
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral Lewis acid-bonded picolinaldehyde enables enantiodivergent carbonyl catalysis in the Mannich/condensation reaction of glycine ester†
Xia
Zhong
 a,
Ziwei
Zhong
a,
Zhikun
Wu
a,
Zhen
Ye
b,
Yuxiang
Feng
b,
Shunxi
Dong
a,
Xiaohua
Liu
*a,
Qian
Peng
*b and
Xiaoming
Feng
*a
a,
Ziwei
Zhong
a,
Zhikun
Wu
a,
Zhen
Ye
b,
Yuxiang
Feng
b,
Shunxi
Dong
a,
Xiaohua
Liu
*a,
Qian
Peng
*b and
Xiaoming
Feng
*a
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn; Web: http://www.scu.edu.cn/chem_asl/
bState Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, 94 Weijin Road, Tianjin 300071, P. R. China. E-mail: qpeng@nankai.edu.cn
First published on 26th January 2021
Abstract
A new strategy of asymmetric carbonyl catalysis via a chiral Lewis acid-bonded aldehyde has been developed for the direct Mannich/condensation cascade reaction of glycine ester with aromatic aldimines. The co-catalytic system of 2-picolinaldehyde and chiral YbIII-N,N′-dioxides was identified to be efficient under mild conditions, providing a series of trisubstituted imidazolidines in moderate to good yields with high diastereo- and enantioselectivities. Enantiodivergent synthesis was achieved via changing the sub-structures of the chiral ligands. The reaction could be carried out in a three-component version involving glycine ester, aldehydes, and anilines with equally good results. Based on control experiments, the X-ray crystal structure study and theoretical calculations, a possible dual-activation mechanism and stereo-control modes were provided to elucidate carbonyl catalysis and enantiodivergence.
Introduction
Asymmetric carbonyl catalysis,1 inspired by enzyme catalysis,2 represents a type of useful organo-covalent-activation method via formation of various species, such as hemiacetal,3 or imine,4 as well as dioxirane from chiral ketone for epoxidation.5 Substantial efforts in developing aldehyde-based chiral organocatalysts have been made after pioneering reports by Kuzuhara,6 Breslow and co-workers.7 Excellent contributions have been documented in recent years by Beauchemin,8 Guo,9 Zhao and Yuan10et al. (Scheme 1a), and the reactions involve Cope-type hydroamination,8 alkylation,9d transamination,10a and addition reactions of glycine esters.9a,b,10b For instance, chiral N-quaternized pyridoxal analogue A5 containing a chiral axis and a carbon stereogenic center allowed a biomimetic asymmetric Mannich reaction of tert-butyl glycine ester and N-diphenylphosphinyl imines (Scheme 1b).10b Cooperative imine/hydrogen-bond bifunctional activation guaranteed high activity and stereoselectivity. On top of that, the dual-catalysis by Lewis acid-bonded picolinaldehyde has been explored by Aron's group in the synthesis of chiral amino acids.11 They found that the metal ion chelated quinonoid intermediate facilitated racemization and Tsuji–Trost allylation, and enantiomerically enriched amino acids could be obtained in concert with Alacase11a or a chiral palladium catalyst.11b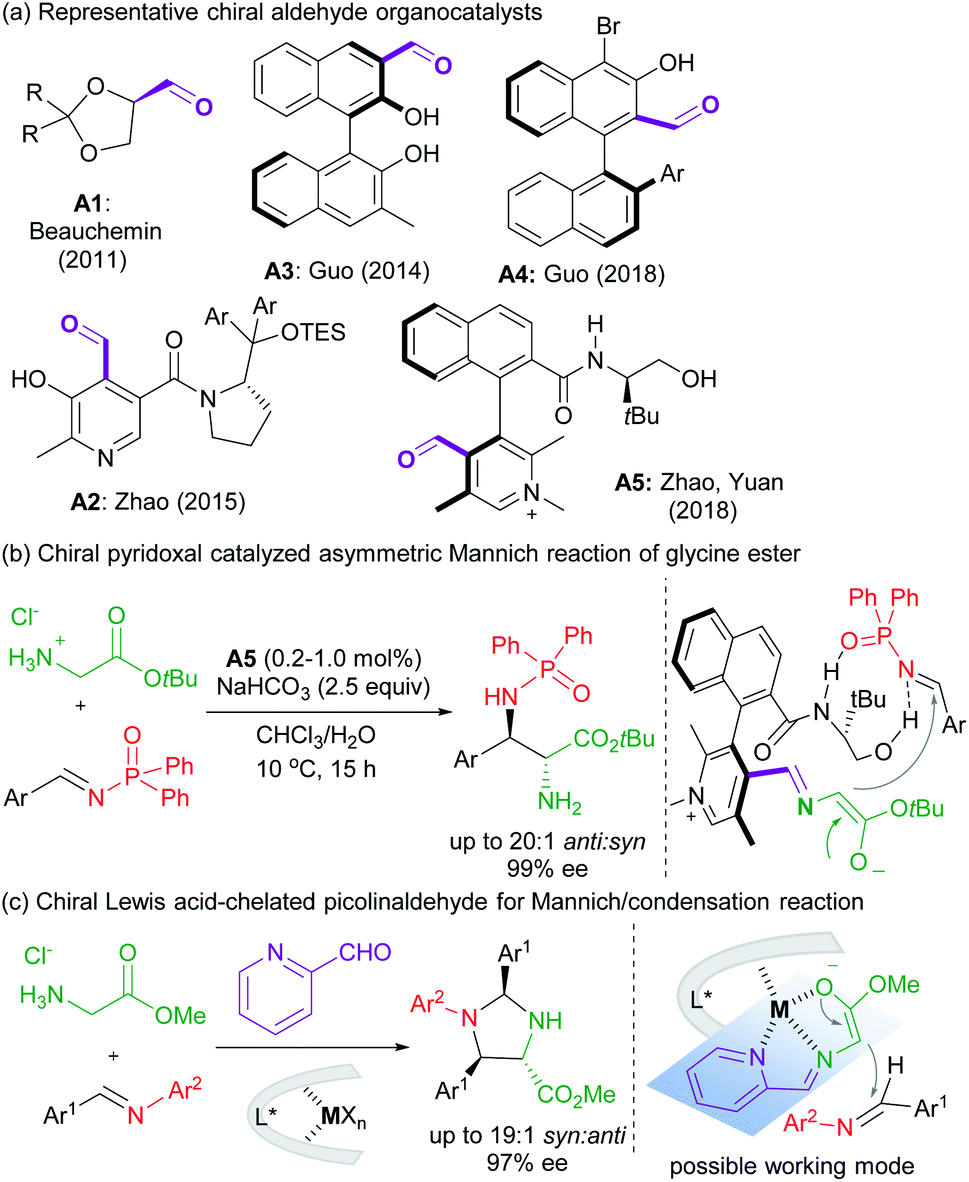 | ||
| Scheme 1 Carbonyl catalysts and selected examples of α-functionalization of glycine esters. | ||
These studies, together with the ingenious imine-based transient directing group strategy in transition metal-catalyzed C–H activation,12 intrigue us to develop chiral Lewis acid-bonded aldehyde as a new route for asymmetric carbonyl catalysis in α-functionalization of glycine esters,13 which might compensate for the design and synthesis of chiral aldehyde organocatalysts de novo. Our research group focused on chiral Lewis acid catalysts of N,N′-dioxides which are capable of a number of asymmetric transformations due to their easy preparation and structural modification.14,15 It is anticipated that the stereo-environment created by the chiral Lewis acid could deliver to the bonded-aldehyde for asymmetric carbonyl catalysis, for instance, the asymmetric nucleophilic addition of the azomethine ylides, generated from picolinaldehyde and glycine ester (Scheme 1c). The study by Wang's group reported the utilization of metallated azomethine ylides in [3 + 2] cycloadditions with a variety of electron deficient alkenes.16 The improved reactivity and efficient enantiocontrol from metal-bonded amino esters shed light on our new chiral imine-bonded activation method for the Mannich-type reaction of glycine ester. Herein, we wish to disclose a new co-catalytic system of 2-picolinaldehyde and a chiral YbIII complex of N,N′-dioxides,17 which was optimized to be efficient to catalyze the diastereo- and enantioselective Mannich/condensation cascade reaction of glycine ester with aromatic aldimines under mild conditions (Scheme 1c). Various enantioenriched imidazolidines were provided in good yields with high stereoselectivities, even in the three-component version with glycine ester, aldehydes and alinines. Interestingly, changing the sub-structure of Feng N,N′-dioxides enabled enantiodivergent synthesis.18 Two different working modes were provided to give a rationale for carbonyl catalysis and enantiodivergence on the basis of the X-ray crystal structures of chiral Yb(III) complexes and DFT calculations.
Results and discussion
Initially, methyl glycine ester hydrochloride (1a) and N-phenyl phenylmethanimine (2a)19 were chosen as the model substrates to optimize the reaction conditions. In the preliminary screening, the chiral complex of Yb(OTf)3 with Feng N,N′-dioxide L-Ra(OiBu)2 was employed as the Lewis acid catalyst. NaOtBu (1.0 equivalent) was used as the base to generate methyl glycine ester from 1ain situ, and 2-picolinaldehyde C1 (10 mol%) was loaded as the carbonyl co-catalyst. The reaction took place smoothly in EtOAc at 35 °C, affording imidazolidine 3a as the final product in 40% yield, 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 dr, and 95% ee after 24 hours (Table 1, entry 1), which is generated from the Mannich reaction of aldimine 2a with glycine ester, following condensation with benzaldehyde (see Fig. 1 for details). The comparison experiments in the absence of 2-picolinaldehyde or in the presence of other aldehydes confirmed that both pyridine and aldehyde groups were indispensable. No reaction occurred without the addition of 2-picolinaldehyde (entry 2) or in the presence of benzaldehyde C2 (entry 3), or isonicotinaldehyde C3 (entry 4). Although the addition of 2-formylpyridine N-oxide C4 benefitted the generation of imidazolidine 3a, the enantioselectivity dropped dramatically (entry 5, 9% ee vs. 95% ee). We rationalized that the stronger chelated 2-picolinaldehyde and the resultant azomethine ylide were involved in initiating the Mannich reaction. We also identified other critical parameters for this asymmetric transformation (see ESI Tables S1–S8† for details). Subsequent investigation of Feng N,N′-dioxides with different chiral backbones indicated that the (S)-2-pipecolic acid-derived L-Pi(OiBu)2 exhibited comparable activity with slightly lower stereoselectivity, while L-perindopril-derived L-Pe(OiBu)2 showed lower activity with similar stereoselectivity (entries 6 and 7). Notably, the introduction of (S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid-derived N,N′-dioxide ligand L-TQtBu, led to the enantiodivergent synthesis of the corresponding product ent-3a in 34% yield, and 86:14 dr with 87% ee (entry 8). With the increase of the amount of co-catalyst C1 to 20 mol%, the product was isolated in a slightly higher yield with comparable stereoselectivity (entry 9, 44% yield, 93:7 dr, 95% ee). Besides, the addition of EtOH as a protonic additive (entry 10), and increasing the amount of aldimine 2a (entry 11) resulted in the formation of 3a with 62% yield, 95:5 dr and 97% ee. Under such conditions, the product ent-3a could be afforded in 50% yield, 88:12 dr and 95% ee by the use of ligand L-TQtBu instead (entry 12). Due to the occurence of several side reactions, the attempt to improve the yield of the product was unsuccessful (see the ESI† and control experiments below for details).
:7 dr, and 95% ee after 24 hours (Table 1, entry 1), which is generated from the Mannich reaction of aldimine 2a with glycine ester, following condensation with benzaldehyde (see Fig. 1 for details). The comparison experiments in the absence of 2-picolinaldehyde or in the presence of other aldehydes confirmed that both pyridine and aldehyde groups were indispensable. No reaction occurred without the addition of 2-picolinaldehyde (entry 2) or in the presence of benzaldehyde C2 (entry 3), or isonicotinaldehyde C3 (entry 4). Although the addition of 2-formylpyridine N-oxide C4 benefitted the generation of imidazolidine 3a, the enantioselectivity dropped dramatically (entry 5, 9% ee vs. 95% ee). We rationalized that the stronger chelated 2-picolinaldehyde and the resultant azomethine ylide were involved in initiating the Mannich reaction. We also identified other critical parameters for this asymmetric transformation (see ESI Tables S1–S8† for details). Subsequent investigation of Feng N,N′-dioxides with different chiral backbones indicated that the (S)-2-pipecolic acid-derived L-Pi(OiBu)2 exhibited comparable activity with slightly lower stereoselectivity, while L-perindopril-derived L-Pe(OiBu)2 showed lower activity with similar stereoselectivity (entries 6 and 7). Notably, the introduction of (S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid-derived N,N′-dioxide ligand L-TQtBu, led to the enantiodivergent synthesis of the corresponding product ent-3a in 34% yield, and 86:14 dr with 87% ee (entry 8). With the increase of the amount of co-catalyst C1 to 20 mol%, the product was isolated in a slightly higher yield with comparable stereoselectivity (entry 9, 44% yield, 93:7 dr, 95% ee). Besides, the addition of EtOH as a protonic additive (entry 10), and increasing the amount of aldimine 2a (entry 11) resulted in the formation of 3a with 62% yield, 95:5 dr and 97% ee. Under such conditions, the product ent-3a could be afforded in 50% yield, 88:12 dr and 95% ee by the use of ligand L-TQtBu instead (entry 12). Due to the occurence of several side reactions, the attempt to improve the yield of the product was unsuccessful (see the ESI† and control experiments below for details).
|
|
||||
|---|---|---|---|---|
| Entry | Variation | Yieldb (%) | drc | eec (%) |
|
a Unless otherwise noted; all reactions were carried out with 1a (0.10 mmol), 2a (2.0 equiv.), NaOtBu (1.0 equiv.), carbonyl catalyst (10 mol%) and ligand/Yb(OTf)3 (1:1, 10 mol%) in EtOAc (0.17 M) at 35 °C for 24 hours. N.R. = no reaction.
b Isolated yield of 3a based on 1a.
c Determined by HPLC on a chiral stationary phase.
d EtOH (8.0 equiv.).
e
2a (3.0 equiv.) for 48 hours.
|
||||
| 1 | None | 40 | 93:7 |
95(+) |
| 2 | Without C1 | N.R. | — | — |
| 3 | C2 | N.R. | — | — |
| 4 | C3 | N.R. | — | — |
| 5 | C4 | 45 | 81:19 |
9(+) |
| 6 | L-Pi(OiBu)2 | 43 | 90:10 |
91(+) |
| 7 | L-Pe(OiBu)2 | 34 | 92:8 |
97(+) |
| 8 | L-TQtBu | 34 | 86:14 |
87(−) |
| 9 | C1 (20 mol%) | 44 | 93:7 |
95(+) |
| 10d | C1 (20 mol%), EtOH | 46 | 94:6 |
97(+) |
| 11d,e | C1 (20 mol%), EtOH | 62 | 95:5 |
97(+) |
| 12d,e | L-TQtBu, C1 (20 mol%) | 50 | 88:12 |
95(−) |

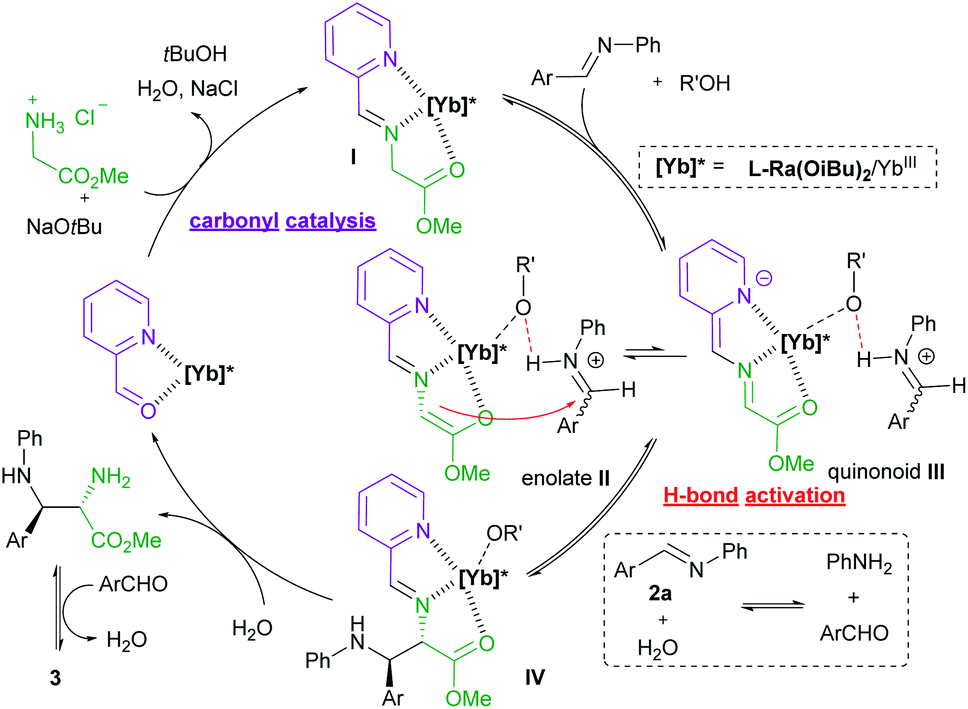 | ||
| Fig. 1 Proposed catalytic cycle. | ||
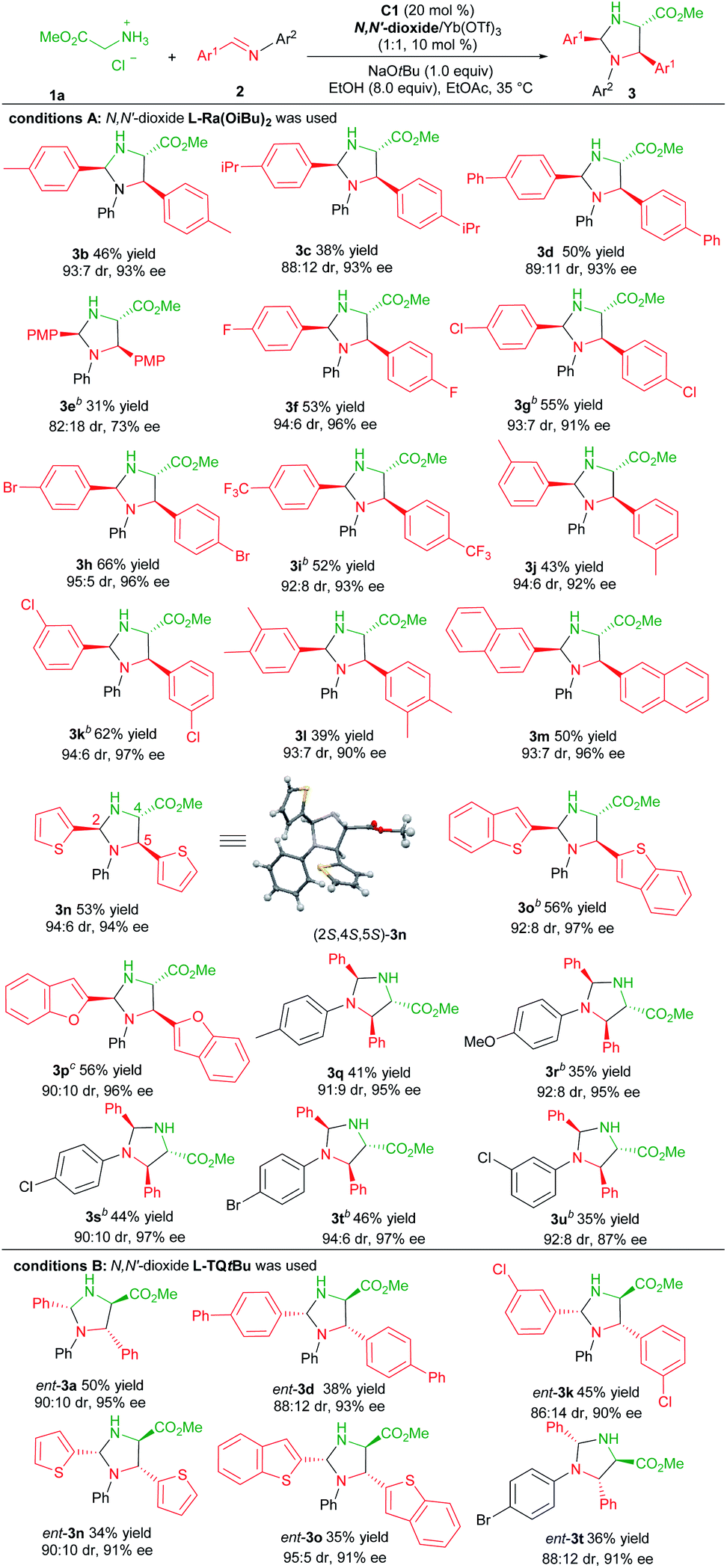
With the optimized reaction conditions established (Table 1, entry 11), the generality of the aldimines was explored (Table 2, conditions A). Firstly, a series of aromatic aldehyde-derived imines were investigated (3b–3p). Regardless of the electronic nature or steric hindrance of the substituents on the phenyl ring (Ar1), the imidazolidine products 3b–3k were obtained in 31–66% yields with excellent stereoselectivities (82:18–95:5 dr, 73–97% ee). Aldimine derived from 3,4-dimethylbenzaldehyde was tolerated (3l, 39% yield, 93:7 dr and 90% ee). The reaction of aldimines derived from 2-naphthaldehyde or heteroaryl aldehydes proceeded well, affording 3m–3p with comparative results (50–56% yields, 90:10–94:6 dr, 94–97% ee). The following screening of the N-aryl substituent (Ar2) of aldimines showed that para-substituted aniline based aldimine had a limited effect on the reaction (3q–3t, 35–46% yields, 90:10–94:6 dr, 95–97% ee). The 4-methoxyaniline derived one resulted in a high enantioselectivity with a decreased yield (3r, 35% yield, 92:8 dr, 95% ee). It should be noted that for several products, the use of the ligand L-Pi(OiBu)2 or L-Pe(OiBu)2 instead was necessary to get higher yields without the change of stereo-preference. The absolute configuration of the product 3n was determined to be (2S,4S,5S)-isomer by X-ray single crystal analysis.20 The stereochemistry of other products was assigned by comparing their CD spectra with those of the product 3n.
|
a Reactions were carried out with 1a (0.10 mmol), 2 (3.0 equiv.), NaOtBu (1.0 equiv.), C1 (20 mol%), EtOH (8.0 equiv.) and L/Yb(OTf)3 (1:1, 10 mol%) in EtOAc (0.17 M) at 35 °C for 48 hours. The isolated yield of 3 based on 1a. The dr value was detected by 1H NMR analysis, and the ee value was determined by HPLC on a chiral stationary phase.
b
L-Pi(OiBu)2 was used as the ligand.
c
L-Pe(OiBu)2 was used as the ligand.
|
|---|
|
Next, we proceeded to prepare the antipodes of the product 3 by the employment of the chiral L-TQtBu/Yb(OTf)3 complex as the Lewis acid catalyst. Representative aldimines were examined and the results are depicted in Table 2, condition B. Generally, these reactions occurred smoothly to deliver the corresponding products ent-3 with high enantio- and diastereoselectivity in slightly diminished yields (34–50% yields, 86:14–95:5 dr, 90–95% ee).
In order to improve the practicability of the reaction, we investigated the three-component synthesis of imidazolidines, from glycine ester, aldehyde and aniline. As shown in Table 3, the three-component reaction was accomplished under slightly modified conditions (see ESI Table S9† for details), and an array of aldehydes 4 and anilines 5 were surveyed. In comparison with the two-component reaction system, similar results were given (39–55% yields, 88:12–94:6 dr, 88–97% ee). By using this protocol, the product 3v was yielded in moderate yield with good diastereo- and enantioselectivity (Table 3, entry 6), which is not possible from the two-component reaction of N-m-tolyl substituted aldimine due to the separation and purification problem of aldimine. Unfortunately, aliphatic amines and aldehydes were not suitable in the current system.
|
|
||||
|---|---|---|---|---|
| Entry | Ar1/Ar2 | Yield (%) | dr | ee (%) |
|
a Reactions were carried out with 1a (0.10 mmol), 4 (3.0 equiv.), 5 (3.0 equiv.), NaOtBu (0.10 mmol), C1 (20 mol%), 4 Å MS (20.0 mg), and Yb(OTf)3/L-Ra(OiBu)2 (1:1, 10 mol%) in EtOAc (0.17 M) at 35 °C for 48 hours. The isolated yield of 3 based on 1a. The dr value was detected by 1H NMR analysis and the ee value was determined by HPLC on a chiral stationary phase.
b With L-Pi(OiBu)2.
c With L-Pe(OiBu)2.
|
||||
| 1 | Ph/Ph (3a) | 55 | 93:7 |
96 |
| 2 | 4-MeC6H4/Ph (3b) | 40 | 88:12 |
88 |
| 3b | 4-ClC6H4/Ph (3g) | 53 | 94:6 |
91 |
| 4 | Ph/4-MeC6H4 (3q) | 39 | 94:6 |
93 |
| 5b | Ph/4-ClC6H4 (3s) | 44 | 94:6 |
97 |
| 6c | Ph/3-MeC6H4 (3v) | 41 | 92:8 |
87 |
To evaluate the synthetic potential of the catalytic system, a scale-up preparation of imidazolidine 3h was carried out (Scheme 2a). Upon treatment of 3.0 mmol of 1a with 9.0 mmol of 2h under the optimized reaction condition A, the desired product 3h was obtained in 62% yield (0.97 g), with 92:8 dr and 93% ee after 60 hours. Furthermore (Scheme 2b), the product 3a was successfully converted into the chiral vicinal diamine product 6avia TsOH-mediated hydrolysis in 51% yield, 93:7 dr and 98% ee after two steps. The change of the ester group had an obvious effect on the transformation, and ethyl glycine ester 1b led to a reduced yield (6b, 30% yield), and benzyl glycine ester 1c provided a lower diastereoselectivity (6c, 78:22 dr, 91%/93% ee). Similarly, ent-6b diamine was obtained in comparable yield when L-TQtBu was used (29%, 82:18 dr, 91% ee). Noteworthily, the syn-diamine derivatives 6 were dominant in the current case (Scheme 2b), which is different from the chiral pyridoxal analogue A5 based catalytic system.10b Reduction of the ester to a primary alcohol led to the imidazolidine derivative 7a in good yield with maintained stereoselectivity. In addition, chiral 2-imdazoline 8a was produced by oxidation with DDQ (Scheme 2c).
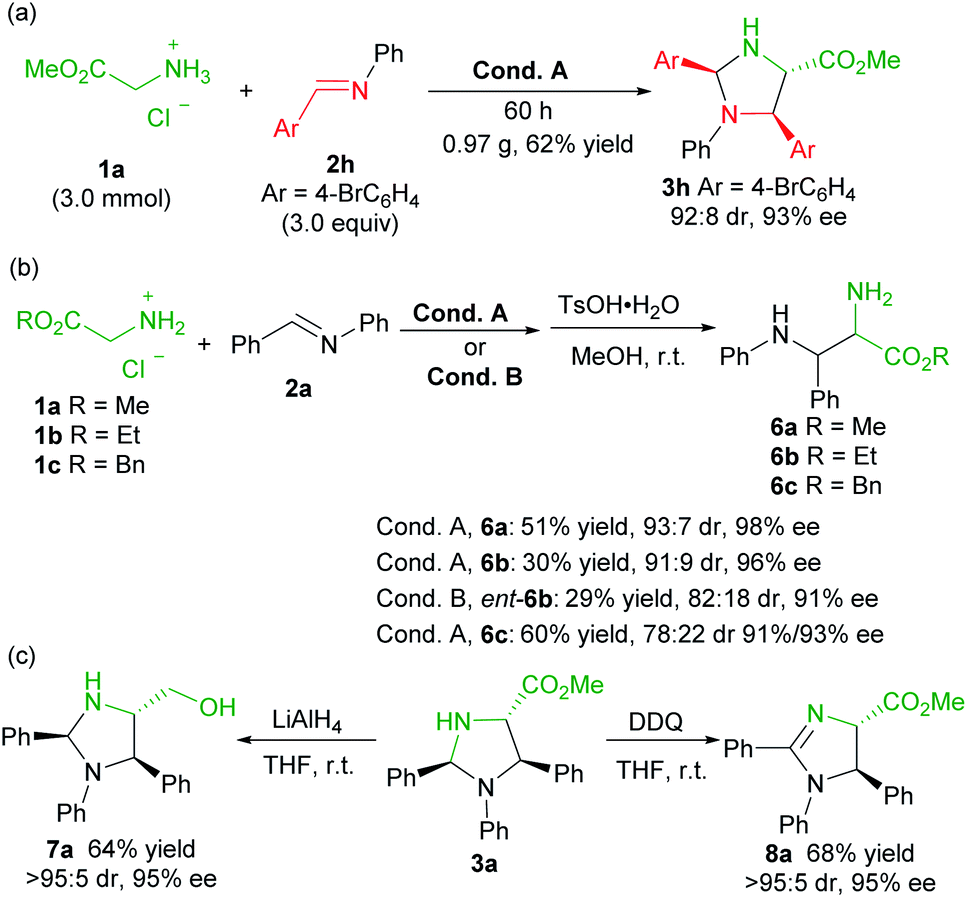 | ||
| Scheme 2 Gram-scale synthesis and transformations. | ||
To get insight into the mechanism of the reaction, a series of control experiments were conducted (Scheme 3). Treatment of chiral diamine 6a with aldimine 2a in the presence of the L-Ra(OiBu)2/Yb(OTf)3 complex delivered the imidazolidine 3a in 51% yield with maintained diastereoselectivity and enantioselectivity (eqn (1)). In contrast, a yield of 90% was obtained if benzaldehyde was used for the formation of imidazolidine 3a (eqn (2)). These results clearly indicated that the final imidazolidine product 3a was generated via a stepwise Mannich/condensation cascade reaction with aldehyde released from the decomposition of aldimine 2a. Therefore, this reaction was different from the previous direct [3 + 2] cycloaddition of azomethine ylides with aldimines.21 When the Schiff base 9 of 2-picolinaldehyde C1 and methyl glycine ester was used instead of 2-picolinaldehyde, the product 3a was isolated in 42% yield with 92:8 dr and 96% ee (eqn (3)). It confirmed that 2-picolinaldehyde probably serves as the carbonyl catalyst to generate the Schiff base intermediate for the subsequent addition reaction. According to HRMS analysis of the reaction mixture, Schiff base 10 and picolinaldehyde-derived imine 11 might exist in the reaction system (eqn (4) and (5)). However, these species were unreactive under the current reaction conditions (see ESI page S24–S27 for detail†). These side reactions along with the instability of products were partly responsible for the moderate yields of the titled process.
 | ||
| Scheme 3 Control experiments. | ||
The influence of the concentration of each component on the reaction rate was detected from operando IR profiles (see ESI page S19–S22 for details†). The kinetic study showed that the initial rate of the reaction was first-order depending on the chiral L-Ra(OiBu)2/Yb(OTf)3 complex, glycine ester, 2-picolinaldehyde C1 and aldimine 2, indicating that these species are involved in the rate-determining process. Furthermore, the HRMS spectra of the mixture of L-Ra(OiBu)2, Yb(OTf)3, C1, 1a, and NaOtBu (1:1:1:1:1) in MeOH exhibited the ion peak at m/z 1470.4521, referring to [L-Ra(OiBu)2 + Yb3+ + 2OTf− + 9]+ species (calculated m/z 1470.4516). It confirmed that chiral Lewis acid bonded 2-picolinaldehyde condenses with methyl glycine ester to generate the metal-chelated Schiff base I (Fig. 1).
Based on the aforementioned results, a plausible catalytic cycle is rationalized for the current reaction (Fig. 1). Initially, chiral ytterbium-bonded picolinaldehyde reacts with glycine ester to yield chiral cation bonded Schiff base species I. In view of the large and variable coordination number of the ytterbium ion, the tridentate coordination of Schiff base 9 and another anion is anticipated to enhance the stability and reactivity of the corresponding enolate species, as well as the quinonoid tautomer. Then, H-bond activated aldimine species undergoes the Mannich reaction via the enolate intermediate II to afford the intermediate IV. Then, hydrolysis of the intermediate IV produces the chiral diamine 6, regenerating the active catalytic species Ligand/YbIII bonded-picolinaldehyde. Eventually, the final product imidazolidine 3 was yielded by the condensation of vicinal diamine and aldehyde.
The X-ray crystal structures of the chiral Lewis acid complexes L-RaEt2/Yb(OTf)3 and L-TQtBu/Yb(OTf)3 (ref. 22) provide interesting spatial information (see ESI Fig. S2 and S3† for full pictures), which might account for the enantiodivergent outcomes. To unveil the different stereochemical control of L-RaEt2 (ref. 23) and L-TQtBu in the formation of chiral intermediate IV, preliminary density functional theory (DFT) calculations were performed at the SMD-B3LYP-D3/MWB59/6-311+G(d,p) level of theory. Due to different thermodynamic stabilities, −OtBu is more favorable to coordinate with YbIII for about −10.2 kcal mol−1 rather than the triflate anion. Moreover, based on the binding orientation of the tridentate Schiff base 9 in different YbIII-chiral N,N′-dioxides complexes, −OtBu always locates at the Si-face or Re-face site of the Schiff base for L-RaEt2/YbIII or L-TQtBu/YbIII, respectively, which would block their opposite sites by substrate/ligand interactions and avoid the steric hindrance of the bulky ligand (bicycle or tert-butyl amide subunits). As shown in L-RaEt2/YbIII of Fig. 2a, the steric effect of the bicycle ring on the ligand would lead to relatively weak coordination 2.60 Å between ligands ON and YbIII in L-RaEt2/Yb-Re, and thus its thermodynamic free energy is 2.2 kcal mol−1 higher than that of L-RaEt2/Yb-Si. On the other hand, the tert-butyl amide in L-TQtBu/YbIII displays major steric hindrance with −OtBu that results in unfavorable coordination in L-TQtBu/Yb-Si.
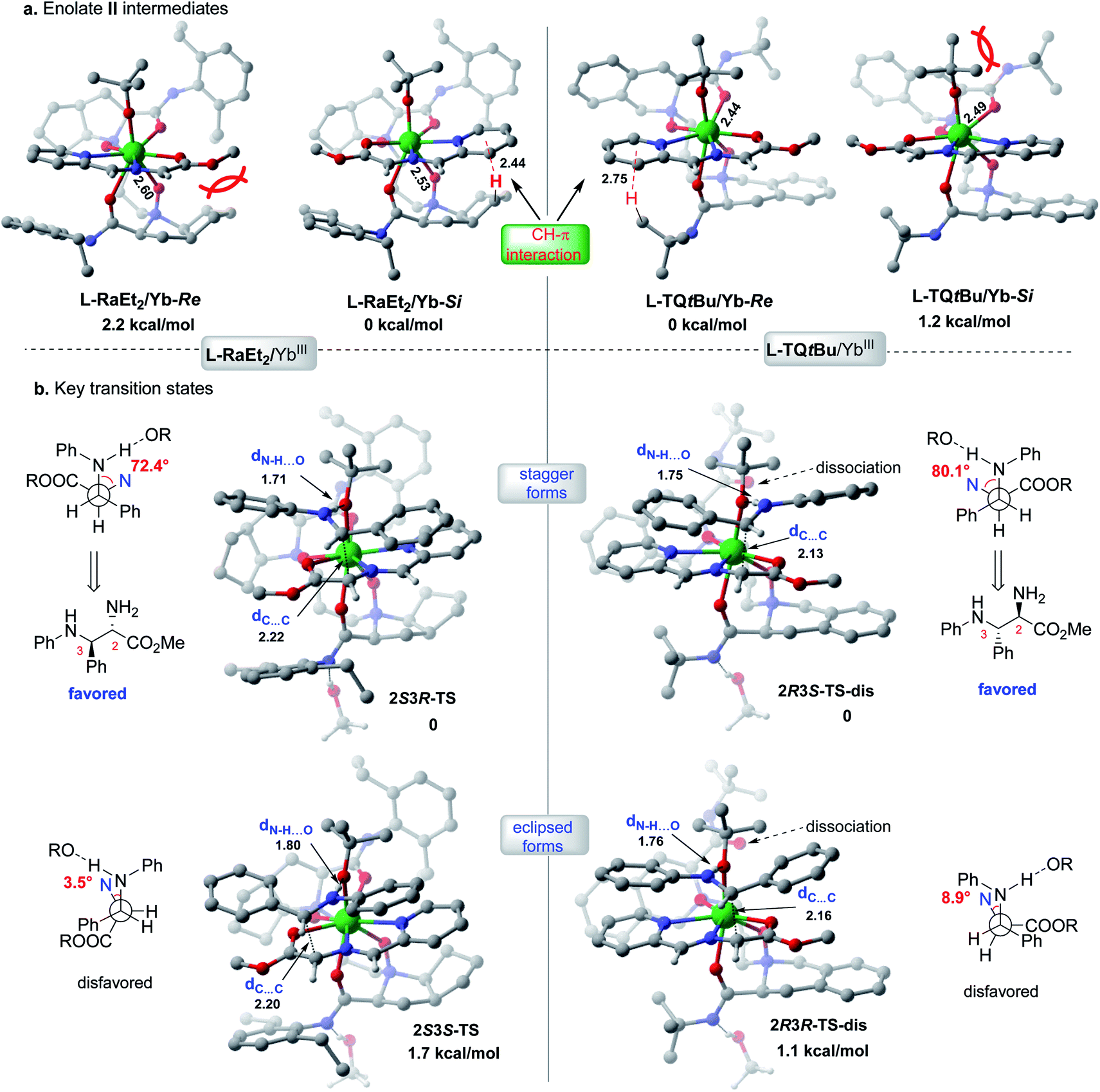 | ||
| Fig. 2 Calculated stereo-models of transition states and intermediates for L-RaEt2/YbIII or L-TQtBu/YbIII complexes. | ||
Further discussion will depend on the most favorable YbIII/chiral N,N′-dioxide complexes with the Schiff base substrate and −OtBu ligand (Fig. 2b). To control the stereoselectivity, an efficient inner-sphere hydrogen bond (around 1.7 Å) between the coordinated −OtBu and protonated aldimine would drive the nucleophilic attacking to form stable transition states in the following Mannich reaction, compared to the outer-sphere hydrogen bond interaction that affords 6.1 kcal mol−1 free energy higher in the transition state 2S3R-TS_1 (see ESI Table S10†). For both the L-RaEt2/YbIII and L-TQtBu/YbIII complexes, the stagger forms of transition states were favored via Si–Si (2S3R-TS) and Re–Re (2R3S-TS-dis) facial selectivity, resulting in (2S,3R)-6a and (2R,3S)-6a respectively, which are in agreement with experimental observations (Fig. 2b). In contrast, the eclipsed forms of transition states via Si–Re and Re–Si facial attacking were calculated to be about 1–2 kcal mol−1 energy higher than those of the most stable ones, indicating the important substrate interactions between the Schiff base and protonated aldimine under each chiral space, which account for the diastereoselectivity. Furthermore, the transient ligand dissociation of the L-TQtBu/YbIII complex at one carbonyl group of amide was inspected in the most stable transition states 2R3S-TS-dis and 2R3R-TS-disvia Re–Re and Re–Si facial selectivities, respectively, and the relative free energies are slightly favorable to the full coordinated transition states (see ESI Fig. S5–S7†), suggesting that the large steric hindrance of transition states in this ligand would reduce the coordination number also supported by the crystal structure of L-TQtBu/Yb(OTf)3. However, the calculated ligand dissociation can be recovered when the protonated aldimine is released from the YbIII center displaying dynamic coordination.
Conclusions
In summary, a new asymmetric carbonyl-catalysis strategy was developed for the Mannich/condensation of glycine ester with aldimine under mild conditions. The chiral N,N′-dioxide/Yb(OTf)3 complex bonded aldehyde enabled carbonyl activation of glycine ester for α-addition transformation. This protocol provided facile and feasible access to a variety of synthetically useful chiral imidazolidine derivatives in moderate yields (up to 66% yield), and excellent diastereo- and enantioselectivities (up to 95:5 dr, 97% ee). The reaction could be performed in either two- or three-component versions. Interestingly, enantiodivergent synthesis was accessible by modulating the sub-structure of the Feng N,N′-dioxide ligand in connection with a multi-coordinated ytterbium metal ion. Theoretical calculations suggested that the steric hindrance and CH–π interaction between the substrate and ligand were responsible for this reversal. And the inner-sphere hydrogen bond and the stagger model of reaction substrates enable stabilization of key transition states leading to high stereoselectivity. Further application of this co-catalytic system in other reactions is under investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (No. 21890723, 21625205, 21890722 and 21950410519), Natural Science Foundation of Tianjin City (19JCJQJC62300, and 18JCYBJC21400), and Tianjin Research Innovation Project for Postgraduate Students (2019YJSB081) for financial support. We thank Dr Yuqiao Zhou at Sichuan University for X-ray single crystal analysis.Notes and references
- For reviews, see: (a) B.-J. Li, C. El-Nachef and A. M. Beauchemin, Chem. Commun., 2017, 53, 13192 RSC; (b) U. Orcel and J. Waser, Chem. Sci., 2017, 8, 32 RSC; (c) Q. Wang, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 6818 CrossRef CAS.
- For reviews of enzymatic reactions, see: (a) P. Clapés and X. Garrabou, Adv. Synth. Catal., 2011, 353, 2263 CrossRef; (b) N. Dückers, K. Baer, S. Simon, H. Gröger and W. Hummel, Appl. Microbiol. Biotechnol., 2010, 88, 409 CrossRef; (c) S. E. Franz and J. D. Stewart, Adv. Appl. Microbiol., 2014, 88, 57 Search PubMed. For selected examples, see: (d) T. Kimura, V. P. Vassilev, G.-J. Shen and C.-H. Wong, J. Am. Chem. Soc., 1997, 119, 11734 CrossRef CAS; (e) K. Hernandez, I. Zelen, G. Petrillo, I. Usón, C. M. Wandtke, J. Bujons, J. Joglar, T. Parella and P. Clapés, Angew. Chem., Int. Ed., 2015, 54, 3013 CrossRef CAS; (f) Q. Chen, X. Chen, J. Feng, Q. Wu, D. Zhu and Y. Ma, ACS Catal., 2019, 9, 4462 CrossRef CAS; (g) J. Cao and T. K. Hyster, ACS Catal., 2020, 10, 6171 CrossRef CAS.
- For a review, see: (a) K. L. Tan, ACS Catal., 2011, 1, 877 CrossRef CAS. For selected examples, see: (b) T. Sammakia and T. B. Hurley, J. Am. Chem. Soc., 1996, 118, 8967 CrossRef CAS; (c) T. Sammakia and T. B. Hurley, J. Org. Chem., 1999, 64, 4652 CrossRef CAS; (d) S.-S. Weng, H.-C. Li and T.-M. Yang, RSC Adv., 2013, 3, 1976 RSC; (e) U. Orcel and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 5250 CrossRef CAS.
- For selected examples, see: (a) K. Toth, T. L. Amyes, J. P. Richard, J. P. G. Malthouse and M. E. NíBeillú, J. Am. Chem. Soc., 2004, 126, 10538 CrossRef CAS; (b) K. Toth, L. M. Gaskell and J. P. Richard, J. Org. Chem., 2006, 71, 7094 CrossRef CAS; (c) D. A. Schichl, S. Enthaler, W. Holla, T. Riermeier, U. Kragl and M. Beller, Eur. J. Org. Chem., 2008, 2008, 3506 CrossRef.
- (a) Y. Shi, Acc. Chem. Res., 2004, 37, 488 CrossRef CAS; (b) D. Yang, Acc. Chem. Res., 2004, 37, 497 CrossRef CAS; (c) O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958 CrossRef CAS.
- (a) H. Kuzuhara, N. Watanabe and M. Ando, J. Chem. Soc., Chem. Commun., 1987, 95 RSC; (b) M. Ando, J. Watanabe and H. Kuzuhara, Bull. Chem. Soc. Jpn., 1990, 63, 88 CrossRef CAS.
- J. T. Koh, L. Delaude and R. Breslow, J. Am. Chem. Soc., 1994, 116, 11234 CrossRef CAS.
- (a) M. J. MacDonald, D. J. Schipper, P. J. Ng, J. Moran and A. M. Beauchemin, J. Am. Chem. Soc., 2011, 133, 20100 CrossRef CAS; (b) N. Guimond, M. J. MacDonald, V. Lemieux and A. M. Beauchemin, J. Am. Chem. Soc., 2012, 134, 16571 CrossRef CAS; (c) M. J. MacDonald, C. R. Hesp, D. J. Schipper, M. Pesant and A. M. Beauchemin, Chem.–Eur. J., 2013, 19, 2597 CrossRef CAS; (d) S. Chitale, J. S. Derasp, B. Hussain, K. Tanveer and A. M. Beauchemin, Chem. Commun., 2016, 52, 13147 RSC.
- (a) B. Xu, L.-L. Shi, Y.-Z. Zhang, Z.-J. Wu, L.-N. Fu, C.-Q. Luo, L.-X. Zhang, Y.-G. Peng and Q.-X. Guo, Chem. Sci., 2014, 5, 1988 RSC; (b) W. Wen, L. Chen, M.-J. Luo, Y. Zhang, Y.-C. Chen, Q. Ouyang and Q.-X. Guo, J. Am. Chem. Soc., 2018, 140, 9774 CrossRef CAS; (c) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159 CrossRef CAS; (d) G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 11464 CrossRef CAS.
- (a) L. Shi, C. Tao, Q. Yang, Y. E. Liu, J. Chen, J. Chen, J. Tian, F. Liu, B. Li, Y. Du and B. Zhao, Org. Lett., 2015, 17, 5784 CrossRef CAS; (b) J. Chen, X. Gong, J. Li, Y. Li, J. Ma, C. Hou, G. Zhao, W. Yuan and B. Zhao, Science, 2018, 360, 1438 CrossRef CAS; (c) S. Li, X.-Y. Chen and D. Enders, Chem, 2018, 4, 2026 CrossRef CAS; (d) J. F. Chen, Y. E. Liu, X. Gong, L. M. Shi and B. G. Zhao, Chin. J. Chem., 2019, 37, 103 CAS.
- (a) A. E. Felten, G. Zhu and Z. D. Aron, Org. Lett., 2010, 12, 1916 CrossRef CAS; (b) P. Fang, M. R. Chaulagain and Z. D. Aron, Org. Lett., 2012, 14, 2130 CrossRef CAS.
- For a review, see: (a) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef CAS; (b) M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898 CrossRef CAS. For selected examples, see: (c) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS; (d) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS; (e) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CrossRef CAS; (f) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073 CrossRef CAS; (g) H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755 CrossRef CAS; (h) H. Song, Y. Li, Q.-J. Yao, L. Jin, L. Liu, Y.-H. Liu and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 6576 CrossRef CAS.
- For selected reviews of α-functionalization of amine, see: (a) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS; (b) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS; (c) M. Waser and J. Novacek, Angew. Chem., Int. Ed., 2015, 54, 14228 CrossRef CAS; (d) H. Noda and M. Shibasaki, Eur. J. Org. Chem., 2020, 2350 CrossRef CAS. For selected examples, see: (e) J. Nakano, K. Masuda, Y. Yamashita and S. Kobayashi, Angew. Chem., Int. Ed., 2012, 51, 9525 CrossRef CAS; (f) Y. Wu and L. Deng, J. Am. Chem. Soc., 2012, 134, 14334 CrossRef CAS; (g) M. Hut'ka, T. Tsubogo and S. Kobayashi, Adv. Synth. Catal., 2013, 355, 1561 CrossRef; (h) Y. Wu, L. Hu and L. Deng, Nature, 2015, 523, 445 CrossRef CAS.
- (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (e) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3 Search PubMed; (f) M.-Y. Wang and W. Li, Chin. J. Chem., 2020, 38 DOI:10.1002/cjoc.202000508. For select examples, see: (g) T. F. Kang, L. Z. Hou, S. Ruan, W. D. Cao, X. H. Liu and X. M. Feng, Nat. Commun., 2020, 11, 3869 CrossRef CAS; (h) X. Y. Zhang, W. B. Wu, W. D. Cao, H. Yu, X. Xu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2020, 59, 4846 CrossRef CAS; (i) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1423 CrossRef; (j) J. Xu, Z. W. Zhong, M. Y. Jiang, Y. Q. Zhou, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1894 CrossRef.
- For reviews and books on co-catalysis, see: (a) B. A. Arndtsen and L.-Z. Gong, Topics in Current Chemistry, Springer, 2020 Search PubMed; (b) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365 CrossRef CAS; (c) S. P. Sancheti, Urvashi, M. P. Shah and N. T. Patil, ACS Catal., 2020, 10, 3462 CrossRef CAS.
- For a review, see: (a) L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084 CrossRef CAS. For selected examples, see: (b) Q.-H. Li, L. Wei, X. Chen and C.-J. Wang, Chem. Commun., 2013, 49, 6277 RSC; (c) H.-M. Wu, Z. Zhang, F. Xiao, L. Wei, X.-Q. Dong and C.-J. Wang, Org. Lett., 2020, 22, 4852 CrossRef CAS; (d) X. Cheng, D. Yan, X.-Q. Dong and C.-J. Wang, Asian J. Org. Chem., 2020, 9, 1567 CrossRef CAS.
- For selected examples, see: (a) Y. Zhang, Y. T. Liao, X. H. Liu, Q. Yao, Y. H. Zhou, L. L. Lin and X. M. Feng, Chem.–Eur. J., 2016, 22, 15119 CrossRef CAS; (b) G. J. Wang, Y. Tang, Y. Zhang, X. H. Liu, L. L. Lin and X. M. Feng, Chem.–Eur. J., 2017, 23, 554 CrossRef CAS; (c) Q. Yao, H. Yu, H. Zhang, S. X. Dong, F. Z. Chang, L. L. Lin, X. H. Liu and X. M. Feng, Chem. Commun., 2018, 54, 3375 RSC; (d) B. Shen, W. Liu, W. D. Cao, X. H. Liu and X. M. Feng, Org. Lett., 2019, 21, 4713 CrossRef CAS; (e) D. Zhang, Z. S. Su, Q. W. He, Z. K. Wu, Y. Q. Zhou, C. J. Pan, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2020, 142, 15975 CrossRef.
- For reviews of enantiodivergent synthesis, see: (a) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef CAS; (b) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC; (c) W. D. Cao, X. M. Feng and X. H. Liu, Org. Biomol. Chem., 2019, 17, 6538 RSC. For selected examples, see: (d) Y. L. Liu, D. J. Shang, X. Zhou, Y. Zhu, L. L. Lin, X. H. Liu and X. M. Feng, Org. Lett., 2010, 12, 180 CrossRef CAS; (e) Z. Wang, Z. G. Yang, D. H. Chen, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928 CrossRef CAS; (f) Y. L. Zhang, N. Yang, X. H. Liu, J. Guo, X. Y. Zhang, L. L. Lin, C. W. Hu and X. M. Feng, Chem. Commun., 2015, 51, 8432 RSC.
- Other protecting groups of imine, such as Ts, Boc and hydroxyphenyl groups were examined as well; however, no better results were obtained. Other amino acid esters and amino ethyl cyanide were examined but with poor chiral control. For more details, see ESI Table S10.†.
- CCDC 1983265 for the X-ray crystal structure of product 3n.†.
- For selected examples of [3 + 2] cycloaddition of azomethine ylides with aldimines, see: (a) W.-J. Liu, X.-H. Chen and L.-Z. Gong, Org. Lett., 2008, 10, 5357 CrossRef CAS; (b) B. Yu, X.-F. Bai, J.-Y. Lv, Y. Yuan, J. Cao, Z.-J. Zheng, Z. Xu, Y.-M. Cui, K.-F. Yang and L.-W. Xu, Adv. Synth. Catal., 2017, 359, 3577 CrossRef CAS; (c) S. Mukhopadhyay and S. C. Pan, Chem. Commun., 2018, 54, 964 RSC.
- CCDC 2011003 for the X-ray crystal structure of L-RaEt2/Yb(OTf)3 and CCDC 2011004 for the X-ray crystal structure of L-TQtBu/Yb(OTf)3.†.
- Switching the ligand L-Ra(OiBu)2 to L-RaEt2, comparable results (42% yield, 88:12 dr, 92%/90% ee vs. 40% yield, 93:7 dr, 95%/91% ee) were afforded after 24 h (see ESI Table S5, line 25†). Currently, we only obtained the X-ray crystal structure of the L-RaEt2/Yb(OTf)3 complex. Therefore, the L-RaEt2/Yb(OTf)3 complex was used for the following computational studies.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR spectra, HPLC spectra, and CD spectra (PDF). X-ray crystallographic data for 3n, L-RaEt2/Yb(OTf)3 and L-TQtBu/Yb(OTf)3 (CIF). CCDC 1983265, 2011003 and 2011004. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc07052a |
| This journal is © The Royal Society of Chemistry 2021 |