
Selective degradation-inducing probes for studying cereblon (CRBN) biology†
Chelsea E.
Powell‡
 ab,
Guangyan
Du‡
ab,
Jonathan W.
Bushman‡
ab,
Zhixiang
He
ab,
Tinghu
Zhang
ab,
Eric S.
Fischer
ab and
Nathanael S.
Gray
*ab
ab,
Guangyan
Du‡
ab,
Jonathan W.
Bushman‡
ab,
Zhixiang
He
ab,
Tinghu
Zhang
ab,
Eric S.
Fischer
ab and
Nathanael S.
Gray
*ab
aDepartment of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA
bDepartment of Cancer Biology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, USA. E-mail: Nathanael_Gray@dfci.harvard.edu
First published on 6th July 2021
Abstract
Targeted protein degradation represents a rapidly growing area in drug discovery and development. Moreover, small molecules that induce the targeted degradation of a given protein also represent an important addition to the chemical probes toolbox as these compounds can achieve selective protein knockdown, thus providing an approach that is orthogonal to genetic knockdowns. In order to develop degradation-inducing chemical probes for studying cereblon (CRBN) biology, we generated six CRBN–CRBN (homo-PROTAC) degraders and six CRBN–VHL (hetero-PROTAC) degraders. From these compounds we identified two potent and selective CRBN degraders (ZXH-4-130 and ZXH-4-137), both of which are CRBN–VHL compounds. We characterized these lead degraders by quantitative proteomics in five cell lines (MM1.S, Kelly, SK-N-DZ, HEK293T, and MOLT-4) and observed high selectivity for CRBN in all cell lines. Furthermore, we directly compared our compounds to current lead CRBN degraders and demonstrated how these probes can be used as chemical knockdown reagents for studying CRBN-dependent processes. Overall, our work provides a roadmap for thorough degrader characterization by combination western and proteomic analysis, as illustrated by the identification of ZXH-4-130 and ZXH-4-137 as CRBN-knockdown tool compounds suitable for cell-based studies.
Introduction
A large amount of knowledge about the function of gene products has been derived through genetic modifications using tools such as RNA interference,1 transcription activator-like effector nucleases (TALENs),2,3 and the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system.4,5 However, there are limitations to these genetic knockdown strategies, including the high expense and time costs of developing animal models, particularly in nonhuman primates, and the potential for genetic compensations or spontaneous mutations in animal models which can obscure the biological consequences of the genetic perturbation.6,7 Therefore, orthogonal methods are needed to overcome some of the pitfalls of genetic knockdown strategies, and small molecule tool compounds have emerged as useful in this context. A recent addition to the chemical probe tool box are small molecule degraders, compounds that bind and recruit an E3 ubiquitin ligase to a target protein, resulting in target ubiquitination and proteasomal degradation. These degrader molecules lead to chemically induced protein-knockdown and thus represent a valuable tool for studying biological processes.Cereblon (CRBN) is a substrate receptor subunit of the E3 ubiquitin ligase complex CRL4, which also includes damaged DNA binding protein 1 (DDB1), cullin-4A (CUL4A), and RING-box protein 1 (RBX1 or ROC1). Although CRBN was identified more than 15 years ago,8 critical details about its endogenous biological function remain unknown. Individual studies have suggested that CRBN may play a role in metabolic regulation or a protective role against DNA damage-induced apoptosis, but these hypotheses have not been fully tested.9,10
More recently CRBN has gained popularity as an E3 ubiquitin ligase that can be hijacked by small molecule degraders and used for targeted protein degradation. The utility of CRBN in this context stems from the discovery that it binds immunomodulatory drugs (IMiDs), such as thalidomide, lenalidomide, and pomalidomide,11 resulting in recruitment of the zinc finger transcription factors (TFs) Ikaros (IKZF1) and Aiolos (IKZF3), and their subsequent ubiquitination and proteasomal degradation.12–14 Additionally, IMiDs can be incorporated into bifunctional degrader molecules (also called PROTACs), which can induce the degradation of target proteins beyond TFs by bringing them into proximity of an E3 ligase.15–17 These compounds are composed of an E3 ligase recruiting element, such as IMiDs that recruit CRBN, and a binding motif for a target of interest. Bifunctional degraders function in the same way as IMiDs, by facilitating formation of the ubiquitination-competent ternary complex. In addition to CRBN, bifunctional degrader molecules that hijack other ligases, such as the von-Hippel Lindau (VHL) E3 ligase, have been reported. Both the CRBN and VHL E3 ligases have been successfully recruited by small molecule degraders for a range of targets, including BRD4, BRD9, TRIM24, BCR-ABL, CDK9, Aurora A, FAK, FER, BTK, ALK, and other kinases.15,18–25 In addition to offering a novel pharmacological strategy with translational implications, the use of selective degraders has yielded important insights into the biology of these targets, such as the identification of TRIM24 as a novel dependency in acute leukemia.24
Previous work has shown that VHL may be recruited to degrade itself,26 and that CRBN degradation may be induced through recruitment of itself or VHL.27–30 To enable further studies of CRBN, here we aimed to develop selective small molecule degraders of CRBN as tool compounds to probe CRBN biology. We generated two different sets of degraders (CRBN–CRBN and CRBN–VHL) and examined their ability to induce selective CRBN degradation. This led to the identification of two CRBN–VHL degraders as potent and selective inducers of CRBN degradation. Our work is in agreement with the recently reported observation that CRBN degradation can be achieved via VHL E3 ubiquitin ligase activity (hetero-PROTACs).27,28,30 Additionally, we observe that degraders that use IMiD scaffolds as both the E3 ligase recruiting moiety and the target binding arm (homo-PROTACs) are not as effective as CRBN–VHL compounds. Using a workflow of western blot analysis to expression proteomics, we thoroughly characterize the selectivity profiles of two highly selective CRBN degraders, ZXH-4-130 and ZXH-4-137, including proteomic analysis of ZXH-4-130 in five different cell lines. These compounds may represent useful tools for achieving chemical knockdown of CRBN as a strategy for studying its biology.
Results and discussion
The chemical synthesis of all compounds discussed is presented in full in the ESI.†CRBN–CRBN degraders (homo-PROTACs)
We designed three CRBN–CRBN degraders that linked two pomalidomide groups together with all carbon linkers of varying lengths (Fig. 1A). All three compounds induced potent CRBN degradation at a dose range of 0.01–0.1 μM after 4 h treatments in the multiple myeloma cell line MM1.S (Fig. 1B). At a higher dose range of 1–10 μM the degraders displayed the “hook effect” that has been previously reported as a trait of small molecule degraders, where at higher doses degrader potency is lost.31,32 In Fig. 1B the cause of the hook effect is well illustrated, with CRBN degradation decreasing as Ikaros degradation increases, indicating that the degraders are out-competing themselves for binding and behaving similar to single agent pomalidomide. These pomalidomide derived CRBN–CRBN degraders were able to induce degradation as early as 1 h and as late as 24 h after 50 nM treatment, with the most potent degradation seen roughly between 4–8 h (Fig. 1C).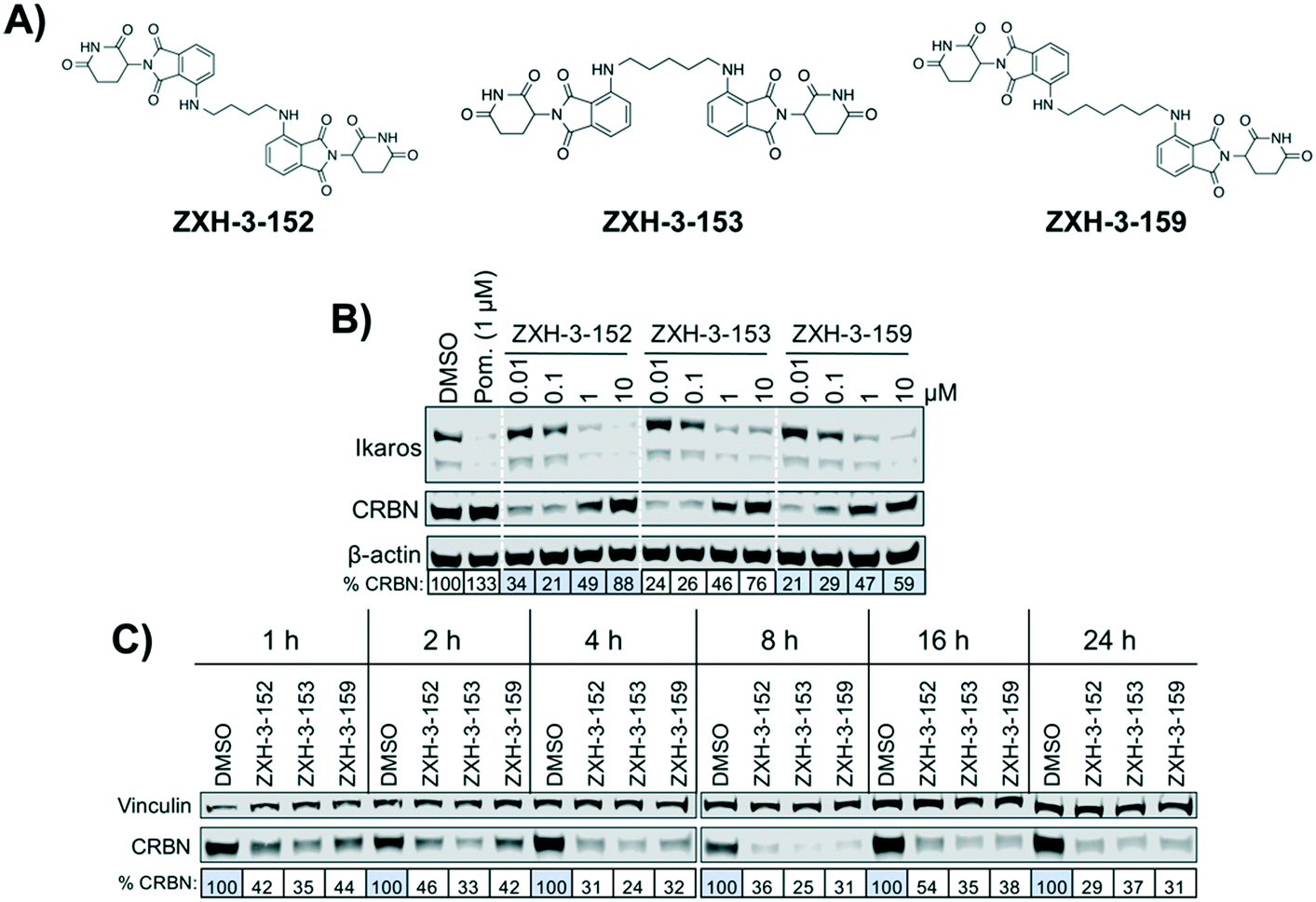 | ||
| Fig. 1 Pomalidomide CRBN–CRBN degrader behavior in MM1.S cells. (A) Structures of pomalidomide derived CRBN–CRBN degraders. (B) Immunoblot after 4 h of treatment with dose titrations of degraders. Pom. is pomalidomide. Dotted white lines provided only as visual aids and do not indicate cuts. (C) Immunoblot after treatment with 50 nM of compound for the indicated amount of time. Quantification shown as percentage of DMSO control normalized to β-actin or vinculin. | ||
We used expression proteomics to examine the selectivity profile of ZXH-3-159 in the leukemia cell line MOLT-4 (50 nM dose for 6 h) and observed that this compound induced down regulation of IKZF1 (Ikaros), ZFP91, and ZNF653 in addition to CRBN (Fig. 2). These off-targets are all known IMiD neo-substrates.12–14,33,34 In an effort to increase the selectivity profile of the CRBN–CRBN degraders, we generated three CRBN–CRBN (homo-PROTAC) compounds that use a tricyclic analog of thalidomide that has been previously reported by our group as a strategy to dial out degradation of IMiD neo-substrates, such as IKZF1, while gaining superior affinity for CRBN compared to pomalidomide (Fig. 3A).35 Although these compounds were selective for CRBN, they induced less potent degradation than the pomalidomide–pomalidomide compounds (Fig. 3B and C).
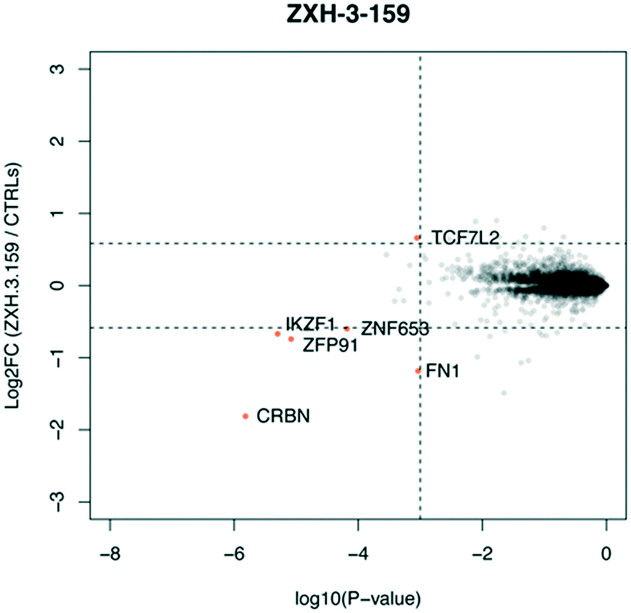 | ||
| Fig. 2 Expression proteomics in MOLT-4 cells after 6 h treatment with 50 nM of ZXH-3-159 or DMSO (singlicate analysis of compound; triplicate analysis of DMSO control). | ||
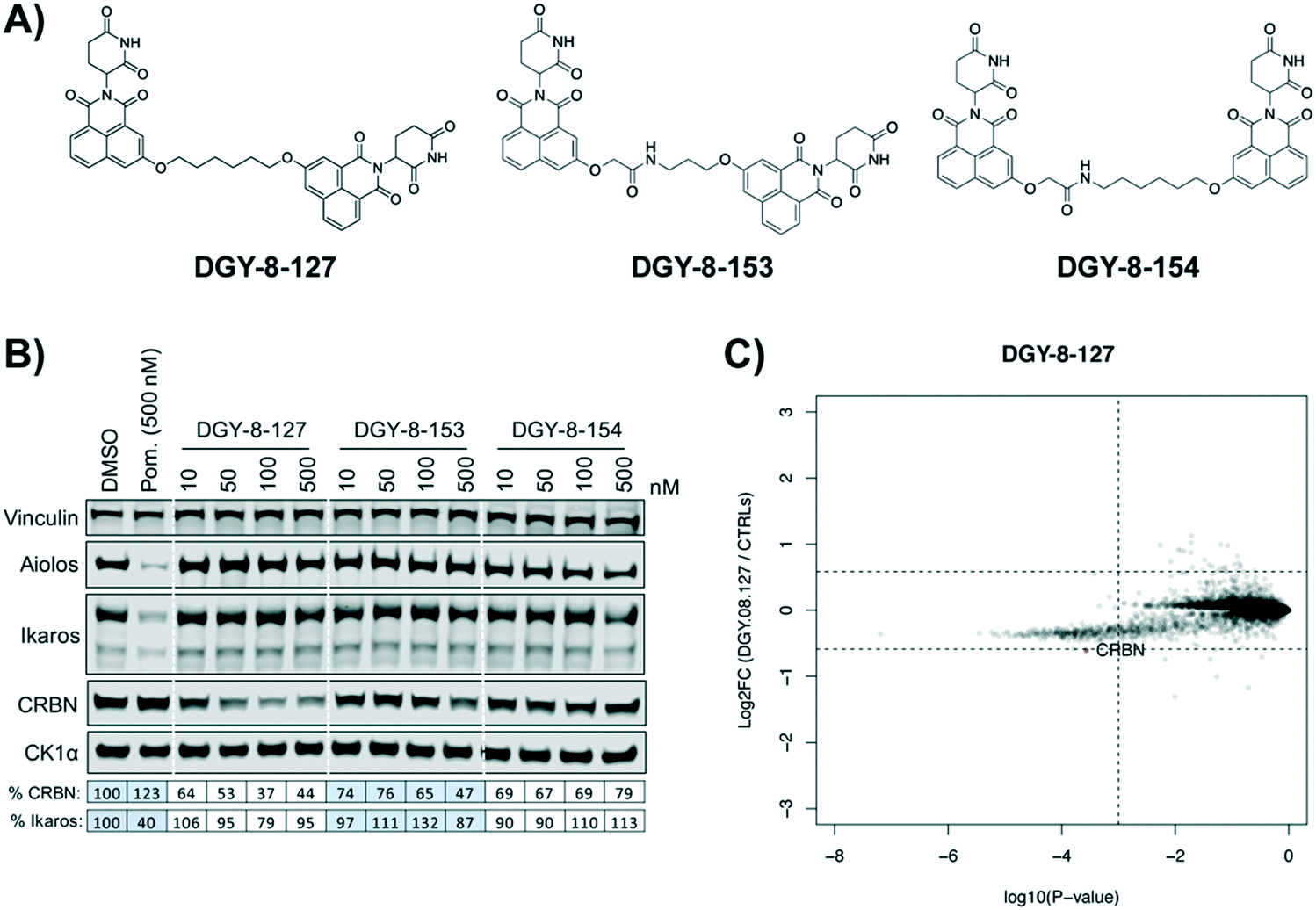 | ||
| Fig. 3 Tricyclic thalidomide analog CRBN–CRBN degrader behavior. (A) Structures of tricyclic thalidomide analog derived CRBN–CRBN degraders. (B) Immunoblot after 4 h of treatment with dose titrations of degraders in MM1.S cells. Vinculin representative of 3 blots. Quantification shown as percentage of DMSO control normalized to vinculin. Pom. is pomalidomide. Dotted white lines provided only as visual aids and do not indicate cuts. (C) Expression proteomics in MOLT-4 cells after 6 h treatment with 100 nM of DGY-8-127 or DMSO (singlicate analysis of compound; triplicate analysis of DMSO control). | ||
CRBN–VHL degraders (hetero-PROTACs)
Our next effort towards developing a selective CRBN degrader focused on examining whether degraders that recruit the VHL E3 ligase show improvement. We generated six pomalidomide–VHL ligand based CRBN–VHL degraders using varying linkers (Fig. 4A). As seen from 4 h dose titration treatments in MM1.S cells, ZXH-4-130 and ZXH-4-137 were highly potent and selective degraders of CRBN (Fig. 4B). These two compounds use the same 11-carbon linker, with the VHL ligand used in ZXH-4-137 being a methylated analog of the VHL ligand in ZXH-4-130. Interestingly, ZXH-4-133 and ZXH-4-136 also share a linker (4-carbon) with ZXH-4-136 containing a methylated VHL ligand. For the ZXH-4-133/ZXH-4-136 pair the methylated VHL ligand improves potency, with ZXH-4-136 being the more potent degrader. However, for the ZXH-4-130/ZXH-4-137 pair the potencies are equivalent. ZXH-4-132 which has a 2-carbon linker induced weak CRBN degradation, likely due to the short linker being less amenable to formation of the ternary complex. While slightly more potent than ZXH-4-132, ZXH-4-135 which contains a 4-polyethylene glycol (PEG) linker also induced weak CRBN degradation. It is possible that since both the CRBN and VHL ligands are fairly hydrophilic, the longer alkyl linker used in ZXH-4-130 and ZXH-4-137 increases the hydrophobicity and clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of the whole compound, improving cell permeability.
P of the whole compound, improving cell permeability.
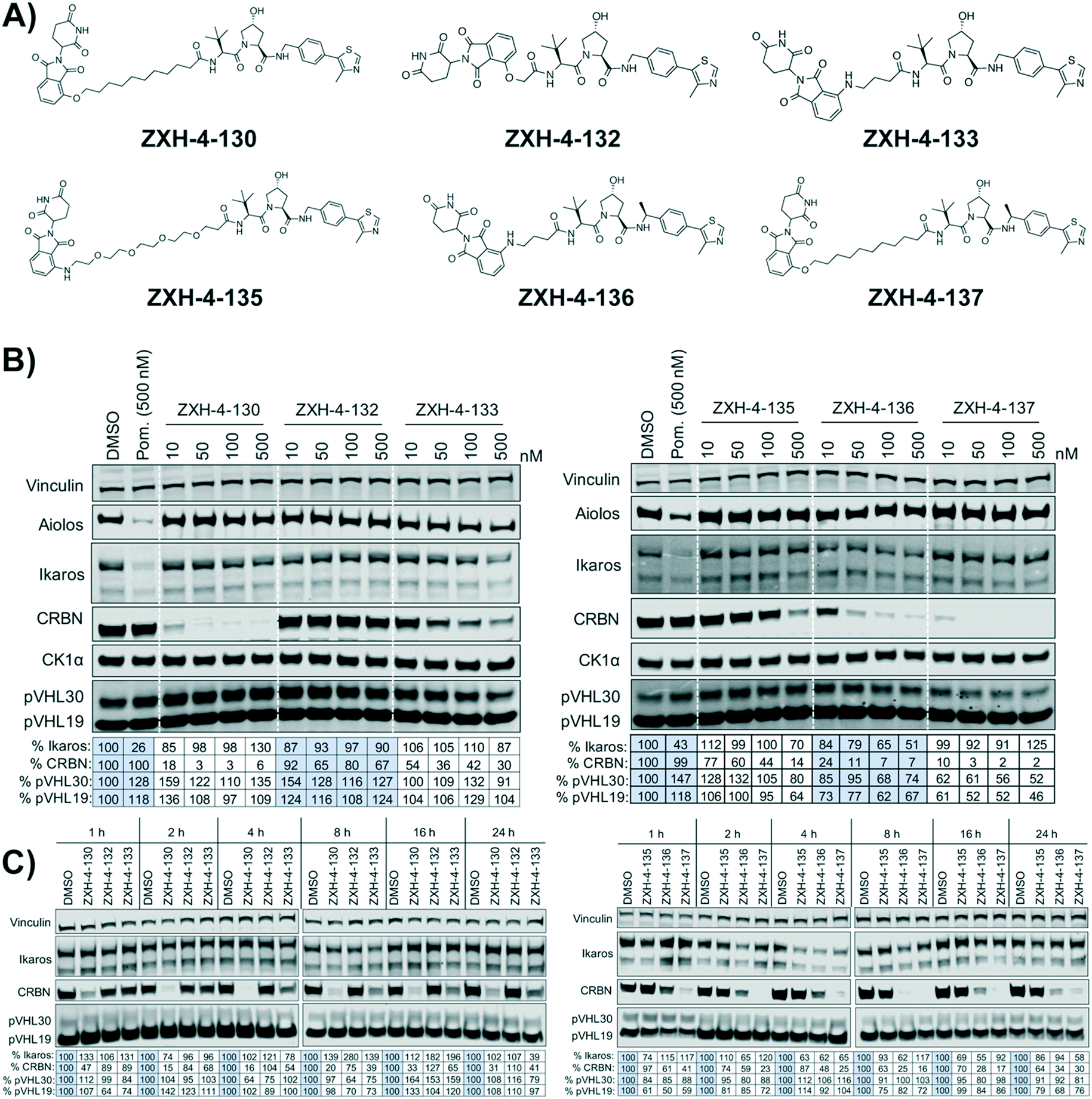 | ||
| Fig. 4 Thalidomide–VHL ligand based CRBN–VHL degrader behavior in MM1.S cells. (A) Structures of thalidomide and VHL ligand derived CRBN–VHL degraders. (B) Immunoblots after 4 h of treatment with dose titrations of degraders. Vinculin representative of 3 blots. Pom. is pomalidomide. Dotted white lines provided only as visual aids and do not indicate cuts. (C) Immunoblots after treatment with 50 nM of compound for the indicated amount of time. Vinculin representative of 2 blots. Quantification shown as percentage of DMSO control normalized to vinculin. | ||
A time course in MM1.S cells with a 50 nM dose demonstrated that ZXH-4-130 was the most potent between 2–8 h, while ZXH-4-137 was the most potent between 2–16 h (Fig. 4C).
Expression proteomics in MM1.S cells after 6 h treatments with 50 nM of ZXH-4-130 or ZXH-4-137 further validated that these degraders are highly selective for CRBN, with CRBN being the only significantly downregulated target (Fig. 5). The high degree of selectivity for ZXH-4-130 was also confirmed by expression proteomics in Kelly, SK-N-DZ, HEK293T, and MOLT-4 cell lines, indicating that this compound may be useful as a tool compound in many cell lines (ESI:† Fig. S1). Additionally, we confirmed by western in MM1.S cells and expression proteomics in MOLT-4 cells that ZXH-4-130 and ZXH-4-137 did not affect the expression of other members of the CRBN E3 ligase complex: DDB1, CUL4A, and ROC1 (Fig. S2 and S3†). This indicates that both of these compounds are promising lead probes for chemically inducing CRBN degradation.
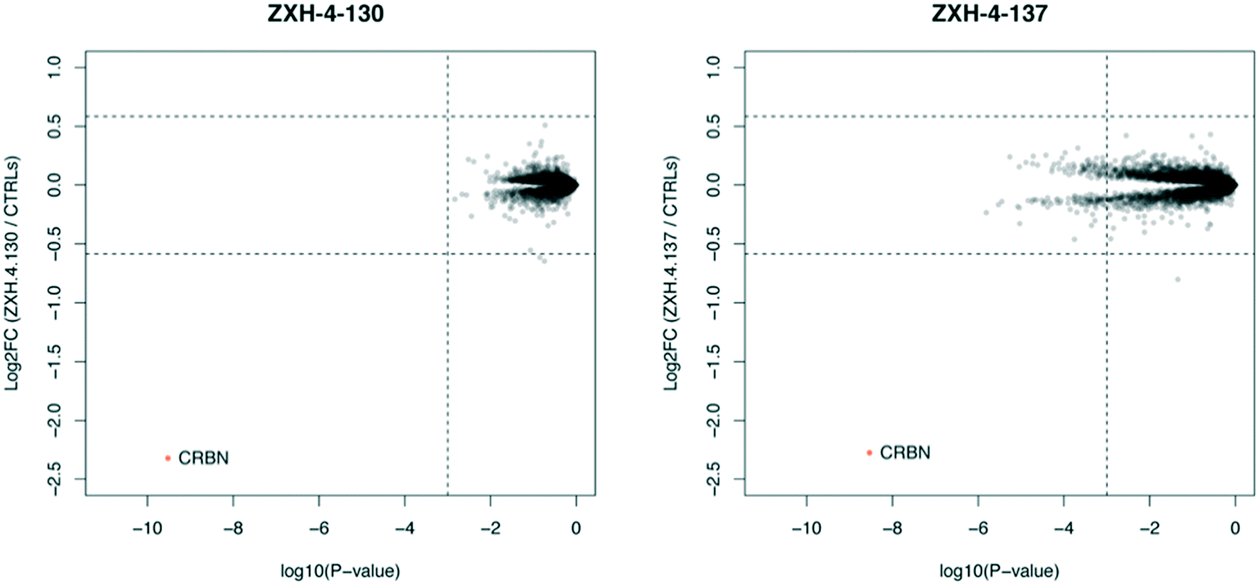 | ||
| Fig. 5 Expression proteomics in MM1.S cells after 6 h treatment with 50 nM of ZXH-4-130, 50 nM of ZXH-4-137, or DMSO (triplicate analysis). | ||
We further assessed the selectivity profile of ZXH-4-130 over time by using expression proteomics in the neuroblastoma cell line SK-N-DZ after 6 h, 12 h, and 24 h treatments of 100 nM compound or DMSO (Fig. 6). ZXH-4-130 remained highly selective for CRBN across these time points (note that proteins F2 and F5 seen at the 6 h time point were not significantly up- or down-regulated at any other time points or in any other cell lines; these secreted proteins are occasionally seen as false positives in cellular proteomics experiments).36
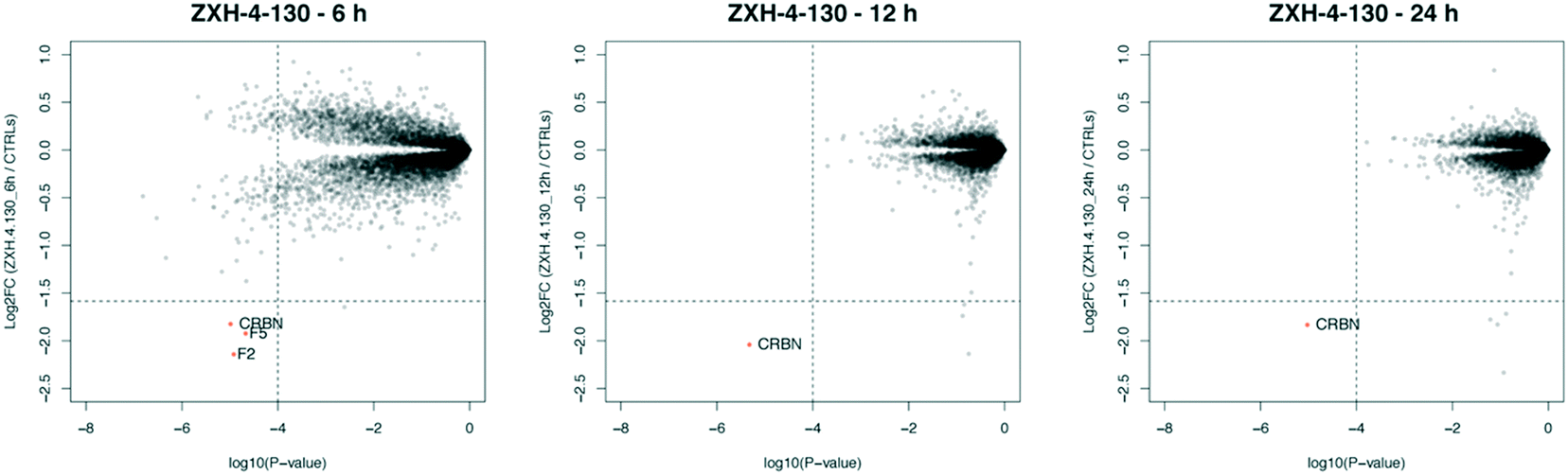 | ||
| Fig. 6 Expression proteomics in SK-N-DZ cells after 100 nM of ZXH-4-130 for various treatment times compared to 24 h DMSO control (duplicate analysis of compound; triplicate analysis of control). | ||
Comparison to previously reported CRBN degraders
We compared our lead CRBN degraders to the pomalidomide based CRBN–CRBN degrader St-15a as well as CRBN-6-5-5-VHL, a potent CRBN targeting thalidomide–VHL ligand based degrader, both of which have been previously reported by Steinebach et al. (Fig. 7A).29,30CRBN-6-5-5-VHL was selected as the CRBN–VHL compound for comparison to our lead degraders due to its higher potency compared to the lead degrader 14a reported by Girardini et al.27 and the lead degrader TD-165 reported by Kim et al.28 Importantly, both ZXH-4-130 and ZXH-4-137 appear more potent against CRBN than 14a and TD-165, which have reported DC50s of >100 nM in HeLa cells and 20.4 nM in HEK293T cells, respectively.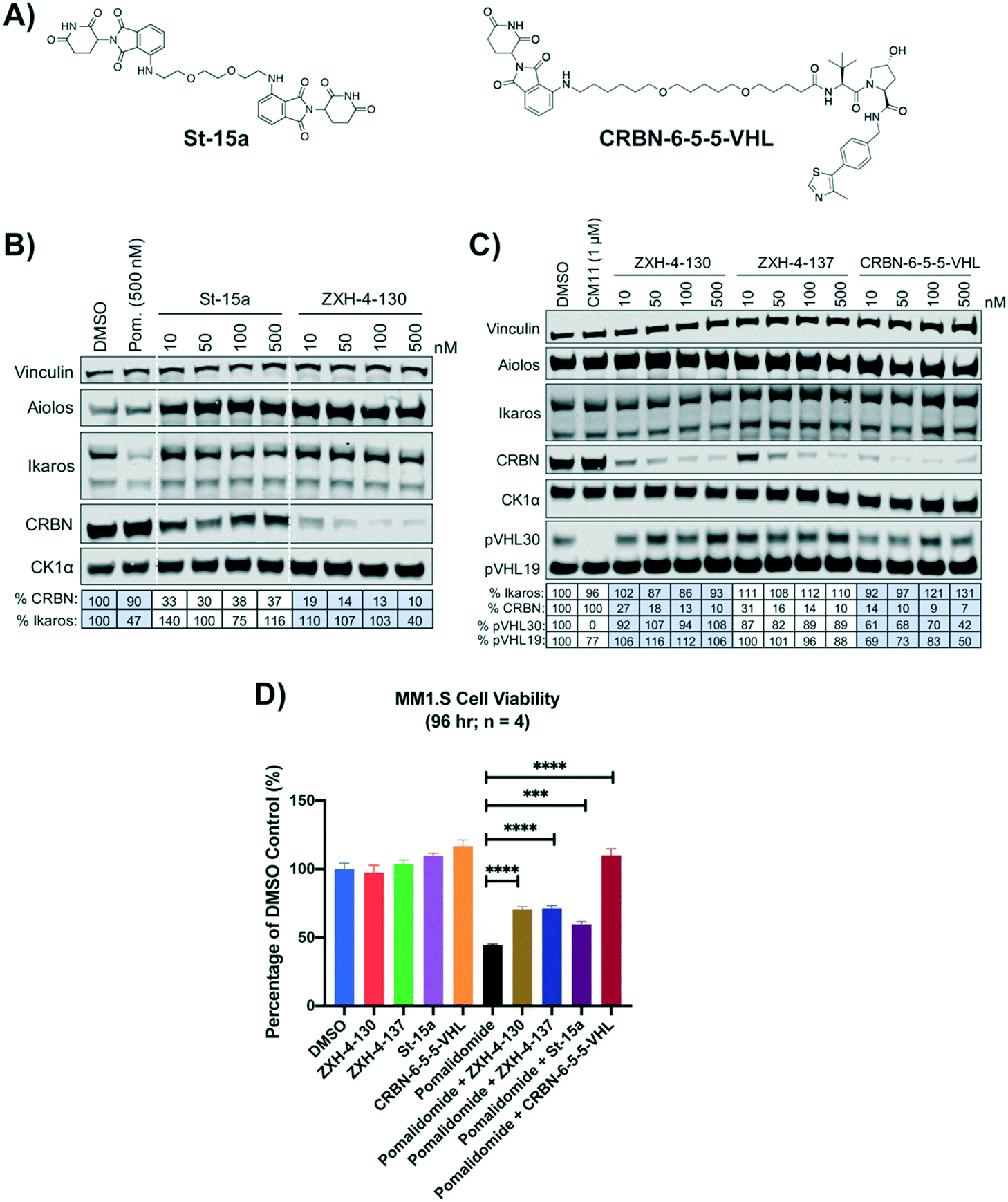 | ||
| Fig. 7 Comparison of lead CRBN degraders to previously reported compounds by Steinebach et al. (A) Structure of pomalidomide based CRBN–CRBN degrader St-15a and thalidomide–VHL ligand based CRBN–VHL degrader CRBN-6-5-5-VHL. (B and C) Immunoblot after 4 h of treatment with dose titrations of degraders in MM1.S cells. Vinculin representative of 4 blots. Quantification shown as percentage of DMSO control normalized to vinculin. Pom. is pomalidomide. The VHL targeting homo-PROTAC CM11 is shown for comparison.26 Dotted white lines provided only as visual aids and do not indicate cuts. (D) MM1.S cell viability after 2 h pre-treatments with 100 nM of ZXH-4-130, ZXH-4-137, St-15a, or CRBN-6-5-5-VHL followed by 96-hour treatment with 1 μM of pomalidomide (four biological replicates; Graphpad Prism 8 software; Welch's t test). | ||
While St-15a appeared similarly selective for CRBN over Ikaros by western, ZXH-4-130 was a much more potent degrader of CRBN at a dose range of 10–500 nM (Fig. 7B). ZXH-4-130 and ZXH-4-137 induced CRBN degradation to a similar extent as CRBN-6-5-5-VHL at a dose range of 10–500 nM (Fig. 7C). All four CRBN degraders prevented pomalidomide cytotoxicity to a significant extent, with St-15a having the least statistically significant amount of prevention and ZXH-4-130, ZXH-4-137, and CRBN-6-5-5-VHL having equally statistically significant amounts of prevention (Fig. 7D).
In order to compare the selectivity profiles of St-15a (CRBN–CRBN), CRBN-6-5-5-VHL (CRBN–VHL), and ZXH-4-130 (CRBN–VHL) we used expression proteomics in MOLT-4 cells (Fig. 8). After 6 h treatments with 50 nM compound or DMSO, all three compounds were highly selective for CRBN. Degradation of CRBN could potentially lead to upregulation of its endogenous substrates. However, known CRBN substrates were not among the proteins upregulated with ZXH-4-130, and we were unable to replicate upregulation of these proteins in an independent MOLT-4 experiment or any other cell lines. Upregulated protein C3 is a member of the complement system, of which several other components (F2, F5, FN1) occasionally appear as false positives in our cellular proteomics experiments; ECM1 also occasionally appears as a false positive.36
 | ||
| Fig. 8 Expression proteomics in MOLT-4 cells after 6 h of 50 nM treatment with St-15a, CRBN-6-5-5-VHL, ZXH-4-130, or DMSO (singlicate analysis of compound; triplicate analysis of DMSO control). | ||
Interestingly, ZXH-4-130 differs only by an exit vector to the compound TD-165 that was reported by Kim et al.; TD-165 uses an amide exit vector from the pomalidomide group instead of an ether.28 Despite this high degree of similarity, TD-165 was reported to be less potent than ZXH-4-130. While TD-165 has a DC50 of 20.4 nM in HEK293T cells, ZXH-4-130 induces ∼80% CRBN degradation at 10 nM in MM1.S cells (n = 3, Fig. 4B, 7B, and C); however, direct cell line comparison is needed. The selectivity profile of TD-165 was not assessed by proteomics, but Kim et al. did report proteomic analysis of a 7-carbon linker analog of TD-165 after 1 μM treatment for 12 h in Jurkat cells. This demonstrated statistically significant downregulation of two proteins in addition to CRBN: CNIH1 and LMBRD2.28
Use of lead CRBN degraders as tool compounds
We examined the potential of using ZXH-4-130 and ZXH-4-137 as probes for studying CRBN biology. We pre-treated MM1.S cells for 2 h with 50 nM of ZXH-4-130 or ZXH-4-137, before exposing them to CC-885, a compound that induces G1 to S phase transition protein 1 (GSPT1) degradation via CRBN E3 ligase recruitment.37 We observed that this pre-treatment with 50 nM of ZXH-4-130 or ZXH-4-137 rescued GSPT1 degradation (Fig. 9A). This prevention of induced degradation was comparable to that observed after pre-treatment with the proteasome inhibitor carfilzomib. Pre-treatment with 50 nM or 10 μM of pomalidomide did not prevent GSPT1 degradation to a similar extent as pre-treatment with ZXH-4-130 or ZXH-4-137, indicating that the ability to prevent induced GSPT1 degradation was due to the CRBN degradative abilities of ZXH-4-130 and ZXH-4-137, not just competition for CRBN binding. We also examined whether or not the chemical knockdown of CRBN by ZXH-4-130 or ZXH-4-137 could prevent the degradative activity of a PROTAC by testing the activity of the cyclin-dependent kinase 9 (CDK9) degrader THAL-SNS-032 in MOLT-4 cells after 2 h pre-treatment with 0.1 μM of the CRBN degraders (Fig. 9B).19ZXH-4-130 and ZXH-4-137 were indeed able to partially prevent CDK9 degradation compared to the DMSO control and 0.1 μM pomalidomide treatment, but to a lesser extent than carfilzomib treatment or 10 μM pomalidomide competition. Of note, both ZXH-4-130 and ZXH-4-137 induced nearly complete CRBN degradation, but THAL-SNS-032's activity against CDK9 was only partially prevented; this speaks to the catalytic power of small molecule degraders.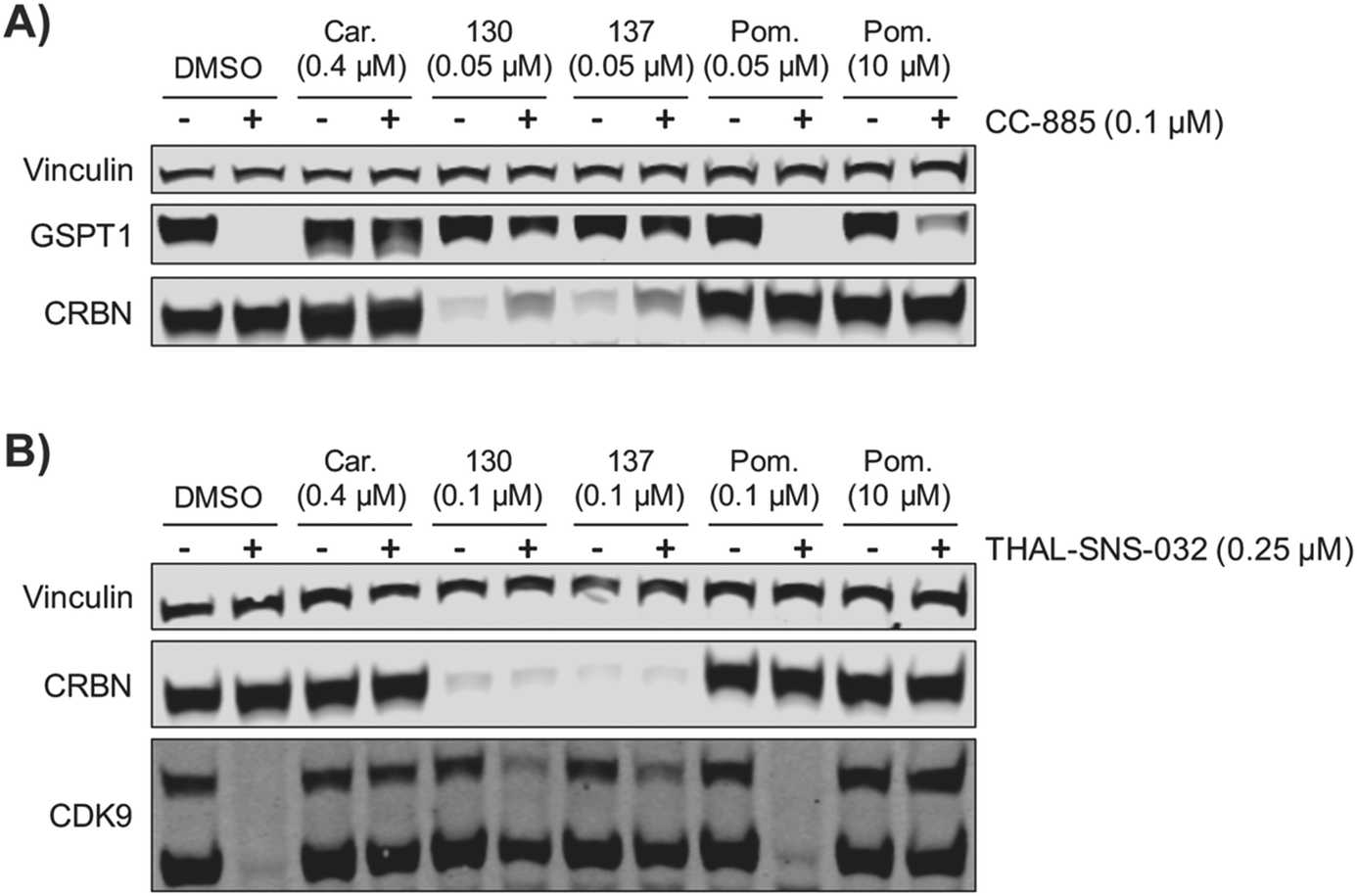 | ||
| Fig. 9 Immunoblots of ZXH-4-130 and ZXH-4-137 as tool compounds for studying CRBN related biology. (A) Immunoblot after 2 h pre-treatments with DMSO, carfilzomib (Car.), ZXH-4-130 (130), ZXH-4-137 (137), or pomalidomide (Pom.) followed by 4 h treatment with CC-885 in MM1.S cells. (B) Immunoblot after 2 h pre-treatments with DMSO, carfilzomib (Car.), ZXH-4-130 (130), ZXH-4-137 (137), or pomalidomide (Pom.) followed by 6 h treatment with THAL-SNS-032 in MOLT-4 cells. | ||
Conclusions
Here we describe the development and characterization of two potent and highly selective degraders of CRBN, ZXH-4-130 and ZXH-4-137. These compounds feature both a CRBN recruiting ligand and a VHL E3 ligase binding ligand, and therefore represent an example of bifunctional degrader molecules that bring two E3 ubiquitin ligases into close proximity. In our studies, this strategy was more successful in achieving CRBN degradation than degraders that featured two CRBN recruiting ligands (homo-PROTACs). These observations were consistent with the literature.30 One possible explanation for the poor performance of homo-PROTACs is that as CRBN is degraded by these compounds, it rapidly reaches a point where the amount of cellular CRBN is lowered to a level such that all remaining CRBN is occupied by one end of a degrader compound, and thus there is no free protein to engage with the second CRBN-recruiting group. This may be considered a variation of the hook effect where lowered protein levels prevent CRBN from being completely degraded by CRBN–CRBN degraders. Hetero-PROTACs can avoid this by recruiting two different E3 ligases, allowing for more complete CRBN degradation.Interestingly, the CRBN–VHL degraders only induced degradation of CRBN and not VHL, which is also consistent with previously published studies on CRBN–VHL degraders.27,28,30 A possible explanation is that the ternary complex formed by the small molecule degraders positions the E3 ligases in a manner where CRBN is more amenable to degradation than VHL. Additionally, it has been suggested by the use of VHL–VHL degraders that VHL in the CRL2-VHL ubiquitin ligase complex may be protected from ubiquitination as selective degradation of pVHL30, a form of VHL that is thought to have ligase independent functions, is seen over degradation of pVHL19, a form of VHL that may preferentially act in the E3 ligase complex.26
Our lead CRBN degraders ZXH-4-130 and ZXH-4-137 performed as expected in our cell-based studies that tested their ability to block the activity of other degraders developed to hijack the CRBN E3 ligase. These results show that CRBN knockdown due to ZXH-4-130 and ZXH-4-137 pre-treatment rescues small molecule-mediated degradation of two different targets (GSPT1 and CDK9). Overall, ZXH-4-130 and ZXH-4-137 are potent and selective CRBN degraders that may assist with new lines of inquiry into CRBN biology.
Experimental
Chemical synthesis
Chemical synthesis and compound characterization are presented in full in the ESI.†Cell culture
MM1.S (CRL-2974), MOLT-4 (CRL-1582), SK-N-DZ (CRL-2149), and HEK293T cells (CRL-3216) were purchased from ATCC. Kelly cells (ECACC 92110411) were purchased from Millipore Sigma.All media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MM1.S and MOLT-4 cells were cultured in RPMI-1640 media containing L-glutamine. Kelly cells were cultured in RPMI-1640 media without L-glutamine. SK-N-DZ cells were cultured in DMEM media containing L-glutamine. HEK293T cells were cultured in DMEM media without L-glutamine. Mycoplasma testing was performed on a monthly basis using the MycoAlert mycoplasma detection kit (Lonza) and all lines were negative.
Cell viability assays
Cell viability was evaluated using the CellTiter-Glo luminescent cell viability assay (Promega) following the manufacturer's standards.Immunoblotting
Cells were washed with PBS before being lysed with cell lysis buffer (Cell Signaling Technology) supplemented with protease and phosphatase inhibitor cocktails (Roche) at 4 °C for 15 minutes. The cell lysate was vortexed before being centrifuged at 14000 × g for 20 min at 4 °C. Protein in cell lysate was quantified by BCA assay (Pierce). Primary antibodies used in this study include β-actin (Cell Signaling Technology, 3700S), CDK9 (Cell Signaling Technology, 2316S), CK1α (Abcam, ab206652), CRBN (Novus Biologicals, NBP1-91810), CUL4A (Cell Signaling Technology, 2699S), DDB1 (Cell Signaling Technology, 5428S), eRF3/GSPT1 (Abcam, ab49878), IKZF1 (Ikaros) (Cell Signaling Technology, 5443S), IKZF3 (Aiolos) (Cell Signaling Technology, 15103S), RBX1 (ROC1) (Cell Signaling Technology, 11922S), VHL (Cell Signaling Technology, 68547S), and vinculin (Abcam, ab130007). Blot quantification was performed using Image Studio 4.0 software, normalizing to loading controls.
Sample preparation for TMT LC-MS3 mass spectrometry
Kelly, MOLT-4, MM1.S, SK-N-DZ, or HEK293T cells were treated for 6, 12, or 24 hours with DMSO in biological triplicates or compound in biological singlicate, duplicate, or triplicate as indicated. Treatment concentrations were as follows: 0.1% DMSO, 50 or 100 nM ZXH-4-130, 50 nM ZXH-4-137, 50 nM ZXH-3-159, 50 nM St-15a, 50 nM CRBN-6-5-5-VHL, 100 nM DGY-8-127, or 50 nM CM11. Cells were harvested by centrifugation and prepared for mass spectrometry as previously described.33,38LC-MS data analysis
Data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) using an MS3-based TMT method.39 Proteome Discoverer 2.2 (Thermo Fisher Scientific) was used for .RAW file processing as previously described.33,38 Reporter ion intensities were normalised and scaled using in-house scripts in the R framework.40 Statistical analysis was carried out using the limma package within the R framework.41Notes
The mass spectrometry datasets generated during this study are available in the Proteomics Identification Database (PRIDE) archive under the project accessions PXD022021, PXD022022, PXD022058, PXD022059, and PXD022060. All other data presented are available in the main text and ESI.†Conflicts of interest
Nathanael S. Gray is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, B2S, Aduro, Jengu and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield and Sanofi. Eric S. Fischer is a founder, science advisory board member and equity holder in Civetta, Jengu (board member), and Neomorph, an equity holder in C4 Therapeutics, and a consultant to Astellas, Novartis, Deerfield, and EcoR1 capital. The Fischer lab receives or has received research funding from Novartis, Astellas, and Deerfield.Acknowledgements
Funding for this work was received from the National Institutes of Health (NIH): Grant R01 CA214608 (E. S. F.) and 5 F31 CA210619-02 (C. E. P.). We would like to thank Nicholas A Eleuteri and Katherine A Donovan (Dana-Farber Cancer Institute) for help with proteomics experiments. We would also like to thank Milka Kostic (Dana-Farber Cancer Institute) for helpful feedback on the manuscript.References
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- B. Wefers, O. Ortiz, W. Wurst and R. Kühn, Methods, 2014, 69, 94–101 CrossRef CAS PubMed.
- Y. Liu, D. Luo, Y. Lei, W. Hu, H. Zhao and C. H. K. Cheng, Methods, 2014, 69, 58–66 CrossRef CAS PubMed.
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Nat. Protoc., 2013, 8, 2281–2308 CrossRef CAS PubMed.
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef PubMed.
- X. Sun, J. Wang, X. Yao, W. Zheng, Y. Mao, T. Lan, L. Wang, Y. Sun, X. Zhang, Q. Zhao, J. Zhao, R.-P. Xiao, X. Zhang, G. Ji and Y. Rao, Cell Discovery, 2019, 5, 10 CrossRef PubMed.
- X. Sun, H. Gao, Y. Yang, M. He, Y. Wu, Y. Song, Y. Tong and Y. Rao, Signal Transduction Targeted Ther., 2019, 4, 64 CrossRef PubMed.
- J. J. Higgins, J. Pucilowska, R. Q. Lombardi and J. P. Rooney, Neurology, 2004, 63, 1927–1931 CrossRef CAS PubMed.
- T. V. Nguyen, J. E. Lee, M. J. Sweredoski, S.-J. Yang, S.-J. Jeon, J. S. Harrison, J.-H. Yim, S. G. Lee, H. Handa, B. Kuhlman, J.-S. Jeong, J. M. Reitsma, C.-S. Park, S. Hess and R. J. Deshaies, Mol. Cell, 2016, 61, 809–820 CrossRef CAS PubMed.
- L. Zhou and G. Xu, Cell Death Discovery, 2019, 10, 1–13 CrossRef PubMed.
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- G. Lu, R. E. Middleton, H. Sun, M. Naniong, C. J. Ott, C. S. Mitsiades, K.-K. Wong, J. E. Bradner and W. G. Kaelin, Science, 2014, 343, 305–309 CrossRef CAS PubMed.
- A. K. Gandhi, J. Kang, C. G. Havens, T. Conklin, Y. Ning, L. Wu, T. Ito, H. Ando, M. F. Waldman, A. Thakurta, A. Klippel, H. Handa, T. O. Daniel, P. H. Schafer and R. Chopra, Br. J. Haematol., 2014, 164, 811–821 CrossRef CAS PubMed.
- J. Kronke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- M. Zengerle, K.-H. Chan and A. Ciulli, ACS Chem. Biol., 2015, 10, 1770–1777 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- A. C. Lai, M. Toure, D. Hellerschmied, J. Salami, S. Jaime-Figueroa, E. Ko, J. Hines and C. M. Crews, Angew. Chem., 2015, 128, 818–821 CrossRef.
- C. M. Olson, B. Jiang, M. A. Erb, Y. Liang, Z. M. Doctor, Z. Zhang, T. Zhang, N. Kwiatkowski, M. Boukhali, J. L. Green, W. Haas, T. Nomanbhoy, E. S. Fischer, R. A. Young, J. E. Bradner, G. E. Winter and N. S. Gray, Nat. Chem. Biol., 2018, 14, 163–170 CrossRef CAS PubMed.
- H.-T. Huang, D. Dobrovolsky, J. Paulk, G. Yang, E. L. Weisberg, Z. M. Doctor, D. L. Buckley, J.-H. Cho, E. Ko, J. Jang, K. Shi, H. G. Choi, J. D. Griffin, Y. Li, S. P. Treon, E. S. Fischer, J. E. Bradner, L. Tan and N. S. Gray, Cell Chem. Biol., 2017, 25, 1–19 Search PubMed.
- C. E. Powell, Y. Gao, L. Tan, K. A. Donovan, R. P. Nowak, A. Loehr, M. Bahcall, E. S. Fischer, P. A. Jänne, R. E. George and N. S. Gray, J. Med. Chem., 2018, 61, 4249–4255 CrossRef CAS PubMed.
- C. H. Kang, D. H. Lee, C. O. Lee, J. D. Ha, C. H. Park and J. Y. Hwang, Biochem. Biophys. Res. Commun., 2018, 505, 1–6 CrossRef PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2017, 25, 1–34 Search PubMed.
- L. N. Gechijian, D. L. Buckley, M. A. Lawlor, J. M. Reyes, J. Paulk, C. J. Ott, G. E. Winter, M. A. Erb, T. G. Scott, M. Xu, H.-S. Seo, S. Dhe-Paganon, N. P. Kwiatkowski, J. A. Perry, J. Qi, N. S. Gray and J. E. Bradner, Nat. Chem. Biol., 2018, 14, 1–13 CrossRef PubMed.
- D. Remillard, D. L. Buckley, J. Paulk, G. L. Brien, M. Sonnett, H.-S. Seo, S. Dastierdi, M. Wühr, S. Dhe-Paganon, S. A. Armstrong and J. E. Bradner, Cancer Res., 2017, 468, 1067 Search PubMed.
- C. Maniaci, S. J. Hughes, A. Testa, W. Chen, D. J. Lamont, S. Rocha, D. R. Alessi, R. Romeo and A. Ciulli, Nat. Commun., 2017, 8, 830 CrossRef PubMed.
- M. Girardini, C. Maniaci, S. J. Hughes, A. Testa and A. Ciulli, Bioorg. Med. Chem., 2019, 27, 2466–2479 CrossRef CAS PubMed.
- K. Kim, D. H. Lee, S. Park, S.-H. Jo, B. Ku, S. G. Park, B. C. Park, Y. U. Jeon, S. Ahn, C. H. Kang, D. Hwang, S. Chae, J. D. Ha, S. Kim, J. Y. Hwang and J.-H. Kim, Sci. Rep., 2019, 9, 19654 CrossRef CAS PubMed.
- C. Steinebach, S. Lindner, N. D. Udeshi, D. C. Mani, H. Kehm, S. Köpff, S. A. Carr, M. Gütschow and J. Krönke, ACS Chem. Biol., 2018, 13, 2771–2782 CrossRef CAS PubMed.
- C. Steinebach, H. Kehm, S. Lindner, L. P. Vu, S. Köpff, Á. L. Mármol, C. Weiler, K. G. Wagner, M. Reichenzeller, J. Krönke and M. Gütschow, Chem. Commun., 2019, 55, 1821–1824 RSC.
- I. Churcher, J. Med. Chem., 2017, 61, 444–452 CrossRef PubMed.
- S. An and L. Fu, EBioMedicine, 2018, 36, 553–562 CrossRef PubMed.
- K. A. Donovan, J. An, R. P. Nowak, J. C. Yuan, E. C. Fink, B. C. Berry, B. L. Ebert and E. S. Fischer, eLife, 2018, 7, e38430 CrossRef PubMed.
- J. An, C. M. Ponthier, R. Sack, J. Seebacher, M. B. Stadler, K. A. Donovan and E. S. Fischer, Nat. Commun., 2017, 8, 1–11 CrossRef PubMed.
- M. de Wispelaere, G. Du, K. A. Donovan, T. Zhang, N. A. Eleuteri, J. C. Yuan, J. Kalabathula, R. P. Nowak, E. S. Fischer, N. S. Gray and P. L. Yang, Nat. Commun., 2019, 10, 3468 CrossRef PubMed.
- K. A. Donovan, F. M. Ferguson, J. W. Bushman, N. A. Eleuteri, D. Bhunia, S. Ryu, L. Tan, K. Shi, H. Yue, X. Liu, D. Dobrovolsky, B. Jiang, J. Wang, M. Hao, I. You, M. Teng, Y. Liang, J. Hatcher, Z. Li, T. D. Manz, B. Groendyke, W. Hu, Y. Nam, S. Sengupta, H. Cho, I. Shin, M. P. Agius, I. M. Ghobrial, M. W. Ma, J. Che, S. J. Buhrlage, T. Sim, N. S. Gray and E. S. Fischer, Cell, 2020, 183, 1714–1731 CrossRef CAS PubMed .e10.
- M. E. Matyskiela, G. Lu, T. Ito, B. Pagarigan, C.-C. Lu, K. Miller, W. Fang, N.-Y. Wang, D. Nguyen, J. Houston, G. Carmel, T. Tran, M. Riley, L. Nosaka, G. C. Lander, S. Gaidarova, S. Xu, A. L. Ruchelman, H. Handa, J. Carmichael, T. O. Daniel, B. E. Cathers, A. Lopez-Girona and P. P. Chamberlain, Nature, 2016, 535, 252–257 CrossRef CAS PubMed.
- J. W. Bushman, K. A. Donovan, N. J. Schauer, X. Liu, W. Hu, A. C. Varca, S. J. Buhrlage and E. S. Fischer, Cell Chem. Biol., 2020, 28, 78–87 CrossRef PubMed .e3.
- G. C. McAlister, D. P. Nusinow, M. P. Jedrychowski, M. Wühr, E. L. Huttlin, B. K. Erickson, R. Rad, W. Haas and S. P. Gygi, Anal. Chem., 2014, 86, 7150–7158 CrossRef CAS PubMed.
- R. D. C. Team, R: A Language and Environment for Statistical Computing, http://www.R-project.org/.
- M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi and G. K. Smyth, Nucleic Acids Res., 2015, 43, e47 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Chemical synthesis methods, compound characterization, and Fig. S1–S3. See DOI: 10.1039/d0md00382d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |