
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bromodomain and BET family proteins as epigenetic targets in cancer therapy: their degradation, present drugs, and possible PROTACs
Mohd. Muddassir a,
Kunjal Sonib,
Chetan B. Sanganib,
Abdullah Alarifia,
Mohd. Afzala,
Naaser A. Y. Abduha,
Yongtao Duanc and
Poonam Bhadja*de
a,
Kunjal Sonib,
Chetan B. Sanganib,
Abdullah Alarifia,
Mohd. Afzala,
Naaser A. Y. Abduha,
Yongtao Duanc and
Poonam Bhadja*de
aDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, KSA
bShri Maneklal M. Patel Institute of Sciences and Research, Kadi Sarva Vishwavidyalaya University, Gandhinagar, Gujarat 382024, India
cHenan Provincial Key Laboratory of Children's Genetics and Metabolic Diseases, Zhengzhou Children's Hospital, Zhengzhou University, Zhengzhou 450018, China
dArthropod Ecology and Biological Control Research Group, Ton Duc Thang University, Ho Chi Minh City, Vietnam
eFaculty of Environment and Labour Safety, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: poonam.bhadja@tdtu.edu.vn
First published on 24th December 2020
Abstract
Alteration in the pattern of epigenetic marking leads to cancer, neurological disorders, inflammatory problems etc. These changes are due to aberration in histone modification enzymes that function as readers, writers and erasers. Bromodomains (BDs) and BET proteins that recognize acetylation of chromatin regulate gene expression. To block the function of any of these BrDs and/or BET protein can be a controlling agent in disorders such as cancer. BrDs and BET proteins are now emerging as targets for new therapeutic development. Traditional drugs like enzyme inhibitors and protein–protein inhibitors have many limitations. Recently Proteolysis-Targeting Chimeras (PROTACs) have become an advanced tool in therapeutic intervention as they remove disease causing proteins. This review provides an overview of the development and mechanisms of PROTACs for BRD and BET protein regulation in cancer and advanced possibilities of genetic technologies in therapeutics.
Introduction
Malignant tumours have been a leading global threat to human health for several decades. Research suggests that approximately 20 million new cases of cancer will be diagnosed every year.1 Notable improvements have been recorded in the field of cancer therapy which include inhibition and inhibitors, monoclonal antibodies, and immunotherapies. Small molecule inhibitors could bind tightly to the target protein to inhibit the enzyme activities and induce cell cycle arrest or apoptosis. However, a target protein within tumour cells tends to restore its activity which leads to acquiring drug resistance by overexpressing or mutations in the target protein.2 Antibody therapies are more and more popular with the advantage of prolonged pharmacokinetic profile and high binding affinity to targets. The main therapeutic route for antibodies is to interrupt the interaction between extracellular protein and protein or ligand. Also, a series of challenges that have to face include poor membrane permeability, enteral administration, and high cost.3–5 RNA interfering molecules often achieve exciting activity to their target protein. Given the catalytic nature, RNAi could work at low exposures because of each siRNA molecule degrading a lot of mRNA transcripts. The shortcomings of current RNAi therapy not only include a lack of oral bioavailability but also poor PK.6 And more, it must be pointed out that the treatment of cancer needs a range of therapeutic strategies. A desirable molecule would be with several combined advantages from the small molecule, RNAi modalities, and antibody such as high selectivity, oral bioavailability, and distributing well into the central nervous system (CNS).7 In the past two decades, more and more researchers have devoted themselves to exploring an effective therapeutic strategy by the regulation of protein levels to modulate protein function. Some small molecules that control protein expression levels instead of affecting protein function have recently been brought into focus. There is no doubt that the most representative compounds of this kind of molecules are proteolysis-targeting chimeric molecules (PROTAC).2,8–11PROTAC is a strategy to target specific proteins and induce their intracellular degradation. Professor Cruise of Yale University was one of the pioneers in the field related to PROTAC.12 Protein knockout induced by PROTAC technology displayed unique advantages over traditional drugs.13 On the one hand, PROTACs may achieve higher potency and efficacy compared to traditional small molecule drugs in vivo.14 Small molecule inhibitors bind to target proteins to achieve an ideal level of therapeutic effects that often-required higher doses and sustained exposure to the target. In contrast, a low dosage of PROTAC could induce tumour regression because of its mechanism which is chemical knockdown rather than by inhibition. On the flip side, the bright calibre to conquer drug resistance that originated through mutation in amino acid binding site.15
The constancy of the intracellular domain is maintained in many ways. One of the main ways is the ubiquitin protease system (UPS, Fig. 1) where PROTAC could bring about targeted protein degradation.10
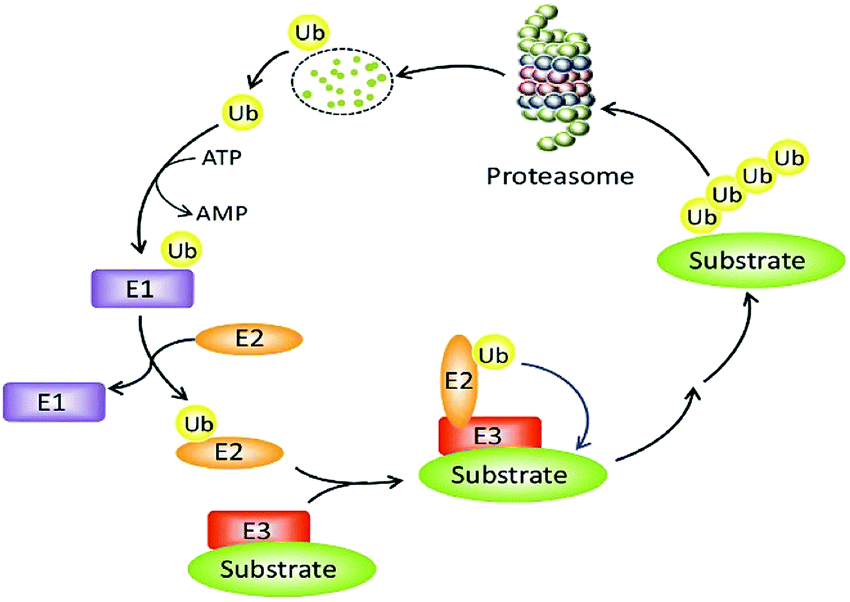 | ||
| Fig. 1 The ubiquitin–proteasome system. | ||
Ubiquitin, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3), proteasome, target proteins all together develop UPS.16 The UPS has an essential role in several vital processes as the protein targets may be cell cycle and apoptosis regulators, transcription factors that regulate cell division and differentiation, growth, signal transduction, and stress response.17
Within the UPS a polypeptide that acts as a molecular label, ubiquitin (Ub) has 76 amino acids in length.18 With seven lysine residues, each UB polypeptide is interacting in multiple Ub polypeptides linking that ultimately forms a chain of polyubiquitin.19 The final fate of protein is decided by the dissemination of ubiquitination patterns, for example, endocytosis, protein sorting, nuclear export of proteins, DNA repair, and transcription regulation have been connected with mono-ubiquitination. Polyubiquitination has been linked to protein degradation, DNA repair, kinase activation, and transcription factor activation.
The formation of protein ubiquitination incorporates three basic modes. Firstly, an E1 ubiquitin-activating enzyme activates Ub at its C-terminus. In the second step, an E2 ubiquitin-conjugating enzyme does conjugation of Ub and in the final third step, an E3 ubiquitin ligase transfers Ub to the substrate protein.20–22
For initiation of proteasomal degradation of a target protein, Ub is one of the vital appliances, although ubiquitin-independent mechanisms have also been reported.23
In 2000, Zhou et al. narrated that by engineered E3 ligases, stable cellular proteins can be degraded in yeast as well as in mammalian cells that leads to PROTAC development.24
Recently, PROTAC has been utilized and developed to target epigenetic proteins. DNA methylation, histone modifications, and chromatin remodelling like epigenetic processes have been affected through many environmental and genetic factors that furnish disease progression.25,26 These processes are being targeted for unique drug development through epigenetic enzymes, known as readers, writers, and erasers.27 Epigenetic investigation and experimentation have tremendous potential in the development of remedy in broad-spectrum disease and oncology. Many small molecules controlling epigenetic mechanisms are known as promising therapeutic agents. Different epigenetic mechanisms have been explored in the past decades such as covalent modifications, RNA transcripts, and nucleosome positioning.28–30 Enzymatic chemical modification or recognition of DNA/histone proteins including regarded as the representative of covalent modifications play central roles in many types of epigenetic inheritance. Several epigenetic protein inhibitors such as vorinostat and azacytidine have been approved for cancer treatment by the FDA.31,32 Except for epigenetic inhibitors, some PROTAC target epigenetic proteins have been reported by hijacking the UPS which may be an efficient strategy.33,34
PROTAC technology; its development and progress in contrast to epigenetic targets, further scope, and provocation of this advanced passage in the application for treatment are highlighted in the current review (Fig. 2).
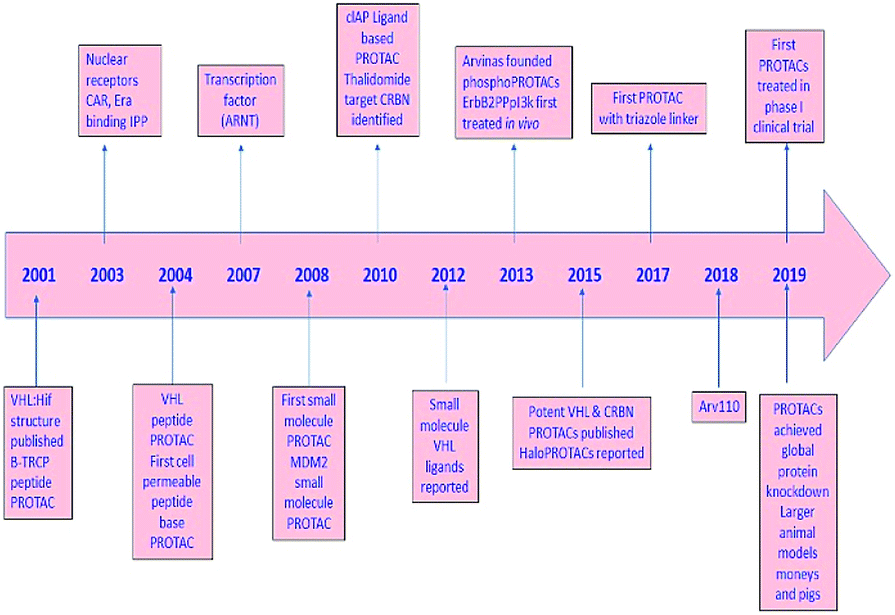 | ||
| Fig. 2 A diagram to demonstrate the development of PROTAC. | ||
BET protein family
In an organism, many different cells present with the same DNA sequences but they are programmed in such a way that they can do distinctive biological functions and retain different phenotypes for the same. The process is identified as cell differentiation and can be obtained through epigenetics.35,36The structurally flexible N- and C-termini of the core histone octamers within chromatin extended out form nucleosomes. They have vast possibilities of post-translational modifications.37 In addition to alterations in DNA methylation, histones with covalent modification are vital apparatus for the epigenetic panorama. Phosphorylation, acetylation, methylation, ubiquitination, and SUMOylation are several kinds of modifications that can be available on histones.37–39
In the cell, for genomic stability integration and gene expression or repression, these sites, and state-specific alterations may act conjointly.39–41
In the human genome changes in DNA and histone proteins comprising chromatin structure are intently connected with gene transcriptional activation or repression. The post-translational modifications of DNA-packaging histones available with the chromatin shaped this complicated and firmly harmonized alliance.42
In cancer, the normal pattern of histone modifications is altered under enzyme deregulations that modify addition, removal, or alters identification of histone markings and mutations.43
The information about ∑-N-acetylation of lysine residues (Kac) on histone tails is connected with an open chromatin engineering and transcriptional activation,44 despite several acetylation marks that have been correlated in place of chromatin compaction45 and with other mechanisms such as, DNA repairing, protein–protein interactions, protein stability, and metabolism46 was discovered about 30 years back.
The highly vigorous alteration, lysine acetylation mainly affects chromatin structure and function and even gene transcription.47–49 Besides, acetylation of lysine has not been limited to histones but it can also be there on various kinds of transcription-associated proteins, which include histone altering enzymes, transcription factors along with chromatin regulators indicating that it may impact as more common protein function regulators above transcriptional governance agnate to phosphorylation.50–52 Acetylation of lysine is one of the essential alterations taking place in histone tails and ambiance of histone code has been extensively investigated.53
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate ∑-N-acetylation of lysine molecules at the amino-terminal end of histones. The previous one is known as “writer”, as it does the addition of acetyl group, while the latter has the function of removing acetyl markings, known as ‘erasers'. In cancers, these enzymes are definitely present having the discomfort of mutations and have chances of other free trade mechanisms.53
The regulation of gene transcription has been done by Bromodomains (BRDs) as they are recognizing this acetyl marking present in histone tails that have been targeted by chromatin-modifying enzymes and other proteins that are site-specific for chromatin.53 Bromodomains (BRDs) are known as “readers” as recognizing this acetyl marking in histone tails (Fig. 3).53
 | ||
| Fig. 3 Overview of bromodomain inhibition. | ||
‘Readers’ of epigenetic marks are structurally diverse proteins each possessing one or more evolutionarily conserved effecter modules, which recognize covalent modifications of histone proteins or DNA.54
The ∑-N-acetylation of lysine molecules can only be specifically verified by Bromodomains (BRD).54
The Bromo- and Extra-terminal (BET) family of proteins, including the ubiquitously expressed BRD2, BRD3, and BRD4 and the testis-specific BRDT, recruit transcriptional regulatory complexes to acetylated chromatin thereby controlling specific networks of genes involved in cellular proliferation and cell cycle progression.55
Alterations in regulation of activities from BET protein, especially BRD4, have been greatly allied with cancer and inflammatory diseases. This makes BET protein as an appealing drug targets.56
Bromodomains
The histone tails having ∑-N-acetylation on lysine molecules have been determined by bromodomains (BDs), as they perform work of reading of acetylated lysine molecules.The bromodomains consist of about 110–120 residues and are structurally conserved. The BDs are present in many chromatin-associated factors, including nuclear histone acetyltransferases (HATs), chromatin remodelling factors, and bromodomains and extra terminal (BET) domains family nuclear proteins (Fig. 4).57
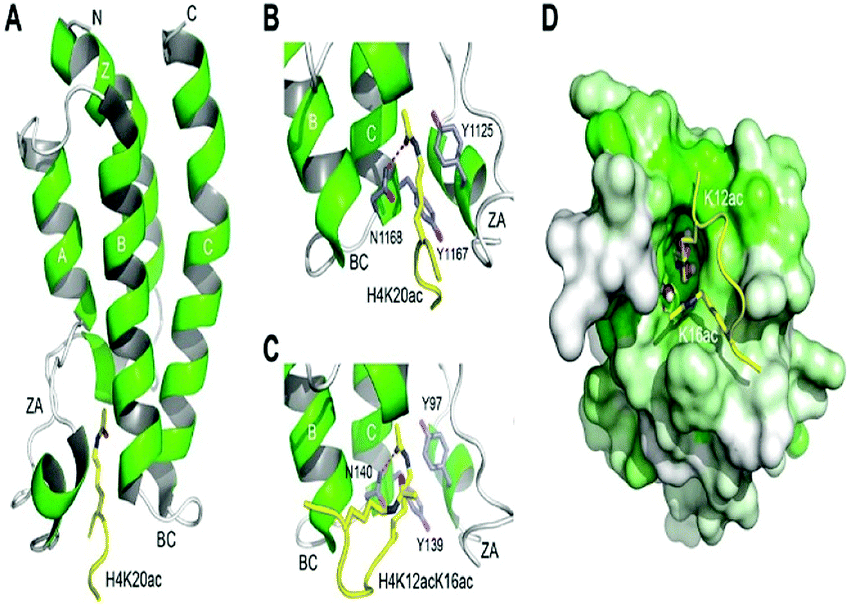 | ||
| Fig. 4 The structural features of the bromodomain as the acetyl-lysine binding domain. | ||
The dynamic role of lysine acetylation is, to some extent, attributed to the bromodomain (BRD), which is the only protein domain whose conserved activity is to function as an acetyl-lysine binding domain.42,57
Several BrD-embracing proteins have been depicted incriminating during disease processes such as cancers, inflammation, and viral replication.42,59–62 In recent years, inhibitors of BrDs based on small molecules have allowed many chemical biology-based investigations for processes of BrDs and resolutely recommend that they can be legitimate drug targets in numerous diseases of human.42,62,63
In the early 1990s, the transformatively preserved pattern of the bromodomain family was distinguished initially in the Brahma gene of Drosophila melanogaster.64
In the human proteome 46 different proteins having a total of 61 bromodomains as per the studies reported.65 Based on their structural arrangement they are grouped into eight subfamilies (Fig. 5).53,65
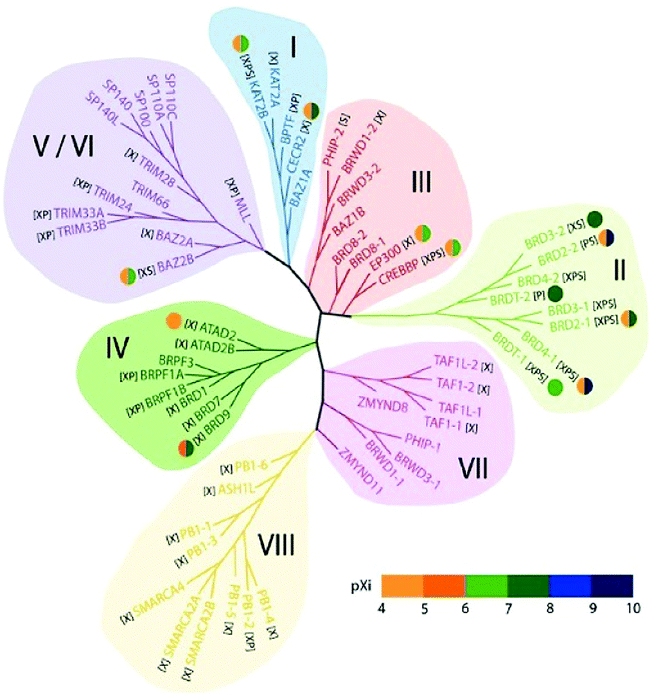 | ||
| Fig. 5 Structure-based phylogeny of the human bromodomains and their inhibitors. | ||
These 46 varying proteins have total 61 BRDs, existing as co-regulators in transcription and in enzymes that do chromatin modification, such as HATs and HAT connected proteins (GCN5, PCAF, BRD9),66,67 chromatin remodelling complexes that are ATP-dependent (BAZ1B),67 helicases (SMARCA),69 SET domain-containing methyl-transferases (MLL and ASH1L),70,71 co-activators of transcription (TRIM/TIF1)72 and mediators (TAF1),73 nuclear scaffolding proteins (PB1)74 and the BET family (Table 1).69,75
| Name | Synonyms | Name | Function | # BrDs |
|---|---|---|---|---|
| ASH1L | ASH1, KMT2H, KIAA1420 | Absent small and homeotic disks protein 1 homolog | Histone-lysine methyltransferase | 1 |
| ATAD2 | ANCCA | ATPase family AAA domain-containing protein 2 | Transcriptional regulator | 1 |
| ATAD2B | KIAA1240 | ATPase family AAA domain-containing protein 2B | Unknown | 1 |
| BAZ1A | ACF1, WCRF180, hWALp1 | Bromodomain adjacent to zinc finger domain protein 1A | Chromatin-remodelling factor | 1 |
| BAZ1B | WBSC10, WBSCR10, WBSCR9, WSTF | Bromodomain adjacent to zinc finger domain protein 1B | Tyrosine-protein kinase; transcriptional regulator | 1 |
| BAZ2A | KIAA0314, TIP5 | Bromodomain adjacent to zinc finger domain protein 2A | Transcriptional repressor | 1 |
| BAZ2B | hWALp4, KIAA1476 | Bromodomain adjacent to zinc finger domain protein 2B | Unknown | 1 |
| BPTF | FAC1, FALZ | Bromodomain and PHD finger- containing transcription factor | Chromatin-remodelling factor | 1 |
| BRD1 | BRL, BRPF2 | Bromodomain-containing protein 1 | Transcriptional regulator | 1 |
| BRD2 | KIAA9001, RING3 | Bromodomain-containing protein 2 | Transcriptional regulator | 2 |
| BRD3 | KIAA0043 RING3L | Bromodomain-containing protein 3 | Transcriptional regulator | 2 |
| BRD4 | HUNK1 | Bromodomain-containing protein 4 | Transcriptional regulator | 2 |
| BRD7 | BP75, CELTIX1 | Bromodomain-containing protein 7 | Transcriptional regulator | 1 |
| BRD8 | SMAP, SMAP2 | Bromodomain-containing protein 8 | Transcriptional regulator | 2 |
| BRD9 | Bromodomain-containing protein 9 | Unknown | 1 | |
| BRDT | Bromodomain testis-specific protein | Chromatin-remodelling factor | 2 | |
| BRPF1 | BR140, Peregrin | Bromodomain and PHD finger- containing protein 1 | Transcriptional activator | 1 |
| BRPF3 | KIAA1286 | Bromodomain and PHD finger- containing protein 3 | Transcriptional regulator | 1 |
| BRWD1 | C21 or f107, WDR9 | Bromodomain and WD repeat-containing protein 1 | Chromatin remodelling factor | 2 |
| BRWD3 | Bromodomain and WD repeat-containing protein 3 | JAK/STAT signalling | 2 | |
| CECR2 | KIAA1740 | Cat eye syndrome critical region protein 2 | Chromatin remodelling factor | 1 |
| CREBBP | CBP, KAT3A | CREB-binding protein | Histone acetyltransferase | 1 |
| EP300 | P300, KAT3B | E1A-associated protein p300 | Histone acetyltransferase | 1 |
| KAT2A | GCN5, GCN5L2, HGCN5 | General control of amino acid synthesis protein 5-like 2 | Histone acetyltransferase | 1 |
| KAT2B | PCAF | P300/CBP-associated factor | Histone acetyltransferase | 1 |
| MLL | KMT2A, ALL1, CXXC7, HRX, HTRX, MLL1, TRX1 | Myeloid/lymphoid or mixed-lineage leukaemia | Histone methyltransferase | 1 |
| PB1 | PBRM1, BAF180 | Polybromo-1 | Transcriptional regulator | 6 |
| PHIP | WDR11 | PH-interacting protein | Insulin signalling | 2 |
| SMARCA2 | BAF190B, BRM, SNF2A, SNF2L2 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2 | Chromatin remodelling factor | 1 |
| SMARCA4 | BAF190A, BRG1, SNF2B, SNF2L4 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 4 | Chromatin remodelling factor | 1 |
| SP100 | Nuclear autoantigen Sp-100 | Transcriptional regulator | 1 | |
| SP110 | Sp110 nuclear body protein | Transcriptional regulator | 1 | |
| SP140 | LYSP100 | Nuclear body protein SP140 | Transcriptional regulator | 1 |
| SP140L | LOC93349 | Nuclear body protein SP140-like protein | Unknown | 1 |
| TAF1 | BA2R, CCG1, CCGS, TAF2A, TAF(II)250 | Transcription initiation factor TFIID subunit 1 | Transcription initiation | 2 |
| TAF1L | TAF(II)210 | Transcription initiation factor TFIID subunit 1-like | Transcription initiation | 2 |
| TRIM24 | RNF82, TIF1, TIF1α | Transcription intermediary factor 1-alpha | Ubiquitin E3 ligase, transcriptional regulator | 1 |
| TRIM28 | KAP1, RNF96, TIF1β | Transcription intermediary factor 1-beta | SUMO E3 ligase, transcriptional regulator | 1 |
| TRIM33 | KIAA1113, RFG7, TIF1γ | Transcription intermediary factor 1-gamma | Ubiquitin E3 ligase, transcriptional regulator | 1 |
| TRIM66 | C11orf29, KIAA0298 | Tripartite motif-containing protein 66 | Transcriptional repressor | 1 |
| ZMYND8 | KIAA1125, PRKCBP1, RACK7 | Zinc finger MYND domain-containing protein 8, protein kinase C-binding protein 1 | Transcriptional regulator | 1 |
| ZMYND11 | BS69 | Zinc finger MYND domain-containing protein 11 | Transcriptional repressor | 1 |
In the first subfamily (I) proteins having acetyl-transferase P300/CBP-associated factor (PCAF),67 amino-acid synthesis general controller 5-like 2 (GCN5L),67 Fatal Alzheimer antigen (FALZ)68 a transcription factor, and cat-eye syndrome chromosome region 2 (CECR2)76 a chromatin remodelling factor all included and present in the nucleus (Table 1).
The subfamily (II) carries bromo and extra terminal (BET) proteins of BRDs, that have a common structural arrangement holds two N-terminal BRDs exhibiting high levels of sequence sustention and also have an extra-terminal (ET) domain and anomalous C-terminal recruitment domain.
BRD2,76 BRD3,77 BRD4,78 and BRDT79 are the four proteins that are included in this subfamily. Intriguingly BET proteins during mitosis recruited on the transcription starting sites80–82 and BRD4, a BET protein has been reported to lead the positive transcription elongation factor (P-TEFb) utilizing specific towards the C-terminus towards the site of transcription.54
The 8B (BRD8B)83 containing transcription regulatory bromodomain, binding protein (CREBBP) and E1A binding protein p300 (EP300)84 having HAT enzymes, the c-terminal domain of chromatin remodelling factors WD repeat domain 9 (WDR9 domain 2),85 adjoining to zinc finger domain 1B (BAZ1B)68 bromodomain, bromodomain-containing protein messing up in leukaemia (BRWD3 domain 2)86 associated with the C-terminal domain of the JAK/STAT pathway and pleckstrin homology domain interacting protein (PHIP domain 2)54,87 connected with the C-terminal domain of the insulin signalling are members of subfamily III of BRDs.
In the subfamily IV bromodomain-containing protein 7 (BRD7)88 a transcription regulator, bromodomain-containing protein 1 (BRD1)78 and (BRPF1)89 composing of PHD finger-containing protein 1, (ATAD2),90 having two AAA domain-containing protein, along with the KIAA1240 protein (KIAA1240) (BRD9) having the KIAA1240 protein (KIAA1240) and (BRPF3)54 that has the bromodomain and PHD finger containing protein 3.
In subfamily V the transcription repressor tripartite motif-66 (TRIM66),91 the tripartite motif-33(TRIM33),92 the regulator of transcription, transcription intermediary factor 1 (TIF1),93 transcription regulators nuclear auto-antigen Sp-100 (SP100),94 antigen Sp-110 (SP110)95 and SP140 nuclear body protein (SP140),96 along with the SP140-like protein (LOC93349), transcription repressing bromodomain contiguous to zinc finger domain 2A (BAZ2A)97 and 2B (BAZ2B)54,98 are categorized in human BRDs.
In subfamily VI of human BRDs acetyl-lysine related interactions have not been found in connection to histone or any other proteins. Subfamily VI has histone methyl-transferase myeloid/lymphoid or mixed-lineage leukaemia (MLL)99 and transcription co-regulating tripartite motif-28(TRIM28).54,100
The transcription repressing zinc finger MYND domain having 11 protein (ZMYND11),101 transcription initiating factors RNA polymerase II TATA box binding protein (TBP)-associated factor (TAF1)102 TAF1-like (TAF1L),103 and the N-terminal BRDs for chromatin remodelling factor WD R9 (WDR9 domain 1),85 the JAK/STAT pathway connected with bromodomain based protein, disorganized during leukaemia (BRWD3 domain 1)86 and insulin signal relevant pleckstrin homology domain interacting protein (PHIP domain 1)54,87 have been grouped in subfamily VII.
Human bromodomain subfamily VIII the last group carries the methyl-transferase ash1 (absent, small, or homeotic)-like (ASH1L),71 chromatin remodelling factor SWI/SNF linked chromatin regulator a2 (SMARCA2)104 that is actin-based and associated with the matrix, chromatin regulator a4(SMARCA4)105 along with the Polybromo 1 (PB1).73,106
BET bromodomains
The family of bromodomain and extra terminal (BET) proteins has been deeply studied. The family includes BRD2, BRD3, BRD4, and BRDT. All of these are ubiquitously expressed, however, BRDT is only expressed in testis.53In cancer, deep associations of BET proteins have been there as they promptly regulate several cancer-related gene expressions, such as c-MYC.53,107
These BET proteins also serve in the regulation of the cell cycle. BRD4 is crucial for the regulation of gene expressions in M to initial G1 phase progression, whereas BRD2 prepares a scaffold on the chromatin that raises the indispensable cell-cycle transcription regulation genes E2F1 and E2F2 (Fig. 6) (Table 2).53
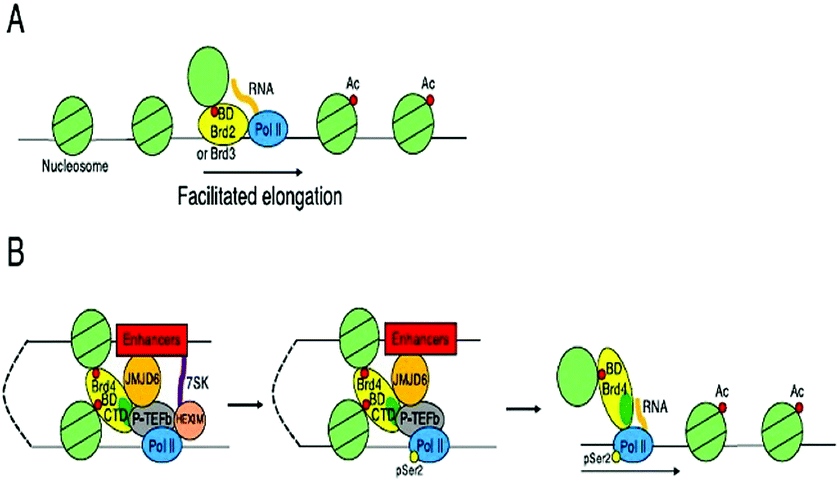 | ||
| Fig. 6 Transcriptional control by BET proteins. | ||
| BET protein | Functions | References |
|---|---|---|
| BRD2 | − Promotion of E2F-dependent cell cycle progression in HeLa and HEK293 cells | 110 and 111 |
| − Closure of the neural tube in mouse embryos | 112 and 113 | |
| − Maintenance of the number of GABAergic neurons in the neocortex and the striatum of mice | 114 | |
| − Assist of transcription in hyperacetylated chromatin (Property of histone-chaperone) | 77 | |
| − Transcriptional activation of HOXA11 and D11 in HEK293 cells | 115 | |
| − Enhancement of GATA1-mediated erythroid gene activation | 116 | |
| − Interaction with LANA of KSHV that mediates episomal replication and persistence of viral genomes | 117 and 118 | |
| BRD3 | − Assist of transcription in hyperacetylated chromatin (Property of histone-chaperone) | 77 |
| − Transcriptional activation of HOXB3, B4, B5, B6, C8, C9, C10, A3, A5, A6, and A7 in HEK293 cells | 115 | |
| − Enhancement of GATA1-mediated erythroid gene activation | 116 | |
| − Carcinogenesis induced by BRD3-NUT fusion protein | 119 | |
| BRD4 | − Stimulation of G2/M transition in HeLa cells | 80 |
| − Cell cycle progression in P19 embryonal carcinoma cells | 81 | |
| − Maintenance of inner cell mass in mouse blastocysts | 120 | |
| − Transcriptional activation of Nanog required for maintaining the pluripotency of ES cells | 121 | |
| − Release from a pause in transcription elongation | 122 and 123 | |
| − Assist of transcription in hyperacetylated chromatin (Property of histone-chaperone) | 124 | |
| − Transcriptional activation of c-Myc and Klf4 in NIH3T3 cells | 124 | |
| − Transcriptional activation of HOXB2, B3, B4, B5, B6, B7, B8, A4, and C5 in HEK293 cells | 115 | |
| − Transcriptional regulation of genes involved in learning and memory in mice | 125 | |
| − Enhancement of INF-induced gene transcription | 126 | |
| − Signal transducer of the cellular response to oxidative stress | 127 | |
| − Prevention of splicing inhibition in heat stress-induced cells | 128 | |
| − A gene bookmark for transcriptional reactivation in post-mitotic cells | 129 and 130 | |
| − Carcinogenesis induced by BRD4-NUT fusion protein | 119 and 131 | |
| − Interaction with LANA of KSHV that mediates episomal replication and persistence of viral genomes | 132 and 133 | |
| − Tethering of BPV genome to host mitotic chromosomes | 134 | |
| − Transcriptional regulation of E2 that mediates episomal maintenance and DNA replication of HPV genome | 135–137 | |
| BRDT | − Transcriptional regulation of genes responsible for meiotic progression during spermatogenesis | 126 |
| − Splicing machinery in testicular cells | 138 | |
| − Chromatin remodelling in MEL, 3T3, and COS7 cells | 139–141 | |
| − Histone replacement at post-meiotic stages during spermatogenesis | 125 |
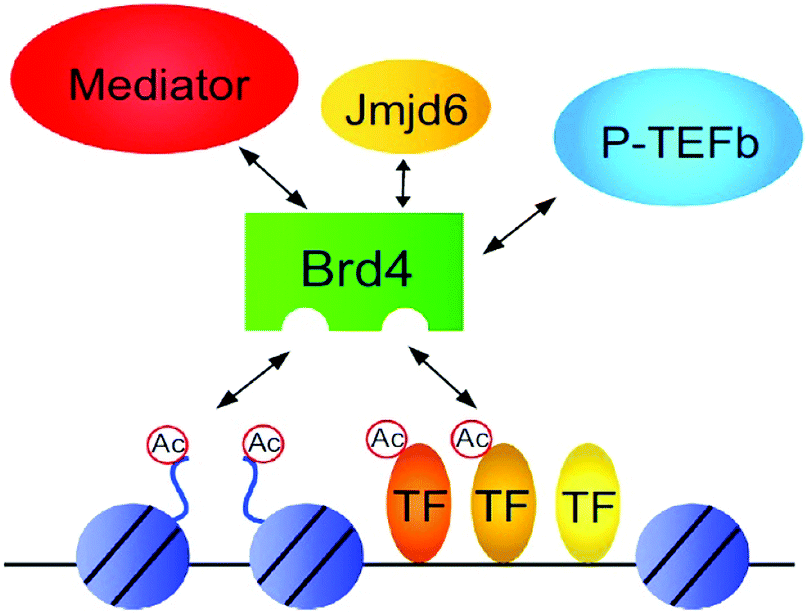
BRD4 is a universal gene transcription regulator so the inhibition of BRD4 would be predicted to attempt universal down-regulation of gene functionality. Inhibition of BRD4 is of prime importance as it is regulating several hundred genes essential for tumorigenesis (Fig. 7–10).53
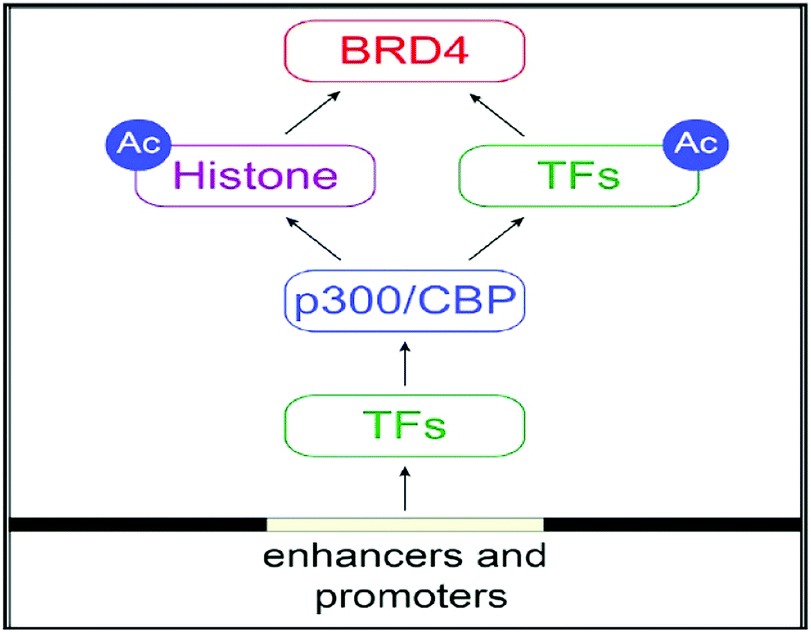 | ||
| Fig. 7 The BET protein BRD4 required for the functional output of an ensemble of lineage-specific transcription factors. | ||
 | ||
| Fig. 8 Binding to acetylated histones. | ||
 | ||
| Fig. 9 Protein–protein Interactions. | ||
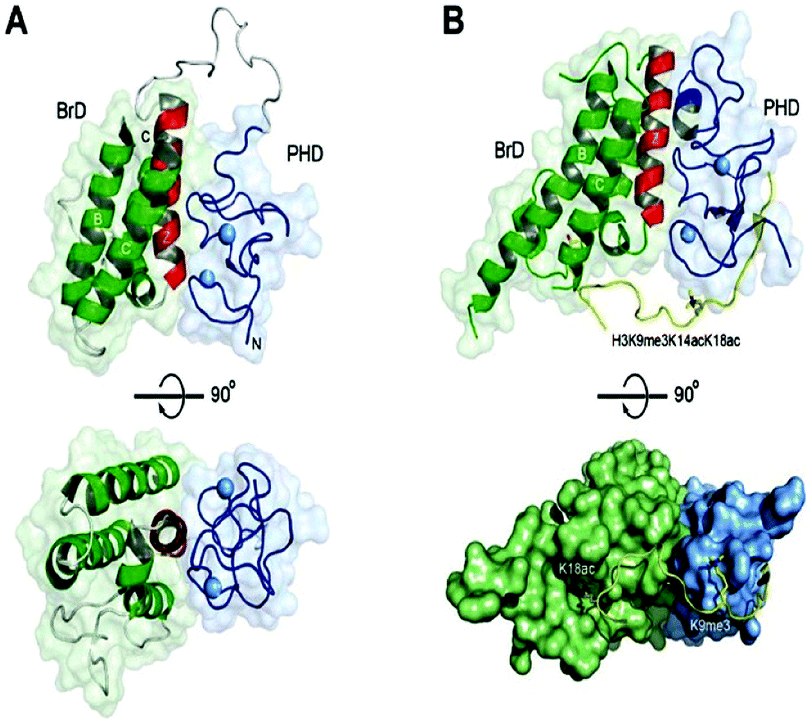 | ||
| Fig. 10 Structures of tandem modules of epigenome reader domains. | ||
Other bromodomain proteins (BRD9 and TRIM24)
As transcriptional co-activators such as tripartite motif-containing proteins (TRIMS) and TBP-associated factors (TAFs) bromodomains show their presence.53An identified chromatin-remodelling BAF SNF/SWI complex component is (BRD9) bromodomain-containing protein 9. Less information has been available about the functionality of it however essentiality of it in cancer has been documented. Recently, in a study, it has been described that for sustaining MYC transcription AML cells need BRD9 and through that proliferation has been increased. Almost similar to BTD9 bromodomain-containing protein 7 (BRD7) is also a subunit of PBAF SWI/SNF. In several reports, it has been documented that as a tumour suppressor gene, BRD7 either partially or completely downregulate several cancers such as small-cell lung cancer, ovarian, colorectal and breast cancers, endometrial carcinoma, and hepatocellular carcinoma where it is part of BRCA1.54
An epigenetic way has been proposed to attempt CRPC for bromodomain and extra-terminal (BET) family protein suppression. In tumour models of CRPC, growth retardation has resulted through BET inhibitors.142–144
The selectively binding to acetylated lysine is done by bromodomain family proteins, the third type of proteins that do epigenetic regulation and based on that acts as “readers” of the acetylated lysine.58
A subset of 46 bromodomain-containing proteins available only in the genome of human.145 Bromodomain containing protein 2 (Brd2), Brd3, Brd4, and testis-specific protein (BrdT) all four together constitute BET protein family.
BRD4 through binding with Kac residues present on histone tails regulates gene expression. This regulation is done by recruitment of positive transcription elongation factor b (p-TEFb) on (RNA pol II) RNA polymerase II enzyme having phosphorylation.108,109
BET proteins, especially Brd4 deregulated and this has been involved in diverse diseases, such as cancer formation and progression. Zuber et al. disclosed that in the preservation of c-Myc gene expression and stimulation of deviant self-renewal of AML cells, Brd4 has a vital role.146
HDAC family
The enzyme class that has histone deacetylases (HDAC), doing acetyl groups (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3) removal from an ε-N-acetyl-lysine amino acid located on histone, permits tighter DNA wrapping by histones.52
C–CH3) removal from an ε-N-acetyl-lysine amino acid located on histone, permits tighter DNA wrapping by histones.52
From yeast originated enzymes based on their homology of sequence and organization of domain, HDAC thus grouped into four classes, class I, II, III, and IV.244
The class of HDACs that consists of a zinc-dependent active site and can be controlled by trichostatin A (TSA) is designated as a “classical” family of HDAC that has class I, II, and IV. On the other hand, class III of the HDAC enzyme family have sirtuins that can be impacted by TSA as they are NAD+-dependent proteins.147 From the yeast-based reports, the homologous of these three classes of HDACs have been names as reduced potassium dependency 3 (Rpd 3) that correlated with the group I, class II connected with histone deacetylase 1 (HDAC1), and class III enzymes interrelated with silent information regulator (Sir2). With only one isoform (HDAC11) that is not truly homologous with any of Rpd3 or HDAC1 enzymes of yeast so HDAC11 has been assigned to its class IV. The class III enzymes have the deviating mode of action and are NAD+-dependent since other classes of enzyme HDACs are dependent on Zn2+, a cofactor.148,149
Present epigenetic target drugs (inhibitors and degraders)
The resultant effect of Brd4 knockdown through shRNAs or by small-molecule based pharmacologic suppression of Brd4 showed consecration of terminal differentiation and eradication of leukemic stem cells. It has also been reported about effective anti-leukaemia potential in numerous AML cell lines and initial patient-derived cells from human.146,150The expansion of tamoxifen-resistive breast cancer cells can effectively be retarded by BET protein suppression as reported by Malley et al.151 In melanoma types of cancer growth, Brd4 has been found highly-strung even at initial and metastatic tissues that have melanoma phase. Prompt inhibition of key cell-cycle genes, having SKP2, ERK1, and c-Myc, can be achieved through Brd3 inhibitor therapy. In vitro melanoma cell spread and in vivo tumour development and metastatic representation has been effectively attenuated through Brd4 inhibitor therapy. The impactive anti-leukemic properties of brd4 inhibitor mediated suppression have been epitomized by the silencing of Brd4 on an individual basis.152
Brd4 silencing can further be recognized Brd4 as a target for therapeutic designing and leads to emphasis discoveries for validation of Brd4 as a druggable target. Two immensely homologous bromodomains on amino-terminal loci absolute assignment of nucleosomes by attaching at distinct acetylated lysines (Kac) on histone tails are critical for the functioning of BET proteins.68
Brd4 inhibitors
Based on interactive modules between BDs and inhibitors two classes of Brd4 inhibitors constituted: monovalent and bivalent. Each bromodomain of Brd4 protein has been separately targeted for binding by valent type Brd4 inhibitors while both bromodomains concurrently joined with bivalent Brd4 inhibitors as they have such proficiency.152(a) Monovalent Brd4 inhibitors Fig. 11(a–h).
 | ||
| Fig. 11 Monovalent Brd4 inhibitors. | ||
(i) Triazolo azepine-based Brd4 inhibitors.153–165
(ii) Isoxazole-based Brd4 inhibitors.70,106,142,153–155,166–179
(iii) Pyridone-based Brd4 inhibitors.174,180–185
(iv) Tetrahydroquinoline-based Brd4 inhibitors.143,186–191
(v) Triazolo pyrazine-based Brd4 inhibitors.192–194
(vi) 4-Acyl pyrrole-based Brd4 inhibitors.195–197
(vii) 2-Thiazolidinone-based Brd4 inhibitors.198
(viii) Other reported inhibitors.199–204
(b) Bivalent Brd4 inhibitors (Fig. 12).205–210
 | ||
| Fig. 12 Bivalent Brd4 inhibitors. | ||
 | ||
| Fig. 13 Brd4 degraders. | ||
Brd4 degraders (Fig. 13)
Based on recent pieces of work it is reported that Brd4 inhibitors prompt settlement in terms of Brd4 protein accumulation during several types of cancers such as lung and prostate cancer and Burkitt's lymphoma due to unimpacted c-Myc retardation, tuned apoptotic initiation, and antiproliferative processes,211,212 although Brd4 inhibitors have shown their auspicious capabilities in numerous C-Myc-driven malignancies. Besides, drug resistivity against triazolo azepine based Brd4 inhibitors I-BET762 and (+)-JQ1 has also been demonstrated.213In consideration of the channel between cancer and Brd4 expression, Brd4 has been recognized as a bright therapeutic target in various kinds of malignancies.214,215 Notable attempts have been invented to flourish pharmacological inhibitors of Brd4 and several Brd4 inhibitors have upgraded to clinical and preclinical assessment.216,217
HDAC inhibitors
Valproic acid-like histone deacetylase inhibitors (HDIs) have been utilized since long as mood-stabilizing agents and antiepileptics drugs in neurological disorders and psychiatric therapeutics.218,219Histone deacetylase inhibitors (HDIs) have a long history of use in psychiatry and neurology as mood stabilizers and anti-epileptics, for example, valproic acid.218,219 At the current time phase, numerous endeavours have been done to establish HDIs as a cancer remedy. In 2006, Vorinostat (SAHA) got permission to use in the treatment of cutaneous instances in patients that have concomitant T cell lymphoma and have been failed to be cured in earlier therapies.220,221
Another HDI, istodax has been endorsed in 2009 for the patients with CTCL4. The exact mode of actions of these molecules that may trade is uncertain, perhaps epigenetic pathways are intended. Further, the effectiveness of valproic acid on the latent pools of HIV that are influenced in persons is under clinical investigation.222 HDIs at present are also being reviewed as chemosensitizers for radiation or cytotoxic chemotherapy, or in companionship with DNA methylation inhibitors-based harmony in vitro phase. Non-histone proteins linked to acetylation and can shift the degree of acetylation can be affected by HDI molecules and thus step up or down their activity.
SIRT2, as a member of the Class III HDAC family, is an NAD+-dependent enzyme. It could interact with a range of proteins and then remove acyl groups which played an important role in many cellular functions.223
The development of PROTACs
During the last two decades, a good sort of work has been done in the field of PROTAC development. Initially described by Craig Crews and Ray Deshaies in 2001, MetAP-2 was degraded by protein targeting chimeric molecule 1 (Protac-1) which recruited MetAP-2 to SCF binding phospho-peptide and small-molecule ovalicin.224 It is in 2004 that the first cell-permeable PROTAC was found which induced androgen receptor degradation.225 Subsequently, PROTACs have entered into a phase with rapid development, and some other kinds of proteins including MetAP-2 and estrogen receptors were displayed to be knocked out in a range of cell lines.226 However, peptide-based PROTACs show shortcomings on unstable peptide bonds, high molecular weight, and poor cell penetration. The disadvantages mentioned above make it poor pharmaceutical candidates. To overcome these weaknesses of PROTACs including peptide 8,227 small-molecule PROTACs were designed and synthesized and are more easily absorbed than peptides by the human body. MDM2, cIAP, VHL, and cereblon were selected as E3 ligases.228A peptide moiety in the form of E3 ligase ligand was present in all first-generation PROTACs. However, due to limited physicochemical properties including less cell permeability, little intracellular stability, and poor applicability in therapeutic development as a chemical molecule are the resultant impacts of a high peptide containing all PROTACs of first-generation.261
It was in 2008 when Crews described all-small molecule PROTAC having a heterobifunctional ability that made up of a PEG-based linker, an androgen receptor (AR) ligand, and an MDM2 ligand (nutlin) all together can initiate ubiquitination and then proteasome based degradation.229 Nutlin (MDM2 ligand) is a group of imidazoline derivatives, which bind to MDM2 for blocking the interaction between MDM2 and p53.230 However, promising as a critical first step away from peptide-based PROTACs, this initial small molecular degradation inducer is less effective than its peptide analogues. Hashimoto research group reported bestatin-based PROTACs binding cIAP1 could induce the degradation of nuclear receptors including AR, ER, and retinoic acid receptor.231 It is a pity that this kind of PROTACs presented serious off-target effects. The study from Science found that thalidomide binds to E3 ligase cereblon to induce degradation of ikzf1 and IKZF3, suggesting thalidomide and its derivatives as initial ligands targeting E3 ligase CRBN. ARV-825, consisting of OTX015 linked to pomalidomide via alkyl group, degraded almost complete BRD4 protein at 10 nm within 6 h.232 Since 2015, the publication of a series of papers on small molecular VHL-based PROTACs reached its peak little by little.233,234 PROTAC technology has been applied by several drug discovery labs. Yale University licensed the PROTAC technology to Arvinas in 2013. What excites people is that the U.S. Approval for the first phase clinical trial of Arvinas has been obtained from FDA to investigate whether ARV110 can be used as a therapy for patients suffering from metastatic prostate cancer that has castration-resistivity. The trial, scheduled to begin by April, will investigate the safety and tolerability of ARV110 in mCRPC patients whose disease progressed after being treated with a minimum of two standards of care therapies.235
Application of PROTACS for epigenetic targets
Besides downregulation of targeted protein, selectively initiated target protein degradation emerged as a novel strategy in drug discoveries.236,237By designing proteolysis-targeting chimeras (PROTACs), to degrade target protein is one of the convicting approaches at present and has highly attracted medicinal chemists and pharmaceutical firms.236,238–240,244
Proteins marked for proteasomal degradation are tagged via covalent attachment of ubiquitin to surface lysine.241,242 Inherited or acquired diseases are often based on abnormal protein functioning, which is currently targeted using a predominantly occupancy-based pharmaceutical strategy; inhibitors bind to disease-implicated proteins and the longer protein function is blocked by inhibitors, the larger the clinical benefit achieved. Therefore, high local inhibitory concentrations (IC90–95) need to be maintained at all times to ensure therapeutic efficacy.243
A heterobifunctional small compound was initially proposed by Deshaies et al.,244 about 15 years back. It consists of three subunits, an E3 ubiquitin ligase binder, a target protein-specific ligand, and a linker or connector that is connected with these two mechanisms (Fig. 14a).
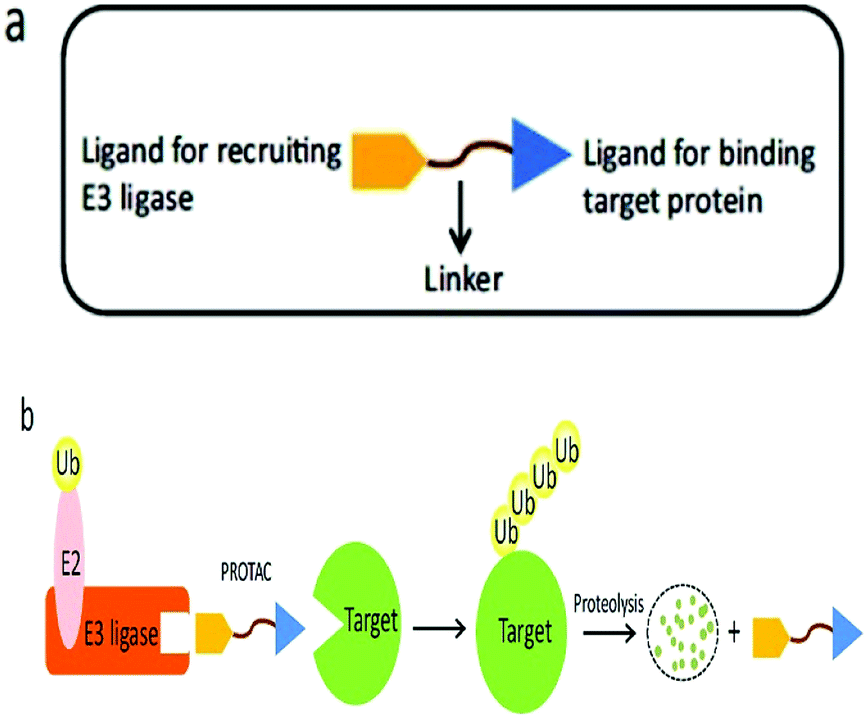 | ||
| Fig. 14 (a) A PROTAC molecule consists of a ligand for recruiting an E3 ubiquitin ligase, a linker, and a ligand binding to the target protein. (b) The PROTAC binds with both the target protein and the E3 ligase simultaneously to induce the formation of a ternary complex. The target protein is then polyubiquitinated and undergoes proteolysis. | ||
Deshaies244 and co-workers synthesized the first-ever proteolysis-targeting chimeric molecule (PROTAC), that can hijack the (UPS) for protein degradation on broad-spectrum during post-translational levels.36
For the alimentation of cellular homeostasis, one of the essential mechanisms that do protein degradation is UPS. It consists of three enzymes, designated molecules, intracellular target proteins, proteasome, and ubiquitin that is responsible for playing a vital role for numerous biological functions such as signal transduction, genome integrity conservation, tumorigenesis, and cell cycling.246
An ATP-incidental enzymatic process, protein ubiquitination, is accomplished by a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3) (Fig. 1).
Disease-generating proteins are degraded by the UPS hijacking instead of inhibition mechanism as in the traditional way by small-molecule based inhibitors. This contributes to a more advanced potent approach.249 Furthermore, as any concerned protein can be targeted by PROTAC, it expressed a more convincing technological aspect in drug discovery as it is not restrained to UPS dependent substrates only.10
A tertiary complex is formed during the binding of target protein, PROTAC, and the E3 ligase. In the further event, ubiquitin can be shifted to protein target as recruitment of an E3 ubiquitin ligase has been done and the target protein has been degraded out through the proteasome (Fig. 14b).243,245,246,248–252
For treating cancer, protein inhibition has given low preference than oncoprotein degradation through PROTACs in theoretical aspects. At the initial level entire protein removal is likely to be more adequate than its inhibition at its active site as remaining protein structure and domains are yet active or functional, next, PROTACs can act enzymatically for the degradation of any targeted protein, and finally, transcription factors like “undruggable” proteins can also be intended.253,254
Sakamoto et al.,36 has prepared the first-ever PROTAC, that is having phosphopeptide (DRHDpSGLDpSM) imitated form NF-κB inhibitor-α (IκBα) to obtain SCFβ-TrCP E3 ligase; ovalicin (OVA), that can bind covalently to methionine aminopeptidase-2 (MetAP-2) active site (His-231) and ubiquitinated MetAP-2 and a linker that can join phosphopeptide and OVA.10
Seven-amino-acid sequence (ALAPYIP), as primitive PROTAC of in vivo, substituted the IκBα-phosphopeptide component coupled with artificial ligand (AP21998) for targeting (F36V) FKBP12 proteins.255
This minimal amino acid sequencing the hypoxia-inducible factor 1α (HIF1α) can be identified by the von Hippel-Lindau tumour suppressor protein (VHL), a known ingredient of CRL2VHL E3 ubiquitin ligase.254
Another advantage is the carboxy terminus of ALAPYIP has an eight-poly-D-arginine tag, that enhances cell permeability and restricts nonspecific proteolysis.256,257
The target proteins have been eradicated in the cellular ambiance by cell-permeable PROTACs. The materialization of cell-permeable PROTACs has been a momentous invention in PROTAC technology and accommodates opportunities for in vivo disease-causing protein targeting.10 This moves towards auspicious administration in the field of cell biology and more precisely for drug development.
Currently, tyrosine kinases,111 estrogen receptor α,112 CDK9,113 Bcr-Abl,114,115 and many other kinds of proteins are degraded by PROTACs. An offbeat epigenetics-based therapy for cancers and have been proposed for provocative antitumor efficiency.152
At present, for targeting enzymes, regulating proteins, transcription factors, skeleton proteins, etc. can be targeted through PROTAC technology.247,258
More advantages have been reported by small molecule-based PROTACs beyond peptide-based PROTAC.259 Interestingly, small molecule-based PROTAC has better capabilities for utilization as a drug smaller molecule can easily be absorbed in the human body over a peptide.258
At present, above 30 proteins were found as a target in disease initiation and progression with primary endeavour over proteins in the therapy of cancer.10,260,261
Several combinations are possible for PROTACs as (1) many types and kinds of ligands can be utilized for specific binding with target proteins and to initiate these proteins on the E3 ligase, and (2) over 600 different ligases encoded in the human genome allowing a broad spectrum of PROTACs based drug invention.10
Small-molecule based BET protein degraders have been developed by some researchers no long ago.262 “Proteolysis targeting chimeras” (PROTACs), are heterobifunctional molecules via trimeric binding complexes permitting ubiquitination and on later stage proteasome-based degradation of target protein (Fig. 15).244
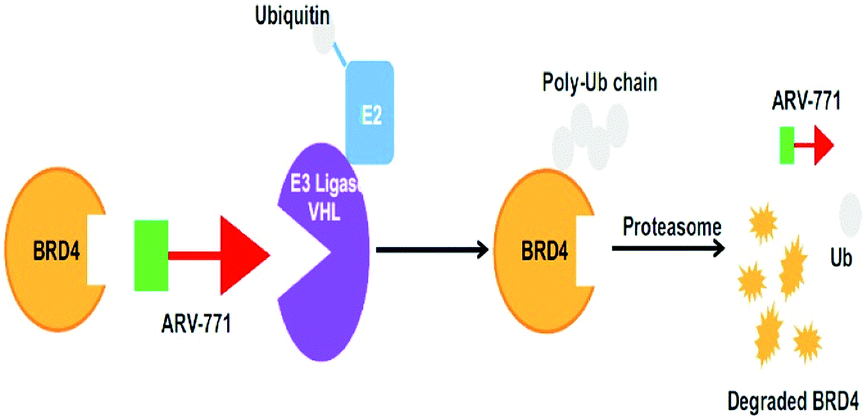 | ||
| Fig. 15 BRD4 PROTAC schematic. | ||
A BET protein that utilizes the E3 ligase cereblon (CRBN) ends up in impressive BET degradation and uninterrupted restraint of downstream signalling in cell lines of Burkitt lymphoma, reported by Raina et al.234
An eminently strong degrader (ARV-825) of the epigenetic regulator BRD4 has been emerged from CRBN-recruiting pomalidomide in mingling with the bromodomain-containing protein 4 (BRD4) inhibitor OTX015.243,262
The protein levels of BRD2, BRD3, and BRD4 has been diminished by most impactive BRD PROTAC, dBET1, with less nanomolar efficacy and surpassed JQ1 for activating of apoptosis in cell lines of AML and lymphoma.243
In the recent past, degraders for the BRD isoform 7 and 9, which plays a vital role in tumour development have been investigated by Zoppi et al.263
BRD7 inactivation makes tumour cells become targetable for T-cell-mediated killing. Because of this, PROTAC selective for BRD7/9 can be used as a chemical probe to study recognition of target and further possibilities for therapeutic invasion (Fig. 16–18).264
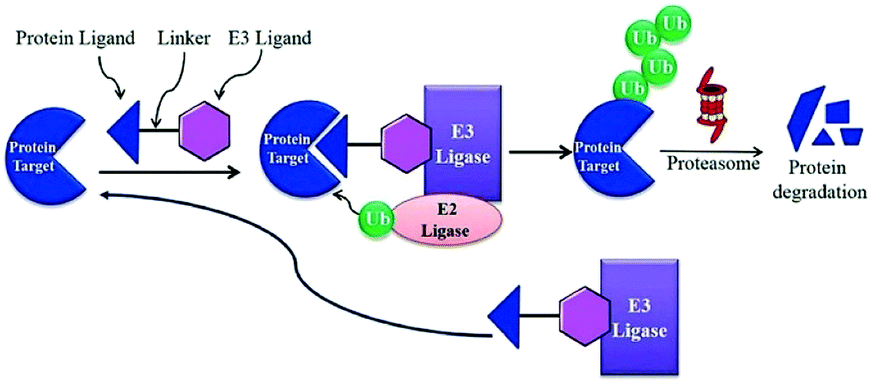 | ||
| Fig. 16 Mechanism of protein degradation by PROTACs. | ||
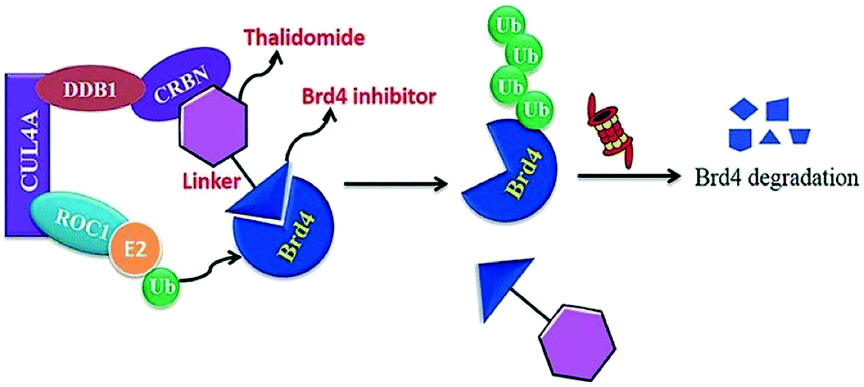 | ||
| Fig. 17 Mechanism of Brd4 degradation by CRL4 CRBN E3-based Brd4 degraders. | ||
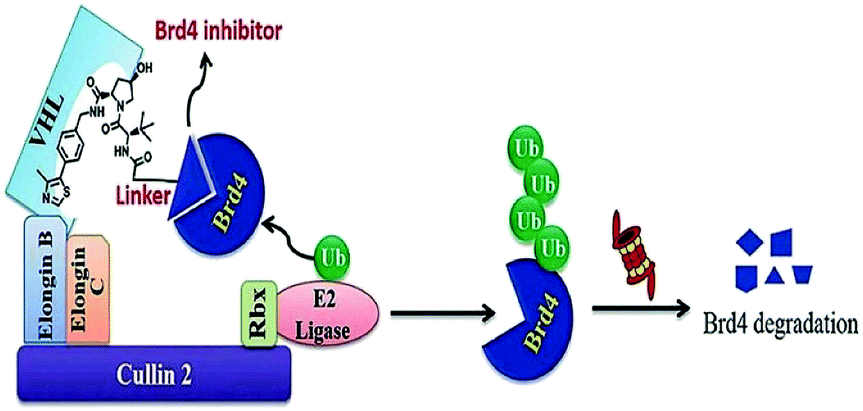 | ||
| Fig. 18 Mechanism of Brd4 degradation by CRL2VHL E3-based Brd4 degraders. | ||
In 2008, Manfred Jung and co-workers first reported chemically induced degradation of Sirtuin 2 by PROTAC based on conjugation SirReals and thalidomide (Fig. 19).268
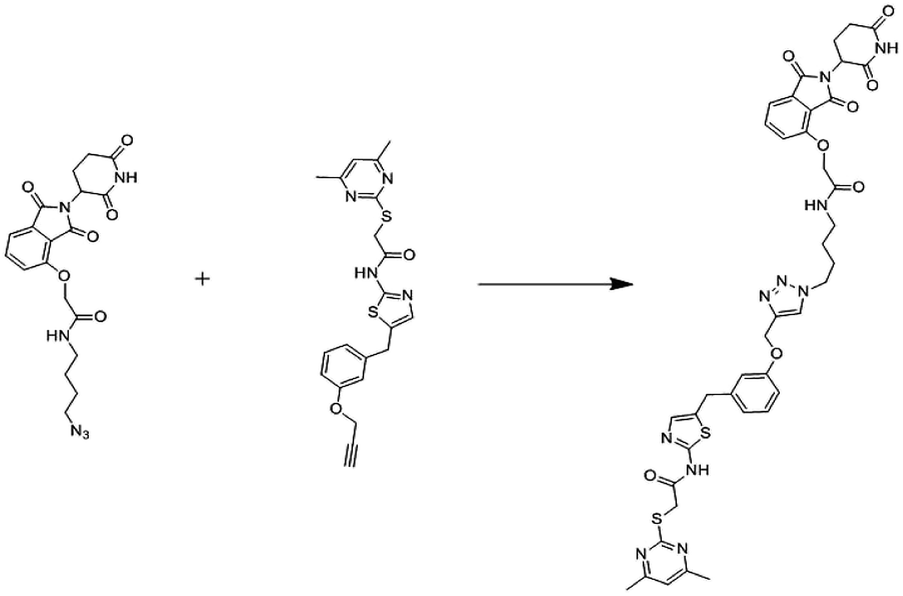 | ||
| Fig. 19 Reported sirt2 PROTAC. | ||
This designed PROTAC (compound Y) presented highly selective SIRT2 inhibitory activity (SIRT2, IC50 = 0.25 μM; SIRT1 and SIRT3, IC50 > μ mM). However, it is unclear whether the degradation of SIRT2 by PROTAC can be used as a strategy for the treatment of tumours.
In humans, an enzyme Histone deacetylase 6 (HDAC6) concealed with the HDAC6 gene is belonging to HDAC family class II and is found in the cytoplasm.265 Contradictory to other nuclear histone targeting HDACs, HDAC6 is effective against transcription and translation as it regulates (Hsp90) the heat-shock protein 90 and stress granules (SGs), in accordingly. It is also acknowledged for binding with ubiquitinated proteins with great compatibility and has a role and involution in SG protein formation.266 Small molecule-based PROTACs against zinc-dependent HDAC6 associated inhibitors of HDAC in compulsion with pomalidomide have been firstly reported by the Tang research group (Fig. 20A).267
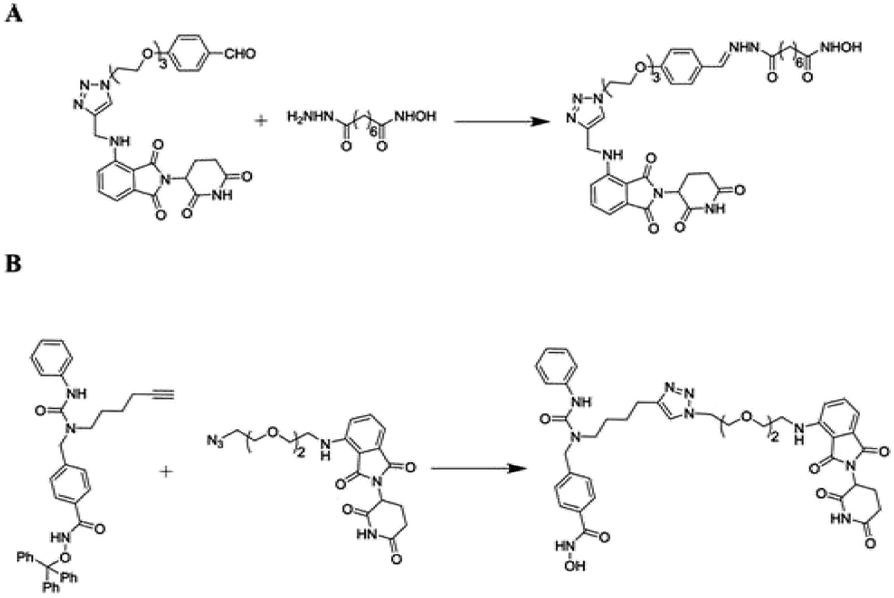 | ||
| Fig. 20 Reported HDAC6 PROTAC. | ||
And more, better selectivity and potential against HDAC6 are ongoing and will be reported in due course from the author's statement.
Subsequently, Rao designed novel HDAC6-targeting PROTACs connecting HDAC6 inhibitor nexturastat A and pomalidomide by the aliphatic chain (Fig. 20B). Compound X induces HDAC6 degradation at 100 nmol L−1 and shows the most potent activities against a range of cell lines. It is gratifying that further functional research of compound X in vivo is now underway in his laboratory.
Conclusions
Cellular functions of BET proteins and their necessity in several malignancies as well as diseases like cardiovascular problems, HIV infection and inflammation have been reported in earlier decades. In addition to functional inhibition by small molecules, Brd4 has been successfully targeted for degradation using PROTACs. Many possibilities have been found in this novel approach of PROTAC development as it proved its selective target degradation abilities, however, some issues such as physicochemical properties, scarce small E3 ligands, are destinations that should be overcome to make PROTACs boost into the clinic.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (No. 2019M652586), Postdoctoral Research Grant in Henan Province (Nos. 1902001 and 19030008) and Henan Medical Science and Technology Program (2018020601). We are also grateful to Shri Maneklal M. Patel Institute of Sciences and Research; and Kadi Sarva Vishwa Vidyalaya University, Gandhinagar, Gujarat for their support and encouragement. The authors extend their appreciation to the Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research work through the Project no. (IFKSURP-05).Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2018, 68, 394–424, DOI:10.3322/caac.21492.
- J. Salami and C. M. Crews, Waste Disposal—An Attractive Strategy for Cancer Therapy, Science, 2017, 355(6330), 1163–1167, DOI:10.1126/science.aam7340.
- H. M. Shepard, G. L. Phillips, C. D. Thanos and M. Feldmann, Developments in Therapy with Monoclonal Antibodies and Related Proteins, Clin. Med., 2017, 17(3), 220–232, DOI:10.7861/clinmedicine.17-3-220.
- J. E. Amengual, R. Lichtenstein, J. Lue, S. Ahmed, C. Deng, E. Lichtenstein, K. Khan, L. Atkins, A. Rada, H. A. Kim, C. Chiuzan, M. Kalac, E. Marchi, L. Falchi, M. A. Francescone, L. Schwartz, S. Cremers, A. Owen and O'Connor, A Phase I Study of Romidepsin and Pralatrexate Reveals Marked Activity in Relapsed and Refractory T-cell Lymphoma, Blood, 2017 DOI:10.1182/blood-2017-09-806737.
- M. Kijanka, B. Dorresteijn, S. Oliveira and P. M. van Bergen en Henegouwen, Nanobody-Based Cancer Therapy of Solid Tumors, Nanomedicine, 2015, 10(1), 161–174, DOI:10.2217/nnm.14.178.
- K. J. Pasi, S. Rangarajan, P. Georgiev, T. Mant, M. D. Creagh, T. Lissitchkov, D. Bevan, S. Austin, C. R. Hay, I. Hegemann, R. Kazmi, P. Chowdary, L. Gercheva-Kyuchukova, V. Mamonov, M. Timofeeva, C.-H. Soh, P. Garg, A. Vaishnaw, A. Akinc, B. Sørensen and M. V. Ragni, Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy, N. Engl. J. Med., 2017, 377(9), 819–828, DOI:10.1056/NEJMoa1616569.
- C. S. Nabzdyk, L. Pradhan-Nabzdyk and F. W. LoGerfo, RNAi Therapy to the Wall of Arteries and Veins: Anatomical, Physiologic, and Pharmacological Considerations, J. Transl. Med., 2017, 15(1), 164, DOI:10.1186/s12967-017-1270-0.
- I. Churcher, Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?, J. Med. Chem., 2018, 61(2), 444–452, DOI: DOI:.
- T. K. Neklesa, J. D. Winkler and C. M. Crews, Targeted Protein Degradation by PROTACs, Pharmacol. Ther., 2017, 174, 138–144, DOI:10.1016/j.pharmthera.2017.02.027.
- S. Gu, D. Cui, X. Chen, X. Xiong and Y. Zhao, PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery, BioEssays, 2018, 40(4), 1700247, DOI:10.1002/bies.201700247.
- Y. Itoh, Chemical Protein Degradation Approach and Its Application to Epigenetic Targets, Chem. Rec., 2018, 18(12), 1681–1700, DOI:10.1002/tcr.201800032.
- M. Toure and C. M. Crews, Small-Molecule PROTACS: New Approaches to Protein Degradation, Angew. Chem., Int. Ed., 2016, 55(6), 1966–1973, DOI:10.1002/anie.201507978.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead, Cell Chem. Biol., 2018, 25(1), 78–87, DOI:10.1016/j.chembiol.2017.09.010.
- C. M. Crews, G. Georg and S. Wang, Inducing Protein Degradation as a Therapeutic Strategy, J. Med. Chem., 2016, 59(11), 5129–5130, DOI:10.1021/acs.jmedchem.6b00735.
- Y. Sun, X. Zhao, N. Ding, H. Gao, Y. Wu, Y. Yang, M. Zhao, J. Hwang, Y. Song, W. Liu and Y. Rao, PROTAC-Induced BTK Degradation as a Novel Therapy for Mutated BTK C481S Induced Ibrutinib-Resistant B-Cell Malignancies, Cell Res., 2018, 28(7), 779–781, DOI:10.1038/s41422-018-0055-1.
- A. Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A.-M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P.-K. Chan, Y.-C. Juang, M.-C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu, A Strategy for Modulation of Enzymes in the Ubiquitin System, Science, 2013, 339(6119), 590–595, DOI:10.1126/science.1230161.
- G. Kleiger and T. Mayor, Perilous Journey: A Tour of the Ubiquitin–Proteasome System, Trends Cell Biol., 2014, 24(6), 352–359, DOI:10.1016/j.tcb.2013.12.003.
- Y. Leestemaker and H. Ovaa, Tools to Investigate the Ubiquitin Proteasome System, Drug Discovery Today: Technol., 2017, 26, 25–31, DOI:10.1016/j.ddtec.2017.11.006.
- M. Guharoy, P. Bhowmick and P. Tompa, Design Principles Involving Protein Disorder Facilitate Specific Substrate Selection and Degradation by the Ubiquitin-Proteasome System, J. Biol. Chem., 2016, 291(13), 6723–6731, DOI:10.1074/jbc.R115.692665.
- M. J. Clague, C. Heride and S. Urbé, The Demographics of the Ubiquitin System, Trends Cell Biol., 2015, 25(7), 417–426, DOI:10.1016/j.tcb.2015.03.002.
- D. J. Klionsky and B. A. Schulman, Dynamic Regulation of Macroautophagy by Distinctive Ubiquitin-like Proteins, Nat. Struct. Mol. Biol., 2014, 21(4), 336–345, DOI:10.1038/nsmb.2787.
- Q. Dou and J. Zonder, Overview of Proteasome Inhibitor-Based Anti-Cancer Therapies: Perspective on Bortezomib and Second-Generation Proteasome Inhibitors versus Future Generation Inhibitors of Ubiquitin-Proteasome System, CCDT, 2014, 14(6), 517–536, DOI:10.2174/1568009614666140804154511.
- D. S. Hewings, J. A. Flygare, M. Bogyo and I. E. Wertz, Activity-Based Probes for the Ubiquitin Conjugation-Deconjugation Machinery: New Chemistries, New Tools, and New Insights, FEBS J., 2017, 284(10), 1555–1576, DOI:10.1111/febs.14039.
- P. Zhou, R. Bogacki, L. McReynolds and P. M. Howley, Harnessing the Ubiquitination Machinery to Target the Degradation of Specific Cellular Proteins, Mol. Cell, 2000, 6(3), 751–756, DOI:10.1016/S1097-2765(00)00074-5.
- M. A. Barbara, Y. Abdilla and J. Calleja-Agius, An Introduction to Epigenetics, Neonatal Netw., 2017, 36(3), 124–128, DOI:10.1891/0730-0832.36.3.124.
- T. Kosciuk, M. Wang, J. Y. Hong and H. Lin, Updates on the Epigenetic Roles of Sirtuins, Curr. Opin. Chem. Biol., 2019, 51, 18–29, DOI:10.1016/j.cbpa.2019.01.023.
- B. Huang, C. Jiang and R. Zhang, Epigenetics: The Language of the Cell?, Epigenomics, 2014, 6(1), 73–88, DOI:10.2217/epi.13.72.
- M. Arif, S. Sadayappan, R. C. Becker, L. J. Martin and E. M. Urbina, Epigenetic Modification: A Regulatory Mechanism in Essential Hypertension, Hypertens. Res., 2019 DOI:10.1038/s41440-019-0248-0.
- S. Morales, M. Monzo and A. Navarro, Epigenetic Regulation Mechanisms of MicroRNA Expression, Biomol. Concepts, 2017, 8(5–6), 203–212, DOI:10.1515/bmc-2017-0024.
- Q. Yao, Y. Chen and X. Zhou, The Roles of MicroRNAs in Epigenetic Regulation, Curr. Opin. Chem. Biol., 2019, 51, 11–17, DOI:10.1016/j.cbpa.2019.01.024.
- T. Luu, K. Kim, S. Blanchard, B. Anyang, A. Hurria, L. Yang, J. H. Beumer, G. Somlo and Y. Yen, Phase IB Trial of Ixabepilone and Vorinostat in Metastatic Breast Cancer, Breast Cancer Res. Treat., 2018, 167(2), 469–478, DOI:10.1007/s10549-017-4516-x.
- E. Galanis, S. K. Anderson, C. R. Miller, J. N. Sarkaria, K. Jaeckle, J. C. Buckner, K. L. Ligon, K. V. Ballman, D. F. Moore, M. Nebozhyn, A. Loboda, D. Schiff, M. S. Ahluwalia, E. Q. Lee, E. R. Gerstner, G. J. Lesser, M. Prados, S. A. Grossman, J. Cerhan, C. Giannini and P. Y. Wen, Alliance for Clinical Trials in Oncology and ABTC. Phase I/II Trial of Vorinostat Combined with Temozolomide and Radiation Therapy for Newly Diagnosed Glioblastoma: Results of Alliance N0874/ABTC 02, Neurooncology, 2018, 20(4), 546–556, DOI:10.1093/neuonc/nox161.
- M. Scheepstra, K. F. W. Hekking, L. van Hijfte and R. H. A. Folmer, Bivalent Ligands for Protein Degradation in Drug Discovery, Comput. Struct. Biotechnol. J., 2019, 17, 160–176, DOI:10.1016/j.csbj.2019.01.006.
- N. Gao, Y.-X. Chen, Y.-F. Zhao and Y.-M. Li, Chemical Methods to Knock Down the Amyloid Proteins, Molecules, 2017, 22(6), 916, DOI:10.3390/molecules22060916.
- C. H. Waddington, Preliminary notes on the development of the wings in normal and mutant strains of Drosophila, Proc. Natl. Acad. Sci. U. S. A., 1939, 25(7), 299–307, DOI:10.1073/pnas.25.7.299.
- R. Holliday, The inheritance of epigenetic defects, Science, 1987, 238(4824), 163–170, DOI:.
- T. Kouzarides, Chromatin Modifications and Their Function, Cell, 2007, 128(4), 693–705, DOI:10.1016/j.cell.2007.02.005.
- B. M. Turner, Reading Signals on the Nucleosome with a New Nomenclature for Modified Histones, Nat. Struct. Mol. Biol., 2005, 12(2), 110–112, DOI:10.1038/nsmb0205-110.
- M. Tan, H. Luo, S. Lee, F. Jin, J. S. Yang, E. Montellier, T. Buchou, Z. Cheng, S. Rousseaux, N. Rajagopal, Z. Lu, Z. Ye, Q. Zhu, J. Wysocka, Y. Ye, S. Khochbin, B. Ren and Y. Zhao, Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification, Cell, 2011, 146(6), 1016–1028, DOI:10.1016/j.cell.2011.08.008.
- T. Jenuwein, Translating the Histone Code, Science, 2001, 293(5532), 1074–1080, DOI:10.1126/science.1063127.
- K. P. Nightingale, L. P. O'Neill and B. M. Turner, Histone Modifications: Signalling Receptors and Potential Elements of a Heritable Epigenetic Code, Curr. Opin. Genet. Dev., 2006, 16(2), 125–136, DOI:10.1016/j.gde.2006.02.015.
- R. Sanchez, J. Meslamani and M.-M. Zhou, The Bromodomain: From Epigenome Reader to Druggable Target, Biochim. Biophys. Acta, Gene Regul. Mech., 2014, 1839(8), 676–685, DOI:10.1016/j.bbagrm.2014.03.011.
- S. L. Berger, T. Kouzarides, R. Shiekhattar and A. Shilatifard, An Operational Definition of Epigenetics, Genes Dev., 2009, 23(7), 781–783, DOI:10.1101/gad.1787609.
- K. Marushige, Activation of Chromatin by Acetylation of Histone Side Chains, Proc. Natl. Acad. Sci., 1976, 73(11), 3937–3941, DOI:10.1073/pnas.73.11.3937.
- M. Shogren-Knaak, Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions, Science, 2006, 311(5762), 844–847, DOI:10.1126/science.1124000.
- I. Celic, H. Masumoto, W. P. Griffith, P. Meluh, R. J. Cotter, J. D. Boeke and A. Verreault, The Sirtuins Hst3 and Hst4p Preserve Genome Integrity by Controlling Histone H3 Lysine 56 Deacetylation, Curr. Biol., 2006, 16(13), 1280–1289, DOI:10.1016/j.cub.2006.06.023.
- V. G. Allfrey, R. Faulkner and A. E. Mirsky, Acetylation and methylation of histones and their possible role in the regulation of rna synthesis, Proc. Natl. Acad. Sci. U. S. A., 1964, 51(5), 786–794, DOI:10.1073/pnas.51.5.786.
- B. M. Turner, Histone Acetylation as an Epigenetic Determinant of Long-Term Transcriptional Competence, Cell. Mol. Life Sci., 1998, 54(1), 21–31, DOI:10.1007/s000180050122.
- S. L. Berger, Histone Modifications in Transcriptional Regulation, Curr. Opin. Genet. Dev., 2002, 12(2), 142–148, DOI:10.1016/S0959-437X(02)00279-4.
- S. Zhao, W. Xu, W. Jiang, W. Yu, Y. Lin, T. Zhang, J. Yao, L. Zhou, Y. Zeng, H. Li, Y. Li, J. Shi, W. An, S. M. Hancock, F. He, L. Qin, J. Chin, P. Yang, X. Chen, Q. Lei, Y. Xiong and K.-L. Guan, Regulation of Cellular Metabolism by Protein Lysine Acetylation, Science, 2010, 327(5968), 1000–1004, DOI:10.1126/science.1179689.
- T. Kouzarides, Acetylation: a regulatory modification to rival phosphorylation?, EMBO J., 2000, 19(6), 1176–1179 CrossRef CAS.
- C. Choudhary, C. Kumar, F. Gnad, M. L. Nielsen, M. Rehman, T. C. Walther, J. V. Olsen and M. Mann, Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions, Science, 2009, 325(5942), 834–840, DOI:10.1126/science.1175371.
- M. Pérez-Salvia and M. Esteller, Bromodomain Inhibitors and Cancer Therapy: From Structures to Applications, Epigenetics, 2017, 12(5), 323–339, DOI:10.1080/15592294.2016.1265710.
- P. Filippakopoulos and S. Knapp, The Bromodomain Interaction Module, FEBS Lett., 2012, 586(17), 2692–2704, DOI:10.1016/j.febslet.2012.04.045.
- D. Gallenkamp, K. A. Gelato, B. Haendler and H. Weinmann, Bromodomains and their pharmacological inhibitors, ChemMedChem, 2014, 9, 438–464 CrossRef CAS.
- B. Padmanabhan, S. Mathur, R. Manjula and S. Tripathi, The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins, Int. J. Mol. Sci., 2016, 17, 1849, DOI:10.3390/ijms17111849.
- L. Zeng and M.-M. Zhou, Bromodomain: An Acetyl-Lysine Binding Domain, FEBS Lett., 2002, 513(1), 124–128, DOI:10.1016/S0014-5793(01)03309-9.
- C. H. Arrowsmith, C. Bountra, P. V. Fish, K. Lee and M. Schapira, Epigenetic Protein Families: A New Frontier for Drug Discovery, Nat. Rev. Drug Discovery, 2012, 11(5), 384–400, DOI:10.1038/nrd3674.
- I. Barbieri, E. Cannizzaro and M. A. Dawson, Bromodomains as Therapeutic Targets in Cancer, Briefings Funct. Genomics, 2013, 12(3), 219–230, DOI:10.1093/bfgp/elt007.
- D. Boehm, R. Conrad and M. Ott, Bromodomain Proteins in HIV Infection, Viruses, 2013, 5(6), 1571–1586, DOI:10.3390/v5061571.
- R. K. Prinjha, J. Witherington and K. Lee, Place Your BETs: The Therapeutic Potential of Bromodomains, Trends Pharmacol. Sci., 2012, 33(3), 146–153, DOI:10.1016/j.tips.2011.12.002.
- G. Zhang, R. Sanchez and M.-M. Zhou, Scaling the Druggability Landscape of Human Bromodomains, a New Class of Drug Targets, J. Med. Chem., 2012, 55(17), 7342–7345, DOI:10.1021/jm3011977.
- J. W. Tamkun, R. Deuring, M. P. Scott, M. Kissinger, A. M. Pattatucci, T. C. Kaufman and A. Kennison, brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2, Cell, 1992, 68(3), 561–572, DOI:10.1016/0092-8674(92)90191-E.
- P. Filippakopoulos, S. Picaud, M. Mangos, T. Keates, J.-P. Lambert, D. Barsyte-Lovejoy, I. Felletar, R. Volkmer, S. Müller, T. Pawson, A.-C. Gingras, C. H. Arrowsmith and S. Knapp, Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family, Cell, 2012, 149(1), 214–231, DOI:10.1016/j.cell.2012.02.013.
- C. Dhalluin, J. E. Carlson, L. Zeng, C. He, A. K. Aggarwal and M.-M. Zhou, Structure and Ligand of a Histone Acetyltransferase Bromodomain, Nature, 1999, 399, 491–496 CrossRef CAS.
- X. J. Yang, V. V. Ogryzko, J. Nishikawa, B. H. Howard and Y. A. Nakatani, p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A, Nature, 1996, 382, 319–324 CrossRef CAS.
- E. Cavellán, P. Asp, P. Percipalle and A.-K. Ö. Farrants, The WSTF-SNF2h Chromatin Remodeling Complex Interacts with Several Nuclear Proteins in Transcription, J. Biol. Chem., 2006, 281(24), 16264–16271, DOI:10.1074/jbc.M600233200.
- K. W. Trotter and T. K. Archer, The BRG1 transcriptional coregulator, Nucl. Recept. Signaling, 2008,(6), 1–12, DOI:10.1621/nrs.06004.
- G. D. Gregory, C. R. Vakoc, T. Rozovskaia, X. Zheng, S. Patel, T. Nakamura, E. Canaani and G. A. Blobel, Mammalian ASH1L Is a Histone Methyltransferase That Occupies the Transcribed Region of Active Genes, Mol. Cell. Biol., 2007, 27(24), 8466–8479, DOI:10.1128/MCB.00993-07.
- S. Malik and S. R. Bhaumik, Mixed Lineage Leukemia: Histone H3 Lysine 4 Methyltransferases from Yeast to Human: H3K4 Methyltransferases from Yeast to Human, FEBS J., 2010, 277(8), 1805–1821, DOI:10.1111/j.1742-4658.2010.07607.x.
- L. Venturini, J. You, M. Stadler, R. Galien, M. H. Koken, M. G. Mattei, A. Ganser, P. Chambon and H. de TheÂ, TIF1g, a Novel Member of the Transcriptional Intermediary Factor 1 Family, Oncogene, 1999, 18, 1209–1217 CrossRef CAS.
- R. H. Jacobson, A. G. Ladurner, D. S. King and R. Tjian, Structure and function of a human TAFII250 double bromodomain module, Science, 2000, 288, 1422–1425 CrossRef CAS.
- Y. Xue, J. C. Canman, C. S. Lee, Z. Nie, D. Yang, G. T. Moreno, M. K. Young, E. D. Salmon and W. Wang, The Human SWI/SNF-B Chromatin-Remodeling Complex Is Related to Yeast Rsc and Localizes at Kinetochores of Mitotic Chromosomes, Proc. Natl. Acad. Sci., 2000, 97(24), 13015–13020, DOI:10.1073/pnas.240208597.
- V. Brès, S. M. Yoh and K. A. Jones, The Multi-Tasking P-TEFb Complex, Curr. Opin. Cell Biol., 2008, 20(3), 334–340, DOI:10.1016/j.ceb.2008.04.008.
- M. Magrane and U. Consortium, UniProt Knowledgebase: A Hub of Integrated Protein Data, Database, 2011, 2011, bar009, DOI:10.1093/database/bar009.
- N. A. sFairbridge, C. E. Dawe, F. H. Niri, M. K. Kooistra, K. King-Jones and H. E. McDermid, Cecr2 Mutations Causing Exencephaly Trigger Misregulation of Mesenchymal/Ectodermal Transcription Factors, Birth Defects Res., Part A, 2010, 88(8), 619–625, DOI:10.1002/bdra.20695.
- G. LeRoy, B. Rickards and S. J. Flint, The Double Bromodomain Proteins Brd2 and Brd3 Couple Histone Acetylation to Transcription, Mol. Cell, 2008, 30(1), 51–60, DOI:10.1016/j.molcel.2008.01.018.
- M. Ullah, N. Pelletier, L. Xiao, S. P. Zhao, K. Wang, C. Degerny, S. Tahmasebi, C. Cayrou, Y. Doyon, S.-L. Goh, N. Champagne, J. Côté and X.-J. Yang, Molecular Architecture of Quartet MOZ/MORF Histone Acetyltransferase Complexes, Mol. Cell. Biol., 2008, 28(22), 6828–6843, DOI:10.1128/MCB.01297-08.
- J. Morinière, S. Rousseaux, U. Steuerwald, M. Soler-López, S. Curtet, A.-L. Vitte, J. Govin, J. Gaucher, K. Sadoul, D. J. Hart, J. Krijgsveld, S. Khochbin, C. W. Müller and C. Petosa, Cooperative Binding of Two Acetylation Marks on a Histone Tail by a Single Bromodomain, Nature, 2009, 461(7264), 664–668, DOI:10.1038/nature08397.
- A. Dey, J. Ellenberg, A. Farina, A. E. Coleman, T. Maruyama, S. Sciortino, J. Lippincott-Schwartz and K. Ozato, A Bromodomain Protein, MCAP, Associates with Mitotic Chromosomes and Affects G2-to-M Transition, Mol. Cell. Biol., 2000, 20(13), 6537–6549 CrossRef CAS.
- A. Dey, F. Chitsaz, A. Abbasi, T. Misteli and K. Ozato, The Double Bromodomain Protein Brd4 Binds to Acetylated Chromatin during Interphase and Mitosis, Proc. Natl. Acad. Sci., 2003, 100(15), 8758–8763, DOI:10.1073/pnas.1433065100.
- T. Kanno, Y. Kanno, R. M. Siegel, M. K. Jang, M. J. Lenardo and K. Ozato, Selective Recognition of Acetylated Histones by Bromodomain Proteins Visualized in Living Cells, Mol. Cell, 2004, 13(1), 33–43, DOI:10.1016/S1097-2765(03)00482-9.
- Y. Cai, J. Jin, C. Tomomori-Sato, S. Sato, I. Sorokina, T. J. Parmely, R. C. Conaway and J. W. Conaway, Identification of New Subunits of the Multiprotein Mammalian TRRAP/TIP60-Containing Histone Acetyltransferase Complex, J. Biol. Chem., 2003, 278(44), 42733–42736, DOI:10.1074/jbc.C300389200.
- E. Kalkhoven, CBP and P300: HATs for Different Occasions, Biochem. Pharmacol., 2004, 68(6), 1145–1155, DOI:10.1016/j.bcp.2004.03.045.
- H. Huang, I. Rambaldi, E. Daniels and M. Featherstone, Expression of TheWdr9 Gene and Protein Products during Mouse Development, Dev. Dyn., 2003, 227(4), 608–614, DOI:10.1002/dvdy.10344.
- P. Müller, D. Kuttenkeuler, V. Gesellchen, M. P. Zeidler and M. Boutros, Identification of JAK/STAT Signalling Components by Genome-Wide RNA Interference, Nature, 2005, 436(7052), 871–875, DOI:10.1038/nature03869.
- A. Podcheko, P. Northcott, G. Bikopoulos, A. Lee, S. R. Bommareddi, J. A. Kushner, J. Farhang-Fallah and M. Rozakis-Adcock, Identification of a WD40 Repeat-Containing Isoform of PHIP as a Novel Regulator of β-Cell Growth and Survival, Mol. Cell. Biol., 2007, 27(18), 6484–6496, DOI:10.1128/MCB.02409-06.
- M. D. Kaeser, A. Aslanian, M.-Q. Dong, J. R. Yates and B. M. Emerson, BRD7, a Novel PBAF-Specific SWI/SNF Subunit, Is Required for Target Gene Activation and Repression in Embryonic Stem Cells, J. Biol. Chem., 2008, 283(47), 32254–32263, DOI:10.1074/jbc.M806061200.
- K. Laue, S. Daujat, J. G. Crump, N. Plaster, H. H. Roehl, Tubingen 2000 Screen Consortium, C. B. Kimmel, R. Schneider and M. Hammerschmidt, The Multidomain Protein Brpf1 Binds Histones and Is Required for Hox Gene Expression and Segmental Identity, Development, 2008, 135(11), 1935–1946, DOI:10.1242/dev.017160.
- M. Ciro, E. Prosperini, M. Quarto, U. Grazini, J. Walfridsson, F. McBlane, P. Nucifero, G. Pacchiana, M. Capra, J. Christensen and K. Helin, ATAD2 Is a Novel Cofactor for MYC, Overexpressed and Amplified in Aggressive Tumors, Cancer Res., 2009, 69(21), 8491–8498, DOI:10.1158/0008-5472.CAN-09-2131.
- K. Khetchoumian, M. Teletin, M. Mark, T. Lerouge, M. Cerviño, M. Oulad-Abdelghani, P. Chambon and R. Losson, TIF1δ, a Novel HP1-Interacting Member of the Transcriptional Intermediary Factor 1 (TIF1) Family Expressed by Elongating Spermatids, J. Biol. Chem., 2004, 279(46), 48329–48341, DOI:10.1074/jbc.M404779200.
- X. Bai, J. Kim, Z. Yang, M. J. Jurynec, T. E. Akie, J. Lee, J. LeBlanc, A. Sessa, H. Jiang, A. DiBiase, Y. Zhou, D. J. Grunwald, S. Lin, A. B. Cantor, S. H. Orkin and L. I. Zon, TIF1γ Controls Erythroid Cell Fate by Regulating Transcription Elongation, Cell, 2010, 142(1), 133–143, DOI:10.1016/j.cell.2010.05.028.
- W.-W. Tsai, Z. Wang, T. T. Yiu, K. C. Akdemir, W. Xia, S. Winter, C.-Y. Tsai, X. Shi, D. Schwarzer, W. Plunkett, B. Aronow, O. Gozani, W. Fischle, M.-C. Hung, D. J. Patel and M. C. Barton, TRIM24 Links a Non-Canonical Histone Signature to Breast Cancer, Nature, 2010, 468(7326), 927–932, DOI:10.1038/nature09542.
- J. S. Yordy, O. Moussa, H. Pei, D. Chaussabel, R. Li and D. K. Watson, SP100 Inhibits ETS1 Activity in Primary Endothelial Cells, Oncogene, 2005, 24(5), 916–931, DOI:10.1038/sj.onc.1208245.
- D. B. Bloch, A. Nakajima, T. Gulick, J.-D. Chiche, D. Orth, S. M. de la Monte and K. D. Bloch, Sp110 Localizes to the PML-Sp100 Nuclear Body and May Function as a Nuclear Hormone Receptor Transcriptional Coactivator, Mol. Cell. Biol., 2000, 20(16), 6138–6146, DOI:10.1128/MCB.20.16.6138-6146.2000.
- R.-T. Zong, C. Das and P. W. Tucker, Regulation of Matrix Attachment Region-Dependent, Lymphocyte-Restricted Transcription through Differential Localization within Promyelocytic Leukemia Nuclear Bodies, EMBO J., 2000, 19(15), 4123–4133, DOI:10.1093/emboj/19.15.4123.
- Y. Zhou, K.-M. Schmitz, C. Mayer, X. Yuan, A. Akhtar and I. Grummt, Reversible Acetylation of the Chromatin Remodelling Complex NoRC Is Required for Non-Coding RNA-Dependent Silencing, Nat. Cell Biol., 2009, 11(8), 1010–1016, DOI:10.1038/ncb1914.
- M. H. Jones, N. Hamana, J. Nezu and M. Shimane, A Novel Family of Bromodomain Genes, Genomics, 2000, 63(1), 40–45, DOI:10.1006/geno.1999.6071.
- Y. Dou, T. A. Milne, A. J. Tackett, E. R. Smith, A. Fukuda, J. Wysocka, C. D. Allis, B. T. Chait, J. L. Hess and R. G. Roeder, Physical Association and Coordinate Function of the H3 K4 Methyltransferase MLL1 and the H4 K16 Acetyltransferase MOF, Cell, 2005, 121(6), 873–885, DOI:10.1016/j.cell.2005.04.031.
- H. M. Rowe, J. Jakobsson, D. Mesnard, J. Rougemont, S. Reynard, T. Aktas, P. V. Maillard, H. Layard-Liesching, S. Verp, J. Marquis, F. Spitz, D. B. Constam and D. Trono, KAP1 Controls Endogenous Retroviruses in Embryonic Stem Cells, Nature, 2010, 463(7278), 237–240, DOI:10.1038/nature08674.
- H. Masselink and R. Bernards, The Adenovirus E1A Binding Protein BS69 Is a Corepressor of Transcription through Recruitment of N-CoR, Oncogene, 2000, 19(12), 1538–1546, DOI:10.1038/sj.onc.1203421.
- D. A. Wassarman and F. Sauer, TAFII250: A Transcription Toolbox, J. Cell Sci., 2001, 114, 2895–2902 CAS.
- P. J. Wang, Functional Substitution for TAFII250 by a Retroposed Homolog That Is Expressed in Human Spermatogenesis, Hum. Mol. Genet., 2002, 11(19), 2341–2346, DOI:10.1093/hmg/11.19.2341.
- K. N. Harikrishnan, M. Z. Chow, E. K. Baker, S. Pal, S. Bassal, D. Brasacchio, L. Wang, J. M. Craig, P. L. Jones, S. Sif and A. El-Osta, Brahma Links the SWI/SNF Chromatin-Remodeling Complex with MeCP2-Dependent Transcriptional Silencing, Nat. Genet., 2005, 37(3), 254–264, DOI:10.1038/ng1516.
- A. Rada-Iglesias, R. Bajpai, T. Swigut, S. A. Brugmann, R. A. Flynn and J. Wysocka, A Unique Chromatin Signature Uncovers Early Developmental Enhancers in Humans, Nature, 2011, 470(7333), 279–283, DOI:10.1038/nature09692.
- B. K. Albrecht, V. S. Gehling, M. C. Hewitt, R. G. Vaswani, A. Côté, Y. Leblanc, C. G. Nasveschuk, S. Bellon, L. Bergeron, R. Campbell, N. Cantone, M. R. Cooper, R. T. Cummings, H. Jayaram, S. Joshi, J. A. Mertz, A. Neiss, E. Normant, M. O'Meara, E. Pardo, F. Poy, P. Sandy, J. Supko, R. J. Sims, J.-C. Harmange, A. M. Taylor and J. E. Audia, Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials, J. Med. Chem., 2016, 59(4), 1330–1339, DOI:10.1021/acs.jmedchem.5b01882.
- J. E. Delmore, G. C. Issa, M. E. Lemieux, P. B. Rahl, J. Shi, H. M. Jacobs, E. Kastritis, T. Gilpatrick, R. M. Paranal, J. Qi, M. Chesi, A. C. Schinzel, M. R. McKeown, T. P. Heffernan, C. R. Vakoc, P. L. Bergsagel, I. M. Ghobrial, P. G. Richardson, R. A. Young, W. C. Hahn, K. C. Anderson, A. L. Kung, J. E. Bradner and C. S. Mitsiades, BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc, Cell, 2011, 146(6), 904–917, DOI:10.1016/j.cell.2011.08.017.
- Z. Yang, J. H. N. Yik, R. Chen, N. He, M. K. Jang, K. Ozato and Q. Zhou, Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4, Mol. Cell, 2005, 19(4), 535–545, DOI:10.1016/j.molcel.2005.06.029.
- Z. Yang, N. He and Q. Zhou, Brd4 Recruits P-TEFb to Chromosomes at Late Mitosis To Promote G1 Gene Expression and Cell Cycle Progression, Mol. Cell. Biol., 2008, 28(3), 967–976, DOI:10.1128/MCB.01020-07.
- G. V. Denis, C. Vaziri, N. Guo and D. V. Faller, RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F, Cell Growth Differ., 2000, 11, 417–424 CAS.
- G. V. Denis, M. E. McComb, D. V. Faller, A. Sinha, P. B. Romesser and C. E. Costello, Identification of Transcription Complexes That Contain the Double Bromodomain Protein Brd2 and Chromatin Remodeling Machines, J. Proteome Res., 2006, 5(3), 502–511, DOI:10.1021/pr050430u.
- E. Shang, X. Wang, D. Wen, D. A. Greenberg and D. J. Wolgemuth, Double Bromodomain-Containing Gene Brd2 Is Essential for Embryonic Development in Mouse, Dev. Dyn., 2009, 238(4), 908–917, DOI:10.1002/dvdy.21911.
- A. Gyuris, D. J. Donovan, K. A. Seymour, L. A. Lovasco, N. R. Smilowitz, A. L. P. Halperin, J. E. Klysik and R. N. Freiman, The Chromatin-Targeting Protein Brd2 Is Required for Neural Tube Closure and Embryogenesis, Biochim. Biophys. Acta, Gene Regul. Mech., 2009, 1789(5), 413–421, DOI:10.1016/j.bbagrm.2009.03.005.
- L. Velíšek, E. Shang, J. Velíšková, T. Chachua, S. Macchiarulo, G. Maglakelidze, D. J. Wolgemuth and D. A. Greenberg, GABAergic Neuron Deficit as An Idiopathic Generalized Epilepsy Mechanism: The Role of BRD2 Haploinsufficiency In Juvenile Myoclonic Epilepsy, PLoS One, 2011, 6(8), e23656, DOI:10.1371/journal.pone.0023656.
- G. LeRoy, I. Chepelev, P. A. DiMaggio, M. A. Blanco, B. M. Zee, K. Zhao and B. A. Garcia, Proteogenomic Characterization and Mapping of Nucleosomes Decoded by Brd and HP1 Proteins, Genome Biol., 2012, 13(8), R68, DOI:10.1186/gb-2012-13-8-r68.
- A. J. Stonestrom, S. C. Hsu, K. S. Jahn, P. Huang, C. A. Keller, B. M. Giardine, S. Kadauke, A. E. Campbell, P. Evans, R. C. Hardison and G. A. Blobel, Functions of BET proteins in erythroid gene expression, Blood, 2015, 125, 2825–2834 CrossRef CAS.
- G. M. Platt, G. R. Simpson, S. Mittnacht and T. F. Schulz, Latent Nuclear Antigen of Kaposi's Sarcoma-Associated Herpesvirus Interacts with RING3, a Homolog of TheDrosophila Female Sterile Homeotic (Fsh) Gene, J. Virol., 1999, 73(12), 9789–9795, DOI:10.1128/JVI.73.12.9789-9795.1999.
- A. Viejo-Borbolla, M. Ottinger, E. Brüning, A. Bürger, R. König, E. Kati, J. A. Sheldon and T. F. Schulz, Brd2/RING3 Interacts with a Chromatin-Binding Domain in the Kaposi's Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 (LANA-1) That Is Required for Multiple Functions of LANA-1, J. Virol., 2005, 79(21), 13618–13629, DOI:10.1128/JVI.79.21.13618-13629.2005.
- C. A. French, C. L. Ramirez, J. Kolmakova, T. T. Hickman, M. J. Cameron, M. E. Thyne, J. L. Kutok, J. A. Toretsky, A. K. Tadavarthy, U. R. Kees, J. A. Fletcher and J. C. Aster, BRD–NUT Oncoproteins: A Family of Closely Related Nuclear Proteins That Block Epithelial Differentiation and Maintain the Growth of Carcinoma Cells, Oncogene, 2008, 27(15), 2237–2242, DOI:10.1038/sj.onc.1210852.
- D. Houzelstein, S. L. Bullock, D. E. Lynch, E. F. Grigorieva, V. A. Wilson and R. S. P. Beddington, Growth and Early Postimplantation Defects in Mice Deficient for the Bromodomain-Containing Protein Brd4, Mol. Cell. Biol., 2002, 22(11), 3794–3802, DOI:10.1128/MCB.22.11.3794-3802.2002.
- W. Liu, P. Stein, X. Cheng, W. Yang, N.-Y. Shao, E. E. Morrisey, R. M. Schultz and J. You, BRD4 Regulates Nanog Expression in Mouse Embryonic Stem Cells and Preimplantation Embryos, Cell Death Differ., 2014, 21(12), 1950–1960, DOI:10.1038/cdd.2014.124.
- M. K. Jang, K. Mochizuki, M. Zhou, H.-S. Jeong, J. N. Brady and K. Ozato, The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription, Mol. Cell, 2005, 19(4), 523–534, DOI:10.1016/j.molcel.2005.06.027.
- W. Liu, Q. Ma, K. Wong, W. Li, K. Ohgi, J. Zhang, A. K. Aggarwal and M. G. Rosenfeld, Brd4 and JMJD6-Associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release, Cell, 2013, 155(7), 1581–1595, DOI:10.1016/j.cell.2013.10.056.
- T. Kanno, Y. Kanno, G. LeRoy, E. Campos, H.-W. Sun, S. R. Brooks, G. Vahedi, T. D. Heightman, B. A. Garcia, D. Reinberg, U. Siebenlist, J. J. O'Shea and K. Ozato, BRD4 Assists Elongation of Both Coding and Enhancer RNAs by Interacting with Acetylated Histones, Nat. Struct. Mol. Biol., 2014, 21(12), 1047–1057, DOI:10.1038/nsmb.2912.
- E. Korb, M. Herre, I. Zucker-Scharff, R. B. Darnell and C. D. Allis, BET Protein Brd4 Activates Transcription in Neurons and BET Inhibitor Jq1 Blocks Memory in Mice, Nat. Neurosci., 2015, 18(10), 1464–1473, DOI:10.1038/nn.4095.
- M. C. Patel, M. Debrosse, M. Smith, A. Dey, W. Huynh, N. Sarai, T. D. Heightman, T. Tamura and K. Ozato, BRD4 Coordinates Recruitment of Pause Release Factor P-TEFb and the Pausing Complex NELF/DSIF To Regulate Transcription Elongation of Interferon-Stimulated Genes, Mol. Cell. Biol., 2013, 33(12), 2497–2507, DOI:10.1128/MCB.01180-12.
- M. Hussong, S. T. Börno, M. Kerick, A. Wunderlich, A. Franz, H. Sültmann, B. Timmermann, H. Lehrach, M. Hirsch-Kauffmann and M. R. Schweiger, The Bromodomain Protein BRD4 Regulates the KEAP1/NRF2-Dependent Oxidative Stress Response, Cell Death Discovery, 2014, 5(4), e1195, DOI:10.1038/cddis.2014.157.
- M. Hussong, C. Kaehler, M. Kerick, C. Grimm, A. Franz, B. Timmermann, F. Welzel, J. Isensee, T. Hucho, S. Krobitsch and M. R. Schweiger, The Bromodomain Protein BRD4 Regulates Splicing during Heat Shock, Nucleic Acids Res., 2017, 45(1), 382–394, DOI:10.1093/nar/gkw729.
- A. Dey, A. Nishiyama, T. Karpova, J. McNally and K. Ozato, Brd4 Marks Select Genes on Mitotic Chromatin and Directs Postmitotic Transcription, Mol. Biol. Cell, 2009, 20(23), 4899–4909, DOI:10.1091/mbc.e09-05-0380.
- R. Zhao, T. Nakamura, Y. Fu, Z. Lazar and D. L. Spector, Gene Bookmarking Accelerates the Kinetics of Post-Mitotic Transcriptional Re-Activation, Nat. Cell Biol., 2011, 13(11), 1295–1304, DOI:10.1038/ncb2341.
- C. A. French, I. Miyoshi, I. Kubonishi, H. E. Grier, A. R. Perez-Atayde and J. A. Fletcher, BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma, Cancer Res., 2003, 63, 304–307 CAS.
- J. You, V. Srinivasan, G. V. Denis, W. J. Harrington, M. E. Ballestas, K. M. Kaye and P. M. Howley, Kaposi's Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen Interacts with Bromodomain Protein Brd4 on Host Mitotic Chromosomes, J. Virol., 2006, 80(18), 8909–8919, DOI:10.1128/JVI.00502-06.
- M. Ottinger, T. Christalla, K. Nathan, M. M. Brinkmann, A. Viejo-Borbolla and T. F. Schulz, Kaposi's Sarcoma-Associated Herpesvirus LANA-1 Interacts with the Short Variant of BRD4 and Releases Cells from a BRD4- and BRD2/RING3-Induced G1 Cell Cycle Arrest, J. Virol., 2006, 80(21), 10772–10786, DOI:10.1128/JVI.00804-06.
- J. You, J. L. Croyle, A. Nishimura, K. Ozato and P. M. Howley, Interaction of the Bovine Papillomavirus E2 Protein with Brd4 Tethers the Viral DNA to Host Mitotic Chromosomes, Cell, 2004, 117(3), 349–360, DOI:10.1016/S0092-8674(04)00402-7.
- S.-Y. Wu, Brd4 Links Chromatin Targeting to HPV Transcriptional Silencing, Genes Dev., 2006, 20(17), 2383–2396, DOI:10.1101/gad.1448206.
- M.-R. Schweiger, J. You and P. M. Howley, Bromodomain Protein 4 Mediates the Papillomavirus E2 Transcriptional Activation Function, J. Virol., 2006, 80(9), 4276–4285, DOI:10.1128/JVI.80.9.4276-4285.2006.
- M. G. McPhillips, J. G. Oliveira, J. E. Spindler, R. Mitra and A. A. McBride, Brd4 Is Required for E2-Mediated Transcriptional Activation but Not Genome Partitioning of All Papillomaviruses, J. Virol., 2006, 80(19), 9530–9543, DOI:10.1128/JVI.01105-06.
- B. D. Berkovits, L. Wang, P. Guarnieri and D. J. Wolgemuth, The Testis-Specific Double Bromodomain-Containing Protein BRDT Forms a Complex with Multiple Spliceosome Components and Is Required for MRNA Splicing and 3′-UTR Truncation in Round Spermatids, Nucleic Acids Res., 2012, 40(15), 7162–7175, DOI:10.1093/nar/gks342.
- S. Dhar, A. Thota and M. R. S. Rao, Insights into Role of Bromodomain, Testis-Specific (Brdt) in Acetylated Histone H4-Dependent Chromatin Remodeling in Mammalian Spermiogenesis, J. Biol. Chem., 2012, 287(9), 6387–6405, DOI:10.1074/jbc.M111.288167.
- C. Pivot-Pajot, C. Caron, J. Govin, A. Vion, S. Rousseaux and S. Khochbin, Acetylation-Dependent Chromatin Reorganization by BRDT, a Testis-Specific Bromodomain-Containing Protein, Mol. Cell. Biol., 2003, 23(15), 5354–5365, DOI:10.1128/MCB.23.15.5354-5365.2003.
- K. Sasaki, T. Ito, N. Nishino, S. Khochbin and M. Yoshida, Real-Time Imaging of Histone H4 Hyperacetylation in Living Cells, Proc. Natl. Acad. Sci., 2009, 106(38), 16257–16262, DOI:10.1073/pnas.0902150106.
- I. A. Asangani, V. L. Dommeti, X. Wang, R. Malik, M. Cieslik, R. Yang, J. Escara-Wilke, K. Wilder-Romans, S. Dhanireddy, C. Engelke, M. K. Iyer, X. Jing, Y.-M. Wu, X. Cao, Z. S. Qin, S. Wang, F. Y. Feng and A. M. Chinnaiyan, Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer, Nature, 2014, 510(7504), 278–282, DOI:10.1038/nature13229.
- A. Wyce, G. Ganji, K. N. Smitheman, C. Chung, S. Korenchuk, Y. Bai, O. Barbash, B. Le, P. D. Craggs, M. T. McCabe, K. M. Kennedy-Wilson, L. V. Sanchez, R. L. Gosmini, N. Parr, C. F. McHugh, D. Dhanak, R. K. Prinjha, K. R. Auger and P. J. Tummino, BET Inhibition Silences Expression of MYCN and BCL2 and Induces Cytotoxicity in Neuroblastoma Tumor Models, PLoS One, 2013, 8(8), e72967, DOI:10.1371/journal.pone.0072967.
- G. Civenni, S. Pedrani, S. Allegrini, A. Bruccoleri, D. Albino, S. Pinton, R. Garcia-Escudero, L. 'H. Ouafik, E. Cvitković, G. Carbone and C. Catapano, Abstract 2625: Targeting prostate cancer stem cells (CSCs) with the novel BET bromodomain (BRD) protein inhibitor OTX015, Cancer Res., 2015, 75, 2625, DOI:.
- E. Ferri, C. Petosa and C. E. McKenna, Bromodomains: Structure, Function and Pharmacology of Inhibition, Biochem. Pharmacol., 2016, 106, 1–18, DOI:10.1016/j.bcp.2015.12.005.
- S. Imai, C. M. Armstrong, M. Kaeberlein and L. Guarente, Transcriptional Silencing and Longevity Protein Sir2 Is an NAD-Dependent Histone Deacetylase, Nature, 2000, 403(6771), 795–800, DOI:10.1038/35001622.
- X.-J. Yang and E. Seto, The Rpd3/Hda1 Family of Lysine Deacetylases: From Bacteria and Yeast to Mice and Men, Nat. Rev. Mol. Cell Biol., 2008, 9(3), 206–218, DOI:10.1038/nrm2346.
- A. J. M. de Ruijter, A. H. van Gennip, H. N. Caron, S. Kemp and A. B. P. van. Kuilenburg, Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family, Biochem. J., 2003, 370(3), 737–749, DOI:10.1042/bj20021321.
- J. Zuber, J. Shi, E. Wang, A. R. Rappaport, H. Herrmann, E. A. Sison, D. Magoon, J. Qi, K. Blatt, M. Wunderlich, M. J. Taylor, C. Johns, A. Chicas, J. C. Mulloy, S. C. Kogan, P. Brown, P. Valent, J. E. Bradner, S. W. Lowe and C. R. Vakoc, RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia, Nature, 2011, 478(7370), 524–528, DOI:10.1038/nature10334.
- P. Valent and J. Zuber, BRD4: A BET(Ter) Target for the Treatment of AML?, Cell Cycle, 2014, 13(5), 689–690, DOI:10.4161/cc.27859.
- Q. Feng, Z. Zhang, M. J. Shea, C. J. Creighton, C. Coarfa, S. G. Hilsenbeck, R. Lanz, B. He, L. Wang, X. Fu, A. Nardone, Y. Song, J. Bradner, N. Mitsiades, C. S. Mitsiades, C. K. Osborne, R. Schiff and B. W. O'Malley, An Epigenomic Approach to Therapy for Tamoxifen-Resistant Breast Cancer, Cell Res., 2014, 24(7), 809–819, DOI:10.1038/cr.2014.71.
- Y. Duan, Y. Guan, W. Qin, X. Zhai, B. Yu and H. Liu, Targeting Brd4 for Cancer Therapy: Inhibitors and Degraders, MedChemComm, 2018, 9(11), 1779–1802, 10.1039/c8md00198g.
- A. Chaidos, V. Caputo, K. Gouvedenou, B. Liu, I. Marigo, M. S. Chaudhry, A. Rotolo, D. F. Tough, N. N. Smithers, A. K. Bassil, T. D. Chapman, N. R. Harker, O. Barbash, P. Tummino, N. Al-Mahdi, A. C. Haynes, L. Cutler, B. Le, A. Rahemtulla, I. Roberts, M. Kleijnen, J. J. Witherington, N. J. Parr, R. K. Prinjha and A. Karadimitris, Potent Antimyeloma Activity of the Novel Bromodomain Inhibitors I-BET151 and I-BET762, Blood, 2014, 123(5), 697–705, DOI:10.1182/blood-2013-01-478420.
- A. Stathis, E. Zucca, M. Bekradda, C. Gomez-Roca, J.-P. Delord, T. de La Motte Rouge, E. Uro-Coste, F. de Braud, G. Pelosi and C. A. French, Clinical Response of Carcinomas Harboring the BRD4–NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628, Cancer Discovery, 2016, 6(5), 492–500, DOI:10.1158/2159-8290.CD-15-1335.
- C. Berthon, E. Raffoux, X. Thomas, N. Vey, C. Gomez-Roca, K. Yee, D. C. Taussig, K. Rezai, C. Roumier, P. Herait, C. Kahatt, B. Quesnel, M. Michallet, C. Recher, F. Lokiec, C. Preudhomme and H. Dombret, Bromodomain Inhibitor OTX015 in Patients with Acute Leukaemia: A Dose-Escalation, Phase 1 Study, Lancet Haematol., 2016, 3(4), e186–e195, DOI:10.1016/S2352-3026(15)00247-1.
- Clinical-Trials.gov, https://clinicaltrials.gov/ct2/show/NCT02308761?%20term=TEN-10%26rank=1, December 11, 2017.
- J. Endo, H. Hikawa, M. Hamada, S. Ishibuchi, N. Fujie, N. Sugiyama, M. Tanaka, H. Kobayashi, K. Sugahara, K. Oshita, K. Iwata, S. Ooike, M. Murata, H. Sumichika, K. Chiba and K. Adachi, A Phenotypic Drug Discovery Study on Thienodiazepine Derivatives as Inhibitors of T Cell Proliferation Induced by CD28 Co-Stimulation Leads to the Discovery of a First Bromodomain Inhibitor, Bioorg. Med. Chem. Lett., 2016, 26(5), 1365–1370, DOI:10.1016/j.bmcl.2016.01.084.
- M. Boi, E. Gaudio, P. Bonetti, I. Kwee, E. Bernasconi, C. Tarantelli, A. Rinaldi, M. Testoni, L. Cascione, M. Ponzoni, A. A. Mensah, A. Stathis, G. Stussi, M. E. Riveiro, P. Herait, G. Inghirami, E. Cvitkovic, E. Zucca and F. Bertoni, The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-Cell Tumor Models and Synergizes with Targeted Drugs, Clin. Cancer Res., 2015, 21(7), 1628–1638, DOI:10.1158/1078-0432.CCR-14-1561.
- N. Schmees, J. Kuhnke, B. Haendler, P. Lienau, A. E. Fernandez-Montalvan, P. Lejeune, S. Siegel and W. Scott, WO2013030150, 2013.
- N. Schmees, J. Kuhnke, B. Haendler, R. Neuhaus, P. Lejeune, S. Siegel, M. Krüger, A. E. Fernandez-Montalvan, H. Künzer and D. Gallenkamp, WO2014048945A1, 2014.
- S. Amorim, A. Stathis, M. Gleeson, S. Iyengar, V. Magarotto, X. Leleu, F. Morschhauser, L. Karlin, F. Broussais, K. Rezai, P. Herait, C. Kahatt, F. Lokiec, G. Salles, T. Facon, A. Palumbo, D. Cunningham, E. Zucca and C. Thieblemont, Bromodomain Inhibitor OTX015 in Patients with Lymphoma or Multiple Myeloma: A Dose-Escalation, Open-Label, Pharmacokinetic, Phase 1 Study, Lancet Haematol., 2016, 3(4), e196–e204, DOI:10.1016/S2352-3026(16)00021-1.
- S. B. Landau and M. KageyWO2016069578, 2016.
- O. Mirguet, R. Gosmini, J. Toum, C. A. Clément, M. Barnathan, J.-M. Brusq, J. E. Mordaunt, R. M. Grimes, M. Crowe, O. Pineau, M. Ajakane, A. Daugan, P. Jeffrey, L. Cutler, A. C. Haynes, N. N. Smithers, C. Chung, P. Bamborough, I. J. Uings, A. Lewis, J. Witherington, N. Parr, R. K. Prinjha and E. Nicodème, Discovery of Epigenetic Regulator I-BET762: Lead Optimization to Afford a Clinical Candidate Inhibitor of the BET Bromodomains, J. Med. Chem., 2013, 56(19), 7501–7515, DOI:10.1021/jm401088k.
- N. Schmees, B. Buchmann, B. Haendler, R. Neuhaus, P. Lejeune, M. Krüger, A. E. Fernandez-Montalvan and H. Künzer, WO2014128070A1, 2014.
- S. Hsiegel, S. Bäurle, A. Cleve, B. Haendler, A. M. Fernández-Montalván, U. Mönning, S. Krause, P. Lejeune, M. Busemann and J. Kuhnke, WO2014128067, 2014.
- B. Ren, C. Y. Zhou and H. Wang, WO2014173241, 2014.
- D. P. Slassi Abdelmalik, WO2016123709, 2016.
- D. S. Hewings, M. Wang, M. Philpott, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, C. Vuppusetty, B. Marsden, S. Knapp, S. J. Conway and T. D. Heightman, 3,5-Dimethylisoxazoles Act As Acetyl-Lysine-Mimetic Bromodomain Ligands, J. Med. Chem., 2011, 54(19), 6761–6770, DOI:10.1021/jm200640v.
- D. S. Hewings, O. Fedorov, P. Filippakopoulos, S. Martin, S. Picaud, A. Tumber, C. Wells, M. M. Olcina, K. Freeman, A. Gill, A. J. Ritchie, D. W. Sheppard, A. J. Russell, E. M. Hammond, S. Knapp, P. E. Brennan and S. J. Conway, Optimization of 3,5-Dimethylisoxazole Derivatives as Potent Bromodomain Ligands, J. Med. Chem., 2013, 56(8), 3217–3227, DOI:10.1021/jm301588r.
- J. Seal, Y. Lamotte, F. Donche, A. Bouillot, O. Mirguet, F. Gellibert, E. Nicodeme, G. Krysa, J. Kirilovsky, S. Beinke, S. McCleary, I. Rioja, P. Bamborough, C.-W. Chung, L. Gordon, T. Lewis, A. L. Walker, L. Cutler, D. Lugo, D. M. Wilson, J. Witherington, K. Lee and R. K. Prinjha, Identification of a Novel Series of BET Family Bromodomain Inhibitors: Binding Mode and Profile of I-BET151 (GSK1210151A), Bioorg. Med. Chem. Lett., 2012, 22(8), 2968–2972, DOI:10.1016/j.bmcl.2012.02.041.
- L. Wang, X. Wu, R. Wang, C. Yang, Z. Li, C. Wang, F. Zhang and P. Yang, BRD4 Inhibition Suppresses Cell Growth, Migration and Invasion of Salivary Adenoid Cystic Carcinoma, Biol. Res., 2017, 50, 19, DOI:10.1186/s40659-017-0124-9.
- M. C. Hewitt, Y. Leblanc, V. S. Gehling, R. G. Vaswani, A. Côté, C. G. Nasveschuk, A. M. Taylor, J.-C. Harmange, J. E. Audia, E. Pardo, R. Cummings, S. Joshi, P. Sandy, J. A. Mertz, R. J. Sims, L. Bergeron, B. M. Bryant, S. Bellon, F. Poy, H. Jayaram, Y. Tang and B. K. Albrecht, Development of Methyl Isoxazoleazepines as Inhibitors of BET, Bioorg. Med. Chem. Lett., 2015, 25(9), 1842–1848, DOI:10.1016/j.bmcl.2015.03.045.
- M. R. McKeown, D. L. Shaw, H. Fu, S. Liu, X. Xu, J. J. Marineau, Y. Huang, X. Zhang, D. L. Buckley, A. Kadam, Z. Zhang, S. C. Blacklow, J. Qi, W. Zhang and J. E. Bradner, Biased Multicomponent Reactions to Develop Novel Bromodomain Inhibitors, J. Med. Chem., 2014, 57(21), 9019–9027, DOI:10.1021/jm501120z.
- P. C. C. Liu, X. S. M. Liu, M. C. Stubbs, T. Maduskuie, R. Sparks, N. Zolotarjova, J. Li, X. M. Wen, M. Favata, P. Feldman, A. Volgina, D. DiMatteo, R. Collins, N. Falahatpisheh, P. Polam, Y. Li, M. Covington, S. Diamond-Fosbenner, R. Wynn, T. Burn, K. Vaddi, S. Yeleswaram, A. P. Combs, W. Q. Yao, R. Huber, P. Scherle and G. Hollis, Discovery of a novel BET inhibitor INCB054329, Cancer Res., 2015, 75, 3523 Search PubMed.
- V. S. Gehling, M. C. Hewitt, R. G. Vaswani, Y. Leblanc, A. Côté, C. G. Nasveschuk, A. M. Taylor, J.-C. Harmange, J. E. Audia, E. Pardo, S. Joshi, P. Sandy, J. A. Mertz, R. J. Sims, L. Bergeron, B. M. Bryant, S. Bellon, F. Poy, H. Jayaram, R. Sankaranarayanan, S. Yellapantula, N. Bangalore Srinivasamurthy, S. Birudukota and B. K. Albrecht, Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors, ACS Med. Chem. Lett., 2013, 4(9), 835–840, DOI:10.1021/ml4001485.
- X. Ran, Y. Zhao, L. Liu, L. Bai, C.-Y. Yang, B. Zhou, J. L. Meagher, K. Chinnaswamy, J. A. Stuckey and S. Wang, Structure-Based Design of γ-Carboline Analogues as Potent and Specific BET Bromodomain Inhibitors, J. Med. Chem., 2015, 58(12), 4927–4939, DOI:10.1021/acs.jmedchem.5b00613.
- Y. Zhao, C.-Y. Yang and S. Wang, The Making of I-BET762, a BET Bromodomain Inhibitor Now in Clinical Development, J. Med. Chem., 2013, 56(19), 7498–7500, DOI:10.1021/jm4014407.
- O. Mirguet, Y. Lamotte, F. Donche, J. Toum, F. Gellibert, A. Bouillot, R. Gosmini, V.-L. Nguyen, D. Delannée, J. Seal, F. Blandel, A.-B. Boullay, E. Boursier, S. Martin, J.-M. Brusq, G. Krysa, A. Riou, R. Tellier, A. Costaz, P. Huet, Y. Dudit, L. Trottet, J. Kirilovsky and E. Nicodeme, From ApoA1 Upregulation to BET Family Bromodomain Inhibition: Discovery of I-BET151, Bioorg. Med. Chem. Lett., 2012, 22(8), 2963–2967, DOI:10.1016/j.bmcl.2012.01.125.
- H. G. Ozer, D. El-Gamal, B. Powell, Z. A. Hing, J. S. Blachly, B. Harrington, S. Mitchell, N. R. Grieselhuber, K. Williams, T.-H. Lai, L. Alinari, R. A. Baiocchi, L. Brinton, E. Baskin, M. Cannon, L. Beaver, V. M. Goettl, D. M. Lucas, J. A. Woyach, D. Sampath, A. M. Lehman, L. Yu, J. Zhang, Y. Ma, Y. Zhang, W. Spevak, S. Shi, P. Severson, R. Shellooe, H. Carias, G. Tsang, K. Dong, T. Ewing, A. Marimuthu, C. Tantoy, J. Walters, L. Sanftner, H. Rezaei, M. Nespi, B. Matusow, G. Habets, P. Ibrahim, C. Zhang, E. A. Mathé, G. Bollag, J. C. Byrd and R. Lapalombella, BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor, Cancer Discovery, 2018, 8(4), 458–477, DOI:10.1158/2159-8290.CD-17-0902.
- D. Liu, J. Pratt, L. Wang, L. A. Hasvold and A. BogdanUS20140256710, 2014.
- H. Engelhardt, L. Martin and C. Smethurst, WO2015022332, 2015.
- H. Engelhardt, WO2015169962, 2015.
- K. F. McDaniel, L. Wang, T. Soltwedel, S. D. Fidanze, L. A. Hasvold, D. Liu, R. A. Mantei, J. K. Pratt, G. S. Sheppard, M. H. Bui, E. J. Faivre, X. Huang, L. Li, X. Lin, R. Wang, S. E. Warder, D. Wilcox, D. H. Albert, T. J. Magoc, G. Rajaraman, C. H. Park, C. W. Hutchins, J. J. Shen, R. P. Edalji, C. C. Sun, R. Martin, W. Gao, S. Wong, G. Fang, S. W. Elmore, Y. Shen and W. M. Kati, Discovery of N -(4-(2,4-Difluorophenoxy)-3-(6-Methyl-7-Oxo-6,7-Dihydro-1 H -Pyrrolo2,3- c Pyridin-4-Yl)Phenyl)Ethane sulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extra terminal Domain (BET) Family Bromodomain Inhibitor, J. Med. Chem., 2017, 60(20), 8369–8384, DOI:10.1021/acs.jmedchem.7b00746.
- L. Wang, J. K. Pratt, T. Soltwedel, G. S. Sheppard, S. D. Fidanze, D. Liu, L. A. Hasvold, R. A. Mantei, J. H. Holms, W. J. McClellan, M. D. Wendt, C. Wada, R. Frey, T. M. Hansen, R. Hubbard, C. H. Park, L. Li, T. J. Magoc, D. H. Albert, X. Lin, S. E. Warder, P. Kovar, X. Huang, D. Wilcox, R. Wang, G. Rajaraman, A. M. Petros, C. W. Hutchins, S. C. Panchal, C. Sun, S. W. Elmore, Y. Shen, W. M. Kati and K. F. McDaniel, Fragment-Based, Structure-Enabled Discovery of Novel Pyridones and Pyridone Macrocycles as Potent Bromodomain and Extra-Terminal Domain (BET) Family Bromodomain Inhibitors, J. Med. Chem., 2017, 60(9), 3828–3850, DOI:10.1021/acs.jmedchem.7b00017.
- S. Vadivelu S. Rajagopal M. Chinnapattu P. K. Gondrala and D. Sivanandhan, WO2016157221, 2016.
- C. Chung, A. W. Dean, J. M. Woolven and P. Bamborough, Fragment-Based Discovery of Bromodomain Inhibitors Part 1: Inhibitor Binding Modes and Implications for Lead Discovery, J. Med. Chem., 2012, 55(2), 576–586, DOI:10.1021/jm201320w.
- D. Amans, S. J. Atkinson, L. A. Harrison, D. J. Hirst, R. P. Law, M. Lindon, A. Preston, J. T. Seal and C. R. Wellaway, WO2014140076, 2014.
- K. W. Bair, T. Herbertz, G. S. Kauffman, K. J. Kayser-Bricker, G. P. Luke, M. W. Martin, D. S. Millan, S. R. Schiller and A. C. Talbot, WO2015074064, 2015.
- N. Schmees, B. Haendler, D. Stöckigt, D. Gallenkamp, R. A. Bissell and R. A. BouglasWO2015004075, 2015.
- P. V. Fish, P. Filippakopoulos, G. Bish, P. E. Brennan, M. E. Bunnage, A. S. Cook, O. Federov, B. S. Gerstenberger, H. Jones, S. Knapp, B. Marsden, K. Nocka, D. R. Owen, M. Philpott, S. Picaud, M. J. Primiano, M. J. Ralph, N. Sciammetta and J. D. Trzupek, Identification of a Chemical Probe for Bromo and Extra C-Terminal Bromodomain Inhibition through Optimization of a Fragment-Derived Hit, J. Med. Chem., 2012, 55(22), 9831–9837, DOI:10.1021/jm3010515.
- R. Gosmini, V. L. Nguyen, J. Toum, C. Simon, J.-M. G. Brusq, G. Krysa, O. Mirguet, A. M. Riou-Eymard, E. V. Boursier, L. Trottet, P. Bamborough, H. Clark, C. Chung, L. Cutler, E. H. Demont, R. Kaur, A. J. Lewis, M. B. Schilling, P. E. Soden, S. Taylor, A. L. Walker, M. D. Walker, R. K. Prinjha and E. Nicodème, The Discovery of I-BET726 (GSK1324726A), a Potent Tetrahydroquinoline ApoA1 Up-Regulator and Selective BET Bromodomain Inhibitor, J. Med. Chem., 2014, 57(19), 8111–8131, DOI:10.1021/jm5010539.
- A. F. Abdel-Magid, Inhibitors of BRD4 as Potential Cancer Therapy, ACS Med. Chem. Lett., 2016, 7(8), 728–729, DOI:10.1021/acsmedchemlett.6b00259.
- H. Engelhardt, D. Gianni and C. Smethurst, US20160129001, 2016.
- J. Blank, V. Bordas, S. Cotesta, V. Guagnano, H. Rueeger and A. Vaupel, US20140349990, 2014.
- L. A. Hasvold, D. Liu, C. H. Park, J. K. Pratt, G. S. Sheppard and L. WangWO2013158952, 2013.
- M. Hügle, X. Lucas, G. Weitzel, D. Ostrovskyi, B. Breit, S. Gerhardt, O. Einsle, S. Günther and D. Wohlwend, 4-Acyl Pyrrole Derivatives Yield Novel Vectors for Designing Inhibitors of the Acetyl-Lysine Recognition Site of BRD4(1), J. Med. Chem., 2016, 59(4), 1518–1530, DOI:10.1021/acs.jmedchem.5b01267.
- X. Lucas, D. Wohlwend, M. Hügle, K. Schmidtkunz, S. Gerhardt, R. Schüle, M. Jung, O. Einsle and S. Günther, 4-Acyl Pyrroles: Mimicking Acetylated Lysines in Histone Code Reading, Angew. Chem., Int. Ed., 2013, 52(52), 14055–14059, DOI:10.1002/anie.201307652.
- L. Zhao, D. Cao, T. Chen, Y. Wang, Z. Miao, Y. Xu, W. Chen, X. Wang, Y. Li, Z. Du, B. Xiong, J. Li, C. Xu, N. Zhang, J. He and J. Shen, Fragment-Based Drug Discovery of 2-Thiazolidinones as Inhibitors of the Histone Reader BRD4 Bromodomain, J. Med. Chem., 2013, 56(10), 3833–3851, DOI:10.1021/jm301793a.
- A. M. Ayoub, L. M. L. Hawk, R. J. Herzig, J. Jiang, A. J. Wisniewski, C. T. Gee, P. Zhao, J.-Y. Zhu, N. Berndt, N. K. Offei-Addo, T. G. Scott, J. Qi, J. E. Bradner, T. R. Ward, E. Schönbrunn, G. I. Georg and W. C. K. Pomerantz, BET Bromodomain Inhibitors with One-Step Synthesis Discovered from Virtual Screen, J. Med. Chem., 2017, 60(12), 4805–4817, DOI:10.1021/acs.jmedchem.6b01336.
- B. Raux, Y. Voitovich, C. Derviaux, A. Lugari, E. Rebuffet, S. Milhas, S. Priet, T. Roux, E. Trinquet, J.-C. Guillemot, S. Knapp, J.-M. Brunel, A. Yu. Fedorov, Y. Collette, P. Roche, S. Betzi, S. Combes and X. Morelli, Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-Terminal Domain (BET) Proteins, J. Med. Chem., 2016, 59(4), 1634–1641, DOI:10.1021/acs.jmedchem.5b01708.
- E. von Schaper, Roche Bets on Bromodomains, Nat. Biotechnol., 2016, 34(4), 361–362, DOI:10.1038/nbt0416-361.
- L. Ouyang, L. Zhang, J. Liu, L. Fu, D. Yao, Y. Zhao, S. Zhang, G. Wang, G. He and B. Liu, Discovery of a Small-Molecule Bromodomain-Containing Protein 4 (BRD4) Inhibitor That Induces AMP-Activated Protein Kinase-Modulated Autophagy-Associated Cell Death in Breast Cancer, J. Med. Chem., 2017, 60(24), 9990–10012, DOI:10.1021/acs.jmedchem.7b00275.
- X. Xue, Y. Zhang, Z. Liu, M. Song, Y. Xing, Q. Xiang, Z. Wang, Z. Tu, Y. Zhou, K. Ding and Y. Xu, Discovery of Benzo Cd Indol-2(1 H)-Ones as Potent and Specific BET Bromodomain Inhibitors: Structure-Based Virtual Screening, Optimization, and Biological Evaluation, J. Med. Chem., 2016, 59(4), 1565–1579, DOI:10.1021/acs.jmedchem.5b01511.
- H.-J. Zhong, L. Lu, K.-H. Leung, C. C. L. Wong, C. Peng, S.-C. Yan, D.-L. Ma, Z. Cai, H.-M. David Wang and C.-H. Leung, An Iridium (III)-Based Irreversible Protein–Protein Interaction Inhibitor of BRD4 as a Potent Anticancer Agent, Chem. Sci., 2015, 6(10), 5400–5408, 10.1039/C5SC02321A.
- G. W. Rhyasen, M. M. Hattersley, Y. Yao, A. Dulak, W. Wang, P. Petteruti, I. L. Dale, S. Boiko, T. Cheung, J. Zhang, S. Wen, L. Castriotta, D. Lawson, M. Collins, L. Bao, M. J. Ahdesmaki, G. Walker, G. O'Connor, T. C. Yeh, A. A. Rabow, J. R. Dry, C. Reimer, P. Lyne, G. B. Mills, S. E. Fawell, M. J. Waring, M. Zinda, E. Clark and H. Chen, AZD5153: A Novel Bivalent BET Bromodomain Inhibitor Highly Active against Hematologic Malignancies, Mol. Cancer Ther., 2016, 15(11), 2563–2574, DOI:10.1158/1535-7163.MCT-16-0141.
- I. L. AZD5153 in Patients with Relapsed or Refractory Solid Tumors, Clinical-Trials.gov, https://clinicaltrials.gov/ct2/show/NCT03205176?term=azd-5153%26rank=1, July 2, 2017.
- M. Tanaka, J. M. Roberts, H.-S. Seo, A. Souza, J. Paulk, T. G. Scott, S. L. DeAngelo, S. Dhe-Paganon and J. E. Bradner, Design and Characterization of Bivalent BET Inhibitors, Nat. Chem. Biol., 2016, 12(12), 1089–1096, DOI:10.1038/nchembio.2209.
- R. H. Bradbury, D. G. Acton, N. L. Broadbent, A. N. Brooks, G. R. Carr, G. Hatter, B. R. Hayter, K. J. Hill, N. J. Howe, R. D. O. Jones, D. Jude, S. G. Lamont, S. A. Loddick, H. L. McFarland, Z. Parveen, A. A. Rabow, G. Sharma-Singh, N. C. Stratton, A. G. Thomason, D. Trueman, G. E. Walker, S. L. Wells, J. Wilson and J. M. Wood, Discovery of AZD3514, a Small-Molecule Androgen Receptor Downregulator for Treatment of Advanced Prostate Cancer, Bioorg. Med. Chem. Lett., 2013, 23(7), 1945–1948, DOI:10.1016/j.bmcl.2013.02.056.
- R. H. Bradbury, R. Callis, G. R. Carr, H. Chen, E. Clark, L. Feron, S. Glossop, M. A. Graham, M. Hattersley, C. Jones, S. G. Lamont, G. Ouvry, A. Patel, J. Patel, A. A. Rabow, C. A. Roberts, S. Stokes, N. Stratton, G. E. Walker, L. Ward, D. Whalley, D. Whittaker, G. Wrigley and M. J. Waring, Optimization of a Series of Bivalent Triazolopyridazine Based Bromodomain and Extraterminal Inhibitors: The Discovery of (3 R)-4-2-4-1-(3-Methoxy-1,2,4Triazolo4,3-b Pyridazin-6-Yl)-4-PiperidylPhenoxyEthyl-1,3-Dimethyl-Piperazin-2-One (AZD5153), J. Med. Chem., 2016, 59(17), 7801–7817, DOI:10.1021/acs.jmedchem.6b00070.
- S. A. Loddick, S. J. Ross, A. G. Thomason, D. M. Robinson, G. E. Walker, T. P. J. Dunkley, S. R. Brave, N. Broadbent, N. C. Stratton, D. Trueman, E. Mouchet, F. S. Shaheen, V. N. Jacobs, M. Cumberbatch, J. Wilson, R. D. O. Jones, R. H. Bradbury, A. Rabow, L. Gaughan, C. Womack, S. T. Barry, C. N. Robson, S. E. Critchlow, S. R. Wedge and A. N. Brooks, AZD3514: A Small Molecule That Modulates Androgen Receptor Signaling and Function In Vitro and In Vivo, Mol. Cancer Ther., 2013, 12(9), 1715–1727, DOI:10.1158/1535-7163.MCT-12-1174.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4, Chem. Biol., 2015, 22(6), 755–763, DOI:10.1016/j.chembiol.2015.05.009.
- T. Shimamura, Z. Chen, M. Soucheray, J. Carretero, E. Kikuchi, J. H. Tchaicha, Y. Gao, K. A. Cheng, T. J. Cohoon, J. Qi, E. Akbay, A. C. Kimmelman, A. L. Kung, J. E. Bradner and K.-K. Wong, Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non-Small Cell Lung Cancer, Clin. Cancer Res., 2013, 19(22), 6183–6192, DOI:10.1158/1078-0432.CCR-12-3904.
- C. Y. Fong, O. Gilan, E. Y. N. Lam, A. F. Rubin, S. Ftouni, D. Tyler, K. Stanley, D. Sinha, P. Yeh, J. Morison, G. Giotopoulos, D. Lugo, P. Jeffrey, S. C.-W. Lee, C. Carpenter, R. Gregory, R. G. Ramsay, S. W. Lane, O. Abdel-Wahab, T. Kouzarides, R. W. Johnstone, S.-J. Dawson, B. J. P. Huntly, R. K. Prinjha, A. T. Papenfuss and M. A. Dawson, BET Inhibitor Resistance Emerges from Leukaemia Stem Cells, Nature, 2015, 525(7570), 538–542, DOI:10.1038/nature14888.
- P. Filippakopoulos and S. Knapp, Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation, Nat. Rev. Drug Discovery, 2014, 13(5), 337–356, DOI:10.1038/nrd4286.
- M. Jung, K. A. Gelato, A. Fernández-Montalván, S. Siegel and B. Haendler, Targeting BET Bromodomains for Cancer Treatment, Epigenomics, 2015, 7(3), 487–501, DOI:10.2217/epi.14.91.
- Z. Liu, P. Wang, H. Chen, E. A. Wold, B. Tian, A. R. Brasier and J. Zhou, Drug Discovery Targeting Bromodomain-Containing Protein 4, J. Med. Chem., 2017, 60(11), 4533–4558, DOI:10.1021/acs.jmedchem.6b01761.
- M. A. Glozak and E. Seto, Histone Deacetylases and Cancer, Oncogene, 2007, 26(37), 5420–5432, DOI:10.1038/sj.onc.1210610.
- T. A. Miller, D. J. Witter and S. Belvedere, Histone Deacetylase Inhibitors, J. Med. Chem., 2003, 46(24), 5097–5116, DOI:10.1021/jm0303094.
- E. Hahnen, J. Hauke, C. Tränkle, I. Y. Eyüpoglu, B. Wirth and I. Blümcke, Histone Deacetylase Inhibitors: Possible Implications for Neurodegenerative Disorders, Expert Opin. Invest. Drugs, 2008, 17(2), 169–184, DOI:10.1517/13543784.17.2.169.
- M. R. Acharya, A. Sparreboom, J. Venitz and W. D. Figg, Rational Development of Histone Deacetylase Inhibitors as Anticancer Agents: A Review, Mol. Pharmacol., 2005, 68(4), 917–932, DOI:10.1124/mol.105.014167.
- T. Beckers, C. Burkhardt, H. Wieland, P. Gimmnich, T. Ciossek, T. Maier and K. Sanders, Distinct Pharmacological Properties of Second Generation HDAC Inhibitors with the Benzamide or Hydroxamate Head Group, Int. J. Cancer, 2007, 121(5), 1138–1148, DOI:10.1002/ijc.22751.
- C. Schwartz, S. Bouchat, C. Marban, V. Gautier, C. Van Lint, O. Rohr and V. Le Douce, On the Way to Find a Cure: Purging Latent HIV-1 Reservoirs, Biochem. Pharmacol., 2017, 146, 10–22, DOI:10.1016/j.bcp.2017.07.001.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation, Proc. Natl. Acad. Sci., 2001, 98(15), 8554–8559, DOI:10.1073/pnas.141230798.
- J. S. Schneekloth, F. N. Fonseca, M. Koldobskiy, A. Mandal, R. Deshaies, K. Sakamoto and C. M. Crews, Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation, J. Am. Chem. Soc., 2004, 126(12), 3748–3754, DOI:10.1021/ja039025z.
- H. Lee, D. Puppala, E.-Y. Choi, H. Swanson and K.-B. Kim, Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach: A Useful Chemical Genetic Tool, ChemBioChem, 2007, 8(17), 2058–2062, DOI:10.1002/cbic.200700438.
- K. C. Carmony and K.-B. Kim, PROTAC-Induced Proteolytic Targeting, Ubiquitin Family Modifiers and the Proteasome, Methods in Molecular Biology, ed. R. J. Dohmen and M. Scheffner, Humana Press, Totowa, NJ, 2012, vol. 832, pp 627–638, DOI:10.1007/978-1-61779-474-2_44.
- S. An and L. Fu, Small-Molecule PROTACs: An Emerging and Promising Approach for the Development of Targeted Therapy Drugs, EBioMedicine, 2018, 36, 553–562, DOI:10.1016/j.ebiom.2018.09.005.
- A. R. Schneekloth, M. Pucheault, H. S. Tae and C. M. Crews, Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics, Bioorg. Med. Chem. Lett., 2008, 18(22), 5904–5908, DOI:10.1016/j.bmcl.2008.07.114.
- O. Shuvalov, A. Kizenko, A. Shakirova, O. Fedorova, A. Petukhov, N. Aksenov, E. Vasileva, A. Daks and N. Barlev, Nutlin Sensitizes Lung Carcinoma Cells to Interferon-Alpha Treatment in MDM2-Dependent but P53-Independent Manner, Biochem. Biophys. Res. Commun., 2018, 495(1), 1233–1239, DOI:10.1016/j.bbrc.2017.11.118.
- Y. Itoh, M. Ishikawa, M. Naito and Y. Hashimoto, Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins, J. Am. Chem. Soc., 2010, 132(16), 5820–5826, DOI:10.1021/ja100691p.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation, Science, 2015, 348(6241), 1376–1381, DOI:10.1126/science.aab1433.
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs, Nat. Chem. Biol., 2015, 11(8), 611–617, DOI:10.1038/nchembio.1858.
- K. Raina, J. Lu, Y. Qian, M. Altieri, D. Gordon, A. M. K. Rossi, J. Wang, X. Chen, H. Dong, K. Siu, J. D. Winkler, A. P. Crew, C. M. Crews and K. G. Coleman, PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(26), 7124–7129, DOI:10.1073/pnas.1521738113.
- T. Neklesa, L. B. Snyder, R. R. Willard, N. Vitale, K. Raina, J. Pizzano, D. A. Gordon, M. Bookbinder, J. Macaluso, H. Dong, Z. Liu, C. Ferraro, G. Wang, J. Wang, C. M. Crews, J. Houston, A. P. Crew and I. Taylor, An oral androgen receptor PROTAC degrader for prostate cancer, J. Clin. Oncol., 2018, 36(6_suppl), 381, DOI:10.1200/JCO.2018.36.6_suppl.381.
- J. Qi and G. Zhang, Proteolysis-Targeting Chimeras for Targeting Protein for Degradation, Future Med. Chem., 2019, 11(7), 723–741, DOI:10.4155/fmc-2018-0557.
- S. Crunkhorn, Selectively Targeting Proteins for Degradation, Nat. Rev. Drug Discovery, 2015, 14(7), 459, DOI:10.1038/nrd4670.
- J. A. Doudna and E. Charpentier, The New Frontier of Genome Engineering with CRISPR-Cas9, Science, 2014, 346(6213), 1258096, DOI:10.1126/science.1258096.
- N. Ohoka, N. Shibata, T. Hattori and M. Naito, Protein Knockdown Technology: Application of Ubiquitin Ligase to Cancer Therapy, Curr. Cancer Drug Targets, 2016, 16(2), 136–146, DOI:.
- K. R. Chi, Drug Developers Delve into the Cell's Trash-Disposal Machinery, Nat. Rev. Drug Discovery, 2016, 15(5), 295–297, DOI:10.1038/nrd.2016.86.
- F. Zhang and S. Ma, Disrupting Acetyl-lysine Interactions: Recent Advance in the Development of BET Inhibitors, Curr. Drug Targets, 2018, 19(10), 1148–1165, DOI:.
- D. Komander and M. Rape, The Ubiquitin Code, Annu. Rev. Biochem., 2012, 81(1), 203–229, DOI:10.1146/annurev-biochem-060310-170328.
- R. Yau and M. Rape, The Increasing Complexity of the Ubiquitin Code, Nat. Cell Biol., 2016, 18(6), 579–586, DOI:10.1038/ncb3358.
- P. M. Cromm and C. M. Crews, Targeted Protein Degradation: From Chemical Biology to Drug Discovery, Cell Chem. Biol., 2017, 24(9), 1181–1190, DOI:10.1016/j.chembiol.2017.05.024.
- R. J. Deshaies, Prime Time for PROTACs, Nat. Chem. Biol., 2015, 11(9), 634–635, DOI:10.1038/nchembio.1887.
- A. Hershko and A. Ciechanover, The Ubiquitin System, Annu. Rev. Biochem., 1998, 67, 425–469 CrossRef CAS.
- D. L. Buckley and C. M. Crews, Small-Molecule Control of Intracellular Protein Levels through Modulation of the Ubiquitin Proteasome System, Angew. Chem., Int. Ed., 2014, 53(9), 2312–2330, DOI:10.1002/anie.201307761.
- D. P. Bondeson and C. M. Crews, Targeted Protein Degradation by Small Molecules, Annu. Rev. Pharmacol. Toxicol., 2017, 57(1), 107–123, DOI:10.1146/annurev-pharmtox-010715-103507.
- G. M. Burslem and C. M. Crews, Small-Molecule Modulation of Protein Homeostasis, Chem. Rev., 2017, 117(17), 11269–11301, DOI:10.1021/acs.chemrev.7b00077.
- K. Raina and C. M. Crews, Targeted Protein Knockdown Using Small Molecule Degraders, Curr. Opin. Chem. Biol., 2017, 39, 46–53, DOI:10.1016/j.cbpa.2017.05.016.
- P. Ottis, M. Toure, P. M. Cromm, E. Ko, J. L. Gustafson and C. M. Crews, Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation, ACS Chem. Biol., 2017, 12(10), 2570–2578, DOI:10.1021/acschembio.7b00485.
- A. C. Lai and C. M. Crews, Induced Protein Degradation: An Emerging Drug Discovery Paradigm, Nat. Rev. Drug Discovery, 2017, 16(2), 101–114, DOI:10.1038/nrd.2016.211.
- P. Ottis and C. M. Crews, Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy, ACS Chem. Biol., 2017, 12(4), 892–898, DOI:10.1021/acschembio.6b01068.
- E. S. Fischer, K. Böhm, J. R. Lydeard, H. Yang, M. B. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. M. Lingaraju, R. B. Tichkule, M. Schebesta, W. C. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. E. J. Beckwith, J. W. Harper, J. L. Jenkins and N. H. Thomä, Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide, Nature, 2014, 512(7512), 49–53, DOI:10.1038/nature13527.
- J. S. Lazo and E. R. Sharlow, Drugging Undruggable Molecular Cancer Targets, Annu. Rev. Pharmacol. Toxicol., 2016, 56(1), 23–40, DOI:10.1146/annurev-pharmtox-010715-103440.
- W.-C. Hon, M. I. Wilson, K. Harlos, T. D. W. Claridge, C. W. Pugh, P. H. Maxwell, P. J. Ratcliffe, D. I. Stuart and E. Y. Jones, Structural Basis for the Recognition of Hydroxyproline in HIF-1a by PVHL, Nature, 2002, 417(4), 975 CrossRef CAS.
- H. Brooks, B. Lebleu and E. Vives, Tat Peptide-Mediated Cellular Delivery: Back to Basics, Adv. Drug Delivery Rev., 2005, 57(4), 559–577, DOI:10.1016/j.addr.2004.12.001.
- C. Bechara and S. Sagan, Cell-Penetrating Peptides: 20 Years Later, Where Do We Stand?, FEBS Lett., 2013, 587(12), 1693–1702, DOI:10.1016/j.febslet.2013.04.031.
- Y. Zou, D. Ma and Y. Wang, The PROTAC Technology in Drug Development: The PROTAC Technology in Drug Development, Cell Biochem. Funct., 2019, 37(1), 21–30, DOI:10.1002/cbf.3369.
- C. M. Crews, Inducing Protein Degradation as a Therapeutic Strategy, J. Med. Chem., 2018, 61(2), 403–404, DOI:10.1021/acs.jmedchem.7b01333.
- K. M. Sakamoto, Protacs for Treatment of Cancer, Pediatr. Res., 2010, 67(5), 505–508, DOI:10.1203/PDR.0b013e3181d35017.
- M. Zengerle, K.-H. Chan and A. Ciulli, Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4, ACS Chem. Biol., 2015, 10(8), 1770–1777, DOI:10.1021/acschembio.5b00216.
- D. L. Buckley, K. Raina, N. Darricarrere, J. Hines, J. L. Gustafson, I. E. Smith, A. H. Miah, J. D. Harling and C. M. Crews, HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins, ACS Chem. Biol., 2015, 10(8), 1831–1837, DOI:10.1021/acschembio.5b00442.
- V. Zoppi, S. J. Hughes, C. Maniaci, A. Testa, T. Gmaschitz, C. Wieshofer, M. Koegl, K. M. Riching, D. L. Daniels, A. Spallarossa and A. Ciulli, Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7, J. Med. Chem., 2019, 62(2), 699–726, DOI:10.1021/acs.jmedchem.8b01413.
- G. Eren, A. Bruno, S. Guntekin-Ergun, R. Cetin-Atalay, F. Ozgencil, Y. Ozkan, M. Gozelle, S. G. Kaya and G. Costantino, Pharmacophore Modeling and Virtual Screening Studies to Identify Novel Selective SIRT2 Inhibitors, J. Mol. Graphics Modell., 2019, 89, 60–73, DOI:10.1016/j.jmgm.2019.02.014.
- M. Schiedel, D. Herp, S. Hammelmann, S. Swyter, A. Lehotzky, D. Robaa, J. Oláh, J. Ovádi, W. Sippl and M. Jung, Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals), J. Med. Chem., 2018, 61(2), 482–491, DOI:10.1021/acs.jmedchem.6b01872.
- H. M. Hesham, D. S. Lasheen and K. A. M. Abouzid, Chimeric HDAC Inhibitors: Comprehensive Review on the HDAC-Based Strategies Developed to Combat Cancer, Med. Res. Rev., 2018, 38(6), 2058–2109, DOI:10.1002/med.21505.
- C. Seidel, M. Schnekenburger, M. Dicato and M. Diederich, Histone Deacetylase 6 in Health and Disease, Epigenomics, 2015, 7(1), 103–118, DOI:10.2217/epi.14.69.
- K. Yang, Y. Song, H. Xie, H. Wu, Y.-T. Wu, E. D. Leisten and W. Tang, Development of the First Small Molecule Histone Deacetylase 6 (HDAC6) Degraders, Bioorg. Med. Chem. Lett., 2018, 28(14), 2493–2497, DOI:10.1016/j.bmcl.2018.05.057.
| This journal is © The Royal Society of Chemistry 2021 |