
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticolor polymeric carbon dots: synthesis, separation and polyamide-supported molecular fluorescence†
Bo
Zhi‡§
 a,
Xiaoxiao
Yao§
a,
Meng
Wu
b,
Arielle
Mensch
c,
Yi
Cui¶
c,
Jiahua
Deng
d,
Juan J.
Duchimaza-Heredia
d,
Kasidet Jing
Trerayapiwat
d,
Thomas
Niehaus
e,
Yoshio
Nishimoto
f,
Benjamin P.
Frank
g,
Yongqian
Zhang
h,
Riley E.
Lewis
a,
Elaine A.
Kappel
a,
Robert J.
Hamers
h,
Howard D.
Fairbrother
g,
Galya
Orr
c,
Catherine J.
Murphy
b,
Qiang
Cui
di and
Christy L.
Haynes
*a
a,
Xiaoxiao
Yao§
a,
Meng
Wu
b,
Arielle
Mensch
c,
Yi
Cui¶
c,
Jiahua
Deng
d,
Juan J.
Duchimaza-Heredia
d,
Kasidet Jing
Trerayapiwat
d,
Thomas
Niehaus
e,
Yoshio
Nishimoto
f,
Benjamin P.
Frank
g,
Yongqian
Zhang
h,
Riley E.
Lewis
a,
Elaine A.
Kappel
a,
Robert J.
Hamers
h,
Howard D.
Fairbrother
g,
Galya
Orr
c,
Catherine J.
Murphy
b,
Qiang
Cui
di and
Christy L.
Haynes
*a
aDepartment of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant Street SE, Minneapolis, Minnesota 55455, USA. E-mail: chaynes@umn.edu
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, Illinois 61801, USA
cEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, 3335 Innovation Boulevard, Richland, Washington 99352, USA
dDepartment of Chemistry, Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, USA
eUniv Lyon, Université Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622 Lyon, France
fGraduate School of Science, Kyoto University, Kyoto 606-8502, Japan
gDepartment of Chemistry, Johns Hopkins University, Baltimore, MD 21218, USA
hDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA
iDepartments of Physics and Biomedical Engineering, Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, USA
First published on 22nd December 2020
Abstract
Multicolor carbon dots (CDs) have been developed recently and demonstrate great potential in bio-imaging, sensing, and LEDs. However, the fluorescence mechanism of their tunable colors is still under debate, and efficient separation methods are still challenging. Herein, we synthesized multicolor polymeric CDs through solvothermal treatment of citric acid and urea in formamide. Automated reversed-phase column separation was used to achieve fractions with distinct colors, including blue, cyan, green, yellow, orange and red. This work explores the physicochemical properties and fluorescence origins of the red, green, and blue fractions in depth with combined experimental and computational methods. Three dominant fluorescence mechanism hypotheses were evaluated by comparing time-dependent density functional theory and molecular dynamics calculation results to measured characteristics. We find that blue fluorescence likely comes from embedded small molecules trapped in carbonaceous cages, while pyrene analogs are the most likely origin for emission at other wavelengths, especially in the red. Also important, upon interaction with live cells, different CD color fractions are trafficked to different sub-cellular locations. Super-resolution imaging shows that the blue CDs were found in a variety of organelles, such as mitochondria and lysosomes, while the red CDs were primarily localized in lysosomes. These findings significantly advance our understanding of the photoluminescence mechanism of multicolor CDs and help to guide future design and applications of these promising nanomaterials.
Since the accidental discovery of luminescent carbon fragments in 2004,1 carbon dots (CDs) have attracted great research interest due to the diverse synthetic methods, tunable luminescence, and applicability in a broad range of fields, including bio-imaging,2–4 sensing,5,6 and light emitting diodes (LEDs).7,8 Typically, CDs are fluorescent carbon nanostructures of sizes less than 10 nm, composed of carbon, oxygen, and nitrogen.9–12 CDs can be produced through bottom-up methods, which involve small molecular precursors like citric acid, malic acid, urea, ethylenediamine, and so on.13–15 In a high temperature reaction, polymerization and dehydration occur among various functional groups, and the resulting products are usually a mixture of small molecule residues, oligomers, and long chain polymers.16 The unclear fluorescence mechanisms and poorly understood internal structure of CDs limit the ability to understand, tune, and fully exploit their fluorescence properties.
Fortunately, in recent years, breakthrough syntheses of multicolor CDs have been achieved.17–19 Several different multicolor CDs have been synthesized with aromatic compounds such as phenylenediamine.4,20–22 However, it should be noted that precursors such as aniline and phenol may have toxic effects on human health and the environment,23,24 and thus should be avoided where possible. Syntheses of colorful CDs from non-aromatic compounds such as citric acid and urea often employ solvothermal methods. Utilizing different solvents such as formamide and dimethylformamide have been shown to play a significant role in tuning CD emission.25,26 In addition, chromatographic post-treatment of as-made CDs plays a critical role in obtaining different colored fractions, using techniques such as anion-exchange column chromatography,26 normal phase silica chromatography,27 and reversed phase silica chromatography.15 Compared with high performance liquid chromatography (HPLC), the aforementioned column chromatography techniques help to separate CDs on a larger scale. These separations are based on charge26 or polarity,21 and are efficient in isolating the desired fractions with distinct colors so that detailed structural characterization can be performed.
To gain insight into the fluorescence mechanism of these multicolor CDs, researchers have considered three hypotheses: quantum size effects,28 the inclusion of molecular fluorophores,29 and surface state-induced emission.30 For example, Rogach and coworkers developed solid-state CDs with tunable fluorescence via the seeded growth method. They attributed the tunable emission to the size of π-conjugated domains.31 Yang and coworkers synthesized CDs by hydrothermal treatment of citric acid and ethylenediamine. They identified a small molecule fluorophore, IPCA (1,2,3,5-tetrahydro-5-oxo-imidazo[1,2-α]pyridiine-7-carboxylic acid) from CD column separation fractions, which contributed to the blue fluorescence.13 Xiong and coworkers synthesized CDs from urea and p-phenylenediamine that emitted a range of colors and separated them with silica column chromatography. They found the degree of carbon oxidation increased as the emission redshifted and thus, they endorsed the surface state hypothesis.21 In addition to the above mechanisms, computational methods such as density functional theory (DFT) have also been applied to analyze the fluorescence origins of CDs. The charge transfer between functional groups on the polymeric unit of CDs made from citric acid and ethylenediamine was found to facilitate blue emission.16
The goal of present work is to understand the fluorescence origin of multicolor CDs. The model multicolor CDs were obtained by reacting citric acid and urea in formamide via a microwave-assisted hydrothermal treatment. An automated chromatographic apparatus was employed to separate as-made CD mixtures into distinct color fractions. The individual separation process took around 20 minutes, and the obtained CD fractions exhibit discrete illumination-induced emission throughout the visible region of the spectrum. Interestingly, the sizes of separated CD fractions are not statistically different from one another, suggesting that the quantum size effects are not the source of differential emission. Solvatochromism experiments showed that the blue and green fractions have similar fluorescence behavior as a function of solvent polarity, but the red fraction behaved differently. Using computational simulations, three models of the fluorescence origin were constructed and evaluated, showing that the formation of small blue fluorescent molecules is likely and pyrene analogs could be the origins for various emission colors. Moreover, two representative CD fractions, the blue- and red-emitting fractions, were chosen for subsequent cell imaging experiments. The localization pattern for the CD fractions differed: blue-emitting CDs were observed in a wide range of organelles, while red-emitting CDs were primarily enclosed in lysosomes. Understanding the origin and the sensitivity of CD emission will improve their utility in bioimaging applications.
Results and discussion
Multicolor CDs were prepared by reacting citric acid and urea in formamide through a microwave-assisted solvothermal treatment. The reaction conditions were optimized for producing red-emissive CDs (see the experimental section and Fig. S1a†). The optimization shows that higher temperature favors the formation of red-emitting fractions (Fig. S1b†). Ultimately, the final reaction conditions consisted of a molar ratio of citric acid to urea of 0.7 and a reaction temperature of 200 °C for 1 h. The crude product was filtered and separated on a C18 reversed-phase column. After three rounds of separation, the obtained CD fractions exhibited seven discrete colors under 365 nm UV illumination, covering the entire visible spectrum (Fig. 1a). The UV-Vis extinction spectra for blue, green, and red fractions (CD-B, G, and R) are depicted in Fig. 1b–d. Conventionally, typical π–π* transitions appear in the 250 nm region which originate from sp2 carbon in the core.21 The n–π* transitions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O that show up at 375 nm are usually related to doping and surface functional group of CDs.32,33 Particularly, for CD-G and R, low energy bands are observable in the region of 450–600 nm. These low energy bands are attributed to n–π* transitions of the extended conjugation system that includes CO and CN.34 Excitation–emission matrix (EEM) measurements were performed to investigate the emission behavior of the CD fractions. Fig. 1e–g represent the global emission peaks for CD-B, G, and R, and it can be deduced that for CD-B, G, and R, the excitation/emission maxima can be achieved at (370 nm, 440 nm), (440 nm, 530 nm), and (560 nm, 600 nm), respectively. We also acquired the excitation/emission maxima for the rest of the CD fractions, as summarized in Table 1. These CD fractions emitted differently upon optimized excitation, covering most of the visible spectrum in the 1D fluorescence spectra (Fig. 1h). Moreover, based on the emission data, the chromaticity of CD fractions can be determined using the CIE 1931 diagram (Fig. 1i), which quantitatively defines the color of their emission wavelengths. It is worthwhile to mention that, even with the aid of an automated chromatographic instrument, it is extremely challenging to completely separate as-made CDs because the physicochemical properties of components are highly similar to one another. Therefore, it is understandable that the peak width for each CD fraction (Table 1) is quite broad (i.e., larger than 70 nm).
O that show up at 375 nm are usually related to doping and surface functional group of CDs.32,33 Particularly, for CD-G and R, low energy bands are observable in the region of 450–600 nm. These low energy bands are attributed to n–π* transitions of the extended conjugation system that includes CO and CN.34 Excitation–emission matrix (EEM) measurements were performed to investigate the emission behavior of the CD fractions. Fig. 1e–g represent the global emission peaks for CD-B, G, and R, and it can be deduced that for CD-B, G, and R, the excitation/emission maxima can be achieved at (370 nm, 440 nm), (440 nm, 530 nm), and (560 nm, 600 nm), respectively. We also acquired the excitation/emission maxima for the rest of the CD fractions, as summarized in Table 1. These CD fractions emitted differently upon optimized excitation, covering most of the visible spectrum in the 1D fluorescence spectra (Fig. 1h). Moreover, based on the emission data, the chromaticity of CD fractions can be determined using the CIE 1931 diagram (Fig. 1i), which quantitatively defines the color of their emission wavelengths. It is worthwhile to mention that, even with the aid of an automated chromatographic instrument, it is extremely challenging to completely separate as-made CDs because the physicochemical properties of components are highly similar to one another. Therefore, it is understandable that the peak width for each CD fraction (Table 1) is quite broad (i.e., larger than 70 nm).
 | ||
| Fig. 1 (a) Photograph of obtained CD fractions illuminated by UV light, (b)–(d) UV-Vis extinction spectra and (e)–(g) 2D EEM of CD-B, CD-G, and CD-R, (h) the normalized fluorescence spectra and (i) the chromaticity diagram of CD fractions (CD-Y and CD-O overlap). | ||
| Ex/Em (nm, nm) | FWHM (nm) | CIE 1931 coordinates (x, y) | QY (%) | |
|---|---|---|---|---|
| CD-V | 372, 443 | 77 | 0.167, 0.128 | 60.1 |
| CD-B | 370, 440 | 77 | 0.156, 0.107 | 49.7 |
| CD-C | 400, 480 | 81 | 0.170, 0.271 | 4.5 |
| CD-G | 440, 530 | 109 | 0.338, 0.542 | 15.1 |
| CD-Y | 470, 560 | 86 | 0.463, 0.528 | 13.1 |
| CD-O | 468, 564 | 87 | 0.461, 0.528 | 12.3 |
| CD-R | 560, 600 | 92 | 0.633, 0.366 | 9.8 |
TEM experiments were conducted to investigate the morphology and sizes of CD fractions. In general, all CDs are apparently spherical, and there are no discernible crystal lattice fringes, indicative of amorphous carbon (Fig. 2 and S2†). The average diameter of CD-B, G, and R fractions are 14.3 ± 8.9 nm, 14.1 ± 6.0 nm, and 13.9 ± 6.4 nm, respectively. As revealed by subsequent one-way ANOVA tests, there is no significant difference among these particle sizes (Fig. S3†). DLS was also applied to measure the hydrodynamic sizes of CD products (Fig. S4†).
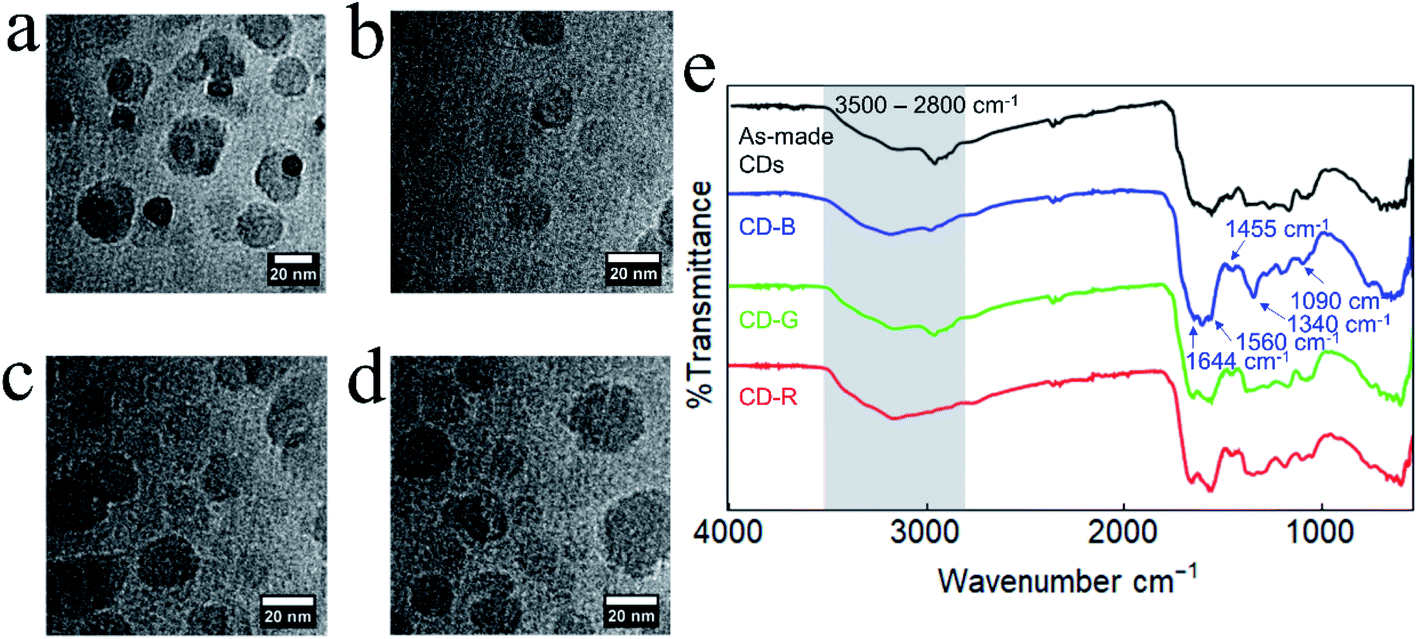 | ||
| Fig. 2 TEM images of (a) as-made CDs, (b) CD-B, (c) CD-G, and (d) CD-R. (e) FT-IR spectra of as-made CDs and CD-B, CD-G, and CD-R. | ||
The as-made CDs and separated CD-B, CD-G, and CD-R exhibit similar Fourier-transform infrared (FT-IR) spectra (Fig. 2e). The broad feature between ≈3500–2800 cm−1 contains contributions from N–H, O–H, and C–H stretching modes.35 The sharper feature between 1600–1500 cm−1 contains peaks associated with CO stretching (1644 cm−1) and N–H bending (1560 cm−1) that are typically observed for carbon dots.36 At lower wavenumber energies, the peaks at 1455 cm−1 and 1090 cm−1 can be attributed to C–N stretching35 and O–C–O bending modes, respectively. Although the FT-IR peaks are generally similar, slight changes are observed between the different fractions and between the fractions and the as-made CDs. For example, the as-made CDs feature a resolvable C–H stretching mode centered at 2965 cm−1 which becomes progressively less intense in green, blue, and red fractions. In CD-B, the peak at 1590 cm−1, which is tentatively assigned as an N–H bending mode, is noticeably more intense than in other fractions.36,37 CD-B also displays a pronounced peak at 1340 cm−1, a region characteristic of C–N stretching.36
Raman spectroscopy also shows similar peaks in as-made CDs and in CD-B, CD-G, and CD-R, with all spectra containing prominent D and G bands at 1373 cm−1 shift and 1645 cm−1 shift, respectively (Fig. S5a†). The D and G bands indicate the presence of sp3 and sp2 hybridized carbon.38 Additional features are present at 1490, 1110, and 980 cm−1 shift, indicating a disordered system.39 Based on the precursors, the 1110 cm−1 shift band is most likely due to an NH2 symmetric rocking mode, and the 1490 and 980 cm−1 shift bands are attributable to additional C–C stretching modes.38,40–42 Similarities in Raman and FT-IR spectra indicate CD structures are comparable across color fractions, suggesting the different fluorescent properties are not from large structural deviations. Raman spectra of the CDs as they age indicate that separated and as-made CDs may degrade at different rates (Fig. S5b†). As-made CDs and CD-B show minimal shifts in D and G band locations, while CD-G and CD-R show a red shift and broadening in the G band over time. Discrepancies in G band shifts and broadening point to CD-G and CD-R experiencing a change in the number and type of functional groups surrounding graphitic sp2 carbons.43–45 It is plausible that the CD-R fraction initially contains the greatest number of functional groups proximate to sp2 carbons as this effect is most prominent in CD-R spectra (Fig. S5c†).44,45
Elemental analysis of as-made CDs shows that the formula is C6.00H6N2.06O5.26. X-ray photoelectron spectroscopy (XPS) analysis of the CD-B, CD-G, and CD-R shows that there is a slight increase in nitrogen content as the emission red shifts (Table S1†). The high-resolution XPS spectra (Fig. 3) reveal that the CD-G has a distinct and more complex composition than CD-B and CD-R. The carbon in the CD-G is dominated by the C–O/C–N peak, and both graphite nitrogen and amide nitrogen are significant. Yet oxygen presents more in the form of O–C than OC. In addition, the peak areas of OC–O and C–O/C–N are higher in red fractions than in blue fractions.
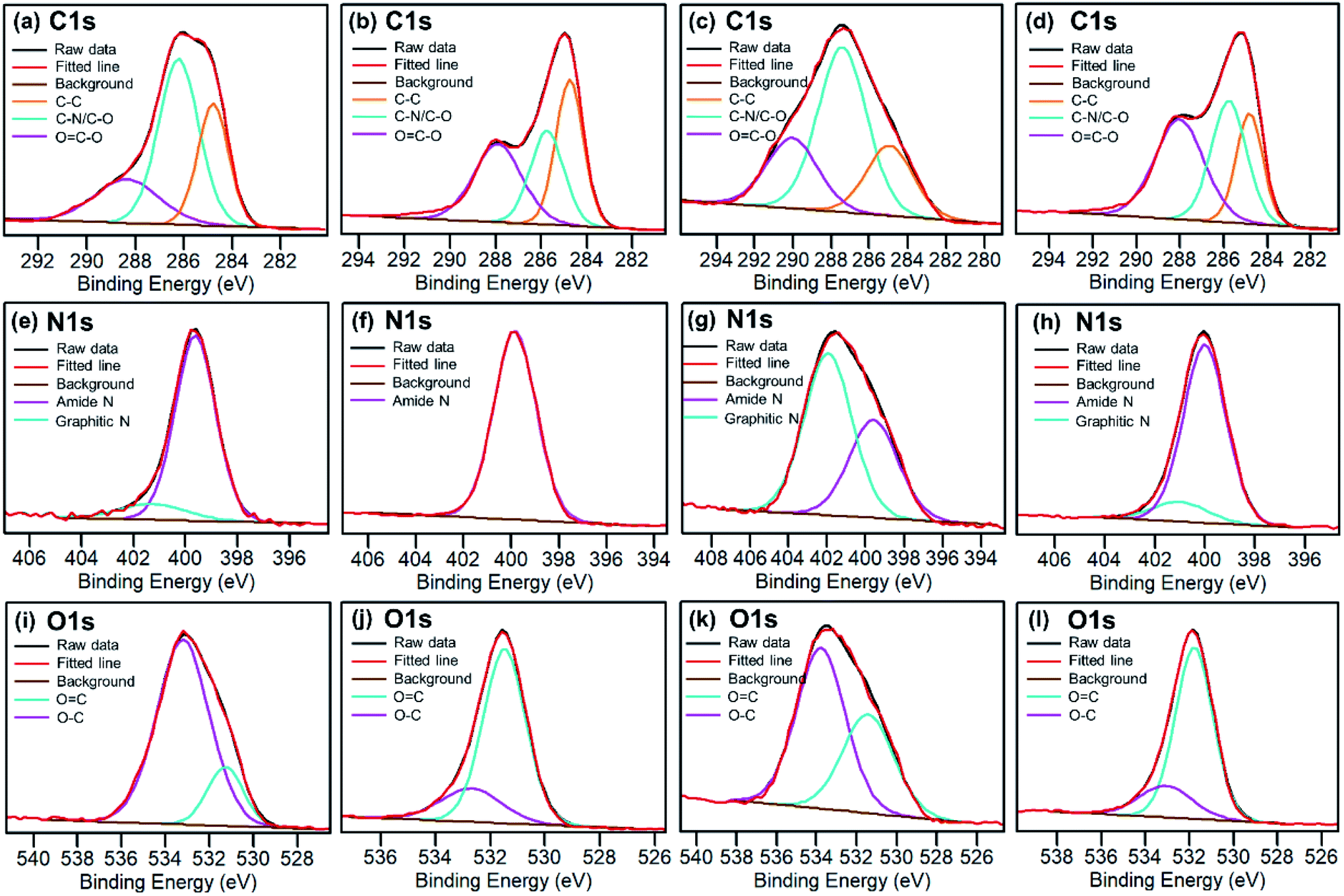 | ||
| Fig. 3 High resolution C 1s, N 1s and O 1s XPS spectra for raw as-synthesized CDs (a, e and i), blue CDs (b, f and j), green CDs (c, g and k) and red CDs (d, h and l). | ||
Fig. 4 shows the 1H NMR spectra of as-made and blue, green, red fractions of CDs in DMSO-d6. We observed both broad and sharp peaks in 1H NMR spectra before dialysis (Fig. 4a). The peaks between 1 to 2 ppm correspond to protons in aliphatic chains. The peaks between 2 to 4 ppm correspond to the protons of hydroxyl groups and/or carbonyl α-hydrogens. The peaks between 6 to 9 ppm correspond to the amide protons, aromatic and vinylic protons. The peaks beyond 10 ppm correspond to protons on aldehydes and carboxylic acids. Compared to small molecules, large CDs tumble slowly in solution, which significantly broadens the NMR signal of the proton species on the CD backbone.37,46 The sharp peaks likely come from free small molecules in the solution and mobile small molecules within the CD structure. The 1H NMR, 13C NMR (Fig. S6†) and 1H–13C HSQC spectra (Fig. S7†) of the CD-B are significantly different than those of the CD-G and CD-R in terms of the sharp peaks. Small molecules can be removed from the carbon dot structures by dialysis using a dialysis membrane of 10k molecular weight cut-off (MWCO). The fluorescence of CDs remained with decreased fluorescence intensity. We observed a significantly decreased number of sharp peaks in 1H NMR spectra after dialysis (Fig. 4b), suggesting that small molecules are mostly removed by dialysis. The remaining broad peaks do not show significant differences among the as-made CDs, CD-B, CD-G and CD-R, suggesting that the CD backbone structures are similar across different fractions (Fig. S8†).
 | ||
| Fig. 4 1H NMR spectra of as made CDs, CD-B, CD-G, and CD-R in DMSO-d6 before dialysis (a) and after dialysis (b). | ||
The solvatochromism of CDs was explored to observe the nature of shifts in the absorption or emission spectra in different solvents (Fig. 5). These studies aim to reveal the hydrogen bonding or dipole–dipole interaction nature of the luminophores responsible for the varied emission characteristics.47 CD-B, G, and R were dissolved in a series of polar aprotic or polar protic solvents: acetone, methanol, ethanol, water, DMF, and DMSO (solvent parameters listed in Table S2†). Similar to previous reports,47–50 there is a shift in the fluorescence spectra when CDs are dispersed in solvents of different polarity. A maximum red shift of 0.1567 eV (1263 cm−1) was observed from CD-B when going from acetone to water. CD-B and CD-G have a similar trend in fluorescence shift as the solvent polarity changes. Fig. 5d shows the correlation of Stokes shift to ENT, a normalized parameter taking account of the overall solvation capacity of solvents, including non-specific interactions (e.g. dipole–dipole) and specific interactions (e.g. hydrogen bonding).51 CD-B and CD-G have a clear dependence on ENT, suggesting a similar structure of the fluorescence center. Their Stokes shifts also show some correlation to α (hydrogen bond donating ability), with R2 values of 0.65 and 0.85 for CD-B and CD-G, respectively (Fig. S9†). However, there is no correlation between the Stokes shift and ENT for CD-R. In addition, CD-R has poor correlation to α, β (hydrogen bond accepting ability), π* (polarizability), and dielectric constants (Fig. S9†). The solvatochromic experiments show that the fluorescence center of CD-R probably has a very different structure or local environment than the other two fractions, and as a consequence, CD-R are least affected by solvents. This conclusion is supported by dialyzing filtered raw product through film of MWCO 500 Da and 3500 Da. The fluorescence spectra (Fig. S10†) show small molecules of MW less than 500 Da could contribute to the blue and green fluorescence of CDs, while the red fluorescence probably comes from a chemical structure of MW 500 Da to 3500 Da. The CDs left inside the dialysis bags still have multicolor fluorescence.
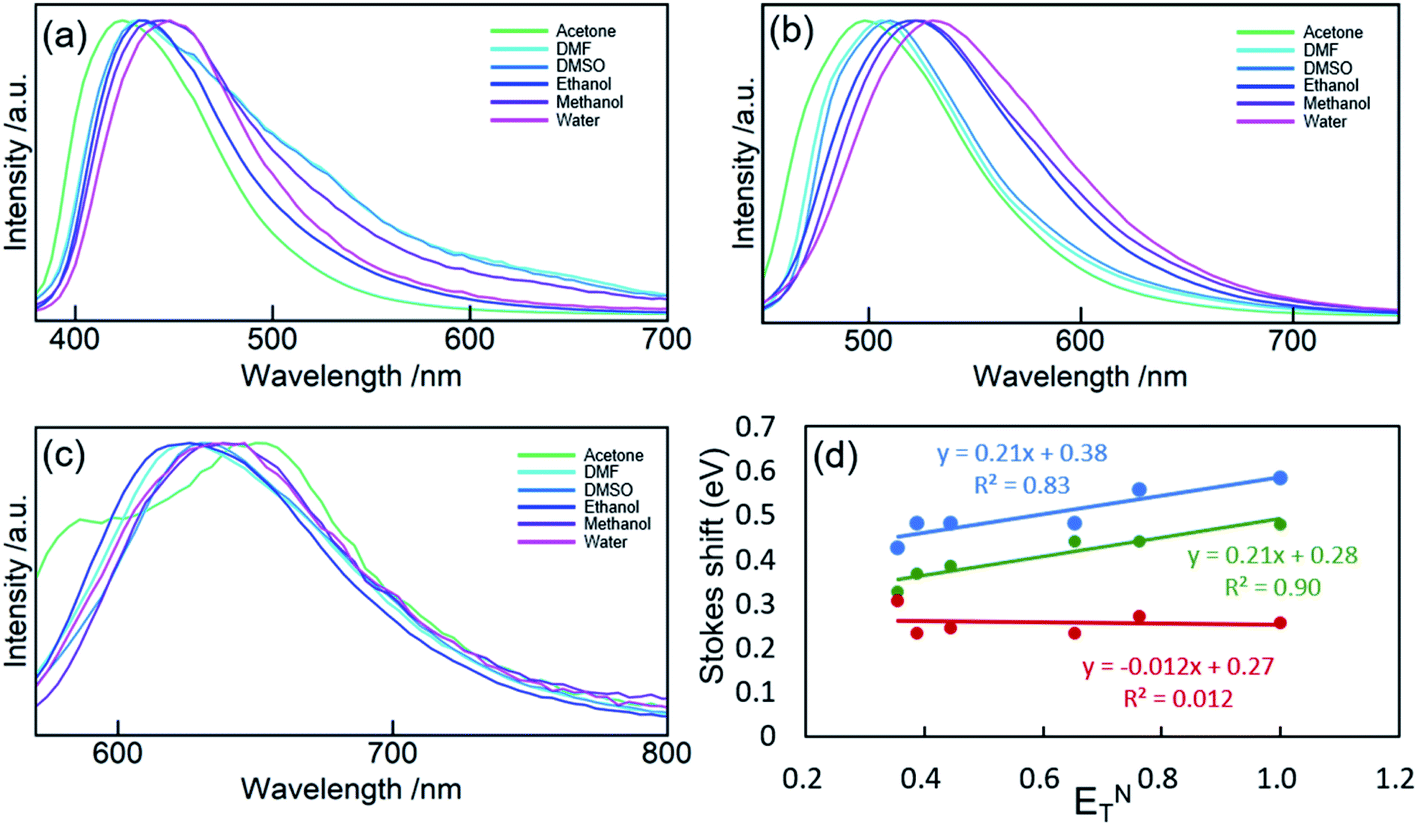 | ||
| Fig. 5 PL spectra of CD-B (a), CD-G (b), and CD-R (c) dispersed in acetone, DMF, DMSO, ethanol, methanol, and water. (d) Stokes shift of CD-B, CD-G, and CD-R plotted against ENT. | ||
Characterization via NMR, FT-IR, and other methods demonstrates that CD-B, CD-G, and CD-R have similar structures. However, their fluorescence behaviors are different in solvents with different polarity. Since CD-B and CD-G share a similar trend and most likely derive from molecules of 100–500 Da, small aromatic molecules were modeled, and extensive geometry optimization, molecular dynamics, and excited state calculations were conducted. The fluorescence origin of CD-R is least affected by solvent polarity and is attributed to a larger structure. Therefore, extended π systems with doped graphitic nitrogen as pyrene analogs26 were modeled to find out if the emission could occur at these long wavelengths. In addition, polyamides are thought to be important components of CD structures and have been proposed in precedent work to be the luminescence source.16 DFT calculations were also conducted to evaluate this possibility. While these models have been studied computationally in previous work,16,26 analysis here involves considerably more extensive conformational sampling and aims to compare different models with consistent computational methodologies.
Small molecular chromophores have been proposed to form through condensation reactions in previous studies of carbon dots formed with citric acid and amine precursors.13,52,53 Following a similar reaction scheme, we propose that several small aromatic molecules might be produced with citric acid, urea, and formamide under the optimized synthesis conditions (Fig. S11†). As shown in Table S3,† while the formation of these small molecules is enthalpically unfavorable, the reactions are entropically favored (due to release of water during dehydration). Under the high-temperature condition, therefore, the formation of these small molecule chromophores is, in fact, favored and competitive with polyamide formation (Table S4†).
TD-DFT calculations were performed for the three small molecular chromophores in the gas phase (Tables S5–S7†). As summarized in Fig. 6, both small molecules 1 and 3 show strong absorption and emission around ∼350 nm and ∼450 nm, respectively. Such features are consistent with the π → π* nature of the excitation (as reflected by the NTOs shown in Fig. 6) and are qualitatively similar to the IPCA molecule discussed in previous study of carbon dots formed with citric acid and amines.13 By comparison, small molecule 2 shows weaker absorption and emission in the relevant energy range. A few other small molecules are also considered in Fig. S12 and S13,† with similar absorption/emission spectra as those shown in Fig. 6.
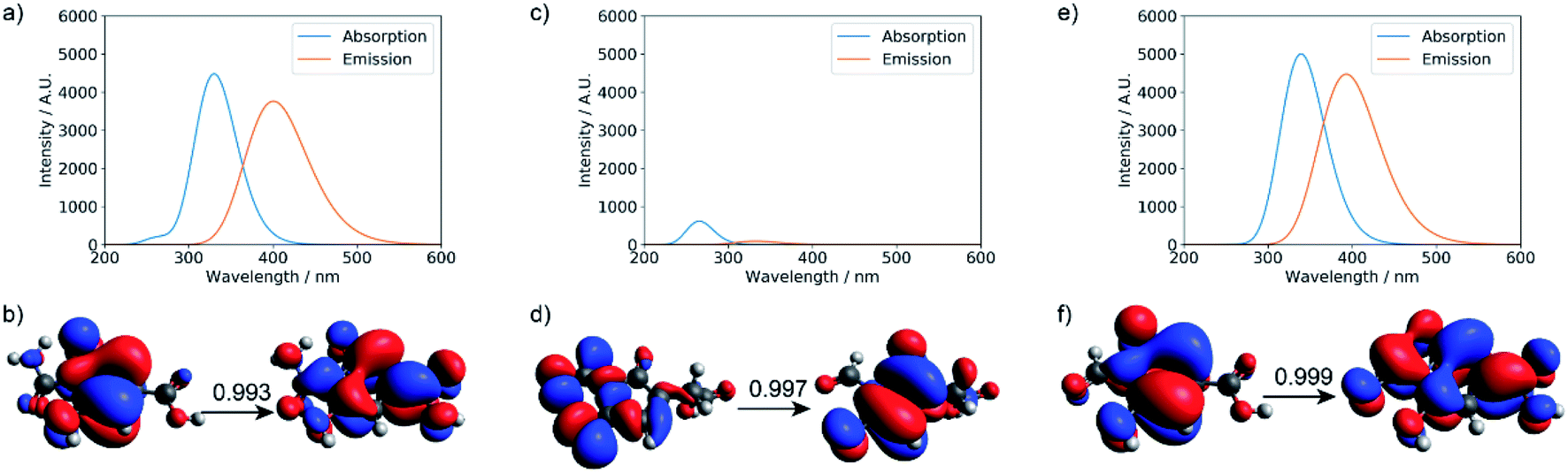 | ||
| Fig. 6 Calculated UV-Vis absorption, fluorescence spectra, and natural transition orbitals (NTOs) of candidate small molecule chromophores (for their structures, see Fig. S11†). The absorption and fluorescence spectra were calculated at the level of TD-ωB97XD/6-31+g(d,p) for (a) product 1, (c) product 2, and (e) product 3; (b, d, and f) are the corresponding dominant NTO pairs. Negative values correspond to holes in red, and positive values correspond to particles in blue. To generate the absorption/emission line shapes in this and subsequent figures, a line width of 0.4 eV (default in Gaussview) was used; for absorption, we consider the three lowest excited states, while for emission, we consider only the first excited state. | ||
As summarized in Fig. 7, explicit hydrogen-bonding interactions or stacking effects may have a notable impact on the absorption and emission of the small molecular chromophore. The largest observed effect is on the order of 30 nm on the emission, with tails extending into the green region of the electromagnetic spectrum. Compared to monomer 1, the absorption peak of the stacking dimer red-shifts by about 18 nm, and the calculated fluorescence spectra show a weak emission peak at 807 nm.
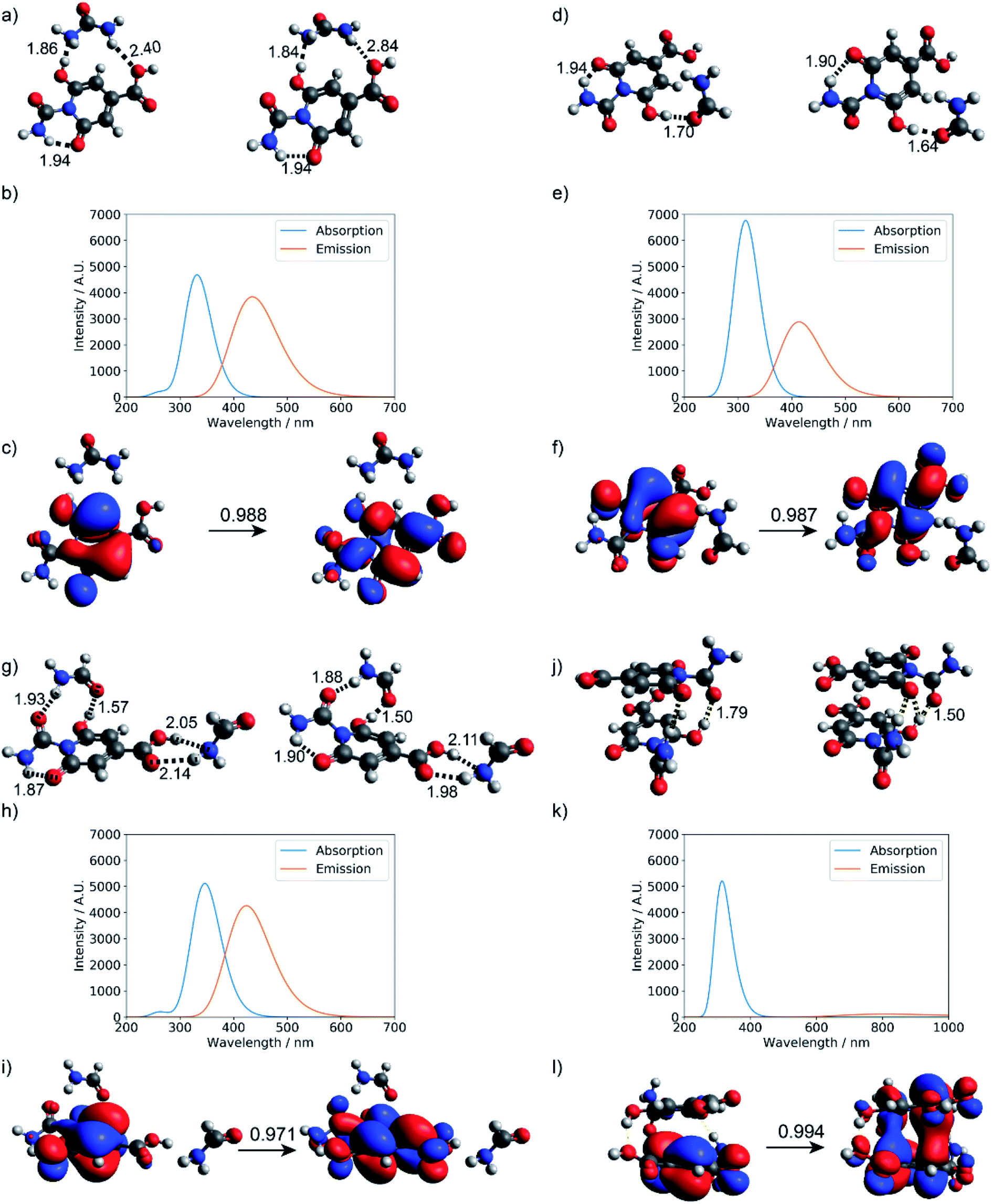 | ||
| Fig. 7 Hydrogen-bonding effect on the photoactivity of small molecule chromophore 1 (see Fig. S11† for structure). Optimized geometries at the ground state and first excited state: (a) urea and product 1, (d) formamide and product 1, (g) two formamide and product 1, and (j) stacked dimer of product 1; (b, e, h, and k) are corresponding absorption and fluorescence spectra computed at the level of TD-ωB97XD/6-31+g(d,p); (c, f, i and l) are corresponding dominant NTO pairs. Negative values correspond to holes in red, and positive values correspond to particles in blue. | ||
Calculations have also been conducted to explore whether larger spectral shifts can be observed when the small molecular chromophore is engaged in more extensive interactions with polyamide models of different lengths (dimer and decamer). In each case, ten structures were collected from a replica exchange molecular dynamics simulation of at least one nanosecond. Photoactivities of these molecules were then evaluated at the TD-DFTB and TD-DFT levels. As shown in Tables S8 and S9† for the dimer model and Table S10† for the decamer model, interactions with the polyamide lead to red-shifts in the emission to ∼470 nm, still in the blue spectral range. As illustrated in Fig. 8, the excitation remains local π → π* in nature, despite extensive hydrogen bonding interactions with the polyamide.
 | ||
| Fig. 8 An example for the structure of a small molecule chromophore surrounded by a decamer polyamide. Also shown is the nature of the excitation, which largely involves excitation from HOMO−1 to LUMO with an amplitude of 0.957. Calculations at LC-TD-DFTB2 and TD-ωB97XD levels are consistent in the nature of excitation (see Table S10† for details). | ||
In short, our calculations suggest that the small molecule chromophores can be formed from citric acid, urea, and formamide through condensation under the synthesis conditions, and they exhibit intense absorption/emission in the relevant UV-Vis energy range. Hydrogen-bonding and stacking interactions with other small molecules or polyamides can lead to further red-shift of the emission spectra to the region of 470 nm. Therefore, small molecular chromophores embedded in a polyamide matrix can indeed contribute significantly to the blue fluorescence observed in the CDs. Considering the similar solvatochromic properties of CD-B and CD-G, it's highly likely that green fluorescence also originates from small molecules of similar structures.
With all the computational models considered so far, no strong emission is observed in the red region. Therefore, the red fluorescence observed in the carbon dots synthesized here is likely from a different origin. Indeed, with the local excitations observed so far, it is difficult to lower the emission to less than 2.5 eV. For the longer-wavelength fluorescence, more extended π systems are likely involved and Holá et al. proposed that pyrene analogs that contain graphitic nitrogen are plausible candidates.26 While the reaction mechanisms that lead to the formation of these analogs starting from citric acids, urea, and formamide are not clear, we estimated the thermodynamic stability of the four pyrene-type of compounds (NPx, x = 1–4)26 with the same level of DFT calculations as for polyamides and the small molecular chromophores. As shown in Table S11,† the formation of these pyrene-type of compounds is thermodynamically favorable. The stability of the four doped NPx decreases progressively as x increases from 1 to 4. Similar to the calculations reported,26 TD-DFT calculations (Tables S12 and S13†) suggest that the pyrene analogs indeed have strong absorption and emission in the relevant UV-vis energy ranges (Fig. 9). For example, with the model compound NP4, there are strong absorptions in the 200–360 nm range and emission around 690 nm; the nature of the excitation is π → π*, as illustrated in Fig. 9.
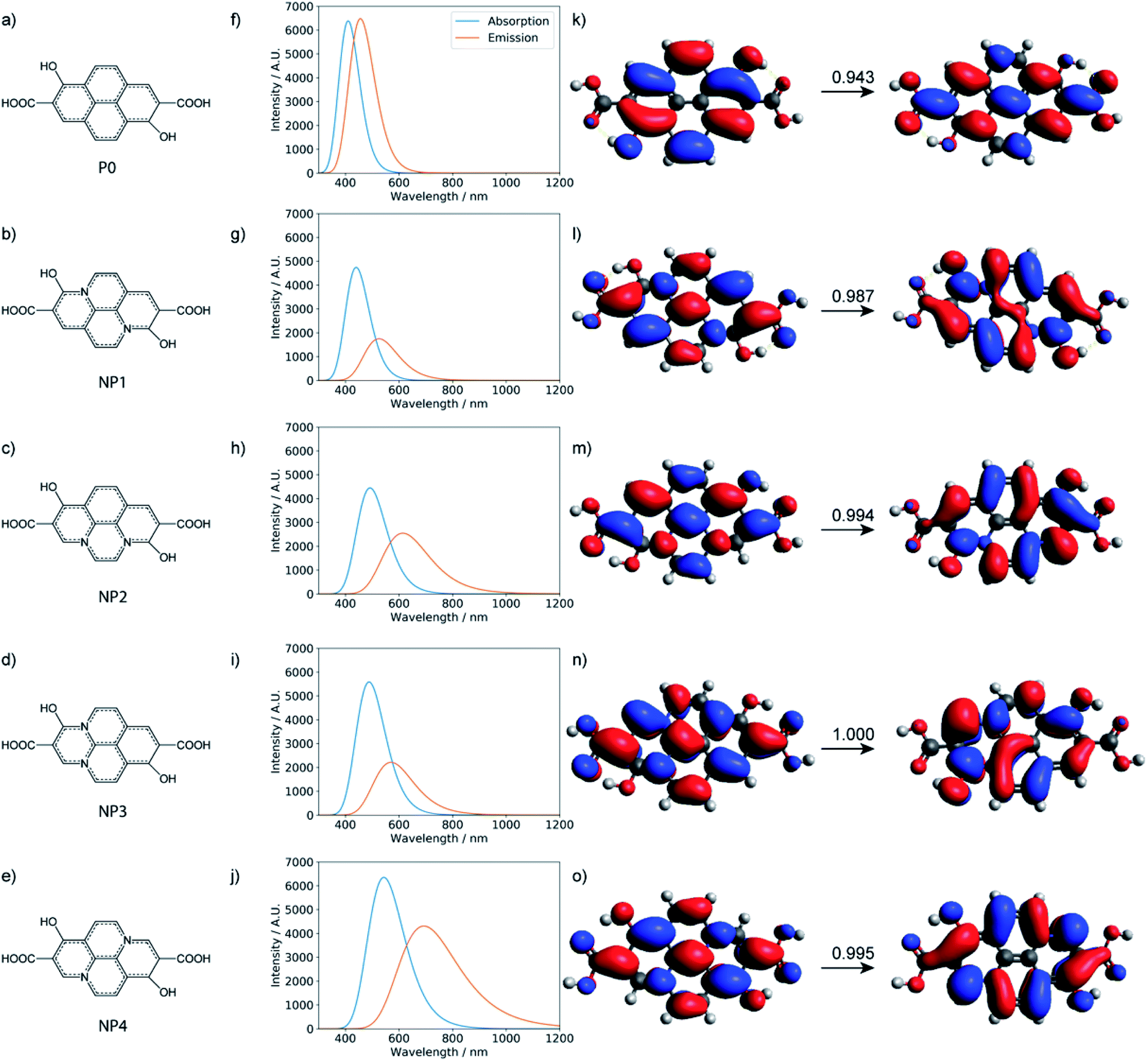 | ||
| Fig. 9 Chemical structures, calculated UV-Vis absorption, fluorescence spectra, and natural transition orbitals (NTOs) for the pyrene-based model. (a–e) The chemical structures of the nitrogen-free system (P0) and nitrogen-doped models (NP1, NP2, NP3, NP4). (f–j) The corresponding absorption and fluorescence spectra calculated at the level of TD-ωB97XD/6-31+g(d,p). (k–o) The corresponding dominant NTOs. | ||
In addition to the above, polyamides have also been proposed to form when citric acid and ethylenediamine undergo condensation reactions.16 Here, we anticipate that polyamides are also formed with citric acid and urea, as depicted in Fig. 10. As shown by the DFT results in Table S4,† the condensation reaction between citric acid and urea is indeed thermodynamically favored in the synthesis conditions. The subsequent dehydration is enthalpically unfavorable but entropically favored, leading to an overall drop in free energy of ∼5 kcal mol−1. The polyamides have absorptions in the range of 250–270 nm (at the ωB97XD level of theory, Table S14†), while emission from S1 occurs in the range of 420–520 nm. With the dehydrated polyamide model (Fig. S14, Tables S15 and S16†), substantially red-shifted emission is observed in some cases. However, the oscillator strength for both absorption and emission is extremely low (typically 10−3 or less) for all the cases analyzed (also see Table S17†). This observation is consistent with the fact that the nature of the electronic excitation is n → π* (Fig. S15†), which is known to have a small transition dipole due to the poor overlap of the relevant orbitals. Along this line, the recent study by Vallan et al. suggested that the low-energy excitation in polyamide is charge transfer in nature.16 However, this finding was likely due to the use of B3LYP in the TD-DFT calculations, which are known to underestimate the energy of charge transfer states due to the delocalization error of commonly used functionals. Indeed, as shown in Table S18 and Fig. S16,† the nature of excitation in S1 changes from charge transfer to local n → π* excitation once range-separated functionals are used. Therefore, charge transfer in polyamides is unlikely to contribute to the blue fluorescence.
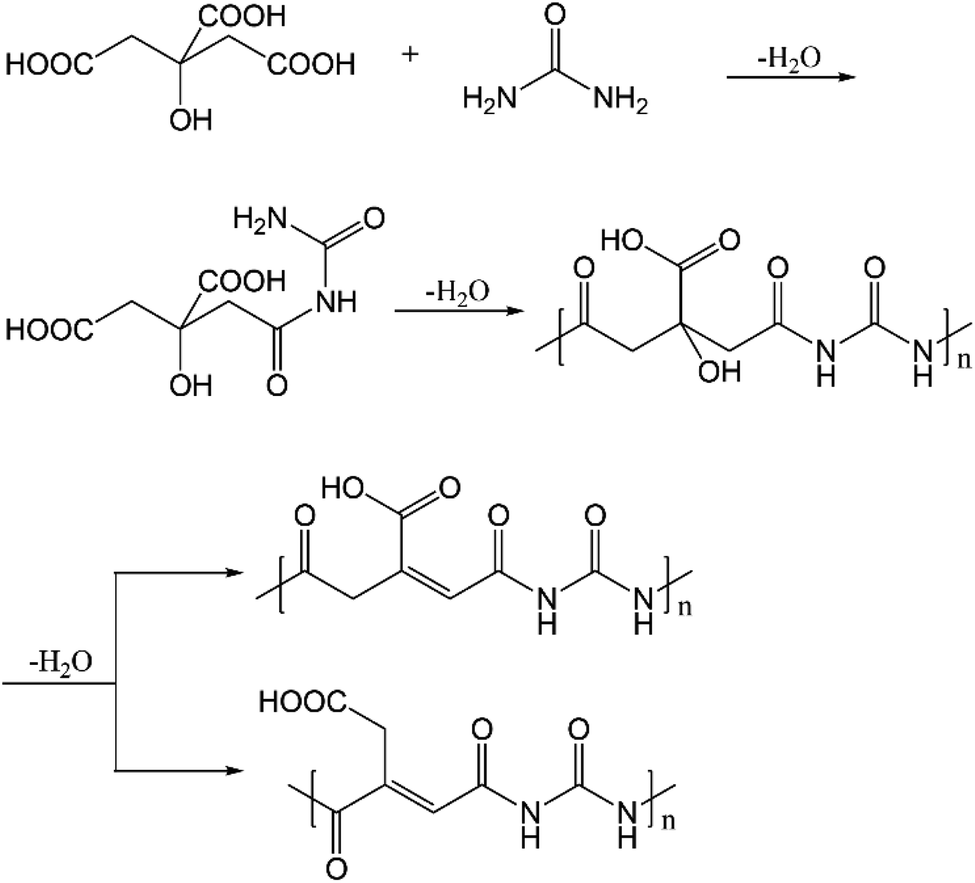 | ||
| Fig. 10 Proposed formation pathway for polyamide and dehydrated polyamide. | ||
One major use of brightly-emitting, water stable nanomaterials is bioimaging. Because it appears that there are some chemical and functional distinctions between the varied carbon dot fractions, the uptake and distribution of multi-color CDs were evaluated in rainbow gill trout epithelial cells. The as-made CDs were incubated with gill cells for 2 h, and it was noted that abundant as-made CDs were taken up by cells. As shown in Fig. 11, when excited with different laser wavelengths, the emitted fluorescence signals localized to distinct intracellular positions, which indicates that as-made CDs of different excitation/emission properties might transport to different locations upon uptake. As-made CDs excited with shorter wavelengths (blue color in Fig. 11) appeared to be more diffuse than CDs excited with longer wavelengths (red color in Fig. 11) inside cells. In light of this finding, we further used CD fractions to investigate their distribution patterns. Two groups of CD fractions with separate spectra were selected, namely CD-B (excited by 405 nm laser) and CD-R (excited by 561 nm laser). In addition, we also introduced a variety of organelle-specific stains to probe possible co-localization with CDs. As shown in Fig. S17,† the red dots mainly co-localized with lysosomes. In comparison, part of the CD-B co-localized with lysosomes (Fig. S18†) while part of them co-localized with mitochondria (Fig. S19†). Moreover, upon exposure to CD-B, enlarged lysosomes were formed in cells (Fig. S18†), suggesting impaired lysosomal fusion–fission balance and higher toxicity induced by the blue-emitting CDs.54 CD-B, albeit at higher concentrations than used for imaging (100 μg mL−1), was the only fraction of CDs to induce a significant decrease in cell viability (Fig. S20†). We reported before that CDs made from malic acid were found predominantly in mitochondria after cell internalization.55 Taken together, we conclude that the different CD fractions localize in distinct subcellular regions where some particles tend to penetrate into more organelles (e.g., lysosomes and mitochondria) and the rest are primarily trapped in lysosomes. We propose the localization difference is a result of hydrodynamic size, as CD-B has a smaller hydrodynamic size, while CD-R tend to aggregate and thus has a larger size as measured by DLS (Fig. S3†). The measured toxicity may arise from the broader intracellular distribution of CD-B and damaged lysosome functions.
 | ||
| Fig. 11 Distribution of as-synthesized multicolor CDs in living gill cells. | ||
Conclusions
In summary, multicolor CDs were synthesized with simple solvothermal treatment of citric acid and urea. The mixture was separated with reversed-phase column chromatography into fractions with varied emission wavelength. TEM shows that there is no statistical difference in size among different fractions. Elemental analysis shows the formula is C6.00H6N2.06O5.26, and there is slight increase in nitrogen percentage from blue to red. By dispersing these CDs into a series of solvents, CD-B and CD-G show similar solvatochromic effects on luminescent emission. CD-R, however, shows strong deviation in solvatochromic behavior. By dialyzing, it was found that small molecules of MW 100–500 Da have blue and green fluorescence, and some structures of MW less than 3500 Da have red fluorescence. Computational modeling reveals that small molecules are possible contributors to blue fluorescence. Pyrene analogs could be responsible for emissions of a broader range, however, the exact chemical structures are not yet obvious. In addition, super-resolution imaging shows the difference in CD localization in cells for the blue- and red-emitting CD fractions. Our combined experimental and theoretical work suggests that the quantum confinement effect is less likely to be the source of polymeric CD fluorescence. The more likely source are fluorescent molecules formed and integrated into the polymer backbone. Nevertheless, it remains challenging to assert that the small molecules analyzed here computationally are the only dominant chromophores for the fluorescence. Therefore, moving forward, it is of critical importance to fully analyze the structures of those small molecules and nanostructures by additional experimental and computational studies, thereby elucidating the relationship between the fluorophores and the polymer structure that make up the CDs.Methods
Materials
Citric acid (Sigma-Aldrich, ≥99.5%), urea (Mallinckrodt, ≥99.6%), formamide (Alfa Aesar, 99%) were used without further purification. DI water was from Direct-Q® 3, 5, 8 laboratory water purification system by MilliporeSigma.Synthesis
The as-made CDs that contain multicolor components were synthesized by fine tuning the reactants ratio, reaction temperature, and reaction duration in a CEM Discover® SP microwave reactor. Citric acid and urea were mixed in molar ratios of 0.1, 0.3, 0.5, 0.7, and 0.9, in 15 mL formamide. The extent of carbonization was controlled by adjusting reaction temperatures from 120 °C to 200 °C, incremented by 20 °C. The reaction time was held at 10 min, 30 min, 1 h, 2 h, or 4 h. As such, the total screening process involved 125 reactions. After each reaction, the UV-Vis and fluorescence spectra were measured, and the blue, green, and red emission peaks were integrated. A summary chart of the fluorescence of the raw products can be found in Fig. S1.† As the red-emissive CDs will provide better tissue penetration depth in imaging experiments, the reaction conditions were optimized for the highest red peak integration ratio. The as-made CDs used for separation were obtained by reacting 7 mmol citric acid with 10 mmol urea in 15 mL of formamide at 200 °C for 1 hour. The products were further filtered to remove bulk carbon residue before column chromatographic separation was performed.To achieve an efficient separation, a CombiFlash® automated chromatography instrument was utilized in combination with a reversed-phase silica column, RediSep® Rf C18 Aq (size: 50 g). Specifically, after a default column equilibration, 1 mL of as-made CD solution was injected into the column, and the separation program started automatically. At the beginning of the separation, pure water (solvent A) was used as the mobile phase and as the separation proceeded, the volume% of acetone (solvent B) increased from 0% to 40% in 14 minutes when all colored components were collected. After the separation, the column was washed with pure acetone and pure water, successively, to completely remove non-fluorescent residue. In some cases, additional separations were performed on collected CD fractions once or twice more to further purify the components.
Material characterization of CDs
The particle size distribution of carbon dots was determined by TEM (FEI Tecnai G2 F30 TEM at 300 kV) and DLS (Microtrac NANO-flex® 180° DLS System). UV-Vis extinction spectra were obtained using a Mikropack DH-2000 UV-Vis-NIR spectrometer. Fluorescence spectra and EEMs were measured with a PTI QuantaMaster™ 400 and ISS K2™ multifrequency cross-correlation phase and modulation fluorometer, respectively. ATR-FTIR spectra were obtained using a Thermo Scientific Nicolet iS5 Fourier Transform Infrared Spectrometer with an iD5 Attenuated Total Reflectance attachment. XPS was measured with PHI 5000 VersaProbe II. CHN elemental analysis was performed on an Exeter Analytical CE440 CHN Analyzer. For NMR analysis, the as-made and red, green, blue fractions of carbon dots were dialyzed against nanopure water (10k MWCO, Thermo Fisher Slide-A-Lyzer dialysis cassettes), lyophilized, and redispersed in 600 μL of DMSO-d6 before the NMR measurement. 1H NMR spectra were recorded at 25 °C on a Carver-Bruker 500 NMR spectrometer. 1H NMR spectra were referenced using the solvent signal. Raman spectra were recorded on a Witec Alpha 300R confocal Raman microscope using a frequency-doubled Nd:YAG 532 nm laser with UHTS300 spectrometer and DV401 CCD detector. Power was 25 mW or less, with 2 accumulations and integration times between 5–15 seconds.Computational methods
The energetics for the formation of different polyamide models, small molecular chromophores, and pyrene analogs starting from citric acid, urea and/or formamide are done with density functional theory (DFT)56 calculations following standard procedures. Geometries are fully optimized at the B3LYP57–59-D3 (ref. 60)/Def2TZVPP61,62 level of theory; vibrational frequencies are computed at the same level of theory and used to estimate the vibrational contributions to enthalpy and Gibbs free energy within the harmonic-oscillator-rigid-rotor framework.63To obtain structural models for the polyamides (e.g., dimer and decamer) formed from citric acid and urea, molecular dynamics simulations are conducted at the DFTB3-D3/3OB64–66 level, which avoids the need to parameterize classical force fields for these polymers. To enhance conformational sampling, replica exchange molecular dynamics (REMD67) simulations in the NVT ensemble are conducted using 16 replicas with a temperature profile exponentially spaced between 300 K and 900 K. In the REMD simulations, a time step of 1 fs is used, and exchanges are attempted every 1000 MD steps, which leads to an acceptance ratio of about 0.3; simulations are conducted typically for one nanosecond.
One example of exchange profiles is plotted in Fig. S21† for the case of a decamer polyamide. The radius of gyration for the replica at the lowest temperature (300 K) is shown in Fig. S22† which illustrates that the polyamide rapidly collapses from an extended structure to a compact structure with the final radius of gyration of 7.26 Å. For subsequent excited state calculations, cluster analysis is conducted to identify ten structures, which are then optimized locally prior to time-dependent density functional tight binding (TD-DFTB) calculations.
For the small molecule models (polyamide monomer/dimers and small molecule chromophores), excited state calculations are conducted with TD-DFT68 methods using a range of functionals (B3LYP, ωB97XD and CAM-B3LYP) and basis sets (6-31+G(d,p) and Def2TZVPP) (see, for example, captions of Tables S14 and S15† in the ESI†); as shown in Table S5,† the effect of basis set on the computed absorption and emission energies is very modest, thus most of the TD-DFT calculations are conducted with the 6-31+G(d,p) basis set. For absorption, structures are first optimized with the respective functional in the ground state and vertical excitation energies are computed with the standard linear-response and adiabatic approximations. For emission, structures are optimized for S1 and the vertical de-excitation energies are recorded.
For larger model systems, such as the case with a small molecule chromophore interacting with polyamide models, structures are first optimized at the LC-DFTB2 (ref. 69 and 70) level and then single point energy calculations are conducted at both LC-TD-DFTB2 (ref. 71) and TD-DFT levels for absorption. The use of the LC variant of the TD-DFTB approach is essential here as otherwise the low-lying excited states are contaminated with charge transfer states. Even then, comparisons in the ESI† indicate that the lowest bright excited state with LC-TD-DFTB2 is usually not S1, in contrast to TD-DFT calculations with a reliable functional (e.g., ωB97XD72). Nevertheless, the results for the lowest bright excited state at the LC-TD-DFTB2 level compare favorably with TD-ωB97XD in terms of energetics, nature of excitation, and oscillator strength. Therefore, for probing emission, we first optimize the structure at the LC-TD-DFTB2 level for the bright state, which is often not S1 (see, for example, Table S8†); single point vertical de-excitation energy calculations are then conducted at the TD-ωB97XD level of theory. For the small molecule-polyamide dimer model, we compare this protocol with TD-ωB97XD S1 optimization and the results are largely consistent, as shown in Tables S9 and S10.†
All MD simulations are done using CHARMM73 v44. DFT and TD-DFT calculations are done using Gaussian 16 Rev. B.74 For the LC-TD-DFTB2 calculations, GAMESS(US)75 with the latest TD-DFTB implementation76 is used; in the LC-TD-DFTB2 calculations, the ω parameter is set to 0.30 unless otherwise indicated.
Toxicity and imaging experiments
Oncorhynchus mykiss (rainbow trout) epithelial gill cells (ATCC, CRL-2523) were cultured with Leibovitz's L-15 media supplemented with 10% fetal bovine serum and 1% antibiotics (complete media). Cells were grown at 19 °C in ambient atmosphere; subcultures from passage 5–10 were used for all imaging experiments. After reaching 75% confluence, cells were incubated with 5 μg mL−1 CDs (as-synthesized or fractions) in phenol-red free medium for 2 h (Thermo Fisher Scientific, A14291DJ). For co-localization analyses, the following staining reagents were used: LysoTracker Blue (Invitrogen L7525), LysoTracker Red (Invitrogen L7528), MitoTracker Deep Red (Invitrogen M22426). The staining process followed the manufacturer's protocols. Images were taken with a Zeiss LSM710 confocal microscope and a 60× (NA 1.4) oil immersion objective. 1–5 mW laser power was used for live-cell imaging.The cell viability of the different fractions of CDs was determined using the MTS proliferation assay (Promega, G3580). Trout gill epithelial cells were seeded in 96 well plates at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in 200 μL of complete media. Cells reached 100% confluency overnight. A stock solution of each type of carbon dot was prepared by bath sonicating the carbon dots for 30 min in 18 MΩ cm water (Advantage A10, Millipore). CDs were diluted in complete media and cells were exposed to a range of concentrations of CDs for either 1 or 24 h. Control cells were exposed to media diluted with nanopure water equivalent to the amount used for exposure to carbon dots. For negative controls, lysis solution (Promega) was added 45 min before the end of the carbon dot exposure period. Following the exposure period, cells were rinsed 3× with 100 μL of media without FBS or phenol red, which were found to interfere with the absorbance measurements, to remove any unbound carbon dots. Cells were incubated with 20 μL of MTS solution in 100 μL of media (without FBS or phenol red) for 4 h at 19 °C in ambient atmosphere and then the absorbance at 485 nm was determined using a Beckman Coulter DTX 880 Multimode Detector.
000 cells per well in 200 μL of complete media. Cells reached 100% confluency overnight. A stock solution of each type of carbon dot was prepared by bath sonicating the carbon dots for 30 min in 18 MΩ cm water (Advantage A10, Millipore). CDs were diluted in complete media and cells were exposed to a range of concentrations of CDs for either 1 or 24 h. Control cells were exposed to media diluted with nanopure water equivalent to the amount used for exposure to carbon dots. For negative controls, lysis solution (Promega) was added 45 min before the end of the carbon dot exposure period. Following the exposure period, cells were rinsed 3× with 100 μL of media without FBS or phenol red, which were found to interfere with the absorbance measurements, to remove any unbound carbon dots. Cells were incubated with 20 μL of MTS solution in 100 μL of media (without FBS or phenol red) for 4 h at 19 °C in ambient atmosphere and then the absorbance at 485 nm was determined using a Beckman Coulter DTX 880 Multimode Detector.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under the NSF Center for Sustainable Nanotechnology, CHE-2001611. The CSN is part of the Centers for Chemical Innovation Program. Part of this work was done at the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by DOE's office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. The TD-DFTB computations were supported by the grant from the NIH to QC (R01 GM106443). Computational resources from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number ACI-1548562, are greatly appreciated. Part of the computational work was performed on the Shared Computing Cluster which is administered by Boston University's Research Computing Services (URL: http://www.bu.edu/tech/support/research/). Part of this work utilized the CEM Discover SP microwave that was purchased with funding provided by the NSF Center for Sustainable Polymers, CHE-1901635. We thank Shuyi Xie in Prof. Timothy P. Lodge's group (Department of Chemistry, University of Minnesota) for his help in using the vacuum oven. We also thank Jiayi He from Christy L. Haynes' group for making the gold substrate used for Raman measurements.References
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, Electrophoretic Analysis and Purification of Fluorescent Single-Walled Carbon Nanotube Fragments, J. Am. Chem. Soc., 2004, 126(40), 12736–12737 CrossRef CAS.
- H. Tao, K. Yang, Z. Ma, J. Wan, Y. Zhang, Z. Kang and Z. Liu, In Vivo NIR Fluorescence Imaging, Biodistribution, and Toxicology of Photoluminescent Carbon Dots Produced from Carbon Nanotubes and Graphite, Small, 2012, 8(2), 281–290 CrossRef CAS.
- X. Huang, F. Zhang, L. Zhu, K. Y. Choi, N. Guo, J. Guo, K. Tackett, P. Anilkumar, G. Liu, Q. Quan, H. S. Choi, G. Niu, Y.-P. Sun, S. Lee and X. Chen, Effect of Injection Routes on the Biodistribution, Clearance, and Tumor Uptake of Carbon Dots, ACS Nano, 2013, 7(7), 5684–5693 CrossRef CAS.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Red, Green, and Blue Luminescence by Carbon Dots: Full-Color Emission Tuning and Multicolor Cellular Imaging, Angew. Chem., Int. Ed., 2015, 54(18), 5360–5363 CrossRef CAS.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and S. X.-A. Zhang, Carbon Dots with Continuously Tunable Full-Color Emission and Their Application in Ratiometric pH Sensing, Chem. Mater., 2014, 26(10), 3104–3112 CrossRef CAS.
- H. Li, C. Sun, R. Vijayaraghavan, F. Zhou, X. Zhang and D. R. MacFarlane, Long lifetime photoluminescence in N, S co-doped carbon quantum dots from an ionic liquid and their applications in ultrasensitive detection of pesticides, Carbon, 2016, 104, 33–39 CrossRef CAS.
- F. Wang, Y.-H. Chen, C.-Y. Liu and D.-G. Ma, White light-emitting devices based on carbon dots’ electroluminescence, Chem. Commun., 2011, 47(12), 3502–3504 RSC.
- C. Sun, Y. Zhang, S. Kalytchuk, Y. Wang, X. Zhang, W. Gao, J. Zhao, K. Cepe, R. Zboril, W. W. Yu and A. L. Rogach, Down-conversion monochromatic light-emitting diodes with the color determined by the active layer thickness and concentration of carbon dots, J. Mater. Chem. C, 2015, 3(26), 6613–6615 RSC.
- Y. Choi, Y. Choi, O.-H. Kwon and B.-S. Kim, Carbon Dots: Bottom-Up Syntheses, Properties, and Light-Harvesting Applications, Chem.–Asian J., 2018, 13(6), 586–598 CrossRef CAS.
- V. Mishra, A. Patil, S. Thakur and P. Kesharwani, Carbon dots: emerging theranostic nanoarchitectures, Drug Discovery Today, 2018, 23(6), 1219–1232 CrossRef CAS.
- B. Zhi, M. J. Gallagher, B. P. Frank, T. Y. Lyons, T. A. Qiu, J. Da, A. C. Mensch, R. J. Hamers, Z. Rosenzweig, D. H. Fairbrother and C. L. Haynes, Investigation of phosphorous doping effects on polymeric carbon dots: fluorescence, photostability, and environmental impact, Carbon, 2018, 129, 438–449 CrossRef CAS.
- A. Cayuela, M. L. Soriano, C. Carrillo-Carrión and M. Valcárcel, Semiconductor and carbon-based fluorescent nanodots: the need for consistency, Chem. Commun., 2016, 52(7), 1311–1326 RSC.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, Investigation from chemical structure to photoluminescent mechanism: a type of carbon dots from the pyrolysis of citric acid and an amine, J. Mater. Chem. C, 2015, 3(23), 5976–5984 RSC.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, Unravelling the Multiple Emissive States in Citric-Acid-Derived Carbon Dots, J. Phys. Chem. C, 2016, 120(2), 1252–1261 CrossRef CAS.
- B. Zhi, Y. Cui, S. Wang, B. P. Frank, D. N. Williams, R. P. Brown, E. S. Melby, R. J. Hamers, Z. Rosenzweig, D. H. Fairbrother, G. Orr and C. L. Haynes, Malic Acid Carbon Dots: From Super-resolution Live-Cell Imaging to Highly Efficient Separation, ACS Nano, 2018, 12(6), 5741–5752 CrossRef CAS.
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, Supramolecular-Enhanced Charge Transfer within Entangled Polyamide Chains as the Origin of the Universal Blue Fluorescence of Polymer Carbon Dots, J. Am. Chem. Soc., 2018, 140(40), 12862–12869 CrossRef CAS.
- D. Shen, Y. Long, J. Wang, Y. Yu, J. Pi, L. Yang and H. Zheng, Tuning the fluorescence performance of carbon dots with a reduction pathway, Nanoscale, 2019, 11(13), 5998–6003 RSC.
- Y. Fanglong, W. Zhibin, L. Xiaohong, L. Yunchao, T. Zhan'ao, F. Louzhen and Y. Shihe, Bright Multicolor Bandgap Fluorescent Carbon Quantum Dots for Electroluminescent Light-Emitting Diodes, Adv. Mater., 2017, 29(3), 1604436 CrossRef.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Synthesis of Carbon Dots with Multiple Color Emission by Controlled Graphitization and Surface Functionalization, Adv. Mater., 2018, 30(1), 1704740 CrossRef.
- Y. Jiao, X. Gong, H. Han, Y. Gao, W. Lu, Y. Liu, M. Xian, S. Shuang and C. Dong, Facile synthesis of orange fluorescence carbon dots with excitation independent emission for pH sensing and cellular imaging, Anal. Chim. Acta, 2018, 1042, 125–132 CrossRef CAS.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, Full-Color Light-Emitting Carbon Dots with a Surface-State-Controlled Luminescence Mechanism, ACS Nano, 2016, 10(1), 484–491 CrossRef CAS.
- K. Jiang, X. Feng, X. Gao, Y. Wang, C. Cai, Z. Li and H. Lin, Preparation of Multicolor Photoluminescent Carbon Dots by Tuning Surface States, Nanomaterials, 2019, 9(4), 529 CrossRef CAS.
- F. Jenkins, J. Robinson, J. Gellatly and G. Salmond, The no-effect dose of aniline in human subjects and a comparison of aniline toxicity in man and the rat, Food Cosmet. Toxicol., 1972, 10(5), 671–679 CrossRef CAS.
- G. Pedersen, J. Brynskov and T. Saermark, Phenol Toxicity and Conjugation in Human Colonic Epithelial Cells, Scand. J. Gastroenterol., 2002, 37(1), 74–79 CrossRef CAS.
- M. Xiang, Q. Dan, Y. Dongxue, N. Bing, Z. Yikang, F. Hongyou and S. Zaicheng, Synthesis of Carbon Dots with Multiple Color Emission by Controlled Graphitization and Surface Functionalization, Adv. Mater., 2018, 30(1), 1704740 CrossRef.
- K. Holá, M. Sudolská, S. Kalytchuk, D. Nachtigallová, A. L. Rogach, M. Otyepka and R. Zbořil, Graphitic Nitrogen Triggers Red Fluorescence in Carbon Dots, ACS Nano, 2017, 11(12), 12402–12410 CrossRef.
- H. Ding, S. B. Yu, J. S. Wei and H. M. Xiong, Full-Color Light-Emitting Carbon Dots with a Surface-State-Controlled Luminescence Mechanism, ACS Nano, 2016, 10(1), 484–491 CrossRef CAS.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. A. Tan, A. Chen, M. Jin and S. Yang, Engineering triangular carbon quantum dots with unprecedented narrow bandwidth emission for multicolored LEDs, Nat. Commun., 2018, 9(1), 2249 CrossRef.
- L. Xiao and H. Sun, Novel properties and applications of carbon nanodots, Nanoscale Horiz., 2018, 3(6), 565–597 RSC.
- M. Shamsipur, A. Barati, A. A. Taherpour and M. Jamshidi, Resolving the Multiple Emission Centers in Carbon Dots: From Fluorophore Molecular States to Aromatic Domain States and Carbon-Core States, J. Phys. Chem. Lett., 2018, 9(15), 4189–4198 CrossRef CAS.
- J. Zhu, X. Bai, X. Chen, H. Shao, Y. Zhai, G. Pan, H. Zhang, E. V. Ushakova, Y. Zhang, H. Song and A. L. Rogach, Spectrally Tunable Solid State Fluorescence and Room-Temperature Phosphorescence of Carbon Dots Synthesized via Seeded Growth Method, Adv. Opt. Mater., 2019, 7(9), 1801599 CrossRef.
- S. Hu, R. Tian, Y. Dong, J. Yang, J. Liu and Q. Chang, Modulation and effects of surface groups on photoluminescence and photocatalytic activity of carbon dots, Nanoscale, 2013, 5(23), 11665–11671 RSC.
- C. Reckmeier, J. Schneider, A. Susha and A. Rogach, Luminescent colloidal carbon dots: optical properties and effects of doping, Opt. Express, 2016, 24(2), A312–A340 CrossRef CAS.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Synthesis of Carbon Dots with Multiple Color Emission by Controlled Graphitization and Surface Functionalization, Adv. Mater., 2018, 30(1), 1704740 CrossRef.
- D. Bhattacharya, M. K. Mishra and G. De, Carbon Dots from a Single Source Exhibiting Tunable Luminescent Colors through the Modification of Surface Functional Groups in ORMOSIL Films, J. Phys. Chem. C, 2017, 121(50), 28106–28116 CrossRef CAS.
- W. Liu, C. Li, Y. Ren, X. Sun, W. Pan, Y. Li, J. Wang and W. Wang, Carbon dots: surface engineering and applications, J. Mater. Chem. B, 2016, 4(35), 5772–5788 RSC.
- X. Luo, W. Zhang, Y. Han, X. Chen, L. Zhu, W. Tang, J. Wang, T. Yue and Z. Li, N,S co-doped carbon dots based fluorescent “on-off-on” sensor for determination of ascorbic acid in common fruits, Food Chem., 2018, 258, 214–221 CrossRef CAS.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, Raman spectroscopy on amorphous carbon films, J. Appl. Phys., 1996, 80(1), 440–447 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Raman Spectra of Graphite Oxide and Functionalized Graphene Sheets, Nano Lett., 2008, 8(1), 36–41 CrossRef CAS.
- P. Tarakeshwar and S. Manogaran, Ground state vibrations of citric acid and the citrate trianion—an ab initio study, Spectrochim. Acta, Part A, 1994, 50(14), 2327–2343 CrossRef.
- J. Jehlička, H. G. M. Edwards and A. Culka, Using portable Raman spectrometers for the identification of organic compounds at low temperatures and high altitudes: exobiological applications, Philos. Trans. R. Soc., A, 2010, 368(1922), 3109–3125 CrossRef.
- K. Itoh and T. Shimanouchi, Vibrational spectra of crystalline formamide, J. Mol. Spectrosc., 1972, 42(1), 86–99 CrossRef CAS.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, Control of sp2/sp3 Carbon Ratio and Surface Chemistry of Nanodiamond Powders by Selective Oxidation in Air, J. Am. Chem. Soc., 2006, 128(35), 11635–11642 CrossRef CAS.
- M. Shams, L. M. Guiney, L. Huang, M. Ramesh, X. Yang, M. C. Hersam and I. Chowdhury, Influence of functional groups on the degradation of graphene oxide nanomaterials, Environ. Sci.: Nano, 2019, 6(7), 2203–2214 RSC.
- W.-C. Hou, I. Chowdhury, D. G. Goodwin, W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Photochemical Transformation of Graphene Oxide in Sunlight, Environ. Sci. Technol., 2015, 49(6), 3435–3443 CrossRef CAS.
- Z. Hens and J. C. Martins, A Solution NMR Toolbox for Characterizing the Surface Chemistry of Colloidal Nanocrystals, Chem. Mater., 2013, 25(8), 1211–1221 CrossRef CAS.
- S. S. Jones, P. Sahatiya and S. Badhulika, One step, high yield synthesis of amphiphilic carbon quantum dots derived from chia seeds: a solvatochromic study, New J. Chem., 2017, 41(21), 13130–13139 RSC.
- A. Pramanik, S. Biswas and P. Kumbhakar, Solvatochromism in highly luminescent environmental friendly carbon quantum dots for sensing applications: conversion of bio-waste into bio-asset, Spectrochim. Acta, Part A, 2018, 191, 498–512 CrossRef CAS.
- F. Arshad, A. Pal, M. A. Rahman, M. Ali, J. A. Khan and M. P. Sk, Insights on the solvatochromic effects in N-doped yellow-orange emissive carbon dots, New J. Chem., 2018, 42(24), 19837–19843 RSC.
- N. Basu and D. Mandal, Solvatochromic Response of Carbon Dots: Evidence of Solvent Interaction with Different Types of Emission Centers, J. Phys. Chem. C, 2018, 122(32), 18732–18741 CrossRef CAS.
- C. Reichardt, Solvatochromic dyes as solvent polarity indicators, Chem. Rev., 1994, 94(8), 2319–2358 CrossRef CAS.
- W. Kasprzyk, S. Bednarz, P. Żmudzki, M. Galica and D. Bogdał, Novel efficient fluorophores synthesized from citric acid, RSC Adv., 2015, 5(44), 34795–34799 RSC.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Hausler, P. Kasak, W. Schnick and A. L. Rogach, Aggregated Molecular Fluorophores in the Ammonothermal Synthesis of Carbon Dots, Chem. Mater., 2017, 29(24), 10352–10361 CrossRef CAS.
- H. Xu and D. Ren, Lysosomal physiology, Annu. Rev. Physiol., 2015, 77, 57–80 CrossRef CAS.
- B. Zhi, Y. Cui, S. Wang, B. P. Frank, D. N. Williams, R. P. Brown, E. S. Melby, R. J. Hamers, Z. Rosenzweig, D. H. Fairbrother, G. Orr and C. L. Haynes, Malic acid carbon dots: from super-resolution live-cell imaging to highly efficient separation, ACS Nano, 2018, 12(6), 5741–5752 CrossRef CAS.
- R. G. Parr and W. T. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- D. A. McQuarrie, Statistical Mechanics, University Science Books, 2000 Search PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(11), 7260–7268 CrossRef CAS.
- M. Gaus, A. Goez and M. Elstner, Parametrization and Benchmark of DFTB3 for Organic Molecules, J. Chem. Theory Comput., 2012, 9, 338–354 CrossRef.
- M. Gaus, Q. Cui and M. Elstner, DFTB-3: Extension of the self-consistent-charge density-functional tight-binding method SCC-DFTB, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS.
- Y. Sugita and Y. Okamoto, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- N. T. Maitra, Perspective: Fundamental aspects of time-dependent density functional theory, J. Chem. Phys., 2016, 144, 220901 CrossRef.
- V. Q. Vuong, J. A. Kuriappan, M. Kubillus, J. J. Kranz, T. Mast, T. A. Niehaus, S. Irle and M. Elstner, Parametrization and Benchmark of Long-Range Corrected DFTB2 for Organic Molecules, J. Chem. Theory Comput., 2018, 14, 115–125 CrossRef CAS.
- V. Lutsker, B. Aradi and T. A. Niehaus, Implementation and benchmark of a long-range corrected functional in the density functional based tight-binding method, J. Chem. Phys., 2015, 143, 184107 CrossRef CAS.
- J. J. Kranz, M. Elstner, B. Aradi, T. Frauenheim, V. Lutsker, A. D. Garcia and T. A. Niehaus, Time-Dependent Extension of the Long-Range Corrected Density Functional Based Tight-Binding Method, J. Chem. Theory Comput., 2017, 11, 1737–1747 CrossRef.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, CHARMM: The Biomolecular Simulation Program, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS.
- M. J. Frisch; G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, General Atomic and Molecular Electronic Structure System, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- Y. Nishimoto, Time-Dependent Long-Range-Corrected Density-Functional Tight-Binding Method Combined with the Polarizable Continuum Model, J. Phys. Chem. A, 2019, 123, 5649–5659 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05743f |
| ‡ Current address: Advanced Performance Material Lab, Research and Development Center, ZEON Corporation, Kanagawa 210-9507, Japan. |
| § These two authors contributed equally to this work. |
| ¶ Current address: MIT Media Lab, Cambridge, MA 02139, United States. |
| This journal is © The Royal Society of Chemistry 2021 |