 These results suggested that the apoptosis induced by peptide 1 and HSA(cRGD)-Au was responsible for cell death. Interestingly, the synergistic cytotoxicity was not observed in three other cell lines, HeLa S3 (cervical cancer), A549 (lung cancer) and MCF-10A (non-tumorigenic mammary epithelial cells), even under the same evaluation conditions (Fig. S6†). The distinctive features of SW620 cells, which were established from metastatic cancer24 might contribute to the cytotoxic function of peptide 1. Although the detailed cytotoxic mechanism of peptide 1 remains unclear, we succeeded in obtaining a proapoptotic peptide that is toxic only when covalently attached to the targeted cell surface. Accordingly, this peptide would be suitable for cancer SeCT therapy.
Based on these results, we applied the combination of proapoptotic peptide 1 and HSA(cRGD)-Au to in vivo cancer therapy of mice bearing tumors of xenografted SW620 cells. The indicated compounds [Saline, HSA(cRGD)-Au, peptide 1, or combination of peptide 1 and HSA(cRGD)-Au] were intravenously administered every day for 10 consecutive days. Co-treatment with peptide 1 and HSA(cRGD)-Au significantly inhibited tumor growth (Fig. 3e, yellow), whereas treatment with peptide 1 or HSA(cRGD)-Au alone exerted only mild inhibition (Fig. 3e, gray and orange, respectively). These differences were clearer in the survival data; co-treated mice lived ∼13 days longer than mice in the other groups (Fig. 3f). The body weight of all groups remained stable during treatment, indicating that our compounds had no serious side effects (Fig. 3g). These findings demonstrate that the combination of peptide 1 and HSA(cRGD)-Au is applicable in vivo and could be used in a SeCT strategy for cancer therapy.
Ruthenium catalyzed alkylation of cell surface using the benzyl fluoride substrate
Although the proapoptotic peptide and Au-catalyzed system yielded good results in cancer therapy, there were still weaknesses; namely, the compounds were individually slightly toxic to tumors without synergy, and daily dosing was necessary (see the in vivo data with single dose injection, Fig S7†). Additionally, the sole example of the SeCT therapy that has succeeded in vivo to date was carried out using our previous Au/PE chemistry.11,12 Therefore, to improve the therapeutic efficacy in vivo via SeCT therapy, development of a new catalytic system is still in high demand. Hence, we focused on the biocompatible ruthenium catalyst Ru1, which can catalyze a deprotection reaction of allyloxycarbonyl (Alloc) group25 and (2 + 2 + 2) cycloaddition to construct the benzene ring.26 The catalytic deprotection of Ru1 has been utilized for fluorescence imaging25,27 and a prodrug strategy25,28 in cell-based experiments, but to date no study has described Ru1-catalyzed protein or cell tagging reactions or their in vivo applications.We envisioned and developed a reactive substrate that can be activated by Alloc deprotection, p-Alloc-protected aminobenzyl fluoride (BnF), for protein or cell modification (Fig. 4a). In this system, the reactive quinone methide imine is generated by deprotection of the Alloc group in a reaction catalyzed by Ru1 following 1,6-elimination. The intermediate is expected to react with adjacent nucleophilic structures such as thiol, alcohol, and amino groups in biomolecules.29 While there are many kinds of catalysts for Alloc deprotection,30 Ru1 that have been applied to cellular experiments should be suitable for this tagging system.25–28
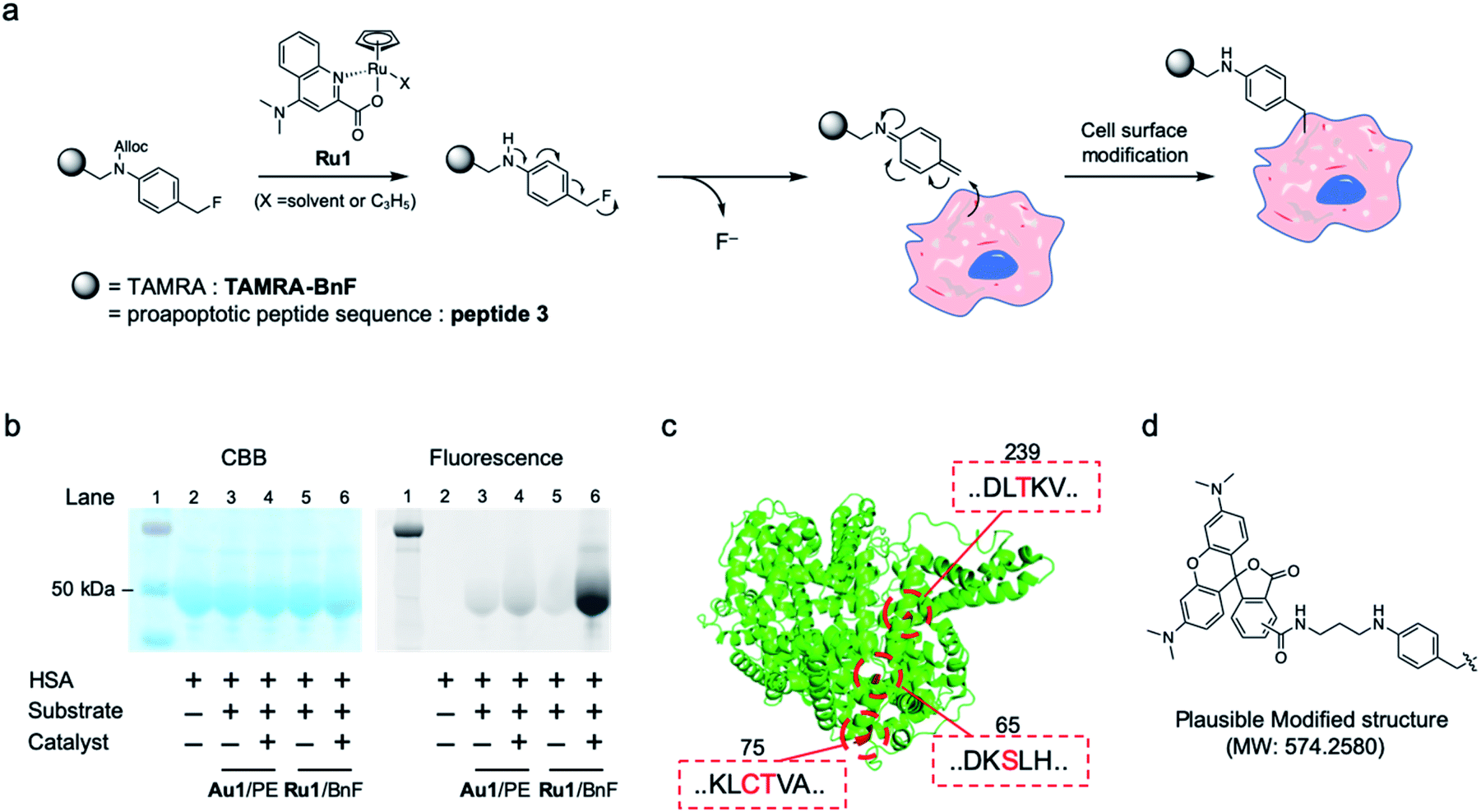 | ||
| Fig. 4 (a) Cell surface modification reaction by Ru1-catalyzed alkylation using benzyl fluoride substrate (Ru/BnF). (b) Model reaction of Ru/BnF with HSA analyzed by SDS-PAGE: protein ladder (lane 1), HSA (lane 2), after reaction of Au1/TAMRA-PE (lanes 3–4) or Ru1/TAMRA-BnF (lanes 5–6). (c) The modified amino acids on HSA-TAMRA (lane 6 in b), as determined by the MS/MS analysis. The ribbon diagram of HSA (PDB: 1AO6) and tagged amino acids are colored in green and red, respectively. (d) Plausible modified structure of amino acids. | ||
The tagging of HSA protein was performed using the synthesized substrate TAMRA-BnF and the Ru1 catalyst. In the SDS-PAGE analysis, the fluorescence band of TAMRA was detected at the same spot as the HSA band, indicating that the Ru1-catalyzed protein tagging was successful (Fig. 4b, lane 6). Furthermore, the fluorescence band was clearer when we used Ru1/TAMRA-BnF than when we used Au1/TAMRA-PE (Fig. 4b, lane 4 vs. 6, and Fig. S8†). In the other words, the protein tagging efficiency of Ru/BnF was superior to that of Au/PE. The tagging reaction of HSA with Au/PE was less effective than the cell surface tagging results (Fig. S4†) due to the different environments between the protein and cell surface. Then, the TAMRA-tagging HSA by Ru/BnF was analyzed by MS-MS analysis after enzymatic digestion (Fig. S9–S11†). The peptide fragment with the tagged form depicted in Fig. 4d (molecular weight change: 574.2580) was detected, and four amino acids were determined as the main tagged sites: Ser65, Cys75, Thr76, and Thr239; these residues are colored red in the crystal structure of HSA (Fig. 4c, PDB: 1AO6 (ref. 31)). The quinone methide structure can react with many kinds of nucleophilic amino acids,29a but the modification sites we detected were hydroxyl and thiol groups. These results suggested that deprotection of Alloc, leading to quinone methide imine formation, proceeded smoothly in our catalytic tagging system. In addition, the key unit of substrate, the benzyl fluoride structure, was stable against the major nucleophile glutathione (Fig. S12†). Therefore, the developed Ru-catalyzed protein modification reaction could be used for an in vivo reaction such as SeCT therapy.
For the in vivo experiment, the complex HSA(cRGD)-Ru was prepared by mixing the HSA(cRGD) and Ru-Cou to generate the targeting system for the catalyst (Fig. 5a). As with HSA(cRGD)-Au, the increased fluorescence of DEAC indicated complex formation between HSA(cRGD) and Ru-Cou (Fig. 5b and S13†). The Ru-Cou binding was determined as site I with a Kd value of 19.6 μM (Fig. S19–S23†), which is stronger than Au-Cou.
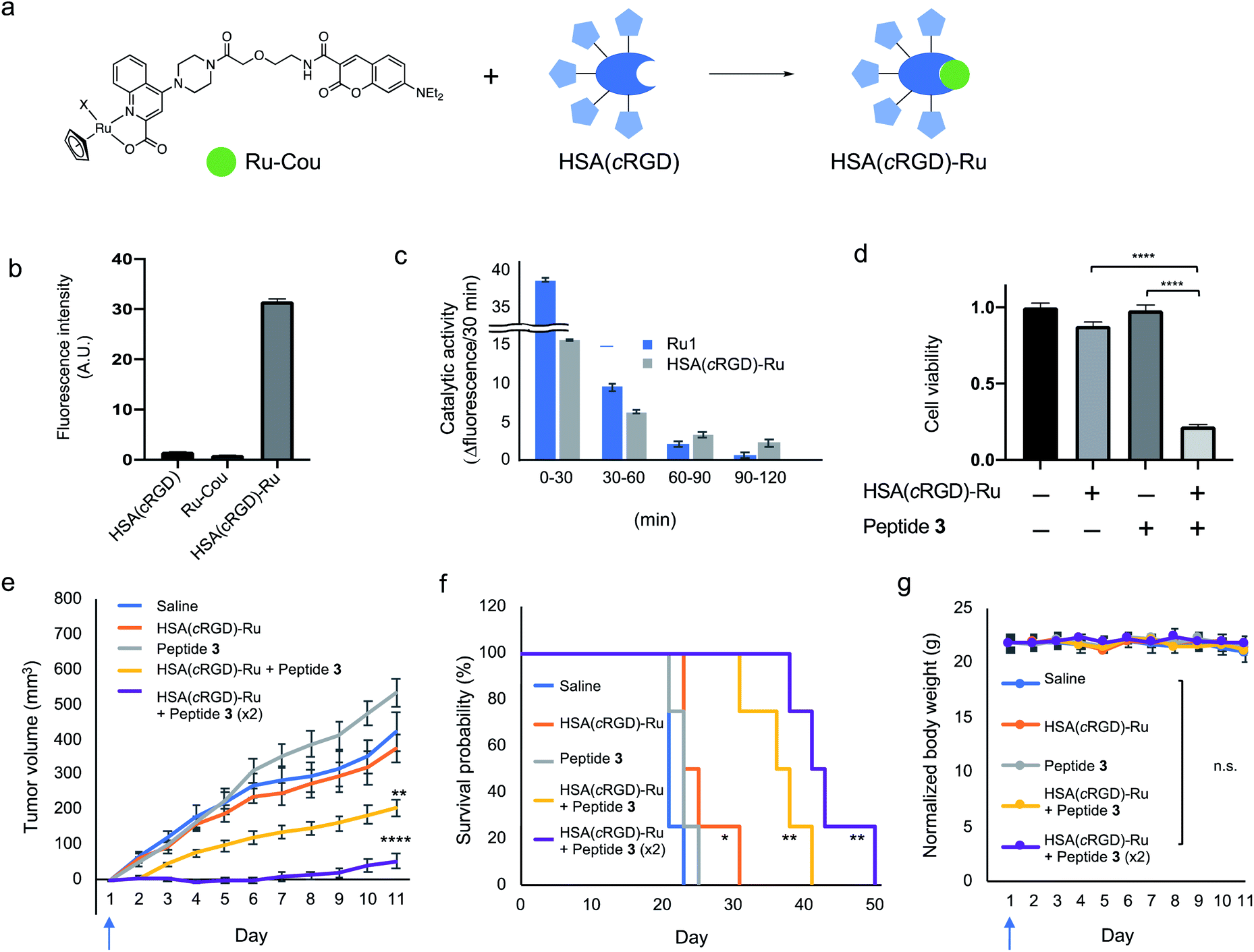 | ||
| Fig. 5 (a) HSA(cRGD)-Ru complex formation by Ru-Cou and HSA(cRGD). (b) Fluorescence change at the 7-diethylaminocoumarin moiety (ex: 425 nm/em: 465 nm) in the preparation of HSA(cRGD)-Ru. (c) Catalytic activity of Ru1 and HSA(cRGD)-Ru catalysts using Alloc-protected 7-amino-4-methylcoumarin (ex: 375 nm/em: 450 nm). (d) SW620 cell viability with the HSA(cRGD)-Ru (20 μM) and proapoptotic peptide 3 (600 μM). In vivo antitumor activity of peptide 3 and HSA(cRGD)-Ru against SW620 xenografted tumor. (e) Tumor volume (n = 8), (f) survival rate (n = 4), and (g) body weight (n = 4) change of various treatments group mice; vehicle (blue), HSA(cRGD)-Ru (69.0 mg kg−1, orange), peptide 3 (11.8 mg kg−1, gray), and peptide 3 + HSA(cRGD)-Ru (11.8 or 23.6 mg kg−1 and 69.0 or 138.0 mg kg−1, yellow or purple, respectively). Blue arrows under the horizontal axis indicate the day of treatment with the compounds. Data are shown as means ± SE. n.s.: not significant, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001. | ||
The catalytic activity of Ru1 and HSA(cRGD)-Ru was evaluated in 2% acetone-d6 in water condition using an Alloc-protected 7-amino-4-methylcoumarin (Cou-Alloc) as the reaction substrate; this compound became fluorescent after removal of the Alloc group (Fig. S14†). Interestingly, we found that the catalytic activity of Ru1 almost disappeared within 2 h under our evaluation conditions (Fig. 5c). Furthermore, the catalytic activity of Ru1 significantly decreased by preincubation in H2O for 30 min even in the absence of glutathione (Fig. S15b†). The instability of Ru1 indicates that the catalyst itself would not tolerate in vivo circulation, so it cannot be used in an in vivo experiment. On the contrary, the complex HSA(cRGD)-Ru was still active after incubating with glutathione or PBS (Fig. 5c and S15c†), because the bound Ru catalyst was protected by the hydrophobic pocket. In addition, the activity of HSA(cRGD)-Ru was surprisingly increased in the presence of glutathione. The series of Ru catalysts have both activation25 and deactivation effects30b by thiol compounds. Our results suggest that HSA not only protects the bound Ru catalyst, but also the catalytic process of the HSA(cRGD)-Ru complex is activated by glutathione, presumably by scavenging ally moiety. The HSA(cRGD)-Ru acts as a catalyst against the model substrate 12 with following kinetic values; KM = 11.5 mM, kcat = 0.605 min−1, kcat/KM = 0.0522 mM−1 min−1, turn over number (TON) = 7.4 (Fig. S24†). Taken together, the Ru complex, HSA(cRGD)-Ru, is a more suitable catalyst for in vivo application than the naked Ru1 catalyst.
Biological evaluation of HSA(cRGD)-Ru and proapoptotic peptide 3
Cytotoxicity against SW620 cells was evaluated using HSA(cRGD)-Ru and proapoptotic peptide 3 with the BnF structure at the C-terminus. Initially, the cytotoxicity of HSA(cRGD)-Ru was lower than that of HSA(cRGD)-Au (Fig. S16†), meaning that the Ru catalyst was more biocompatible. Due to its low toxicity, HSA(cRGD)-Ru could be used at a higher concentration in this system. In addition, peptide 3 exerted significant cytotoxicity against SW620 cells in the presence of HSA(cRGD)-Ru catalyst (Fig. 5d) under the same conditions as HSA(cRGD)-Au/peptide 1. In addition, we observed DNA fragmentation when cells were co-treated with HSA(cRGD)-Ru and peptide 3, whereas the DNA remained unfragmented in the presence of Z-VAD-FMK (Fig. S17†). Although the modification of the cell surface should be different than when Au/PE is used, the cytotoxicity was observed in this Ru catalytic system. On the basis of these results, the peptide sequence Ac-GGKLFG-X (X = reactive group) should be applicable to other kinds of cell-surface tagging methods for the tumor therapy.Finally, we applied the combination of HSA(cRGD)-Ru and peptide 3 to in vivo tumor therapy, again using mice bearing SW620 tumors. The compounds [Saline, HSA(cRGD)-Ru, peptide 3, or combination of peptide 3 and HSA(cRGD)-Ru] were intravenously injected only on the first day, in contrast to the experiment using HSA(cRGD)-Au and peptide 1 (Fig. 3e–g), which required 10 injections. Tumor growth was significantly suppressed by co-injection of HSA(cRGD)-Ru and peptide 3 (Fig. 5e, yellow line), whereas peptide or catalyst alone did not inhibit tumor growth (gray and orange lines, respectively), indicating a synergistic effect of peptide 3 and HSA(cRGD)-Ru (p < 0.005, two-way ANOVA) (The photograph of tumor at day 1 and day 8 is shown in Fig. S18†). This approach yielded a much stronger therapeutic effect than Au/PE chemistry with everyday injection, probably due to the higher reactivity of Ru/BnF (Fig. 4b) and lower toxicity of the HSA(cRGD)-Ru catalyst (Fig. S16†). Furthermore, an increasing dose of peptide 3 and HSA(cRGD)-Ru only a single intravenous injection led to stronger suppression of tumor growth (purple line), and the survival time improved from 23 days (saline) to 50 days (co-administration of HSA(cRGD)-Ru and peptide 3) (Fig. 5f). The combination of catalyst and peptide significantly damaged the tumor at the first injection, and hence the tumor growth was effectively suppressed even after 50 days. Considering solely the Kd value of Ru-Cou to albumin, some metal catalyst may exist at an unbound state in the blood. The unbound catalyst could cause off-target reaction hence leading to cytotoxicity, while such adverse effects were not observed in this study. Nevertheless, the high-affinity or covalent binding of the metals to HSA could be more preferred for wider application of the method. This is the first successful example of an in vivo application of Ru1, which was achieved by the amelioration of targeting ability and catalyst susceptibility with HSA(cRGD). These results expand the applicability of metal catalysts to tumor tagging strategies, and demonstrate that this approach represents a promising alternative to cancer therapy.
Conclusions
Here, we have described an advanced application of SeCT therapy for cancer treatment based on a proapoptotic peptide that is covalently attached to the cell surface (Ac-GGKLFG-X, X = reactive moiety) and a Ru-catalyzed tagging reaction. This is the first report of such a peptide, although many kinds of proapoptotic peptides have been reported.21 The features of the proapoptotic peptide were suited to the SeCT concept, and the activation of apoptotic pathways is a promising strategy for cancer therapy. In addition, the cell tagging reaction using the Ru1 catalyst yielded better results than the previous gold-catalyzed reaction. In particular, the combination of HSA(cRGD)-Ru and proapoptotic peptide yielded an excellent therapeutic effect against in vivo tumors after only a single intravenous injection. This is the first report describing application of the Ru1 catalyst in vivo, and the results suggest that the other sensitive metal catalysts could be used for in vivo SeCT therapy, taking advantage of the protective effect of the hydrophobic pocket of HSA. Furthermore, the SeCT therapy has wide applicability, not only for inhibition of cell adhesion or prodrug activation12 but also for tumor therapy by direct killing of tumor cells using a covalent tagging strategy.SeCT therapy for cancer therapy would consist of three components (catalyst, reactive substrate, and metal carrier), and the appropriate combination of these would achieve a high therapeutic index. We anticipate that our results will provide an alternative way to treat cancer in the future. Moreover, the application of the selective tagging concept is not limited to tumor treatment, but could also be used against various other diseases.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: K. Tanaka, formal analysis, P. A. and K. M.; funding acquisition: K. M., T. C. C. and K. Tanaka, investigation: P. A., K. M., T. C. C., S. T., K. Tsubokura, Y. E., T. S. and N. D.; project administration: N. D., Y. N. and K. Tanaka; supervision: K. Tanaka; visualization: P. A., and K. M., writing–original draft: K. M. and K. Tanaka, writing–review and editing: P. A., K. M., T. C. C. and K. Tanaka.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of RIKEN and approved by the Animal Ethics Committee of RIKEN (W2019-2-049). This work was financially supported by the AMED Grant JP15KM0908001, a research grant from the Astellas Foundation, Mizutani Foundation for Glycoscience, and JSPS KAKENHI Grant Numbers, JP19J00396 (to K. M.), JP20K15968 (to K. M.), JP19K15708 (to T. C. C.), JP21H02065 (to K. Tanaka), JP21K19042 (to K. Tanaka). This work was also funded by the subsidy allocated to Kazan Federal University for state assignment in the sphere of scientific activities (0671-2020-0063), with the support of the Kazan Federal University Strategic Academic Leadership Program.Notes and references
- (a) D. Y. Lee, B. H. Cha, M. Jung, A. S. Kim, D. A. Bull and Y. W. Won, J. Biol. Eng., 2018, 12, 28 CrossRef CAS PubMed; (b) J. Park, B. Andrade, Y. Seo, M. J. Kim, S. C. Zimmerman and H. Kong, Chem. Rev., 2018, 118, 1664 CrossRef CAS PubMed; (c) L. Liu, H. He and J. Liu, Polymers, 2019, 11, 2017 CrossRef CAS PubMed.
- (a) D. Sarkar, P. K. Vemula, G. S. Teo, D. Spelke, R. Karnik, L. Y. Wee and J. M. Karp, Bioconjugate Chem., 2008, 19, 2105 CrossRef CAS PubMed; (b) S. A. Kim and J. S. Peacock, J. Immunol. Methods, 1993, 158, 57 CrossRef CAS PubMed.
- (a) J. L. Contreras, D. Xie, J. Mays, C. A. Smyth, C. Eckstein, F. G. Rahemtulla, C. J. Young, J. A. Thompson, G. Bilbao, D. T. Curiel and D. E. Eckhoff, Surgery, 2004, 136, 537 CrossRef PubMed; (b) G. Digilio, V. Menchise, E. Gianolio, V. Catanzaro, C. Carrera, R. Napolitano, F. Fedeli and S. Aime, J. Med. Chem., 2010, 53, 4877 CrossRef CAS PubMed; (c) A. G. Torres and M. J. Gait, Trends Biotechnol., 2012, 30, 185 Search PubMed.
- L. K. Swee, S. Lourido, G. W. Bell, J. R. Ingram and H. L. Ploegh, ACS Chem. Biol., 2015, 10, 460 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS PubMed.
- (a) D. Rabuka, M. B. Forstner, J. T Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 5947 CrossRef CAS PubMed; (b) Y. W. Won, A. N. Patel and D. A. Bull, Biomaterials, 2014, 35, 5627 CrossRef CAS PubMed.
- (a) Z. Cheng, L. Ou, X. Zhou, F. Li, X. Jia, Y. Zhang, X. Liu, Y. Li, C. A. Ward, L. G. Melo and D. Kong, Mol. Ther., 2008, 16, 571 CrossRef CAS PubMed; (b) D. M. Barrett, S. A. Grupp and C. H. June, J. Immunol., 2015, 195, 755 CrossRef CAS PubMed.
- (a) R. J. Ernst, T. P. Krogager, E. S Maywood, R. Zanchi, V. Beránek, T. S Elliott, N. P. Barry, M. H. Hastings and J. W. Chin, Nat. Chem. Biol., 2016, 12, 776 CrossRef CAS PubMed; (b) Y. Chen, J. Ma, W. Lu, M. Tian, M. Thauvin, C. Yuan, M. Volovitch, Q. Wang, J. Holst, M. Liu, S. Vriz, S. Ye, L. Wang and D. Li, Cell Res., 2017, 27, 294 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341 CrossRef CAS PubMed.
- (a) J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873 CrossRef CAS PubMed; (b) A. A. Neves, H. Stöckmann, Y. A. Wainman, J. C.-H. Kuo, S. Fawcett, F. J. Leeper and K. M. Brindle, Bioconjugate Chem., 2013, 24, 934 CrossRef CAS PubMed; (c) H. Wang, R. Wang, K. Cai, H. He, Y. Liu, J. Yen, Z. Wang, M. Xu, Y. Sun, X. Zhou, Q. Yin, L. Tang, I. T. Dobrucki, L. W. Dobrucki, E. J. Chaney, S. A. Boppart, T. M. Fan, S. Lezmi, X. Chen, L. Yin and J. Cheng, Nat. Chem. Biol., 2017, 13, 415 CrossRef CAS PubMed.
- K. Tsubokura, K. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS PubMed.
- K. Vong, T. Tahara, S. Urano, I. Nasibullin, K. Tsubokura, Y. Nakao, A. Kurbangalieva, H. Onoe, Y. Watanabe and K. Tanaka, Sci. Adv., 2021, 7, eabg4038 CrossRef CAS PubMed.
- (a) E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697 CrossRef CAS PubMed; (b) J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9 CrossRef CAS PubMed; (c) M. Pfaff, K. Tangemann, B. Müller, M. Gurrath, G. Müller, H. Kessler, R. Timpl and J. Engel, J. Biol. Chem., 1994, 269, 20233 CrossRef CAS; (d) M. Aumailley, M. Gurrath, G. Muller, J. Calvete, R. Timpl and H. Kessler, FEBS Lett., 1991, 291, 50 CrossRef CAS PubMed; (e) Y. Cheng and Y. Ji, Eur. J. Pharm. Sci., 2019, 128, 8 CrossRef CAS PubMed.
- (a) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (b) J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106 CrossRef CAS PubMed; (c) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- (a) S. Eda, I. Nasibullin, K. V. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. T. Tanaka, Nat. Catal., 2019, 2, 780 CrossRef CAS; (b) T. C. Chang, K. Vong, T. Yamamoto and K. Tanaka, Angew. Chem., Int. Ed., 2021, 60, 2 CrossRef.
- (a) A. Ogura, T. Tahara, S. Nozaki, K. Morimoto, Y. Kizuka, S. Kitazume, M. Hara, S. Kojima, H. Onoe, A. Kurbangalieva, N. Taniguchi, Y. Watanabe and K. Tanaka, Sci. Rep., 2016, 6, 21797 CrossRef CAS PubMed; (b) A. Ogura, T. Tahara, S. Nozaki, H. Onoe, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Bioorg. Med. Chem. Lett., 2016, 26, 2251 CrossRef CAS PubMed; (c) A. Ogura, S. Urano, T. Tahara, S. Nozaki, R. Sibgatullina, K. Vong, T. Suzuki, N. Dohmae, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Chem. Commun., 2018, 54, 8693 RSC; (d) L. Latypova, R. Sibgatullina, A. Ogura, K. Fujiki, A. Khabibrakhmanova, T. Tahara, S. Nozaki, S. Urano, K. Tsubokura, H. Onoe, Y. Watanabe, A. Kurbangalieva and K. Tanaka, Adv. Sci., 2016, 4, 1600394 CrossRef PubMed; (e) I. Smirnov, R. Sibgatullina, S. Urano, T. Tahara, P. Ahmadi, Y. Watanabe, A. R. Pradipta, A. Kurbangalieva and K. Tanaka, Small, 2020, 16, 2004831 CrossRef CAS PubMed; (f) L. Gao, Y. Zhang, L. Zhao, W. Niu, Y. Tang, F. Gao, P. Cai, Q. Yuan, X. Wang, H. Jiang and X. Gao, Sci. Adv., 2020, 6, eabb1421 CrossRef CAS PubMed; (g) S. Ichimizu, H. Watanabe, H. Maeda, K. Hamasaki, K. Ikegami, V. T. G. Chuang, R. Kinoshita, K. Nishida, T. Shimizu, Y. Ishima, T. Ishida, T. Seki, H. Katsuki, S. Futaki, M. Otagiri and T. Maruyama, J. Control. Release, 2019, 304, 156 CrossRef CAS PubMed; (h) K. Chen, J. Xie and X. Chen, Mol. Imag., 2009, 8, 65 CrossRef CAS.
- (a) A. El-Sayed, W. Bernhard, K. Barreto, C. Gonzalez, W. Hill, L. Pastushok, H. Fonge and C. R. Geyer, Theranostics, 2018, 8, 4856 CrossRef CAS PubMed; (b) Y. Koide, Y. Urano, K. Hanaoka, W. Piao, M. Kusakabe, N. Saito, T. Terai, T. Okabe and T. Nagano, J. Am. Chem. Soc., 2012, 134, 5029 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387 CAS.
- G. Jones II, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294 CrossRef.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38 CrossRef CAS PubMed.
- (a) H. M. Ellerby, W. Arap, L. M. Ellerby, R. Kain, R. Andrusiak, G. D. Rio, S. Krajewski, C. R. Lombardo, R. Rao, E. Ruoslahti, D. E. Bredesen and R. Pasqualini, Nat. Med., 1999, 5, 1032 CrossRef CAS PubMed; (b) K. A. Min, P. Maharjan, S. Ham and M. C. Shin, Arch Pharm. Res., 2018, 41, 594 CrossRef CAS PubMed; (c) J. C. Mai, Z. Mi, S. Kim, B. Ng and P. D. Robbins, Cancer Res., 2001, 61, 7709 CAS; (d) C. Borghouts, C. Kunz and B. Groner, J. Pept. Sci., 2005, 11, 713 CrossRef CAS PubMed.
- M. J. I. Andrews, C. McInnes, G. Kontopidis, L. Innes, A. Cowan, A. Platera and P. M. Fischera, Org. Biomol. Chem., 2004, 2, 2735 RSC.
- H. Sun and Y. Wang, Cell Res., 1995, 5, 181 CrossRef.
- (a) C. Slater, J. A. De La Mare and A. L. Edkins, Oncol. Lett., 2018, 15, 8516 Search PubMed; (b) A. Leibovltz, J. C. Stlnson, W. B. McCombs III, C. E. McCoy, K. C. Mazur and N. D. Mabry, Cancer Res., 1976, 36, 4562 Search PubMed.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- J. Miguel-Ávila, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2020, 59, 17628–17633 CrossRef PubMed.
- (a) T. Heinisch, F. Schwizer, B. Garabedian, E. Csibra, M. Jeschek, J. Vallapurackal, V. B. Pinheiro, P. Marlière, S. Panke and T. R. Ward, Chem. Sci., 2018, 9, 5383–5388 RSC; (b) E. Bartolami, D. Basagiannis, L. Zong, R. Martinent, Y. Okamoto, Q. Laurent, T. R. Ward, M. Gonzalez-Gaitan, N. Sakai and S. Matile, Chem.–Eur. J., 2019, 25, 4047–4051 CrossRef CAS PubMed; (c) H. T. Hsu, B. M. Trantow, R. M. Waymouth and P. A. Wender, Bioconjugate Chem., 2016, 27, 376–382 CrossRef CAS PubMed; (d) M. Szponarski, F. Schwizer, T. R. Ward and K. Gademann, Commun. Chem., 2018, 1, 84 CrossRef; (e) Y. Cheng, L. Zong, J. López-Andarias, E. Bartolami, Y. Okamoto, T. R. Ward, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2019, 58, 9522 CrossRef CAS PubMed.
- (a) Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, Nat. Commun., 2018, 9, 1943 CrossRef PubMed; (b) M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed; (c) T. Völker and E. Meggers, ChemBioChem, 2017, 18, 1083 CrossRef PubMed.
- (a) J. Liu, S. Li, N. A. Aslam, F. Zheng, B. Yang, R. Cheng, N. Wang, S. Rozovsky, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2019, 141, 9458 CrossRef CAS PubMed; (b) V. Ahmed, Y. Liu and S. D. Taylor, ChemBioChem, 2009, 10, 1457 CrossRef CAS PubMed; (c) N. W. Polaske, B. D. Kelly, J. Ashworth-Sharpe and C. Bieniarz, Bioconjugate Chem., 2016, 27(3), 660 CrossRef CAS PubMed; (d) E. Modica, R. Zanaletti, M. Freccero and M. Mella, J. Org. Chem., 2001, 66(1), 41 CrossRef CAS PubMed.
- (a) F. Guibe, Tetrahedron, 1997, 53, 13509 CrossRef CAS; (b) N. Kamo, T. Kujirai, H. Kurumizaka, H. Murakami, G. Hayashi and A. Okamoto, Chem. Sci., 2021, 12, 5926 RSC.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01784e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |