
Recent advances in the preparation of semifluorinated polymers
Joseph A.
Jaye
and
Ellen M.
Sletten
 *
*
Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: Sletten@chem.ucla.edu
First published on 3rd November 2021
Abstract
Synthesis of semifluorinated polymers containing fluorous groups on the backbone or as side chains is an increasingly popular field of research. Introduction of fluorine in polymers offers many potential benefits such as decreased surface energy, modification of thermal properties, and enhanced self-assembly. Preparation of novel semifluorinated polymers offers an interesting challenge, as fluorous monomers are less common, often need to be synthesized, and can have different reactivities than their hydrocarbon analogues. Alternatively, polymers can be fluorinated as a post-polymerization modification, however achieving a balance between fluorination and polymer degradation remains a challenge. In this mini-review, we will explore recent methods in the post-polymerization fluorination of commodity polymers as well as the polymerization of less common fluorous monomers.
Introduction
Fluorinated polymers have widespread utility as a result of the high chemical and thermal stability imparted by the C–F bond.1–8 In addition, C–F bonds bestow other beneficial properties on materials such as decreased surface energies and increased phase-separation in both solution and solid states.9–16 This can be observed with the comparison between high density polyethylene (HDPE, 1), poly(vinylidene difluoride) (PVDF, 2), and the most widely produced fluoropolymer, poly(tetrafluoroethylene, 3), better known as Teflon™ (Fig. 1A). With increasing weight percent fluorine (wt% fluorine), melting point and decomposition temperature are increased.8,17–19 Water contact angle also increases from HDPE to PTFE due to the extreme hydrophobicity of perfluorocarbons; however, the dipoles within PVDF prevent the same trend between contact angle and wt% fluorine.11,12,14 As a result of the unique properties imparted by fluorine, fluorinated polymers have been incorporated into materials such as electronics, weatherable clothing, non-stick pans, dental floss, and insulators.6,20–22 | ||
| Fig. 1 Overview of development of fluoropolymers. (A) Comparison of thermal properties and surface energies of commercial polymers. (B) Non-selective post-polymerization fluorination of polymers and the process of “upcycling” commercial polymers. (C) Architecture control of fluorinated polymers through copolymerization of fluorinated and non-fluorinated monomers. | ||
Although an essential component to many modern-day devices, PTFE has some significant drawbacks. The high crystallinity of the polymer, coupled with its aversion to organic and fluorous solvents, have made it difficult to process into advanced materials.23–25 Furthermore, its monomer, tetrafluoroethylene, is an explosive and toxic gas.26–28 Due to emulsion polymerization being standard for the production of fluoropolymers, fluorinated surfactants are required to achieve maximum molar mass. These fluorinated surfactants have received intense scrutiny for their toxicity and bioaccumulation,29–32 leading to an expanding field of research in perfluoroalkylated substance (PFAS) remediation.33–35 The processability and safety challenges apparent with PTFE are also encountered in other commercial fluoropolymers with tetrafluoroethylene-derived monomers.22,36
Alternative polymerization methods are necessary to overcome the long-standing issues in the preparation of fluorinated polymers. There has been significant progress towards the controlled polymerization of vinylidene difluoride, yielding semi-fluorinated polymers with improved compositional control.37–40 After tetrafluoroethylene and vinylidene fluoride, two common monomers to obtain (semi)fluorinated polymers are fluorinated acrylates41,42 and styrenes.43–45 Other routes include polymerization of perfluorovinyl ethers via a [2 + 2] cycloaddition to give poly(perfluorocyclobutyl) ethers or use of fluorous diols and isocyanate monomers to generate fluorinated polyurethanes. These methodologies have already been discussed in detail in recent reviews,23,46 and will not be covered here. Inorganic fluoropolymers such as those containing phosphorus and silicon have also been previously highlighted47,48 and are beyond the scope of this review. In recent years, there have been multiple extensive reviews on perfluorinated and semi-fluorinated polymers for biomedical and commercial purposes, all with a heavy emphasis on the polymerization of fluorous olefins,7,22,49–51 as well as a review summarizing the solution self-assembly of an array of fluoropolymers.15
Here we highlight two more recent approaches to the creation of semifluorinated polymers: post-polymerizaiton fluorination of commodity polymers (Fig. 1B) and the polymerization of new fluorinated monomers (Fig. 1C). The first approach leverages new chemistries for installation of perfluorocarbons. Imparting unique, advanced properties into common industrial polymers has been termed “upcycling”. Upcycling has the advantage of widespread availability of a polymer starting material but lacks homogeneity in the final materials. To create defined semifluorinated polymers, new fluorinated monomers with unique reactivity are necessary to overcome the explosive nature of tetrafluoroethylene and complement fluorinated styrene, acrylate, and vinyl ether monomers. We cover the recent additions of fluorinated lactide, oxazoline, norbornene, and diiodoperfluoroalkane monomers to create an array of semifluorinated polymers with pendant perfluorinated chains and/or perfluorination installed directly in the backbone. The new monomers allow for more precise control of polymer physical properties and architectures with the trade-off of generally higher cost of synthesis.
Post-polymerization fluorination of commodity polymers
Post-polymerization fluorination of aromatic group-containing polymers
Commercial polymers containing an aromatic ring, such as polystyrenes (5a–5c), numerous polycarbonates (7), and poly(ethylene terephthalate) (9) (Fig. 2A–C), make up a significant portion of plastics. The ability to tune the properties of these polymers is of great interest due to their overwhelming availability, either as freshly produced polymer or from recycling feedstocks. Specific interest is in increasing thermal stability, chemical resistance, water repulsion, and/or processability. Post-polymerization fluorination represents avenues to improve the properties of, i.e. upcycle, commercial polymers. | ||
| Fig. 2 Trifluoromethylation of various aromatic containing commercial polymers and the resulting polymer properties. (A) Fluorination of polystyrene and polystyrene derivatives (5a–5c) to give fluorinated styrenes (6a–6d). Trifluoromethylation via ruthenium catalyst (green text) was slightly more efficient than via phenazine organic photocatalyst (blue text). Reproduced from ref. 56 with permission from the Royal Society of Chemistry, Copyright 2019. (B) Fluorination of poly(BPA) (7) to give fluorinated poly(BPA) (8). (C) Fluorination of poly(ethylene terephthalate) (9) to give fluorinated PET (10). (D) A table summarizing thermal properties of polymers as well as surface energy through contact angle measurement against water. | ||
The fluorination of polystyrene was originally studied by Margrave and Lagow in 1974, by exposure of finely powdered polystyrene to fluorine gas.52 Through elemental analysis and IR spectroscopy of the resulting polymer, it was theorized that perfluorination had been achieved. The perfluorinated polystyrene was significantly more thermally stable than the parent polystyrene, with degradation temperature increasing to 175–250 °C from 120 °C in an atmosphere of air. Although fluorination could be achieved, highly toxic and corrosive fluorine gas was used as a fluorine source, and further characterization was limited, leaving questions regarding chain scission or crosslinking events. Following this report, perfluoroalkylation of the aromatic groups in poly(α-methyl styrene) and poly(phenyl methacrylate) were observed by Shuyama in 1985. Treatment of these aromatic polymers with (perfluoroalkyl)phenyliodinium trifluoromethylsulfonates allowed 14–74% of repeat units to be modified, with polymer scission also observed.53 This work was expanded by Zhao and coworkers in 1996 through the reaction of polystyrene with perfluorodiacyl peroxides.54 Finally, Sawada and coworkers studied the trifluoromethylation of polystyrene, poly(diphenylacetylene), and poly(2,6-dimethyl-p-phenylene oxide) utilizing similar chemistry in 2001.55 Trifluoromethylation was successful on all polymers, with 16–100% of aromatic repeat units being modified. As previously observed, contact angle increased with higher levels of fluorination, but size exclusion chromatography (SEC) analysis showed increased dispersity of the polymers, indicative of possible polymer scission or cross-linking events.
After a modest gap in research, Leibfarth and coworkers have recently developed a photocatalytic method towards the fluorination of aromatic moieties in polymer chains (Fig. 2A).56 Through use of a ruthenium catalyst and pyridine-N-oxide oxidant, aromatic rings could be fluorinated via radicals generated from trifluoroacetic anhydride (4a), or other fluorous anhydrides. Fluorous acyl chloride (4b) could be used for C7F15 addition (Fig. 2A). The degree of fluorination could be controlled through the equivalents of fluorinating agent, with multiple polystyrene derivatives (5a–c, Fig. 2A), poly(bisphenol A carbonate) (7, Fig. 2B), and poly(ethylene terephthalate) (9, Fig. 2C) all being successfully modified to give fluorinated analogues 6a–6d, 8, and 10. Polystyrene could be modified with 20–110 fluorous groups per 100 styrene repeating units. In contrast to older reports, polymer dispersity was not significantly affected by fluorination. In most cases, degradation temperature (Td) remained similar between starting polymer and fluorinated polymer, with a lowering of the glass transition temperature (Tg) (Fig. 2D, table entries 1–7).57,58 The introduction of fluorous character was assayed by the wettability of the polymer films. The authors found that the contact angle of water increased with increasing levels of fluorination and length of fluorous chain (Fig. 2D, table entries 1–3). Soon after this report, a similar transformation was successful with a phenazine organic photocatalyst, albeit with a slightly lower fluorination efficiency (Fig. 2A, blue text).59
Post-polymerization fluorination of poly(ether ether ketones)
Semi-crystalline aromatic poly(ether ether ketones) are produced on an industrial scale as high-performance thermoplastics. Containing both aromatic rings and electrophilic carbonyls, these polymer scaffolds have attracted attention for post-polymerization modification in bulk and on surface level films.60 Although the synthesis of fluorine containing poly(ether ether ketones) has been investigated,61,62 there are few cases of post-polymerization fluorination. In 1995, Badyal and Hopkins studied the fluorination of poly(ether ether ketone) (PEEK) through the use of CF4 plasma.63 Elemental analysis provided evidence of high levels of fluorination, but further polymer analysis was not performed. Noiset and coworkers also reported surface level fluorination of PEEK through reduction of polymer ketones to alcohols with subsequent fluorination via diethylaminosulfur trifluoride in 1997.64 Most recently, Liu and coworkers have developed methods for surface level fluorination of PEEK via activation with argon plasma, followed by treatment with aqueous HF.65 Fluorinated PEEK was shown to be a promising polymer for biomedical inserts.Building from the work performed by the Liu group, Colquhoun and coworkers adapted small molecule chemistry for the trifluoromethylation of poly(ether ether ketone) 11 in solution phase (Fig. 3A).66,67 Following generation of a trifluoromethyl anion, nucleophilic addition occurred quantitatively at the carbonyl sites in the polymer backbone. Subsequent alcohol deprotection gave quaternary carbons containing both the hydrophobic trifluoromethyl group and the hydrophilic alcohol on polymer 12. Having amphiphilic functionality allowed for polymer dissolution in methanol and ethanol while retaining similar physical and surface properties of the original THF-soluble polymer. Similar to the modification of polystyrene, the glass transition temperature was lowered with trifluoromethylation (Fig. 3D, entries 1–2). The degradation temperature was also lowered, likely due to the presence of labile benzylic alcohol groups.
 | ||
| Fig. 3 Post-polymerization modifications of commercial polymers. (A) Synthetic scheme showing trifluoromethylation of the electrophilic ketone of a synthesized poly(ether ether ketone) (11), followed by desilylation to give the fluorinated polymer (12). (B) Addition of fluorous groups to a statistical copolymer of polystyrene and polyisoprene (13) via triazolinediones (14a and 14b) giving a mixture of isomers on polymers 15a and 15b. (C) Addition of fluorinated ester 17 to polyisoprene 16 giving a mixture of regioisomers on fluorous polymer 18. (D) A table summarizing thermal properties of polymers as well as surface energy through contact angle measurement against water. | ||
Post-polymerization fluorination of polydienes
Polydienes represent another important class of commercial polymers, with polybutadiene and polyisoprene being the hallmark members of this family. The polydienes contain units of unsaturation in the polymer chain or incorporation of an allyl side group, which are convenient handles for direct functionalization of the polymer. Like polystyrene, original attempts of fluorination required fluorine gas. Two research groups used F2 to fluorinate either polyisoprene or poly(sulfone-butadiene) copolymers in 1995, although further characterization of the polymers was limited.63,68 In 1998, Hillmyer and coworkers introduced a mild fluorination technique from a fluorous carbene generated through hexafluoropropylene oxide as a fluorine source.69–71 Quantitative fluorination could be achieved on polyisoprene, polybutadiene, and polydimethylbutadiene. In all cases, Td was slightly lowered due to the incorporation of labile gem-difluorocyclopropane moiety, but Tg was significantly increased, as well as the water contact angle. Further work by the Hillmyer group in 2001 led to addition of a perfluoroalkyl iodide to polydienes,72 showing similar thermal and contact angle changes as their previous report. The addition of fluorous carbenes has also been applied to poly(1,3-cyclohexadiene) by Mays and coworkers in 2008.73Since these seminal reports, there has been a resurgence in the addition of fluorous groups to polydienes. Barner-Kowollik and coworkers applied multi-component reactions for the addition of bromide and an alcohol across the double bond of polydienes using N-bromosuccinimide (NBS) and a perfluoroaryl acid in THF.74 Quantitative loss of olefin was reported, and further post-polymerization modifications could be performed on the alkyl bromide, such as displacement by sodium azide.
Du Prez and coworkers have also adapted a method for polydiene (13) functionalization via triazolinediones (Fig. 3B).75 Fluorous triazolinediones (14a, 14b) could be synthesized in three steps from an amine, and upon mixing with poly(styrene-isoprene-styrene) block copolymer, fluorination was achieved in as little as 10 minutes. When a long fluorous chain was used, 15a was observed with 34% efficiency. In the case of a perfluorinated aryl moiety, 15b was generated with 99% efficiency. The level of fluorination could be tuned through the percent of 14a or 14b in the reaction mixture, and as seen with previous cases, fluorination of the polymer greatly increased the static contact angle against water (Fig. 3D, entries 3–5). In this study, the thermal properties were not recorded so comments cannot be made on the variation of the Td or Tg.
Most recently, Tsarevsky and coworkers have modified polyisoprene (16) through various techniques utilizing hypervalent iodine compounds with fluorine-containing ligands (Fig. 3C).76 Fluoride atoms, trifluoromethyl groups, and fluorinated esters could be added concurrently with halides. Specifically, the addition of fluorous esters alpha to an alkyl iodide was accomplished through reaction of 16 with hypervalent iodine intermediate 17. The fluorinated polymer (18) had significantly increased contact angle of water, but the electrophilic fluorous esters and alkyl iodide lowered the thermal stability of the polymer (Fig. 3D, entries 6 and 7).
Synthesis of fluorous polymers through fluorinated monomers
Uncontrolled polymerization of monomers with fluorous sidechains
While upcycling and post-polymerization fluorination approaches are able to alter the bulk properties of a material, fine control over the chemical composition remains poor. Although not having the same widespread availability as commercial polymers for fluorination, there is increased interest in the development of new fluoropolymer scaffolds, with control over the monomer composition for tuning of fluorination and bulk polymer properties. With PTFE and its derivative polymers being highly crystalline and insoluble, there has been development of alkenes containing longer fluoroalkyl chains. By the addition of bulky fluoroalkyl chains, crystallinity can be lowered giving a more easily processed polymer.36 In particular, perfluorohexylethylene (PFHE, 19b, Fig. 4A) is of interest due to the long hexyl chain. Although resistant to homopolymerization, PFHE was successfully copolymerized with non-fluorous olefins such as vinyl acetate (Vac, 19a, Fig. 1A) in 1985 giving polymer 20. This copolymerization was further studied by Sen and Borkar via free and controlled radical polymerization, along with copolymerization of PFHE with methyl methacrylate via atom transfer radical polymerization (ATRP) in 2005.77,78 A patent was granted for the copolymerization of PFHE with tetrafluoroethylene for the development of fluorous membranes and films in 2009, due to its potential as an alternative to Teflon-AF.79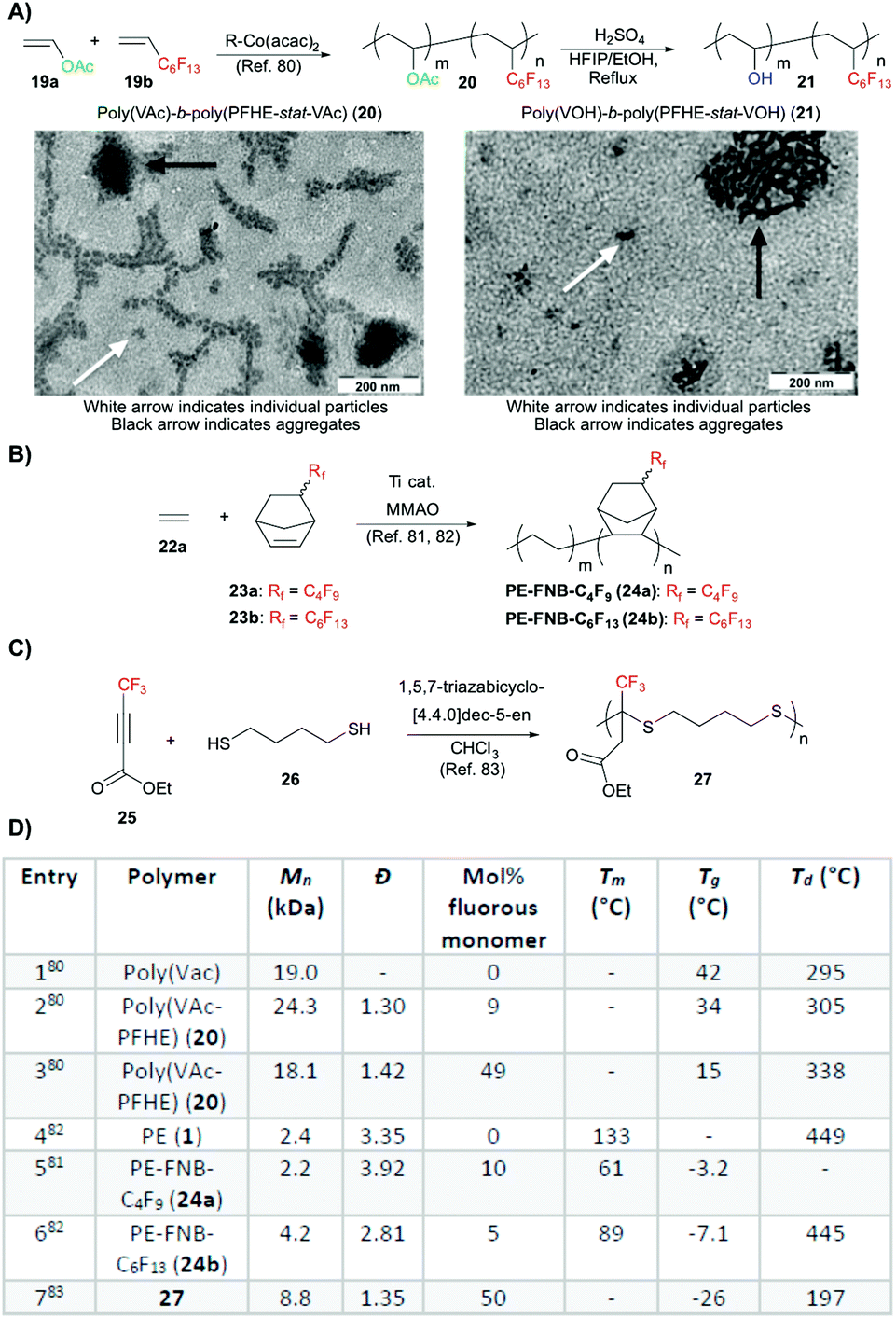 | ||
| Fig. 4 Use of fluoroalkyl alkenes to give semi-fluorinated polymers. (A) Top: Copolymerization of perfluorohexylethylene (PFHE, 19a) and vinyl acetate (Vac, 19b) to give polymer 20 with subsequent hydrolysis of acetate giving polymer 21. R = (VAc)4–C(CH3)(CN)CH2C(CH3)2OCH3 Bottom: TEM images of polymers 20 and 21 demonstrating increased polymer aggregation with generation of alcohol functionalities. Reproduced from ref. 80 with permission from the American Chemical Society, Copyright 2017. (B) Copolymerization of ethylene (22a) with fluorinated norbornenes (FNB, 23a or 23b) to give polymer 24a or 24b. (C) Step-growth polymerization of fluorinated acetylenecarboxylate 25 with dithiol 26 to give polymer 27. Representative dithiol shown, extended alkyl, ether, and aromatic containing dithiols were also demonstrated. (D) Table of polymer properties comparing homopolymers to those containing fluoroalkyl side chains. | ||
Most recently, Detrembleur and coworkers have optimized a cobalt mediated radical copolymerization of PFHE and VAc (Fig. 4A, top).80 PFHE could be incorporated into the copolymer in up to 49 mol% of the statistical mixture with low dispersity. As the ratio of PFHE monomer was increased there was also an increase in dispersity of the resultant polymer (Fig. 4D, table entries 2 and 3). As predicted, increasing the fluoroalkyl monomer feed decreased the Tg of the polymer, but the thermal stability was improved (Fig. 4D, table entries 1–3). Contact angles of the polymers were not measured in this study, but it had been previously observed that the incorporation of perfluorohexyl chain in PVAc increased water contact angle to 104°,78 while perfluorooctyl and perfluorodecyl groups gave increases to 111° and 114°, respectively.
The poly(PFHE-stat-VAc) copolymer, 20, could be further modified through hydrolysis with a strong acid with full conversion from ester to alcohol giving poly(PFHE-stat-VA) copolymers, 21. TEM images display increased aggregation of 21 when compared to 20, likely due to the hydrogen bonding of the alcohol generated upon hydrolysis of the acetate groups (Fig. 4A, bottom).
Another strategy for fluorine incorporation in polymers is to separate the reactive monomer functionality from the fluorous group to preserve monomer reactivity. The Cai and Tang groups have independently developed methods for the copolymerization of ethylene (22a) and fluorinated norbornenes (FNB) 23a or 23b as a statistical copolymer, 24a or 24b, respectively (Fig. 4B).81,82 Both groups utilized titanium and MMAO catalysts, with reaction times between two and fifteen minutes. Fluorous norbornene incorporation could be varied between 1 and 5 mol%. In both reports, the degradation temperatures (Td) of the copolymers were unchanged by fluorous chain addition (Fig. 4D, table entries 4–6), while the melting points (Tm) were decreased with increasing fluorous chain incorporation. The Cai group also demonstrated enhanced mechanical properties (increased % elongation at break) with the addition of the fluorous chains.
Towards a new scaffold, a step-growth polymerization between a trifluoromethyl acetylenecarboxylate (25) and dithiol (26) was presented by Durmaz and coworkers (Fig. 4C).83 Trifluoromethyl containing polythioether (27), as well as other alkyl, ether, or aromatic containing polythioethers, could be easily synthesized at room temperature with polymer of moderate molar mass being generated in as little as one minute (Fig. 4D, entry 7). The thermal properties of these polymers could be readily modified through dithiol selection, although contact angle of water remained consistent across all polymer films due to preservation of the trifluoromethyl group.
Controlled polymerization of monomers with fluorous side chains
While uncontrolled polymerizations of fluorinated monomers are adequate to control the overall wt% fluorine within a polymer, fine control over the polymer architecture remains poor. In many instances, defined architectures that allow for self-assembled structures or uniform surface coverage are desirable. For these applications, controlled polymerization are ideal. Fluoropolymers with a controlled architecture can be prepared using fluorinated acrylate monomers in standard RAFT and ATRP polymerization, providing fluorinated poly(acrylate)s which have been the subject of previous reviews.41,42 Here we highlight the controlled or living polymerization of alternative fluorous monomers, including lactides, oxazolines, and acetylenes.Poly(lactides) can be formed through a controlled ring-opening polymerization from natural monomers. Their biocompatibility and biodegradability have rendered them popular biomaterials.84 Methods to tune both the degradation properties as well as the glass transition temperature have prompted the introduction of fluorinated monomers.85 In 1998, McKie and coworkers prepared the first poly(trifluoromethyl lactic acid).86 Twenty years later, Boydston and coworkers revisited trifluoromethylated poly(lactide) and prepared trifluoromethyl lactide monomer 28 (Fig. 5A, top scheme).87 Using a tin catalyst and benzyl alcohol as an initiator, a trifluoromethyl polylactic acid (PLA) could be synthesized with excellent conversion and low dispersity (29a). Both the Tg and Td were lowered by the trifluoromethylation of lactide monomer, consistent with other fluorinated polymers. When polyethylene glycol (PEG) was used as an initiator (Fig. 5A, bottom scheme), triblock copolymer (29b) with controlled block lengths could be synthesized. In water, polymer 29b self-assembled into vesicles with ∼300 nm diameter with a 2.3 nm polymer shell (Fig. 5B). The methods reported by Boydston and coworkers, to prepare fluorinated poly(lactic acid)s have been reproduced by others where contact angle and protein adsorption have been characterized.88 The contact angle of fluorous PLA 29a, synthesized by Ratner and coworkers, was significantly increased to 88° in comparison to 70° from standard PLA. It was also demonstrated that fluorous PLA has a higher rate of protein adsorption which is hypothesized to improve thromboresistance, a promising property for the application of these materials.
 | ||
| Fig. 5 Synthesis and self-assembly of polylactide copolymers. (A) Top: Homopolymerization of trifluoromethyl lactide monomer 28 resulting in polymer 29a. Bottom: Polymerization of trifluoromethyl lactide 28 with bifunctional polyethylene glycol (PEG) to give ABA block copolymer 29b. (B) Self-assembly of fluorous lactide ABA block copolymer 29b in water. Reproduced from ref. 87 with permission from the American Chemical Society, Copyright 2018. | ||
Poly(oxazoline)s are another polymer scaffold with growing biomaterials interests that are obtained through a controlled ring opening polymerization.89,90 While it is poly(methyl-2-oxazoline) and poly(ethyl-2-oxazoline) that have garnered the most attention as poly(ethylene glycol) replacements,91 the polymerization of hydrophilic, hydrophobic and fluorous oxazoline monomers (30a–g, Fig. 6A) allows for the creation of amphiphiles and the tuning of the thermoresponsive behaviour.92 Although the polymerization of oxazolines has been known since 1966, incorporation of a fluorous group on the oxazoline scaffold (30c) was not performed until 1988 by Saegusa and coworkers (Fig. 6A).93 In this study, it was found that the electron withdrawing nature of the fluorous group greatly hampered polymerization kinetics, with most conditions only providing oligomeric product 31a (Fig. 6B, top). Shortly after, Sogah and coworkers found that addition of an ethyl spacer between the oxazoline heterocycle and fluorous group (30e) greatly improved polymerization giving 31b (Fig. 6B, bottom).94 Building from this finding, Papadakis and coworkers synthesized the first hydrophilic/fluorous block copolymers using oxazoline monomers 30a and 30f and observed micelle formation in an aqueous environment.95 They found that the fluorous core formed an elongated micelle in comparison to the spherical micelle formed from a lipophilic core (synthesized from 30a and 30b), demonstrating that incorporation of fluorine has a unique effect on self-assembly likely, a due to the increased rigidity of perfluoroalkyl chains.
 | ||
| Fig. 6 Oxazoline monomers, polymers, and materials. (A) Select hydrophilic, hydrophobic, and fluorophilic oxazoline monomers 30a–g. (B) Comparison of the polymerization of oxazolines with fluorous groups directly attached to the oxazoline scaffold (30c) vs. those with an ethyl spacer (30e). (C) Left: Comparison of polymerization kinetics between oxazoline monomers containing different alkyl spacers between the fluorous group and oxazoline heterocycle (30c–e). Right: Size exclusion chromatography (SEC) traces of the 30e homopolymer demonstrating low dispersity. Reproduced from ref. 96 with permission from the American Chemical Society, Copyright 2018. (D) Di and triblock copolymers (31c and 31d) synthesized from methyl, alkyl, or fluorous oxazoline monomers. (E) Triblock polyoxazoline 31e and the stability of the fluorous emulsions generated from it. | ||
To better understand the effect of fluorine on oxazoline polymerization, the Hoogenboom group has thoroughly studied the addition of alkyl spacers between the oxazoline heterocycle and fluorous group (Fig. 6C, left).96 The polymerization rates of 2-trifluoromethyl-2-oxazoline (30c), 2-(2,2,2-trifluoroethyl)-2-oxazoline (30d), and 2-(2,2,2-trifluoropropyl)-2-oxazoline (30e) monomers were compared. It was found that when 2-trifluoromethyl-2-oxazline was used as a monomer, very little polymerization occurred (Fig. 6C, left, blue line). Alternatively, the use of a methyl spacer allowed for polymerization, but the robust living kinetics of CROP were not observed (Fig. 6C, left, black line). Extending to an ethyl spacer provided enough of a shield from the electron-withdrawing nature of the fluorous group that polymerization proceeded readily with living kinetics, yielding polymers with a dispersity under 1.2 (Fig. 6C, left, red line, and Fig. 6C, right). Hydrophilic-fluorophilic diblock and hydrophilic-lipophilic-fluorophilic triblock polymers could be prepared, with morphology being observed via TEM. In efforts to increase the fluorous content of the poly(2-oxazolines), the ethyl spacer was retained and a longer perfluorohexyl group was appended in place of the trifluoromethyl group (oxazoline 30g) to give polymers 31c and 31d.97 Reaction kinetics were comparable for 30e and 30g, allowing highly fluorinated di and tri block copolymers to be prepared through similar methodology (Fig. 6D). Notably, polymer 31d was found to self-assemble in both water and DMSO, with DMSO self-assembly only successful when a fluorinated block was present. We have employed a similar fluorinated oxazoline monomer to prepare custom, functionalizable amphiphiles for the stabilization of perfluorocarbon nanoemulsions (Fig. 6E). The incorporation of fluorous character into the amphiphile (polymer 31e) lead to more stable nanoemulsions over sixty days, as compared to a hydrocarbon variant.98
Other controlled polymerizations to achieve polymers with pendant fluorous chains
Seki and coworkers have studied the self assembly of low dispersity, unsymetrical, alkyl-fluoroalkyl side chain containing polyacetylenes.99 Living cyclopolymerization of functional diynes gave a mixture of cis and trans olefins in the polymer backbone, forming predominantly coil architectures. Photoisomerization of the olefin isomers yielded a trans-rich polymer backbone, which transitioned to a rod assembly. It was found that fluorinated polyacetylenes self-assembled further into aggregated rods upon cooling, whereas no further assembly was observed on the hydrocarbon analog. Ihara and coworkers have recently developed a palladium initiated polymerization of fluoroalkyldiazo acetates.100 This polymerization led to poly(substituted methylene)s of modest molar mass, which could be quantitatively modified through reactions with primary amines.Synthesis of polymers with a fluorinated backbone
An important distinction of semi-fluorinated polymers is between those containing fluorous side chains or fluorous backbones. Thus far, we have highlighted incorporation of fluorous character onto the side chains. While there are many commercial polymers with fluorine installed directly on the backbone, most are derived from fluorinated olefins which have significant safety concerns.26–28 Other approaches to incorporate fluorine directly into the backbone are the use of telechelic functional poly(perfluoroethers) or leveraging difunctional perfluorinated monomers. The former approach has been recently reviewed by Améduri and Friesen.50 We will highlight the latter approach below.Diiodoperfluoroalkanes (DIPFAs, 32) are a convenient starting material for the incorporation of fluorine into a polymer backbone as they have unique reactivity with unsaturated carbons.101,102 These monomers are industrially produced via the telomerization of tetrafluoroethylene (TFE) in the presence of iodine, and many lengths are commercially available (e.g. I(CF2)xI, x = 2, 4, 6, 8). One of the main advantages of the polymerization of diiodoperfluoroalkane monomers with dienes or diynes is the installation of alkyl or vinyl iodide functionality across the polymer backbone, respectively (Fig. 7A and E). Due to the close resemblance to the thiol–ene or thiol–yne reactions, this polymerization has been named the iodo-ene or iodo-yne polymerization. It should be noted that iodine and fluorine atoms have also been placed on polymer backbones through post-polymerization modification of polyisoprene.74
 | ||
| Fig. 7 Recently developed methods for the synthesis and derivatization of semi-fluorinated iodo-ene and idoo-yne polymers. (A) A reaction scheme demonstrating polymerization of diiodoperfluoroalkanes (DIPFAs) (32) with dienes or diynes (33a or 33b) to give iodo-ene (34) or iodo-yne (35) polymers. (B) Modification of iodo-ene polymer (34) via nucleophile or reductant (36, top) or elimination with strong base (37, bottom). (C) Table of different initiators, monomer combinations, and modifications. (D) Table summarizing thermal and surface properties of iodo-ene polymers. (E) Structure of iodo-yne polymer (37) and the resulting modifications to give 38. | ||
The first published use of DIPFA to form iodo-ene polymers was by Wilson and Griffin in 1993 (Fig. 7A and C, table entry 1) to study the mesophase separation of fluorous and hydrocarbon blocks in the solid state.103 In this study, AIBN was chosen as the free radical initiator for the addition of DIPFAs of different lengths to hydrocarbon dienes in the bulk phase. Polymers of modest molar mass were produced (34, Fig. 7A), and it was found that the resulting alkyl iodides could easily be reduced via tributyltin hydride, either as a separate reaction or sequentially in the initial polymerization reaction (36, R2 = H, Fig. 7B, top). Interestingly, the fluorocarbon-hydrocarbon block copolymers were found to have multiple melting transitions between 80 °C and 150 °C due to phase separation of the fluorous and alkyl backbones. Following this seminal report, similar iodo-ene polymers were synthesized by the Percec group utilizing a palladium catalyzed polymerization (Fig. 7A and C, table entry 2).104
The development of iodo-ene polymers then largely remained dormant until revitalization by the Zhu group in 2016.105 The notable advance in this work was the use of blue light and a ruthenium catalyst for the polymerization, with recent work transitioning to organic photocatalysts (Fig. 7A and C, table entry 3).106–108 The improved reaction conditions allowed for the scope of the diene monomers to be increased and polymers up to 20 kDa could be synthesized, twice as large as previous work. Zhu and coworkers also found that the degradation temperature of the iodo-ene polymers was enhanced significantly upon iodine reduction with Td changes from 224 °C (34, Fig. 7A and B) to 400 °C (36, R2 = H, Fig. 7B, top). In their most recent work with iodo-ene polymers, polymer 34 with terminal fluorous iodide functionality could be used as a macro-initiator to form well defined block copolymers.109
We have also advanced the iodo-ene polymerization through optimization of the polymerization conditions. We found that the use of sodium dithionite in aqueous acetonitrile yielded iodo-ene polymers (34) exceeding 100 kDa with sonication as an energy input (Fig. 7A and C, table entry 4).110 The molar mass could also be controlled through use of mono-functional monomers to end-cap the polymer. We further analysed the thermal and surface properties of iodo-ene polymers with varying fluorous block lengths (Fig. 7D, table entries 1–5). In most cases, degradation temperature and glass transition temperature were similar due to inclusion of bulky and labile alkyl iodides. Notably, although having lower wt% fluorine, all had an increased contact angle against water in comparison to PVDF, which we attribute to the increased succession of fluorous methylene units (Fig. 7D, table entries 1–5 and 7). We also further increased the weight percent fluorine in the semi-fluorinated polymers by the use of a fluorous diene (Fig. 7D, table entry 5). The alkyl iodide installed in the polymer backbone during the iodo-ene polymerization provides a convenient functional handle for post-polymerization modification and crosslinking. Previous work had simply reduced the iodine demonstrating increased stability; however, we expanded the scope of modifications to include SN2 displacement by thiols and azide, homolytic reactivity to install allyl groups (36, Fig. 7B, top), and elimination (37, Fig. 7B, bottom). These chemistries allowed for modification of thermal properties, covalent crosslinking, and surface modification. We have also recently developed a polymerization method utilizing diiodoperfluoroalkanes and diynes in place of dienes, named the iodo-yne polymerization (Fig. 7A and E).111 This polymerization produces vinyl iodide groups along the backbone (35), which can be cross-coupled through multiple metal-catalyzed reactions, such as Sonogashira, Suzuki, Stille, and Kumada couplings to give the general polymer structure (38). The vinyl iodide could also be eliminated to give electron deficient alkynes, which could undergo cycloadditions with azides at elevated temperatures.
In our studies, we found that iodo-ene polymers (34) could be photocrosslinked in the presence of 2,2-dimethoxyphenyl acetophenone (DMPA) photo-initiator to give semifluorinated gel 39 (Fig. 8A). Another approach to semifluorinated crosslinked iodo-ene polymers is to employ triene and tetraene monomers in place of dienes (Fig. 8B). Both the Kloxin and Bowman labs have demonstrated rapid formation of cross-linked networks via tetra-ene ethers and tri-ene phosphates, respectively.112,113 In both cases, diiodoperfluoroalkane consumption occurred in under five minutes when 254 nm light was used as an energy input. Notably, the Bowman group found that alkyl iodide could be eliminated without the use of base, by annealing iodo-ene polymer films at 80 °C for 1 hour, yielding polymer 40. The newly generated alkenes provided the polymer with enhanced shape memory at room temperature which could be reset upon further annealing (Fig. 8B). The alkenes on this cross-linked network were also further modified through thiol–ene chemistry to alter surface wettability.
 | ||
| Fig. 8 Cross-linked iodo-ene polymers. (A). The use of 2,2-dimethoxyphenyl acetophenone (DMPA) and 365 nm light to cross-link iodo-ene polymers into fluorinated gels (39). (B) Annealed cross-linked iodo-ene polymer 40 demonstrating enhanced shape memory and facile deformation. Reproduced from ref. 113 with permission from the American Chemical Society, Copyright 2019. | ||
Polymerizations of activated perfluoroaryl monomers
Aside from the use of DIPFAs, other functionalities are being incorporated for step-growth fluorinated polymers. Although having different properties than their linear perfluoroalkyl counterparts, perfluoroaryl moieties are finding use. Perfluoroaryl azides are being increasingly used for the generation of polymers with a fluorinated backbone (Fig. 9). Due to the electron withdrawing effect of the perfluoroaryl groups, the azides (41) are highly active towards click reactions. This was demonstrated in 2013 by Tang and coworkers, who polymerized 4,4′-diazidoperfluorobenzophenone (41) with various diynes (42) at elevated temperature to give triazole- containing polymers (43) reaching 34.0 kDa in up to 95% yield.114 The Tang group built upon this work in 2017 to develop aggregate induced emission (AIE) based polymers through reactions with aryl alkynes (Fig. 9A).115 Through similar chemistry, Yan and coworkers could react bis(perfluoroaryl azides) (41) with bis(diphenyl phosphines) (44) to give fluorinated polyphosphazines (45) with molar masses up to 59 kDa and dispersities from 1.1–1.4 promoted by Staudinger reactions (Fig. 9B).116 Impressively, the polymerization occurred at room temperature in as little as 15–30 minutes. | ||
| Fig. 9 Activated perfluoroaryl azide polymerizations. Thermal polymerization of a perfluoroaryl azide monomer (41) with aromatic or alkyl diynes (42) to yield triazole containing polymers 43 (Top) and Staudinger polymerization of perfluoroaryl azide monomer (41) with bis-phosphines (44) giving polyphosphazine polymers (45) (Bottom). | ||
Conclusions and outlook
In the last few years, new approaches to incorporate fluorine into polymers to achieve the advantageous stability and hydro/lipophobicity have been developed to complement the ubiquitous approaches of fluorinated acrylate or olefin monomers. The more modern methods to incorporate fluorine employ a variety of chemistries and starting materials. There is no single best approach and instead one should consider the needs of the material when choosing how to incorporate fluorine. Starting from fluorinated monomers allows for more control over the polymer backbone, which is critical for self-assembly, or the ability to introduce co-monomers with additional functionality for polymer derivatization. Even inclusion of small amounts of fluorous monomer in uncontrolled polymerizations can give polymers enhanced thermal stability or greater water repulsion, while increasing workability by lowering the glass transition or melting points. If cost and scale are a concern, post-polymerization fluorination methods are desirable as they can be performed on recycled commodity polymers. While these approaches provide heterogenous samples, in many cases they improve thin film water contact angle while lowering the glass transition of rigid polymers.Looking forward, there is much room for growth in fluoropolymer synthesis. Upcycling commodity polymers into fluoropolymers has only recently gained traction and there are many small molecule chemistry methodologies that can be applied to recycled materials. There are opportunities for new fluorinated monomers for controlled polymerizations. An exciting development would be an approach that allows for fluorination installed directly in the backbone to be obtained with living reaction kinetics. Finally, we anticipate that the convergence of advances in functional, dynamic polymer materials to continue to merge with fluoropolymers to provide next-generation materials with enhanced stability and function.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr. Heidi van de Wouw, Dr. Rachael Day, Dr. Daniel Estabrook, Irene Lim, and Joseph Garcia for their assistance in preparation of this review.Notes and references
- H. Teng, Appl. Sci., 2012, 2, 496–512 CrossRef.
- J. G. Drobny, Technology of Fluoropolymers, CRC Press, 2008 Search PubMed.
- D. W. Smith, S. T. Iacono and S. S. Iyer, Handbook of Fluoropolymer Science and Technology, John Wiley & Sons, 2014 Search PubMed.
- Fluorinated Polymers: Volume 1: Synthesis, Properties, Processing and Simulation, ed. B. Ameduri and H. Sawada, Royal Society of Chemistry, 2016 Search PubMed.
- S. L. Madorsky, E. Hart, S. Straus and V. A. Sedlak, J. Res. Natl. Inst. Stand. Technol., 1953, 51, 327–333 CAS.
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications, Elsevier, 2004 Search PubMed.
- B. Ameduri, Chem. – Eur. J., 2018, 24, 18830–18841 CrossRef CAS PubMed.
- M. Gauthier, in Eng. Mater. Handb. Desk Ed, ASM International, 1995 Search PubMed.
- W. A. Zisman, in Contact Angle, Wettability, and Adhesion, American Chemical Society, 1964, vol. 43, ch. 1, pp 1–51 Search PubMed.
- F. J. du Toit, R. D. Sanderson, W. J. Engelbrecht and J. B. Wagener, J. Fluor. Chem., 1995, 74, 43–48 CrossRef CAS.
- S. Lee, J. Park and T. R. Lee, Langmuir, 2008, 24, 4817–4826 CrossRef CAS PubMed.
- A. C. Zettlemoyer, J. Colloid Interface Sci., 1968, 28, 343–369 CrossRef CAS.
- P. F. Rios, H. Dodiuk, S. Kenig, S. McCarthy and A. Dotan, J. Adhes. Sci. Techol., 2007, 21, 227–241 CrossRef CAS.
- S. Wu, J. Polym. Sci., Part C: Polym. Symp., 1971, 34, 19–30 CrossRef.
- M. Guerre, G. Lopez and B. Améduri, Mona Semsarilar and Vincent Ladmiral, Polym. Chem., 2021, 12, 3852–3877 RSC.
- M. P. Krafft, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4251–4258 CrossRef CAS.
- L. Zhao, P. Song, Z. Cao, Z. Fang and Z. Guo, J. Nanomater., 2012, 2012, 340962 Search PubMed.
- T. Nguyen, Polym. Rev., 1985, 25, 227–275 Search PubMed.
- J. A. Conesa and R. Font, Polym. Eng. Sci., 2001, 41, 2137–2147 CrossRef CAS.
- H. Ebnesajjad, Introduction to Fluoropolymers, Elsevier, 2013 Search PubMed.
- Fluorinated Polymers: Volume 2: Applications, ed. B. Ameduri and H. Sawada, Royal Society of Chemistry, 2016 Search PubMed.
- B. Améduri, Macromol. Chem. Phys., 2020, 221, 1900573 CrossRef.
- T. Tervoort, J. Visjager and P. J. Smith, Fluor. Chem., 2002, 114, 133–137 CrossRef CAS.
- M. Li, W. Zhang, C. Wang and H. Wang, J. Appl. Polym. Sci., 2012, 123, 1667–1674 CrossRef CAS.
- O. Olabisi and K. Adewale, Handbook of thermoplastics, CRC Press, 2015 Search PubMed.
- D. A. Hercules, C. A. Parrish, T. S. Sayler, K. T. Tice, S. M. Williams, L. E. Lowery, M. E. Brady, R. B. Coward, J. A. Murphy, T. A. Hey, A. R. Scavuzzo, L. M. Rummler, E. G. Burns, A. V. Matsnev, R. E. Fernandez, C. D. McMillen and J. S. Thrasher, J. Fluor. Chem., 2017, 196, 107–116 CrossRef CAS.
- E. De Rademaeker, B. Fabiano, S. S. Buratti, F. Ferrero, R. Zeps, M. Kluge, V. Schröder and T. Spoormaker, Chem. Eng. Trans., 2013, 13, 817–822 Search PubMed.
- D. A. Hercules, D. D. DesMarteau, R. E. Fernandez, J. L. Clark and J. S. Thrasher, in Handbook of Fluoropolymer Science and Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2014, pp. 413–431 Search PubMed.
- M. P. Krafft and J. G. Riess, Curr. Opin. Colloid Interface Sci., 2015, 20, 192–212 CrossRef CAS.
- P. Wang, Y. Lu, T. Wang, J. Meng, Q. Li, Z. Zhu, Y. Sun, R. Wang and J. P. Giesy, J. Hazard. Mater., 2016, 307, 55–63 CrossRef CAS PubMed.
- C. Lau, J. L. Butenhoff and J. M. Rogers, Toxicol. Appl. Pharmacol., 2004, 198, 231–241 CrossRef CAS PubMed.
- K. Kannan, S. Corsolini, J. Falandysz, G. Fillmann, K. Kumar, B. Loganathan, M. Mohd, J. Olivero, N. Wouwe, J. Yang and K. Aldous, Environ. Sci. Technol., 2004, 38, 4489–4495 CrossRef CAS PubMed.
- E. Kumarasamy, I. M. Manning, L. B. Collins, O. Coronell and F. A. Leibfarth, ACS Cent. Sci., 2020, 6, 487–492 CrossRef CAS PubMed.
- L. Xiao, Y. Ling, A. Alsbaiee, C. Li, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 7689–7692 CrossRef CAS PubMed.
- M. J. Klemes, Y. Ling, C. Ching, C. Wu, L. Xiao, D. E. Helbling and W. R. Dichtel, Angew. Chem., Int. Ed., 2019, 58, 12049–12053 CrossRef CAS PubMed.
- G. J. Puts, P. Crouse and B. M. Ameduri, Chem. Rev., 2019, 119, 1763–1805 CrossRef CAS PubMed.
- L. Zhang, Z. Zhu, U. Azhar, J. Ma, Y. Zhang, C. Zong and S. Zhang, Ind. Eng. Chem. Res., 2018, 57, 8689–8697 CrossRef CAS.
- V. S. D. Voet, G. Ten Brinke and K. Loos, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2861–2877 CrossRef CAS.
- G. Laruelle, E. Nicol, B. Ameduri, J. F. Tassin and N. Ajellal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3960–3969 CrossRef CAS.
- A. D. Asandei, Chem. Rev., 2016, 116, 2244–2274 CrossRef CAS PubMed.
- H. Gong, Y. Gu and M. Chen, Synlett, 2018, 1543–1551 CAS.
- W. Yao, Y. Li and X. Huang, Polymer, 2014, 55, 6197–6211 CrossRef CAS.
- S. Borkar, K. Jankova, H. W. Siesler and S. Hvilsted, Macromolecules, 2004, 37, 788–794 CrossRef CAS.
- M. N. Wadekar, W. F. Jager, E. J. R. Sudhölter and S. J. Picken, J. Org. Chem., 2010, 75, 6814–6819 CrossRef CAS PubMed.
- N. Audic, P. W. Dyer, E. G. Hope, A. M. Stuart and S. Suhard, Adv. Synth. Catal., 2010, 352, 2241–2250 CrossRef CAS.
- J. Zhou, Y. Tao, X. Chen, X. Chen, L. Fang, Y. Wang, J. Sun and Q. Fang, Mater. Chem. Front., 2019, 3, 1280–1301 RSC.
- H. Peng, Polym. Rev., 2019, 59, 739–757 CrossRef CAS.
- M. Wehbi, A. Mehdi, C. Negrell, G. David, A. Alaaeddine and B. Améduri, ACS Appl. Mater. Interfaces, 2020, 12, 38–59 CrossRef CAS PubMed.
- S. A. Mohammad, S. Shingdilwar, S. Banerjee and B. Ameduri, Prog. Polym. Sci., 2020, 106, 101255 CrossRef CAS.
- C. M. Friesen and B. Améduri, Prog. Polym. Sci., 2018, 81, 238–280 CrossRef CAS.
- V. F. Cardoso, D. M. Correia, C. Ribeiro, M. M. Fernandes and S. Lanceros-Méndez, Polymers, 2018, 10, 161–187 CrossRef PubMed.
- R. J. Lagow and J. L. Margrave, J. Polym. Sci., Polym. Lett. Ed., 1974, 12, 177–184 CrossRef CAS.
- H. Shuyama, J. Fluor. Chem., 1985, 29, 467–470 CrossRef CAS.
- Z. B. Zhou, H. Y. He, Z. Y. Weng, Y. L. Qu and C. X. Zhao, J. Fluor. Chem., 1996, 79, 1–5 CrossRef CAS.
- Y. Hayakawa, N. Terasawa and H. Sawada, Polymer, 2001, 42, 4081–4086 CrossRef CAS.
- S. E. Lewis, B. E. Wilhelmy and F. A. Leibfarth, Chem. Sci., 2019, 10, 6270–6277 RSC.
- B. N. Jang and C. A. Wilkie, Polym. Degrad. Stab., 2004, 86, 419–430 CrossRef CAS.
- S. A. Jabarin and E. A. Lofgren, Polym. Eng., 1984, 24, 1056–1063 CrossRef.
- S. E. Lewis, B. E. Wilhelmy and F. A. Leibfarth, Polym. Chem., 2020, 11, 4914–4919 RSC.
- D. Shukla, Y. S. Negi, J. Sen Uppadhyaya and V. Kumar, Polym. Rev., 2012, 52, 189–228 CrossRef CAS.
- A. A. Goodwin, F. W. Mercer and M. T. McKenzie, Macromolecules, 1997, 30, 2767–2774 CrossRef CAS.
- H. Asghar, A. Ilyas, Z. Tahir, X. Li and A. L. Khan, Sep. Purif. Technol., 2018, 203, 233–241 CrossRef CAS.
- J. Hopkins and J. P. S. Badyal, J. Phys. Chem., 1995, 99, 4261–4264 CrossRef CAS.
- J. Marchand-Brynaert, G. Pantano and O. Noiset, Polymer, 1997, 38, 1387–1394 CrossRef CAS.
- M. Chen, L. Ouyang, T. Lu, H. Wang, F. Meng, Y. Yang, C. Ning, J. Ma and X. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 16824–16833 CrossRef CAS PubMed.
- F. Leroux, R. A. Bennett, D. F. Lewis and H. M. Colquhoun, Macromolecules, 2018, 51, 3415–3422 CrossRef CAS.
- F. O. Oladoyinbo, D. F. Lewis, D. J. Blundell and H. M. Colquhoun, Polym. Chem., 2019, 11, 75–83 RSC.
- A. N. Ozerin, A. V. Rebrov, V. I. Feldman, M. A. Krykin, I. P. Storojuk, A. A. Kotenko and M. N. Tul'skii, React. Funct. Polym., 1995, 26, 167–175 CrossRef CAS.
- Y. Ren, T. P. Lodge and M. A. Hillmyer, J. Am. Chem. Soc., 1998, 120, 6830–6831 CrossRef CAS.
- Y. Ren, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2000, 33, 866–876 CrossRef CAS.
- D. A. Davidock, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2003, 36, 4682–4685 CrossRef CAS.
- Y. Ren, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2001, 34, 4780–4787 CrossRef CAS.
- T. Huang, J. M. Messman and J. W. Mays, Macromolecules, 2008, 41, 266–268 CrossRef CAS.
- C. M. Geiselhart, J. T. Offenloch, H. Mutlu and C. Barner-Kowollik, ACS Macro Lett., 2016, 5, 1146–1151 CrossRef CAS.
- K. De Bruycker, M. Delahaye, P. Cools, J. Winne and F. E. Du Prez, Macromol. Rapid Commun., 2017, 38, 1700122 CrossRef PubMed.
- Y. Cao, K. D. Sayala, P. L. Gamage, R. Kumar and N. V. Tsarevsky, Macromolecules, 2020, 53, 8020–8031 CrossRef CAS.
- S. Borkar and A. Sen, Macromolecules, 2005, 38, 3029–3032 CrossRef CAS.
- S. Borkar and A. Sen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3728–3736 CrossRef CAS.
- E. A. Sabol and R. L. Baillie, WL Gore and Associates, US7531611B2, 2009.
- J. Demarteau, B. Améduri, V. Ladmiral, M. A. Mees, R. Hoogenboom, A. Debuigne and C. Detrembleur, Macromolecules, 2017, 50, 3750–3760 CrossRef CAS.
- Y. Sun, C. Wang, R. Tanaka, T. Shiono and Z. Cai, Macromol. Chem. Phys., 2019, 220, 1900306 CrossRef CAS.
- L. Ji, J.-S. Liu, X.-Y. Wang, J.-F. Li, Z. Chen, S. Liao, X.-L. Sun and Y. Tang, Polym. Chem., 2019, 10, 3604–3609 RSC.
- O. Daglar, E. Cakmakci, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2020, 53, 2965–2975 CrossRef CAS.
- R. Mehta, V. Kumar, H. Bhunia and S. N. Upadhyay, J. Macromol. Sci., Polym. Rev., 2005, 45, 325–349 CrossRef.
- R. Bhardwaj and A. K. Mohanty, Biomacromolecules, 2007, 8, 2476–2484 CrossRef CAS PubMed.
- D. B. McKie and S. Lepeniotis, Chemometrics and Intelligent Laboratory Systems, Elsevier, 1998, vol. 41, pp. 105–113 Search PubMed.
- C. U. Lee, R. Khalifehzadeh, B. Ratner and A. J. Boydston, Macromolecules, 2018, 51, 1280–1289 CrossRef CAS.
- R. Khalifehzadeh and B. D. Ratner, Biomater. Sci., 2019, 7, 3764–3778 RSC.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS.
- O. Sedlacek, B. D. Monnery, S. K. Filippov, R. Hoogenboom and M. Hruby, Macromol. Rapid Commun., 2012, 33, 1648–1662 CrossRef CAS PubMed.
- M. Grube, M. N. Leiske, U. S. Schubert and I. Nischang, Macromolecules, 2018, 51, 1905–1916 CrossRef CAS.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2016, 8, 24–40 RSC.
- M. Miyamoto, K. Aoi and T. Saegusa, Macromolecules, 1988, 21, 1880–1883 CrossRef CAS.
- J. M. Rodríguez-Parada, M. Kaku and D. Y. Sogah, Macromolecules, 1994, 27, 1571–1577 CrossRef.
- R. Ivanova, T. Komenda, T. B. Bonné, K. Lüdtke, K. Mortensen, P. K. Pranzas, R. Jordan and C. M. Papadakis, Macromol. Chem. Phys., 2008, 209, 2248–2258 CrossRef CAS.
- L. I. Kaberov, B. Verbraeken, A. Riabtseva, J. Brus, Y. Talmon, P. Stepanek, R. Hoogenboom and S. K. Filippov, ACS Macro Lett., 2018, 7, 7–10 CrossRef CAS.
- L. I. Kaberov, B. Verbraeken, A. Riabtseva, J. Brus, A. Radulescu, Y. Talmon, P. Stepanek, R. Hoogenboom and S. K. Filippov, Macromolecules, 2018, 51, 6047–6056 CrossRef CAS.
- D. A. Estabrook, A. F. Ennis, R. A. Day and E. M. Sletten, Chem. Sci., 2019, 10, 3994–4003 RSC.
- Y. Motomura, Y. Hattori, T. Sakurai, S. Ghosh and S. Seki, Macromolecules, 2019, 52, 4916–4925 CrossRef CAS.
- H. Shimomoto, T. Kudo, S. Tsunematsu, T. Itoh and E. Ihara, Macromolecules, 2018, 51, 328–335 CrossRef CAS.
- M. Shinmen, K. Sasahara, S. Nakamura, T. Kanbara and T. Yajima, J. Fluor. Chem., 2020, 229, 109417 CrossRef CAS.
- R. G. Closser, M. Lillethorup, D. S. Bergsman and S. F. Bent, ACS Appl. Mater. Interfaces, 2019, 11, 21988–21997 CrossRef CAS PubMed.
- L. M. Wilson and A. C. Griffin, Macromolecules, 1993, 26, 6312–6314 CrossRef CAS.
- V. Percec, D. Schlueter and G. Ungar, Macromolecules, 1997, 30, 645–648 CrossRef CAS.
- T. Xu, H. Yin, X. Li, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2017, 38, 1600587 CrossRef PubMed.
- T. Xu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2017, 8, 3910–3920 RSC.
- T. Xu, K. Tu, J. Cheng, Y. Ni, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2018, 39, 1800151 CrossRef PubMed.
- T. Xu, L. Zhang, Z. Cheng and X. Zhu, RSC Adv., 2017, 7, 17988–17996 RSC.
- J. Cheng, K. Tu, E. He, J. Wang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2020, 11, 7497–7505 RSC.
- J. A. Jaye and E. M. Sletten, ACS Cent. Sci., 2019, 5, 982–991 CrossRef CAS PubMed.
- J. A. Jaye and E. M. Sletten, ACS Macro Lett., 2020, 9, 410–415 CrossRef CAS.
- T. F. Scott, J. C. Furgal and C. J. Kloxin, ACS Macro Lett., 2015, 4, 1404–1409 CrossRef CAS.
- J. Sinha, B. D. Fairbanks, H. B. Song and C. N. Bowman, ACS Macro Lett., 2019, 8, 213–217 CrossRef CAS.
- Q. Wang, H. Li, Q. Wei, J. Z. Sun, J. Wang, X. A. Zhang, A. Qin and B. Z. Tang, Polym. Chem., 2013, 4, 1396–1401 RSC.
- Y. Wu, B. He, C. Quan, C. Zheng, H. Deng, R. Hu, Z. Zhao, F. Huang, A. Qin and B. Z. Tang, Macromol. Rapid Commun., 2017, 38, 1700070 CrossRef PubMed.
- M. Sundhoro, J. Park, B. Wu and M. Yan, Macromolecules, 2018, 51, 4532–4540 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |