
Selective visible-light driven highly efficient photocatalytic reduction of CO2 to C2H5OH by two-dimensional Cu2S monolayers†
Shiyan
Wang
,
Xiaowan
Bai
,
Qiang
Li
 ,
Yixin
Ouyang
,
Li
Shi
and
Jinlan
Wang
*
,
Yixin
Ouyang
,
Li
Shi
and
Jinlan
Wang
*
School of Physics, Southeast University, Nanjing 211189, China. E-mail: jlwang@seu.edu.cn
First published on 22nd May 2021
Abstract
Solar-driven highly-efficient photocatalytic reduction of CO2 into value-added fuels has been regarded as a promising strategy to assuage the current global warming and energy crisis, but developing highly product-selective, long-term stable and low-cost photocatalysts for C2 production remains a grand challenge. Herein, we demonstrate that two-dimensional β- and δ-phase Cu2S monolayers are promising photocatalysts for the reduction of CO2 into C2H5OH. The calculated potential-limiting steps for the CO2 reduction reaction (CO2RR) are less than 0.50 eV, while those for the hydrogen evolution reaction are as high as 1.53 and 0.87 eV. Most strikingly, the C–C coupling only needs to overcome an ultra-low kinetic barrier of ∼0.30 eV, half of that on the Cu surface, indicating that they can boost the C2H5OH conversion efficiency greatly. Besides, these catalysts also exhibit satisfactory band edge positions and suitable visible light absorption, rendering them ideal for the visible light driven CO2RR. Our work not only provides a promising photocatalyst for achieving the efficient and selective CO2RR, but also brings new opportunities for advanced sustainable C2H5OH product.
New conceptsCapture and conversion of CO2 into value-added fuels has been regarded as a promising strategy to assuage the current global warming and energy crisis, but developing highly product-selective, long-term stable and low-cost photocatalysts for C2 production remains a grand challenge. Based on the fact that low-coordinated Cu+ surface atoms enhance CO* binding and S atoms suppress the HER, they hold promising prospects for boosting both the activity and product selectivity. Herein, for the first time, we report a two-dimensional Cu+ based catalyst, Cu2S monolayers, as a very compelling photocatalyst for the CO2RR into C2H5OH by means of state-of-the-art DFT calculations. The calculated potential-limiting step for the CO2RR is less than 0.50 eV, while that for the HER is higher than 0.87 eV. Meanwhile, the C–C coupling only needs to overcome an ultra-low kinetic barrier of ∼0.30 eV, half of that on the Cu surface, indicating that the catalyst can boost the C2H5OH conversion efficiency greatly. Besides, the catalysts also exhibit satisfactory band edge positions and suitable visible light absorption, rendering them ideal for visible light driven CO2RR. Our work not only provides a promising photocatalyst for achieving the efficient and selective CO2RR, but also provides new insights into the development of novel photocatalysts. |
Introduction
The development of artificial photosynthesis that captures and converts carbon dioxide (CO2) into fuels and value-added chemicals by solar energy has drawn increasing attention due to the energy crisis and global warming.1–3 Recently, conversion of CO2 to gaseous carbon products of CO,4,5 CH46,7 and C1 liquid products, such as HCOOH8,9 and CH3OH,10,11 has been achieved in photocatalytic strategies with high reaction rate. In contrast, reports on the photocatalytic CO2RR to C2 products are very limited. In fact, the C2 liquid alcohol products are more desirable for a wide range of applications in transportation and the chemical industry due to their higher energy density and commercial value than C1 products.12–14 However, the selectivity of the products of the CO2RR is uncontrollable15–17 and it suffers from the competing hydrogen evolution reaction (HER)18,19 as well. Besides, the catalytic CO2RR to C2 products with a low kinetic barrier is still a challenge because the C–C coupling is very difficult. Meanwhile, to be a good photocatalyst, satisfactory visible-light absorption and suitable band edge positions are required while current photocatalysts are often suffering from high electron–hole recombination rates and poor visible-light absorption in the wide-bandgap semiconductor materials.20 Therefore, the development of highly active semiconductor photocatalysts with high product selectivity to achieve efficient photocatalytic CO2RR is very desirable but rather challenging.During the CO2RR process, it is hard to achieve a significantly low free energy barrier because the scaling relation of the adsorption energies between CO* and CHO* intermediates imposes a fundamental limitation on the catalytic efficiency.21 Some approaches to break this limitation have been proposed: (i) introducing a covalent character of p-orbitals of main-group elements,22 (ii) transition-metal atom doping,23 (iii) designing a special interfacial catalyst motif,24 (iv) alloying metal catalysts,25 and (v) adopting a different mechanism without formation of CO*/CHO*.26 By removing this limitation, the CHO* is independently stabilized over CO*, thus deviating from the scaling relation of transition-metals. To date, copper is one of the most up and coming candidates for the CO2RR into C2 hydrocarbons and oxygenates.27,28 However, the high overpotential and the C–C coupling kinetic barrier cause the low faradaic efficiency and the poor selectivity of C2 products. One of the most promising approaches to boost both the efficiency and selectivity for the CO2RR to multi-carbon alcohols is mostly based on copper(I) species. The surface Cu+ could modify the surface structure of copper(I)-based catalysts and promote the CO*–CO* hydrogenation13,29,30 and C–C coupling reaction,31 thereby improving the faradaic efficiency for C2 products.
Very recently, an ultrathin two-dimensional (2D) β-phase Cu2S bilayer has been successfully synthesized in the laboratory32–34 and a 2D δ-phase Cu2S monolayer has also been predicted theoretically35,36 with superior oxidation resistance, ultrahigh carrier mobility and excellent thermodynamic stability. Importantly, the p-type semiconducting Cu2S bulk can act as the light absorber, which has been widely employed in photovoltaics, photocatalysis and solar energy conversion.34,37–41 In particular, the great difference of the mobilities between electrons and holes of 2D Cu2S35 can dramatically reduce the possibility of the photo-induced carrier recombination and improve the photocatalytic efficiency of the CO2RR.42 Meanwhile, copper sulfide compounds could balance the binding strengths of the H* and CO* intermediates, respectively, i.e. the low-coordinated Cu surface atoms enhance CO* binding and S atoms suppress the HER, which hold promising prospects for boosting both the activity and product selectivity.43,44
In this work, we demonstrate that 2D β-Cu2S and δ-Cu2S monolayers are two promising photocatalysts for the CO2RR into C2H5OH under visible light irradiation. Both catalysts possess strong adsorption ability for CHO* and CO* intermediates, resulting in a large deviation from the scaling relation of the transition-metal. Moreover, the strong CO* adsorption promotes an increase of local CO* coverage, which significantly reduces the limiting potential. The calculated potential-limiting steps for the whole CO2RR process are no more than 0.50 eV, while those for the HER are as high as 1.53 and 0.87 eV. More importantly, the CHO–CO coupling only needs to overcome an ultra-low kinetic barrier of ∼0.3 eV, half of that on the Cu surface,45,46 which facilitates the formation of CHOCO* and high selectivity for C2H5OH products. Besides, these catalysts also exhibit satisfactory visible light absorption and suitable band edge positions, which might be applied to CO2 reduction under visible light irradiation.
Computational methods
All the density functional theory (DFT) calculations in this work were carried out using the Vienna Ab Initio Simulation Package (VASP).47 The projector augmented wave potentials48 was employed to describe the electron–ion interactions in the periodic system. The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)49 was utilized to treat the exchange–correlation interactions and the cutoff energy of 500 eV was selected for the plane-wave basis.35,36 The convergence criteria for the electronic self-consistent iteration was set to 10−5 eV and the ionic relaxation continued until the maximum force on each atom was less than 0.02![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV Å−1, which was updated by the conjugate gradient approach. In order to avoid the interaction between two periodic units, a 20 Å vacuum layer was added to all the slab models. The DFT-D3 method by Grimme et al.50 was performed for the van der Waals correction. Ab initio molecular dynamics (AIMD) simulations were employed by using a 4 × 4 × 1 supercell under the canonical ensemble lasting for 10 ps with a time step of 2.0 fs. The temperature was set to 800 K by using the Nóse–Hoover method.51 Bader charge was calculated by using the Bader Charge Analysis script written by Henkelman and co-workers.52 The electronic band structures and optical properties of β-Cu2S and δ-Cu2S monolayers were calculated accurately by using the Heyd–Scuseria–Ernzerhof hybrid (HSE06) functional.53 All the C–C coupling barrier calculations were employed using the climbing image nudged elastic band (CI-NEB)54 method. All the vibrational frequencies are positive except for one negative one, indicating that the transition state is a reliable first order saddle point. More details are given in the ESI.†
eV Å−1, which was updated by the conjugate gradient approach. In order to avoid the interaction between two periodic units, a 20 Å vacuum layer was added to all the slab models. The DFT-D3 method by Grimme et al.50 was performed for the van der Waals correction. Ab initio molecular dynamics (AIMD) simulations were employed by using a 4 × 4 × 1 supercell under the canonical ensemble lasting for 10 ps with a time step of 2.0 fs. The temperature was set to 800 K by using the Nóse–Hoover method.51 Bader charge was calculated by using the Bader Charge Analysis script written by Henkelman and co-workers.52 The electronic band structures and optical properties of β-Cu2S and δ-Cu2S monolayers were calculated accurately by using the Heyd–Scuseria–Ernzerhof hybrid (HSE06) functional.53 All the C–C coupling barrier calculations were employed using the climbing image nudged elastic band (CI-NEB)54 method. All the vibrational frequencies are positive except for one negative one, indicating that the transition state is a reliable first order saddle point. More details are given in the ESI.†
Results and discussion
Binding energies of the key reduction intermediates of CO* and CHO* scaling have been shown to be the bottleneck in CO2RR activity for metallic catalysts.21 By deviating from the above scaling relationship, CHO* can be stabilized over CO*, which can greatly reduce the potential determining step (PDS) for the CO* reduction into CHO*.23,24Fig. 1a depicts the high free energy change caused by the scaling relation between CHO* and CO*. The strong CO* adsorption promotes an increase of CO* coverage, which will significantly reduce the PDS by stabilizing CHO* and CO* on Cu2S monolayers (Fig. 1b). Fig. 1c presents the binding energies of CHO* versus CO* on Cu2S monolayers, as well as transition-metal (100) scaling relations. The detailed adsorption structure and binding energies of CHO* and CO* on Cu(100), β-Cu2S and δ-Cu2S are shown in Fig. S1 (ESI†). Clearly, both β-Cu2S and δ-Cu2S deviate from the scaling relation of the transition-metal (100),21 which is expected to achieve dramatic reduction in PDS.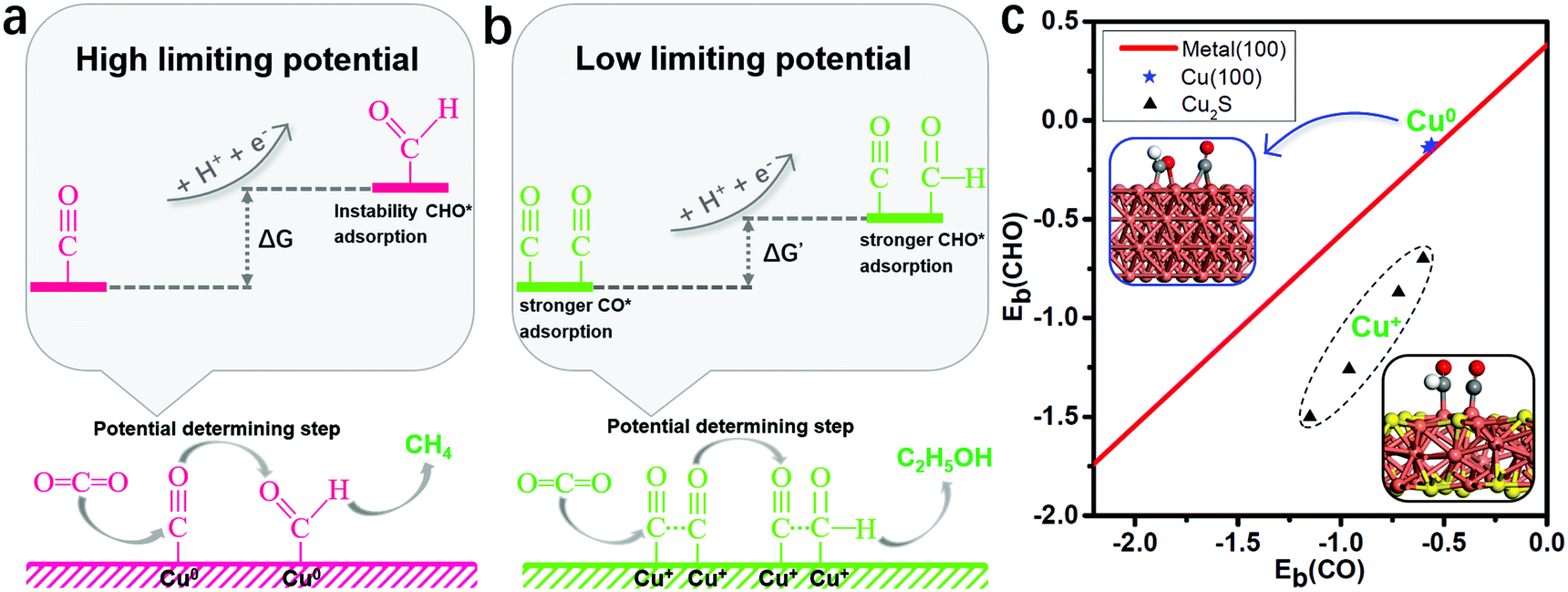 | ||
| Fig. 1 (a) Schematic diagram of high free energy change caused by the scaling relationship between CHO* and CO*. (b) Reducing the free energy change by stabilizing CO* and CHO* on Cu2S. (c) Binding energies of CHO* [Eb(CHO)] versus CO* [Eb(CO)] on Cu2S, as well as the transition-metal (100) scaling relation. Cu, orange; S, yellow; C, grey; O, red and H, white. The detailed binding energy data of CHO* and CO* are given in the ESI.† | ||
During the CO2RR into CO*, two proton/electron pairs will be consumed through proton-coupled electron transfer with COOH* intermediate. However, the formation of HCOOH* and CO* products is competitive, because the COOH* intermediate can also be directly protonated into HCOOH*. Thus, it is very necessary to compare the product selectivity of HCOOH* and CO* when two proton/electron pairs are transferred. The adsorbed structures of COOH* and HCOO* intermediates are shown in Fig. 2a, including carbon coordination and oxygen coordination. In Fig. 2b, the CO2 protonation has two different pathways: (i) CO2 → COOH* → CO*; (ii) CO2 → HCOO*→ HCOOH*. The optimized structures of the intermediates and corresponding hydrogenation free energies are plotted in Fig. S2 and Table S2 (ESI†), respectively. Clearly, the rate-determining step of HCOO* → HCOOH* needs to overcome a higher free energy barrier than that of CO2 → COOH* on δ-Cu2S (0.46 vs. 0.21 eV, Fig. 2c) and β-Cu2S (0.43 vs. 0.08 eV, Fig. 2d), then CO* can easily form due to the downhill free energy difference of ΔGCOOH*→CO*. In addition, the free energy of ΔGCO* is significantly lower than that of ΔGHCOOH* because of the much stronger binding of CO on δ-Cu2S and β-Cu2S (the black line displayed in Fig. 2c and d). Therefore, the β-Cu2S and δ-Cu2S monolayers are desirable Cu-based catalysts for the CO2RR with better selectivity to CO* rather than HCOOH*.
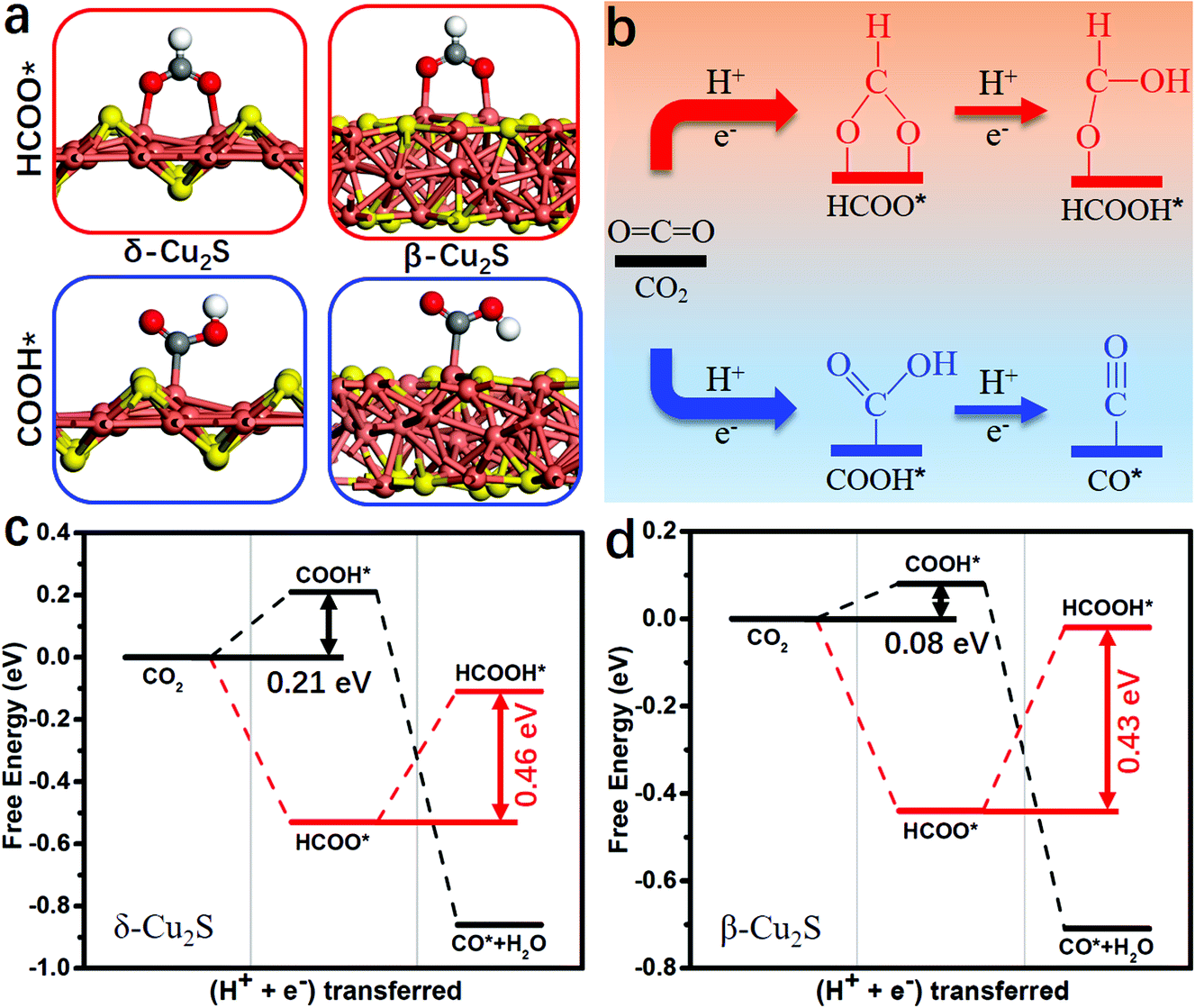 | ||
| Fig. 2 (a) Optimized geometry of the intermediates of HCOO* and COOH* on δ-Cu2S and β-Cu2S. Cu, orange; S, yellow; C, grey; O, red and H, white. (b) The CO2 protonation has two different pathways: (i) CO2 → COOH* → CO* (bottom path); (ii) CO2 → HCOO* → HCOOH* (top path). The free energy profiles of CO2 hydrogenation into CO* and HCOOH* on (c) δ-Cu2S and (d) β-Cu2S when two proton/electron pairs (2H+/2e−) are transferred. | ||
As the adsorption of intermediates is affected by the surface coverage of CO*, we explore the binding energies of these two catalysts under different CO* coverage. Fig. S3 (ESI†) shows the varying relationship between the CO* coverage ranging from 1/9 to 6/9 and the binding energies, and the catalysts binding 2CO* are the most stable (detailed adsorption configurations are shown in Fig. S4, ESI†). Under 2CO* coverage, the PDS for the CO* reduction into CHO* is significantly reduced to 0.39 and 0.50 eV (see Fig. 3a) on β-Cu2S and δ-Cu2S, respectively. This indicates that the stronger adsorption strength of CO* promotes the CO* coverage, and thereby reduces the PDS remarkably, which agrees well with the previous report on Cu4@g-C3N4.55
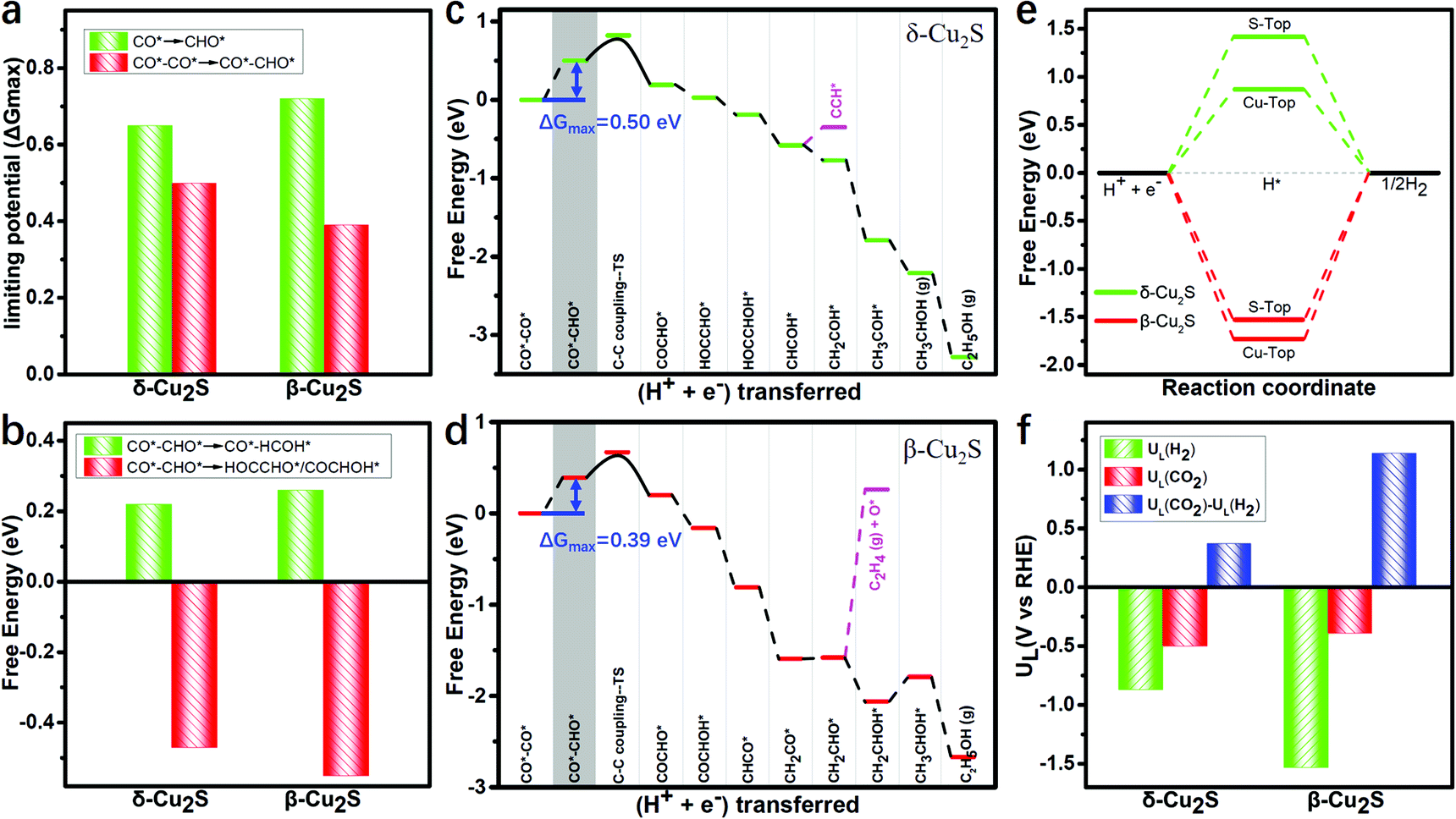 | ||
| Fig. 3 Free energy of the hydrogenation step of (a) CO*–CO* → CO*–CHO* and CO* → CHO*, (b) CO*–CHO* → CO*–HCOH* and CO*–CHO* → HOCCHO*/COCHOH*. Free energy diagrams of CO*–CO* hydrogenation to C2H5OH on (c) δ-Cu2S and (d) β-Cu2S. Limiting potential is obtained from the maximum free energy change (ΔGmax) of the whole reaction process, which is marked by blue lines. (e) Free energy profiles of the HER on δ-Cu2S (green line) and β-Cu2S (red line). (f) Limiting potentials for the CO2RR UL(CO2), HER UL(H2) and difference UL(CO2) − UL(H2) on δ-Cu2S and β-Cu2S monolayers. | ||
Generally, CO*–CHO* is a vital intermediate in determining the selectivity between the C1 and C2 products, i.e. CH3OH and C2H5OH. For C1 products, the hydrogenation of CHO* → HCOH* only needs to overcome very low free energy barriers of 0.22 and 0.26 eV, which are much lower than that of CO* → CHO* (0.99 and 0.84 eV) on δ-Cu2S and β-Cu2S (Fig. S5, ESI†), respectively. In other words, the reduction of CHO* is relatively facile because the CHO* (sp2 hybridization) has more p-character than the sp hybridized CO* (Fig. S6, ESI†). For C2 products, the free energies of the hydrogenation of CO*–CHO* into HOCCHO*/COCHOH* on δ-Cu2S and β-Cu2S are −0.47 and −0.55 eV (Fig. 3b), respectively. Therefore, the formation of C2 products becomes more favorable than that of the C1 product since the former occurs spontaneously, while the latter is endothermic. In general, the energy of C sp3 hybrid orbitals increases with the increase of p-character (see Fig. S6, ESI†), which makes the hydrogenation of CO*–CHO* → HOCCHO*/COCHOH* much easier than that of CO*–CHO* → CO*–HCOH* (Fig. S7, ESI†).
The full free energy profiles of the CO*–CO* hydrogenation to C2H5OH on δ-Cu2S and β-Cu2S are displayed in Fig. 3c and d, respectively, and the detailed adsorption structures of the intermediates are plotted in Fig. S8 (ESI†). The C–C coupling is crucial for C2 products, which normally largely affects the faradaic efficiency for a C2H5OH product. Generally, C–C coupling has two major pathways: (i) CO–CO coupling; (ii) CO–CHO coupling. For the CO–CO coupling pathway, OCCO* is automatically split into two separated CO* after full structure optimization, suggesting that the OCCO* is energetically unfavorable. For CO–CHO coupling, the calculated kinetic barriers are only 0.28 and 0.32 eV on β-Cu2S and δ-Cu2S (Fig. 4a), respectively, half of that on the Cu(100) surface45,46 (0.74 eV). From the atomic structures of the initial, transition and final states of CHO–CO coupling, we can clearly see that the CHO*, CO* and OCCHO* are stabilized on Cu top sites due to the upshift of the Cu d-band center on the Cu2S surface relative to the Cu(100) surface (Fig. 4b). In addition, Bader charge analysis reveals substantial charge, i.e., 0.34e, transfer from each copper to sulfur, resulting in a positively charged copper oxidation state. As mentioned above, the surface Cu+ can modify the surface structure of copper(I)-based catalysts, which leads to the change of binding strength between the adsorbate and catalyst, promoting the CHO–CO coupling reaction. The adsorption strength of CO* on Cu2S (−0.6 to −1.15 eV) is relatively lower than that of Ni, Pt, Pd and Rh (−1.65 to −1.88 eV).21 The mild adsorption strength may probably avoid CO poisoning and thereby promote the subsequent reaction of CO. Subsequently, the C2H5OH is easily formed due to the downhill free energy of COCHO* → C2H5OH on δ-Cu2S, while the step of the CH2CHOH* → CH3CHOH* only needs to overcome a particularly low free energy barrier of 0.26 eV on β-Cu2S. During the CO2RR process, once the CH2CHO* is formed, the reaction route bifurcates into ethylene and ethanol. As plotted in Fig. 3c and d, the C2 species are more favorable to be reduced to ethanol than ethylene as the calculated free energy barrier of the latter is significantly higher than that of the former, indicating that the ethanol is the main product of the CO2RR. Moreover, we find that the free energy barrier decreases more for the formation of CH2COH*/CH2CHOH* (ethanol route) than that of CCH*/C2H4 (ethylene route) with increasing CO* coverage ranging from 2/9 to 6/9 ML (Fig. S9, ESI†). Therefore, the catalysts have relatively high selectivity for ethanol over ethylene.
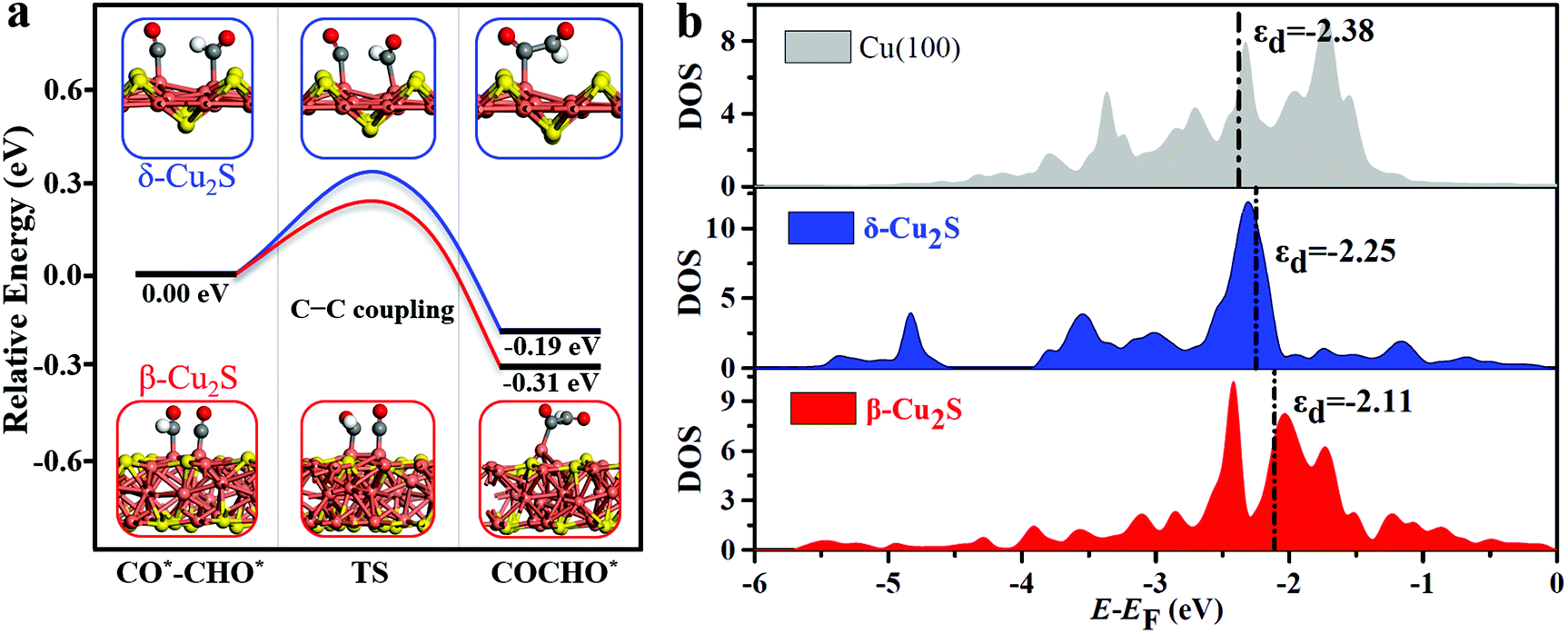 | ||
| Fig. 4 (a) Relative energy diagram of the CHO–CO coupling processes on δ-Cu2S and β-Cu2S. Cu, orange; S, yellow; C, grey; O, red and H, white. (b) Density of states (DOS) of the d-band center (εd) on δ-Cu2S, β-Cu2S and Cu(100). | ||
Note that the above conclusions have been drawn based on the computational hydrogen electrode model, in which the effects of solvents are not explicitly considered. The effects of solvents may play an essential role in the largely affected chemical reactivity and the product selectivity of CO2 reduction.56 Besides, it can also greatly reduce the C–C coupling kinetic barriers and improve the faradaic efficiency for C2 products.57 In the near future, we will further consider the influence of the effects of solvents to design efficient CO2 reduction catalysts.
The above conclusion only considers the ground state DFT calculations. In order to further explore how the photoexcitation process drives the different reaction routes and thus the selectivity, the excited state DFT calculations were taken into account. We used the delta self-consistent field (ΔSCF) method for constrained DFT calculations58 by promoting five spin-down electrons from the highest occupied to the spin-up lowest unoccupied molecular orbital at each K point.59,60 As shown in Fig. S10 (ESI†), the Cu2S catalysts have relatively high selectivity for ethanol over ethylene under excited state conditions, consistent with the ground state results. Therefore, the photoexcitation process has not changed the reaction routes and selectivity of the CO2 reduction on Cu2S.
Additionally, as the major competing reaction during the CO2RR, the HER, which normally largely affects the faradaic efficiency for reduction reactions like CO2RR and NRR, should also be considered. The H* structures of two different adsorption sites and the corresponding free energy diagram of the HER are shown in Fig. S11 (ESI†) and Fig. 3e, respectively. Clearly, the Gibbs free energy barriers of the HER (1.53 eV and 0.87 eV) are significantly higher than those of the potential determining step barriers of CO2RR (0.39 eV and 0.50 eV) on β-Cu2S and δ-Cu2S, indicating the high suppressing effect on the HER during the CO2RR. The difference [ΔUL(CO2) − UL(H2)] between the limiting potentials for CO2RR UL(CO2) and HER UL(H2) can reasonably describe the CO2RR selectivity, where a more positive UL(CO2) − UL(H2) value means a higher selectivity toward the CO2RR over the HER.61,62 As shown in Fig. 3f, the β-Cu2S and δ-Cu2S have positive UL(CO2) − UL(H2) values, indicative of the higher selectivity for the CO2RR against the HER.
As a good photocatalyst, the catalyst should also have a satisfactory band gap value to efficiently absorb visible light. As shown in Fig. 5a and b, the direct band gap of β-Cu2S at the Γ point is 1.45 eV, which is slightly larger than the value of 1.24 eV for δ-Cu2S. The satisfactory band gap values indicate that these two catalysts can effectively absorb visible (VI) light and ultraviolet (UV) light. As plotted in Fig. 5c, the absorption spectra of β-Cu2S and δ-Cu2S show remarkably high absorption coefficients (105 cm−1) under the irradiation of the full spectrum. For β-Cu2S, there is a high intensity of absorption VI and UV light. In particular, the first and second absorption peaks are located at 1.95 eV and 3.89 eV, respectively, in good accordance with the experimental results.38,63 In addition, the δ-Cu2S monolayer with a wide optical absorption range can be utilized and the intense absorption band is centered at 1.50 eV in the infrared (IR) region. Moreover, previous studies showed that β-Cu2S and δ-Cu2S both have fairly large electron mobilities,35 suggesting that the photo-generated electron and hole pairs will quickly reach the surface of these two catalysts to participate in the CO2RR. Meanwhile, the great difference between the electron and hole effective mass on the catalysts will dramatically reduce the possibility of photo-induced carrier recombination as well, which should further facilitate the photocatalytic efficiency of the CO2RR.
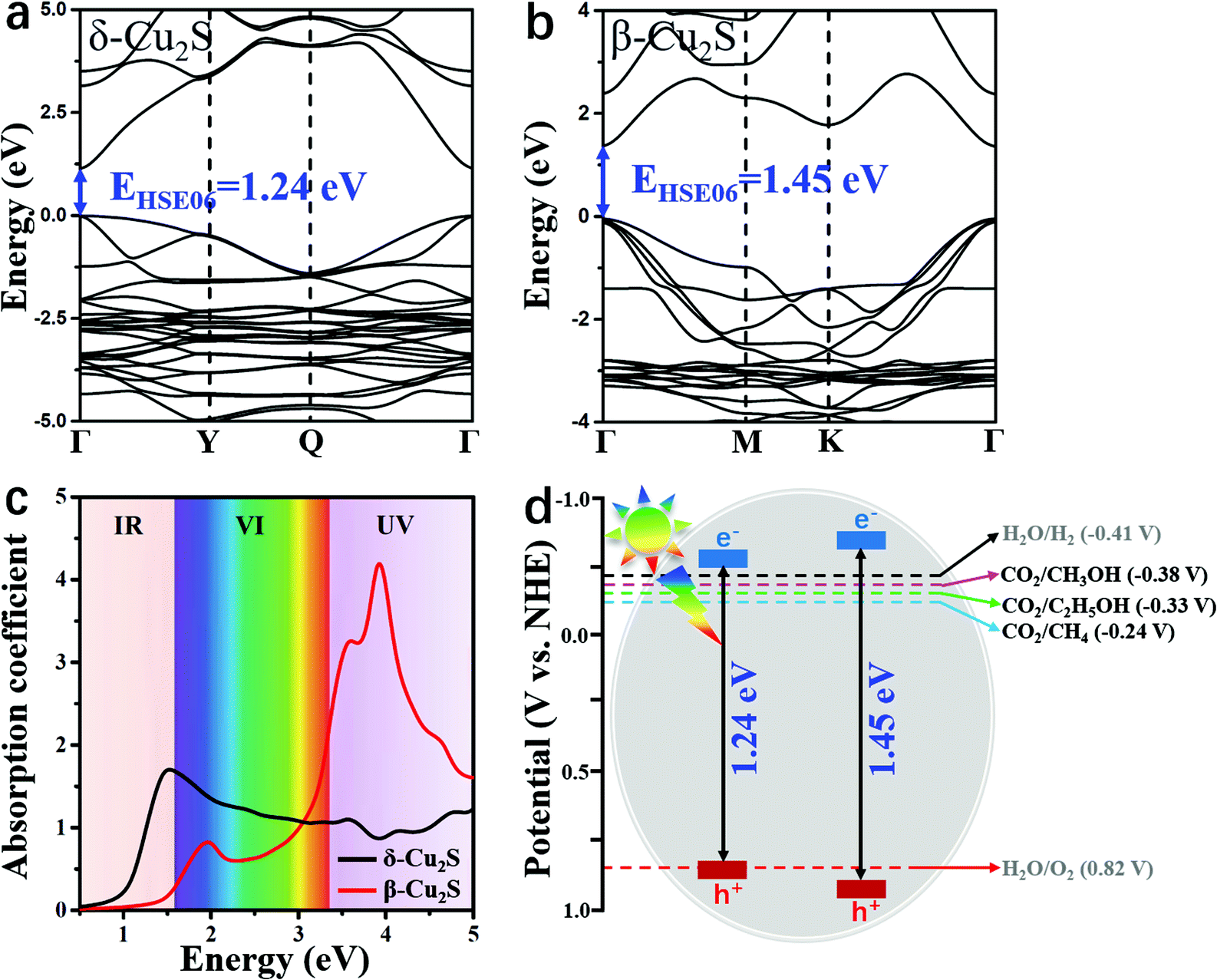 | ||
| Fig. 5 Electronic band structure of (a) δ-Cu2S and (b) β-Cu2S with a direct band gap at the Γ point. (c) Optical absorption spectra of β-Cu2S and δ-Cu2S are indicated by red and black lines, respectively. (d) Schematic illustration of the band edge positions of β-Cu2S and δ-Cu2S relative to NHE at pH = 7. The calculations were performed by HSE06. | ||
As a photocatalyst for CO2 reduction, the semiconductor should have satisfactory band edge positions to match the potentials of CO2 reduction and H2O oxidation as well. According to the above obtained band gaps (Fig. 5a and b), the electronic band structures versus normal hydrogen electrode (NHE) potential at pH = 7 can be elucidated. As manifested in Fig. 5d, the conduction band minima (CBMs) of β-Cu2S and δ-Cu2S are more negative than the redox potentials of CO2/C2H5OH (−0.33 V vs. NHE),42 suggesting that photo-generated electrons have sufficiently high reducing power and can be utilized as photocatalysts for the CO2RR to C2H5OH. Meanwhile, the valence band maximum (VBM) is below the H2O/O2 potential3 (0.82 V vs. NHE) and the high oxidation ability of the hole carriers enables H2O splitting and generates H+ for the CO2RR. These results indicate that β- and δ-Cu2S monolayers possess suitable band gaps as well as appropriate band edge positions, and can act as promising photocatalysts for the CO2RR into C2H5OH. In fact, previous studies have shown that the Cu2S bulk is excellent for oxygen evolution reaction activity and durability, and has been widely used in water splitting to produce H+.64–66 Therefore, we deduce that β-Cu2S and δ-Cu2S monolayers that have the merits of satisfactory band edge position and visible light absorption properties, and highly active surface are promising photocatalysts for the CO2RR.
In addition, we have further evaluated the stability of the catalysts. The δ-Cu2S and β-Cu2S have tetragonal and hexagonal structures, respectively, as shown in Fig. S12a and b (ESI†). In AIMD simulations, in which the time step was set to be 2.0 fs for a total period of 10 ps, the energy and temperature fluctuations with time evolution oscillate near the equilibrium state and the atomic configurations remain very well at the experimental synthesis temperature of β-Cu2S (∼800 K)33 (Fig. S12c and d, ESI†). In fact, as pointed out above, the ultrathin 2D β-phase Cu2S bilayer has been successfully synthesized in the laboratory32–34 and a 2D δ-phase Cu2S monolayer has also been predicted theoretically with superior oxidation resistance in the air and moisture atmosphere.35 More importantly, during the process of photochemical CO2RR, the structure still maintains stability under the existence of the solvent and potential (see Fig. S13, ESI†). These results suggest the high structural stability and a large experimental feasibility for Cu2S based catalysts.
Conclusions
In summary, we have demonstrated that 2D β-Cu2S and δ-Cu2S monolayers, as promising highly efficient photocatalytic semiconductor materials, can efficiently achieve the hydrogenation of CO2 to C2H5OH under the irradiation of visible-light. Our DFT calculations suggest that the β-Cu2S and δ-Cu2S photocatalysts can not only greatly suppress the competing HER, but also break the scaling relations in the multi-intermediates to increase CO* coverage and reduce the potential-limiting steps of CO* → CHO*. More importantly, the CHO–CO coupling only needs to overcome a very low kinetic barrier of 0.28 and 0.32 eV, and COCHO* is recognized as the key intermediate for C2 formation, resulting in a high selectivity for C2H5OH on both β-Cu2S and δ-Cu2S. Moreover, these two catalysts also exhibit satisfactory band edge positions and visible light absorption for the photocatalytic CO2RR and high thermodynamic stability over 800 K. Low limiting potential, high product selectivity, good thermal stability and strong visible light absorbance make Cu2S a very attractive photocatalyst for the CO2RR, which may provide new insights into the development of novel semiconductor photocatalysts.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was financially supported by the Natural Science Foundation of China (22033002, 21525311, 21773027 and 21703032) and the Fundamental Research Funds for the Central Universities (2242021k10009). We acknowledge the computational resources from Big Data Center of Southeast University and National Supercomputing Center of Tianjin.References
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 Search PubMed.
- Y. Zhao, G. I. N. Waterhouse, G. Chen, X. Xiong, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Soc. Rev., 2019, 48, 1972–2010 Search PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 Search PubMed.
- J. Di, et al. , Nat. Commun., 2019, 10, 2840 Search PubMed.
- X. Li, L. Liang, Y. Sun, J. Xu, X. Jiao, X. Xu, H. Ju, Y. Pan, J. Zhu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 423–430 Search PubMed.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 3169 Search PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 Search PubMed.
- J. Chen, J. Yin, X. Zheng, H. Ait Ahsaine, Y. Zhou, C. Dong, O. F. Mohammed, K. Takanabe and O. M. Bakr, ACS Energy Lett., 2019, 4, 1279–1286 Search PubMed.
- B. Zhou, et al. , Energy Environ. Sci., 2019, 12, 2842–2848 Search PubMed.
- T. Yan, L. Wang, Y. Liang, M. Makaremi, T. E. Wood, Y. Dai, B. Huang, A. A. Jelle, Y. Dong and G. A. Ozin, Nat. Commun., 2019, 10, 2521 Search PubMed.
- X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5267–5274 Search PubMed.
- C. G. Morales-Guio, et al. , Nat. Catal, 2018, 1, 764–771 Search PubMed.
- T.-T. Zhuang, et al. , Nat. Catal, 2018, 1, 421–428 Search PubMed.
- E. Bertheussen, et al. , Angew. Chem., Int. Ed., 2016, 128, 1472–1476 Search PubMed.
- S. Sorcar, Y. Hwang, C. A. Grimes and S.-I. In, Mater. Today, 2017, 20, 507–515 Search PubMed.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 Search PubMed.
- Z. Jiang, et al. , Energy Environ. Sci., 2018, 11, 2382–2389 Search PubMed.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 Search PubMed.
- K.-H. Liu, H.-X. Zhong, S.-J. Li, Y.-X. Duan, M.-M. Shi, X.-B. Zhang, J.-M. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64–111 Search PubMed.
- I. Shown, et al. , Nano Lett., 2014, 14, 6097–6103 Search PubMed.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 Search PubMed.
- H.-K. Lim, H. Shin, W. A. Goddard, Y. J. Hwang, B. K. Min and H. Kim, J. Am. Chem. Soc., 2014, 136, 11355–11361 Search PubMed.
- X. Hong, K. Chan, C. Tsai and J. K. Nørskov, ACS Catal., 2016, 6, 4428–4437 Search PubMed.
- S. B. Varandili, J. Huang, E. Oveisi, G. L. De Gregorio, M. Mensi, M. Strach, J. Vavra, C. Gadiyar, A. Bhowmik and R. Buonsanti, ACS Catal., 2019, 9, 5035–5046 Search PubMed.
- J. Zeng, W. Zhang, Y. Yang, D. Li, X. Yu and Q. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 33074–33081 Search PubMed.
- A. Bhowmik, H. A. Hansen and T. Vegge, ACS Catal., 2017, 7, 8502–8513 Search PubMed.
- H. Mistry, et al. , Nat. Commun., 2016, 7, 12123 Search PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 Search PubMed.
- Z. Q. Liang, et al. , Nat. Commun., 2018, 9, 3828 Search PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 Search PubMed.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 Search PubMed.
- B. Li, L. Huang, G. Zhao, Z. Wei, H. Dong, W. Hu, L. W. Wang and J. Li, Adv. Mater., 2016, 28, 8271–8276 Search PubMed.
- F. B. Romdhane, O. Cretu, L. Debbichi, O. Eriksson, S. Lebegue and F. Banhart, Small, 2015, 11, 1253–1257 Search PubMed.
- R. Shahzad, T. Kim, J. Mun and S. W. Kang, Nanotechnology, 2017, 28, 505601 Search PubMed.
- Y. Guo, Q. Wu, Y. Li, N. Lu, K. Mao, Y. Bai, J. Zhao, J. Wang and X. C. Zeng, Nanoscale Horiz., 2019, 4, 223–230 Search PubMed.
- J. Yu, T. Li, G. Nie, B. P. Zhang and Q. Sun, Nanoscale, 2019, 11, 10306–10313 Search PubMed.
- A. B. Wong, S. Brittman, Y. Yu, N. P. Dasgupta and P. Yang, Nano Lett., 2015, 15, 4096–4101 Search PubMed.
- Q. Cao, R. Che and N. Chen, Appl. Catal., B, 2015, 162, 187–195 Search PubMed.
- J.-Y. Li, L. Yuan, S.-H. Li, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2019, 7, 8676–8689 Search PubMed.
- A. Manzi, T. Simon, C. Sonnleitner, M. Doblinger, R. Wyrwich, O. Stern, J. K. Stolarczyk and J. Feldmann, J. Am. Chem. Soc., 2015, 137, 14007–14010 Search PubMed.
- P. Kar, S. Farsinezhad, X. Zhang and K. Shankar, Nanoscale, 2014, 6, 14305–14318 Search PubMed.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 Search PubMed.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 Search PubMed.
- Y. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, ACS Appl. Mater. Interfaces, 2018, 10, 28572–28581 Search PubMed.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 Search PubMed.
- H. Zhang, Y. Zhang, Y. Li, S. Ahn, G. T. R. Palmore, J. Fu, A. A. Peterson and S. Sun, Nanoscale, 2019, 11, 12075–12079 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 Search PubMed.
- L. A. Burns, A. Vazquez-Mayagoitia, B. G. Sumpter and C. D. Sherrill, J. Chem. Phys., 2011, 134, 084107 Search PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 Search PubMed.
- X. Bai, Q. Li, L. Shi, X. Niu, C. Ling and J. Wang, Small, 2020, 16, 1901981 Search PubMed.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 Search PubMed.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 Search PubMed.
- J. Gavnholt, T. Olsen, M. Engelund and J. Schiøtz, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075441 Search PubMed.
- Y. Zhao, et al. , Adv. Mater., 2018, 30, 1803127 Search PubMed.
- W. Gao, et al. , Chem., 2018, 4, 2917–2928 Search PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Norskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 Search PubMed.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1706617 Search PubMed.
- Y. Wu, C. Wadia, W. Ma, B. Sadtler and A. P. Alivisatos, Nano Lett., 2008, 8, 2551–2555 Search PubMed.
- Y. He, et al. , Nat. Mater., 2019, 18, 1098–1104 Search PubMed.
- L. He, D. Zhou, Y. Lin, R. Ge, X. Hou, X. Sun and C. Zheng, ACS Catal., 2018, 8, 3859–3864 Search PubMed.
- L. An, P. Zhou, J. Yin, H. Liu, F. Chen, H. Liu, Y. Du and P. Xi, Inorg. Chem., 2015, 54, 3281–3289 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00196e |
| This journal is © The Royal Society of Chemistry 2021 |