
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMimicking transition metals in borrowing hydrogen from alcohols†‡
Ananya
Banik
 ,
Jasimuddin
Ahmed
,
Swagata
Sil
and
Swadhin K.
Mandal
*
,
Jasimuddin
Ahmed
,
Swagata
Sil
and
Swadhin K.
Mandal
*
Department of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata, Mohanpur, 741246, India. E-mail: swadhin.mandal@iiserkol.ac.in
First published on 7th May 2021
Abstract
Borrowing hydrogen from alcohols, storing it on a catalyst and subsequent transfer of the hydrogen from the catalyst to an in situ generated imine is the hallmark of a transition metal mediated catalytic N-alkylation of amines. However, such a borrowing hydrogen mechanism with a transition metal free catalytic system which stores hydrogen molecules in the catalyst backbone is yet to be established. Herein, we demonstrate that a phenalenyl ligand can imitate the role of transition metals in storing and transferring hydrogen molecules leading to borrowing hydrogen mediated alkylation of anilines by alcohols including a wide range of substrate scope. A close inspection of the mechanistic pathway by characterizing several intermediates through various spectroscopic techniques, deuterium labelling experiments, and DFT study concluded that the phenalenyl radical based backbone sequentially adds H+, H˙ and an electron through a dearomatization process which are subsequently used as reducing equivalents to the C–N double bond in a catalytic fashion.
Introduction
Borrowing hydrogen from alcohols, and utilization of the harvested hydrogen for further reduction of in situ generated imines is the hallmark of a transition metal mediated catalytic process which leads to the synthesis of N-alkylated amines. It may be noted that N-alkylated amines have widespread applications in chemistry and biology. These amines are extensively utilized as universal building blocks for organic synthesis, pharmaceuticals, agrochemicals, drugs, materials science, the polymer industry, and various natural products.1–5 Traditional methods for the preparation of substituted amines followed the reductions of imines6 and amides,7,8 hydroamination,9,10 reductive amination11 and also, there are straight forward methods such as the N-alkylation of amines.12,13 The conventional methods for the alkylation of amines involve the use of highly genotoxic alkylating agents such as alkyl halides or sulfonate esters. However, these methods suffer from the generation of stoichiometric amounts of halide waste, and the chemoselectivity issue for the formation of over alkylated by-products. In this regard, the use of an alcohol as a benign alkylating agent has drawn considerable attention due to its remarkable abundance in biomass as well as for the generation of green waste (water as the sole by-product). Thus, the use of an alcohol for N-alkylation of amines is considered as a green, sustainable and environmentally safe process. In this regard, Winans and Adkins14 have discovered this reaction and later Grigg15 and Watanabe16 have reported the first homogeneous catalysis towards the alkylation of amines using an alcohol, which has been termed ‘Borrowing Hydrogen or Hydrogen Autotransfer (BH/HA)’. In this methodology, an alcohol is dehydrogenated to a more reactive carbonyl compound, where a proton and a hydride are transferred to the transition metal-based catalyst and typically stored as the metal dihydride (Scheme 1). Next, the carbonyl compound undergoes a condensation reaction with an amine generating an imine which subsequently undergoes reduction to an N-alkylated amine through the transfer of the stored hydride and proton when the catalyst is regenerated. This method involves the use of precious metals such as ruthenium,17,18 rhodium19 and iridium20-based catalysts and was extensively used in early studies. In more recent years, the alkylation of amines through the borrowing hydrogen strategy was dominated by inexpensive, earth-abundant metal-based catalysts21–35 replacing the expensive noble metal catalysts. In this regard, substantial progress has been demonstrated by the group of Feringa and Barta,21,22 Beller,23 Milstein,24 Kempe,25–27 and others28–31 using early transition metals such as iron, manganese, cobalt, nickel and chromium-based catalysts to make the processes more environmentally benign. Towards the sustainable synthesis of alkylated amines, the need for a transition metal-free catalyst is inevitable. In this regard, a bioinspired catalytic combination such as NADH-ADH36,37 or transition metal free combination using pyridine/KOtBu38 evolved recently for the alkylation of amines, but they differ conceptually from the borrowing hydrogen mechanism followed typically by a transition metal based catalyst. The transition metal based catalyst stores both proton and hydride in its backbone (Scheme 1), which in turn can deliver a hydrogen molecule for further reductions. On the other hand, the existing metal-free catalysts are not capable of mimicking such a role as they can store only hydride while proton is stored in a different molecule. Although a few other studies have been reported where a purely base mediated39–43N-alkylation by an alcohol is achieved using KOH, CsOH and NaOH respectively, they do not follow the borrowing hydrogen pathway as observed in transition metal based catalysis. Such an observation in the existing literature prompted us to strategize a transition metal-free catalyst, which can conceptually mimic the role of a transition metal in borrowing hydrogen mediated N-alkylation of amines involving the storage and transfer of hydrogen molecules from the catalyst backbone.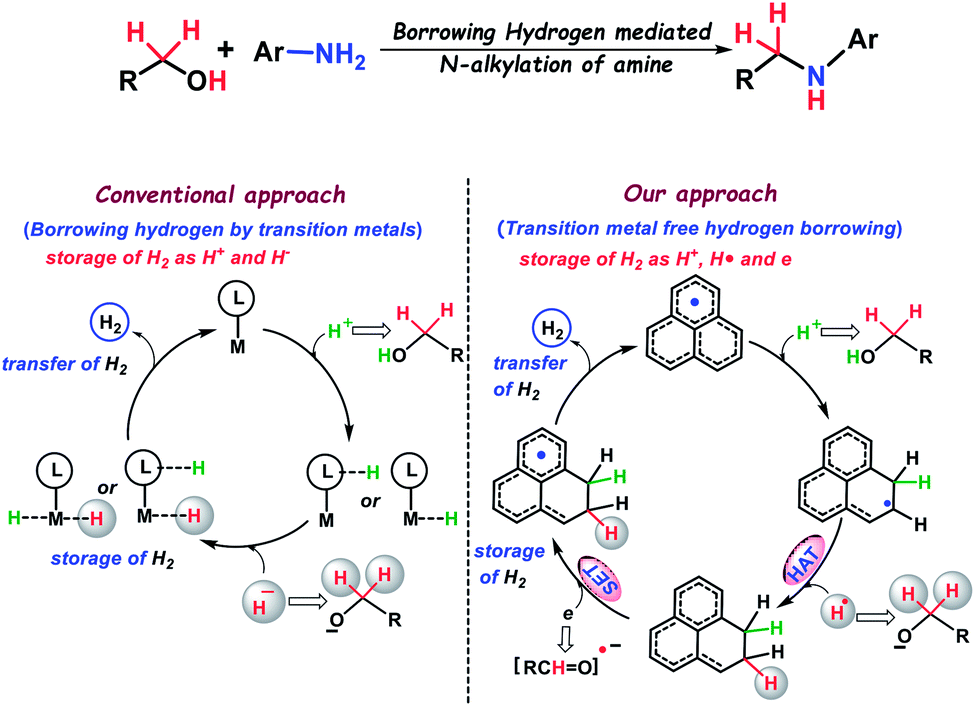 | ||
| Scheme 1 Alkylation of amines by alcohols via hydrogen borrowing: conventional approach and our strategy. | ||
Herein, we present the alkylation of anilines by various alcohols using a redox non-innocent phenalenyl (PLY) based ligand as a transition metal-free catalyst. It is well established that PLY based molecules can access three redox active states, namely a closed shell cation, an open shell radical, and a closed shell anion using the non-bonding molecular orbital (NBMO).44 Over the past few decades, a remarkable advance has been envisaged in the field of quantum spin simulators,45–48 organic molecular conductors,49–52 molecular batteries,53 and spin electronic devices54 by the group of Haddon, Nakasuzi, Takui, Morita, and Kubo55 using the radical state of PLY molecules. The catalytic applications using in situ generated PLY based radicals have only been recently developed.56 In this regard, we earlier demonstrated that PLY can store and transfer redox equivalents in the form of C–H bonds during multi-electron reduction processes.57 Very recently, we reported that phenalenyl ligands can be reduced by two electrons and can subsequently trap two protons via dearomatization of one of the phenyl rings of PLY.58 Such observations57,58 made us curious whether the PLY moiety can store both hydride and proton together thus imitating the role of a metal in a typical borrowing hydrogen mechanism followed by its transfer for subsequent reduction integrated in a catalytic fashion.
In the current study, we have established that the PLY based catalyst can store the borrowed hydrogen molecule by addition of H+, H˙ and an electron sequentially from the alcohol partner and stores in the form of two C–H bonds through a dearomatization process which are subsequently transferred to the in situ generated imines to realize the corresponding alkylated aromatic amines (Scheme 1). This study presents an alternative strategy for replacing the transition metal based catalysts in the alkylation of aromatic amines through the borrowing hydrogen methodology.
Results and discussion
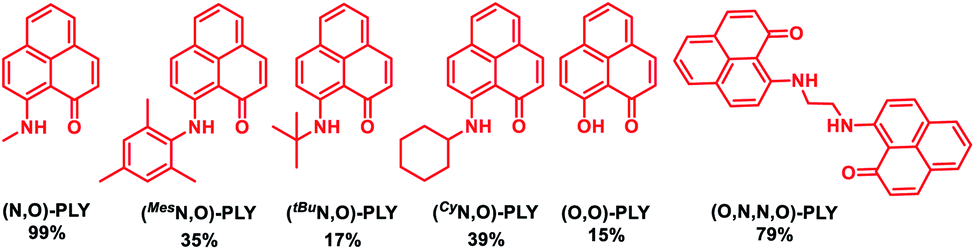
At first, several redox non-innocent PLY based molecules [(O,O)-PLY, various (N,O)-PLY and (O,N,N,O)-PLY] were synthesized according to literature methods.59–62 The preliminary investigation of transition metal-free alkylation of amines started with probing the reaction of aniline and benzyl alcohol in the presence of a catalytic amount of these PLY molecules with 1.2 equivalents of base, KOtBu, to realize the corresponding N-benzylaniline (Table 1, also see ESI, Table S1‡). The reaction of aniline, 1a (0.3 mmol), and benzyl alcohol, 2a (0.33 mmol), in the presence of a catalytic amount of (N,O)-PLY (5.0 mol%) with KOtBu (1.2 equivalents) in toluene at 130 °C afforded 43% yield of the N-alkylated amine, 3a (N-benzylaniline) within 18 hours (entry 1, Table S1, ESI‡). Further increasing the loading of (N,O)-PLY (10.0 mol%) and scrutinizing different PLY molecules (Table 1), the maximum NMR yield (99%) of N-alkylated amine, 3a, was realized (entries 3–8, Table S1, ESI‡). Moreover, a drastically reduced yield was observed when the temperature or amount of KOtBu was lowered (entries 9–11, Table S1, ESI‡). Notably, with the help of further trials using different bases, solvents, and durations of the reaction, optimized conditions were accomplished in the presence of 10.0 mol% (N,O)-PLY and 1.2 equivalents of KOtBu in toluene at 130 °C within 18 hours, providing 91% isolated yield of N-benzylaniline (entry 3, Table S1, ESI‡). Control experiments unarguably validated that the alkylation of amines could not materialize in the absence of base, and only a trace (5%) amount of N-benzylaniline, 3a, was realized in the absence of (N,O)-PLY.|
|
|---|
| a The reactions were carried out using aniline (0.3 mmol), benzyl alcohol (0.33 mmol), different PLY based molecules (10 mol%), KOtBu (1.2 equivalents), and toluene (1.5 mL). NMR yields. |
|
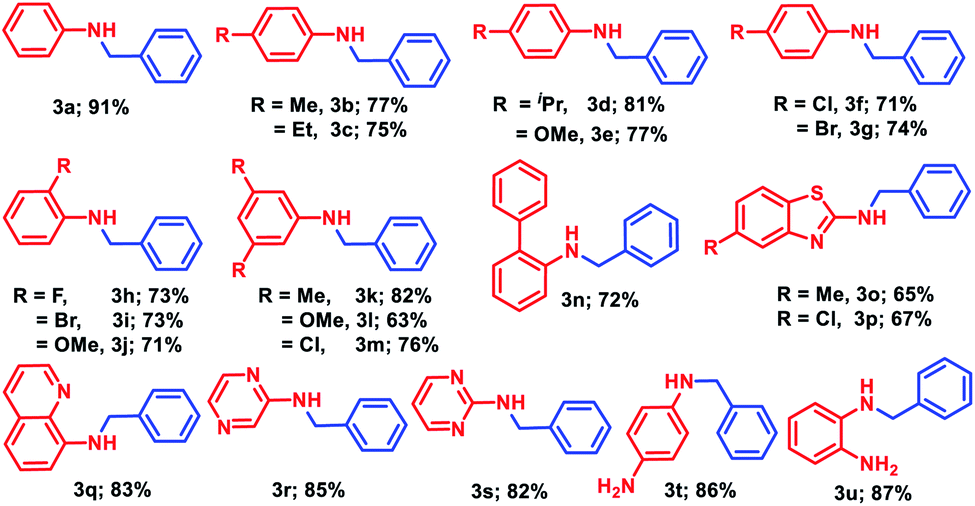
Having the optimized reaction conditions in hand (entry 3, Table S1, ESI‡), we investigated the efficacy of this methodology for the selective monoalkylation of various anilines (1a–1u) with benzyl alcohol, 2a, and the derivatives of N-benzylaniline (3a–3u) were isolated through column chromatography (Table 2). Under the standard reaction conditions, aniline (1a) and para-substituted anilines (1b–1g) containing both electron donating or electron withdrawing groups such as methyl, ethyl, isopropyl, methoxy, chloro, and bromo afforded good to excellent yield (71–91%) of the corresponding N-benzylanilines (3a–3g). Next, ortho-substituted anilines containing fluoro, bromo, and methoxy substituents were tested for the alkylation, and very good isolated yields (71–73%) of the corresponding N-alkylated products (3h–3j) were obtained. Under similar reaction conditions, 3,5-disubstituted anilines (1k–1m) furnished good to excellent isolated yields (63–82%) of the corresponding substituted amines (3k–3m). The standard catalytic reaction protocol was also well applicable for biarylated amine, i.e., [1,1′-biphenyl]-2-amine, which yielded 72% of the N-benzyl-[1,1′-biphenyl]-2-amine (3n). Given the importance of heterocyclic amines for the production of pharmaceuticals and natural products, we endeavoured several heteroaryl anilines such as 5-substituted benzothiazole, quinoline, pyrazine, and pyrimidine containing amines for the alkylation with benzyl alcohol. Under the optimized reaction conditions, very good to excellent yields (65–85%) of the corresponding N-alkylated products (3o–3s) were realized. To our delight, when different diamines such as benzene-1,4-diamine or benzene-1,2-diamine was subjected to N-alkylation with benzyl alcohol, only the formation of mono-alkylated products (3t and 3u) was observed with excellent yield (86% and 87%, respectively).
|
|
|---|
| a The reactions were carried out using anilines (0.3 mmol), alcohols (0.33 mmol), (N,O)-PLY (0.03 mmol), KOtBu (0.36 mmol), and toluene (1.5 mL) at 130 °C for 18 h. Average isolated yields from two catalytic runs are presented. Characterized by 1H and 13C NMR spectroscopy. |
|
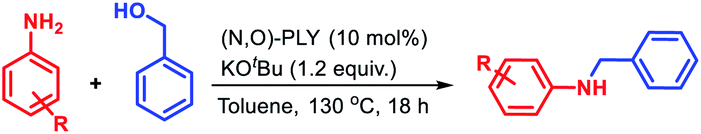
Furthermore, the catalytic alkylation of aniline was investigated using different alcohols to realize substituted N-benzyl anilines (Table 3). In this study, the effect of the various alcohols was evaluated using a series of substituted benzyl alcohols, naphthalen-1-ylmethanol, and substituted diphenylmethanol. Preferably, we tested different para substituted benzyl alcohols containing halide functionality for the alkylation, affording 76–87% isolated yield of the corresponding products without any functionalization of the halide moieties (4b–4e). Under the standard reaction conditions, electron donating methyl, isopropyl and methoxy groups containing 4-substituted benzyl alcohol provided moderate to good yield (55–70%) of alkylated amines (4f–4h). The more sterically encumbered 2-methyl, 3,5-dimethoxy, and 3,4,5-trimethoxy benzyl alcohol delivered good yield (53–70%, 4i–4k) as well as strong electron withdrawing 3-trifluoromethyl benzyl alcohol also afforded the desired alkylated product with 78% yield (4l). Under similar reaction conditions, naphthalen-1-ylmethanol, 2m, reacts with aniline to give 87% yield of N-(naphthalen-1-ylmethyl)aniline (4m). Next, the versatility of this catalytic protocol was established by using various secondary alcohols in the reaction medium, whereas diphenylmethanol and di-para-tolylmethanol afforded 89% and 85% yield of the corresponding N-alkylated amines 4n and 4o, respectively.
|
|
|---|
| a The reactions were carried out using anilines (0.3 mmol), alcohols (0.33 mmol), (N,O)-PLY (0.03 mmol), KOtBu (0.36 mmol), and toluene (1.5 mL) at 130 °C for 18 h. Average isolated yields from two catalytic runs are presented. Characterized by 1H and 13C NMR spectroscopy. Parentheses indicate the GC yield. |
|
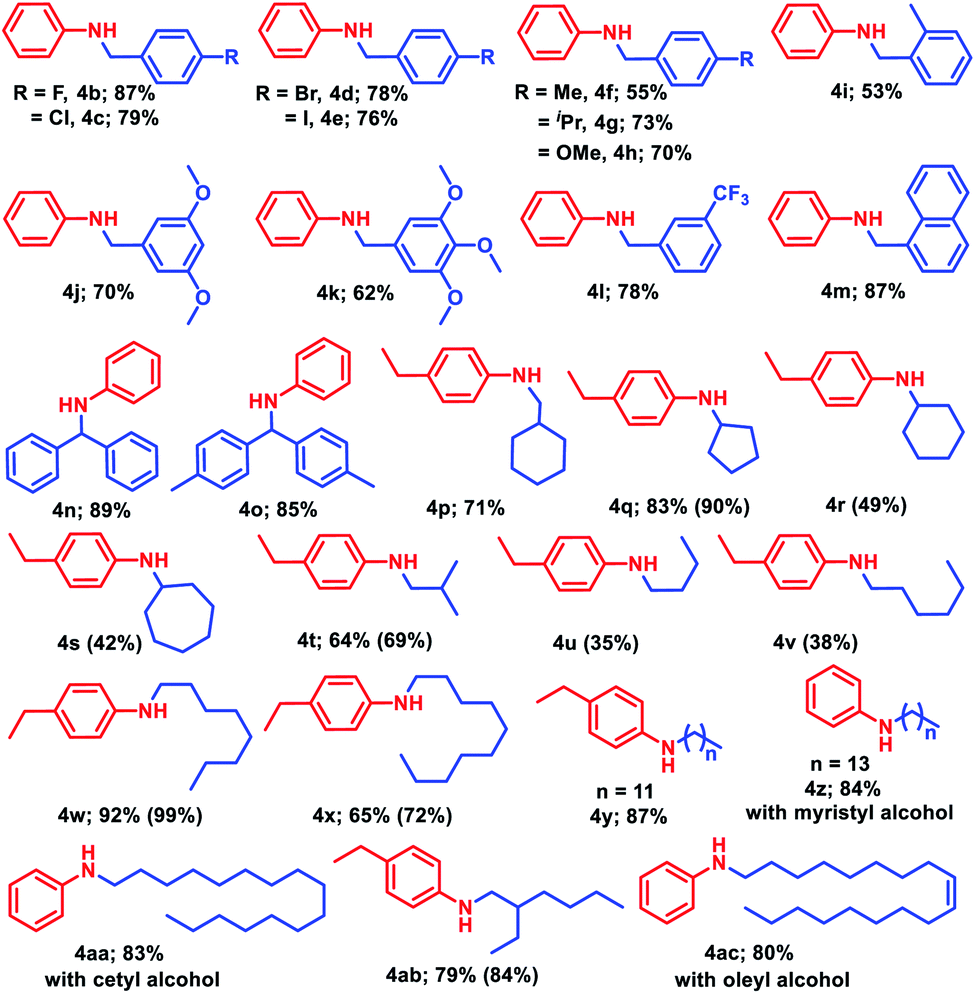
Next, to realize the general applicability of the catalytic protocol, more inert aliphatic alcohols were explored. To our delight, cyclohexylmethanol, 2p, reacted with 4-ethylaniline under the standard reaction conditions to accomplish 71% isolated yield of the corresponding product (4p). Moreover, other cyclic alcohols such as cyclopentanol, cyclohexanol and cycloheptanol provided 42–90% yield of 4q–4s, respectively. Isobutanol, 2t, delivered moderate isolated yield (64%) of 4-ethyl-N-isobutylaniline (4t), whereas 1-butanol and 1-hexanol displayed lower yields of corresponding N-alkylated aromatic amines, 4u–4v (35–38%). Biologically active long chain alcohols were also tested successfully for N-alkylation of aromatic amines, when 1-octanol or 1-decanol or 1-dodecanol was charged with 4-ethylaniline to achieve excellent isolated yields (65–87%) of the corresponding products (4w–4y). Similarly, 1-tetradecanol (myristyl alcohol) and 1-hexadecanol (cetyl alcohol) furnished 84% and 83% isolated yield of the corresponding products 4z and 4aa, respectively when reacted with aniline. Under the standard reaction conditions, 4-ethyl-N-(2-ethylhexyl)aniline (4ab) was obtained in 79% yield with 2-ethyl-1-hexanol, whereas oleyl alcohol delivered 80% yield of (Z)-N-(octadec-9-en-1-yl)aniline (4ac).
The broad substrate scope of this reaction prompted us to further study the applicability of this transition metal free borrowing hydrogen methodology for the preparation of pharmaceutical molecules or natural products without discomforting any other functionality. We could successfully functionalize the derivative of (±)-α-tocopherol, i.e., vitamin E, by introducing a benzyl group as an N-benzylated amine with the help of our standard catalytic protocol, where 69% yield of the corresponding amine was achieved (3v, Scheme 2). Moreover, gram scale synthesis of the product, 3v, was accomplished, where 65% yield of 3v was obtained. Another commercially available drug, adapalene, which is a third-generation topical retinoid and used for the treatment of acne, could be functionalized after reduction in carboxylic acid functionality. The corresponding reduced alcohol (2ad) when charged with aniline (1a) under the standard catalytic protocol provides 72% monoalkylated amine (4ad, Scheme 2).
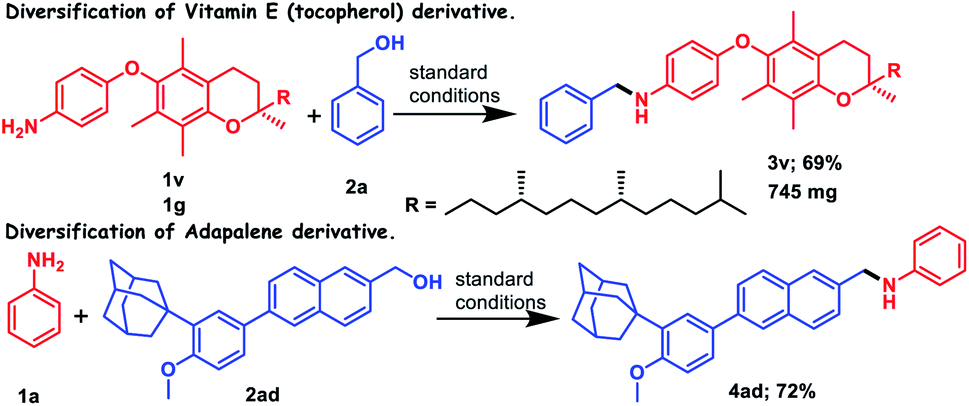 | ||
| Scheme 2 Applications of (N,O)-PLY catalyzed N-alkylation towards the diversification of biologically important molecules. | ||
The successful catalytic N-alkylation of aromatic amines using redox active PLY molecules encouraged us to delve the mechanistic details based on a series of control experiments and DFT study. Transition metal catalyzed N-alkylation of amines has been well documented in the literature and typically followed the process of alcohol dehydrogenation, condensation, and imine hydrogenation, where a transition metal catalyst can store hydrogen via borrowing it from alcohols and further transfers to in situ generated imines for the production of N-alkylated amines. However, the transition metal-free borrowing of hydrogen from alcohols which mimics transition metals by storing it in a catalyst has not been realized although such attempts have been reported in the literature in which the hydride was stored in the transition metal free catalyst backbone while the proton was stored separately.36–38 Nevertheless, the signature of the borrowing hydrogen mechanism considers the formation of aldehyde and imine as intermediates in the reaction medium. Interestingly, we noticed the formation of aldehyde and imine intermediates during time dependent NMR studies (supported by 1H NMR peaks at δ 10.02 ppm and δ 8.46 ppm, Fig. S110, ESI‡) rather than the generation of carbocations or ethers as reported for other metal-free N-alkylation mechanisms.63,64 Such an observation indicates that the borrowing hydrogen mechanism is operative. Furthermore, to understand the role of KOtBu, we have treated benzyl alcohol, 2a, with 1 equivalent N-benzylideneaniline, 5a, along with 10 mol% (N,O)-PLY and only 20 mol% KOtBu resulting in 88% N-benzylaniline, 3a, after isolation (Scheme 3a). This experiment suggests that only 0.2 equivalent base suffices the imine hydrogenation step while the additional equivalent of base (total 1.2 equivalent base is used in the catalytic reaction) is required in the imine generation step. Next, we have performed deuterium labelling experiments using deuterium enriched 4-chloro benzyl alcohol (4-Cl-C6H4-CD2-OH, 2c-d2) and aniline to afford a mixture of deuterium labelled isotopomers of N-alkylated anilines (Scheme S5, ESI‡). Such an observation indicates that the source of hydrogen in this reaction is benzyl alcohol.
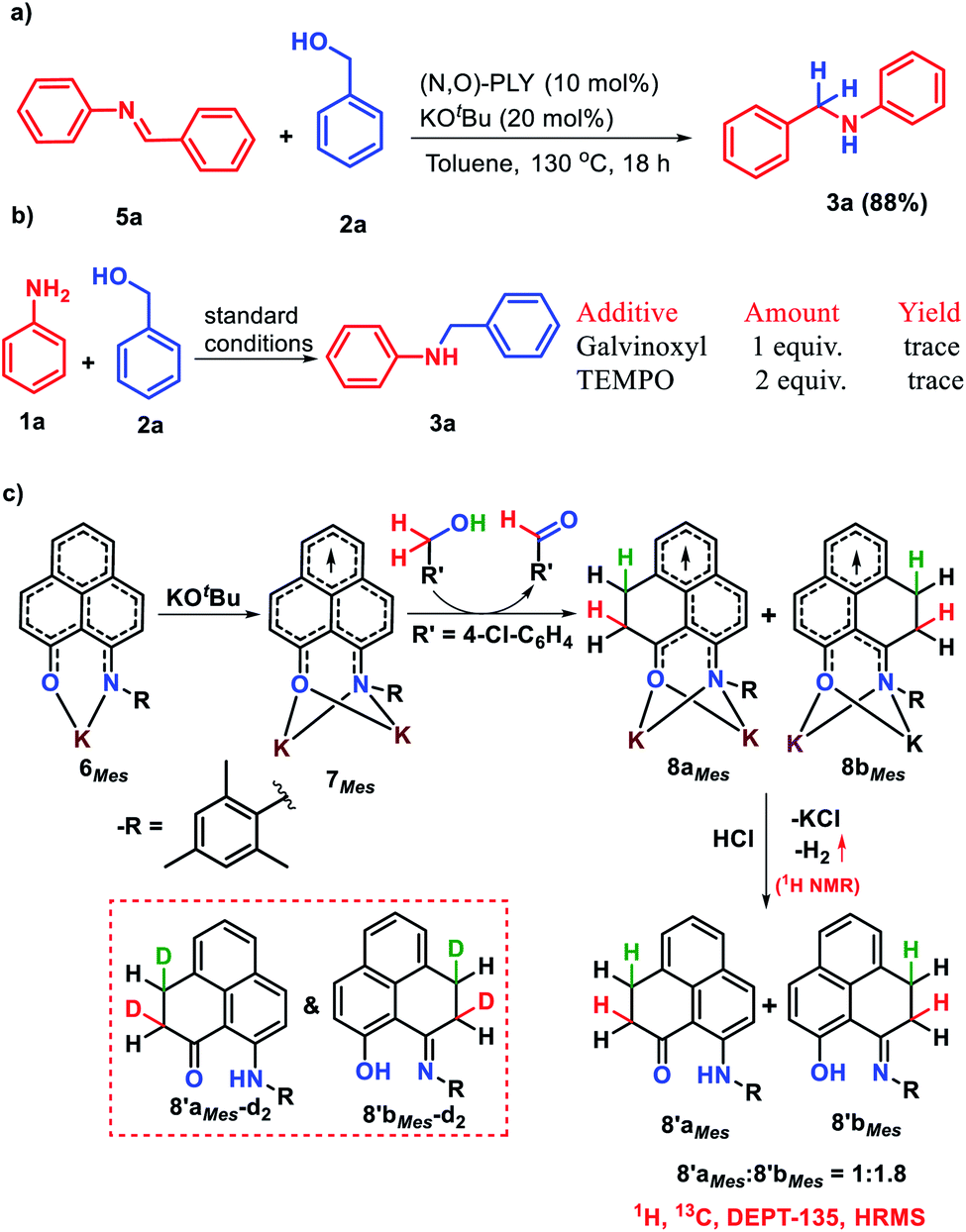 | ||
| Scheme 3 Control experiments. (a) Reduction of imine by an alcohol with a catalytic amount of base. (b) Quenching of the catalytic reaction in the presence of radical inhibitors. (c) Storing of borrowed hydrogen from the alcohol on the PLY backbone via dearomatization (inset indicates deuterium incorporation from the deuterium enriched alcohol). | ||
As discussed earlier, phenalenyl (PLY) based moieties can store and transfer a redox equivalent through its backbone.57 The efficacious catalytic alkylation of amines supported by PLY based ligands, encouraged us to demonstrate how a borrowed hydrogen molecule from the alcohol partner is stored in the PLY backbone and transferred to the in situ generated imine. Accordingly, we planned a series of stoichiometric reactions. At first, to establish whether the alkylation of amines followed a radical pathway or not, we examined the effect of 2,2,6,6-tetramethylpiperidinoxyl (TEMPO) as well as galvinoxyl radical for the alkylation of aniline (1a) using benzyl alcohol (2a) under standard reaction conditions (Scheme 3b). The reduced yield (56%) of N-benzylaniline was realized upon charging of 1 equivalent TEMPO, whereas a trace amount of N-benzylaniline was observed when 2 equivalents of TEMPO or 1 equivalent of galvinoxyl free radical was used. These experiments suggest that the catalytic alkylation of amine proceeds through a radical pathway. Keeping this information in mind, we proceeded to react the bidentate (MesN,O)-PLY based ligand with one equiv. KOtBu which produces a K-(MesN,O)-PLY complex 6Mes. The formation of such a complex was earlier established and was characterized by single crystal X-ray crystallography.58 Next, the addition of another equivalent of KOtBu to K-coordinated (MesN,O)-PLY (6Mes) produces the radical anion species (7Mes) upon acceptance of an electron from KOtBu and such a radical anion species was also recently characterized by single crystal X-ray and spectroscopic measurements.58 Interestingly, the radical anion species 7Mes can next accept the alcoholic proton from benzyl alcohols which was supported by DFT calculation (vide infra).
Next, to corroborate whether the borrowed hydrogen from the alcohol is stored in the PLY backbone or not, we performed a control reaction between (MesN,O)-PLY radical anion species (7Mes) and benzyl alcohol in toluene (Scheme 3c). On treatment of (MesN,O)-PLY with two equivalents of KOtBu and one equivalent of 4-chlorobenzyl alcohol (2c) in toluene at 130 °C, a color change from orange to wine red was observed. The resulting reaction mixture was subsequently charged with aqueous HCl to leach the coordinated K affording a deep green reaction mixture when hydrogen gas evolution was noted and confirmed by GC-MS analysis (Fig. S131, ESI‡) as well as 1H NMR spectroscopy (Fig. S130, ESI‡) (δ 4.46 ppm in toluene-d8).57 On scrutiny of the 1H NMR spectrum of the isolated products after HCl treatment (by column chromatography), two triplets in the region δ 2.63 ppm and 3.12 ppm (Fig. S117, ESI‡) along with a multiplate at δ 4.19–4.28 ppm (Fig. S117, ESI‡) were observed, suggesting the dearomatization of the PLY ring.57,58 The treatment of DCl in place of HCl also resulted in similar dearomatized products as confirmed by NMR analysis, which suggests that the two new C–H bonds in 8′aMes/8′bMes result from the borrowed hydrogen molecule from the alcohol which is stored in the PLY molecule. Furthermore, two peaks at δ 15.90 ppm and 13.29 ppm in the 1H NMR spectrum were observed for the purified product of such a reaction which can be assigned to –OH and –NH functional groups, respectively, as shown in 8′aMes and 8′bMes (Scheme 3c). Such findings suggest the formation of two isomers (8′aMes and 8′bMes) in a 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (from 1H NMR) as shown in Scheme 3c. Further 13C NMR (Fig. S118, ESI‡) and DEPT-135 (Fig. S119, ESI‡) NMR spectroscopy support our assumption of proposed dearomatized products as depicted in Scheme 3c. In the high resolution mass spectrometry spectra of the isolated products, only one peak at m/z 316.1697 amu (Fig. S122, ESI‡) refers to the presence of two isomers when correlated with the NMR spectroscopic results. Moreover, when (MesN,O)-PLY was charged with two equivalents of KOtBu and one equivalent of deuterium analogue of 4-chlorobenzylalcohol (4-Cl-C6H4-CD2-OD, 2c-d3) in another set of reactions, after leaching with aqueous HCl, to our delight, we observed the formation of dideuterated dearomatized PLY molecules such as 8′aMes-d2 and 8′bMes-d2 (Scheme 3c, inset) along with other isotopomers (Fig. S128, ESI‡). The formation of such dideuterated dearomatized isotopomers was confirmed by high-resolution mass spectrometry when a peak at 318.1861 was observed (see Fig. S128, ESI‡). Such an observation unarguably establishes the source of the harvested D2 originating from the deuterated benzyl alcohol. Furthermore, we have performed deuterium labelling experiments with 4-chlorobenzylalcohol (4-Cl-C6H4-CD2-OH), (2c-d2) when the relative concentration of dideuterated dearomatized isotopomers (8′aMes-d2 and 8′bMes-d2) decreased as expected (HRMS, Fig. S131, ESI‡), while the use of DCl in place of HCl did not alter the relative intensity of such a dideuterated dearomatized isotopomer (Table S2, ESI‡) as observed from HRMS (Fig. S129, ESI‡). All these control experiments further strengthen the proposal that the borrowed hydrogen molecule from the alcohol is stored on the PLY moiety in the form of two C–H bonds via a dearomatization process.
:1 ratio (from 1H NMR) as shown in Scheme 3c. Further 13C NMR (Fig. S118, ESI‡) and DEPT-135 (Fig. S119, ESI‡) NMR spectroscopy support our assumption of proposed dearomatized products as depicted in Scheme 3c. In the high resolution mass spectrometry spectra of the isolated products, only one peak at m/z 316.1697 amu (Fig. S122, ESI‡) refers to the presence of two isomers when correlated with the NMR spectroscopic results. Moreover, when (MesN,O)-PLY was charged with two equivalents of KOtBu and one equivalent of deuterium analogue of 4-chlorobenzylalcohol (4-Cl-C6H4-CD2-OD, 2c-d3) in another set of reactions, after leaching with aqueous HCl, to our delight, we observed the formation of dideuterated dearomatized PLY molecules such as 8′aMes-d2 and 8′bMes-d2 (Scheme 3c, inset) along with other isotopomers (Fig. S128, ESI‡). The formation of such dideuterated dearomatized isotopomers was confirmed by high-resolution mass spectrometry when a peak at 318.1861 was observed (see Fig. S128, ESI‡). Such an observation unarguably establishes the source of the harvested D2 originating from the deuterated benzyl alcohol. Furthermore, we have performed deuterium labelling experiments with 4-chlorobenzylalcohol (4-Cl-C6H4-CD2-OH), (2c-d2) when the relative concentration of dideuterated dearomatized isotopomers (8′aMes-d2 and 8′bMes-d2) decreased as expected (HRMS, Fig. S131, ESI‡), while the use of DCl in place of HCl did not alter the relative intensity of such a dideuterated dearomatized isotopomer (Table S2, ESI‡) as observed from HRMS (Fig. S129, ESI‡). All these control experiments further strengthen the proposal that the borrowed hydrogen molecule from the alcohol is stored on the PLY moiety in the form of two C–H bonds via a dearomatization process.
To gain more insight into the reaction mechanism, the alkylation of aniline (1a) with benzyl alcohol (2a) was investigated by density functional theory (DFT) for a full mechanistic cycle by the m062x method employing the 6-31+g(d) basis set with the SMD solvent model for toluene, as elucidated in Fig. 1. The catalytic alkylation was studied with the N-Me substituted phenalenyl [(N,O)-PLY] radical anion, 7. The radical anion 7 upon reaction with benzyl alcohol accepts the alcoholic –OH proton at one of the spin-bearing positions of PLY via a transition state TS1 with an activation energy barrier, ΔG‡1 = 12.4 kcal mol−1, to form the intermediate 7Int where one unpaired electron is situated on the adjacent carbon atom of the phenalenyl moiety. Notably, the interaction of the alkoxide anion with the potassium ion in 7Int favors the benzylic hydrogen atom transfer (HAT) from alkoxide to the radical carbon center of PLY through the transition state TS2. Such a hydrogen atom transfer mechanism was further supported by a series of control experiments which enabled us to trap the ketyl radical intermediates by TEMPO and characterize them. For example, we have been able to trap the ketyl radical generated from 4-fluoro benzyl alcohol (2b) upon reaction with 2 equivalents of TEMPO and was characterized by high resolution mass spectroscopy (m/z 282.1870, Fig. S107, ESI‡). The trapped ketyl radicals with 4-nitrobenzyl alcohol (2r) and 4-trifluoromethylbenzyl alcohol (2s) were also characterized through GC-MS (Fig. S108 and S109, ESI‡).
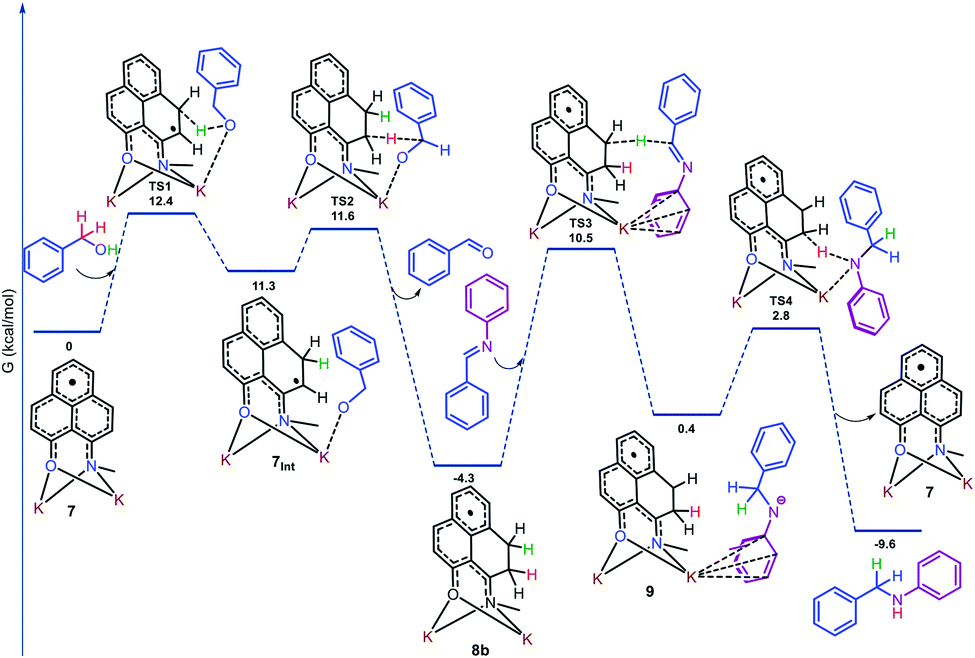 | ||
| Fig. 1 Energy profile diagram along the reaction coordinate for the catalytic N-alkylation, obtained from DFT calculation. Only one isomer is represented for the sake of simplicity. | ||
The transition state energy barrier for this HAT process is only 0.3 kcal mol−1 (ΔG‡2), which results in the formation of dearomatized phenalenyl radical intermediate 8 (consists of 8a and 8b), where both alcoholic O–H and benzylic C–H hydrogens were stored as two C–H bonds via dearomatization. Interestingly, the formation of intermediate 8 through the transition state, TS2, involves two concerted steps. At the first step, the HAT from the benzylic position of alkoxide to the catalyst produced a new C–H bond and results in the formation of a benzylic radical. In the very next step, this newly generated ketyl radical anionic species transfers an electron to the dearomatized phenalenyl catalyst backbone to release the aldehyde. The aldehyde and aniline undergo KOtBu mediated imine formation, and the TS calculation shows that this process involves two transition states TS1i (ΔG‡1i = 1.5 kcal mol−1) and TS2i (ΔG‡2i = 21.4 kcal mol−1), and both the transition states consider interaction with K counter ions (Fig. S133, ESI‡). This calculation excludes the possibility of catalyst's involvement in the imine formation step. Next, the stored hydrogens in the form of C–H bonds get transferred to the imine intermediate to yield the corresponding amines. The transfer of stored hydrogens follows a two-step process, where a hydride is transferred from intermediate 8 to the imine to generate an anion over the nitrogen centre of the imine, which is stabilized by delocalization over the aromatic ring (9) through the transition state TS3 with an activation energy barrier of 14.8 kcal mol−1. Next, the final step is accomplished by abstracting a proton from the phenalenyl catalyst backbone by the nitrogen center of anilido to produce the desired alkylated product through transition state TS4 with a transition state energy barrier of 3.2 kcal mol−1 (ΔG‡4). This final step regenerates the active radical anionic phenalenyl catalyst 7 along with the product formation.
Accounting all these trapped and in situ generated intermediates based on several control experiments and DFT calculations, we depicted the plausible mechanism as a borrowing hydrogen methodology in Scheme 4 where sequentially, H+, a H-atom and an electron are stored in the PLY backbone via dearomatization and subsequently transferred to the in situ generated imine functionality thus mimicking the metal based borrowing hydrogen mechanism. At first, the active radical catalyst 7 is formed by addition of two equivalents of KOtBu to (N,O)-PLY. Next, upon interacting with benzyl alcohol, the –OH proton and the benzylic hydrogen atom are transferred to the PLY backbone followed by an electron transfer process, which results in the dearomatization of the PLY backbone and the hydrogen molecule is stored in the form of two C–H bonds as shown in 8 (consists of 8a and 8b). It may be noted that in the first step, the resulting alkoxide after proton transfer can bind with the K ion as shown in 7Int for its stabilization. Subsequently, the benzylic hydrogen atom can be transferred to the dearomatized PLY through a HAT process, which generates a radical on the benzylic carbon. The benzylic radical species recombines with the anionic oxygen to release aldehyde and an electron is transferred to the PLY species generating 8. Next, the in situ generated aldehyde, 2′, reacts with aniline to produce the corresponding imine. Finally, the dearomatized intermediate 8 transfers the stored hydrogens sequentially to the imine. Upon transfer of one hydride from 8 to the imine, it generates an anionic K-coordinated complex, 9, where anionic charge is distributed over the aryl ring and is coordinated with K. Further acceptance of another proton from the dearomatized PLY backbone produces the desired alkylated product 3a along with the regeneration of the active catalyst, 7.
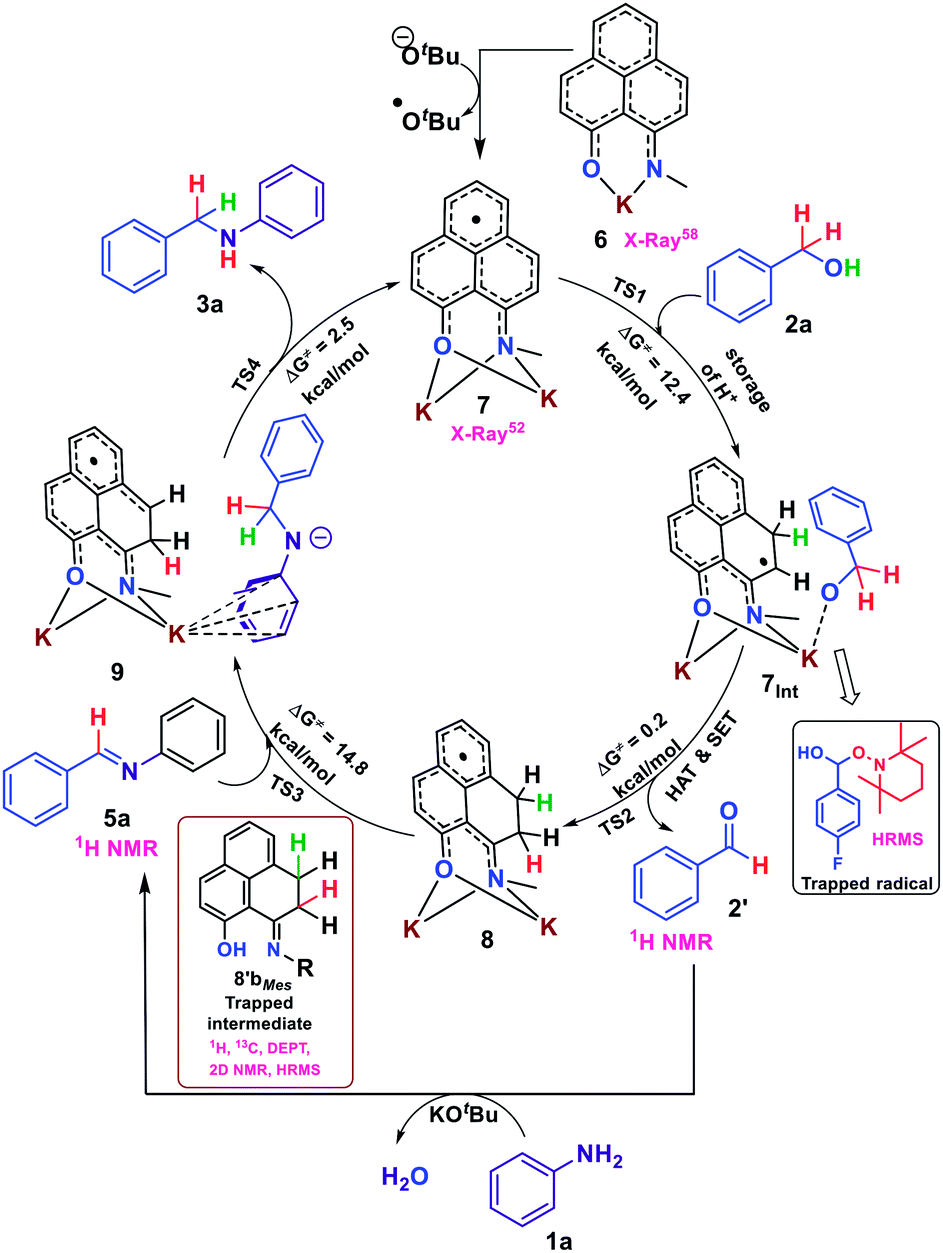 | ||
| Scheme 4 Plausible mechanism for the transition metal free catalytic N-alkylation of amines. | ||
Conclusions
In conclusion, we have demonstrated an efficient method for the storage of a hydrogen molecule harvested from an alcohol in a redox non-innocent organic molecule and transferred it to an in situ generated imine for its reduction in a catalytic fashion. This catalytic protocol for the alkylation of aromatic amines affords good to excellent yield of N-alkylated amines and it is applicable for the diversification of natural products such as the derivatives of vitamin E in gram scale and adapalene derivative. Based on several control experiments, deuterium labelling experiments, time dependent NMR experiments, DFT study and isolation/trapping of several intermediates along the catalytic cycle, we infer that the borrowed hydrogen from the alcohol is stored in the redox active PLY backbone in the form of two C–H bonds upon its dearomatization. Such a hydrogen molecule stored as C–H bonds was further transferred to the in situ generated imine resulting in its reduction and regenerates the active catalyst. Thus, this mechanism conceptually resembles the transition metal catalyzed borrowing hydrogen mediated reactions. This study opens up the possibility of exploring various other transition metal free catalytic Borrowing Hydrogen/Hydrogen Autotransfer (BH/HA) mediated reactions.Experimental section
General considerations
Different phenalenyl (PLY) ligands were prepared following reported literature procedures.59–62 All solvents were distilled from Na/benzophenone or calcium hydride prior to use. All chemicals were purchased and used as received unless otherwise mentioned. Liquid anilines were distilled under vacuum before using in catalytic reactions. The 1H and 13C{1H} NMR spectra were recorded on 400 and 500 MHz spectrometers in CDCl3 with residual undeuterated solvent (CDCl3, 7.26/77.0) as an internal standard using tetramethylsilane as a reference. Chemical shifts (δ) are given in ppm, and J values are given in Hz. Chemical shifts (δ) downfield from the reference standard were assigned positive values. Column chromatography and thin layer chromatography (TLC) were performed on silica gel (Merck silica gel 100–200 mesh). Potassium tert-butoxide was purchased from Sigma Aldrich. High-resolution mass spectrometry (HRMS) was performed on a Bruker maXis impact.General procedure for the N-alkylation of amines
An oven dried 15 mL tube was charged with amines, 1a–v (0.3 mmol), alcohols, 2a-ad (0.33 mmol), (N,O)-PLY (6.3 mg, 0.03 mmol, 10 mol%), and KOtBu (40.4 mg, 0.36 mmol, 1.2 equivalents) along with 1.5 mL toluene inside a nitrogen-filled glovebox and sealed prior to bring out from the glovebox. Next, the reaction mixture was allowed to stir at 130 °C for 18 h. After completion of the reaction, the crude reaction mixture was passed through Celite using 25 mL ethyl acetate (EtOAc) and dried over anhydrous sodium sulphate. The solvent was removed under reduced pressure and the crude product was purified by column chromatography using a neutral alumina and hexane/EtOAc mixture as an eluent to yield the pure desired product which was further characterized by NMR spectroscopy.Radical quenching with radical inhibitors
An oven dried 15 mL tube was charged with aniline, 1a (27.4 μL, 0.3 mmol), benzyl alcohol, 2a (34.3 μL, 0.33 mmol), (N,O)-PLY (6.3 mg, 0.03 mmol, 10 mol%), KOtBu (40.4 mg, 0.36 mmol, 1.2 equivalents), and TEMPO (93.6 mg, 0.6 mmol) or galvinoxyl, free radical (126.5 mg, 0.3 mmol) along with 1.5 mL toluene inside a nitrogen-filled glovebox and sealed prior to bring out from the glovebox. Subsequently, the reaction mixture was allowed to stir at 130 °C for 18 h. Next, the reaction mixture was cooled down and the crude reaction mixture was passed through Celite using 25 mL ethyl acetate (EtOAc) and dried over anhydrous sodium sulphate. The solvent was removed under reduced pressure and the NMR yields were determined by 1H NMR spectroscopy using 1,4-dimethoxybenzene as the internal standard.Leaching experiment with HCl
An oven dried 15 mL tube was charged with (MesN,O)-PLY (31.4 mg, 0.1 mmol) and KOtBu (22.4 mg, 0.2 mmol, and 20 mol%) along with 1.5 mL toluene inside a nitrogen-filled glovebox and sealed prior to bring out from the glovebox. Subsequently, the reaction mixture was allowed to stir at 130 °C for 6 h and a sharp color change from orange to wine red was observed. After 6 hours, the reaction mixture was cooled down and 4-chloro benzyl alcohol, 2c (14.2 mg, 0.1 mmol), was added inside a nitrogen filled glovebox. Again, the reaction mixture was stirred at 130 °C for another 8 h. After completion of the reaction, 1.0 mL of 12 (M) aqueous HCl was added dropwise to the reaction mixture and stirred at room temperature for 1 h and the crude organic product was extracted in Et2O. The solvent was removed under reduced pressure and the dearomatized PLY (8′aMes and 8′bMes) was isolated through column chromatography using a hexane:ethyl acetate mixture as the eluent (10:1). Upon successful isolation, the oily product was characterized through NMR spectroscopy and mass spectrometry (HRMS).
Computational details
Theoretical calculations were performed with the Gaussian16 program suite.65 All theoretical calculations were carried out using the density functional theory (DFT) method with the m062x method66 employing the 6-31+g(d) basis set67 for all atoms for the geometry optimization for all molecules including transition states and intermediates. Single point energy calculations were used for carrying out the m062x method66 with the 6-31+g(d) basis set considering SMD solvent model in toluene.68Author contributions
SKM originated the idea of the work. AB and SKM planned the experiments. AB carried out the catalytic reactions and control experiments. JA conducted all the theoretical calculations. SS carried out the optimization reactions. SKM supervised the work. AB and SKM analyzed the data and prepared the manuscript.Conflicts of interest
There is no conflict to declare.Acknowledgements
We thank SERB (DST), India (Grant No. EMR/2017/000772) for financial support. A. B. and J. A. thank IISER Kolkata and S. S. thanks CSIR India for research fellowships.Notes and references
- S. A. Lawerence, Amines: Synthesis, Properties and Applications, Cambridge University, Cambridge, 2004 Search PubMed.
- H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Industrial Organic Chemicals, Wiley-Interscience, New York, 2nd edn, 2004 Search PubMed.
- J. L. Mcguire, Pharmaceuticals: Classes, Therapeutic Agents, Areas of Application, Wiley-VCH, 2000, vol. 1–4 Search PubMed.
- C. Hansch, P. G. Sammes and J. B. Taylor, Comprehensive Medicinal Chemistry, Pergamon Press, Oxford, UK, 1990, ch. 7.1, vol. 2 Search PubMed.
- A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS PubMed.
- Y.-Q. Zou, S. Chakraborty, A. Nerush, D. Oren, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 8014–8019 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- V. I. Tararov and A. Borner, Synlett, 2005, 2, 203–211 CrossRef.
- J. F. Hartwig, Synlett, 2005, 9, 1283–1294 Search PubMed.
- S. L. Buchwald, C. Mauger, G. Mignani and U. Scholz, Adv. Synth. Catal., 2006, 348, 23–39 CrossRef CAS.
- C. F. Winans and H. Adkins, J. Am. Chem. Soc., 1932, 54, 306–312 CrossRef CAS.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612 RSC.
- Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS.
- G. Chelucci, Coord. Chem. Rev., 2017, 331, 1–36 CrossRef CAS.
- P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2006, 47, 6787–6789 CrossRef CAS.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc. Chem. Commun., 1987, 248–249 RSC.
- O. Saidi, A. J. Blacker, G. W. Lamb, S. P. Marsden, J. E. Taylor and J. M. J. Williams, Org. Process Res. Dev., 2010, 14, 1046–1049 CrossRef CAS.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed.
- T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381–388 CrossRef CAS.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. B. David, N. A. E. Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed.
- S. Rosler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- R. Fertig, T. Irrgang, F. Freitag, J. Zander and R. Kempe, ACS Catal., 2018, 8, 8525–8530 CrossRef CAS.
- F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 11789–11793 CrossRef CAS PubMed.
- M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Chem. Commun., 2019, 55, 6213–6216 RSC.
- K. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2013, 3, 112–117 CrossRef CAS.
- M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- A. K. Bains, A. Kundu, S. Yadav and D. Adhikari, ACS Catal., 2019, 9, 9051–9059 CrossRef CAS.
- Y. Wu, Y. Huang, X. Dai and F. Shi, ChemSusChem, 2019, 12, 3185–3191 CrossRef CAS PubMed.
- S. P. Midya, J. Pitchaimani, V. G. Landge, V. Madhu and E. Balaraman, Catal. Sci. Technol., 2018, 8, 3469–3473 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- S. L. Montgomery, J. Mangas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 10491–10494 CrossRef CAS PubMed.
- R. Pothikumar, V. T. Bhat and K. Namitharan, Chem. Commun., 2020, 56, 13607–13610 RSC.
- Q.-Q. Li, Z.-F. Xiao, C.-Z. Yao, H.-X. Zheng and Y.-B. Kang, Org. Lett., 2015, 17, 5328–5331 CrossRef CAS PubMed.
- Q. Xu, Q. Li, X. Zhu and J. Chen, Adv. Synth. Catal., 2013, 355, 73–80 CrossRef CAS.
- C. Wang, C. Chen, J. Han, J. Zhang, Y. Yao and Y. Zhao, Eur. J. Org. Chem., 2015, 2972–2977 CrossRef CAS.
- X.-H. Lu, Y.-W. Sun, X.-L. Wei, C. Peng, D. Zhou and Q.-H. Xia, Catal. Commun., 2014, 55, 78–82 CrossRef CAS.
- A. Porcheddu and G. Chelucci, Chem. Rec., 2019, 19, 2398–2435 CrossRef CAS PubMed.
- D. H. Reid, Q. Rev., Chem. Soc., 1965, 19, 274–302 RSC.
- R. C. Haddon, Aust. J. Chem., 1975, 28, 2343–2351 CrossRef CAS.
- A. Ueda, S. Suzuki, K. Yoshida, K. Fukui, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2013, 52, 4795–4799 CrossRef CAS PubMed.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- R. G. Hicks, Nat. Chem., 2011, 3, 189–191 CrossRef CAS PubMed.
- M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443–1445 CrossRef CAS PubMed.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281–284 CrossRef CAS PubMed.
- K. Goto, T. Kubo, K. Yamamoto, K. Nakasuji, K. Sato, D. Shiomi, T. Takui, M. Kubota, T. Kobayashi, K. Yakusi and J. A. Ouyang, J. Am. Chem. Soc., 1999, 121, 1619–1620 CrossRef CAS.
- Y. Ikabata, Q. Wang, T. Yoshikawa, A. Ueda, T. Murata, K. Kariyazono, M. Moriguchi, H. Okamoto, Y. Morita and H. Nakai, npj Quantum Mater., 2017, 2, 27 CrossRef.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS PubMed.
- K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, P. Lazic, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blugel, M. Munzenberg and J. S. Moodera, Nature, 2013, 493, 509–513 CrossRef CAS PubMed.
- K. Uchida, Z. Mou, M. Kertesz and T. Kubo, J. Am. Chem. Soc., 2016, 138, 4665–4672 CrossRef CAS PubMed.
- A. Mukherjee, S. C. Sau and S. K. Mandal, Acc. Chem. Res., 2017, 50, 1679–1691 CrossRef CAS PubMed.
- M. Bhunia, S. R. Sahoo, B. K. Shaw, S. Vaidya, A. Pariyar, G. Vijaykumar, D. Adhikari and S. K. Mandal, Chem. Sci., 2019, 10, 7433–7441 RSC.
- J. Ahmed, P. Datta, A. Das, S. Jomy and S. K. Mandal, Chem. Sci., 2021, 12, 3039–3049 RSC.
- R. C. Haddon, R. Rayford and A. M. Hirani, J. Org. Chem., 1981, 46, 4587–4588 CrossRef CAS.
- S. K. Mandal, M. E. Itkis, X. Chi, S. Samanta, D. Lidsky, R. W. Reed, R. T. Oakley, F. S. Tham and R. C. Haddon, J. Am. Chem. Soc., 2005, 127, 8185–8196 CrossRef CAS PubMed.
- R. Paira, B. Singh, P. K. Hota, J. Ahmed, S. C. Sau, J. P. Johnpeter and S. K. Mandal, J. Org. Chem., 2016, 81, 2432–2441 CrossRef CAS PubMed.
- A. Banik, R. Paira, B. K. Shaw, G. Vijaykumar and S. K. Mandal, J. Org. Chem., 2018, 83, 3236–3244 CrossRef CAS PubMed.
- S.-S. Meng, X. Tang, X. Luo, R. Wu, J.-L. Zhao and A. S. C. Chan, ACS Catal., 2019, 9, 8397–8403 CrossRef CAS.
- M. M. Guru, P. R. Thorve and B. Maji, J. Org. Chem., 2020, 85, 806–819 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Christian Bruneau. |
| ‡ Electronic supplementary information (ESI) available: Detailed experimental procedures, NMR and HRMS spectra, and coordinates of the computed structures. See DOI: 10.1039/d1sc01681d |
| This journal is © The Royal Society of Chemistry 2021 |