
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unified mechanistic interpretation of amine-assisted silica synthesis methods to enable design of more complex materials
Joseph R. H.
Manning
 *ab,
Carlos
Brambila
a and
Siddharth V.
Patwardhan
*a
*ab,
Carlos
Brambila
a and
Siddharth V.
Patwardhan
*a
aGreen Nanomaterials Research Group, Department of Chemical and Biological Engineering, University of Sheffield, Mappin Street, Sheffield S1 3JD, UK. E-mail: s.patwardhan@sheffield.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: joe.manning@ucl.ac.uk
First published on 14th January 2021
Abstract
The design of porous sol–gel silica materials is a thriving research field, owing to silica's diversity of properties and potential applications. Using a variety of additives, most commonly amine-based organic molecules, several families of silica materials have been developed including silica nanospheres, zeolites, mesoporous silicas, and bioinspired silicas with controlled particle and pore morphology on multiple length scales. Despite the wide range of study into these materials, and similarity in terms of reagents and additive compounds, none can recreate the features and complexity present within naturally occurring biosilica materials. This is due in part to a lack of ‘joined-up’ thinking during research into silica synthesis strategies and methodology. Specifically, mechanistic insights gained for one set of conditions or additive structures (i.e. material types) are not translated to other material types. In order to improve the structural complexity available in synthetic silica materials, as well as to improve both understanding and synthesis methods for all silica types, a unified approach to mechanistic understanding of formation in amine-assisted silica synthesis is required. Accordingly, in this review we analyse contemporary investigations into silica synthesis mechanism as a function of (amine) additive structure, analysing how they imprint varying levels of order into the eventual silica structure. We identify four fundamental driving forces through which additives control silica structure during synthesis: (i) controlling rates of silica precursor hydrolysis and condensation; (ii) forming charge-matched adducts with silicate ions in solution; (iii) self-assembling into mesophases to physically template pores; and (iv) confining the location of synthesis into specifically shaped vesicles. We analyse how each of these effects can be controlled as a function of additive structure, and highlight recent developments where multiple effects have been harnessed to form synthetic silica materials with further structural complexity than what was previously possible. Finally, we suggest further avenues of research which will lead to greater understanding of the structure–function relationship between amine additives and final materials, hence leading to more complex and high-value silica and other materials.
 Joe Manning | Dr. Joe Manning is a postdoctoral researcher in inorganic nanomaterials design in the group of Dr. Gemma-Louise Davies at UCL. Prior to this he worked as a postdoctoral researcher with Prof. Tina Düren at the University of Bath, studying host–guest interactions in MOF materials to understand their process chemistry. He received an MSci in Chemistry and Chemical Engineering from the University of Strathclyde, Scotland (2014), and a PhD in the design and process intensification of bioinspired silica materials from the University of Sheffield (2019) under the supervision of Prof. Siddharth Patwardhan. |
 Carlos Brambila | Dr Carlos Brambila is a postdoctoral researcher in green chemistry in the group of Prof Siddharth Patwardhan. His work seeks to implement sustainable manufacturing of mesoporous silicas at industrial scale. He obtained a BSc in Chemical Engineering in Materials from Universidad Autonoma de Queretaro, Mexico (2012), followed by 2 years as a Technical Adviser within the Mexican public service. He received an MSc in Solid State Chemistry from the University of Sheffield (2015), where he went on to obtain a PhD studying the formation and 3D-microstructure of porous nano-oxides under the supervision of Dr Günter Möbus. |
 Siddharth | Siddharth, a chemical engineer and a materials chemist, is a Fellow of the EPSRC and the RSC. He leads the Green Nanomaterials Research group, with a vision of developing sustainable, scalable and economical routes to functional nanomaterials. He recently authored the book on Green Nanomaterials covering emerging bioinspired green methods for nanomaterials. Applying the excellence in green chemistry to nanomaterials manufacturing, the group is currently commercialising novel materials suitable for diverse applications. He is a recipient of multiple awards including the Dedicated Outstanding Mentor award four-times, “SuperVisionary” award twice and Teaching Excellence award twice. |
Design, System, ApplicationSeveral families of porous silica materials have been developed including recent bioinspired silicas. Despite the wide range of studies into these materials, and similarity in terms of reagents and additive compounds, none can recreate the features and complexity present within naturally occurring biosilica materials. This is due in part to a lack of ‘joined-up’ thinking during research into silica synthesis strategies and methodology. For the first time, we present a unified approach to mechanistic understanding of formation in amine-assisted silica synthesis. Using our proposed approach, mechanistic insights gained from one family of materials can be translated to other material types. This has the potential to improve the structural complexity available in synthetic silica materials, as well as to improve both understanding and synthesis methods for all silica types. Utilising this unified knowledge, we open-up future avenues of research including experiments and computations, spanning multiple length-scales and production scales, which will lead to greater understanding of the structure–function relationship between amine additives and final materials, hence leading to more complex and high-value silica and other materials. |
1. Background
As process engineering evolves, so do the requirements on the materials used. With increasingly prominent need for carbon capture and sequestration,1,2 advanced drug delivery systems,3–6 and next-generation catalysts and supports,7,8 more complex material structures and surface chemistries are required to meet the growing and specific demands. In order to achieve this, new synthesis methods for porous materials are required and are the subject of significant scientific interest. An excellent example of this is porous silicas (either amorphous precipitated silicas or crystalline zeolites), a widely used industrial material whose versatility enables application in sectors ranging from polymer fillers, to acid–base catalysts, pharmaceutical excipients, filtration media and pollutant sorbents.As with most metal oxides, silicas are generally formed using sol–gel methods. In general, a monomeric precursor e.g. tetraethylorthosilicate (TEOS) is hydrolysed into a silicate anion, which then undergoes dehydration polymerisation into dimers and trimers, small oligomers, and finally macromolecular colloidal particles.9 This process is particularly versatile, providing a rich parameter space for researchers and manufacturers to control and improve the properties of the materials. Such modifications are mostly achieved by introducing organic molecules (termed ‘additives’) into the process, which assist in the formation and nanostructuring of the materials. A schematic representation of several silica materials and the additives used to synthesise them is shown in Fig. 1. As can be observed, the change in additive structure can result in an entirely different product, even when otherwise similar processing conditions are used. Based on such modifications, the following major families of materials have emerged:
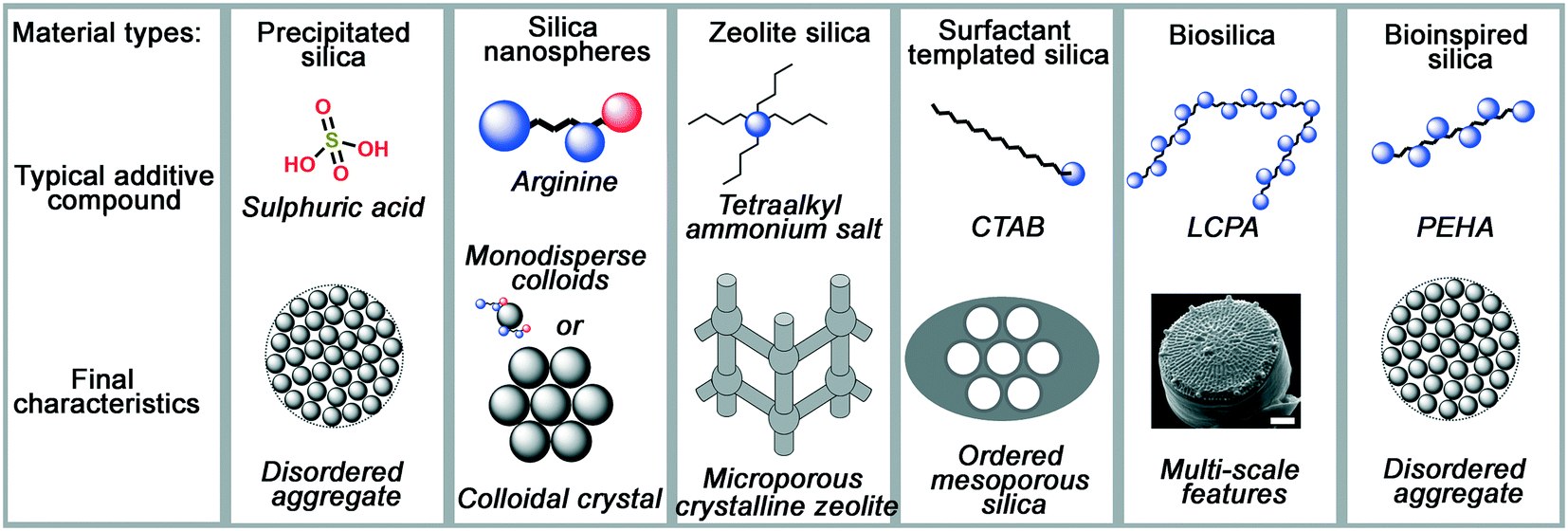 | ||
| Fig. 1 A summary of typical additive compounds for each silica category, as divided into their families, and the key features of silica materials made through their strategies. Blue circles in additives represent amine moieties, red circles represent carboxylate moieties. Diatom image reproduced from ref. 10. | ||
• Colloidal silica nanospheres: silica nanospheres, make use of small additives like ammonia or monopeptides to carefully control the rate of silica polymerisation and make monodisperse silica colloids.
• Zeolites: in a similar fashion to colloidal silicas, templated silica zeolites often use small, charged additives like tetraalkylammonium salts to template small pores within crystalline silicate.
• Surfactant-templated silicas: more complex porosity is introduced in surfactant-templated amorphous silicas, which take advantage the amphiphilic nature of the surfactant additive (commonly cetyl trimethyl ammonium bromide, CTAB) to imprint larger, liquid-crystal-like pores into the final structure.
• Biological silicas: highly complex structures, not seen in any of the above examples, are imprinted in biological silicas, which use the polyfunctionality of proteins and biogenic long-chain polyamines (LCPAs) to imprint more complex hybrid functionality into silica materials than artificial organic compounds can provide.
• Bioinspired silica: these materials apply a reductionist approach to biological additives, using simpler but still polyfunctional organic amine additives which retain most advantages of biomolecule templates while improving compatibility with industrial processing capabilities.
None of the artificial strategies described approaches the complexity imprinted into biosilicas, however. For synthetic templating strategies to become as complex as biosilica templating (or even to understand their formation mechanisms better), a more holistic view of template structure–function relationships is required. We propose that the driving forces present in additive structure direction throughout these different classes of materials are identical and therefore insights from any additive-silica system can provide insight into any other.
1.1. Structure of this article
In section 2 we will survey how organic additives modify and direct silica synthesis during each of the pathways summarised in Fig. 1, sorted according to the level of organisation imprinted on the eventual silica material. The chemical function of these additive molecules will be discussed, connecting their molecular structure to catalytic or templating behaviour observed. In section 3, we will present a combined perspective on additive structure–function relationships, categorising the structure-directing activity of templates into four fundamental pathways. The specific chemical moieties which enable this activity are then identified, and the benefits of a unified approach to additive structure in material design are discussed. Finally, in section 4, we will suggest avenues for future research which we believe will provide important new insight into the synthesis of designer silica materials.2. Structural organisation imprinted by a template
A distinct indicator of the dissociation across silica studies is shown by the nomenclature used to identify the additive molecules that facilitate silica precipitation. Such constituents are present across all silica syntheses (bar notable exceptions), however reports of each silica family use a different term used to denote these analogous species. In the case of colloidal silicas, ammonia is referred to as a catalyst. In zeolite synthesis, the additive is commonly termed an organic structure directing agent (OSDA)11,12 or a template. The terms template or surfactant can refer to the same type of constituent in mesoporous silicas.13 Biomolecules used in biological or biomimetic silica synthesis are often named after their biological function, such as proteins, peptides, or post-translationally modified polyamines.14,15 Finally, organic molecules used to synthesise bioinspired silicas are conventionally referred to as additives. The latter is the term used throughout the present work to refer to all such synthesis-assisting molecules as we believe such designation best encapsulates the complex role of organic molecules during synthesis, which can simultaneously include catalysis, stabilisation, aggregation and structure-direction.16,17Analogous to the similarities between silica families identified, there also exist parallels between the additives used in silica syntheses. Such similarities have been made evident by studies of additive structure–function relationships during silica formation (discussed in depth in section 2). Such studies have evidenced that amine functional groups in the additive aid in synthesis by providing one or several of the following: proton-transfer catalysis; hydrogen bonding; and organisation of building blocks, oligomers or precursors through charge–charge interactions. On the basis of these functions, four key features controlling the behaviour of additives can be identified:
1. The number of functional groups present in the additive molecule.17–19
2. The degree of separation between the functional groups.20
3. The level of substitution (e.g. methylation) of each functional group.17,19,21,22
4. The presence of large sidechains on the molecule (e.g. long aliphatic tails providing amphiphilic behaviour).23
Despite these findings, only a few studies have explicitly explored the relationship between additive molecular structure and final silica; furthermore, these have largely focused on the bioinspired family of silica materials with no equivalent systematic studies being performed for silica nanospheres, zeolites, or surfactant templated silicas.
The present work addresses the lack of such studies outside bioinspired silicas by surveying previous scientific studies of additive-assisted silica synthesis. Specifically, we outline the different levels of structural organisation which can be imprinted onto the various families of silica materials by organic additives as a result of the features present on the additive itself. Such behaviour results in structural organisation of the eventual silica materials, the dimensionality of which is a result of the additive structure:
1. 0-D involves control over anisotropic morphological properties such as particle size or pore imprinting in the shape of the additive molecule itself.
2. 1-D organisation involves isotropic ordering in a single direction only such as through the creation of oriented pores within otherwise uncontrolled nanoparticles.
3. 2-D control generally takes place by simultaneously combining aspects of 0d and 1d organisation, e.g. by producing ordered porosity in particles of controlled size.
4. 3-D templating consists of control over internal porosity and particle size in multiple directions and length scales.
2.1. No order
Even without recourse to organic molecules to control silica polymerisation and growth, it is possible to modulate the rate of hydrolysis and condensation on a number of length scales by carefully controlling the concentration of inorganic bases in solution. Combined, these studies enable a significant degree of understanding regarding which processes are of critical importance to the overall polymerisation in the absence of organic additives. They also provide a good benchmark for understanding how organic molecules can modulate the polymerisation transition states and oligomerisation kinetics to modify silica structure.
Control of silica polymerisation can be achieved by balancing the rates of precursor hydrolysis (khyd, eqn (1)) and condensation (kcondeqn (2)), enabling growth of either monodisperse spherical silica “sols,” porous gel networks, or porous precipitated aggregates.9 These two reactions are both accelerated by proton-transfer catalysis,27,28i.e. acidic or basic dissolved species. Therefore, control over the extent, timing and relative rates of these two processes are highly dependent on solution conditions during synthesis.
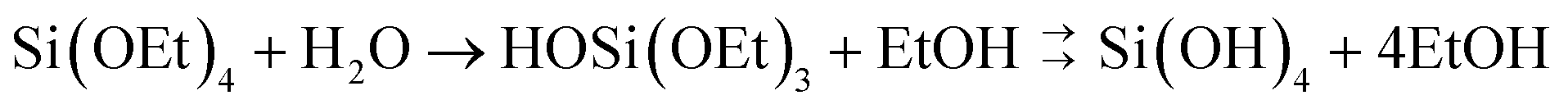 | (1) |
| 2Si(OH)4 → (HO)3Si–O–Si(OH)3 + H2O | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salt species molar ratio, solvent dielectric constant, presence of catalysts, and mixing conditions leads to a wide parameter space.24,33
:salt species molar ratio, solvent dielectric constant, presence of catalysts, and mixing conditions leads to a wide parameter space.24,33
This parameter space is significantly augmented by the inclusion of organic molecules containing basic (i.e. amino) moieties. The ability of organic amines to adopt a range of hard and soft base behaviour dependent on overall molecular structure allows them to simultaneously modify the progression of silica polymerisation reactions and template ordered structural motifs into the resultant materials.
Earlier bioinspired materials were synthesised using biological or synthetic polymers in an attempt to directly replicate the activity of a peptide derived from silaffin proteins isolated from diatom biosilica.36 This was performed by using poly(L-lysine) (PLL) and poly(allylamine) (PAA) as synthetic additives to mimic the length and multifunctionality of biomolecular templates (see Fig. 2a); silica particles were formed in under 5 minutes reminiscent of silaffin-precipitated silica.37,38 For both of these additive molecules, BIS was formed as polydisperse spherical particles with little increase in surface area compared to equivalent NaCl-templated control silicas (albeit with larger pore sizes).39 Although the long-range order observed in diatoms was yet to be achieved, the benefits of biomineralization conditions were maintained when using bioinspired polymeric additives.
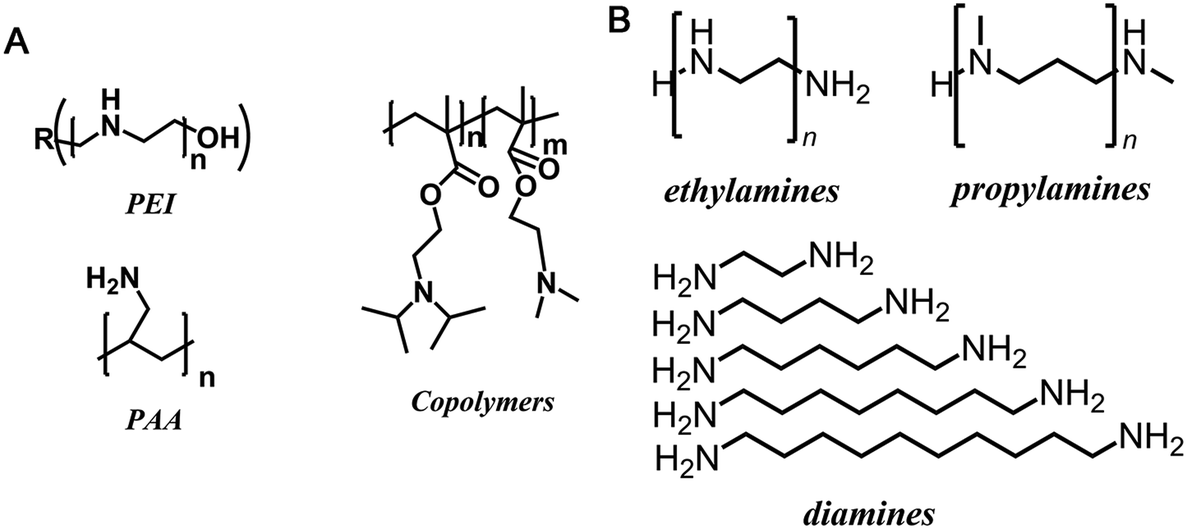 | ||
| Fig. 2 Selected examples of the bioinspired additives used in the silica formation in vitro (A) synthetic polymers and (B) small molecules (n = 1–7). Images reproduced or adapted with permission from ref. 16–18. | ||
These early studies triggered a range of further investigations using polymeric bioinspired/biomimetic additives. These included the use of natural and synthetic polymers, homo- and block co-polymers, polypeptides and dendrimers.16 These studies applied the knowledge of polymer chemistry and synthesis to tailor the chemical functionality and morphology/assembly of polymers such that they could control silica synthesis. This strategy successfully produced a wide range of silica structures and morphologies, with increased control over the organisation, including nanoparticles, porous materials and anisotropic particles.40
Some recent research has focused on small polyamine molecules to reduce the system complexity and generate better additive structure–function relationships. Initially, simplification to oligopeptides containing 1–5 residues enabled investigation of additive catalysis during silica precipitation.41 Further experiments were largely carried out using ethylenediamine (MEDA, NH2CH2CH2NH2) and its homologues containing between 2 and 7 moieties (shown in Fig. 2b where MEDA is n = 1), revealing the effect these species have on silica synthesis. These have demonstrated the importance of molecular length,17,18 functional moiety separation distance,20 degree of amine methylation, and the presence of assisting chemicals such as pH buffers22 on both the initial polymerisation and the interactions between colloidal silica particles.
The key controlling feature that was identified from these investigations was the ionisation/protonation of small amines used. The complexity arising from small differences in protonation of each amine functionality within these multi-amine molecules was interesting. This led to dynamic changes with respect to the changes in the pH and the presence of silica precursor, further resulting in (co-operative) self-assembly (or not) of these additives. The outcomes from these studies enabled the control of porosity and other properties. For example, by changing the length of the additives/number of amines in a single molecule (i.e. from 2 to 7 N atoms,17 and occasionally up to 18 N atoms42), surface area of silica could be modulated from being non-porous to >300 m3 g−1.17 Similarly, the length of the additives and their concentration was used to control the solution stability of silica (driving either silica precipitation or stabilising colloidal suspensions).18
BIS synthesis is both significantly faster than industrial precipitated silica materials and uses significantly milder conditions than those used for templated silica materials. The BIS family of materials bridge the gap between the complex biosilicas and the functional synthetic materials described in the previous sections. Yet, despite tremendous advances, so far only BIS with disordered pores and a broad pore size distribution (mainly microporous) have been reported. There are some reports on the bioinspired or biomimetic synthesis of mesoporous silica using a range of polypeptides, custom synthesised surfactant or polymeric templates.43–46 The materials obtained from these templates have mesoporosity, along with micro- and/or macro-porosity, while with broad pore size distributions and they all remain disordered in nature – neither the silica “walls” nor the pores are organised.
In the example without additives, pH and electrolyte environment can be modified to optimise the sol–gel conditions leading to desired particle properties. Conversely, incorporation of amine additives in the case of bioinspired silica serves chiefly to accelerate the rates of both khyd and kcond at the expense of such precise control over particle properties (e.g. size, polydispersity). This acceleration, based on the mild conditions under which biosilica materials are deposited, reduces reaction times and temperatures during manufacture, leading to environmental and technoeconomical benefits over contemporary sol–gel silica manufacture.
Exploration of different additive chemical structures has also led to control over a wider range of silica properties than are possible in the additive-free system, such as the imprinting of disordered internal mesopores or formation of hollow particles. Notwithstanding the disorder introduced through accelerated khyd and kcond, this finding clearly demonstrates that changing the chemical structure of additives is an effective strategy for tailoring the properties of silica materials. To best take advantage of including amine additives in silica synthesis methods, the connection between additive structure and reaction progression (both in terms of khyd and kcond and other structure-directing effects) must be fully understood.
2.2. 0-Dimensional structural ordering
Whereas BIS materials are generally synthesised at circum-neutral pH with catalytically active amine additives, materials with imprinted order are generally synthesised under much less reactive conditions. In these cases, silica formation from alkoxysilane and other precursors is commonly induced by monofunctional isotropic templates such as ammonia and tetramethylammonium ions. This process can result in two distinct material types, monodisperse colloidal nanospheres and zeolite silicas. In both cases, the properties of the material can be effectively controlled by varying the structure and concentration of the additive as well as the reaction conditions.Fig. 3 shows a schematic representation of the major stages involved in the formation of SNS via the Stöber process.47 The effect of ammonia concentration can be appreciated in the illustration. Initially, alkoxide precursors are hydrolysed by ammonia and water through basic proton transfer catalysis. Once a sufficient amount of free silicic acid is formed, particle nucleation and growth occurs. After this seed induction time (Ti in Fig. 3), at high concentrations the nucleation-growth activity stops and the seeds begin to aggregate, giving place to further monomer addition and monodisperse particle formation. As a catalyst ammonia favours condensation over hydrolysis,48,49 however, should ammonia concentration become too low, the rates of each reaction step (khyd and kcond) are imbalanced and particle nucleation times become nonuniform, leading to continuous formation and growth of seeds (Fig. 3, lower). From this information it would seem that a higher ammonia concentration is advantageous for Stöber silica formation, however higher [NH3] also leads to larger particle sizes.50
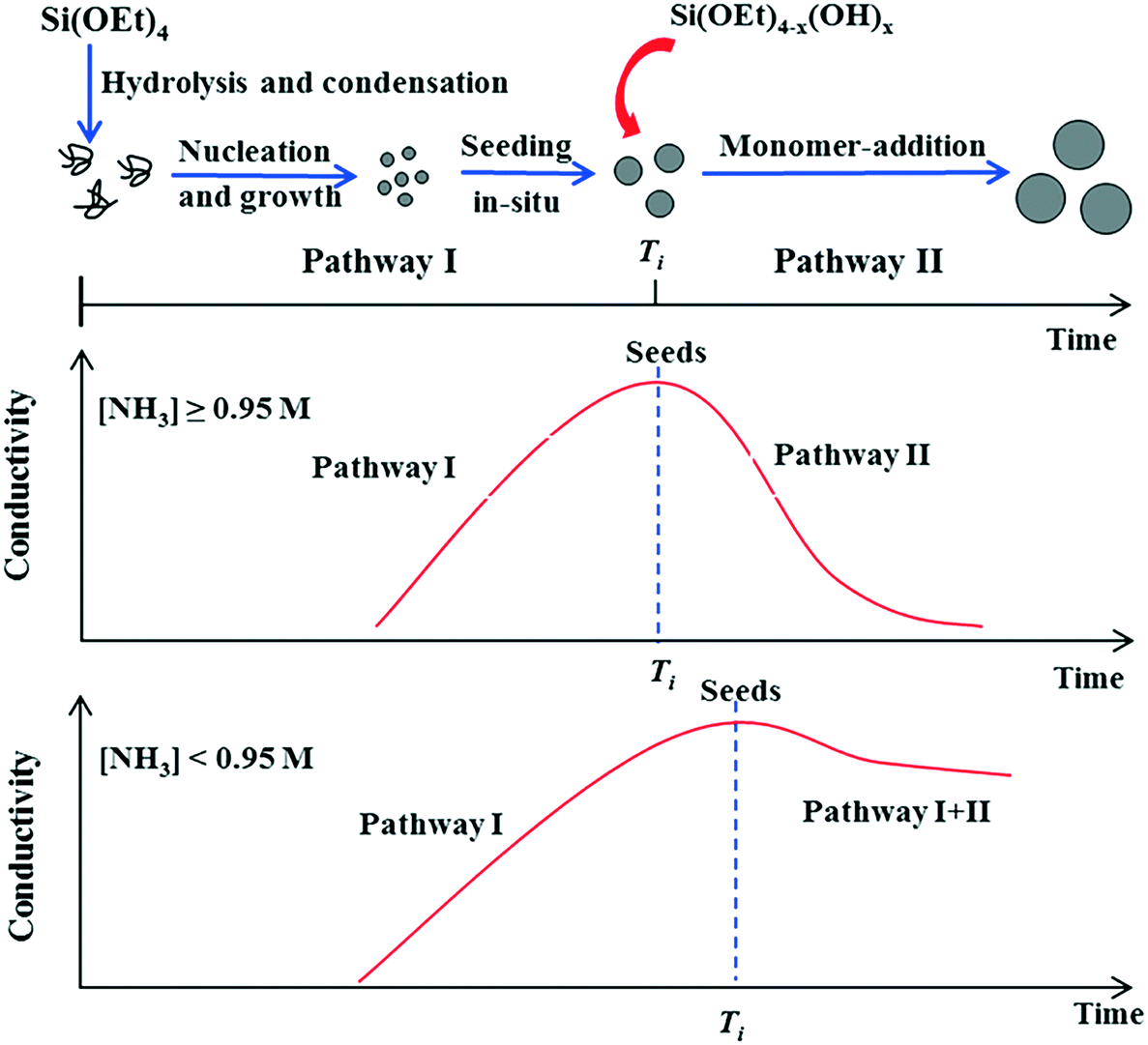 | ||
| Fig. 3 Effect of ammonia concentration on the formation pathway of Stöber SNS. Figure reproduced with permission from ref. 47. | ||
Despite the stringent requirements on relative rates of khyd and kcond, SNS synthesis methods have been developed using more active amine additives (monopeptides and polyazamacrocycles, as shown in Fig. 4A). In order to maintain monodispersity in the face of more active polyfunctional additives, modifications to the Stöber method are necessary. In the case of monopeptides, monodispersity can be controlled either by using an emulsion-based system to control rates of hydrolysis (i.e. using a TEOS-water emulsion to ensure hydrolysis could only be as fast as phase-transfer between the systems),51 or by using bioinspired methods (aqueous, low total [Si], curcum-neutral pH).52 In the former case, the use of emulsion hydrolysis enables lower additive concentrations (hence eventual particle sizes) than in traditional Stöber synthesis without leading to polydispersity due to a slightly modified particle nucleation and growth mechanism.53 In the latter case, the ability to produce monodisperse particles is presumably due to an inability of the azamacrocycles due to their cyclic structure to bridge the electric double layer (EDL) between particles.18
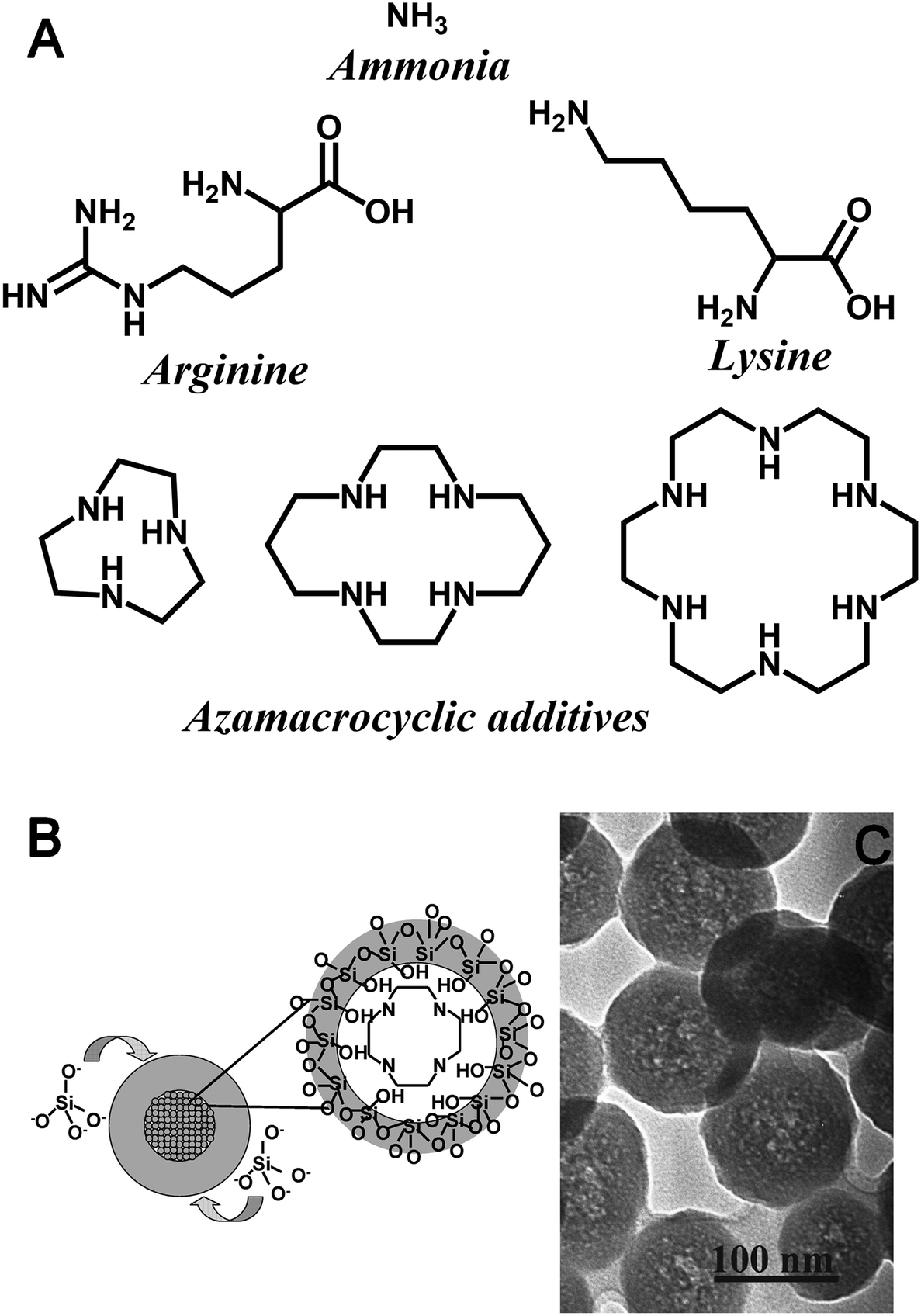 | ||
| Fig. 4 (A) Chemical structures of additives used to impact 0-D geometric control in the formation of colloidal silicas. (B) Schematic illustration and (C) TEM image of core–shell seed formation during silica polymerisation assisted by azamacrocycle amine additives in Stöber-like conditions. Reproduced with permission from ref. 55. | ||
While larger azamacrocycles are also able to synthesize SNS under bioinspired and Stöber-like conditions, these molecules have several distinctions to ammonia or monopeptide additives. In the aqueous bioinspired SNS synthesis, the final pH and azamacrocycle structure were the key determining factors for particle size, which is unique among all SNS synthesis methods.54 Conversely when using the basic, ethanolic synthesis method, particle size remains a function of additive concentration and azamacrocycle structure. However, this additive again has unique aspects – azamacrocyclic additives create adducts with silica precursors during initial stages of synthesis, leading eventually to hollow SNS synthesis (Fig. 4B and C).55,56
Quaternary ammonium cation additives cannot directly catalyse the hydrolysis and condensation of silica precursors as they contain no Lewis base sites. Instead, their inclusion performs two functions during material formation, summarised in Fig. 5.61 First, additives act as a salt counterion to bases (‘mineralizers’ in Fig. 5A), enabling hydrolysis of silica precursors. Once precursors have been hydrolysed, the additives themselves act as a structure-directing agent to template polysilicate ions in solution (Fig. 5B). These complexes organise into a crystalline arrangement (usually under hydrothermal conditions), wherein individual additives are encapsulated within a pore, connected by ‘windows’ made up from silicate rings of varying size dependent on the crystal phase (Fig. 5C). Upon additive removal e.g. by calcination (an ‘activation’ step), uniform porous domains are revealed.
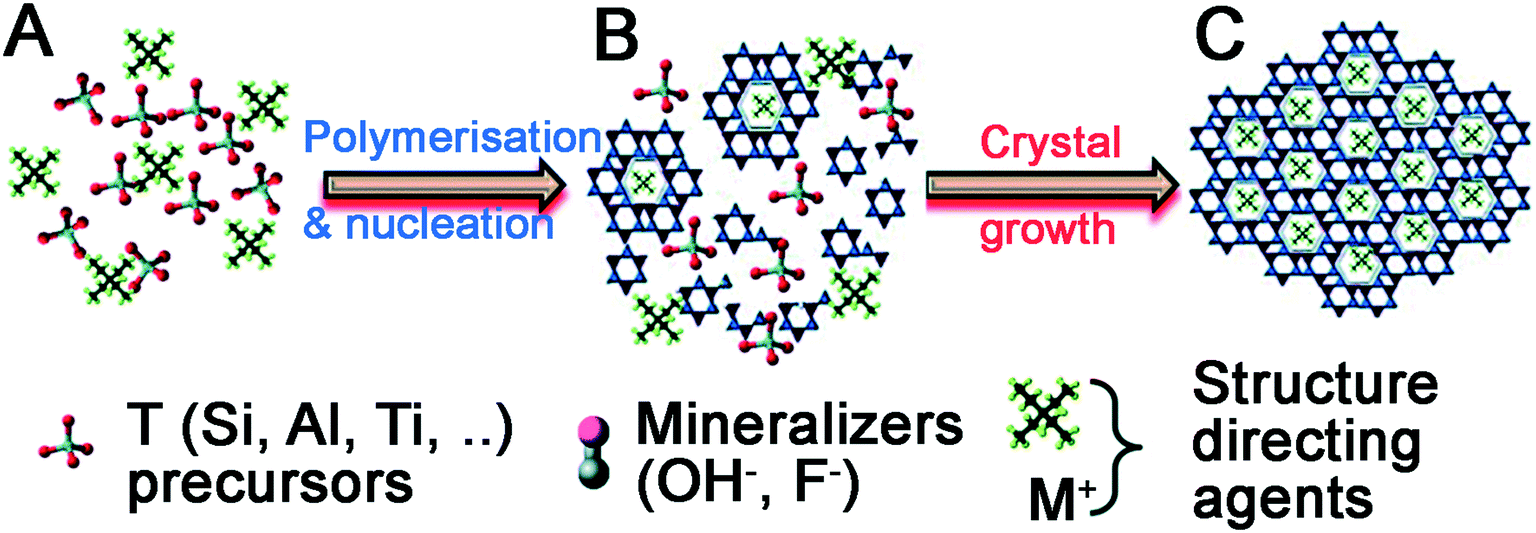 | ||
| Fig. 5 Universal stages of zeolite material formation. (A) Hydrolysis of precursors, (B) arrangement of polysilicate anions around additive ions in solution, (C) organisation of additive-polysilicate adducts into a macroscopic crystal phase. Reproduced with permission from ref. 61. | ||
The first step in zeolite synthesis is not directly related to the additive structure, but instead the concentration of mineralisers is crucial to synthesis progression – should basic species such as OH− be in excess of silica precursors, polysilicate formation and aggregation will be prevented.62 If silica precursors are in excess, hydrolysis can occur as with SNS, and polysilicate structures and silica oligomers begin to form in solution. At this point, the structure of the additive becomes important, templating the synthesis by promoting the formation of specific intermediates.63,64
Impact of additive structure on pore geometry. On the face of it, the impact of additive structure on pore geometry is quite simple, with eventual pore geometry closely matching the Van der Waals radius of the additive compound regardless of additive functional groups.59 Indeed, it is entirely possible to predict the structure–property relationships between organic additives and eventual zeolite pores on this basis, enabling new additive molecular structures to be designed with specific pore geometry in mind.65,66 Accordingly, a broad variety of quaternary ammonium additive molecules have been designed, which have been comprehensively reviewed in ref. 67. This body of research has produced some generalised rules of thumb regarding the behaviour of zeolite additives. For example, the ratio of carbon to nitrogen atoms (C/N+ ratio) is important in determining the density of the resultant zeolite framework as well as the geometry of the pores therein. Specifically, additives with C/N+ > 9 are too large to be encapsulated fully within discrete silicate cages, leading to less dense materials being formed.68 Further, rigid additives are highly selective towards different zeolite crystal phases, but are more likely to fail to provide structural ordering (i.e. creating an amorphous material) compared to more flexible alternatives.69
Given that zeolite pore structure is dependent on the size and shape of the additive molecule, early theories of zeolite formation mechanism concluded that the nature of dissolved additive-silica adducts mirror this shape: monomeric silicate ions surrounding an individual additive ion prior to silica condensation.70,71 However, recent studies using ab initio simulations to observe additive-silicate adduct formation with TMA+, TPA+ and tetrabutylammonium (TBA+) additives indicate that the organic compound cannot fit fully within the key intermediate silica species being formed.72 In the simplest case, TMA+ ions have been shown both experimentally73,74 and computationally75 to stabilise octameric silicate cages.75 As can be seen in Fig. 6, TMA+ ions sit externally to the Q38 structure, located centrally on the outside of each face of the cubic ion.75,76 Similarly, TPA+ and TBA+ externally stabilise bicyclic Si11 structures through nonspecific Van der Waals forces, thus aiding in their dimerization and trimerization, which forms the basis of the macrostructure formation step shown in Fig. 5C.23
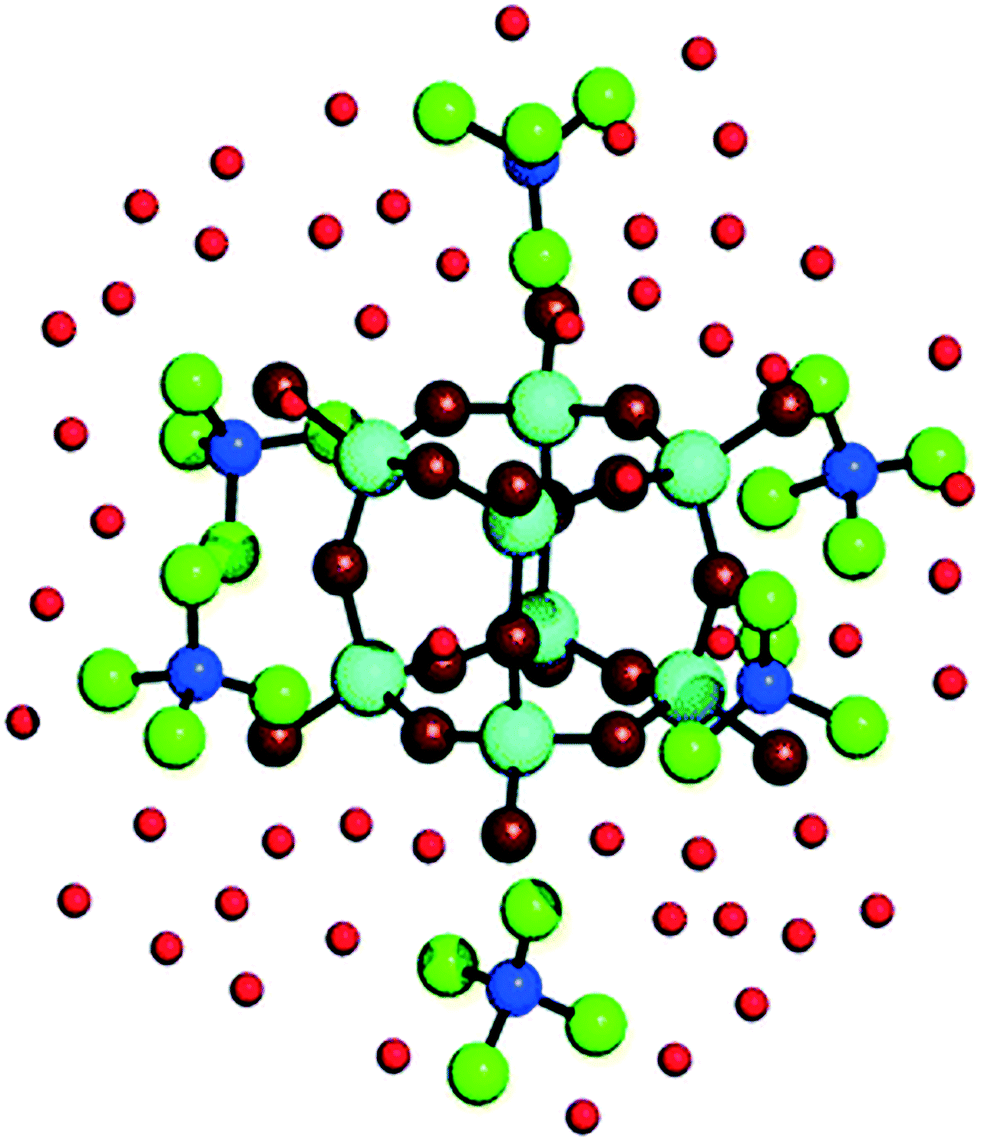 | ||
| Fig. 6 Visualisation of adducts between TMA+ ions and silicate precursors. Dark blue balls represent N atoms, cyan balls represent Si atoms, green balls represent CH3 groups, dark red balls represent O atoms in Si8O12 complexes, light red balls represent O atoms in water. Reproduced with permission from ref. 74. | ||
Impact of additive functional groups. Along with the discovery that the additive size closely matches that of pore cages in zeolites, their insensitivity to chemical functionality of the additive was also discovered.59 Accordingly, a wide variety of additive molecules have been tested (e.g. polyfunctional quaternary ammonium additives, aminoalkanes with varying levels of alkyl substitution, ether, thioether, and azamacrocycle compounds).12,59,77,78 These compounds retain their cationic properties (i.e. highly basic tertiary amine ‘proton sponges’ which are present in a protonated form or organic sulfonium ions), satisfying the need for positive charges around which silicate monomers can assemble. Even in the case of crown ether molecules, the active species during synthesis is still cationic – crown ether molecules scavenge inorganic ions such as Na+ from solution, thus acting as an additive in the form of a cation-crown ether complex.79
Overall, the benefits of using alternative chemical functionality to ammonium moieties within additives for zeolites lie in terms of their secondary characteristics rather than changing their templating behaviour. Specifically, it is possible to enhance additive thermal stability, additive environmental stability, and processability through the choice of different additives.78 As an example, the use of additives containing ketal functionality enables in situ, reversible deconstruction of the additive after synthesis, enabling them to be extracted from the pores under solvothermal conditions and reused for the creation of further zeolite materials rather than thermally decomposing during calcination.77,80
Other uses of additives during zeolite formation. Beyond control of individual pore geometry through pore templating, multifunctional additives can also be used as zeolite growth modifiers. By binding to specific surface crystal planes, polyfunctional additives such as those used in BIS synthesis methods81 can alter the anisotropic rate(s) of crystal growth thus controlling crystal size and habit depending on the additive molecular structure.82
Unlike amino groups, quaternary ammonium ions have no direct effect on silica polymerisation kinetics. Instead, through the formation of specific additive-silicate adducts, these molecules serve to stabilise key intermediates for zeolite pore formation through nonspecific Van der Waals forces. These species are then encapsulated in the eventual material structure, leading to templating of ordered porosity dependent on their Van der Waals radius. Again, polyfunctional amines have recently been introduced into zeolite materials synthesis, acting to bind and prevent crystal growth along specific planes leading to anisotropic growth.
One of the key limitations of using isotropic additives to template porous zeolites is that the eventual pore size is fundamentally limited by the size of the template molecule. Further, ring strain effects prevent the formation of larger pore windows in crystalline materials.11 Accordingly, to create larger pores, an alternative method to using single molecular templates is required, specifically using supramolecular assemblies of anisotropic additives to create noncrystalline mesoporous silicas.
2.3. 1-Dimensional templating through surfactant self-assembly
The additives discussed in section 2.2 provided control over solid structural complexity due to their ability to control nucleation and particle growth or through stabilisation of polysilicate “cages”. These additives were largely isotropic in chemical structure. Conversely, 1-dimensional structural order is achieved through the use of amphiphilic additives such as surfactants or block copolymers. Surfactant compounds are highly anisotropic, consisting of long, hydrophobic carbon “tails” (generally C8–C18)83 with a single hydrophilic moiety e.g. trimethylammonium as the molecule's “headgroup”. In the case of polymers, block co-polymerisation is used to create a hydrophobic centre (e.g. poly(propyleneoxide), PPO) and hydrophilic ends (e.g. poly(ethyleneoxide), PEO).84 Herein we will focus on the former group in order to illustrate the mechanistic role of these molecules.The amphiphilic nature of surfactant compounds leads them to spontaneously self-assemble in solution, creating mesophases around which silica can form (shown in Fig. 7, bottom left). These mesophases have a variety of well-defined morphologies depending on the surfactant concentration, molecular structure, and molecular structure, and solution conditions,85 ranging from individual micelles dispersed in a bulk aqueous phase to rod-like, bicontinuous, or lamellar liquid crystals. Incorporation of surfactants into conventional sol–gel synthesis methods enables corresponding pore geometries to be imprinted into silica materials, most notably in the Mobil Compositions of Matter (MCM) family of porous silicas.86 This strategy towards silica templating is illustrated in Fig. 7 – at lowest surfactant concentrations, rod-shaped micelles assemble into a honeycomb pattern to form MCM-41. Higher surfactant concentrations lead to bicontinuous or lamellar phases, leading to the formation of MCM-48 and -50, respectively.87 After activation (which commonly involves calcination in a parallel to zeolite materials), ordered porosity in the shape of the surfactant mesophase is revealed.
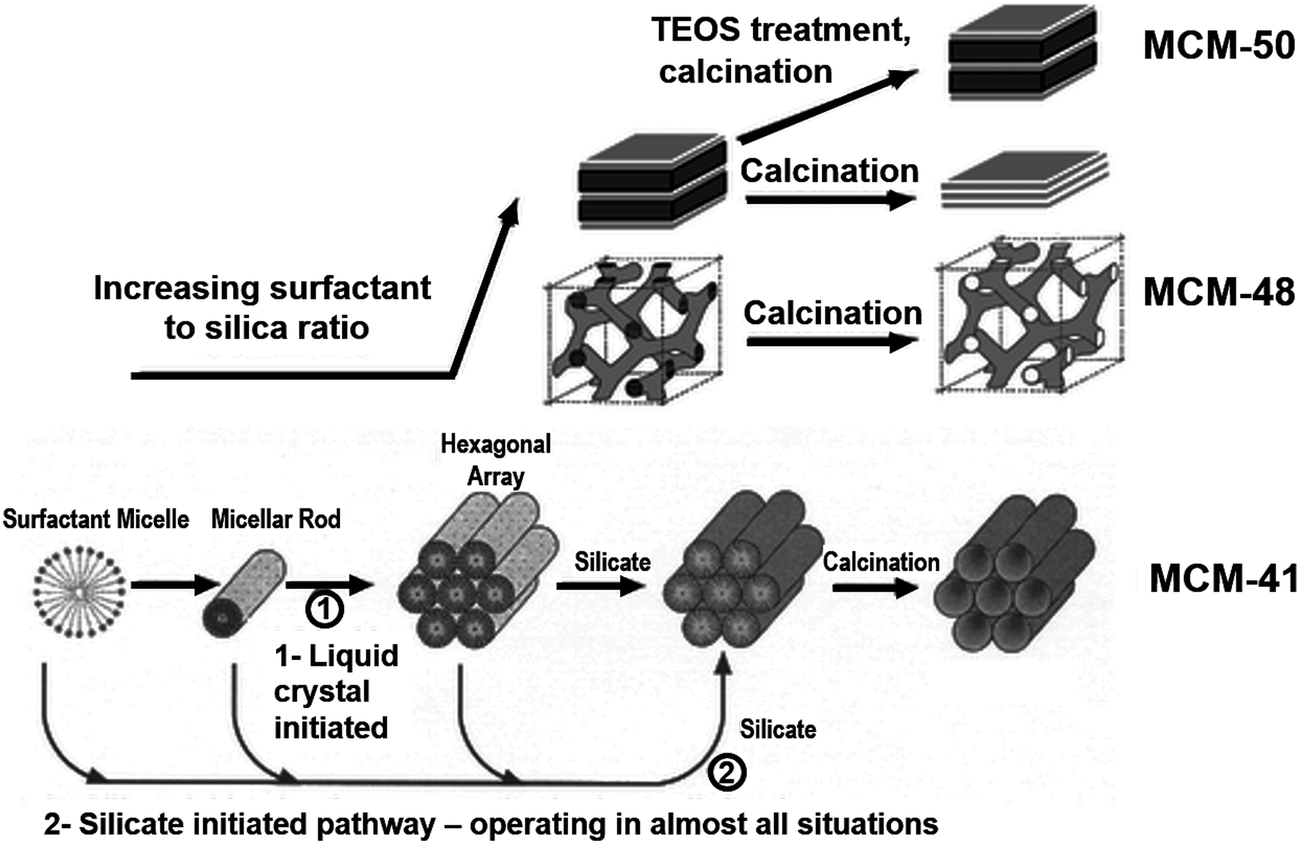 | ||
| Fig. 7 Visualisation of the initial liquid crystal templating mechanism for mesoporous silica synthesis, and the eventual silica structures produced. At low concentrations (bottom) surfactants assemble into spherical and rodlike micelles leading to MCM-41, characterised by hexagonally-packed pores. Higher surfactant concentrations lead to bicontinuous or lamellar phase separation leading to MCM-48 and MCM-5, respectively. Reproduced with permission from ref. 87. | ||
Typical MCM-41 synthesis conditions are well below the surfactant concentrations expected for liquid-crystal formation, however. Many studies have therefore focused on understanding the mechanisms of surfactant self-assembly both through experimental and computational means,88,89 leading to an improved understanding of the cooperative assembly between preformed surfactant–silicate adducts, rather than assembly of pure surfactants, followed by silicate interactions. In-depth discussion of such assembly is outside the scope of the current work and has been excellently reviewed elsewhere,90 however it is important to note that charge–charge interactions between surfactant and silicate precursors was identified as crucial. Accordingly, the variety of effective surfactant headgroup chemistry is dependent on its silicate interaction strength, as discussed below.
Headgroup structure. The most common surfactant additive used in synthesis of MCM and related materials is CTA+ (cetyl trimethyl ammonium ions, commonly as a bromide salt). This is a quaternary ammonium salt surfactant with similar headgroup functionality to the TMA+ additives used in zeolite synthesis. Accordingly, CTA+ does not play a catalytic role in the silica polymerisation, only its spatial organisation during synthesis.91
In solution, CTA+ ions interact with silica precursors either directly (in the case of synthesis proceeding at basic pH) or through intermediary counterions (in the case of synthesis proceeding at acidic pH).92 These mechanisms are termed S+I− or S+X−I+ due to the molecular charge of the surfactant, inorganic, and counterion (X) species, respectively (N.B. silica synthesised using the latter pathway are often named SBA-3);83 although changing mesophase-interface chemistry has no distinct impact on the pore geometry,83,93 it affects the silica surface acidity.92
Similarly to how zeolite templating changes when additive sidechain length increases from TMA+ to TPA+ and TBA+,23 increasing the sidechain length on surfactant headgroups also causes changes in eventual pore structure.94 While the range of available liquid crystal morphologies remains unchanged from Fig. 7, increasing the headgroup size from methyl to ethyl or propyl alters the phase behaviour i.e. micelle surface curvature.95 As a result, pores in these materials pack into a square rather than hexagonal arrangement (Fig. 8A and B).94,96,97 This behaviour contrasts with zeolites, where sidechain chain length can completely change which crystal phase is formed.23 Accordingly, the self-assembly of surfactant additives supersedes any structure-directing activity of the headgroups alone, leading to only relatively minor changes to pore structure.
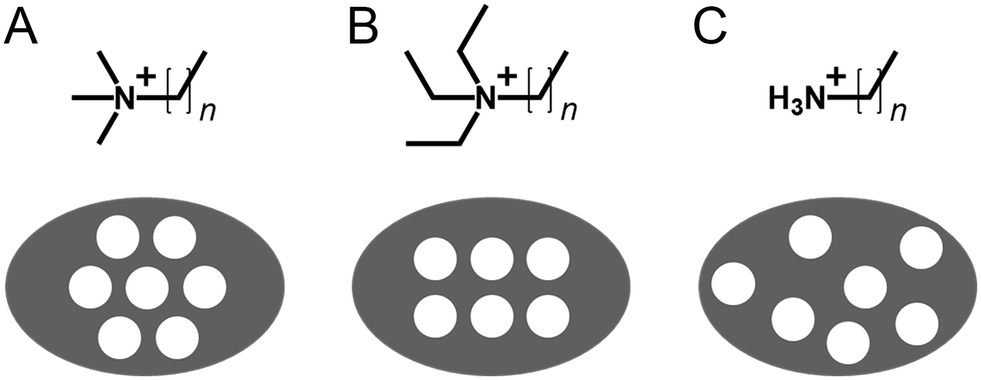 | ||
| Fig. 8 Visualisation of cylindrical pore arrangement within surfactant templated silicas as a function of headgroup chemistry for (A) permthylated, (B) perethylated, and (C) unmodified amine surfactants. N is typically 8–22. | ||
While changing the headgroup structure can lead to changes in how individual pores are arranged, the progression of the synthesis is largely similar for all quaternary ammonium surfactants. Conversely, changing the headgroup moiety from quaternary to primary amines leads to a significantly altered formation pathway. Primary amine surfactants like dodecylamine are similarly capable of imprinting tubular mesopores into silica (named HMS or hexagonal mesoporous silica), albeit with no ordered arrangement of pores like in materials templated by CTA+ and analogous ammonium surfactants (Fig. 8C).98 Initially, it was believed that avoiding permanently charged quaternary amine headgroups would lead to a neutral alternative to the S+I− reaction pathway – a so-called S0I0 route involving hydrogen bonds at the surfactant–silicate interface rather than charge-matching interactions.83 However, recent coarse-grained molecular dynamics simulations have shown that such a pathway does not lead to the expected self-assembly, and rather that dodecylamine additives are temporarily protonated during mesophase formation.99
Finally, the range of available headgroup chemistry allows surfactant choice to be made with other factors in mind than just templating activity: surfactants either partially or fully derived from natural sources (e.g. containing fatty-acid derived tails or amino-acid derived headgroups) can make the synthesis more sustainable.13 This is especially important given the environmental toxicity of CTA+ and dodecylamine-based surfactants, (both listed as very toxic to aquatic life) whose remediation contributes significantly to the cost of silica synthesis scale-up and commercial implementation.100
Tail structure and micelle swelling agents. While additive headgroup structure can modify the orientation of pores within surfactant-templated silicas, it has a relatively small impact on the pore diameter itself;95 this is the domain of the surfactant tail group. Several studies have investigated the impact of surfactant tail length on pore diameter, which has been excellently discussed in ref. 95. Therefore here we will only reiterate the salient points.
Although mesophase shape is dependent on the ratio of surfactant tail volume with headgroup area and length, in practise this ratio is largely unaffected by changes in tail length for CTA+ and its analogues.97 This is because, above a certain number of carbon atoms, the saturated aliphatic tail coils around itself representing a maximum tail length. Conversely, if the number of carbons in the tail group is too low then amphiphilic behaviour of the additive is compromised leading to no clear mesophase formation.83 Therefore in the absence of any further assistance, pore sizes are limited from ca. 1.7 nm (when tails contain 8 carbons)83 to ca 4.2 nm (with 22 carbons in the tail group).101
Covalent tethering of surfactant tails. A special case of surfactant templating comes when the additive and silica source are a single compound – the surfactant tail is modified with an alkoxysilyl functionality (Fig. 9A). Amine-functionalised silica precursors e.g. aminopropylethoxysilane (APTES) are commonly used to post-synthetically modify silica surface chemistry.102 APTES and similar additives are also capable of self-templating however, leading to the formation of lamellar 1-d ordered materials similar to those shown in Fig. 7 (top).103 Alternatively, hollow silica microparticles can be synthesised from mixed APTES/TEOS systems in the absence of any other additives, through the formation of silicate emulsions.104 Therein, amphiphilic APTES is located at the surface of alkoxysilane mesophases, driving silica polymerisation at the surface of the droplet only, leading to the formation of a core–shell morphology.104
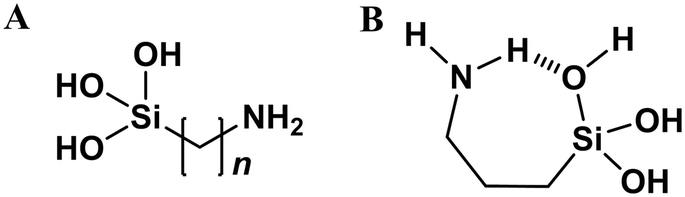 | ||
| Fig. 9 (A) General chemical structure of typical covalently tethered amine-silicate additives (n = 3, 11). (B) Proposed mechanism for intramolecular hydrogen bonding in hydrolysed APTES, stabilising the monomer species, adapted from ref. 105. | ||
Self-organisation of covalently tethered additives becomes particularly pronounced as the aliphatic tail becomes longer and upon introduction of secondary additives which coordinate to the amino headgroups (e.g. CO2, acetic acid, pivalic acid, valproic acid). Respectively, these secondary additives can synthesise silica nanosheets,106 silica nanorods,107 or hexagonal porous structures,108 depending also on the presence of water and TEOS within the reaction system.
The need for a secondary additive in these systems highlights one of the peculiarities of using covalently tethered additives and silica sources. Unlike the other chemical systems discussed in this review, the formation of additive-silicate adducts is counterproductive to material formation – these complexes are predominantly intramolecular, which stabilise the hydrolysed precursor, hampering polymerisation (Fig. 9B).105 These peculiarities of the systems extend the range of possible morphologies beyond what is summarised in Fig. 7 as the system is no longer limited to silica polymerisation at the surfactant headgroup, meaning that all micellar and inverse micellar morphologies are accessible.
In terms of templating effects, templating using surfactants is dominated by their self-assembly rather than the nonspecific Van der Waals interactions present in zeolite templating, despite similarities between headgroup chemistry of surfactants and zeolite additives.70,74 Further, unlike the nonspecific Van der Waals interactions present in zeolite synthesis, charge-matching interactions present between surfactant additives and silicate precursors are crucial to the 1-dimensional templating in surfactant-silica systems. Although this means that zeolites are capable of forming a greater variety of specific crystal phases and structures,109 surfactant-templated silicas are capable of exhibiting a broader range of structural characteristics.
• Surfactants do not play a catalytic role in the hydrolysis or condensation of silica precursors.
• Formation of ordered pores depends on the ability to form strong surfactant–silicate complexes by charge matching; the use of non-permanently charged surfactants only produces ordered pores when surfactants are in charged states.
• Dynamic complexation between silicates and the surfactants changes the self-assembly behaviour of surfactants, reducing the overall micelle curvature and changing the phase space of the self-assembly.
Despite the impressive number of different structures possible to be synthesised by the incorporation of surfactants instead of isotropic templating additives, the structures they can adopt are limited by the self-assembly behaviour. These limitations can be extended by the use of additives covalently tethered to the silica sources – enabling polymerisation at the tail group and freeing up the head group to interact with secondary additives thus extending the range of possible morphologies. Self-assembly of dispersed phases in solution is a mature science however, therefore it is evident that only a few overarching pore geometries will be possible. In order to surpass this, further complexity in the additive structure is required.
2.4. 2-Dimensional structural ordering
We define 2-dimensional ordering as the simultaneous control over two structural or morphological properties. Although countless combinations of properties could be produced for specific applications, their control is always reliant on the additive chosen for their synthesis. To illustrate this correlation, we summarise the three major families of materials prepared with such structural specifications. Namely, hierarchically porous silicas, mesoporous silica nanoparticles and silica morphologies beyond spheres. In all these cases, the approach has been to combine previous methods to control features at different length-scales. This includes the simultaneous use of small amines and surfactants or surfactants and hard spheres to impart micro–meso or meso–macro porosities, respectively. While the activity of these additives has already been discussed, combining more than one together without compromising the activity of either requires some consideration.Alternatively, hierarchical porosity can be included by using additives which combine the activity of both small molecular and liquid-crystal like templating. A key example of this is the block copolymer PEO–PPO–PEO in hierarchically porous SBA-15.114 In this material, PPO cores of the polymer assemble into liquid crystal phases similar to those described in section 2.3, creating mesoporous channels of 4–20 nm diameter.112 PEO groups become encapsulated in pore walls upon condensation of silica around the PPO cores, leading to microporous ‘bridges’ between channels, depending on the conditions under which the additive is removed.114 By bridging between pores, guest transport within the materials is improved compared to surfactant-templated silicas.
Hierarchical porosity is of particular importance for bulk-chemistry applications, where transport of fluids and surface reactions need to be optimised. By combining large pores that lead to smaller ones, the structure is accessible while maintaining a high surface area, reminiscent of the cavernous structure of the lungs.115 Therefore, hierarchically porous materials are studied beyond the mere presence of two or more defined pore sizes. Additionally, researchers seek to characterise and control the interconnectivity of pores in three dimensions. Such demand and complexity have also made hierarchically porous silicas a prevalent material in the benchmarking of three-dimensional electron tomography.116,117
As with all bulk chemistry applications, the synthesis of such materials is required to be inexpensive and scalable. Numerous strategies have been developed towards that end. For instance, Anderson et al. used readily available diatomaceous earth to provide macroporous structure, which was coated with a microporous zeolite.118 Further attempts to accelerate and economise the formation of silicas of hierarchical porosity have been obtained by one-pot synthesis approaches, which take advantage of the aforementioned optimal combination of organic additives.115 At a higher length scale, precise control of macro-/meso-porosity combinations have been achieved by Sel et al. by using block copolymers as templates and controlling the charge of the reaction liquid to avoid phase separation.119
:water solvent ratios for surfactant templated silicas are ca. 1:100,83 in strong contrast with SNS synthesis methods where the ratio is typically 6.5:1.
Initial synthesis methods used a single additive – surfactants such as CTAB – however this necessitated very high dilution (which has been proven to lower condensation rates despite water's proton-catalytic nature120), leading to monodisperse particles but with very low specific yields.121 To synthesise mesoporous SNSs at more typical mesoporous silica synthesis concentrations, a mixed additive system is therefore required, consisting of a surfactant additive and an additive which decelerates rather than accelerates hydrolysis and condensation. To fulfil this role, triethanolamine (TEA) is commonly used.
Being a tertiary amine, TEA is able to act as a base, hydrolysing alkoxysilane precursors in aqueous systems. In anhydrous conditions, TEA is also able to complex with silicates in solution, forming highly stable atrane complexes (Fig. 10).122 Initial syntheses combining TEA and surfactant additives prepared such anhydrous atrane complexes as precursors, forming mesoporous silicas upon introduction of surfactant solutions.123 However such a two-step synthesis method is not required, and direct introduction of TEOS into solutions of TEA and CTA+ lead to synthesis of MSNs with controlled monodisperse particle sizes (Fig. 11B).121,124
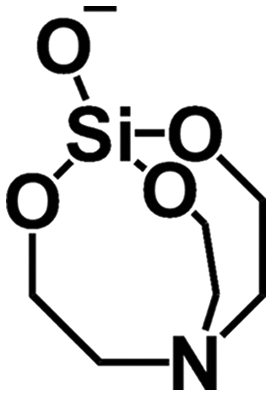 | ||
| Fig. 10 Chemical structure of a silicon atrane complex. N–Si coordination bond omitted for clarity. | ||
 | ||
| Fig. 11 Selected electron micrographs showing the diversity of 2-dimensional structural ordering which has been developed. (A) Surfactant templated mesoporosity combined with hard-sphere-templated macroporosity, reproduced with permission from ref. 113. (B) Stable colloidal MSNs produced using surfactant-templating and TEA to control hydrolysis and condensation, reproduced with permission from ref. 124. (C) Hexagonally-shaped MSNs synthesised with the addition of secondary surfactant NaDC additive, reproduced with permission from ref. 129. Finally, hexagonal platelets and star-like BIS structures produced using PLL in either static (D) or flowing (E) conditions, reproduced with permission from ref. 132. | ||
The need for TEA as an additive in MSN synthesis reveals the potential pitfalls of designing a mixed-additive synthesis system – surfactant additives are far less sensitive to system concentration compared to additives which act through proton-transfer catalysis. The flexibility of silica synthesis mechanisms is also highlighted however – using TEA as an inhibitor to TEOS hydrolysis rather than a catalyst restored the SNS synthesis mechanism to an otherwise incompatible reaction composition. Therefore, through thorough understanding of the reaction mechanisms present and the additive's role therein, it is clearly possible to design much more complex silica materials than otherwise would be available.
As a first example, SBA-15 materials templated by PEO–PPO–PEO block copolymers can be arranged into hexagonal morphologies through control over liquid crystal formation during synthesis. As previously shown in Fig. 7, initial stages of mesoporous silica formation involves co-condensation of silicate ions around surfactant micelles. In the case of polymeric additives, the size of these polymer–silicate complexes is discrete since silica condenses around a single additive (unlike surfactant–silica micelles whose self-assembling behaviour changes during co-condensation90). Importantly, these polymer–silicate complexes can behave as colloids in their own right, with association behaviour affected by temperature and ionic strength.125,126 Therefore, by careful control of temperature and solution ionic conditions during early stages of the reaction, ordered association of polymer–silicate micelles into liquid-crystal phases is possible, leading to hexagonal ‘monocrystalline’ particles with controllable aspect ratio.127,128
This ‘colloidal phase separation mechanism’ can be applied to surfactant templated MSNs using secondary additives. Specifically, the use of binary surfactant mixtures, such as CTAB and sodium deoxycholate (NaDC), has proven successful in producing a variety of MSN shapes (Fig. 11C).129 By varying the ratios of organic additives in the catanionic mixture, reproducible tuning of nanoshapes has been reported. Such studies have been able to predictably produce hexagonal plates, toroidal particles and nanorods.130
In contrast, morphology control is achieved in bioinspired syntheses using chiral polymers such as poly-L-lysine.131 Through the self-assembly of α-helixes during synthesis, flake-like hexagonal silicas can be synthesised (Fig. 11D). Interestingly, morphology can be altered by performing the synthesis under flow (Fig. 11E).132
Finally, macroporous silica microspheres can be introduced by combining covalently-tethered additives with strong surfactant character with TEOS: in the case of APTES additives, reactions with TEOS and water led to the formation of hollow microbeads.104 When the length of the aliphatic tail is extended from 3 carbons to 11 and well mixed, it forms a double-emulsion system with TEOS and water, leading to the formation of macroporous particles.133 Although control over both particle size and macropore diameter have yet to be demonstrated, this interesting new approach shows significant promise for creating a wide variety of 2-dimensional templating behaviour.
2.5. 3-Dimensional structural ordering
A true 3D organisation of silica structures (including pores) in a highly regulated fashion occurs in biological silica as seen in diatoms and sponges. As evident from Fig. 12, biosilica exhibits hierarchical organisation over several length scales.134 Specifically the architectural hierarchy of their porous structures covers three orders of magnitude in size spanning from nm to sub-μm: microscale (of the order of the cell), mesoscale (higher order assemblies of biosilica particles and pores) and nanoscale (primary particles and their aggregates).10,16,135,136 They also exhibit fractal pore architecture (self-replicating structures at multiple length-scales). This degree of structural sophistication is well beyond any of the synthetic silica materials, including those discussed earlier in this review. Further, biosilicification occurs under mild pH and ambient temperatures using naturally occurring silicic acid. In contrast, synthetic approaches to silica and silicates typically involves solution based sol–gel chemistry operating at low temperature (<100 °C) using alkoxysilanes as silica precursors, which are toxic with limited water solubility.9,97,137–139 Significant efforts have been focussed on developing the understanding of the mechanism of biosilica formation such that a reductionist methodology can be applied to take 2D artificial materials to the next level of highly organised 3D structures. As such, the section below provides a brief summary of the learnings from biological silica formation (also noted in ref. 16 and 35), while advanced readers are directed to detailed description of biosilicification.10,140 | ||
| Fig. 12 Electron micrographs of various diatom species, highlighting the diversity and complexity of their structure. Scale bars (in μm) as follows: a and k – 1; b – 5; c, f and l – 10; d – 0.5; e, g, h and j – 2; and i – 50. Reproduced with permission from ref. 10. | ||
The key stages involved in biosilica formation are the cellular uptake of silicon, its intracellular transport, its biochemical transformation and deposition. Each of these stages are under strict spatio-temporal control and involve highly specialised biological molecules to execute the tasks. The transformation and deposition steps are of particular relevance herein as they can offer insights on how to generate complex 3D architectures. Various biochemical mechanisms allow the organisms to uptake and concentrate silicon from highly unsaturated extra-cellular concentration to a (super)saturated, yet stable intra-cellular concentration. This allows the organism to regulate the precursor chemistry and supply, which in turn provides spatio-temporal control over silica formation and deposition.
The functions of biomolecules are much beyond the templating seen in synthetic silicas. These biomolecules provide specific recognition to bind monomeric silicon for transport, form chemical complexes with intermediate silicate oligomers for stabilisation, and catalyse and/or template the formation of biosilica. A number of biomolecules, including proteins have been isolated from biosilica of diatoms, sponges and plants e.g. silaffins (a family of silica forming proteins), SiMat proteins, propylamines, silicalemma-associated proteins (SAPs), silacidin, cingulins, and polysaccharides from diatoms;36,140–145 silicatein and silintaphin proteins from sponges;146–150 and a range of biomolecules from plants151–155 and choanoflagellates.156 Broadly, the functions of these components can be generalised into framework or scaffold, catalytic, and regulatory biomolecules. Orchestrating the timing and location of their expression is crucially important to control biosilica formation and its hierarchical assembly – something beyond synthetic capabilities yet.140
The framework components, which are typically anchored within a membrane, support the catalytic and regulatory components. The catalytic components carry out the chemical transformation of (poly)silicic acids, while the regulatory components control the amount of silica formation for example. Following from previous sections, it is not surprising to note that amine functionalities such as lysine or arginine side groups and propylamines participate in the catalysis of silica as well as the self-assembly of the organic–inorganic. However, there are three main differences between synthetic silicas (discussed above) and biosilica pertaining to the amine chemistry, the use of more than one task-specific biomolecules and a consorted control.
• Firstly, the chemical structure and architecture of amines enables unique activities in catalysing, regulating and templating biosilica formation.142,157 An example of this is the family of silaffin proteins (see Fig. 13) and propylamines associated with diatoms which are polyfunctional. The proteins contain a unique sequence of amino acids and various modifications such as phosphorylation and propylamine functionalisation. These modifications allow control over the self-assembly of these molecules with silicates and other biomolecules.142 The N to N separation by carbons and amine substitution further allows a dynamic (often pH-responsive) protonation and deprotonation of amines, which modulate additive-silicate interactions and ultimately control the catalysis of silicic acids.17
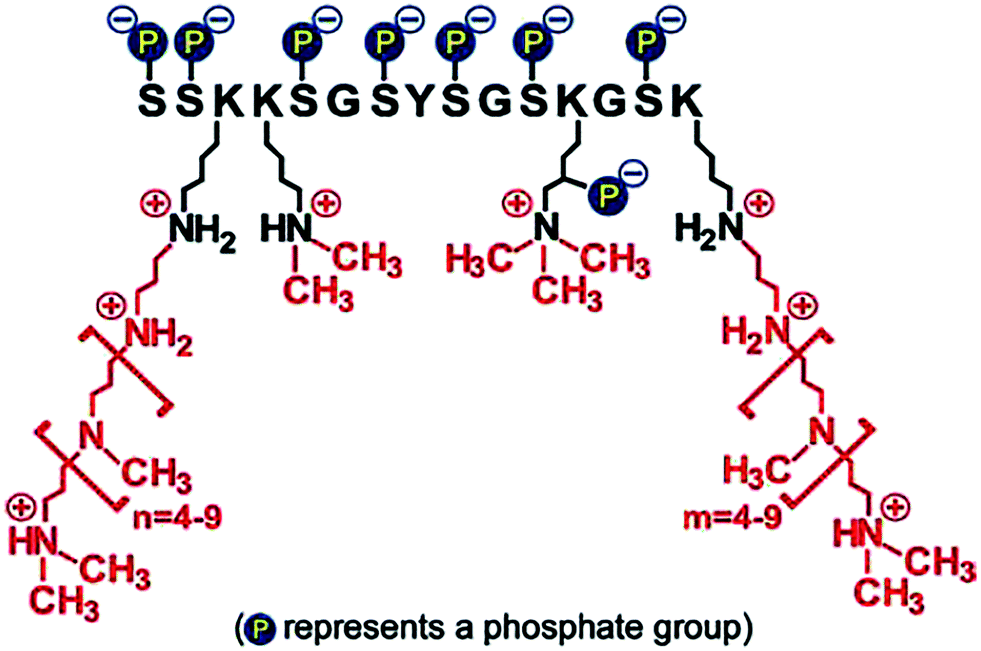 | ||
| Fig. 13 Structure of one of the silaffins, showing three key functionalities. Reproduced with permission from ref. 158. | ||
• Secondly, the presence of more than one biomolecule with specific tasks (self-assembly or regulation). This is very different to the typical in vitro synthesis where one or at the most two additives are used.
• Thirdly, the special control exerted by the framework components as well as other dynamic processes that can move these freshly formed biosilica particles to the desired sites. This level of very complex temporal and spatial control over each step, makes biosilica remarkably distinct.
To demonstrate how these effects combine to form such complex architectures, we present the case study of the silica frustule in Thalassiosira pseudonana (Fig. 12A) where the overall structure takes the shape of a nonporous cylinder ‘girdle’ capped with ‘valves’ of highly ordered hierarchical porosity.159,160 On the largest scales, the separate structure of frustule walls and valves is a result of reaction localisation within the cell – valves are deposited within a specialised silica deposition vesicle (SDV) whose overall shape matches that of the valve structure. Valves are then exocytosed from the diatom, whereupon segments of the girdle begin to form and attach themselves to the extracellular silica structure.145 While the SDV provides a template for the overall structure of the valve, its ordered pore network is formed through complex, synergistic self-assembly of silaffins and other polyamine biomolecules.161 The exact mechanism of the hierarchical pore formation within the SDV is a matter of ongoing research,162 however is it clear that synergistic self-assembly between multiple biomolecules within a confined space is key for the formation of 3-dimensionally ordered frustule valves.
3. A combined perspective on additive function
Given their important applications and fascinating chemistry, each family of silica materials has developed into an independently thriving field of research. Analysis of the reaction conditions and structural properties of additives used to produce different classes of silicas discussed above can lead to a unified correlation between moieties and silica structures they produce (as summarised in Table 1). Hence, we propose design rules for new additives, such that novel silica morphologies exhibiting both desired templating activity and broader properties (e.g. catalytic activity, process compatibility) can be designed.| Silica types → | Bioinspired | Colloids | Zeolites | Surfactant-templated | Biosilica |
|---|---|---|---|---|---|
| Additive features ↓ | |||||
| Structural organisation | None | 0D | 0D | 1D and 2D | 3D |
| No. of functional groups | 2–7, polymeric | 1–4 | 1 | 1–2 | Polymeric |
| Functional group separation (C atoms) | 2–3 C | 0–2 | N/A | 0–3 | 3–4 |
| Level of amine substitution | 1° to 4° | Ammonia to 2° | 4° | 1° to 4° | 1° to 3° |
| Large sidechains | No | No | No | Yes (aliphatic) | Yes (with amines) |
A categorical distinction between these families are the organic additives used during their synthesis. These diverse molecules are selected and studied based on the needs of each individual product and application. In the most elementary of silica-synthesis archetypes, silicas formed with no organic additive provide important insights into the fundamentals of sol–gel growth mechanisms. Bioinspired silica materials, using polyfunctional amine molecules to accelerate synthesis rates and promote coagulation, demonstrate the breadth of available additive structures. At the succeeding level of complexity, small proton-transfer catalysts e.g. ammonia are used for SNS synthesis, providing significant understanding of the fundamentals of silica polymerisation, nucleation and growth. Similarly, small isotropic templates in zeolite synthesis enable the study of how structures can be directed by non-specific, unidirectional interactions. Mesoporous materials templated by polymers and surfactant micelles demonstrate the importance of directional interactions between additives and silica precursors and showcase how such interactions can alter the self-assembly behaviour of templates themselves. Finally, in the most complex of silica syntheses, biomolecules involved in biosilica templating, evidence the capabilities of confinement and polyfunctionality to template silica into incredibly complex patterns.
Interestingly, recent studies show examples of bioinspired additives producing silicas with similar properties to SNSs and mesoporous materials, connecting the mechanistic insight found for those material types. This ability can easily lead to finding the transitions between the behaviour of each of the families defined above, which indicates that the fundamental phenomena underlying each of the above material types are the same. Therefore, a universal theory for amine-assisted silica theory can be developed, and more complex materials designed.
3.1. The universal impacts of additives on silica synthesis
As described above, there are a plethora of different additives and strategies to template silica materials for a variety of pore and particle morphologies. With increasing complexity of additive molecules and template conditions, increasing amount of structural order can be imprinted into the eventual silica matrix. None of the artificial strategies described approaches the complexity imprinted into biosilicas, however. For synthetic templating strategies to become as complex as biosilica templating (or even to understand their formation mechanisms better), a more holistic view of template structure–function relationships is required. The first step towards this lies in analysing what similarities are present between the different templating strategies described before and hence how directly comparable different synthesis methods are.Irrespective of synthesis method, the synthesis methods of all silicas described in this work can be broken down into 3 stages.
1. First, additives and silica precursors are introduced into the reaction mixture together, and its electrolyte environment adjusted as necessary.
2. Second, additives and small silica species (Si1–11) interact, forming additive-silicate complexes which form the basis of templating.
3. Silica species within these complexes then condense (either spontaneously or under some external driving forces), forming structured silica materials.
4. Finally, almost ubiquitously for artificial silica materials, the additive is then removed by an ‘activation’ process.
Fig. 14 graphically demonstrates the similarities between different silica synthesis strategies. Industrial precipitated silica is produced using no structural direction, only silica salts and acids to neutralise them (Fig. 14A). This leads to silica with a range of possible pore sizes depending on how the sol–gel reactions progress, requiring no activation of the pore structure before eventual use (Fig. 14B). Incorporation of polyamine additives to the system at high concentration leads to acceleration of this process (Fig. 14C), however without any clear ordering at circum-neutral pH (where the rate of condensation is maximised, Fig. 14D).
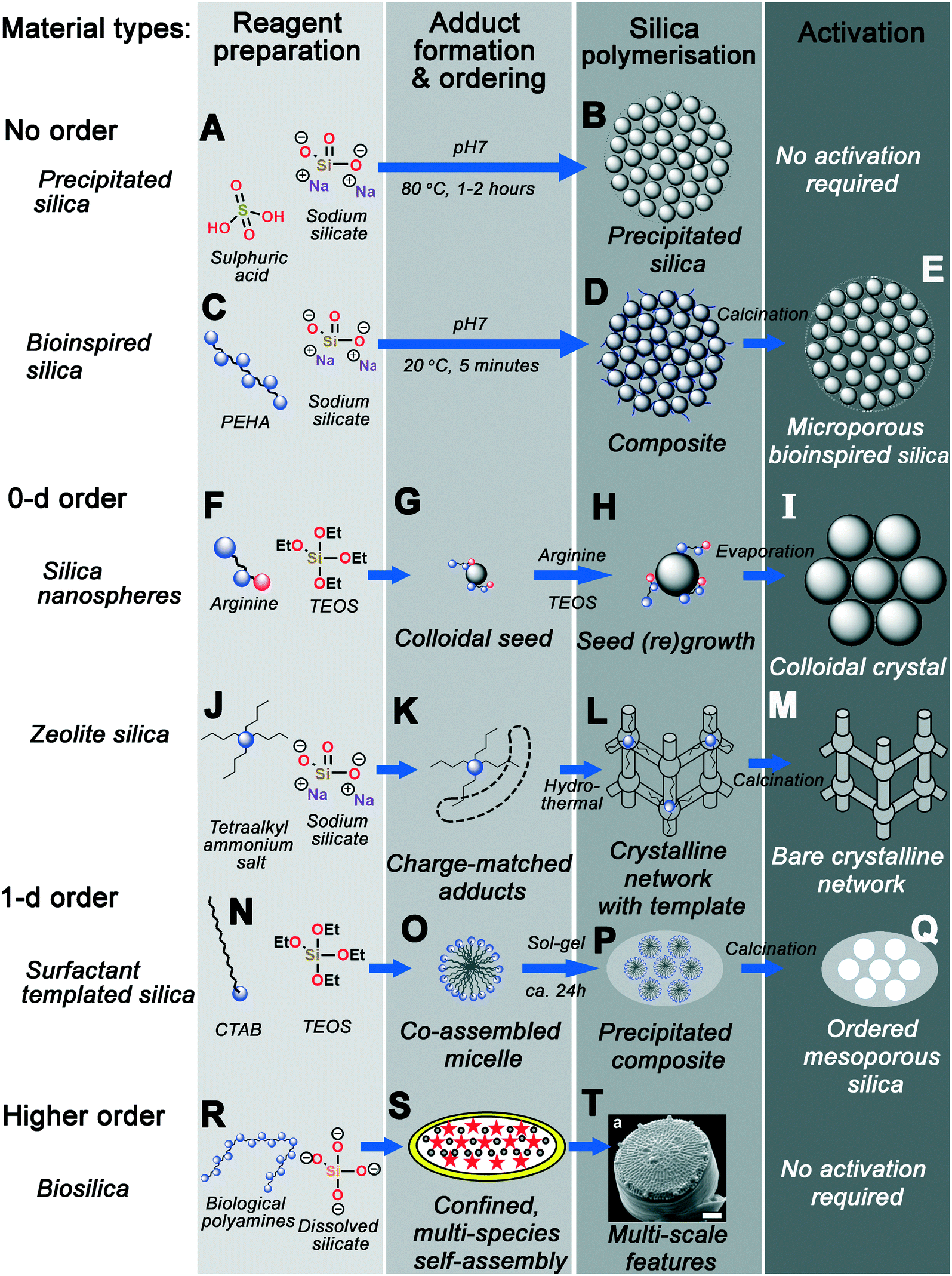 | ||
| Fig. 14 Graphic summary of the formation mechanism of the material types discussed herein. Each process is categorised into the same four overarching stages – reagent preparation, adduct formation and phase ordering, silica polymerisation, and activation. (A and B) precipitated silica synthesised with no additive; (C–E) disordered BIS synthesised using polyamines; (F and G) silica nanospheres produced with ammonia or monopeptides, leading to (H and I) colloidal crystals upon seed regrowth; (J–M) zeolites produced using quaternary ammonium additives; (N–Q) ordered mesoporous silicas synthesised using various surfactants; finally (R–T) heirarchical diatom frustules deposited with biomolecule additives. S and T reproduced from ref. 161 and 10, respectively. | ||
Smaller protonatable additives like ammonia, amino acids, or azamacrocycles leads to controlled nucleation and growth of stable colloidal nanoparticles, depending on a careful balance of TEOS hydrolysis and condensation rates (Fig. 14F). Once homogeneous nucleation has occurred, a monodisperse suspension of stable colloidal silicas can be grown by particle regrowth (Fig. 14G), or ordered crystals seeded from the parent sol by solvent evaporation (Fig. 14H–I).
Substitution of protonatable amine additives with per-substituted ammonium salt additives leads to synthesis of zeolite materials rather than colloidal silicas (Fig. 14J). Rather than controlling the rate of precursor hydrolysis, charge-matched adducts between additives and silicate species form, templating the eventual zeolite pore structure dependent on additive chemical structure (Fig. 14K). Aggregation and ordering of additives–silicate complexes (e.g. under hydrothermal conditions) leads to the formation of crystalline networks with additive-shaped pores (Fig. 14L). These pores are finally revealed through calcination or other activation procedures (Fig. 14M).
Incorporation of amphiphilic moieties e.g. hydrophobic tails into additives permits simultaneous additive self-assembly and additive-silicate adduct formation, imprinting larger pore spaces due to liquid-crystal-like additive co-assembly with silica precursors (Fig. 14N–O). Although a range of surfactant headgroup chemistry is possible, these co-assembled adducts appear to act similarly to those in Fig. 14K – requiring charge-matching interactions for ordered mesophase formation. Sol–gel precipitation of these structures leads to a nonporous composite containing well-ordered organic domains, and again calcination of the organic additive reveals an ordered pore network (Fig. 14P–Q).
Higher-order templating involves the use of mixed additive species as well as other driving forces such as controlled flow or additive confinement (Fig. 14R). For example, in the case of biosilica templating in the diatom T. pseudonana, a mixture of silaffin proteins and long-chain polyamines are postulated to self-assemble inside a silica-deposition vesicle (Fig. 14S).160,161 Polymerisation around this mesophase then occurs, leading to formation of highly patterned silica on a number of length scales (Fig. 14T).
We propose that the mechanistic driving forces shown in Fig. 14 are identical; that is to say specific reaction intermediates identified in section 2 form due to the same fundamental additive-silicate interactions and that differences in the eventual silica structure arise primarily due to variations in the additive molecular structure. As a consequence of our hypothesis, insights from any additive-silica system will be able to provide insight into any other. In the following sections, we justify this hypothesis by highlighting examples of additive-silica systems exhibiting behaviour typically found in other material “families” (as defined in Fig. 1), particularly bioinspired silica (BIS) materials (the system where the most diversity of additive structure has been explored).16 Although within this work BIS materials are classified as providing no structural direction, examples of ‘bioinspired’ techniques and additives can be found throughout: be it the use of peptides51,52,163 and azamacrocycles54–56 to synthesise monodisperse silica nanospheres and colloidal crystals; bio-derived surfactants providing 1d structural direction;13 or flow-induced arrangement of polypeptides into plates.132
From the mechanisms summarised in Fig. 14, 4 key driving forces can be identified:
• Control over relative rates of silica hydrolysis and condensation (hence nucleation).
• Charge-matching silicate-additive adduct formation.
• Self-assembly into liquid-crystal-like mesophases.
• Reaction centre localisation through flow or confinement.
Structural complexity is imprinted into materials when one or more of the above features is present in additive molecules present within the reaction system. When multiple driving forces are present (e.g. in the cases of surfactant additives which charge-match with silicates), or multiple additive species are present, greater structural information can be imprinted. By identifying the 4 key driving forces to ordering regardless of the silica system used, specific moieties connected to each driving force can be identified and hence design rules for systematically introducing more complexity into porous silicas can be made.
3.2. Additive activity – from molecules to moieties
What aspects of each molecule lead to activity or templating? Activity is clearly based around the amine and cationic ammonium moieties, but as can be seen from biosilicas the location of these functional groups on the molecule has a further impact on activity. There have been several studies linking additive molecular structure with activity,17,18,20,21 from which we can infer design rules for future additives.At initial stages of reactions, amine moieties play an important role in facilitating hydrolysis (if present) and condensation of silica precursors. From fundamental computational and NMR-based studies of very early stages of synthesis, a strong correlation between conjugate base concentration and both hydrolysis and condensation rate has been identified.19 Accordingly, lowering the pKa of amine additives can lead to acceleration of silica synthesis (albeit to the detriment of ordered structural direction17). Initial stages of silica condensation proceed with third-order kinetics with respect to [Si], indicating the formation of trimeric silica species from Si1–2 species.41,164,165 On this basis, kinetic investigations into the effect of additive structure on silicate condensation rate have confirmed the positive correlation between conjugate base behaviour and silicate trimerization rates.17
In terms of additive structure, hydrolysis is therefore accelerated through alkylation of amine molecules,17,21,22 additive homologation (i.e. introduction of a larger number of amine moieties),18,41,166 and separation of amine moieties.17,20 Further, reduction of pKa by e.g. using pyridyl additives19 or substituting with electron-donating groups (benzyl or allyl)21 further accelerate silica condensation. We note that even slight structural changes can lead to dramatic increases in catalysis rates – increasing amine separation from 2 bridging carbon atoms to 3, or adding a single methyl group per amine, can lead to large increases in catalysis rates.17 Although few studies have studied the catalytic behaviour of quaternary ammonium additives, it appears that these moieties are even more effective than free-base amine moieties despite the fact that they cannot participate in proton transfer catalysis.21,22 It has been postulated that quaternary ammonium ions instead work by creating areas of higher local [Si] concentration through macroscopic charge-matching effects (coacervation).166
In terms of pore templating, amine moiety separation of more than 6 carbon atoms introduces hydrophobic character into the molecule to induce phase separation behaviour similar to that seen in surfactant additives. This leads to increased pore sizes when used to synthesise BIS, but at the expense of comparatively reduced overall synthesis rates due to the lack of proximate polyfunctionality.135 Increasing hydrophobicity of the templates, by either high homologation or methylation have also shown a tendency to form hollow silica particles. This is due to mesophase formation creating large microdroplets of the additive in solution around which silica shells form, in a parallel to both surfactant-templated and biomineralized silicas.17
Charge-matching-based aggregation between positively charged additives and larger colloidal silica is also important beyond the molecular scale as it can promote or prevent coagulation. Coagulation of colloidal silicas only occurs once interparticle repulsive forces in the form of an electric double layer (EDL) between particles have been overcome. With regards to BIS, the EDL has been shown to be between 0.63 and 0.87 nm in width, corresponding to the length of additives butylenediamine and hexylenediamine. Between these (and the analogous ethylenediamine and its homologue triethylenetetraamine (TETA), which are 0.39 and 1.1 nm in length respectively), there is a step change in observed silica gel times and particle growth rates.20 Additives of this length are able to bridge across the EDL, significantly accelerating the onset of particle coagulation. This has been proposed as the cause behind the reduced reaction times in BIS (and R5-templated biomimetic silica) compared to industrial sol–gel silicas.18
Although ethylenediamine has been shown in ref. 17 to be too short to bridge silica particles' EDL, other studies using this additive have successfully achieved formation of coagulated silica.21,22 These found that increased electropositivity of amine moieties further promotes silica formation despite the inability to bridge the electric double layer,21,22 similar to findings for longer additive chemicals wherein hydrophobicity was correlated with increased activity.17 The apparent contradiction between these studies can be explained by the use of buffers such as phosphate or citrate ions during synthesis.
Phosphate groups, ubiquitous in silaffin biomolecules, are highly important for the self-assembly associated with complex templating in diatoms due to the formation of a coacervate in the silica deposition vesilce.14 Similarly, self-assembly of bioinspired additives around phosphate anions, creating a large ‘liquid precipitate’ around which silica can very easily be precipitated. Beyond enabling short bioinspired additives to bridge the EDL, buffered BIS systems can create hollow silica nanoparticles by using a tri-functional citrate buffer along with the additive PAH; initially, the polymer condenses around the citrate to form a spherical structure (rather than fully elongated as it would be predisposed to be in solution), after which silica precursors can be added to create a shell.167 Interestingly, this is similar to hollow silica particles formed in the absence of counterions wherein highly hydrophobic additives phase separate into stable nanodroplets, creating a similar effect.17
The effects of different additive structures on coagulation of BIS materials are summarised in Fig. 15. In unbuffered systems (Fig. 15A) use of small, positively charged additives such as ethylenediamine lead to stabilisation rather than coagulation of the negatively charged silica colloids, as the additives cannot bridge the electric double layer. Incorporation of multivalent anionic buffers (Fig. 15B) increases the effective length of the additive such that it can bridge the gap. This is equivalent to the activity seen with unbuffered PEHA in Fig. 15C, where longer additives in unbuffered systems naturally bridge the electric double layer, leading to rapid coagulation of BIS. Hence, reports of amine group alkylation lead to similar conclusions for the buffered21,22 and unbuffered17 studies. Similar effects are seen with longer additives that can self-assemble: unbuffered PAH (Fig. 15D) does not self-assemble, producing materials similar to unbuffered PEHA. Incorporation of multivalent anionic buffers leads to coacervation with PAH (Fig. 15E), creating hollow silica spheres.167 Equivalent hollow silica is seen in the unbuffered system if the additive is sufficiently hydrophobic to spontaneously phase-separate (Fig. 15F) e.g. through greater amine–amine separation or high levels of alkylation.
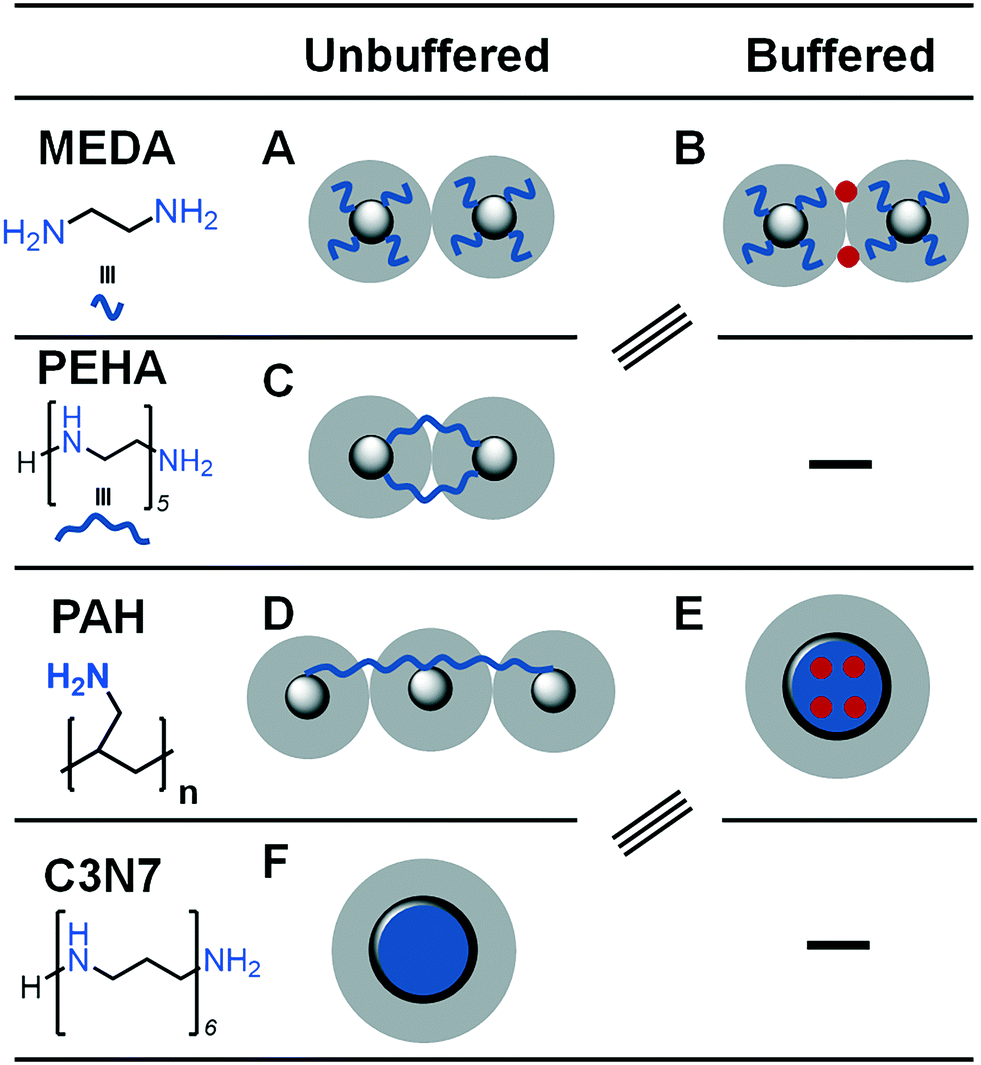 | ||
| Fig. 15 Visualisation of BIS additive self-assembly in the absense (A, C, D and F) and presence (B and E) of inorganic buffer species. Blue lines and circles – amine-containing additives, red circles – buffer ions, dark grey balls – colloidal silica particles, light grey circles – electric double layer around silica. | ||
Fig. 15 highlights the importance of functional group homologation and separation on silica morphology, and how coacervation with anionic buffers leads to further coagulation and self-assembly. In addition to homologation and separation, alkylation of functional groups increases basicity thereby accelerating silica hydrolysis and condensation by lowering the pKa. Peralkylation to create quaternary ammonium functionality leads to both further acceleration of silica condensation and the formation of silicate-additive charge matching adducts. These charge-matching effects can then lead to coacervate mesophase formation with the aid of anionic buffer species, similar to self-assembly in surfactant additive systems. Given this understanding of how specific moieties and additive structural motifs lead to eventual silica templating patterns, bespoke additives can be designed to imprint specific functional properties in silica materials.
3.3. The benefits of a unified perspective
A key inference from the parallel consideration of silica syntheses, as presented throughout this review and summarised in Fig. 14, is that the differences in structure and functionalities of silica materials are chiefly determined by the functional groups of the additives employed to support or catalyse their formation reactions. Developing this correlation between the final material structure and additive functional group, rather than considering the additive as a whole, enables the lines separating the different families of silicas to be narrowed. Existing examples of such a mindset are implicit in the widespread efforts to develop materials that combine properties across different silica families. Therefore, understanding the similarities between their syntheses enables translation between different silica families, from which new additives systems can be designed to fulfil specific functions.Fig. 16 shows examples of how silica synthesis knowledge has already been shared across different silica families, leading to the creation of more complex structural ordering as a result. As each family of materials can be defined by the additive structure typical for each class of materials, the implicit understanding of the similarities between different silicas has already provided promising results in different areas of research. Most importantly, we seek to emphasise the role of organic additives in the design and synthesis of these “crossover” materials.
 | ||
| Fig. 16 Example of silica synthesis methods combining features from more than one category of silica material types, leading to more complex overall structural ordering. | ||
Except for the less common destructive methods, where a preformed zeolite is periodically demetallized to obtain the desired hierarchical porosity,171 mesoporous zeolites are normally achieved via modified surfactant templating. In other words, advances in these materials are derived from advances in the selection and design of the organic additives assisting their synthesis. Such combination methods have produced promising materials such as Ti-MMM-1,172 MTS-5, MTS-8,173 and MTS-9.174 In all cases, the synthesis of the mesoporous zeolites was achieved by mixing an organic surfactant commonly found in mesoporous silica preparations (e.g. CTAB) and a tetra-alkyl ammonium additive used for the synthesis of microporous zeolites (e.g. TMAOH, TPAOH). These additives are carefully added into the same mixture at staggered times with narrowly controlled reaction temperature and pH values.
For the purpose of conferring the products with the desirable properties of both mesoporous and colloidal silicas, their synthesis relies on the combination of their corresponding organic additives. Namely, their long-range uniform porosity is induced and controlled by the same type of surfactants seen in such materials as MCM-41 (e.g., CTAB). Correspondingly, their particle size is controlled in analogous manner to that of Stöber materials, using amine molecules as templates (e.g. triethanolamine).124
The promising in vivo behaviours of MSNs were discovered after mesoporous silica materials encountered several limitations as drug delivery agents, due to their cytotoxicity.178 A great number of nanoparticles have been developed and investigated for biomedical applications, including polymers, ceramics and metals,179 each encountering their own unique set of problems. Particle size is a chiefly determinant factor in their therapeutic and diagnostic applications; if the materials are too small, they may unintentionally diffuse across membranes and into the extracellular matrix, causing issues with unwanted release of their cargo. Conversely, if the particles are too big, they cannot be rejected by the renal system and will bio-accumulate with hazardous results. Therefore, the particle size control capabilities of Stöber processes was an ideal complement to the loading capabilities conferred by mesoporosity. MSNs can be manipulated similarly to stable colloidal silica materials, with their size and porosity being further controlled by particle regrowth, often in the presence of further surfactants, or deposited into ordered colloidal arrays with hierarchical porosity.
For drug delivery applications, the pores of MSNs are filled with drugs or cytotoxins. Their functionalisation capabilities allow the pharmaceutical nanovehicles to be absorbed into targeted cells through endocytosis. By improving cell targeting through porosity, particle size and surface chemistry control, mesoporous silica nanospheres have been pitched as a promising material to be used in revolutionary cancer treatments.180
In the Stöber process described in section 2.2.1, ammonia can be substituted in the reaction by another catalyst. Such versatility is the basis for the introduction of polyamines into the synthesis instead of ammonia. In the case of cyclen and related polyazamacrocycle additives, a significant improvement in control over silica particle was found, for example.56 Furthermore, the incorporation of metal-containing cyclic amines made it possible to synthesise catalytic composites in one step, with the amine essentially acting both as precursor and catalyst.55,56 Other studies have relied on the addition of amino acids into the Stöber reactions. By substituting ammonia for basic amino acids, Yokoi et al. were able to synthesise silica nanospheres below 50 nm without compromising in sphericity or size distribution.51 Such contribution is of great significance to high-tech applications where there is an increasing demand for such specifications. In accordance with the acumen of the present review, such crossover methods essentially rely on using a biomolecule/bioinspired amine as additive or catalyst to an otherwise traditional Stöber process.
Given the problems associated with the use of conventional additives, it is unsurprising that their exemption from zeolite synthesis has long been an objective in zeolite research.182 A great body of research has been devoted to inducing and controlling the polymerisation and growth of such silica nanostructures. By complementing the knowledge of traditional silica precipitation with the effect of composition variables, such as Al2O3 and Na2O contents, researchers have been able to produce industrially demanded zeolites like ZSM-5 without the use of organic additives.183 Such SDA-free zeolites not only avoid the environmental and economic costs of calcination, but their mechanical integrity remains unperturbed by the thermal treatment, which often causes pinholes and cracks in conventional zeolites.184 An unfortunate downside of these synthesis has been their low yields and long crystallisation times. However, more recent developments have incorporated seed surface crystallisation (SSC) mechanisms using small amounts of ZSM-5 and silicalite-1 as seeds to accelerate the formation of zeolite crystals and control the size or micropores.181
3.4. Summary
Although the cases above represent only some limited examples of where lessons from one family of organic-assisted silica is able to influence the science of another, we believe they prove the key relationships between each family, and how unified approaches to their research can handily prevent duplication of effort.An important step in all organic-assisted syntheses is the removal of the organic additive. Therefore, developments in the field of organic additive removal can be shared across material families. Although removal by template deconstruction followed by extraction has been effective for zeolites,77,185 few examples of complete extraction-based removal exists for surfactant-templated silica materials,83,186 by combining surfactants without a permanent charge with acidification. This approach to template removal has inspired a similar extraction approach to additive removal from BIS materials, the first such non-calcination-based approach for these materials.187
Equally, through molecular dynamics simulations of the above materials, modulation of surface charge was identified as the key driving factor in additive extraction from BIS materials, as it was understood that bioinspired additives carry a permanent positive charge due to their multiple pKa values. On the basis of these simulations, further analysis of the surfactant-templated silica system became possible. Molecular dynamics could only reproduce the creation of wormlike porosity in simulations with charged surfactant species, proving that non-permanent charged surfactant species were essential for the templating activity. After backing up with NMR analyses of the reaction mixtures, this BIS-inspired study was therefore able to clarify a 20 year-old postulate of how ‘neutral’ surfactant templating proceeds.99
4. Conclusions and future directions
Amine-assisted silica synthesis methods lead to a large range of porous solids, being found in environments as diverse as hydrocarbon cracking in oil refineries to siliceous cell walls in plankton. The diversity in silica morphology is a result of the structure of the amine additive used during synthesis, which can influence the overall synthesis mechanisms according to 4 key driving forces (summarised schematically in Fig. 17): | ||
| Fig. 17 Schematic representation of the four driving forces identified, which lead to structural organisation at different length scales within silica materials. | ||
1. Controlling rates of initial silica hydrolysis and condensation, leading to controlled particle nucleation and growth.
2. Forming charge-matched adducts between ammonium and silicate ions, leading to control over silica polymerisation sites on a molecular length scale.
3. Additive self-assembly and coacervation into separate mesophases, enabling silica polymerisation at the interface hence control on a macromolecular length scale.
4. Confinement of reaction mixtures into vesicles or by flow, leading to control on larger length scales.
Employing one or more of the above driving forces can lead to the synthesis of materials with specific structural order, the complexity of which is dependent on how many of the 4 mechanisms are present in the reaction system.
An important consequence of identifying these 4 driving forces for structural organisation during silica synthesis is that none are specific to a certain additive molecular structure. Indeed, throughout this work we identified several reaction systems (summarised in Fig. 1) where the same driving force is present in combination with significantly different additive molecules. An excellent example of this is for the ‘bioinspired’ synthesis method, which employs aqueous and circum-neutral reaction conditions along with polyamine additives (in contrast to alcohol/water co-solvent systems at high or low pH values). Although normally producing disordered silicas, recent reports have shown bioinspired silica syntheses exhibiting 3 of the above 4 driving forces.
We believe that explicit investigation into these driving forces as a function of additive structure will greatly enhance the range of artificial silica morphologies which are synthetically available, and further lead to significantly more complex artificial silica morphologies.
4.1. Computational investigations into proton-transfer catalysis and silicate complexing
Ab initio studies have demonstrated that the first driving force can be modulated by controlling the basicity of amine moieties in the organic additives.19 By extending these to wider libraries of additive compounds such as those studied in ref. 17 and 20–22, comprehensive structure–function relationships between additive structure and condensation driving forces can be made. Similarly, exploring the ability of bioinspired additives to perform the second driving force can be achieved by mirroring recent ab initio molecular dynamics76 and in situ NMR74 studies of zeolite precursor formation. By extending the range of additives studied to polyfunctional structures, more complex templating behaviour can be understood and consequently designed into novel silica materials.4.2. Investigation of silica synthesis under controlled flow fields
To control silica morphology at larger length scales, investigating the influence of controlled flow fields on additive self-assembly,132 silica formation within dispersed phases of controlled morphology or confinement will enable artificial reproduction of conditions within diatom silica-deposition vesicles. For example, performing silica syntheses in Taylor–Couette flow cells as a function of both shear rate and additive molecular structure would enable identification of flow-dependence during silica assembly. In addition to providing further mechanistic insight into later stages of the sol–gel process, understanding of how morphology can be controlled through physical effects such as shear rate will provide greater control over silica morphology and enable more complex materials to be designed.4.3. Meta-analysis of previously published silica synthesis methods
Despite the relative dearth of scientific studies connecting additive structure and reaction conditions with eventual silica morphology and structural organisation, the design and synthesis of different silica materials remains a thriving research field. Accordingly, collation of existing studies together and subsequent analysis of the materials produced can provide significant insight into this relationship (as can be seen through existing systematic studies of zeolite synthesis composition in ref. 188). By gathering previous reports of silica synthesis for other material families, the extent to which the parameter space has been explored can be determined. As a result, more universal design rules for different silica materials can be produced, for example by extending the empirical models for SNS particle size developed in ref. 50.4.4. Lifecycle and technoeconomic analyses of silica synthesis methods as a function of additive structure
Finally, in order to best enable translation of silica synthesis methods to larger scales, the environmental and technoeconomic impact of additive molecular structure and reaction conditions must be characterised and understood. As a pertinent example, common additives used in bioinspired silica synthesis such as pentaethylenehexamine have seen wide industrial usage as curing agents in epoxy resins.189–191 Although on the face of it this previous industrial usage would be an advantage, these chemicals are being phased out due to their high environmental toxicity and therefore may represent a barrier to scale-up and wider implementation. By employing thorough environmental impact analysis and technoeconomic assessment, additive choice can be made as both a function of feasibility as well as silica structure, enabling faster and more efficient technology transfer.Conflicts of interest
The authors declare no financial conflicts of interest.Acknowledgements
We thank EPSRC (projects EP/P006892/1 and EP/R025983/1) and the University of Sheffield for the financial support. JRHM thanks University College London and the ERC (grant agreement No 648283 “GROWMOF”) for their financial support.References
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815 RSC.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- S. K. Natarajan and S. Selvaraj, RSC Adv., 2014, 4, 14328–14334 RSC.
- A. Rahikkala, S. A. P. Pereira, P. Figueiredo, M. L. C. Passos, A. R. T. S. Araújo and M. L. M. F. S. Saraiva, Adv. Biosyst., 2018, 2, 1800020 CrossRef.
- R. Narayan, U. Y. Nayak, A. M. Raichur and S. Garg, Pharmaceutics, 2018, 10, 1–49 CrossRef.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277 RSC.
- S. Shylesh, A. Wagner, A. Seifert, S. Ernst and W. R. Thiel, Chemistry, 2009, 15, 7052–7062 CrossRef CAS.
- C. J. Brinker and G. W. Sherer, Sol-gel science: the physics and chemistry of sol–gel processing, Elsevier, 1990 Search PubMed.
- M. Hildebrand, Chem. Rev., 2008, 108, 4855–4874 CrossRef CAS.
- F. Schüth, Angew. Chem., Int. Ed., 2003, 42, 3604–3622 CrossRef.
- O. V. Shvets, N. Kasian, A. Zukal, J. Pinkas and J. Čejka, Chem. Mater., 2010, 22, 3482–3495 CrossRef CAS.
- C. Gérardin, J. Reboul, M. Bonne and B. Lebeau, Chem. Soc. Rev., 2013, 42, 4217–4255 RSC.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS.
- J. I. B. Janairo, T. Sakaguchi, K. Mine, R. Kamada and K. Sakaguchi, Protein Pept. Lett., 2018, 25, 4–14 CrossRef CAS.
- S. V. Patwardhan, Chem. Commun., 2011, 47, 7567–7582 RSC.
- D. J. Belton, S. V. Patwardhan, V. V. Annenkov, E. N. Danilovtseva and C. C. Perry, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5963–5968 CrossRef CAS.
- D. J. Belton, S. V. Patwardhan and C. C. Perry, J. Mater. Chem., 2005, 15, 4629 RSC.
- K. M. Delak and N. Sahai, Chem. Mater., 2005, 17, 4272 CrossRef CAS.
- D. J. Belton, S. V. Patwardhan and C. C. Perry, Chem. Commun., 2005, 3475–3477 RSC.
- D. B. Robinson, J. L. Rognlien, C. A. Bauer and B. A. Simmons, J. Mater. Chem., 2007, 17, 2113 RSC.
- A. Jantschke, K. Spinde and E. Brunner, Beilstein J. Nanotechnol., 2014, 5, 2026–2035 CrossRef.
- B. M. Szyja, P. Vassilev, T. T. Trinh, R. A. Van Santen and E. J. M. Hensen, Microporous Mesoporous Mater., 2011, 146, 82–87 CrossRef CAS.
- A. Issa and A. Luyt, Polymer, 2019, 11, 537 Search PubMed.
- R. K. Iler, The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica, Wiley, 1979 Search PubMed.
- O. Florke, H. A. Graetsch, F. Brunk, S. Paschen, H. H. E. Bergna, W. O. Roberts, W. A. Welsh, D. Chapman, M. Ettlinger, D. Kerner, M. Maier, W. Meon, R. Schmoll, H. Gies, D. Schiffmann, O. W. Flörke, H. A. Graetsch, F. Brunk, L. Benda, S. Paschen, H. H. E. Bergna, W. O. Roberts, W. A. Welsh, C. Libanati, M. Ettlinger, D. Kerner, M. Maier, W. Meon, R. Schmoll, H. Gies and D. Schiffmann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 6th edn, 2008, pp. 22698–22786 Search PubMed.
- H. Hu, H. Hou, Z. He and B. Wang, Phys. Chem. Chem. Phys., 2013, 15, 15027–15032 RSC.
- H. Henschel, A. M. Schneider and M. H. Prosenc, Chem. Mater., 2010, 22, 5105–5111 CrossRef CAS.
- A. A. Issa, M. El-Azazy and A. S. Luyt, Chem. Phys., 2020, 530, 110642 CrossRef CAS.
- J. L. Trompette, J. Phys. Chem. B, 2017, 121, 5654–5659 CrossRef CAS.
- M. Van Der Linden, B. O. Conchúir, E. Spigone, A. Niranjan, A. Zaccone and P. Cicuta, J. Phys. Chem. Lett., 2015, 6, 2881–2887 CrossRef CAS.
- T. Oncsik, G. Trefalt, M. Borkovec and I. Szilagyi, Langmuir, 2015, 31, 3799–3807 CrossRef CAS.
- G. A. Icopini, S. L. Brantley and P. J. Heaney, Geochim. Cosmochim. Acta, 2005, 69, 293–303 CrossRef CAS.
- S. V. Patwardhan, J. R. H. Manning and M. Chiacchia, Curr. Opin. Green Sustain. Chem., 2018, 12, 110–116 CrossRef.
- S. V. Patwardhan and S. S. Staniland, Green Nanomaterials, IOP Publishing, 2019 Search PubMed.
- N. Kroger, R. Deutzmann and M. Sumper, Science, 1999, 286, 1129–1132 CrossRef CAS.
- S. V. Patwardhan, N. Mukherjee and S. J. Clarson, J. Inorg. Organomet. Polym., 2002, 11, 193–198 CrossRef.
- S. V. Patwardhan, N. Mukherjee and S. J. Clarson, Silicon Chem., 2002, 1, 47–54 CrossRef CAS.
- T. Coradin, O. Durupthy and J. Livage, Langmuir, 2002, 18, 2331–2336 CrossRef CAS.
- A. Sugawara-Narutaki, S. Tsuboike, Y. Oda, A. Shimojima, K. B. Landenberger, T. Okubo and S. Aoshima, Langmuir, 2019, 35, 10846–10854 CrossRef CAS.
- D. J. Belton, G. Paine, S. V. Patwardhan and C. C. Perry, J. Mater. Chem., 2004, 14, 2231 RSC.
- A. Bernecker, R. Wieneke, R. Riedel, M. Seibt, A. Geyer and C. Steinem, J. Am. Chem. Soc., 2010, 132, 1023–1031 CrossRef CAS.
- J. N. Cha, G. D. Stucky, D. E. Morse and T. J. Deming, Nature, 2000, 403, 289–292 CrossRef CAS.
- T. Coradin, C. Roux, J. Livage, C. Roux and J. Livage, J. Mater. Chem., 2002, 12, 1242–1244 RSC.
- Q. Sun, E. G. Vrieling, R. A. van Santen and N. A. J. M. Sommerdijk, Curr. Opin. Solid State Mater. Sci., 2004, 8, 111–120 CrossRef CAS.
- J. Li, L. Xu, B. Yang, Z. Bao, W. Pan and S. Li, Mater. Sci. Eng., C, 2015, 55, 367–372 CrossRef CAS.
- Y. Han, Z. Lu, Z. Teng, J. Liang, Z. Guo, D. Wang, M. Y. Han and W. Yang, Langmuir, 2017, 33, 5879–5890 CrossRef CAS.
- S.-L. Chen, P. Dong, G.-H. Yang and J.-J. Yang, Ind. Eng. Chem. Res., 1996, 35, 4487–4493 CrossRef CAS.
- Y. Xu, X. Sun, D. Wu, Y. Sun, Y. Yang, H. Yuan, F. Deng and Z. Wu, J. Solution Chem., 2007, 36, 327–344 CrossRef CAS.
- D. Qi, C. Lin, H. Zhao, H. Liu and T. Lü, J. Dispersion Sci. Technol., 2017, 38, 70–74 CrossRef CAS.
- T. Yokoi, J. Wakabayashi, Y. Otsuka, W. Fan, M. Iwama, R. Watanabe, K. Aramaki, A. Shimojima, T. Tatsumi and T. Okubo, Chem. Mater., 2009, 21, 3719–3729 CrossRef CAS.
- J. R. H. Manning, Sustainable Chemistry & Process Engineering of Bioinspired Silica Materials, PhD Thesis, University of Sheffield, 2019 Search PubMed.
- C. C. M. C. Carcouët, M. W. P. Van De Put, B. Mezari, P. C. M. M. Magusin, J. Laven, P. H. H. Bomans, H. Friedrich, A. C. C. Esteves, N. A. J. M. Sommerdijk, R. A. T. M. Van Benthem and G. De With, Nano Lett., 2014, 14, 1433–1438 CrossRef.
- G. E. Tilburey, T. J. Blundell, S. V. Patwardhan, S. P. Argent and C. C. Perry, Dalton Trans., 2019, 48, 15470–15479 RSC.
- S. Masse, G. Laurent, F. Chuburu, C. Cadiou, I. Déchamps and T. Coradin, Langmuir, 2008, 24, 4026–4031 CrossRef CAS.
- S. Masse, G. Laurent and T. Coradin, Phys. Chem. Chem. Phys., 2009, 11, 10204–10210 RSC.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS.
- M. E. Davis, Chem. Mater., 2014, 26, 239–245 CrossRef CAS.
- H. Gies and B. Marler, Zeolites, 1992, 12, 42–49 CrossRef CAS.
- M. Dusselier and M. E. Davis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS.
- J. Grand, H. Awala and S. Mintova, CrystEngComm, 2016, 18, 650–664 RSC.
- J. M. Fedeyko, D. G. Vlachos and R. F. Lobo, Langmuir, 2005, 21, 5197–5206 CrossRef CAS.
- C. E. A. Kirschhock, R. Ravishankar, F. Verspeurt, P. J. Grobet, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 1999, 103, 4965–4971 CrossRef CAS.
- C. T. G. Knight, S. D. Kinrade, C. E. A. Kirschhock, R. Ravishankar, F. Verspeurt, P. J. Grobet, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 2002, 106, 3329–3334 CrossRef CAS.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- D. W. Lewis, G. Sankar, J. K. Wyles, J. M. Thomas, C. R. A. Callow and D. J. Willock, Angew. Chem., Int. Ed. Engl., 1997, 36, 2675–2677 CrossRef CAS.
- L. Gómez-Hortigüela and M. Á. Camblor, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Springer International Publishing, Cham, 2018, pp. 1–41 Search PubMed.
- S. I. Zones, A. W. Burton, G. S. Lee and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 9066–9079 CrossRef CAS.
- Y. Kubota, M. M. Helmkamp, S. I. Zones and M. E. Davis, Microporous Mater., 1996, 6, 213–229 CrossRef CAS.
- S. L. Burkett and M. E. Davis, Chem. Mater., 1995, 7, 920–928 CrossRef CAS.
- S. L. Burkett and M. E. Davis, J. Phys. Chem., 1994, 98, 4647–4653 CrossRef CAS.
- T. Verstraelen, B. M. Szyja, D. Lesthaeghe, R. Declerck, V. Van Speybroeck, M. Waroquier, A. P. J. Jansen, A. Aerts, L. R. A. Follens, J. A. Martens, C. E. A. Kirschhock and R. A. Van Santen, Top. Catal., 2009, 52, 1261–1271 CrossRef CAS.
- E. J. J. Groenen, A. G. T. G. Kortbeek, M. Mackay and O. Sudmeijer, Zeolites, 1986, 6, 403–411 CrossRef CAS.
- Y. Chen, N. M. Washton, R. P. Young, A. J. Karkamkar, J. J. De Yoreo and K. T. Mueller, Phys. Chem. Chem. Phys., 2019, 21, 4717–4720 RSC.
- S. Caratzoulas, D. G. Vlachos and M. Tsapatsis, J. Phys. Chem. B, 2005, 109, 10429–10434 CrossRef CAS.
- T. T. Trinh, K. Q. Tran, X. Q. Zhang, R. A. Van Santen and E. J. Meijer, Phys. Chem. Chem. Phys., 2015, 17, 21810–21818 RSC.
- H. Lee, S. I. Zones and M. E. Davis, Microporous Mesoporous Mater., 2006, 88, 266–274 CrossRef CAS.
- F. Rey and J. Simancas, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Springer International Publishing, Cham, 2018, pp. 103–138 Search PubMed.
- E. J. P. Feijen, K. De Vadder, M. H. Bosschaerts, J. L. Lievens, J. A. Martens, P. J. Grobet and P. A. Jacobs, J. Am. Chem. Soc., 1994, 116, 2950–2957 CrossRef CAS.
- J. Patarin, Angew. Chem., Int. Ed., 2004, 43, 3878–3880 CrossRef CAS.
- A. I. Lupulescu, W. Qin and J. D. Rimer, Langmuir, 2016, 32, 11888–11898 CrossRef CAS.
- W. Qin, A. Agarwal, M. K. Choudhary, J. C. Palmer and J. D. Rimer, Chem. Mater., 2019, 31, 3228–3238 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Chem. Mater., 1996, 8, 2068–2079 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 2015, 1–22 CrossRef.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson and E. W. Sheppard, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge and W. J. Roth, Chem. Soc. Rev., 2013, 42, 3663–3670 RSC.
- C. Vautier-Giongo and H. O. Pastore, J. Colloid Interface Sci., 2006, 299, 874–882 CrossRef CAS.
- S. C. Chien, G. Pérez-Sánchez, J. R. B. Gomes, M. N. D. S. Cordeiro, M. Jorge, S. M. Auerbach and P. A. Monson, J. Phys. Chem. C, 2017, 121, 4564–4575 CrossRef CAS.
- M. Jorge, A. W. Milne, O. N. Sobek, A. Centi, G. Pérez-Sánchez and J. R. B. B. Gomes, Mol. Simul., 2018, 44, 435–452 CrossRef CAS.
- A. E. Palmqvist, Curr. Opin. Colloid Interface Sci., 2003, 8, 145–155 CrossRef CAS.
- N. Baccile, G. Laurent, C. Bonhomme, P. Innocenzi and F. Babonneau, Chem. Mater., 2007, 19, 1343–1354 CrossRef CAS.
- R. E. Morsi and R. S. Mohamed, R. Soc. Open Sci., 2018, 5, 172021 CrossRef.
- F. Michaux, N. Baccile, M. Impéror-Clerc, L. Malfatti, N. Folliet, C. Gervais, S. Manet, F. Meneau, J. S. Pedersen and F. Babonneau, Langmuir, 2012, 28, 17477–17493 CrossRef CAS.
- A. F. P. de Campos, A. R. O. Ferreira, L. L. da Silva, P. P. M. Neto and D. Cardoso, Catal. Today, 2020, 344, 41–51 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schüth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- Q. Huo, D. I. Margolese and G. D. Stucky, Chem. Mater., 1996, 8, 1147–1160 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CrossRef CAS.
- A. Centi, J. R. H. Manning, V. Srivastava, S. Van Meurs, S. V. Patwardhan and M. Jorge, Mater. Horiz., 2019, 6, 1027–1033 RSC.
- C. Drummond, R. McCann and S. V. Patwardhan, Chem. Eng. J., 2014, 244, 483–492 CrossRef CAS.
- S. K. Jana, A. Mochizuki and S. Namba, Catal. Surv. Asia, 2004, 8, 1–13 CrossRef CAS.
- S. Zhang, C. Chen and W. S. Ahn, Curr. Opin. Green Sustain. Chem., 2019, 16, 26–32 CrossRef.
- Y. Kaneko, N. Iyi, T. Matsumoto, K. Fujii, K. Kurashima and T. Fujita, J. Mater. Chem., 2003, 13, 2058–2060 RSC.
- X. Yang, N. Zhao, Q. Zhou, Z. Wang, C. Duan, C. Cai, X. Zhang and J. Xu, J. Mater. Chem., 2012, 22, 18010–18017 RSC.
- J. Alauzun, E. Besson, A. Mehdi, C. Reyé and R. J. P. Corriu, Chem. Mater., 2008, 20, 503–513 CrossRef CAS.
- J. Alauzun, A. Mehdi, C. Reyé and R. J. P. Corriu, J. Am. Chem. Soc., 2005, 127, 11204–11205 CrossRef CAS.
- R. Besnard, G. Arrachart, J. Cambedouzou and S. Pellet-Rostaing, Langmuir, 2016, 32, 4624–4634 CrossRef CAS.
- R. Besnard, J. Cambedouzou, G. Arrachart, O. Diat and S. Pellet-Rostaing, Langmuir, 2013, 29, 10368–10375 CrossRef CAS.
- A. W. Burton, S. I. Zones and S. Elomari, Curr. Opin. Colloid Interface Sci., 2005, 10, 211–219 CrossRef CAS.
- P. S. Metkar, V. Balakotaiah and M. P. Harold, Chem. Eng. Sci., 2011, 66, 5192–5203 CrossRef CAS.
- Y. Wei, T. E. Parmentier, K. P. De Jong and J. Zečević, Chem. Soc. Rev., 2015, 44, 7234–7261 RSC.
- A. F. Sierra-Salazar, A. Ayral, T. Chave, V. Hulea, S. I. Nikitenko, S. Abate, S. Perathoner and P. Lacroix-Desmazes, Stud. Surf. Sci. Catal., 2019, 178, 377–397 CrossRef CAS.
- Z. Y. Yuan and B. L. Su, J. Mater. Chem., 2006, 16, 663–677 RSC.
- Z. Zhang and I. Melián-Cabrera, J. Phys. Chem. C, 2014, 118, 28689–28698 CrossRef CAS.
- T. Sen, G. J. T. Tiddy, J. L. Casci and M. W. Anderson, Angew. Chem., Int. Ed., 2003, 42, 4649–4653 CrossRef CAS.
- M. Weyland, P. A. Midgley and J. M. Thomas, J. Phys. Chem. B, 2001, 105, 7882–7886 CrossRef CAS.
- W. Wang, S. Schlabach, S.-J. Reich, A. Svidrytski, D. Hlushkou, D. Stoeckel, A. Höltzel, U. Tallarek and C. Kübel, Microsc. Microanal., 2019, 25, 406–407 CrossRef.
- M. W. Anderson, S. M. Holmes, N. Hanif and C. S. Cundy, Angew. Chem., Int. Ed., 2000, 39, 2707–2710 CrossRef CAS.
- O. Sel, D. Kuang, M. Thommes and B. M. Smarsly, Langmuir, 2006, 22, 2311–2322 CrossRef CAS.
- A. A. Issa, M. S. Elazazy and A. S. Luyt, Int. J. Chem. Kinet., 2018, 50, 846–855 CrossRef CAS.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605–612 CrossRef.
- J. G. Verkade, Acc. Chem. Res., 1993, 26, 483–489 CrossRef CAS.
- S. Cabrera, J. El Haskouri, C. Guillem, J. Latorre, A. Beltrán-Porter, D. Beltrán-Porter, M. D. Marcos and P. Amorós, Solid State Sci., 2000, 2, 405–420 CrossRef CAS.
- Y. S. Lin, K. R. Hurley and C. L. Haynes, J. Phys. Chem. Lett., 2012, 3, 364–374 CrossRef CAS.
- Y. Ding, H. Dan, X. Lu, Y. Wu, S. Yuan and X. Mao, Mater. Chem. Phys., 2014, 148, 17–20 CrossRef CAS.
- V. Alfredsson and H. Wennerström, Acc. Chem. Res., 2015, 48, 1891–1900 CrossRef CAS.
- C. Yu, J. Fan, B. Tian and D. Zhao, Chem. Mater., 2004, 16, 889–898 CrossRef CAS.
- P. Linton, J. C. Hernandez-Garrido, P. A. Midgley, H. Wennerström and V. Alfredsson, Phys. Chem. Chem. Phys., 2009, 11, 10973–10982 RSC.
- L. Travaglini and L. De Cola, Chem. Mater., 2018, 30, 4168–4175 CrossRef CAS.
- L. Travaglini, P. Picchetti, A. Del Giudice, L. Galantini and L. De Cola, Microporous Mesoporous Mater., 2019, 279, 423–431 CrossRef CAS.
- S. V. Patwardhan, R. Maheshwari, N. Mukherjee, K. L. Kiick and S. J. Clarson, Biomacromolecules, 2006, 7, 491–497 CrossRef CAS.
- S. V. Patwardhan, N. Mukherjee, M. Steinitz-Kannan and S. J. Clarson, Chem. Commun., 2003, 1122–1123 RSC.
- R. Besnard, G. Arrachart, J. Cambedouzou and S. Pellet-Rostaing, J. Sol-Gel Sci. Technol., 2017, 81, 452–467 CrossRef CAS.
- C. C. Perry, in Biomineralization: Chemical and Biochemical Perspectives, ed. S. Mann, J. M. Webb and R. J. P. Williams, VCH, 1989, pp. 223–257 Search PubMed.
- E. G. Vrieling, T. P. M. Beelen, Q. Sun, S. Hazelaar, R. A. Van Santen and W. W. C. Gieskes, J. Mater. Chem., 2004, 14, 1970–1975 RSC.
- W. E. G. Müller, Silicon biomineralization: biology, biochemistry, molecular biology, biotechnology, 2003 Search PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. G. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- M. Hildebrand, S. J. L. Lerch and R. P. Shrestha, Front. Mar. Sci., 2018, 5, 1–19 CrossRef.
- N. Kröger, R. Deutzmann, C. Bergsdorf and M. Sumper, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14133–14138 CrossRef.
- N. Kröger, S. Lorenz, E. Brunner and M. Sumper, Science, 2002, 298, 584–586 CrossRef.
- N. Poulsen, M. Sumper and N. Kröger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12075–12080 CrossRef CAS.
- N. Poulsen and N. Kröger, J. Biol. Chem., 2004, 279, 42993–42999 CrossRef CAS.
- A. Scheffel, N. Poulsen, S. Shian and N. Kröger, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3175–3180 CrossRef CAS.
- K. A. S. Himizu, J. E. C. Ha and G. A. D. S. Tucky, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6234–6238 CrossRef.
- J. N. Cha, K. Shimizu, Y. Zhou, S. C. Christiansen, B. F. Chmelka, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 361–365 CrossRef CAS.
- Y. Zhou, K. Shimizu, J. N. Cha, G. D. Stucky and D. E. Morse, Angew. Chem., Int. Ed., 1999, 38, 779–782 CrossRef.
- M. Rina, C. Pozidis, K. Mavromatis, M. Tzanodaskalaki, M. Kokkinidis and V. Bouriotis, Eur. J. Biochem., 2000, 267, 1230–1238 CrossRef CAS.
- H. C. Schröder, V. A. Grebenjuk, X. Wang and W. E. G. Müller, Bioinspiration Biomimetics, 2016, 11, 041002 CrossRef.
- C. C. Harrison, Phytochemistry, 1996, 41, 37–42 CrossRef CAS.
- C. C. Perry and T. Keeling-Tucker, Chem. Commun., 1998, 2587–2588 RSC.
- C. C. Perry and T. Keeling-Tucker, Colloid Polym. Sci., 2003, 281, 652–664 CrossRef CAS.
- H. A. Currie and C. C. Perry, Ann. Bot., 2007, 100, 1383–1389 CrossRef CAS.
- H. A. Currie and C. C. Perry, Phytochemistry, 2009, 70, 2089–2095 CrossRef CAS.
- N. Gong, M. Wiens, H. C. Schröder, E. Mugnaioli, U. Kolb and W. E. G. Müller, J. Exp. Biol., 2010, 213, 3575–3585 CrossRef CAS.
- C. Gröger, K. Lutz and E. Brunner, Cell Biochem. Biophys., 2008, 50, 23–39 CrossRef.
- M. Sumper and N. Kröger, J. Mater. Chem., 2004, 14, 2059–2065 RSC.
- N. Poulsen, A. Scheffel, V. C. Sheppard, P. M. Chesley and N. Kro
![[g with combining umlaut]](https://www.rsc.org/images/entities/char_0067_0308.gif) er, J. Biol. Chem., 2013, 288, 20100–20109 CrossRef CAS.
er, J. Biol. Chem., 2013, 288, 20100–20109 CrossRef CAS. - N. Kröger and N. Poulsen, Annu. Rev. Genet., 2008, 42, 83–107 CrossRef.
- N. Kröger and N. Poulsen, Handb. Biominer.: Biol. Aspects Struct. Form., 2008, 1, 43–58 Search PubMed.
- C. Heintze, P. Formanek, D. Pohl, J. Hauptstein, B. Rellinghaus and N. Kröger, BMC Mater., 2020, 2, 11 CrossRef.
- T. Yokoi, J. Jpn. Pet. Inst., 2012, 55, 13–26 CrossRef CAS.
- C. C. Harrison and N. Loton, J. Chem. Soc., Faraday Trans., 1995, 91, 4287–4297 RSC.
- D. J. Belton, O. Deschaume, S. V. Patwardhan and C. C. Perry, J. Phys. Chem. B, 2010, 114, 9947–9955 CrossRef CAS.
- T. Coradin, O. Durupthy and J. Livage, Langmuir, 2002, 18, 2331–2336 CrossRef CAS.
- S. B. Kadali, N. Soultanidis and M. S. Wong, Top. Catal., 2008, 49, 251–258 CrossRef CAS.
- C. Perego and R. Millinib, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- J. Vernimmen, M. Guidotti, J. Silvestre-Albero, E. O. Jardim, M. Mertens, O. I. Lebedev, G. Van Tendeloo, R. Psaro, F. Rodríguez-Reinoso, V. Meynen and P. Cool, Langmuir, 2011, 27, 3618–3625 CrossRef CAS.
- R. Chal, C. Gérardin, M. Bulut and S. VanDonk, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- R. H. P. R. Poladi and C. C. Landry, Microporous Mesoporous Mater., 2002, 52, 11–18 CrossRef CAS.
- K. Lin, Z. Sun, S. Lin, D. Jiang and F. S. Xiao, Microporous Mesoporous Mater., 2004, 72, 193–201 CrossRef CAS.
- X. Meng, D. Li, X. Yang, Y. Yu, S. Wu, Y. Han, Q. Yang, D. Jiang and F. S. Xiao, J. Phys. Chem. B, 2003, 107, 8972–8980 CrossRef CAS.
- S. H. Wu, Y. S. Lin, Y. Hung, Y. H. Chou, Y. H. Hsu, C. Chang and C. Y. Mou, ChemBioChem, 2008, 9, 53–57 CrossRef CAS.
- Q. He, Z. Zhang, F. Gao, Y. Li and J. Shi, Small, 2011, 7, 271–280 CrossRef CAS.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS.
- T. Heikkilä, H. A. Santos, N. Kumar, D. Y. Murzin, J. Salonen, T. Laaksonen, L. Peltonen, J. Hirvonen and V. P. Lehto, Eur. J. Pharm. Biopharm., 2010, 74, 483–494 CrossRef.
- N. Sanvicens and M. P. Marco, Trends Biotechnol., 2008, 26, 425–433 CrossRef CAS.
- I. Slowing, B. G. Trewyn and V. S. Y. Lin, J. Am. Chem. Soc., 2006, 128, 14792–14793 CrossRef CAS.
- N. Ren, J. Bronić, B. Subotić, X. C. Lv, Z. J. Yang and Y. Tang, Microporous Mesoporous Mater., 2011, 139, 197–206 CrossRef CAS.
- R. W. Grose and E. M. Flanigen, US4257885A, 1979.
- H. Kalipcilar and A. Culfaz, Cryst. Res. Technol., 2001, 36, 1197–1207 CrossRef CAS.
- M. Pan and Y. S. Lin, Microporous Mesoporous Mater., 2001, 43, 319–327 CrossRef CAS.
- H. Lee, S. I. Zones and M. E. Davis, J. Phys. Chem. B, 2005, 109, 2187–2191 CrossRef CAS.
- K. Cassiers, P. Van Der Voort and E. F. Vansant, Chem. Commun., 2000, 2489–2490 RSC.
- J. R. H. Manning, T. W. S. Yip, A. Centi, M. Jorge and S. V. Patwardhan, ChemSusChem, 2017, 10, 1683–1691 CrossRef CAS.
- A. Rivas-Cardona, M. Chovanetz and D. F. Shantz, Microporous Mesoporous Mater., 2012, 155, 56–64 CrossRef CAS.
- H. J. White, J. Prot. Coat. Linings, 1989, 6, 47–56 Search PubMed.
- C. Zhang, Y.-X. Wu, X. Xu, Y. Li, L. Feng and G. Wu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 936–946 CrossRef CAS.
- C. Ding and A. S. Matharu, ACS Sustainable Chem. Eng., 2014, 2, 2217–2236 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |