
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and assembly properties of a water-soluble blue naphthalene diimide†
Thomas A.
Welsh
 a,
Olga
Matsarskaia
b,
Ralf
Schweins
b and
Emily R.
Draper
*a
a,
Olga
Matsarskaia
b,
Ralf
Schweins
b and
Emily R.
Draper
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Emily.Draper@glasgow.ac.uk
bInstitut Laue-Langevin, Large Scale Structures Group, 71 Avenue des Martyrs, CS 20156, F-38042 Grenoble CEDEX 9, France
First published on 14th July 2021
Abstract
Herein we report on the synthesis and characterisation of a water soluble deep blue naphthalene diimide, (iPrNH)2NDI–V. The synthesis is performed under mild conditions and careful consideration of the purification of the precuror materials leads to improved results in final yield and purity. The optoelectronic properties are explored through cyclic voltammetry and UV-vis absorption and emission spectroscopy in multiple solvents and the self-assembling properties as part of a multi-component hydrogel are investigated through rheology and small angle neutron scattering.
Introduction
Research into new organic electronic materials has led to rapid developments in photovoltaics,1–4 field-effect transistors,5–7 light-emitting diodes,8–10 sensors,11,12 and batteries.13 Organic materials possess a number of advantages over alternatives such as their lightweight, flexibility, tuneable optoelectronic properties, solution processability, and derivability from renewable feedstocks. Of particular interest to the synthetic chemist are the readily tuneable optoelectronic properties and solution processability of organic materials. Through simple chemical reactions, a wide variety of modifications can be made to the material scaffold that drastically alter optical, electronic, and solubility properties.When considering material practicality, solubility is one of the most important factors. In order to be developed on an industrial scale, materials should be soluble in environmentally friendly, nontoxic solvents.14 Water is the ideal solvent but many organic materials are poorly soluble in water unless specially functionalised. In our research, we synthesise water soluble naphthalene diimides (NDIs) by functionalising at the imide positions with amino acids, which can be deprotonated to make fully water-soluble organic molecules (Scheme 1).15 NDIs are typically used as acceptor moieties in organic electronic devices owing to their ability to form stable radical anions.16,17 These stable radical anions are often accompanied by a large change in the absorption properties which allows for NDI application in electrochromic devices. We have previously worked with water soluble amino acid-functionalised NDIs in hydrogels and found that they display transparent-to-dark electrochromism,15 which makes them promising candidates for electrochromic device applications such as smart windows.18
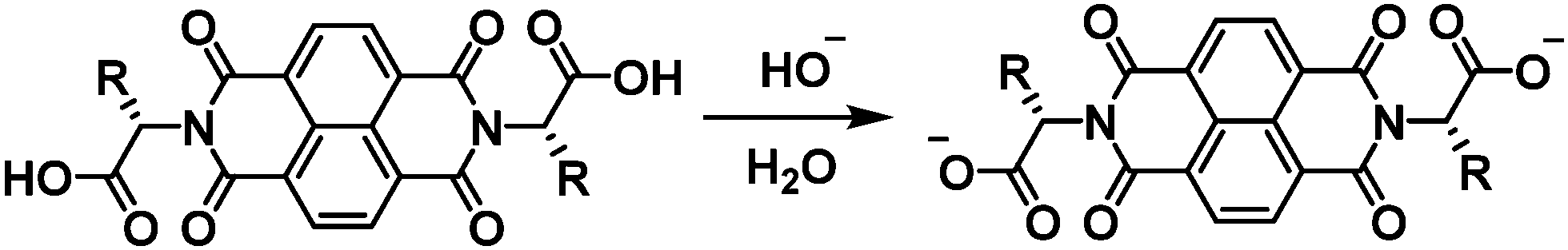 | ||
| Scheme 1 The method by which amino-acid functionalised NDIs are made water soluble. | ||
Naphthalene diimides are common moieties found in many electronic materials applications and a wide variety of functionalisation options are available to them. Aside from the aforementioned imide functionalisation, functionalisation of the naphthalene core is also very common.19 While functionalisation at the imide positions typically only affects the solubility properties, core functionalisation can also affect the optoelectronic properties of the species. Many core-functionalised NDIs exist with a wide variety of colours and applications.19–22 One such functionalisation is the incorporation of amino groups off the core which has been shown to provide NDIs with exceptional photophysical properties including a deep blue colour.22–26 The synthesis can also be done under mild conditions without the need for co-reagents or transition metal catalysts.26 Some of these materials have even been made water soluble. This is typically achieved by using acid to protonate the amino substituents to make water soluble cationic salts, as was reported by Weißenstein et al.23 and Doria et al.,24 which is in contrast to our method of using base to deprotonate amino acid side chains to make water soluble NDIs.15 Another strategy employed by Higginbotham et al.25 is to incorporate hydrophilic neutral functional groups such as glycols. However, this method was not as effective for making water soluble materials as producing charged species for the neutral materials formed micelles in aqueous solution.
Herein, we describe the synthesis of a water-soluble NDI core-functionalised with isopropylamino groups using mild reaction conditions. We investigate the optoelectronic and self-assembly properties via cyclic voltammetry (CV), UV-vis absorption and emission spectroscopy, rheology, and small angle neutron scattering (SANS) and assess its potential applications.
Results and discussion
Synthesis
Obtaining a core-functionalised NDI requires beginning with the unsubstituted naphthalene tetracarboxylic dianhydride (NTCDA). Through our previous research with synthesising NDIs, we have found that the NTCDA supplied by common chemical suppliers is only about 20% pure with the majority of the material composition being the monoanhydride form. This has led to poor reaction outcomes and so we use a method to regenerate and purify the dianhydride by refluxing the crude material in acetic anhydride and recrystallising from N,N-dimethylformamide (DMF). A more detailed account of the purification method can be found in the ESI.† With pure NTCDA, the core-functionalised NDI could be obtained following the procedure outlined in Scheme 2.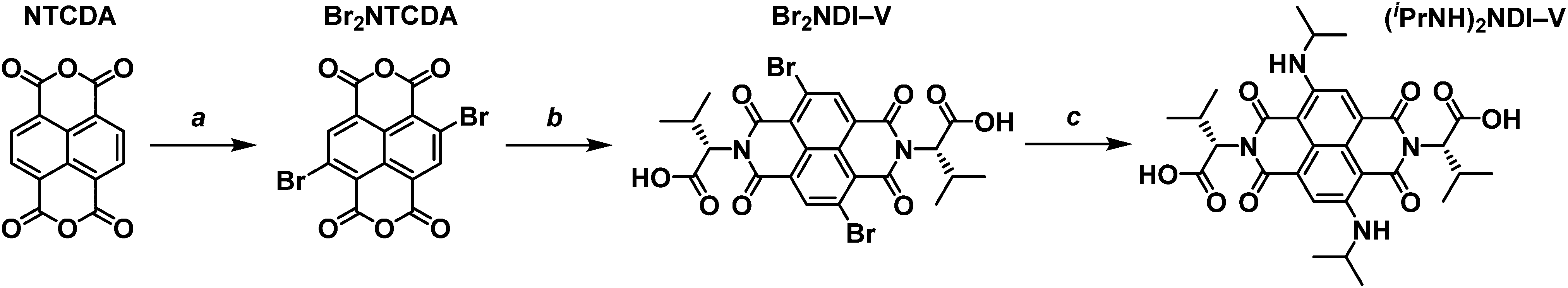 | ||
| Scheme 2 The synthetic method to obtain core-functionalised blue (iPrNH)2NDI–V. (a) Dibromoisocyanuric acid (2.5 eq.), H2SO4·20% SO3, 25 °C, 5 h, 29% yield. (b) L-Valine (2 eq.), glacial acetic acid, 120 °C, 18 h, 76% yield. (c) Isopropylamine, 25 °C, 10 days, 80% yield. | ||
The first step of preparing a core-substituted NDI is to chlorinate or brominate the NTCDA. Such reactions often require harsh conditions and so should be performed before any other functionalisation is done. There are many methods by which to introduce halides to an aromatic core27–29 and we have chosen the more mild route as outlined by Würthner et al.30 “Mild” here is of course a relative term as this method does require the use of fuming sulfuric acid. This reaction results in the bright yellow and highly insoluble dibrominated NTCDA. Due to its insolubility this material is often used in further reactions without purification or even NMR analysis. However, we have found that it is soluble enough in DMSO-d6 to obtain an NMR spectrum (ESI,† Fig. S1). The 1H NMR spectrum of the crude product shows that it is a mixture of species of varying levels of bromination (ESI,† Fig. S2) which can be purified by boiling in acetonitrile and filtering while hot to obtain Br2NTCDA in a 29% yield. Purifying Br2NTCDA in this way leads to improved results in the subsequent reactions.
The Br2NTCDA can be reacted with an amino acid, in this case L-valine, in glacial acetic acid to form the dibrominated NDI, Br2NDI–V. The crude product can be obtained as an orange solid in about a 75% yield by precipitating the reaction mixture in cold water and vacuum filtering. The crude product can be used without further purification, but it can be purified by stirring in acetonitrile at room temperature for a few hours and filtering.
The final reaction involves suspending the orange Br2NDI–V in isopropylamine and stirring at room temperature. According to the literature,26 this reaction leads to a 48% yield of the disubstituted product after 48 hours. The major impurity of this reaction is the monosubstituted form which is magenta in colour. However, we have found that leaving the mixture to stir at room temperature for 10 to 12 days leads to a much higher (80%) yield of the blue disubstituted product after column purification. The final product, (iPrNH)2NDI–V, is isolated as a dark blue solid and characterised by 1H and 13C NMR.
Electronic and optical characterisation
One of our primary interests in water-soluble NDIs lies in their electrochromic properties. The unfunctionalised analogue of (iPrNH)2NDI–V, NDI–V, for instance, displays distinct colourless to dark purple electrochromism upon reduction (Fig. 1). However, (iPrNH)2NDI–V does not display any change from its deep blue colour upon oxidation or reduction, in stark contrast to its analogue NDI–V or our previously reported electrochromic NDI.15 Thus, while isopropylamino-substitution on the naphthalene core does imbue the species with a vibrant blue colour, it also prevents any electrochromism. As a result, (iPrNH)2NDI–V would not be suitable for application in smart window or display technology and would be better suited as a water-soluble dye.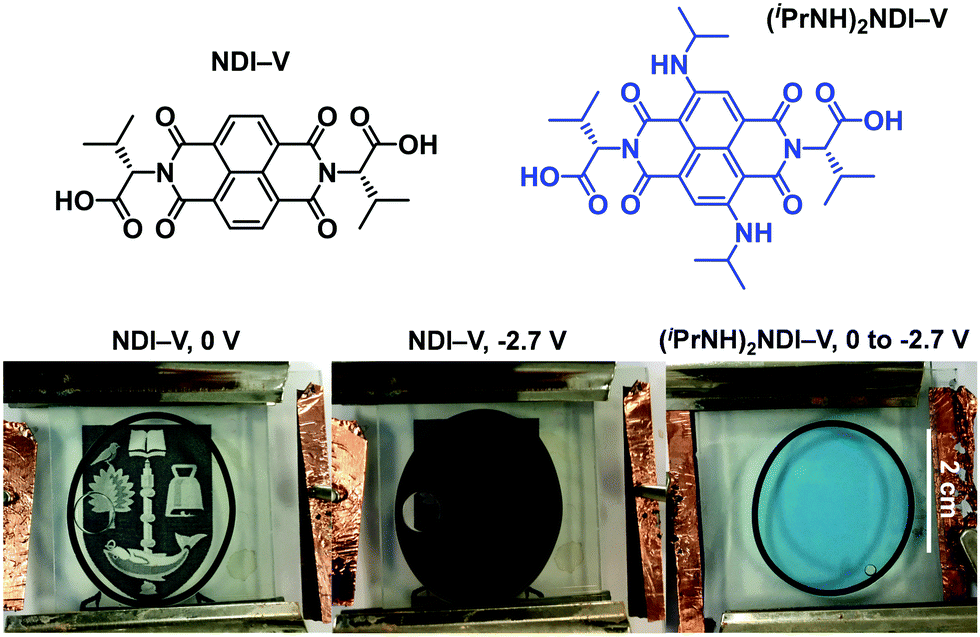 | ||
| Fig. 1 The chemical structures of NDI–V and (iPrNH)2NDI–V along with a display of the electrochromic properties of NDI–V and the lack of electrochromic properties of (iPrNH)2NDI–V. Both species were dissolved in water with 40% v/v 0.1 M NaCl electrolyte at pH 8. | ||
The electronic characteristics of the blue (iPrNH)2NDI–V were characterised via CV. The cyclic voltammogram is shown in Fig. 2a. It was measured in dichloromethane (DCM) with 0.1 M tetrabutylammonium hexafluorphosphate (NBu4PF6) electrolyte. There is a reversible oxidation wave with an E1/2 potential 0.64 V and an onset of 0.55 V which gives an ionization potential of 5.4 eV. There also appears to be two reduction events: an initial irreversible reduction with an onset of −1.01 V and a partially reversible reduction with an E1/2 of −1.58 V. From the onset of the first reduction, the electron affinity is calculated to be 3.8 eV which gives the material an electronic band gap of 1.6 eV. These values are listed in Table 1. Naphthalene diimides are known to be readily reduced to a radical anion and dianion31,32 and so the two reduction events observed in the CV likely correspond to these species. A fully reversible oxidation peak is usually not observed for NDIs but for (iPrNH)2NDI–V the incorporation of the amino substituents on the NDI core has raised the HOMO energy level and lowered the ionization potential thereby enabling reversible oxidation. Density functional theory calculations on similarly functionalised NDIs have shown that the core-amino group contributes significantly to the HOMO energy level.23
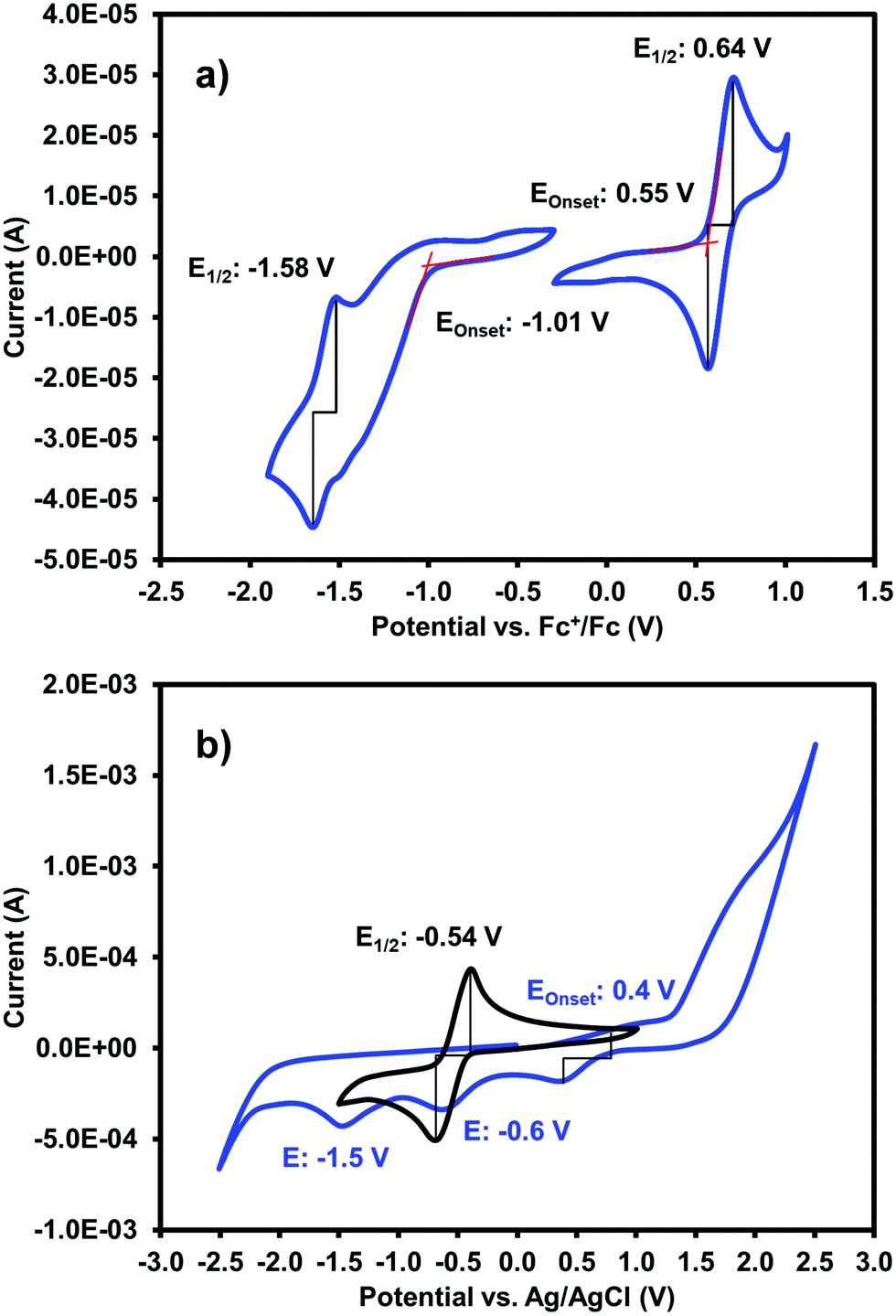 | ||
| Fig. 2 (a) The cyclic voltammogram for (iPrNH)2NDI–V measured in DCM with 0.1 M NBu4PF6 electrolyte. Oxidation and reduction onsets and E1/2 potentials are shown. (b) The cyclic voltammograms for (iPrNH)2NDI–V (blue trace) and NDI–V (black trace) measured in water with a 40% v/v 0.1 M NaCl electrolyte at pH 8. | ||
| Property | Value |
|---|---|
| a Energies were calculated by (onset V + 4.8) where Fc HOMO = 4.8 eV.33 b Stokes shift was calculated by (Eabs − Eems) where (Emax = h × c/λmax; h = Planck's constant, c = speed of light). c Optical band gap was calculated from the onset absorption (700 nm) where (Eopt = h × c/λonset). | |
| Ionization potential, IPa (eV) | 5.4 |
| Electron affinity, EAa (eV) | 3.8 |
| Electronic band gap, Eelec (eV) | 1.6 |
| Absorption maximum, λabs (nm) | 634 |
| Emission maximum, λem (nm) | 681 |
| Molar absorptivity coefficient, ε, 634 nm (M−1 cm−1) | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
| Integrated molar absorptivity, 300–800 nm, (M−1) | 0.197 |
| Quantum yield (%) | 7.5 |
| Stokes shiftb (eV) | 0.31 |
| Optical band gap, Eoptc (eV) | 1.8 |
Due to (iPrNH)2NDI–V is water soluble at high pH the CV was also measured in water with 40% 0.1 M NaCl as an electrolyte and pH adjusted to 8 with 1 M NaOH (Fig. 2b). The CV in aqueous solution is less defined than that in DCM, likely because of the material's aggregation in aqueous media. There appears to be a partially reversible oxidation event with an onset at 0.5 V along with two irreversible reduction events at −0.6 V and −1.5 V which likely correspond to the two reduction events observed in the DCM CV. The CV profile remained consistent for multiple scans and at different scan rates (ESI,† Fig. S8–S10). Unfunctionalised NDI–V, in contrast, displays a clearly reversible reduction wave in aqueous media with an E1/2 potential of −0.54 V (Fig. 2b), which is at similar potential to the first reduction peak observed for (iPrNH)2NDI–V. This indicates that, while the addition of amino substituents to the NDI core has had a significant impact on the HOMO energy level, the LUMO is relatively unaffected. The differences in reversibility for the reduction peaks between NDI–V and (iPrNH)2NDI–V are most likely due to differences in the materials’ self-assembly in aqueous media caused by the additional steric and hydrogen-bonding influences of the amino substituents.
The optical properties of (iPrNH)2NDI–V were characterised via UV-vis absorption and emission spectroscopy. The UV-vis absorption profile, measured in water with 40% v/v 0.1 M NaCl and pH-adjusted to 10 with 1 M NaOH, is shown in Fig. 3 along with the emission profile with excitation at 380 nm. The absorption spectrum displays absorption bands at 350 nm and 367 nm which are typical for naphthalene diimides and are the result of π–π* transition in the NDI core.25 These same bands appear in the UV-vis absorption profile for the unfunctionalised NDI–V (Fig. 3) along with an additional band at 384 nm which is not present in the absorption profile of (iPrNH)2NDI–V. The main feature of the absorption profile of (iPrNH)2NDI–V is a large absorption band from 500 nm to 700 nm with a maximum at 634 nm and a slight shoulder at 590 nm due to vibronic coupling.25 This band is the result of the isopropyl amino substituents raising the HOMO energy level of the material and is commonly observed in other NDIs with similar core substituents.23,25,34 As such, it is not present in the unfunctionalised analogue which displays little to no absorption above 400 nm. The absorption profile also indicates that (iPrNH)2NDI–V is fully dissolved in water for if it were forming aggregates such as micelles we would expect to see significant broadening of the absorption bands.25 The material has a high molar absorptivity coefficient of 13 700 M−1 cm−1 (ESI,† Fig. S22) at 634 nm and gives rise to the distinct deep blue colour of the species. The onset of the absorption at 700 nm gives an optical band gap (i.e. band gap determined from the optical absorbance spectrum) of 1.8 eV, which corresponds well to the electrical band gap of 1.6 eV calculated from the CV. The absorption of (iPrNH)2NDI–V across the visible spectrum is exceptionally high for an NDI species with (iPrNH)2NDI–V having an integrated molar absorptivity of 0.197 M−1 from 300 nm to 800 nm.
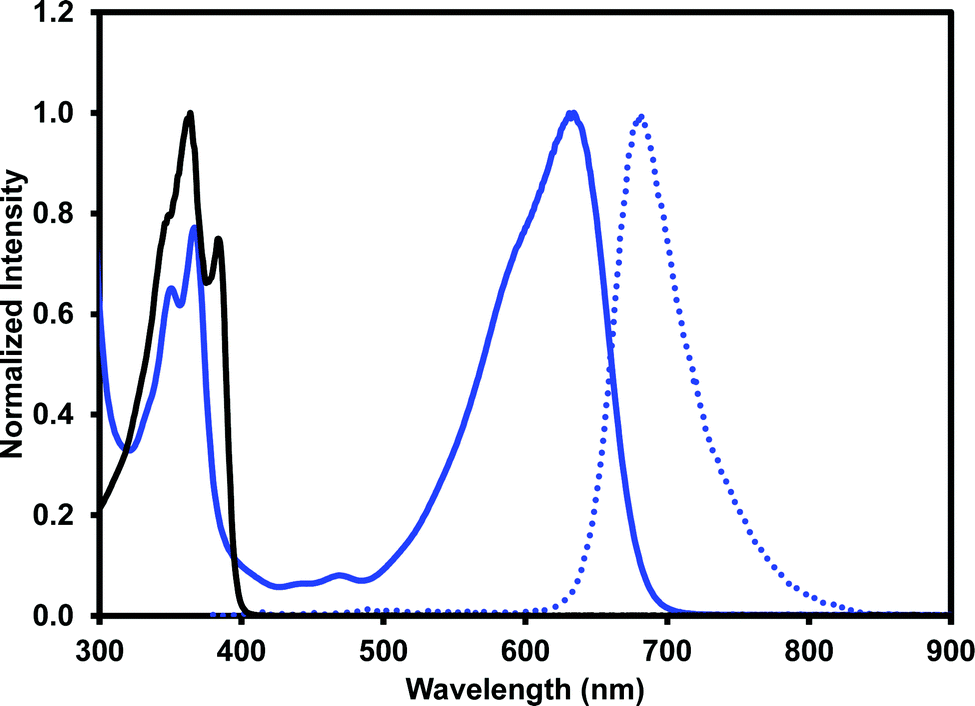 | ||
| Fig. 3 The absorption (solid blue) and emission (dot blue) profiles for (iPrNH)2NDI–V and absorption (solid black) profile for NDI–V. All spectra were measured in water with a 40% v/v 0.1 M NaCl electrolyte at pH 10. Emission profile was obtained with excitation at 360 nm. | ||
Upon excitation at 360 nm, (iPrNH)2NDI–V emits in the red to NIR portion of the spectrum from 620 nm to 820 nm with an emission maximum at 681 nm. An identical emission profile is also obtained upon excitation at 600 nm (ESI,† Fig. S25). The quantum yield of (iPrNH)2NDI–V is 7.5%, which is relatively low but not out of the range of other similarly functionalised NDIs in the literature.23,24 The low quantum yield here is likely due to photo-induced electron transfer occurring from the lone electron pairs of the amino groups to the NDI core thereby quenching emission.23 Protonating the amino side groups has been shown to increase quantum yield of similarly functionalised NDIs23,24 but for (iPrNH)2NDI–V protonation of the amino groups is not possible without also protonating the carboxylate groups and rendering the material insoluble in aqueous media. Although (iPrNH)2NDI–V is not electrochromic, its optical properties do present alternative applications. The vivid and electronically stable colour combined with its water solubility makes (iPrNH)2NDI–V well suited as a water-soluble dye, while the red to NIR emission, despite the low quantum yield in aqueous media, could be useful in sensory applications. The optical properties of (iPrNH)2NDI–V are summarised in Table 1.
Due to (iPrNH)2NDI–V's water solubility is dependent on pH similar to other amino-functionalised NDIs,23,24 the effects of pH on the optical properties of (iPrNH)2NDI–V in aqueous solution were also analysed. As shown in Fig. 4, the optical profiles at high pH values remain unchanged. At lower pH values, starting at pH 5, a drop in absorbance across the spectrum is observed which is an indication of the material precipitating out of solution. There is also a noticeable blue shift in the absorption maxima as well as an increase in the 590 nm shoulder of the broad absorption band relative to the maximum. These changes are most pronounced at pH 3 and 4 and are indicative of a change in aggregation in solution, which is to be expected for this material. As discussed above, these types of NDIs are water-soluble through deprotonation of the amino acid side chains and lowering the pH would lead to protonation of these groups, thereby changing the material's aggregation in aqueous solution and eventually leading to precipitation. Cyclic voltammetry was also performed at different pH values and changing the pH had little to no effect on the electronic properties (ESI,† Fig. S11–S15). This is in contrast to our other systems that have different electronic properties depending on their aggregated state.35–37
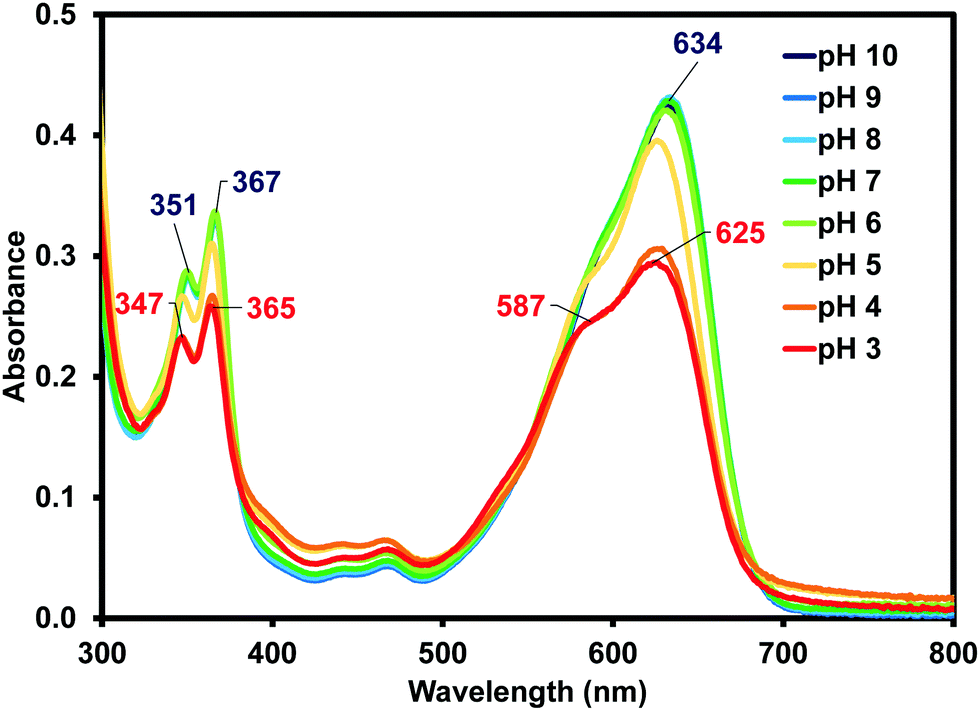 | ||
| Fig. 4 The UV-vis absorption profile for (iPrNH)2NDI–V measured in water with a 40% v/v 0.1 M NaCl at various pH values from pH 10 to pH 3 with absorption maxima shown at pH 10 (blue) and pH 3 (red). | ||
The blue (iPrNH)2NDI–V also displays solvatochromic properties. The absorption profile was measured in acetone, DCM, and methanol, as shown in Fig. 5a. The organic solvents blueshift the absorption profile maxima relative to water with acetone and methanol leading to the largest shift of about 20 nm (634 nm maximum in water vs. 612 nm and 615 nm maxima in acetone and methanol, respectively). DCM causes a smaller blue-shift of about 10 nm. Materials with a larger dipole moment in the ground state than in the excited state will experience greater stabilization of the ground state than the excited state in more polar organic solvents, leading to a larger band gap and absorption at shorter wavelengths.38 This is known as negative solvatochromism. For materials where the excited state has larger dipole moment than the ground state, the excited state is stabilised to a greater degree than the ground state by more polar organic solvents and positive solvatochromism occurs with absorption at longer wavelengths. The organic solvents also give rise to a more pronounced shoulder in the broad absorption band at 575 nm than water. These solvatochromic effects have been observed for similarly functionalised NDIs.25 Unlike in water, deprotonation is not necessary to dissolve (iPrNH)2NDI–V in these organic solvents so the material would be expected to have different aggregation properties which give rise to the changes in the absorption profile and likely account for why absorption in water occurs at longer wavelengths.
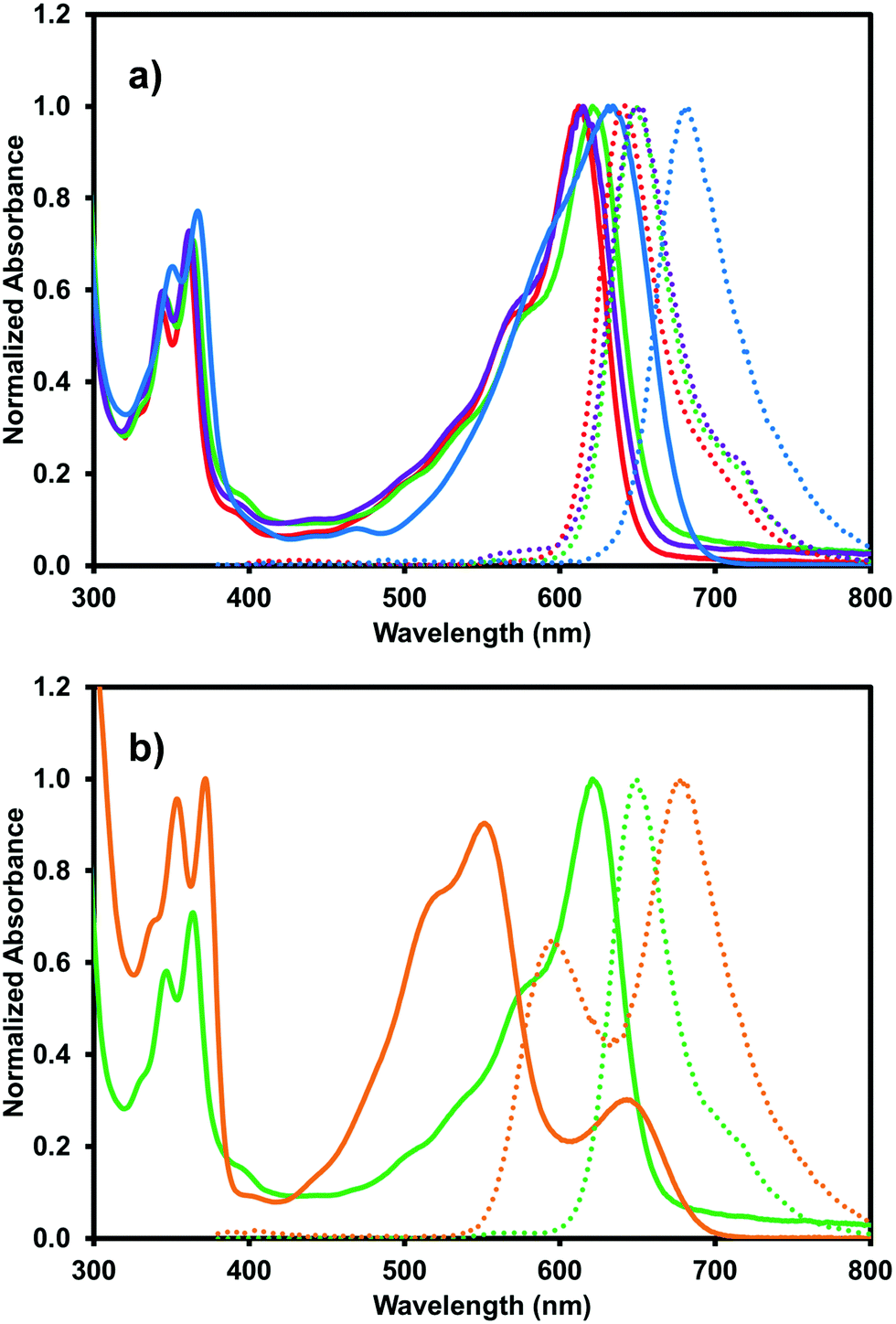 | ||
| Fig. 5 (a) UV-vis absorption (solid) and emission (dot) profiles for (iPrNH)2NDI–V measured in acetone (red), DCM (green), methanol (purple), and water at pH 10 (blue); (b) absorption and emission profiles for (iPrNH)2NDI–V measured in DCM (green) and in DCM with 2.5% v/v TFA (orange). Emission was obtained with excitation at 360 nm. | ||
The effects of acid on the absorption profile can also be explored in organic solvents without the material precipitating from solution. This was done by adding 2.5% v/v trifluoroacetic acid (TFA) to the acetone, DCM, and methanol solutions. The addition of TFA in acetone and methanol had no effect on the absorption profile (ESI,† Fig. S23, S24) but a significant change was observed for the DCM solution (Fig. 5b). A significant blue shift of the broad absorption peak was observed from 620 nm to 551 nm as well as the appearance of a new band at 643 nm. The intensity of the high frequency bands at 364 nm and 347 nm also increased and redshifted to 372 nm and 353 nm, respectively. All of this had the effect of turning the solution from blue to a deep purple. Clearly, the addition of acid on the absorption profile of (iPrNH)2NDI–V in DCM, which is the least polar of the solvents explored, has significant effects on the material's solution aggregation properties. It is most likely that the strong acid TFA is protonating (iPrNH)2NDI–V at the amino positions and leading to new types of intermolecular arrangements.38 The splitting of the absorption band observed in Fig. 5b may indicate that these new intermolecular arrangements are an example of oblique aggregation, or a hybrid of H- and J-aggregates.39,40 Given that these changes are not observed in acetone and methanol indicates that the TFA interaction with (iPrNH)2NDI–V and formation of new intermolecular arrangements are suppressed in these solvents due to their higher polarity relative to DCM. It is also likely that the protonation of the amino groups has affected the energy levels of (iPrNH)2NDI–V which is also contributing to the change in absorption.
The organic solvents also influence the emission profile of (iPrNH)2NDI–V. Once again, a blue shift is observed in organic solvents with the emission maximum shifting to 642 nm, 649 nm, and 649 nm in acetone, dichloromethane, and methanol, respectively. Emission profiles are identical with excitation wavelengths of either 360 nm or 600 nm (ESI,† Fig. S25). The addition of a 2.5% v/v TFA aliquot also drastically changes the emission profile in DCM with much broader emission from 550 nm to 800 nm and two emission maxima occurring at 677 nm and 592 nm. Here, changing the excitation wavelength does change the intensity ratio of the emission maxima (ESI,† Fig. S26). Excitation at 360 nm leads to a higher emission intensity at 677 nm than at 592 nm while excitation at 500 nm and 550 nm leads to higher emission intensity at 592 nm than 677 nm. As mentioned above, protonation of the amino groups in similar functionalised NDIs has been shown to lead to an increase in quantum yield.23,24 The quantum yield for (iPrNH)2NDI–V measured in DCM with 2.5% v/v TFA is 15.5% which has increased compared to 7.5% in water. However, the quantum yield measured in DCM without the addition of TFA was found to be 36.3%. It is likely that the protonation of the amino groups of (iPrNH)2NDI–V leading to new aggregation is suppressing more than enhancing the emission, in contrast to what has previously been reported.23,24 A summary of the solvatochromic properties of (iPrNH)2NDI–V is shown in Table 2.
| Solvent | λ abs max (nm) | λ em max (nm) | QY (%) |
|---|---|---|---|
| Water, pH 10 | 634, 367, 351 | 681 | 7.5 |
| Water, pH 3 | 625, 365, 347 | 681 | NA |
| Acetone | 612, 361, 344 | 642 | NA |
| CH2Cl2 | 620, 364, 347 | 649 | 36.3 |
| Methanol | 615, 361, 344 | 649 | NA |
| CH2Cl2 + TFA | 643, 551, 372, 353 | 677, 592 | 15.5 |
Gel formation and characterisation
Previous research with water soluble NDIs has shown that they can be used to prepare hydrogels when functionalised with certain peptides.15 We saw that the immobilisation of the NDI as a gel was advantageous for device fabrication as immobilisation reduced leakage and enabled material patterning. To that end, we have fabricated a multicomponent hydrogel using the blue (iPrNH)2NDI–V and the dipeptide 1-ThNap-FF (Fig. 6), a material that has been shown to form both single component and multicomponent hydrogels.41–44 This would enable the dye to be immobilised in or as part of a gel network.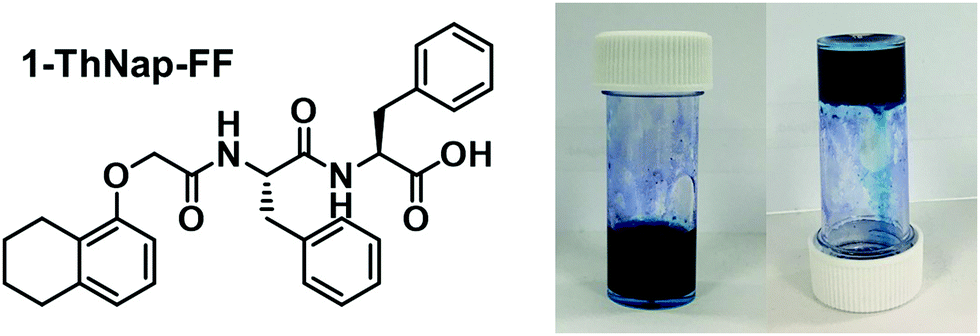 | ||
| Fig. 6 The chemical structure of 1-ThNap-FF and photographs of the blend (iPrNH)2NDI–V (5 mg mL−1) and 1-ThNap-FF (5 mg mL−1) hydrogel. | ||
The strategy employed to form hydrogels involved dissolving (iPrNH)2NDI–V and 1-ThNap-FF in aqueous solutions at pH 11 with 1 M NaOH, combining the solutions in a 1:1 volumetric ratio, and mixing with glucono-δ-lactone (GdL). In aqueous solution, GdL hydrolyses which lowers the pH of the solution thereby triggering gelation. Both (iPrNH)2NDI–V and 1-ThNap-FF were used at 5 mg mL−1 concentrations and 10 mg mL−1 of GdL was used to achieve slow, uniform acidification of the solution to a pH of 3.8. After 18 hours the solutions had become dark blue gels capable of remaining suspended when inverted (Fig. 6). Single-component gels of 1-ThNap-FF at 5 mg mL−1 were also prepared following the same method to study the effects of (iPrNH)2NDI–V incorporation in the blend gels.
The single and multicomponent gels were characterised using rheology. Strain and frequency measurements show that the blend gels behave like typical low molecular weight gels previously studied in our group (ESI,† Fig. S27).15 The multicomponent gels have slightly lower storage modulus (G′) values and loss modulus (G′′) values than the single-component gels. This is likely due to the assembly of (iPrNH)2NDI–V disrupting gel formation mediated by 1-ThNap-FF though not to a sufficient degree to completely inhibit gelation. For both single and multicomponent gels, the gels break at just under 10% strain and G′ and G′′ are independent of frequency.
Time-sweep rheological data was also obtained for single and multicomponent gels and combined with pH measurements to show how lowering the pH triggers gelation. The data (ESI,† Fig. S28, S29) clearly show that a decrease in pH is accompanied by an increase in G′ and G′′, which is consistent with pH-induced gel formation. The self-assembly of the multi-component gel displays a slightly different behaviour. The same stages occur corresponding to the decrease in pH and the assembly of the 1-ThNap-FF, although the data is a little noisier. This may indicate that (iPrNH)2NDI–V is templating onto the gel fibres formed by 1-ThNap-FF rather than remaining freely dispersed throughout the hydrogel or incorporating itself into the gel fibres. Time-sweep rheological and pH data were obtained for the system containing only (iPrNH)2NDI–V and GdL (ESI,† Fig. S30) to show that (iPrNH)2NDI–V does not gel by itself when the pH is lowered. Therefore, gelation in the multicomponent system is solely due to 1-ThNap-FF. A direct comparison of the time-sweep rheological data for both single and multicomponent gels (ESI,† Fig. S31) shows that while gelation seems to initiate faster in the multicomponent than in the single component system, both systems seem to reach a plateau in G′ and G′′ after the same amount of time. However, a direct comparison cannot be made from gelation kinetics or final rheological values due to the different amounts of material present in the single and multicomponent systems and the different amount of GdL needed to reach the same final pH in both systems.
Cyclic voltammetry and UV-vis absorption measurements were performed on the multi-component gels prepared from 5 mg mL−1 (iPrNH)2NDI–V and 5 mg mL−1 1-ThNap-FF to see if gelation had altered the optoelectronic properties of the (iPrNH)2NDI–V in any way (Fig. 7). Additional CV data for gels prepared with 0.25 mg mL−1 (iPrNH)2NDI–V and 5 mg mL−1 1-ThNap-FF is provided in the ESI,† Fig. S18, to more directly compare with the solution CV data, which was measured with 0.25 mg mL−1 (iPrNH)2NDI–V concentration. Although the gel CV shows even less resolution than the aqueous solution measurements, the data does seem to indicate that (iPrNH)2NDI–V possesses the same electronics in solution and in the gel state, again suggesting that the (iPrNH)2NDI–V has not been associated into the 1-ThNap-FF fibres. The lack of resolution in the reduction peaks could be due to the (iPrNH)2NDI–V being effectively immobilised on the 1-ThNap-FF network, and so diffusion is limited. The UV-vis profile of the multicomponent gel is identical to the aqueous solution UV-vis profile, further indicating that gelation has not affected the optoelectronic properties of (iPrNH)2NDI–V. This again suggests that the (iPrNH)2NDI–V is not in the gel pores, as the difference in aggregation would likely lead to differences in the absorption profile, but a part of the network.
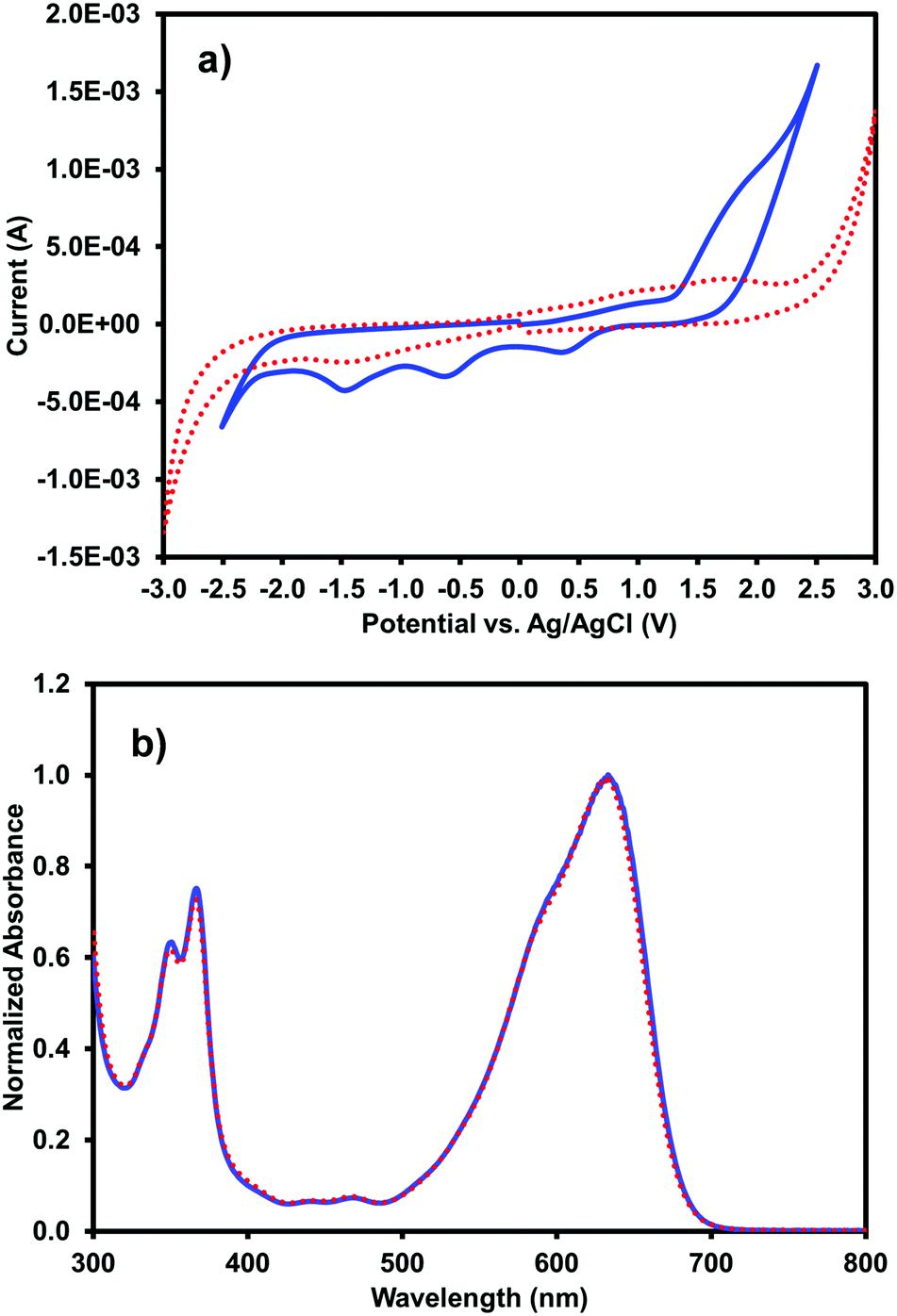 | ||
| Fig. 7 (a) Cyclic voltammograms for (iPrNH)2NDI–V measured as a solution (solid blue line) in water at pH 10 with 40% 0.1 M NaCl electrolyte and as a hydrogel (red dotted line) with 1-ThNap-FF and 40% 0.1 M NaCl electrolyte; (b) UV-vis absorption profiles for (iPrNH)2NDI–V measured as a solution (solid blue line) in water at pH 10 and as a gel with 1-ThNap-FF (red dotted line). | ||
Small angle neutron scattering (SANS) was collected on both the single component and multicomponent systems to assess the gel networks and structures formed at low pH. The scattering data for each system is shown in Fig. 8 and ESI,† Fig. S32–S34. (iPrNH)2NDI–V fitted best to a power law and had the lowest scattering intensity. This was expected as we know from the rheological data (ESI,† Fig. S30) that (iPrNH)2NDI–V does not form an extended network and only aggregates upon lowering the pH. 1-ThNap-FF fitted to a flexible cylinder with a radius of 33 Å combined with a power law, which has been observed before and agrees with it forming a fibrous gel network.41,42 The blend gel also fitted to a flexible cylinder combined with a power law, but with a larger radius of 50 Å and a 0.2 polydispersity. This demonstrates that although the (iPrNH)2NDI–V does not affect the ability of 1-ThNap-FF to form a gel or fibres, it does affect the fibres formed both in uniformity and diameter. This suggests that the network is self-sorted rather than co-assembled, but that perhaps the (iPrNH)2NDI–V has associated itself with the fibres of 1-ThNap-FF giving a larger fibre diameter, due to their hydrophobic nature. This complexity in assembly is also seen in the rheology time-sweep data. Further information and table of fits used for the SANS data can be found in the ESI.†
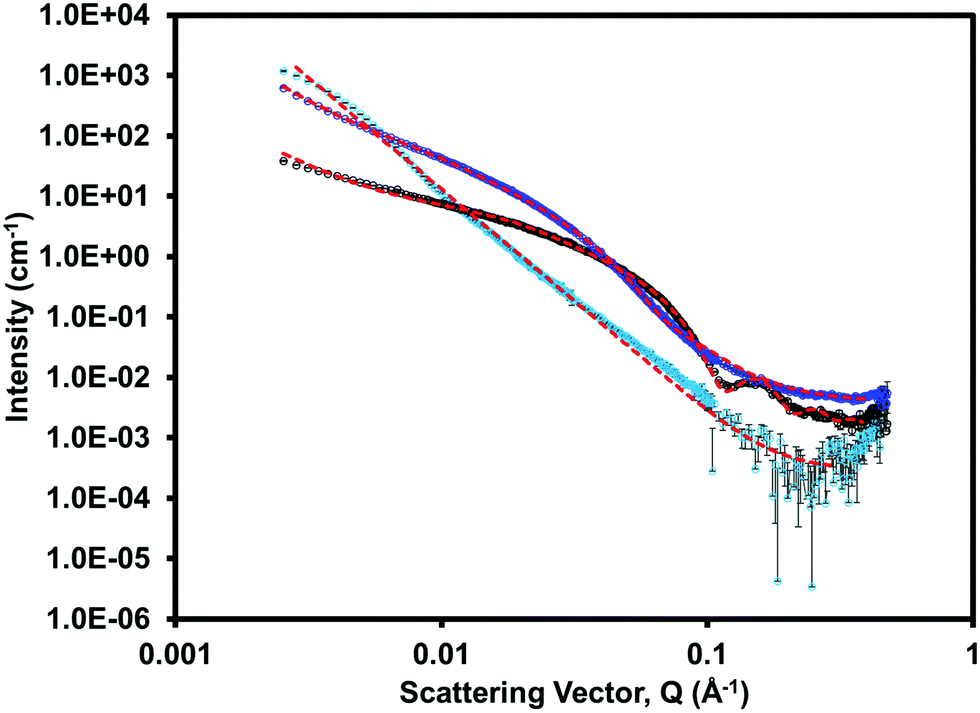 | ||
| Fig. 8 Small angle neutron scattering (SANS) for the single component (iPrNH)2NDI–V solution (light blue), single component 1-ThNap-FF gel (black), and multicomponent 1-ThNap-FF + (iPrNH)2NDI–V gel (dark blue) with fit lines for each shown in red. | ||
Conclusions
Herein we described the synthesis and optoelectronic characterisation of a new water-soluble NDI core-functionalised with isopropylamino substituents, (iPrNH)2NDI–V, which possesses a vibrant blue colour. The material was synthesised following previously established procedures from the literature26,30 and it was found that purifying the starting materials, something that had not been attempted or discussed in prior literature, led to improved results. It was also found that leaving the synthesis to react for 10 to 12 days led to dramatically improved results in favouring the disubstituted product over the monosubstituted product.The optoelectronic properties of (iPrNH)2NDI–V were characterised using CV and UV-vis absorption and emission spectroscopy in both aqueous and organic solvents. Unlike the unfunctionalised NDI–V, (iPrNH)2NDI–V did not possess electrochromic properties, but was highly absorbing in the visible spectrum to give it a vibrant blue colour as well as being emissive in the red to NIR. The (iPrNH)2NDI–V also displayed solvatochromic properties. Dissolving (iPrNH)2NDI–V in common organic solvents led to blue shifts in both the absorption and emission maxima. (iPrNH)2NDI–V was also combined with a known gelator and was found to make strongly coloured blue hydrogels that possessed rheological properties typical for low-molecular weight hydrogels with little change in its absorption or electronic properties. This makes it a promising candidate for use in gel-based electronic devices.
With the synthesis of (iPrNH)2NDI–V, we have provided an efficient framework to make water-soluble organic materials with simple functionalisations that lead to dramatic changes in the optoelectronic properties of the products obtained. Ease of functionalisation and tuneable optoelectronic properties is a well-known advantage of organic materials and synthesising water-soluble organic materials is very important in developing cost-effective and environmentally friendly organic electronic devices. With its unique blue colour and high visible light absorption (iPrNH)2NDI–V has potential application as a dye or acceptor in various organic electronic devices. Future efforts will be devoted to developing other types of water-soluble NDIs with simple core functionalisations to give rise to new and unique optoelectronic properties.
Author contributions
Experiments conceived by ERD and TAW. All authors contributed towards the writing of the paper. Funding was from ERD. TAW performed the synthesis and purification for (iPrNH)2NDI–V and its precursors. TAW also performed the NMR, cyclic voltammetry, optical spectroscopy, and rheology characterisation. SANS data was collected by RS and OM. SANS was processed by ERD using SAS View.45Conflicts of interest
There are no conflicts to declare.Acknowledgements
ERD would like to thank EPSRC for a NIA award (EP/S032673/1) which also funds TAW. ERD also thanks the Leverhulme Trust for an Early Career Fellowship (ECF-2017-223) and the University of Glasgow for a LKAS Leadership Fellowship. We acknowledge beamtime allocation on D11 at Institut Laue-Langevin (ILL) doi:10.5291/ILL-DATA.9-10-1670, Grenoble. This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.References
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed.
- R.-P. Xu, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- A. Rostami and M. S. Taylor, Macromol. Rapid Commun., 2012, 33, 21–34 CrossRef CAS PubMed.
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- L. Gonzalez, C. Liu, B. Dietrich, H. Su, S. Sproules, H. Cui, D. Honecker, D. J. Adams and E. R. Draper, Commun. Chem., 2018, 1, 1–8 CrossRef CAS.
- M. Saha and S. Bandyopadhyay, J. Phys. Chem. C, 2021, 125, 6427–6432 CrossRef CAS.
- M. B. Avinash, K. Swathi, K. S. Narayan and T. Govindaraju, ACS Appl. Mater. Interfaces, 2016, 8, 8678–8685 CrossRef CAS PubMed.
- D. R. Rosseinsky and R. J. Mortimer, Adv. Mater., 2001, 13, 783–793 CrossRef CAS.
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225–4237 RSC.
- S. V. Bhosale, M. B. Kalyankar, S. V. Bhosale, S. J. Langford, E. F. Reid and C. F. Hogan, New J. Chem., 2009, 33, 2409–2413 RSC.
- Y. Hu, Y. Qin, X. Gao, F. Zhang, C. Di, Z. Zhao, H. Li and D. Zhu, Org. Lett., 2012, 14, 292–295 CrossRef CAS PubMed.
- T. D. M. Bell, S. Yap, C. H. Jani, S. V. Bhosale, J. Hofkens, F. C. De Schryver, S. J. Langford and K. P. Ghiggino, Chem. – Asian J., 2009, 4, 1542–1550 CrossRef CAS PubMed.
- A. Weißenstein, V. Grande, C. R. Saha-Möller and F. Würthner, Org. Chem. Front., 2018, 5, 2641–2651 RSC.
- F. Doria, I. Manet, V. Grande, S. Monti and M. Freccero, J. Org. Chem., 2013, 78, 8065–8073 CrossRef CAS PubMed.
- H. F. Higginbotham, S. Maniam, S. J. Langford and T. D. M. Bell, Dyes Pigm., 2015, 112, 290–297 CrossRef CAS.
- R. S. K. Kishore, V. Ravikumar, G. Bernardinelli, N. Sakai and S. Matile, J. Org. Chem., 2008, 73, 738–740 CrossRef CAS PubMed.
- H. Vollmann, H. Becker, M. Corell and H. Streeck, Justus Liebigs Ann. Chem., 1937, 531, 1–159 CrossRef CAS.
- M. Korzec, S. Kotowicz, K. Łaba, M. Łapkowski, J. G. Małecki, K. Smolarek, S. Maćkowski and E. Schab-Balcerzak, Eur. J. Org. Chem., 2018, 1756–1760 CrossRef CAS.
- K. Chen, J. Zhao, X. Li and G. G. Gurzadyan, J. Phys. Chem. A, 2019, 123, 2503–2516 CrossRef CAS PubMed.
- C. Thalacker, C. Röger and F. Würthner, J. Org. Chem., 2006, 71, 8098–8105 CrossRef CAS PubMed.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup, C. P. Woodward, G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- S.-L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428–7448 CrossRef CAS PubMed.
- R. Lundy, E. R. Draper and J. J. Walsh, New J. Chem., 2018, 42, 19020–19025 RSC.
- D. McDowall, B. J. Greeves, R. Clowes, K. McAulay, A. M. Fuentes-Caparrós, L. Thomson, N. Khunti, N. Cowieson, M. C. Nolan, M. Wallace, A. I. Cooper, E. R. Draper, A. J. Cowan and D. J. Adams, Adv. Energy Mater., 2020, 10, 2002469 CrossRef CAS.
- C. L. Smith, L. L. E. Mears, B. J. Greeves, E. R. Draper, J. Doutch, D. J. Adams and A. J. Cowan, Phys. Chem. Chem. Phys., 2019, 21, 26466–26476 RSC.
- F. Würthner, J. Org. Chem., 2021 DOI:10.1021/acs.joc.1c00625.
- M. S. Barclay, S. K. Roy, J. S. Huff, O. A. Mass, D. B. Turner, C. K. Wilson, D. L. Kellis, E. A. Terpetschnig, J. Lee, P. H. Davis, B. Yurke, W. B. Knowlton and R. D. Pensack, Commun. Chem., 2021, 4, 1–11 CrossRef.
- K. J. Thorley and F. Würthner, Org. Lett., 2012, 14, 6190–6193 CrossRef CAS PubMed.
- M. C. Nolan, A. M. F. Caparrós, B. Dietrich, M. Barrow, E. R. Cross, M. Bleuel, S. M. King and D. J. Adams, Soft Matter, 2017, 13, 8426–8432 RSC.
- S. Panja, A. M. Fuentes-Caparrós, E. R. Cross, L. Cavalcanti and D. J. Adams, Chem. Mater., 2020, 32, 5264–5271 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Giant, 2021, 5, 100041 CrossRef CAS.
- S. Panja, B. Dietrich, O. Shebanova, A. J. Smith and D. J. Adams, Angew. Chem., Int. Ed., 2021, 60, 9973–9977 CrossRef CAS PubMed.
- SasView, SasView, https://sasview.github.io/, (accessed 16 April 2021).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj02557k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |