DOI:
10.1039/D1DT02414K
(Communication)
Dalton Trans., 2021,
50, 12800-12805
Anion recognition by halogen bonding and hydrogen bonding bis(triazole)-imidazolium [2]rotaxanes†
Received
20th July 2021
, Accepted 20th August 2021
First published on 24th August 2021
Introduction
Negatively charged species are widely pervasive in a plethora of biological, industrial, and environmental spheres.1 Their importance in a diverse range of fundamental processes has stimulated the development of synthetic host architectures capable of strong and selective anion recognition. To this end, the employment of hydrogen bonding (HB) interactions has been one of the most popular strategies in anion host design.2–4 In contrast, the considered use of halogen bonding (XB) for the purposes of recognition remains in its infancy.5–8 Although recent years have realised considerable promise for solution phase XB-mediated recognition, particularly in the context of anion binding, reports remain surprisingly scarce. As a consequence the scope of XB donor motif design and variation is underdeveloped, particularly in combination with arrays of HB donors.2 Of the commonly employed HB motifs, imidazolium derivatives have featured heavily over the past few decades in anion receptor design, however their continued interest is in no small part due to their ability to bind anions in competitive aqueous media9 and their proficiency as functional based sensors for challenging bio-molecules.10–12 In more recent years, iodo-triazoles have emerged as synthetically accessible and highly potent XB donors, and accordingly have received intense interest in the field of anion recognition.13–28
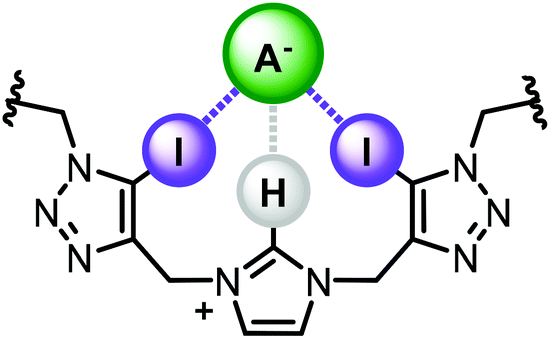
Herein we report a novel, potentially synergistic mixed XB/HB anion recognition motif, comprising of a central imidazolium unit with two adjacent iodotriazole groups (Fig. 1). The receptor design exploits a bidentate XB donor array connected via flexible methylene linkers to a charge-assisted C—H HB donor. Incorporation of the motif, and its HB analogue, into a stoppered axle component enabled the synthesis of [2]rotaxane host architectures via discrete chloride anion templation. 1H NMR anion titration experiments reveal the interlocked architectures are capable of strong halide and oxoanion recognition in highly competitive aqueous–organic solvent mixtures.
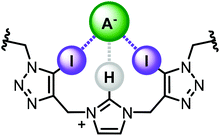 |
| | Fig. 1 Target bis-iodotriazole-imidazolium motif and proposed anion binding mode. | |
Results and discussion
Rotaxane synthesis
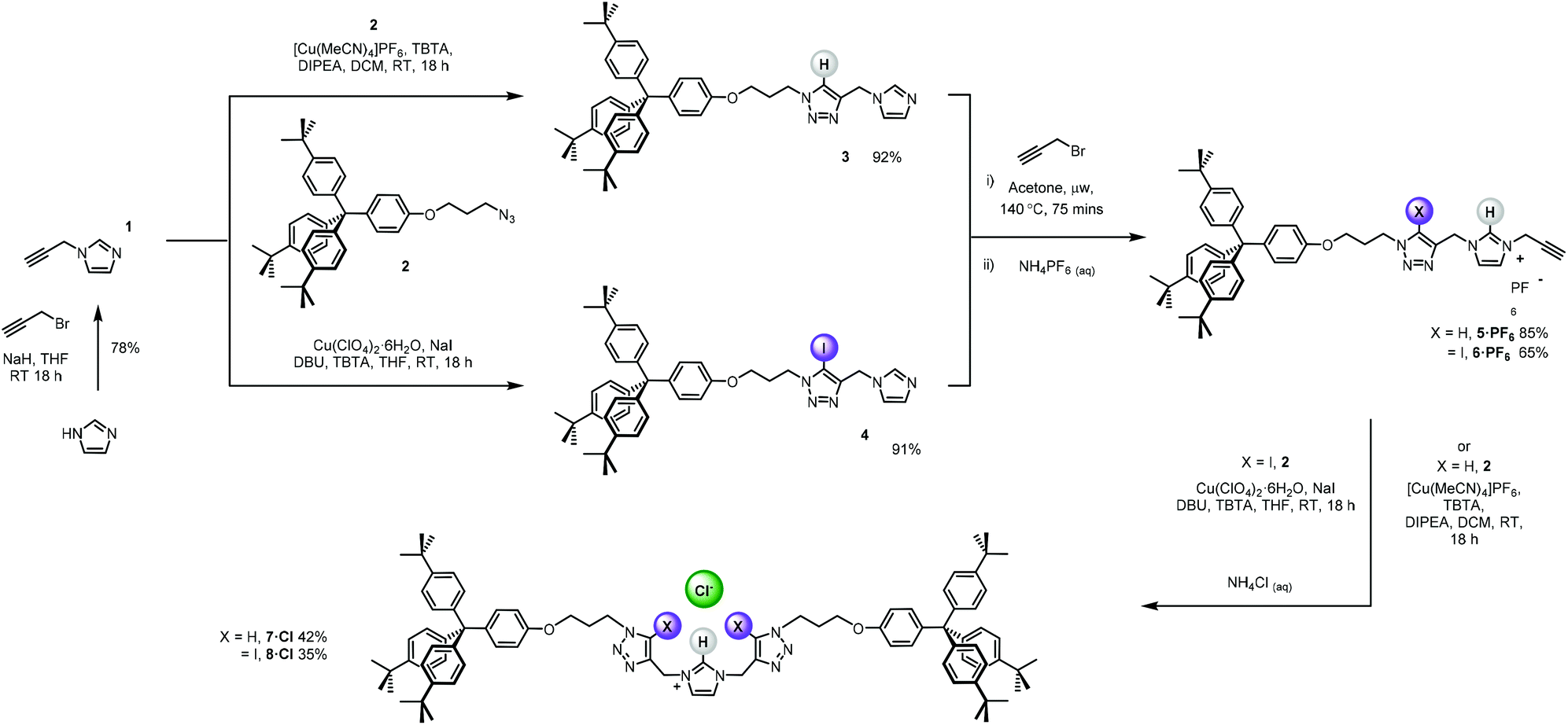
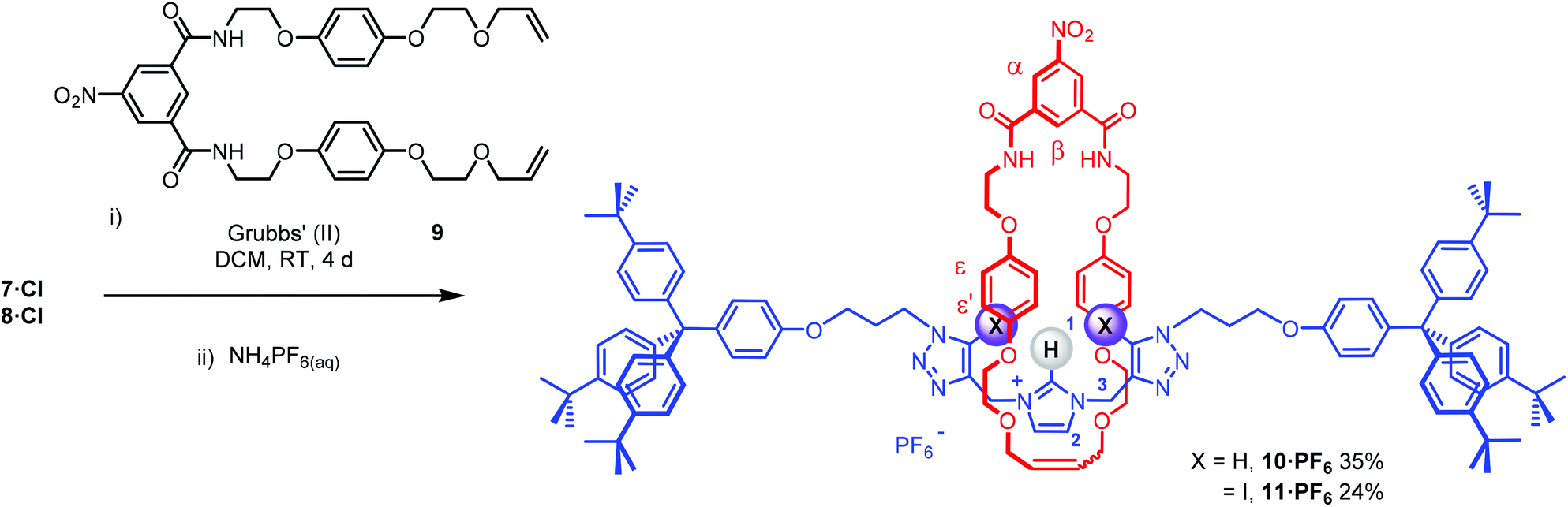
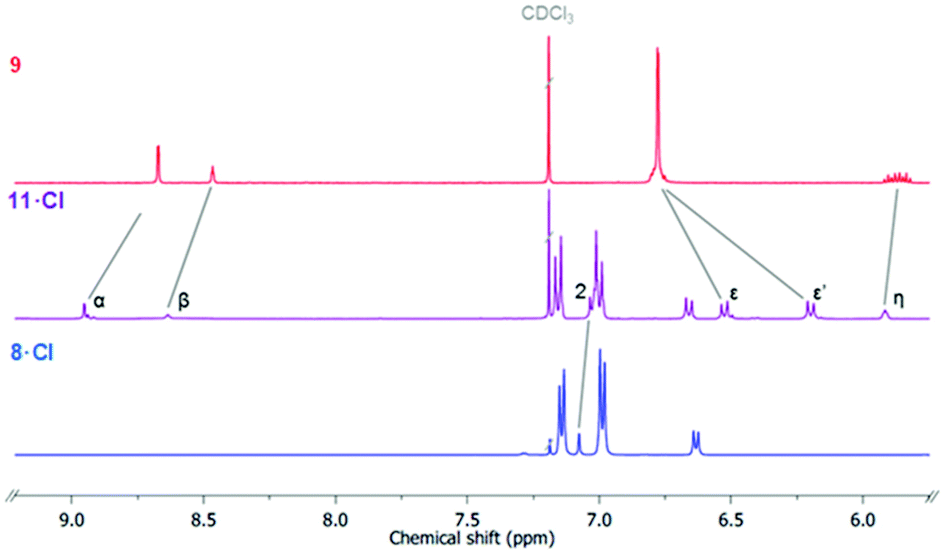
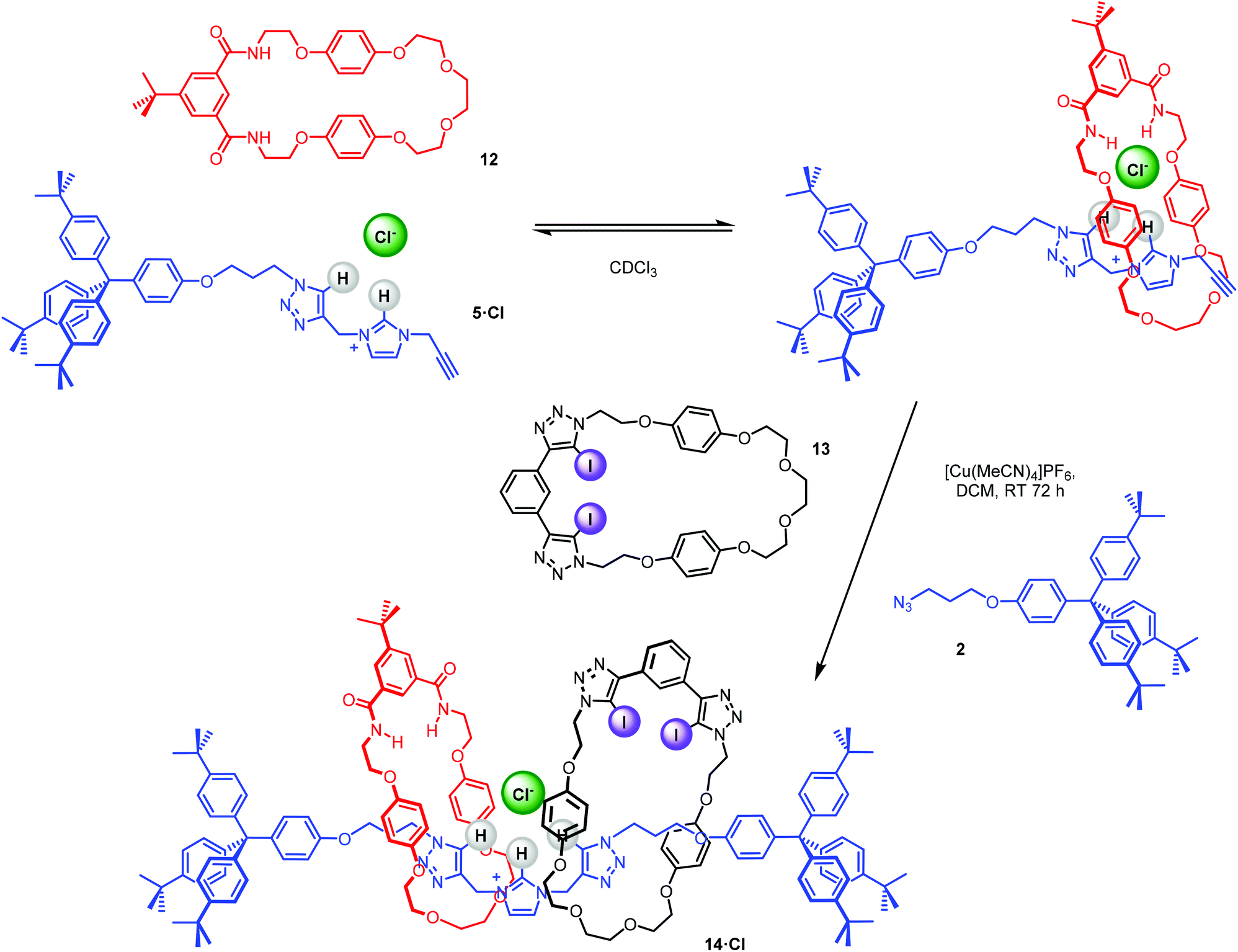
The bis-triazole imidazolium containing axle components were prepared according to a multistep synthetic strategy outlined in Scheme 1. N-Alkylation of imidazole at the N1-position with propargyl bromide produced alkyne 1 in 78% yield.29 A conventional CuAAC reaction with terphenyl stopper azide 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 in the presence of [Cu(MeCN)4]PF6, DIPEA and the rate accelerating ligand TBTA afforded the proto-triazole product 3 in 92% yield. Exploiting the ‘one-pot’ methodology developed by Zhu et al.,31 treatment of 1 with Cu(ClO4)2·H2O, NaI and DBU, TBTA and 2 gave the iodo-triazole appended stopper precursor 4 in 93% yield. Installation of the second alkyne group was achieved through a microwave assisted alkylation of the imidazole nitrogen of 3 and 4 with propargyl bromide affording 5·Br and 6·Br respectively. Subsequent anion exchange to the non-coordinating hexafluorophosphate salt was achieved through iterative washing of a CH2Cl2 solution of the bromide salts with aqueous solutions of NH4PF6 to give 5·PF6 and 6·PF6 in 85% and 65% (over two steps) respectively. The alkyne terminated cationic imidazolium axle precursors 5·PF6 and 6·PF6 were subjected to analogous proto- and iodo-triazole copper(I)-catalysed azide alkyne cycloaddition (CuAAC) reaction forming conditions with azide 2, which after anion exchange gave the desired bis-triazole imidazolium containing axle components as their chloride salts 7·Cl and 8·Cl in 42% and 35% yield respectively. The synthesis of the target HB and XB rotaxanes was achieved through a Grubbs’-catalysed chloride anion template ring closing metathesis reaction of 7·Cl and 8·Cl with bis-vinyl appended macrocycle precursor 932 in anhydrous dichloromethane over the duration of 4 days (Scheme 2). Purification of the crude reaction mixtures by preparative TLC followed by anion exchange to the non-coordinating hexafluorophosphate salts by copious washing with NH4PF6(aq) afforded rotaxanes 10·PF6 and 11·PF6 in respective 35% and 24% isolated yields. Both novel rotaxanes were characterised via high resolution ESI mass spectrometry and 1H and 13C NMR spectroscopy. Evidence for the successful formation of XB rotaxane 11·PF6 is shown through comparison of 1H NMR spectra of the interlocked product, the macrocycle precursor 9 and axle 8·Cl (Fig. 2). Upon mechanical bond formation, an upfield shift of imidazolium proton 2 is observed. The signals associated with the macrocycle component hydroquinone environments (ε,ε′) split and move significantly upfield. These diagnostic shifts are due to the favourable donor–acceptor interactions between the electron-deficient imidazolium axle and the electron-rich hydroquinone groups of the macrocycle, indicating the interlocked nature of the product. Substantial downfield shifts of the aromatic protons (α, β) of the macrocycle are also observed. Furthermore, the resonance associated with the terminal alkene protons η confirmed the ring-closing reaction had been successful. Further evidence for the interlocked nature of 11·PF6 was also provided by 2D ROESY NMR spectroscopy (Fig. S1†). Similar chemical shift differences and observations in the 2D ROESY NMR spectrum were observed in the 1H NMR spectrum of HB rotaxane 10·PF6.
30 in the presence of [Cu(MeCN)4]PF6, DIPEA and the rate accelerating ligand TBTA afforded the proto-triazole product 3 in 92% yield. Exploiting the ‘one-pot’ methodology developed by Zhu et al.,31 treatment of 1 with Cu(ClO4)2·H2O, NaI and DBU, TBTA and 2 gave the iodo-triazole appended stopper precursor 4 in 93% yield. Installation of the second alkyne group was achieved through a microwave assisted alkylation of the imidazole nitrogen of 3 and 4 with propargyl bromide affording 5·Br and 6·Br respectively. Subsequent anion exchange to the non-coordinating hexafluorophosphate salt was achieved through iterative washing of a CH2Cl2 solution of the bromide salts with aqueous solutions of NH4PF6 to give 5·PF6 and 6·PF6 in 85% and 65% (over two steps) respectively. The alkyne terminated cationic imidazolium axle precursors 5·PF6 and 6·PF6 were subjected to analogous proto- and iodo-triazole copper(I)-catalysed azide alkyne cycloaddition (CuAAC) reaction forming conditions with azide 2, which after anion exchange gave the desired bis-triazole imidazolium containing axle components as their chloride salts 7·Cl and 8·Cl in 42% and 35% yield respectively. The synthesis of the target HB and XB rotaxanes was achieved through a Grubbs’-catalysed chloride anion template ring closing metathesis reaction of 7·Cl and 8·Cl with bis-vinyl appended macrocycle precursor 932 in anhydrous dichloromethane over the duration of 4 days (Scheme 2). Purification of the crude reaction mixtures by preparative TLC followed by anion exchange to the non-coordinating hexafluorophosphate salts by copious washing with NH4PF6(aq) afforded rotaxanes 10·PF6 and 11·PF6 in respective 35% and 24% isolated yields. Both novel rotaxanes were characterised via high resolution ESI mass spectrometry and 1H and 13C NMR spectroscopy. Evidence for the successful formation of XB rotaxane 11·PF6 is shown through comparison of 1H NMR spectra of the interlocked product, the macrocycle precursor 9 and axle 8·Cl (Fig. 2). Upon mechanical bond formation, an upfield shift of imidazolium proton 2 is observed. The signals associated with the macrocycle component hydroquinone environments (ε,ε′) split and move significantly upfield. These diagnostic shifts are due to the favourable donor–acceptor interactions between the electron-deficient imidazolium axle and the electron-rich hydroquinone groups of the macrocycle, indicating the interlocked nature of the product. Substantial downfield shifts of the aromatic protons (α, β) of the macrocycle are also observed. Furthermore, the resonance associated with the terminal alkene protons η confirmed the ring-closing reaction had been successful. Further evidence for the interlocked nature of 11·PF6 was also provided by 2D ROESY NMR spectroscopy (Fig. S1†). Similar chemical shift differences and observations in the 2D ROESY NMR spectrum were observed in the 1H NMR spectrum of HB rotaxane 10·PF6.
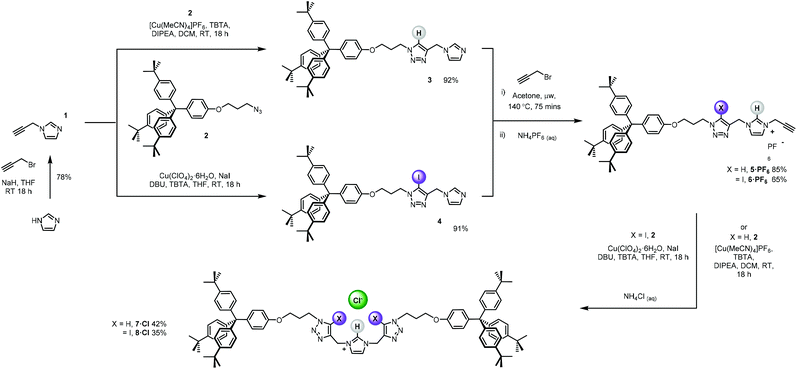 |
| | Scheme 1 Synthesis of the bis-iodo/prototriazole imidazolium axle components. | |
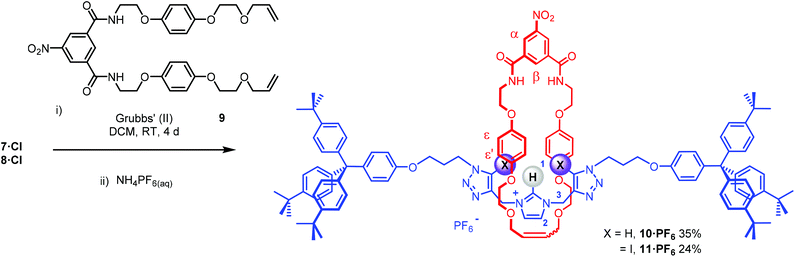 |
| | Scheme 2 Chloride anion templated synthesis of the target XB and HB [2]rotaxanes. | |
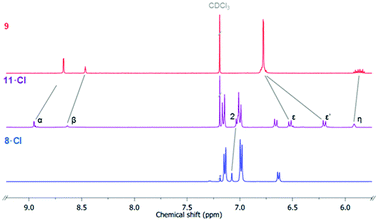 |
| | Fig. 2 Stacked truncated 1H NMR spectra of macrocycle precursors 9 (top), XB [2] rotaxane 11·Cl (middle) and free axle 8·Cl (CDCl3, 500 MHz, 298 K). | |
1H NMR anion binding investigations
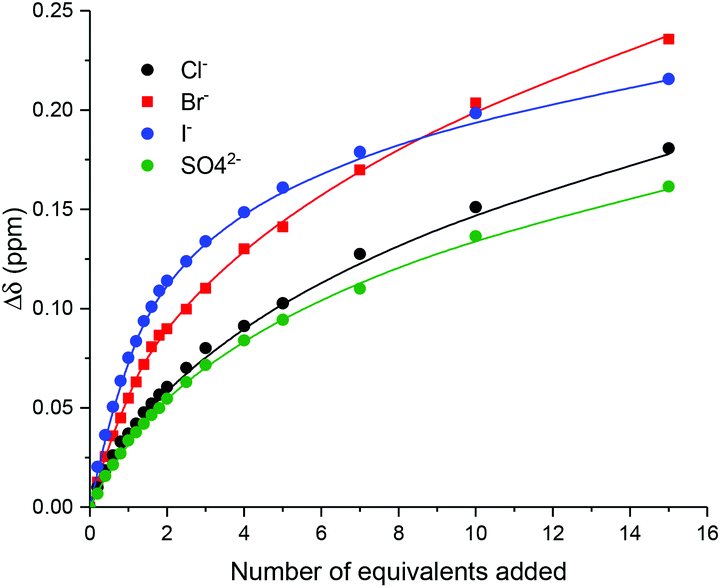
The anion recognition properties of 10·PF6 and 11·PF6 were investigated by 1H NMR titration experiments in the competitive aqueous–organic solvent mixture CDCl3/CD3OD/D2O (45:45:10 v/v). In a typical experiment the addition of halide or sulfate anion, as their tetrabutylammonium salt, induced significant downfield perturbations in signals associated with the interlocked anion binding cavity, namely internal macrocycle proton β (∼0.2–0.3 ppm) and the axle methylene signal 3 (∼0.1 ppm). In the case of the HB rotaxane, 10·PF6, notable downfield shifts of the axle triazole protons were also observed.
Monitoring the shifts of macrocycle proton β, WinEQNMR233 analysis of the titration data (Fig. 3) determined 1:2 host:guest stoichiometric association constants, with K2 values being of much smaller magnitude than K1 (Table 1). The 1:2 host:guest binding stoichiometry may be rationalised by the conformational freedom in the respective rotaxane's axle component by virtue of the methylene linker facilitating, at higher anion equivalents, a second albeit considerably weaker anion binding event, between one iodo- or proto-triazole donor and the C3 and C4 protons of the imidazolium group (Fig. 4).
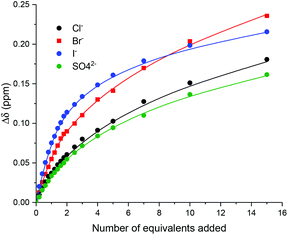 |
| | Fig. 3 Experimental titration data (points) and calculated 1:2 host:guest stoichiometric binding isotherms (lines) for the addition of anions as their TBA salts to XB rotaxane 11·PF6 monitoring internal macrocycle proton β (CDCl3/CD3OD/D2O 45:45:10, 298 K, 500 MHz). | |
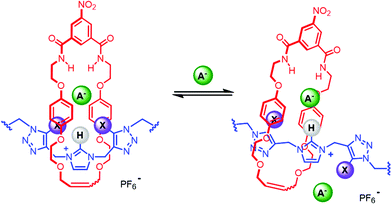 |
| | Fig. 4 Postulated 1:2 anion binding stoichiometry of the HB and XB [2]rotaxanes 10·PF6 and 11·PF6. | |
Table 1 Association constants Ka/M−1 for rotaxanes with various anions in CDCl3/CD3OD/D2O (45:45:10 v/v)a
| Anion |
10·PF6
|
11·PF6
|
|
K
1:1
|
K
1:2
|
K
1:1
|
K
1:2
|
|
Calculated using WinEQNMR2, estimated standard errors (in parentheses). Anions added as TBA salts. [receptor] = 1 mM, 500 MHz, 298 K.
|
| Cl− |
2960 (83) |
41 |
3080 (124) |
51 |
| Br− |
2110 (73) |
77 |
3200 (132) |
41 |
| I− |
2803 (230) |
72 |
2360 (88) |
66 |
| SO42− |
3130 (231) |
67 |
2650 (193) |
61 |
Inspection of Table 1 reveals that both XB and HB rotaxanes exhibited strong halide and sulfate binding in this competitive aqueous–organic solvent mixture. However, it is noteworthy that the XB rotaxane 11·PF6 displayed an enhanced association constant for bromide over the HB rotaxane analogue 10·PF6. Furthermore, the XB rotaxane showed a modest preference for the relatively smaller anions (Cl− = Br− > SO42− > I−), whereas HB rotaxane displayed no pronounced difference in the Ka values for Cl−, I− and SO42−. This can be attributed to possible unfavourable steric hindrance effects on the binding of larger anions imparted by the XB rotaxane's axle iodotriazole motifs. Moreover, it was postulated the imidazolium methylene spacer groups present in the axles of the rotaxanes allow for significant flexibility both within the axle and between the components of the interlocked host, resulting in binding domains capable of accommodating a range of anionic guest species. The flexibility introduced in rotaxanes 10·PF6 and 11·PF6 however, could prove useful for incorporation into [3]rotaxanes, allowing for the binding of larger oxoanions in the binding cavity formed between two macrocycles in a higher order interlocked structure. An attempt to prepare such a [3]rotaxane is discussed in the following section.
Attempted synthesis of hetero[3]rotaxane
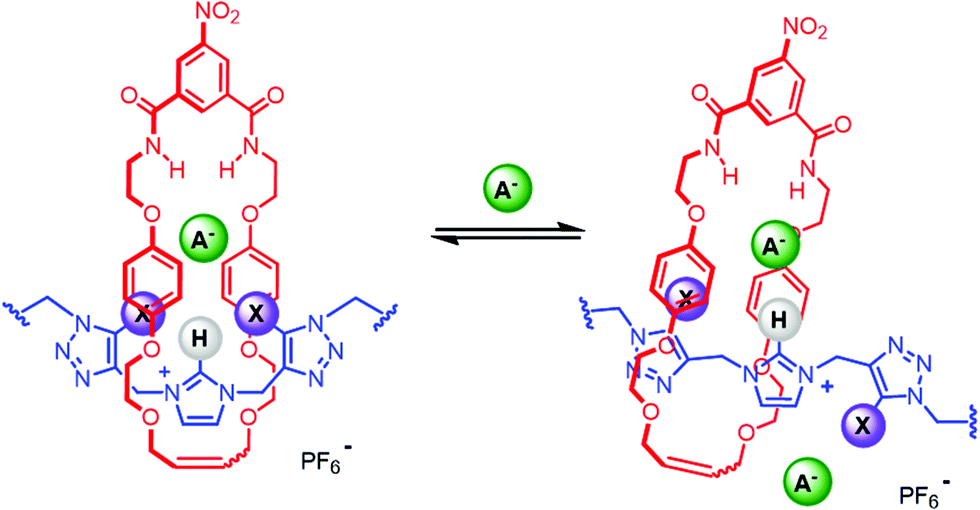
The synthesis of a hetero[3]rotaxane using the active metal template (AMT) method30,34 for mechanical bond formation in combination with the anion template approach was undertaken. Ambitiously, it was envisaged that a chloride-template pseudorotaxane with an axle precursor containing a terminal alkyne, may subsequently be used in an AMT CuAAC ‘click’ reaction with stopper azide and a suitable copper(I) coordinating macrocycle to produce a hetero[3]rotaxane (Fig. 5).
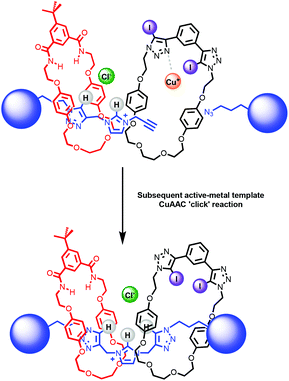 |
| | Fig. 5 Schematic representation of route to hetero[3]rotaxane combining chloride anion – template and active–metal template methodologies. | |
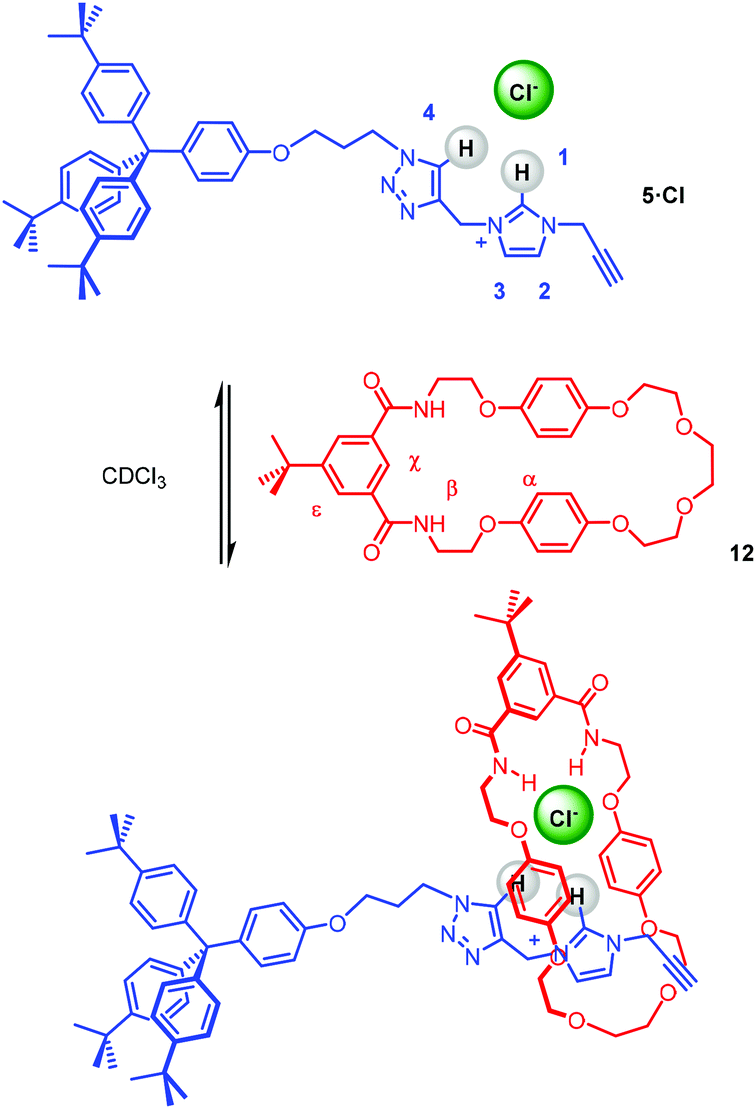
The capability of stopper precursor imidazolium–alkyne 5·Cl to participate in anion–template pseudorotaxane assembly formation, as shown in Scheme 3, was investigated initially through a qualitative 1H NMR titration experiment. Stopper imidazolium–alkyne 5·Br was converted to the chloride salt for the formation of a chloride anion–template pseudorotaxane, by successive washing with NH4Cl(aq).
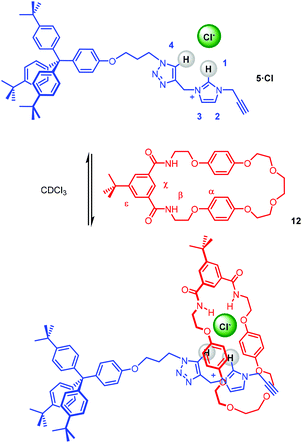 |
| | Scheme 3 Pseudorotaxane formation between imidazolium-alkyne thread 5·Cl and isophthalamide macrocycle 12. | |
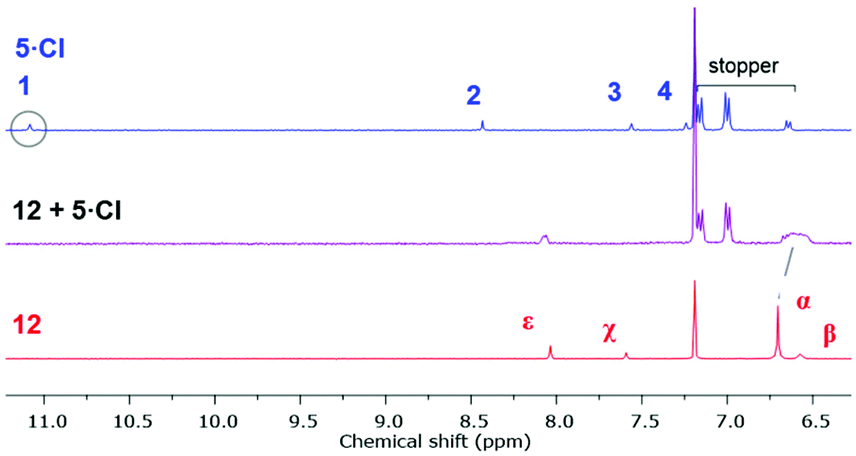
The 1H NMR spectrum of an equimolar mixture of isophthalamide macrocycle 1235 and 5·Cl in CDCl3 revealed evidence of successful pseudorotaxane formation via the upfield shifts and splitting of macrocycle hydroquinone protons α and the disappearance of imidazolium proton 1 (Fig. 6). The splitting of the hydroquinone resonances of the macrocycle were due to π–π donor–acceptor interactions between the aromatic systems of the imidazolium-alkyne thread and macrocycle.
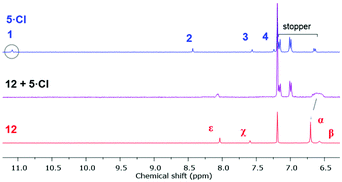 |
| | Fig. 6 Truncated 1H NMR spectra (400 mHz, CDCl3, 298 K) of imidazolium-alkyne 5·Cl (top), an equimolar mixture of macrocycle 12 and 5·Cl (middle), and macrocycle 12 (bottom). | |
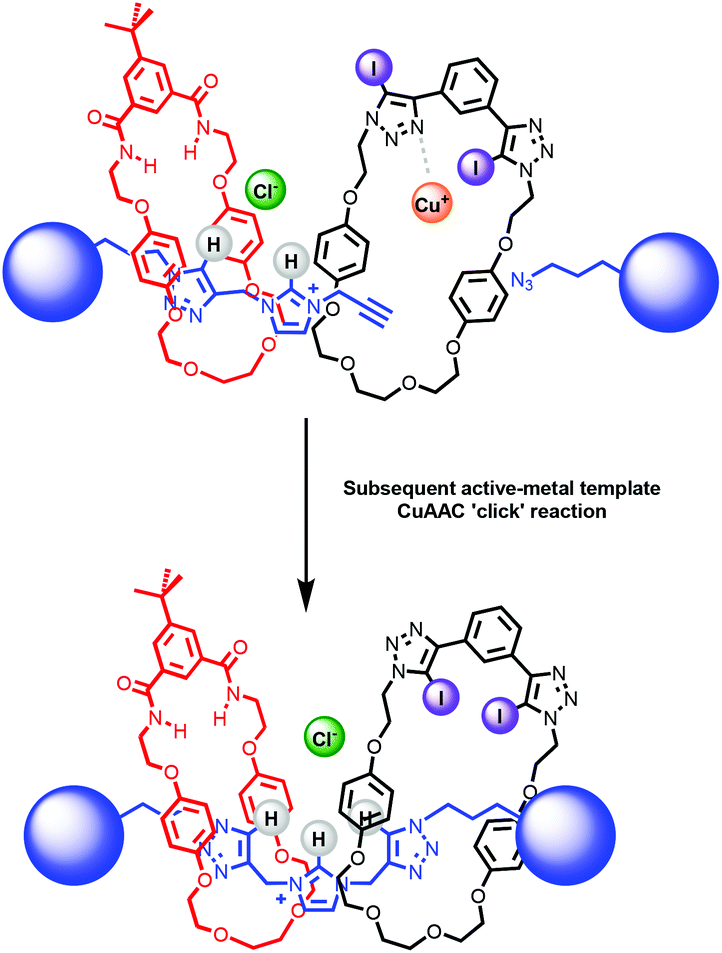
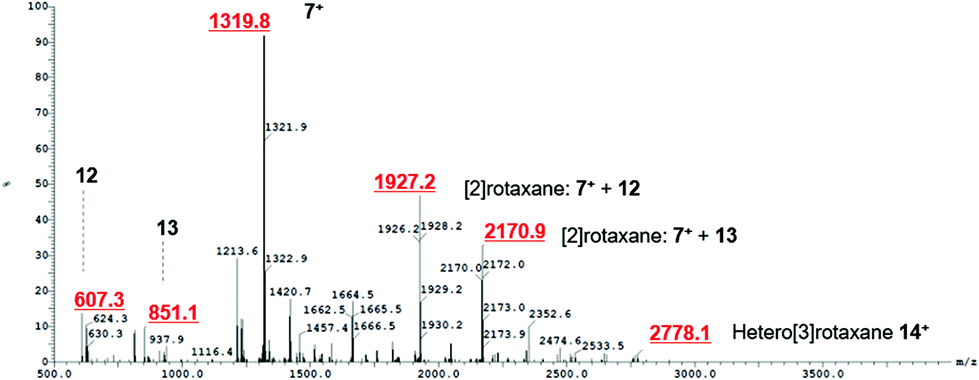
After the successful formation of a pseudorotaxane assembly with imidazolium–alkyne 5·Cl and isophthalamide macrocycle 12, the synthesis of a hetero[3]rotaxane combining active metal- and anion–template methods was then undertaken (Scheme 4). Five equivalents of stopper imidazolium–alkyne 5·Cl and macrocycle 12 were initially added to DCM to allow for the formation of the pseudorotaxane. A DCM solution of one equivalent of macrocycle 13,36 one equivalent of [Cu(MeCN)4][PF6] and five equivalents of stopper precursor azide 2 was subsequently added and the reaction mixture left to stir for three days. Analysis via ESI mass spectroscopy revealed the formation of axle 7 (m/z = 1319.8), a [2]rotaxane containing macrocycle 12 (m/z = 1927.2), a [2]rotaxane with macrocycle 13 (m/z = 2170.9) and most importantly target hetero[3]rotaxane 14·Cl (m/z = 2778.1) (Fig. 7).
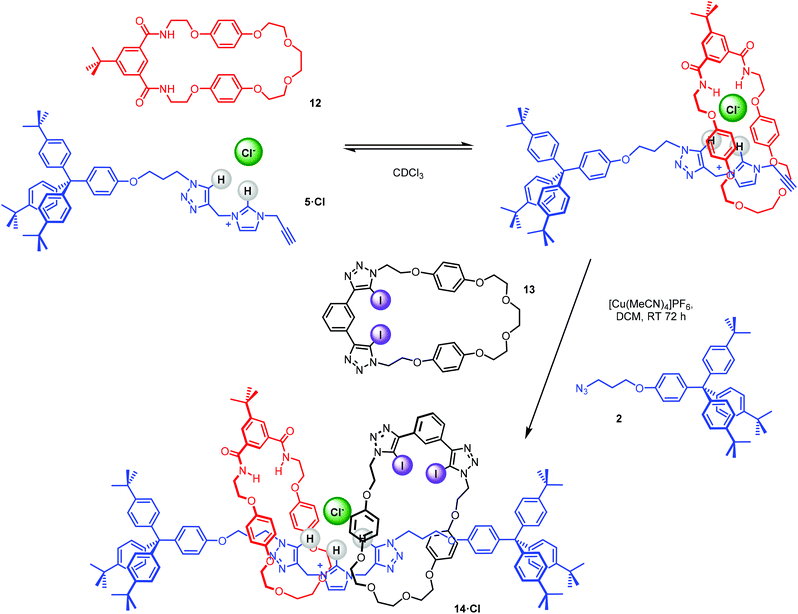 |
| | Scheme 4 Attempted synthesis of hetero[3]rotaxane 14·Clvia sequential anion–template pseudorotaxane formation and active–metal template stoppering rotaxanation reaction. | |
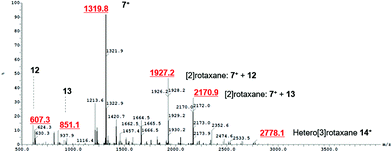 |
| | Fig. 7 ESI mass spectrum of crude reaction mixture showing formation of axle 7+, [2]rotaxane with macrocycle 12, [2]rotaxane with macrocycle 13 and hetero[3]rotaxane 14+. | |
Attempts to isolate the rotaxane products were carried out via preparative TLC, using a range of eluents. However, the similar polarities of the interlocked structures negated the successful isolation of the individual rotaxane products. This was nonetheless a promising proof-of-principle result and demonstrated that combining active metal- and anion–template methodologies was a plausible route for the formation of a hetero[3]rotaxane.
Conclusions
In conclusion, new XB and HB [2]rotaxanes incorporating novel bis-iodotriazole- and bis-prototriazole-imidazolium axle motifs were successfully prepared by an anion template methodology. 1H NMR anion titration experiments conducted in CDCl3/CD3OD/D2O (45:45:10 v/v) reveal both rotaxanes are capable of strong halide and sulfate recognition in competitive aqueous–organic solvent media. In comparison to the HB rotaxane analogue, the anion binding results notably highlight the incorporation of axle iodo-triazole XB donor motifs modulate the anion recognition preference for the lighter halides over iodide and sulfate, possibly as a consequence of steric hindrance effects disfavouring the binding of larger anions. An attempt was also made to prepare a novel hetero[3]rotaxane containing the bis-triazole imidazolium donor motif, via a combined AMT and chloride-template rotaxanation reaction. Although evidence of the target hetero[3]rotaxane was observed in the ESI mass spectrum of the crude reaction mixture, the higher order rotaxane could not be isolated by chromatographic purification. The integration of XB donor motifs into mechanically bonded interlocked host structural frameworks for aqueous anion recognition applications is continuing in our laboratories.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
G.T. and A.D thank the EPSRC for studentships (Grant reference numbers EP/M508111/1 and EP/N509711/1).
References
- M. T. Albelda, J. C. Frías, E. García-España and H.-J. Schneider, Chem. Soc. Rev., 2012, 41, 3859–3877 RSC.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- X. Wu, A. M. Gilchrist and P. A. Gale, Chem, 2020, 6, 1296–1309 CAS.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- G. Berger, P. Frangville and F. Meyer, Chem. Commun., 2020, 56, 4970–4981 RSC.
- Z. Xu, S. K. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 1457–1466 RSC.
- H. N. Kim, E.-H. Lee, Z. Xu, H.-E. Kim, H.-S. Lee, J.-H. Lee and J. Yoon, Biomaterials, 2012, 33, 2282–2288 CrossRef CAS PubMed.
- H. N. Kim, J. Lim, H. N. Lee, J.-W. Ryu, M. J. Kim, J. Lee, D.-U. Lee, Y. Kim, S.-J. Kim, K. D. Lee, H.-S. Lee and J. Yoon, Org. Lett., 2011, 13, 1314–1317 CrossRef CAS PubMed.
- B. Shirinfar, N. Ahmed, Y. S. Park, G.-S. Cho, I. S. Youn, J.-K. Han, H. G. Nam and K. S. Kim, J. Am. Chem. Soc., 2013, 135, 90–93 CrossRef CAS PubMed.
- F. Ostler, D. G. Piekarski, T. Danelzik, M. S. Taylor and O. G. Mancheño, Chem. – Eur. J., 2021, 27, 2315–2320 CrossRef CAS PubMed.
- H. A. Klein and P. D. Beer, Chem. – Eur. J., 2019, 25, 3125–3130 CAS.
- A. Borissov, J. Y. C. Lim, A. Brown, K. E. Christensen, A. L. Thompson, M. D. Smith and P. D. Beer, Chem. Commun., 2017, 53, 2483–2486 RSC.
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS PubMed.
- T. Bunchuay, A. Docker, U. Eiamprasert, P. Surawatanawong, A. Brown and P. D. Beer, Angew. Chem., Int. Ed., 2020, 59, 12007–12012 CrossRef CAS PubMed.
- R. Tepper, B. Schulze, M. Jäger, C. Friebe, D. H. Scharf, H. Görls and U. S. Schubert, J. Org. Chem., 2015, 80, 3139–3150 CrossRef CAS PubMed.
- Y. C. Tse, A. Docker, Z. Zhang and P. D. Beer, Chem. Commun., 2021, 57, 4950–4953 RSC.
- A. Docker, C. H. Guthrie, H. Kuhn and P. D. Beer, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202108591.
- A. Docker, T. Bunchuay, M. Ahrens, A. J. Martinez-Martinez and P. D. Beer, Chem. – Eur. J., 2021, 27, 7837–7841 CrossRef CAS PubMed.
- A. Docker, X. Shang, D. Yuan, H. Kuhn, Z. Zhang, J. J. Davis, P. D. Beer and M. J. Langton, Angew. Chem., Int. Ed., 2021, 60, 19442–19450 CrossRef CAS PubMed.
- L. E. Bickerton, A. Docker, A. J. Sterling, H. Kuhn, F. Duarte, P. D. Beer and M. J. Langton, Chem. – Eur. J., 2021, 27, 11738–11745 CrossRef CAS PubMed.
- T. K. Ghosh, S. Mondal, S. Bej, M. Nandi and P. Ghosh, Dalton Trans., 2019, 48, 4538–4546 RSC.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 213281 CrossRef CAS.
- R. Tepper, B. Schulze, P. Bellstedt, J. Heidler, H. Görls, M. Jäger and U. S. Schubert, Chem. Commun., 2017, 53, 2260–2263 RSC.
- S. C. Patrick, R. Hein, A. Docker, P. D. Beer and J. J. Davis, Chem. – Eur. J., 2021, 27, 10201–10209 CrossRef CAS PubMed.
-
A. Docker and P. D. Beer, in Halog. Bond. Solut, John Wiley & Sons, Ltd, 2021, pp. 83–120 Search PubMed.
- J. Warnan, Y. Pellegrin, E. Blart and F. Odobel, Chem. Commun., 2011, 48, 675–677 RSC.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed.
- W. S. Brotherton, R. J. Clark and L. Zhu, J. Org. Chem., 2012, 77, 6443–6455 CrossRef CAS PubMed.
- M. R. Sambrook, P. D. Beer, M. D. Lankshear, R. F. Ludlow and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 1529–1538 RSC.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- J. D. Crowley, S. M. Goldup, A. L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292–2302 CrossRef CAS PubMed.
- J. Y. C. Lim, T. Bunchuay and P. D. Beer, Chem. – Eur. J., 2017, 23, 4700–4707 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt02414k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*