
Integrating abiotic chemical catalysis and enzymatic catalysis in living cells
Christopher
Adamson
and
Motomu
Kanai
 *
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kanai@mol.f.u-tokyo.ac.jp
First published on 14th October 2020
Abstract
Life emerges from networks of multiple chemical reactions mediated by enzymes. If abiotic chemical catalysis is implanted into the reaction network of life, such an integration would produce organisms generating unique secondary metabolites and value-added materials from feedstocks or even air, or new diagnostics and therapeutics against diseases. In this review, we introduce selected papers in this emerging field of catalysis research.
 Christopher Adamson | Christopher Adamson received his B.Sc. from the University of Alberta in 2011 and his M.Sc. from Simon Fraser University in 2016. This was followed by two years in pharmaceutical process development at Gilead Sciences (Edmonton, Canada). In 2018, Christopher started his Ph.D. studies under the supervision of Motomu Kanai at the University of Tokyo. Christopher's research is focused on live-cell protein modification using synthetic catalysis. |
 Motomu Kanai | Motomu Kanai graduated from The University of Tokyo in 1989. In the middle of his graduate course, he obtained an assistant professor position in Osaka University in 1992. He received his PhD from Osaka University in 1995. After doing a postdoctoral work in University of Wisconsin, USA, he was appointed as an assistant professor in The University of Tokyo in 1997. He was promoted to a full professor in 2010. His research interest is catalysis development linking molecular synthesis and life science. |
Introduction
Achieving co-operativity among abiotic, small-molecule chemical catalysis and cellular metabolism mediated by enzymes represents an exciting frontier with myriad potential applications. For instance, a catalyst might convert a metabolic intermediate into a value-added non-natural product. A catalyst might also complement physiological processes as a treatment for enzyme deficiency (catalysis medicine) or as a tool for epigenome editing. Unfortunately, only a small fraction of the abiotic catalysts reported to date are suitable for biological application. Aside from the aqueous stability requirement, a catalyst must also have a high turnover frequency (TOF) at physiological temperature to cope with low concentrations of catalyst and substrate(s). While an extracellular catalyst may be incorporated into a metabolic pathway,1–5 we will limit our discussion in this review to systems where an abiotic catalyst is used within living cells. While this allows a more direct interaction with cellular machinery, it also presents additional challenges. First, the catalyst must be membrane permeable, which precludes many large (>1000 Da), hydrophilic, or anionic catalysts.6 Second, protein binding, rapid efflux, or thiol metabolites such as glutathione (GSH) may render a catalyst ineffective within cells. Third, the catalyst must be nontoxic toward the host organism. Fourth, it can be difficult to know if a reaction of interest is truly intracellular.7,8 This review will briefly survey recent (2019-) work toward the integration of various abiotic catalyst modalities within cellular metabolism. As we will show, this emerging field is affording new technologies for chemical synthesis and manipulation of cellular function.Transition metal (TM) coordination complexes
A groundbreaking study on ruthenium-catalyzed amine deprotection through allylcarbamate cleavage within cells was reported by Meggers and Streu in 2006.9 In the following years, TM complexes have become a developed modality for artificial catalysis within cells, with notable examples featuring copper10 and palladium11 complexes. However, true integration of TM complexes within metabolism is still in its early stages. In this regard, the Sadler group's development of osmium complexes12 for in-cell asymmetric transfer hydrogenation of pyruvate to D-lactate with potential application to anti-cancer treatments was an important milestone.Catecholamines, such as dopamine, norepinephrine, and epinephrine are hormones and neurotransmitters whose concentrations are tightly regulated in the human body. Their dysregulation is implicated in various disease states including schizophrenia and Parkinson's disease. Selective detection of catecholamines in a complex biological environment is therefore useful for understanding their physiological roles, as well as for developing diagnostic and therapeutic strategies. Au-Yeung and co-workers reported a copper complex for the fluorescent detection of catecholamines within living cells.13 The authors designed this copper complex based on the structure of dopamine β-hydroxylase, a copper monooxygenase. A mechanistic outline for this transformation is shown in Fig. 1. First, O2 and catecholamine 1 act in tandem to convert complex 2 to reactive Cu(II)-peroxo species 3. The exact sequence of events and fate of 1 has not been fully elucidated. Nevertheless, the unique one-electron redox chemistry of 1 is thought to play a key role. Second, intramolecular oxidation of the weak benzylic C–H bond affords a hemiacetal (or ester), and C–O bond scission results in fluorophore activation through decaging the 7-hydroxycoumarin moiety. Critically, while the presence of dopamine, epinephrine, or norepinephrine resulted in time- and O2-dependant fluorophore activation, almost no activation occurred in the presence of many other cellular metabolites and other related small molecules, including catechol. Thus, both the catechol and amine functional groups in catecholamines are essential for this fluorophore activation process.
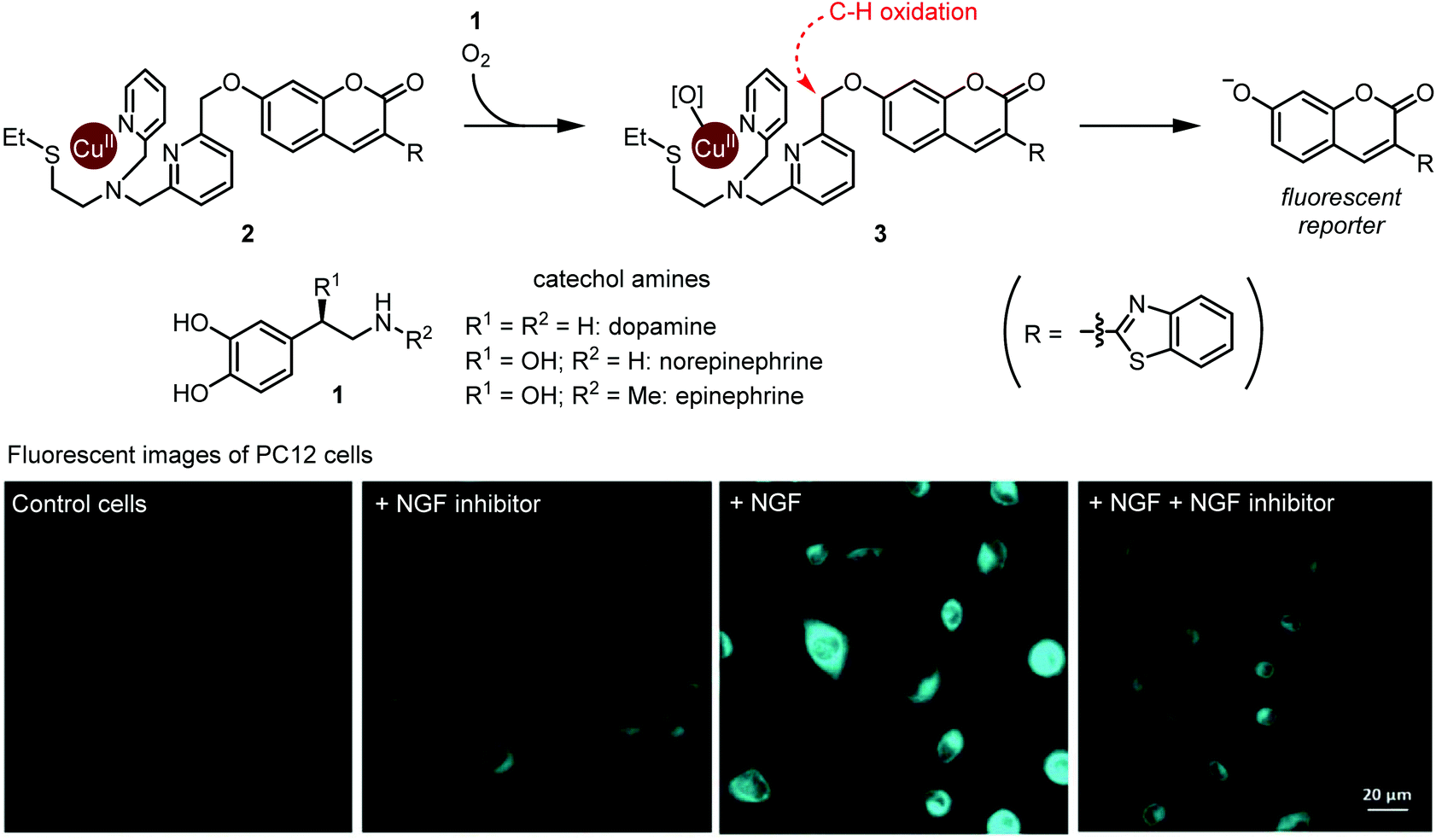 | ||
| Fig. 1 Detection of metabolically synthesized catecholamines in cells via copper-mediated abiotic C–H oxidation. Fluorescent image photos are reproduced from ref. 13 with permission from the Royal Society of Chemistry. | ||
The authors evaluated 2 for visualization of dopamine in the neural cell line PC12. Upon exposure to neural growth factor (NGF), PC12 cells are known to increase dopamine biosynthesis.14 Upon subjecting 20 μM 2, cells treated with NGF displayed increased fluorescence compared with NGF-untreated cells, or with cells treated with NGF and a NGF receptor inhibitor (Fig. 1). These results establish Cu-mediated C–H oxidation as a platform for imaging catecholamines within live cells. We note that since complex 2 completes only one turnover, it is not a true catalyst. Nonetheless, we believe that this work is an important step toward selective metabolite activation with an abiotic catalyst.
Artificial metalloenzymes (ArMs)
Noble metal encapsulation within a protein scaffold has enabled new-to-nature reactivity such as asymmetric catalysis,15 cell surface labelling,16 and prodrug activation.17,18 From the perspective of in-cell application, a current obstacle is the installation of a precious metal cofactor within a scaffold protein. One approach uses genetic code expansion19 to install a bio-orthogonal handle (for instance, an azido group) into the scaffold. Despite several elegant examples of in vitro assembly,20,21 genetic code expansion has to our knowledge not yet enabled in vivo ArM assembly. Pioneered by Whitesides and Wilson in 1978,22 the most common approach for in vivo assembly involves tethering a TM complex to a cell-permeable protein ligand. Upon cellular delivery, the synthetic cofactor noncovalently binds to the scaffold protein. Many elegant studies have leveraged the tight interaction of biotin and streptavidin for ArM assembly.23 However, as biotin resides in a rather shallow binding pocket, the catalytic metal is vulnerable to inactivation by GSH,24 an intracellular thiol present in millimolar concentrations. Thus, streptavidin is typically expressed in the periplasmic space of Gram-negative bacteria25 or at the cell surface.26The Clark, Mukhopadhyay, Keasling, and Hartwig groups recently teamed up to integrate a noble metal catalyst within an anabolic pathway.27 Ir(Me)-mesoporphyrin IX (4; Fig. 2a), when bound to cytochrome P450 protein apo-CYP119, is a potent catalyst for asymmetric olefin cyclopropanation.28,29 However, as this cofactor is large (MW = 770) and anionic, it cannot cross the cell membrane. To overcome this challenge, the authors generated E. coli mutants overexpressing the membrane hemin receptor ChuA in addition to the apo-CYP119 scaffold. As an initial trial, these cells were treated with 4 followed by carvone and ethyl diazoacetate (EDA; Fig. 2b). Critically, the product stereoisomer distribution was consistent with the desired ArM acting as catalyst rather than unbound cofactor 4.
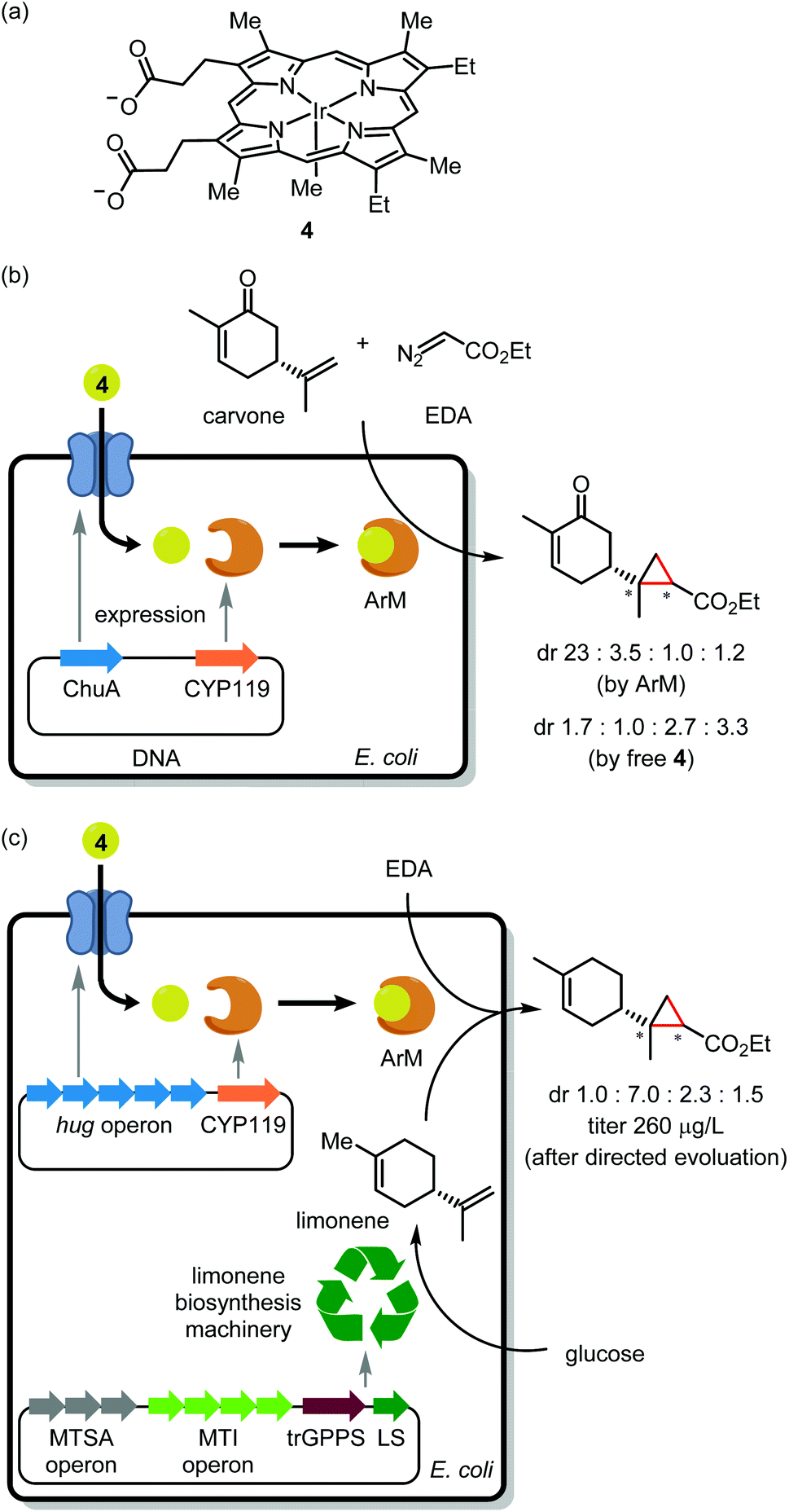 | ||
| Fig. 2 Integration of terpene biosynthesis and Ir-catalyzed cyclopropanation to produce unnatural terpenoids. (a) Structure of Ir–porphyrin complex as a cofactor of ArM. (b) Generation of ArM containing the Ir–porphyrin complex and cytochrome P450 (CYP) in living cells by using a cofactor transport system. ArM-catalyzed cyclopropanation of carvone as a test case. (c) From glucose to artificial terpenoid by integrating terpene biosynthesis, a cofactor transport system, and the artificial Ir–porphyrin complex. | ||
Having demonstrated in vivo ArM assembly, the authors turned their attention to cyclopropanation of an endogenous metabolite. To this end, E. coli mutants harboring genes for de novo limonene synthesis were employed. Unfortunately, apo-CYP119 co-expression severely antagonized limonene production. The apo-CYP119 titer was accordingly dialed down by employing a weaker promoter. To compensate for the reduced scaffold expression, ChuA was replaced with the more productive HUG cofactor transport system.30 Using the resulting E. coli mutants, ArM assembly followed by exposure to EDA afforded cyclopropyl limonene in 250 μg L−1 titer (Fig. 2c). The authors then performed directed evolution of apo-CYP119 to increase cyclopropanation diastereoselectivity. Mutagenesis at R256 then V254 yielded the R256W/V254A mutant,31 which boosted selectivity for the major cyclopropyl limonene isomer to 72% dr as compared with 43% dr for the original scaffold protein. While this work bodes well for the scalable synthesis of value-added natural product analogues, the current reaction yield may limit preparative applications. To address this, further metabolic engineering may be required to generate the ArM at a higher concentration within cells. A further caveat is that endogenous heme competes with 4 for binding to apo-CYP119, which made it necessary to grow cells under low iron conditions to suppress heme biosynthesis. We wonder if the bump-and-hole approach32 might be used to create a cofactor/scaffold pair which is impervious to heme.
Nanoparticle (NP)-supported TM catalysts
Nanoparticles which display a large hydrophobic surface can exploit noncovalent substrate binding to achieve rapid reactions.33 Thus, NP-supported catalysts may be especially suitable for the low concentrations of catalyst and substrate(s) found within cells. Furthermore, NP-supported TM catalysts frequently possess greater biostability and lower toxicity compared with traditional TM catalysts.34 However, due to their large size (ca. 1–200 nm), cytosolic delivery of NPs is often not trivial.35 A pioneering study on polystyrene-supported Pd catalysts was reported by the Bradley group in 2011,36 and many other cell-compatible NP catalysts have since been published.37–41The Wang group reported catalyst 5, which mediates the reduction of NAD+ to NADH within mitochondria (Fig. 3a).425 possesses a Ru-based catalytic moiety which can mediate transfer hydrogenation at low catalyst loadings in aqueous media.43 Further, 5 incorporates a benzyl diphenyl phosphonium moiety, which is a well-known tactic for achieving mitochondrial localization.44 Finally, 5 includes an oligo(p-phenylenevinylene) (OPV) group which provides a handle for fluorescence microscopy while enabling self-assembly in water. Indeed, 5 readily forms nanoparticles with ca. 50 nm in diameter as shown by SEM and DLS. Kinetic measurements of the 5-catalyzed reduction of NAD+ by formate showed that 5 has a ca. 15-fold higher maximal TOF compared with the non-aggregating catalyst Ru(p-cymene)(TsEn). This reactivity boost is attributed to the large, cationic microenvironment presented by 5, which increases the concentration of anionic NAD+ near the Ru centers. 5 proved to be permeable toward ovarian cancer A2780 cells with mitochondrial localization being attained within one hour. Exposure of cells to 5 with inhibitors of various uptake mechanisms implicates clathrin-dependent endocytosis as the initial point of entry. The authors then evaluated the 5-catalyzed reduction of NAD+ in A2780 cells using formate as an externally added reductant (Fig. 3b). NAD+/NADH ratios were reduced in a catalyst- and formate-dependent manner. While this work demonstrates a creative approach for substrate binding, some areas for improvement include the toxicity and rather modest TOF of the catalyst. In eukaryotic cells, mitochondrial ATP production consumes NADH and FADH2, which are in turn supplied by the citric acid cycle. A future research direction might be to create ‘semi-synthetic mitochondria’ which use TM-catalyzed transfer hydrogenation as a replacement for the citric acid cycle.
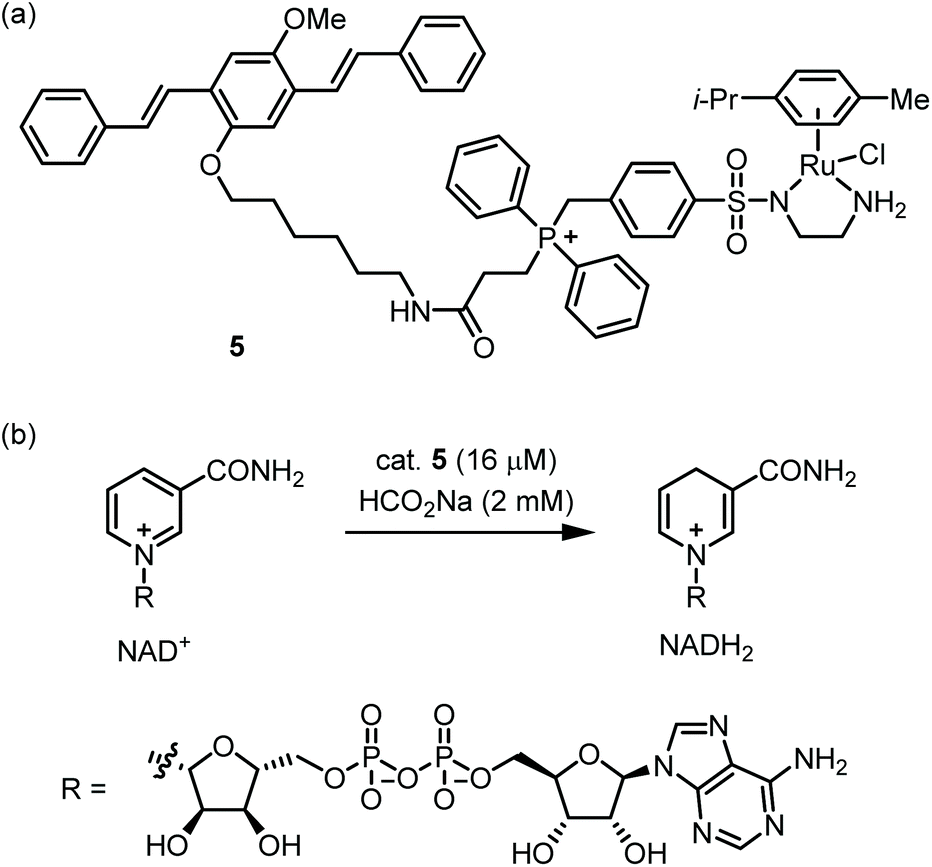 | ||
| Fig. 3 Perturbing intracellular redox state via transfer hydrogenation of mitochondorial NAD+ to NADH catalyzed by NP-forming Ru complex 5 with HCOONa as hydrogen source. (a) Structure of mitochondoria-accumulating, NP-forming Ru complex catalyst. (b) Reduction of NAD+ to NADH catalyzed by 5. | ||
The Zimmerman group reported ruthenium-containing single chain NPs (RuSCPs) for azide reduction within endosomes.45 Acrylamide co-polymer 6 was reacted with a Ru(bpy)3-containing bis-alkyne crosslinker 7 under copper click conditions (Fig. 4a). The resulting RuSCNP 8 is ca. 7 nm in diameter and features a lipophilic interior impregnated with Ru(bpy)3 units and a cationic surface. 8 readily enters HeLa endosomes where it catalyzes the photochemical reduction of the azide function in 9 to rhodamine 110 (10; Fig. 4b). Of note, the reaction exploits the reducing conditions within endosomes and does not require an exogenous reducing agent. The authors then demonstrated that 8 tightly binds β-galactosidase and mediates its transport into endosomes. These two catalysts can then work in parallel to activate two fluorophores or two toxins. These results provide proof-of-concept for repurposing the endosome as a ‘designer organelle’ where an abiotic catalyst and an enzyme can work in parallel or as an assembly line. As an impressive further application, 8 was used to ferry β-galactosidase into E. coli cells, where the two catalysts work in series to activate masked fluorophore 11 (Fig. 4c). Specifically, 8-catalyzed reduction of azide groups in 11 led to deprotection of the galactose moiety to afford 12, which then was hydrolyzed by β-galactosidase to produce a coumarin-type fluorescent reporter.
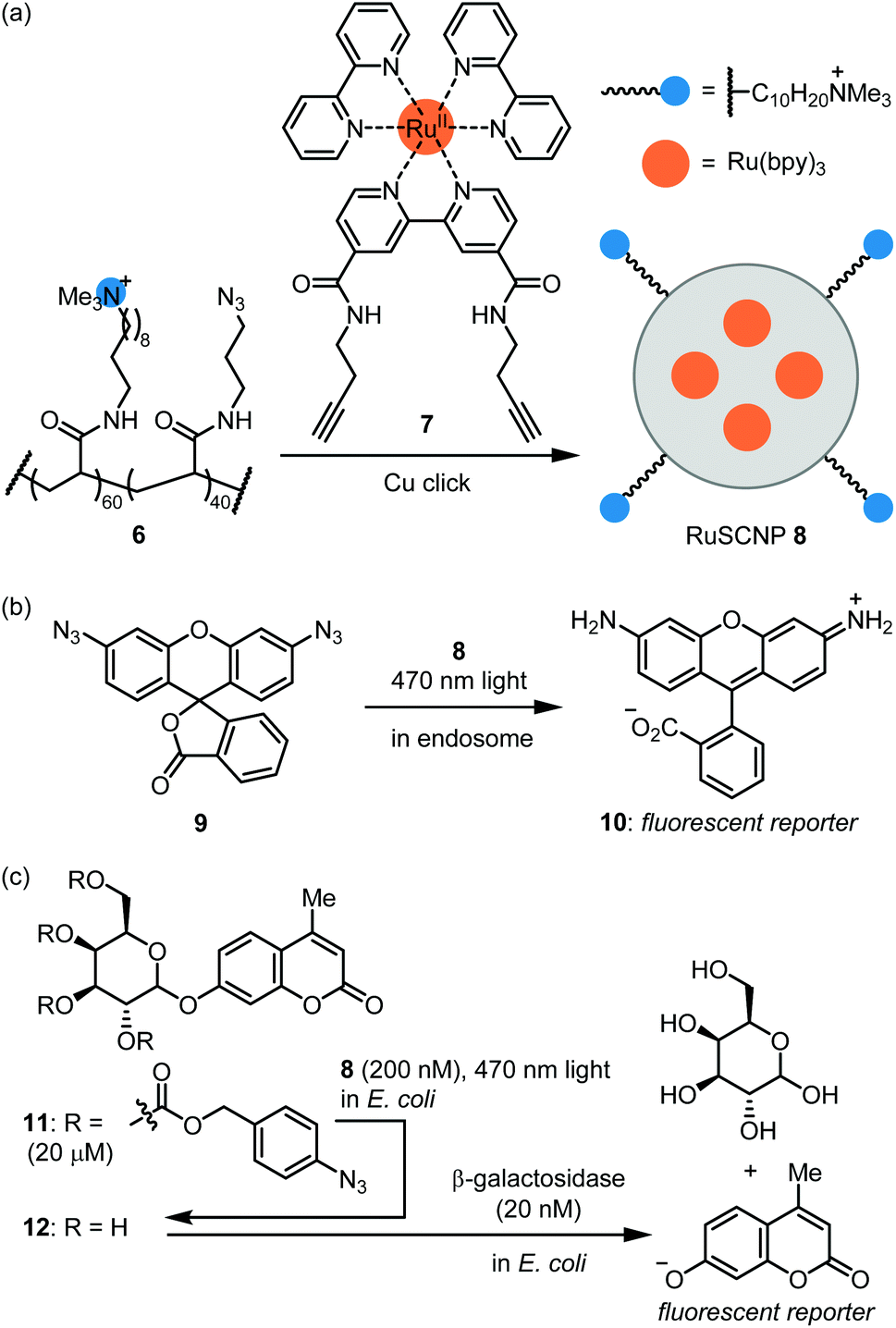 | ||
| Fig. 4 Hybrid catalysis comprising Ru photosensitizer nanoparticle and β-galactosidase. (a) Formation of RuSCNP 8. (b) A fluorogenic reaction by photocatalytic reduction of azide functions to amines proceeds in endosome of HeLa cells. (c) Integration of catalyst 8 and β-galactosidase in E. coli. | ||
Quantum dot (QDs)
In the pursuit of environmental sustainability, bacterial metalloenzymes which couple ATP hydrolysis with the reduction of N2 or CO2 have attracted considerable interest.46,47 The Nagpal group reported a hybrid catalyst system for using light instead of ATP to drive these reactions within living cells (Fig. 5).48 Upon light irradiation, a photoexcited electron is transferred from QD to a metalloenzyme expressed in cells. The reduced enzyme then mediates the reduction of either N2 or CO2, depending on the enzyme used. Finally, an electron donor (D) regenerates the QD and completes the catalytic cycle. An advantage of such nanobiohybrid organisms (or nanorgs) is that the redox potential to activate the enzymes can be tuned by the band gaps of QDs depending on the target reaction.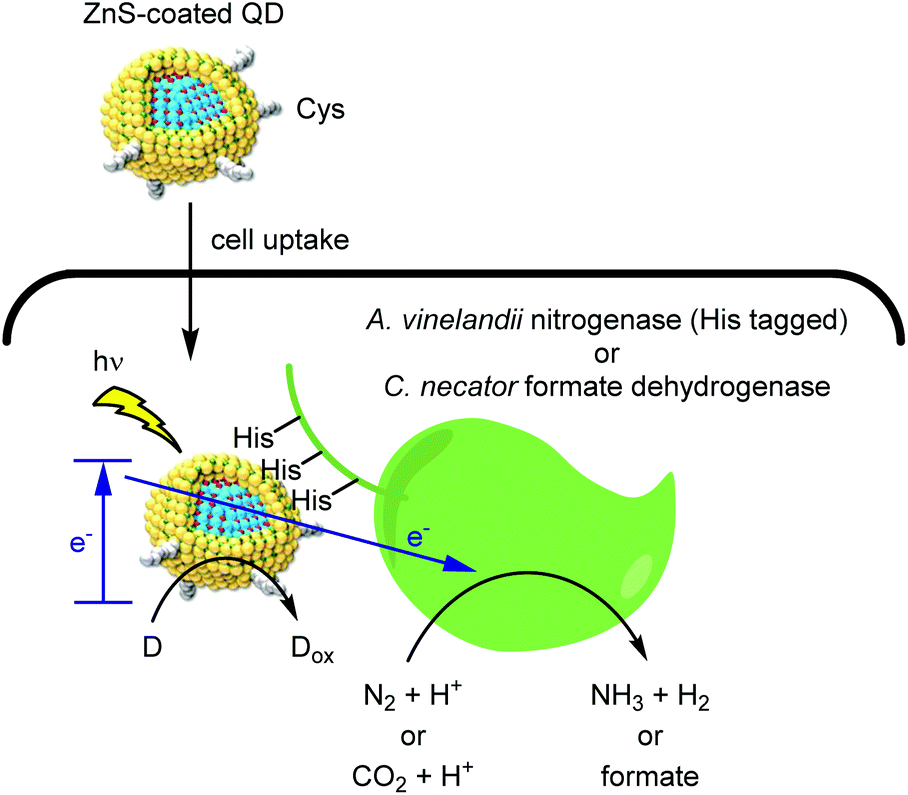 | ||
| Fig. 5 Hybrid catalysis comprising QD and redox enzymes within organisms to generate value-added small molecules from natural aerobic resources. | ||
To realize this process within living cells, two QD design features were critical. First, a double monolayer zinc sulfide shell served as a handle for complexation to the target metalloenzyme. Second, capping the QDs with the zwitterionic cysteine ligand provided cell permeability while avoiding the toxicity frequently associated with polycationic molecules.49 Complexation of QDs with His-tagged molybdenum iron nitrogenase within A. vinelandii cells afforded a N2 reduction catalyst with a TOF of more than 107 h−1. Furthermore, QD binding to the cytosolic formate dehydrogeanse50,51 within C. necator strains enabled the light-driven conversion of CO2 to feedstock chemicals such as polyhydroxybutyrate (PHB), ethylene, and methyl ketones. Indium phosphide QDs permitted the use of visible light, thus enabling gram scale synthesis of PHB using a 4 liter reactor. As a stepping stone toward industrial scale application, the use of living cells is attractive as it eliminates the need for enzyme purification. We propose that continuous flow chemistry52 using immobilized cells might help overcome product inhibition and enable efficient photon delivery.
Organic photosensitizer
Organic photosensitizers are by far the most successful class of small molecule catalyst in terms of clinical translation. Thus far, their most fruitful application is anticancer therapy, and several photosensitizers are represented among approved drugs.53 The Urano group reported a photosensitizer for selective killing of cells expressing β-galactosidase (Fig. 6a).54 SPiDER-killer-βGal 13 is itself cell-permeable and non-phototoxic. After 13 enters lacZ-positive cells, the glycosidic bond is cleaved by β-galactosidase to afford quinone methide intermediate 14. This intermediate then reacts with an endogenous nucleophile, thus trapping the photosensitizer within the cell. Adduct 15 potently generates singlet oxygen upon visible light irradiation, which leads to apoptotic cell death. When a co-culture of HEK-293 (pre-stained green) and HEK-293-lacZ(+) cells (pre-stained blue) was treated with 13, only the HEK-293-lacZ(+) cells underwent apoptosis (Fig. 6b). Importantly, a previously reported photosensitizer which lacks the masked electrophile55 does not exhibit this selectivity as the active phototoxin can leak out of the cell. In further experiments, the GAL-4/UAS system56 was used to generate fruit fly wing discs which express lacZ only in the posterior region. Upon incubation with 13 and light irradiation, the posterior wing disc cells were selectively killed, as indicated by the lack of calcein-AM fluorescence. As the authors note, 13 will provide an alternative to genetically encoded photosensitizers57,58 for selective killing of specific cell types.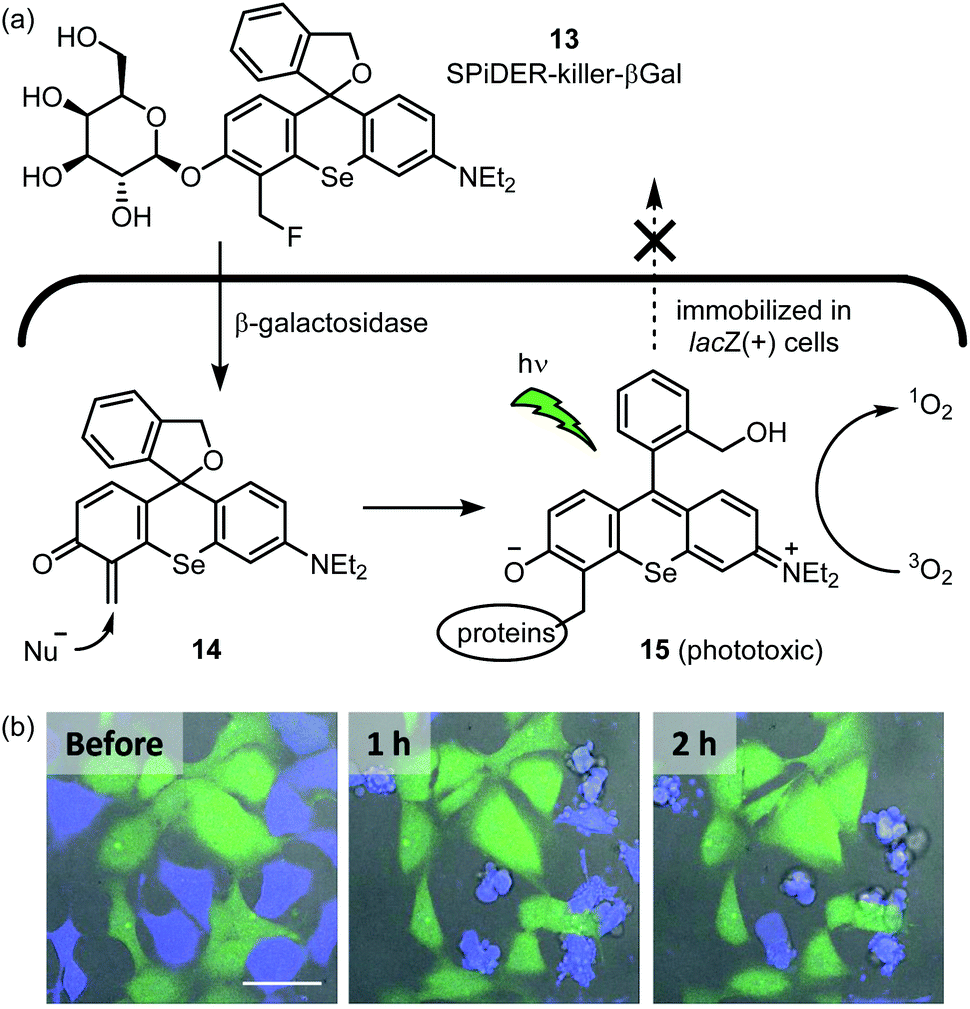 | ||
| Fig. 6 In-cell hybrid catalysis comprising β-galactosidase and activatable photosensitizer. Selective cytotoxicity to lacZ(+) cells expressing β-galactosidase is achieved. (a) Mechanism for enzymatic activation of cell permeable and non-phototoxic 13 and intracellular retention of phototoxic form 15. (b) Time-lapse fluorescence imaging of a coculture of HEK293 (green) and HEK-lacZ(+) (blue) cells. The coculture was incubated with 1 μM 13 for 1 h at 37 °C, followed by light irradiation (ν = 561 nm, 5 min). Immediately thereafter, time-lapse fluorescence imaging was started. Fluorescent image photos are reproduced from ref. 54 with permission from the American Chemical Society (ACS). Further permissions related to the material excerpted should be directed to the ACS. | ||
Organocatalyst
Compared with the modalities described above, in-cell synthetic organocatalysis is much less developed. We propose two reasons for this. First, organocatalyst-mediated reactions frequently suffer from low TOFs and thus require high catalyst loadings. Thus, the catalyst's toxicity and solubility may be of concern. Second, reaction mechanisms which use hydrogen bonding or strong acids/bases (for instance, Brønsted acid or anion binding catalysis) are difficult in aqueous solvent and especially difficult in a complex intracellular environment.59The Kanai group recently reported site-selective lysine acylation of histones within living cells using organocatalyst 16 (Fig. 7a).60 This catalyst is comprised of a nucleosome-localizing N-terminal LANA peptide61 and a prodrugged catalytically-active Lewis base moiety, DSSMe. 16 is reduced prior to the acylation reaction either outside or inside the cells by biocompatible reductant such as tris(2-carboxyethyl)phosphine (TCEP) or GSH, to generate catalytically active PEG–LANA–DSH (17). 17 catalyzes regioselective acyl transfer from cell-permeable donor 18 to histone H2B K120 (Fig. 7b and c). The regioselectivity is defined by proximity of the DSH moiety to the acylated lysine residue (Fig. 7b).62 Although the LANA moiety is unstable toward proteases, attachment of a polyethylene glycol (PEG) ensured sustained chromatin localization in living cells. Increasing the length of the PEG group increased catalyst stability, albeit at the expense of some catalytic activity. This is the first example of introducing physiologically-relevant histone posttranslational modifications (PTMs) in living cells by entirely chemical means without relying on enzymes. Analogous to the blocking of a chemical reaction using protecting group chemistry, synthetic H2B K120 acetylation suppressed enzymatic ubiquitination of this same residue in living cells (Fig. 7c). As H2B K120 ubiquitination regulates cell-cycle progression and DNA damage response,63 proximity-assisted Lewis base catalysis may be a useful tactic for interrogating epigenetic mechanisms. However, a current area for improvement is the requirement for bead-loading64 to achieve catalyst delivery.
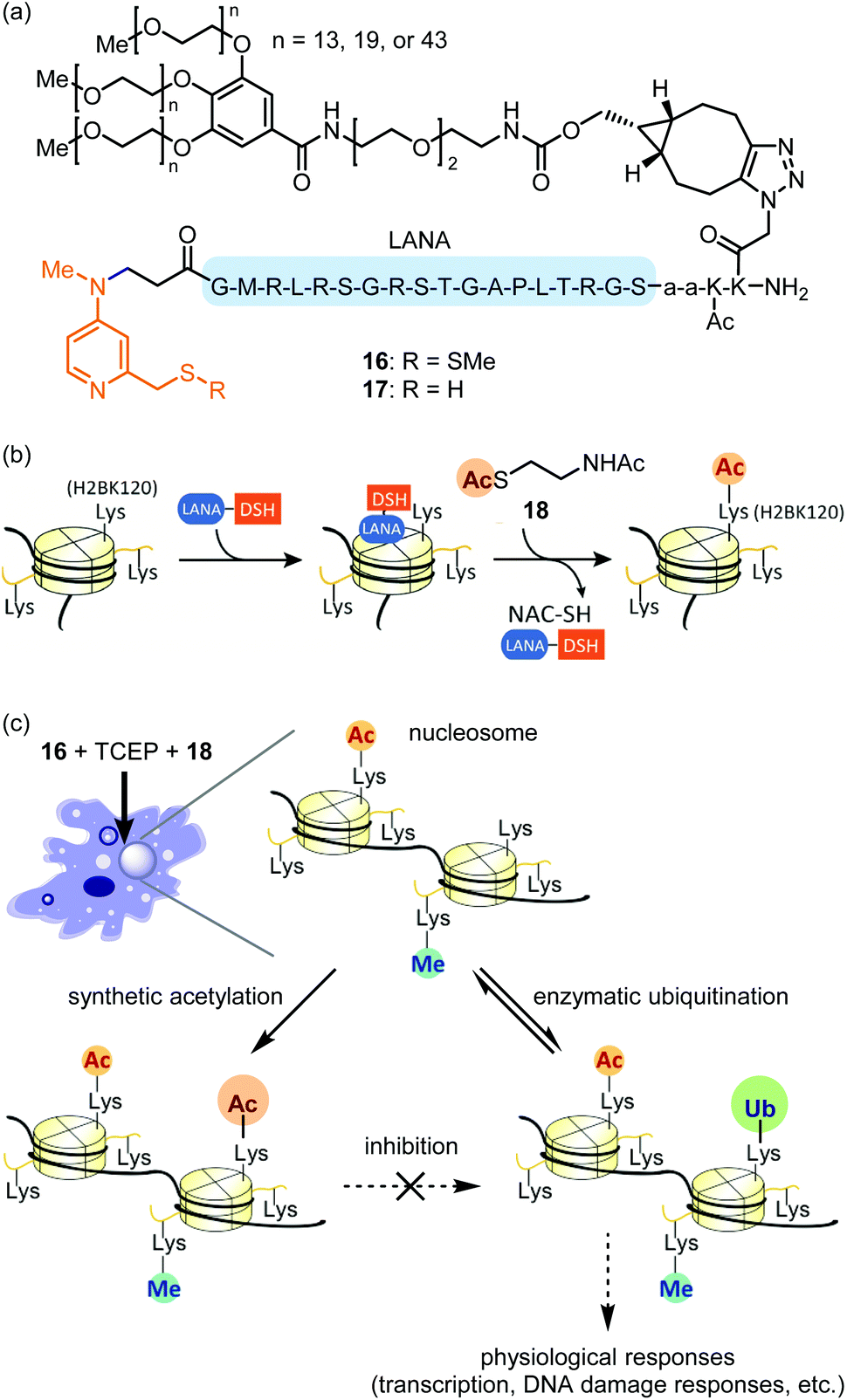 | ||
| Fig. 7 Competition between chemical catalysis and enzymatic catalysis in living cells. (a) Structures of regioselective acylation catalysts of nucleosomal histones in living cells. (b) Concept of ligand-directed catalysis, promoting H2BK120-selective acylation. (c) H2BK120 is regioselectively “protected” by synthetic acetylation from ubiquitin ligase-promoted ubiquitination. H2BK ubiquitination is a physiologically significant epigenome mark. Synthetic acetylation in living cells is conducted with bead-loaded 16 (500 μM), TCEP (2 mM), and 18 (10 mM) at 37 °C for 8 h. After the reaction, cells were incubated for 36 h for enzymatic ubiquitination/deubiquitination. | ||
Conclusions and outlook
As we have shown, many abiotic catalyst modalities can achieve co-operativity with the enzymatic machinery of living cells. However, increased biocompatibility and catalyst activity are required for real-world applications. Improvements in ligand design may be an avenue to enhance the biostability of noble metal-based catalysts. Within the realm of ArMs, recent advances in deep learning for the prediction of protein folding65 foreshadow a greater role for computer-assisted design of scaffold proteins. We propose that intracellular organocatalysis could benefit from the substrate binding provided by a hydrophobic scaffold. Further, halogen bonding66 might be a useful bio-orthogonal interaction to be harnessed by an organocatalyst.The coming years will see increased use of abiotic catalysts within multicellular organisms,16 which present far greater pharmacokinetic obstacles compared with cultured cells. We hope that this work will establish synthetic catalysis as a bona fide pharmaceutical modality (catalysis medicine). Furthermore, we envision that stable integration of synthetic catalyst(s) within cellular metabolism will afford ‘semi-synthetic life’, which combines the desirable features of living organisms (self-replication and -repair, evolution) with the unique reactivity of abiotic catalysts. Such hybrid organisms might be envisioned to convert waste biomass or CO2 to useful feedstocks, or to decompose plastic waste.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank JSPS KAKENHI Grant Numbers JP17H06442 and JP20H00489 for financial support (M. K.). C. A. is the recipient of a Natural Sciences and Engineering Research Council of Canada (NSERC) PGS-D fellowship.Notes and references
- G. Sirasani, L. Tong and E. P. Balskus, A Biocompatible Alkene Hydrogenation Merges Organic Synthesis with Microbial Metabolism, Angew. Chem., Int. Ed., 2014, 53, 7785–7788 CrossRef CAS.
- S. Wallace and E. P. Balskus, Interfacing Microbial Styrene Production with a Biocompatible Cyclopropanation Reaction, Angew. Chem., Int. Ed., 2015, 54, 7106–7109 CrossRef CAS.
- S. Wu, Y. Zhou, D. Gerngross, M. Jeschek and T. R. Ward, Chemo-Enzymatic Cascades to Produce Cycloalkenes from Bio-Based Resources, Nat. Commun., 2019, 10, 5060–5069 CrossRef.
- J. M. Foulkes, K. J. Malone, V. S. Coker, N. J. Turner and J. R. Lloyd, Engineering a Biometallic Whole Cell Catalyst for Enantioselective Deracemization Reactions, ACS Catal., 2011, 1, 1589–1594 CrossRef CAS.
- J. Guo, M. Suástegui, K. K. Sakimoto, V. M. Moody, G. Xiao, D. G. Nocera and N. S. Joshi, Light-Driven Fine Chemical Production in Yeast Biohybrids, Science, 2018, 362, 813–816 CrossRef CAS.
- C. Hannesschlaeger, A. Horner and P. Pohl, Intrinsic Membrane Permeability to Small Molecules, Chem. Rev., 2019, 119, 5922–5953 CrossRef CAS.
- H. T. Hsu, B. M. Trantow, R. M. Waymouth and P. A. Wender, Bioorthogonal Catalysis: A General Method to Evaluate Metal-Catalyzed Reactions in Real Time in Living Systems Using a Cellular Luciferase Reporter System, Bioconjugate Chem., 2016, 27, 376–382 CrossRef CAS.
- Y. Bai, J. Chen and S. C. Zimmerman, Designed Transition Metal Catalysts for Intracellular Organic Synthesis, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- C. Streu and E. Meggers, Ruthenium-Induced Allylcarbamate Cleavage in Living Cells, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Fast, Cell-Compatible Click Chemistry with Copper-Chelating Azides for Biomolecular Labeling, Angew. Chem., Int. Ed., 2012, 51, 5852–5856 CrossRef CAS.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Palladium-Triggered Deprotection Chemistry for Protein Activation in Living Cells, Nat. Chem., 2014, 6, 352–361 CrossRef CAS.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Asymmetric Transfer Hydrogenation by Synthetic Catalysts in Cancer Cells, Nat. Chem., 2018, 10, 347–354 CrossRef CAS.
- K. Y. Tong, J. Zhao, C. W. Tse, P. K. Wan, J. Rong and H. Y. Au-Yeung, Selective Catecholamine Detection in Living Cells by a Copper-Mediated Oxidative Bond Cleavage, Chem. Sci., 2019, 10, 8519–8526 RSC.
- Y. Obara, A. Yamauchi, S. Takehara, W. Nemoto, M. Takahashi, P. J. S. Stork and N. Nakahata, ERK5 Activity Is Required for Nerve Growth Factor-Induced Neurite Outgrowth and Stabilization of Tyrosine Hydroxylase in PC12 Cells, J. Biol. Chem., 2009, 284, 23564–23573 CrossRef CAS.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Artificial Metalloenzymes: Reaction Scope and Optimization Strategies, Chem. Rev., 2018, 118, 142–231 CrossRef CAS.
- K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, In Vivo Gold Complex Catalysis within Live Mice, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS.
- Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. A. Ward, Cell-Penetrating Artificial Metalloenzyme Regulates a Gene Switch in a Designer Mammalian Cell, Nat. Commun., 2018, 9, 1943–1949 CrossRef.
- V. Sabatino, J. G. Rebelein and T. R. Ward, “Close-to-Release”: Spontaneous Bioorthogonal Uncaging Resulting from Ring-Closing Metathesis, J. Am. Chem. Soc., 2019, 141, 17048–17052 CrossRef CAS.
- T. S. Young and P. G. Schultz, Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS.
- I. Drienovská, A. Rioz-Martínez, A. Draksharapu and G. Roelfes, Novel Artificial Metalloenzymes by in Vivo Incorporation of Metal-Binding Unnatural Amino Acids, Chem. Sci., 2015, 6, 770–776 RSC.
- H. Yang, A. M. Swartz, H. J. Park, P. Srivastava, K. Ellis-Guardiola, D. M. Upp, G. Lee, K. Belsare, Y. Gu, C. Zhang, R. E. Moellering and J. C. Lewis, Evolving Artificial Metalloenzymes via Random Mutagenesis, Nat. Chem., 2018, 10, 318–324 CrossRef CAS.
- M. E. Wilson and G. M. Whitesides, Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety, J. Am. Chem. Soc., 1978, 100, 306–307 CrossRef CAS.
- H. J. Davis and T. R. Ward, Artificial Metalloenzymes: Challenges and Opportunities, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS.
- Y. M. Wilson, M. Dürrenberger, E. S. Nogueira and T. R. Ward, Neutralizing the Detrimental Effect of Glutathione on Precious Metal Catalysts, J. Am. Chem. Soc., 2014, 136, 8928–8932 CrossRef CAS.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Directed Evolution of Artificial Metalloenzymes for in Vivo Metathesis, Nature, 2016, 537, 661–665 CrossRef CAS.
- T. Heinisch, F. Schwizer, B. Garabedian, E. Csibra, M. Jeschek, J. Vallapurackal, V. B. Pinheiro, P. Marlière, S. Panke, T. R. Ward and E. Coli, Surface Display of Streptavidin for Directed Evolution of an Allylic Deallylase, Chem. Sci., 2018, 9, 5383–5388 RSC.
- J. Huang, Z. Liu, B. Bloomer, D. Clark, A. Mukhopadhyay, J. Keasling and J. Hartwig, Artificial Biosynthetic Pathway for an Unnatural Terpenoid with an Iridiumcontaining P450, ChemRxiv, 2020 DOI:10.26434/chemrxiv.11955174.v1.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron, Nature, 2016, 534, 534–537 CrossRef CAS.
- H. M. Key, P. Dydio, Z. Liu, J. Y. E. Rha, A. Nazarenko, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclopropanations of Structurally Diverse Alkenes, ACS Cent. Sci., 2017, 3, 302–308 CrossRef CAS.
- D. P. Henderson, E. E. Wyckoff, C. E. Rashidi, H. Verlei and A. L. Oldham, Characterization of the Plesiomonas Shigelloides Genes Encoding the Heme Iron Utilization System, J. Bacteriol., 2001, 183, 2715–2723 CrossRef CAS.
- M. T. Reetz and J. D. Carballeira, Iterative Saturation Mutagenesis (ISM) for Rapid Directed Evolution of Functional Enzymes, Nat. Protoc., 2007, 2, 891–903 CrossRef CAS.
- K. Islam, The Bump-and-Hole Tactic: Expanding the Scope of Chemical Genetics, Cell Chem. Biol., 2018, 25, 1171–1184 CrossRef CAS.
- R. Cao-Milán, L. D. He, S. Shorkey, G. Y. Tonga, L.-S. Wang, X. Zhang, I. Uddin, R. Das, M. Sulak and V. M. Rotello, Modulating the Catalytic Activity of Enzyme-like Nanoparticles through Their Surface Functionalization, Mol. Syst. Des. Eng., 2017, 2, 624–628 RSC.
- K. Neumann, A. Lilienkampf and M. Bradley, Responsive Polymeric Nanoparticles for Controlled Drug Delivery, Polym. Int., 2017, 66, 1756–1764 CrossRef CAS.
- C. M. Beddoes, C. P. Case and W. H. Briscoe, Understanding Nanoparticle Cellular Entry: A Physicochemical Perspective, Adv. Colloid Interface Sci., 2015, 218, 48–68 CrossRef CAS.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Palladium-Mediated Intracellular Chemistry, Nat. Chem., 2011, 3, 239–243 CrossRef CAS.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nano-Palladium Is a Cellular Catalyst for in Vivo Chemistry, Nat. Commun., 2017, 8, 15906–15918 CrossRef CAS.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, In-Cell Dual Drug Synthesis by Cancer-Targeting Palladium Catalysts, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS.
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, A Biocompatible Heterogeneous MOF–Cu Catalyst for In Vivo Drug Synthesis in Targeted Subcellular Organelles, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS.
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, Near-Infrared Light Dual-Promoted Heterogeneous Copper Nanocatalyst for Highly Efficient Bioorthogonal Chemistry in Vivo, ACS Nano, 2020, 14, 4178–4187 CrossRef CAS.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Supramolecular Regulation of Bioorthogonal Catalysis in Cells Using Nanoparticle-embedded Transition Metal Catalysts, Nat. Chem., 2015, 7, 597–603 CrossRef CAS.
- N. Dai, H. Zhao, R. Qi, Y. Chen, F. Lv, L. Liu and S. Wang, Fluorescent and Biocompatible Ruthenium-Coordinated Oligo(p-Phenylenevinylene) Nanocatalysts for Transfer Hydrogenation in the Mitochondria of Living Cells, Chem. – Eur. J., 2020, 26, 4489–4495 CrossRef CAS.
- J. Canivet and G. Süss-Fink, Water-Soluble Arene Ruthenium Catalysts Containing Sulfonated Diamine Ligands for Asymmetric Transfer Hydrogenation of α-Aryl Ketones and Imines in Aqueous Solution, Green Chem., 2007, 9, 391–397 RSC.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS.
- J. Chen, K. Li, J. S. Shon and S. C. Zimmerman, Single-Chain Nanoparticle Delivers a Partner Enzyme for Concurrent and Tandem Catalysis in Cells, J. Am. Chem. Soc., 2020, 142, 4565–4569 CrossRef CAS.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Light-Driven Dinitrogen Reduction Catalyzed by a CdS:Nitrogenase MoFe Protein Biohybrid, Science, 2016, 352, 448–450 CrossRef CAS.
- R. Cai, R. D. Milton, S. Abdellaoui, T. Park, J. Patel, B. Alkotaini and S. D. Minteer, Electroenzymatic C-C Bond Formation from CO2, J. Am. Chem. Soc., 2018, 140, 5041–5044 CrossRef CAS.
- Y. Ding, J. R. Bertram, C. Eckert, R. R. Bommareddy, R. Patel, A. Conradie, S. Bryan and P. Nagpal, Nanorg Microbial Factories: Light-Driven Renewable Biochemical Synthesis Using Quantum Dot-Bacteria Nanobiohybrids, J. Am. Chem. Soc., 2019, 141, 10272–10282 CrossRef CAS.
- B. D. Monnery, M. Wright, R. Cavill, R. Hoogenboom, S. Shaunak, J. H. G. Steinke and M. Thanou, Cytotoxicity of Polycations: Relationship of Molecular Weight and the Hydrolytic Theory of the Mechanism of Toxicity, Int. J. Pharm., 2017, 521, 249–258 CrossRef CAS.
- D. Niks, J. Duvvuru, M. Escalona and R. Hille, Spectroscopic and Kinetic Properties of the Molybdenum-Containing, NAD+-Dependent Formate Dehydrogenase from Ralstonia Eutropha, J. Biol. Chem., 2016, 291, 1162–1174 CrossRef CAS.
- X. Yu, D. Niks, A. Mulchandani and R. Hille, Efficient Reduction of CO2 by the Molybdenum-Containing Formate Dehydrogenase from Cupriavidus Necator (Ralstonia Eutropha), J. Biol. Chem., 2017, 292, 16872–16879 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- R. R. Allison and C. H. Sibata, Oncologic Photodynamic Therapy Photosensitizers: A Clinical Review, Photodiagn. Photodyn. Ther., 2010, 7, 61–75 CrossRef CAS.
- M. Chiba, M. Kamiya, K. Tsuda-Sakurai, Y. Fujisawa, H. Kosakamoto, R. Kojima, M. Miura and Y. Urano, Activatable Photosensitizer for Targeted Ablation of LacZ-Positive Cells with Single-Cell Resolution, ACS Cent. Sci., 2019, 5, 1676–1681 CrossRef CAS.
- Y. Ichikawa, M. Kamiya, F. Obata, M. Miura, T. Terai, T. Komatsu, T. Ueno, K. Hanaoka, T. Nagano and Y. Urano, Selective Ablation of β-Galactosidase-Expressing Cells with a Rationally Designed Activatable Photosensitizer, Angew. Chem., Int. Ed., 2014, 53, 6772–6775 CrossRef CAS.
- J. B. Duffy, GAL4 System in Drosophila: A Fly Geneticist's Swiss Army Knife, Genesis, 2002, 34, 1–15 CrossRef CAS.
- M. E. Bulina, D. M. Chudakov, O. V. Britanova, Y. G. Yanushevich, D. B. Staroverov, T. V. Chepurnykh, E. M. Merzlyak, M. A. Shkrob, S. Lukyanov and K. A. A. Lukyanov, Genetically Encoded Photosensitizer, Nat. Biotechnol., 2006, 24, 95–99 CrossRef CAS.
- X. Shu, V. Lev-Ram, T. J. Deerinck, Y. Qi, E. B. Ramko, M. W. Davidson, Y. Jin, M. H. Ellisman and R. Y. Tsien, A Genetically Encoded Tag for Correlated Light and Electron Microscopy of Intact Cells, Tissues, and Organisms, PLoS Biol., 2011, 9, e1001041 CrossRef CAS.
- M. P. van der Helm, B. Klemm and R. Eelkema, Organocatalysis in Aqueous Media, Nat. Rev. Chem., 2019, 3, 491–508 CrossRef CAS.
- Y. Fujiwara, Y. Yamanashi, Y. Sato, T. Kujirai, H. Kurumizaka, H. Kimura, K. Yamatsugu, S. A. Kawashima and M. Kanai, Live-Cell Epigenome Manipulation by Synthetic Histone Acylation Catalyst System, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12186531.
- A. J. Barbera, J. V. Chodaparambil, B. Kelley-Clarke, V. Joukov, J. C. Walter, K. Luger and K. M. Kaye, The Nucleosomal Surface as a Docking Station for Kaposi's Sarcoma Herpesvirus LANA, Science, 2006, 311, 856–861 CrossRef CAS.
- Y. Amamoto, Y. Aoi, N. Nagashima, H. Suto, D. Yoshidome, Y. Arimura, A. Osakabe, D. Kato, H. Kurumizaka, S. A. Kawashima, K. Yamatsugu and M. Kanai, Synthetic Posttranslational Modifications: Chemical Catalyst-Driven Regioselective Histone Acylation of Native Chromatin, J. Am. Chem. Soc., 2017, 139, 7568–7576 CrossRef CAS.
- B. Zhu, Y. Zheng, A. D. Pham, S. S. Mandal, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Monoubiquitination of Human Histone H2B: The Factors Involved and Their Roles in HOX Gene Regulation, Mol. Cell, 2005, 20, 601–611 CrossRef CAS.
- P. L. McNeil and E. Warder, Glass Beads Load Macromolecules into Living Cells, J. Cell Sci., 1987, 88, 669–678 Search PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Improved Protein Structure Prediction Using Potentials from Deep Learning, Nature, 2020, 577, 706–710 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |