
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT insights into the photocatalytic reduction of CO2 to CO by Re(I) complexes: the crucial role of the triethanolamine “magic” sacrificial electron donor†
Athanassios C.
Tsipis
 * and
Antonia A.
Sarantou
* and
Antonia A.
Sarantou
Department of Chemistry, University of Ioannina, Ioannina 45110, Greece. E-mail: attsipis@uoi.gr
First published on 13th September 2021
Abstract
The reaction mechanism for the photocatalytic reduction of CO2 to CO catalyzed by the [Re(en)(CO)3Cl] complex in the presence of triethanolamine, R3N (R = CH2CH2OH) abbreviated as TEOA, in DMF solution was studied in-depth with the aid of DFT computational protocols by calculating the geometric and free energy reaction profiles for several possible reaction pathways. The reaction pathways studied start with the “real” catalytic species [Re(en)(CO)3], [Re(en)(CO)3]− and/or [Re(en)(CO)2Cl]− generated from the excited triplet T1 state upon single and double reductive quenching by a TEOA sacrificial electron donor or photodissociation of a CO ligand. The first step in all the catalytic cycles investigated involves the capture of either CO2 or the oxidized R2NCH2CH2O˙ radical. In the latter case, the CO2 molecule is captured (inserted) by the Re–OCH2CH2NR2 bond forming stable intermediates. Next, successive protonations (TEOA also acts as a proton donor) lead to the release of CO either from the energy consuming 2e− reduction of [Re(en)(CO)4]+ or [Re(en)(CO)2Cl]+ complexes in the CO2 capture pathways or from the released unstable diprotonated [R2NCH2CH2OC(OH)(OH)]+ species regenerating TEOA and the catalyst. The CO2 insertion reaction pathway is the favorable pathway for the photocatalytic reduction of CO2 → CO catalyzed by the [Re(en)(CO)3Cl] complex in the presence of TEOA manifesting its crucial role as an electron and proton donor, capturing CO2 and releasing CO.
Introduction
It is generally accepted that anthropogenic sources, such as burning of fossil fuels, are mainly responsible for the rise of atmospheric CO2, which in turn accentuates the greenhouse effect.1 Many studies have been devoted to how to tackle this problem and there are continuous ongoing efforts not only to capture CO2 but also to catalytically transform it into fine chemicals.2 Among the various strategies to achieve this goal, the most challenging and the very attractive one is to convert CO2 efficiently into useful compounds using solar light as an energy source.3 Many transition metal-based complexes (Ni, Fe, Re, Cr, Ir, Mo, etc.) have been studied extensively for the homogeneous electrocatalytic and photocatalytic reduction of carbon dioxide.4–16Apaydin et al.7 reported an excellent comprehensive overview on the homogeneous and heterogeneous CO2 reduction catalyzed by organic, organometallic and bioorganic systems. Mechanistic details of the CO2 reduction processes undertaken by electrochemical, bioelectrochemical and photoelectrochemical approaches are thoroughly analyzed. In parallel to the electrochemical CO2 reduction, a plethora of efforts were focused on the photocatalytic CO2 reduction, which relied on systems comprising a light harvesting unit consisting of a photosensitizer (PS) compound and two catalytic sites.8–14 In the oxidation site, a donor provides an electron e− to the PS after its excitation to the triplet excited state (3MLCT) which is subsequently reductively quenched by the reduction site and finally, the e− is transferred to CO2. However, in many cases, the PS acts not only as a photosensitizer but also as a reduction site as well.
Among the mononuclear transition metal complexes developed so far for electro- and photocatalytic CO2 reduction, polypyridyl transition metal complexes constitute the class of molecular catalysts employed in the reduction of CO2 to CO. An excellent overview of the CO2 reduction catalyzed by polypyridyl transition metal complexes has recently been published by Fontecave's group,8 presenting and thoroughly analyzing the proposed catalytic cycles for the CO2 reduction by Re(bpy)CO3(X) investigated by both experimental and computational approaches. Generally, mechanistic studies indicated that, initially a two electron reduction of the Re(bpy)CO3(X) catalyst affords the anionic five-coordinated 17e− [Re(bpy)(CO)3]− complex which captures CO2 forming the [Re(bpy)(CO)3(CO2)]− intermediate, which upon protonation yields the Re(bpy)(CO)3(CO2H) intermediate which upon second protonation and additional electron reduction is converted into [Re(bpy)(CO)4]−. However, a question still remains concerning the release of the CO product from the [Re(bpy)(CO)4]− complex. More recently, Ishitani and co-workers11 presented an excellent discussion of all the reaction mechanisms proposed for the photochemical CO2 reduction catalyzed by Re(I) and Ru(II) complexes. The authors stated that no one from the numerous mechanisms proposed is a universal mechanism. This review article is recommended to the readers and researchers in the field. On the other hand, Cramer's group reported mechanistic details on the proton-dependent electrocatalytic reduction of CO2 to CO by fac-Re(bpy)(CO)3Cl using first principles quantum chemistry.13 The Cramer's group also compared the complete electrocatalytic cycles of CO2 to CO reduction catalyzed by fac-Re(bpy)(CO)3Cl and fac-Mn(bpy)(CO)3Cl catalysts.14
The photo-induced reduction of CO2 to CO in the acetonitrile/water/triethylamine solution in the presence of a [Ru(2,2′-bipyridine)3]2+/Co2+ system was first reported by Lehn and Ziessel.17 In subsequent publications, Lehn's group18,19 showed that the most selective reduction catalysts to produce CO from CO2 are Re(I) octahedral complexes with the general formula [Re(bpy)(CO)3X] (X = Cl, Br). The proposed catalytic cycle for the photoreduction of CO2, catalyzed by the (bpy)Re(CO)3X complexes, comprises excitation to the 3MLCT state and reduction of the Re(I) catalyst using triethanolamine (TEOA) as an electron donor yielding the one electron reduced Re(I) complex (OER-species) that loses the X− ligand forming an unstable 17e− species. Next, the 17e− Re(I) complex could capture CO2via coordination to the rhenium metal center.17–19 However, the mechanism of conversion of CO2 to CO has not yet been fully understood and there are some points that are still under debate and need to be clarified.
The mechanism of the photoinduced reduction of CO2 to CO in a TEOA/DMF/[ReBr(CO)3(bpy)] system has been investigated by Kutal et al.20,21 Reductive quenching of the photoexcited [ReBr(CO)3(bpy)] complex by TEOA affords the reduced [Re′Br-(CO)3(bpy−)] species which can be viewed as a Re’ center bound to a 2,2′-bipyridine radical anion. The TEOA˙+ generated can rapidly abstract a hydrogen atom from another TEOA molecule to produce the strong reducing radical, TEOA˙. Next, the 19e− Re species activates CO2 for reduction to CO, but the composition, structure, or subsequent reactivity of any intermediates formed is unknown.
Although several studies22–29 point towards the existence of the 17e− reactive species, the next step involving capturing CO2 by the catalytic system is unclear. Kubiak and co-workers22–25 investigated the catalytic activity of the Re(bpy)(CO)3X complexes (bpy = 4,4′-dicarboxyl-2,2′-bipyridine, 2,2′-bipyridine, 4,4′-dimethyl-2,2′-bipyridine, 4,4′-di-tert-butyl-2,2′-bipyridine, and 4,4′-dimethoxy-2,2′-bipyridine) and found that Re(bipy-tBu)(CO)3Cl has the most significant catalytic activity for the reduction of CO2 to CO. DFT calculations showed that the geometry of the [Re(bipy-tBu)(CO)3(CO2)]− doubly reduced species converges only upon inclusion of a cation (H, Li, Na, or K). Based on these studies, the authors proposed a catalytic cycle that involves a two-electron reduction of the [Re(bpy-R)(CO)3X] (R = H, Me, tBu, X = halogen, OTf) complex which upon dissociation of the X ligand yields the anionic [Re(bpy-R)(CO)3]− species. Several [Re(bpy-R)(CO)3]− anions were isolated and fully characterized.25 Next, the CO2 molecule is coordinated to the Re metal center yielding a carboxylate [Re(bpy-R)(CO)3(CO2)]− intermediate, which upon protonation forms the carboxylato [Re(bpy-R)(CO)3(CO2H)] complex. Further protonation of the [Re(bpy-R)(CO)3(CO2H)] complex releases H2O and produces the cationic tetracarbonyl species. The release of CO from the [Re(bpy-R)(CO)4]+ complex is catalysed by electron transfer.
On the other hand, Muckerman et al.26 in a prominent theoretical study precluded direct coordination of the Re metal center and instead proposed that the catalytic cycle proceeds via the formation of a Re dimeric intermediate species. In the latter, CO2 acts as a bridging ligand between the two Re metal centers, though the formation of the carboxylate dimeric species is entropically unfavorable. Ishitani et al.27 proposed an alternative route by revealing the crucial role of TEOA in the CO2 capture by the Re(I) catalyst. The authors proposed a catalytic cycle for the photocatalytic CO2 reduction catalyzed by the [Re(bpy)(CO)3(NCS)] complex that involves excitation of the S0 ground state of the [Re(bpy)(CO)3(NCS)] complex to the triplet 3MLCT state, which subsequently is reductively quenched by TEOA, giving the one electron reduced (OER) [Re(bpy)(CO)3(NCS)]−· species. The dissociation of the SCN− ligand from the OER yields a 17e− five-coordinated intermediate which reacts with CO2 to give the CO2 adduct(s). Further electron donation to the OER results in the release of CO and the [Re(bpy)(CO)3]+ complex and regenerates the catalyst. Later Ishitani's group28 performing spectrochemical and cyclic voltammetry measurements on adding TEOA to a solution of fac-[ReI(bpy)(CO)3(CH3CN)]PF6 dissolved in pure DMF provided evidence for the existence of the fac-[ReI(bpy)(CO)3(OCH2CH2NR2)] (R = CH2CH2OH) intermediate in solution. A Re(I) intermediate with the CO2 inserted between the Re(I) metal center and the TEOA anionic ligand was isolated and fully characterized.
The photocatalytic activity of transition metal-based catalysts towards the reduction of CO2 to CO depends upon the structure of the complexes. Attempts to address this issue have already been made. For example, Kurz et al.29 performed an experimental structure–activity study of [(diimine)(CO)3ReX] complexes with respect to their ability to act as photocatalysts for the reduction of CO2 to CO. Also, Ishida et al.30 studied the effect of ligand L on the photocatalytic activity for CO2 conversion in a series of trans-(Cl)-[Ru(L)(CO)2Cl2] complexes, revealing the significance of their reduction potential, Ep. The latter is modulated by the Lowest Unoccupied Molecular Orbital (LUMO) eigenvalue which in turn depends on the nature of L.
In this regard, the main goal that we pursued in this work was to scrutinize the mechanistic details of the photocatalytic conversion of CO2 to CO by the octahedral [(en)(CO)3ReCl] (en = ethylenediamine) complex selected as a model complex, employing DFT methods. Our efforts were directed towards the elucidation of the mechanism of the reaction step related to CO2 capture and CO release in the reduction of CO2 to CO catalyzed by Re(I) complexes in DMF/TEOA solutions.
Computational methods
All stationary points (reactants, transition states, and products) located on the potential energy surfaces (PES) were fully optimized at the wB97XD/Def2-TZVP level of theory as implemented in the Gaussian16 suite of programs.31 The wB97XD functional32–35 containing empirical dispersion terms and long-range corrections provides good descriptions of the reaction profiles, including geometries, heats of reaction, and barrier heights.36 Analytical frequencies were calculated at the same level of theory, and the nature of the stationary points was determined in each case according to the number of the negative eigenvalues of the Hessian matrix. Gibbs free energies were calculated at 298.150 Kelvin and 1 Atm. Solvent effects were accounted for by means of the Polarizable Continuum Model (PCM) with the integral equation formalism variant (IEF-PCM) being the default self-consistent reaction field (SCRF) method.37 DMF was used as the solvent. Natural Bond Orbital (NBO) population analysis was performed using Weinhold's methodology.38,39 Time-dependent density functional theory (TD-DFT)40–42 calculations were performed on the equilibrium ground state geometries in DMF solution employing the wB97XD/Def2-TZVP/PCM computational protocol.Results and discussion
Computation-based catalytic cycles
The photocatalytic CO2 → CO transformation catalyzed by the selected [(en)(CO)3Re(Cl)] model catalyst proceeds through three main reaction steps: (i) step A1 corresponds to the photo-excitation of the [(en)(CO)3Re(Cl)] catalyst accompanied by one electron reduction of the triplet excited state T1 by TEOA, the so-called “magic” sacrificial electron donor,28 yielding very reactive coordinatively unsaturated, 17e− intermediates that are the catalytically active species initiating step B1. (ii) Step B1 involves the interaction of the oxidized TEOA˙+ species formulated as [R2NCH2CH2OH]˙+ (R = CH2CH2OH) with the Re metal center yielding [(en)(CO)3Re(OCH2CH2NR2)] intermediates capable of capturing CO2 by insertion into the Re–O bond. Step B1 has thoroughly been investigated both experimentally and theoretically by Ishitani's group28 employing the fac-[ReI(bpy)(CO)3(CH3CN)]PF6 catalyst. (iii) Step C starts with the [R2NCH2CH2C(O)OH]˙+ species released upon protonation of the [(en)(CO)3ReO(O)(COCH2CH2NR2)] intermediate and its conversion upon further protonation to the desired CO molecule, H2O and regenerating TEOA. Step C manifests the crucial role of TEOA in the photocatalytic reduction of CO2 → CO, thus clarifying, the “magic” sacrificial electron donor features of TEOA.28The reaction steps for the photocatalytic reduction of CO2 to CO catalyzed by the [(en)(CO)3ReCl] (en = ethylenediamine) catalyst, calculated at the wB97XD/def2-TZVP level, in a DMF solution are depicted schematically in the ESI (Fig. S1†). The alternative reaction steps B1′ and B1′′ starting with the coordination of CO2 to the Re metal center of ImA1 and/or ImA1′ (one electron reduced ImA1 species) intermediates yielding the [Re(en)(CO)3(CO2)] and/or [Re(en)(CO)3(CO2)]− intermediates, which upon successive protonations afford the tetracarbonyl [Re(en)(CO)4]2+ and [Re(en)(CO)4]+ complexes respectively will be discussed separately and compared with step B1. We also investigated alternative reaction pathways starting with a five-coordinated [(en)(CO)2ReCl]− intermediate that resulted from the dissociation of one of the equatorial CO ligands of the excited T1 triplet state (step A2) which will be discussed later on.
Reaction steps A1 and A2: photophysics of the [(en)(CO)3ReCl] catalyst
Let us examine first steps A1 and A2 concerning the photo-excitation of the [(en)(CO)3ReCl] catalyst. Since excitation of the [(en)(CO)3ReCl] complex is a prerequisite in order to initiate the photocatalytic reduction of CO2 to CO, we set out to study the photophysical properties by means of Time Dependent DFT (TDDFT) electronic structure calculations. The simulated absorption spectrum of the [(en)(CO)3ReCl] complex calculated by the TDDFT/wB97XD/Def2-TZVP/PCM computational protocol shows absorption bands spanning between 200 and 300 nm with the most intense among them peaking at 211, 231 and 273 nm. The peak at 211 nm exhibits a complex nature and could be assigned as Metal to Ligand/Ligand to Ligand Charge Transfer (MLCT/LL′CT). The same assignment holds also true for the electronic transitions absorbing at 231 and 273 nm as well.The equilibrium geometries of the [(en)(CO)3ReCl] complex in the ground S0 and excited triplet T1 states along with the catalytically active five-coordinated [(en)(CO)3Re] and [(en)(CO)2ReCl] species formed in photo-physical steps A1 and A2 respectively are shown in Fig. 1. Calculations predicted that the fac-[(en)(CO)2ReCl] isomer is more stable than the mer-[(en)(CO)2ReCl] isomer in both the S0 and T1 states by 24.1 and 6.8 kcal mol−1 respectively.
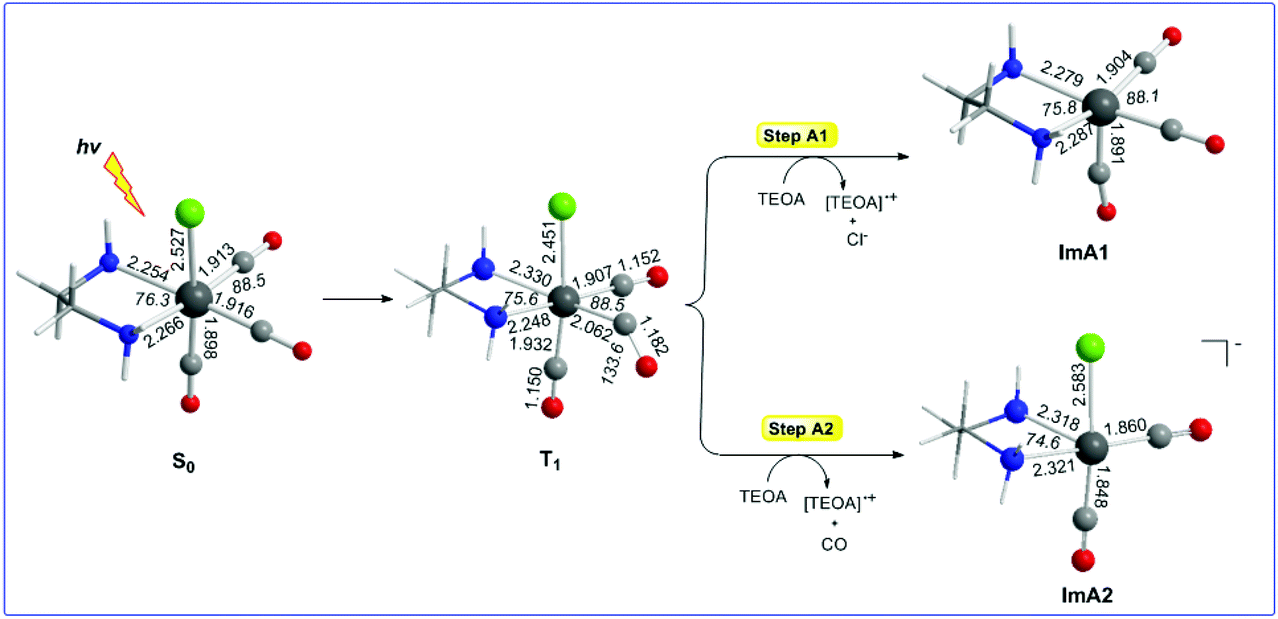 | ||
| Fig. 1 Equilibrium geometries of the [(en)(CO)3ReCl] complex in the ground S0 and excited triplet T1 states along with the catalytically active five-coordinated [(en)(CO)3Re], ImA1 and [(en)(CO)2ReCl], ImA2 species formed in photo-physical steps A1 and A2 respectively calculated at the wB97XD/Def2-TZVP level of theory in a DMF solution. | ||
It can be seen that one electron transfer from TEOA to the T1 state of [(en)(CO)3ReCl] promotes the cleavage of either the Re–Cl or the Re–CO bond in the reduced 19e− transient OER species formed, yielding the very reactive 17e− [(en)(CO)3Re], ImA1 and [(en)(CO)2ReCl]−, ImA2 intermediates respectively. Noteworthily in T1 one of the equatorial CO ligands forms a weaker Re–CO bond (Re–CO bond length of 2.062 Å) compared to the Re–CO bonds of the other two CO ligands in the equatorial and axial coordination sites with bond lengths of 1.907 and 1.932 Å respectively. Interestingly the Re–C–O bond angle of the weakly bonded CO ligand is 133.6°. Scheme 1 shows the 3D plots of the frontier molecular orbitals (FMOs) and the spin density distribution of T1, along with the estimated Wiberg Bond Indices (WBOs) for the Re–Cl and Re–CO bonds in T1 (the entry in the right side of Scheme 1).
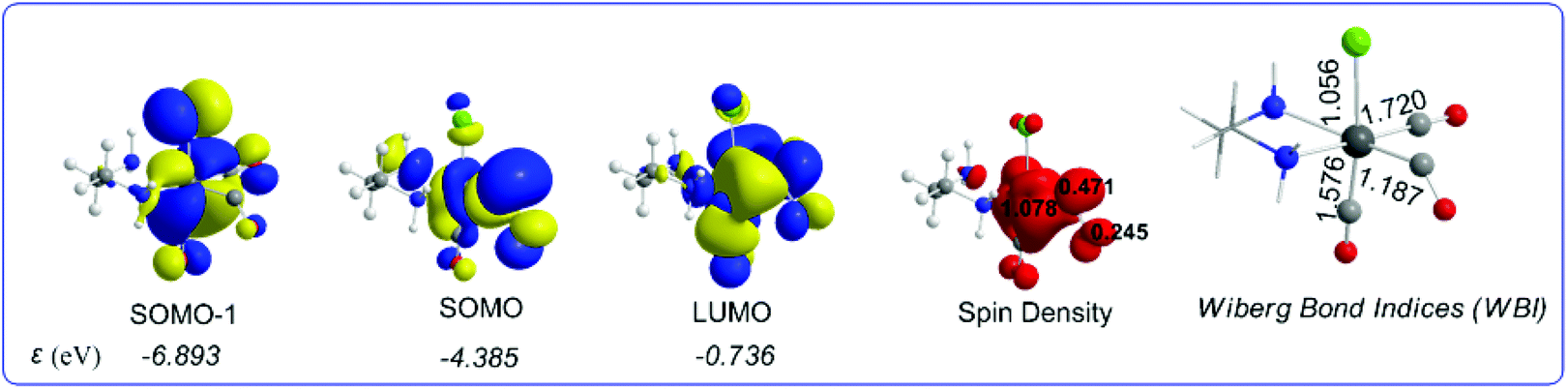 | ||
| Scheme 1 3D plots of the frontier molecular orbitals (FMOs), spin density distribution and Wiberg Bond Indices (WBI) of T1, calculated at the wB97XD/Def2-TZVP level of theory. | ||
It can be seen that the Single Occupied Molecular Orbital (SOMO) in T1 is mainly localized on the weakly bonded CO ligand. The SOMO is composed of 19.1% Re spd, 45.2% C sp hybrid orbitals and 18.9% O p orbitals. The Lowest Unoccupied Molecular Orbital (LUMO) is composed of 25.8% Re spd and 24.0% C sp hybrid orbitals. The spin density is distributed on the Re–CO bond region, 1.078|e| on Re and 0.716 on the CO ligand. The estimated WBI(Re–CO) shows also their bond strengths, being 1.187 for the weaker bonded CO ligand and 1.720 and 1.576 for the CO ligands coordinated at the equatorial and axial coordination sites respectively.
Reaction step B1: CO2 insertion
The geometric and free energy reaction profiles calculated for step B1 are shown in Fig. 2.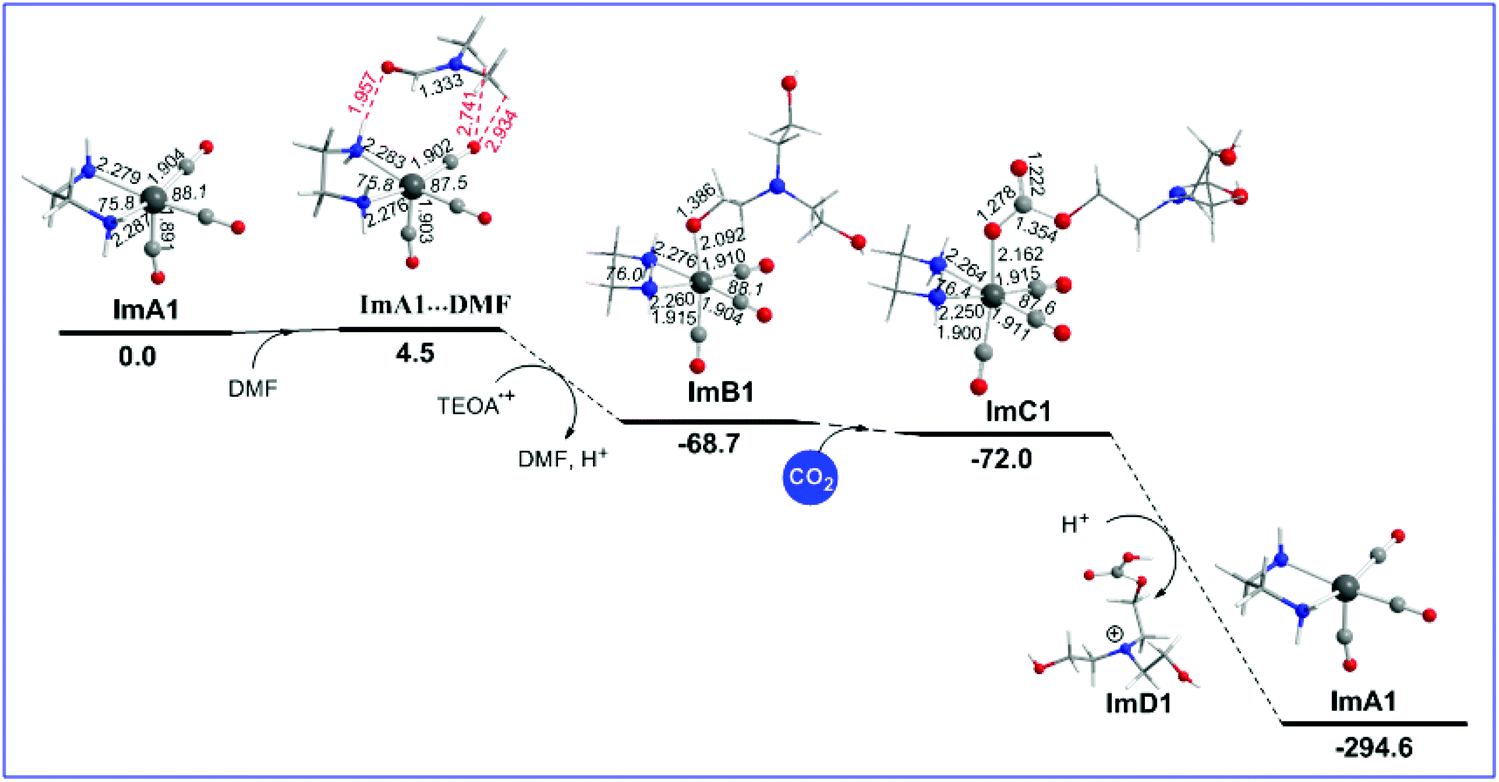 | ||
| Fig. 2 Geometric and free energy, ΔG (in kcal mol−1), reaction profiles for step B of the reduction of CO2 to CO catalyzed by the [(en)(CO)3ReCl] catalyst calculated at the wB97XD/Def2-TZVP level in the DMF solution. The sum of GImA1 + GDMF at an infinite distance was considered to be 0.00. | ||
Initially, a DMF molecule interacts with the very reactive 17e− five coordinate intermediate ImA1, with the estimated interaction energy (IE) being −7.4 kcal mol−1 forming a loose association through an endergonic process (ΔG = 4.5 kcal mol−1). The ImA1⋯DMF interactions correspond to non-covalent interactions clearly shown in Scheme 2.
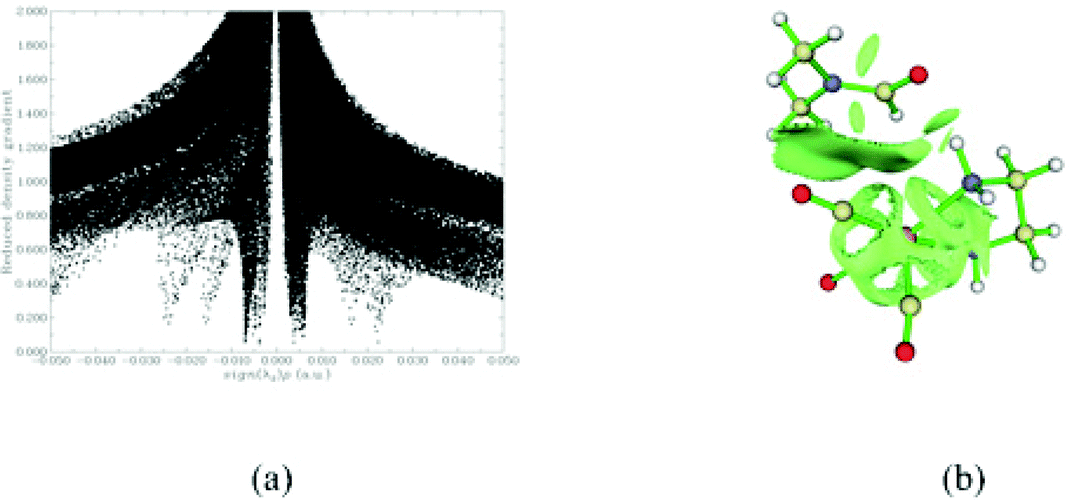 | ||
| Scheme 2 Scatter graphs of the RDG vs. sign(λ2)ρ(r) correlations (a) along with the 3D RDG isosurfaces (isosurface = 0.70 au) for the ImA1⋯DMF intermediate calculated at the wB97XD/Def2-TZVP level of theory. | ||
Next, DMF is substituted by an OCH2CH2NR2 ligand yielding an ImB1′ intermediate, in line with the experimental observations by Ishitani's group28 that upon adding TEOA to a solution of the fac-[ReI(bpy)(CO)3(DMF)]+ complex (Im_DMF), a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 thermodynamically equilibrated mixture of Im_DMF with the fac-[ReI(bpy)(CO)3(OCH2CH2NR2)] (R = CH2CH2OH) (ImB) intermediate is obtained. The equilibrium between Im_DMF and ImB intermediates was further verified by IR spectroscopy.11 The substitution reaction is exergonic (ΔG = −68.7 kcal mol−1).
:2 thermodynamically equilibrated mixture of Im_DMF with the fac-[ReI(bpy)(CO)3(OCH2CH2NR2)] (R = CH2CH2OH) (ImB) intermediate is obtained. The equilibrium between Im_DMF and ImB intermediates was further verified by IR spectroscopy.11 The substitution reaction is exergonic (ΔG = −68.7 kcal mol−1).
Natural Bond Orbital (NBO) population analysis revealed that the OCH2CH2NR2 ligand is coordinated to the Re metal centre of the [(en)(CO)3Re] catalytic species, ImA1, forming a relatively weak Re–O bond with an estimated WBI(Re–O) of 0.550. Note that both the Re metal centre and the O donor atom of the coordinated OCH2CH2NR2 ligand acquire negative natural atomic charges of −0.592 and −0.779|e| respectively. The bonding σ(Re–O) NBO is constructed from the interaction of sp2.69d2.88 hybrid orbitals (40.8% p and 43.7% d character) of Re with an sp1.82 hybrid (64.5% p-character) on the oxygen donor atom and is described as σ(Re–O) = 0.395hRe + 0.919hO. The occupancy or the σ(Re–O) NBO is 1.939|e|.
Next, a CO2 molecule is inserted into the Re–OCH2CH2NR2 bond transforming the OCH2CH2NR2 to the OC(O)OCH2CH2NR2 ligand yielding intermediate ImC1. The insertion process is slightly exergonic by only −3.3 kcal mol−1 (Fig. 2). ImC1 could be subjected to protonations at the O atoms of the coordinated OC(O)OCH2CH2NR2 ligand marked as Oa and Ob protonation sites (Fig. S1†). The natural atomic charges at the Oa and Ob atoms are −0.759 and −0.708|e| respectively. The protonation either at Oa or Ob of the coordinated [R2N–CH2CH2O–C(O)O] ligand forces the rupture of the Re–O bond which yields ImD1 formulated as [R2N–CH2CH2O–C(O)OH]+ and regenerates the five coordinated 17e−ImA1 catalytic species, thus closing the catalytic cycle. The protonation accompanied by ligand dissociation corresponds to a strongly exergonic process (ΔG = −222.6 kcal mol−1).
Reaction step C
ImD is the species initiating reaction step C that delivers the desired CO product. The geometric and free energy reaction profiles calculated for step C are shown in Fig. 3. As in the case of ImC1 (vide supra), there are two possible protonation sites in ImD (marked as a and b in Fig. S1†). The natural atomic charges on the Oa and Ob atoms of ImD are −0.668 and −0.659|e| respectively. Thus, the protonation at the Oa site is the most favoured one yielding ImF which upon further protonation and one electron reduction delivers the CO product and regenerates TEOA. The ImD → ImF transformation is strongly exergonic (ΔG = −107.5 kcal mol−1). ImF can also be obtained upon protonation at the Ob site in ImD forming a transition state TS (νi = −1888 cm−1) surmounting an activation barrier of 41.1 kcal mol−1 (Fig. 1). The normal coordinate vectors (arrows) of the vibrational modes corresponding to the imaginary frequency of TS show that the dominant motions reflect the structural changes leading to the formation of ImF. The relatively high activation barrier indicates that the ImD → TS → ImF is not a favoured pathway. Noteworthily reaction step C is a key step that manifests the crucial role of the TEOA sacrificial electron donor in the photocatalytic reduction of CO2 to CO catalyzed by Re(I) complexes.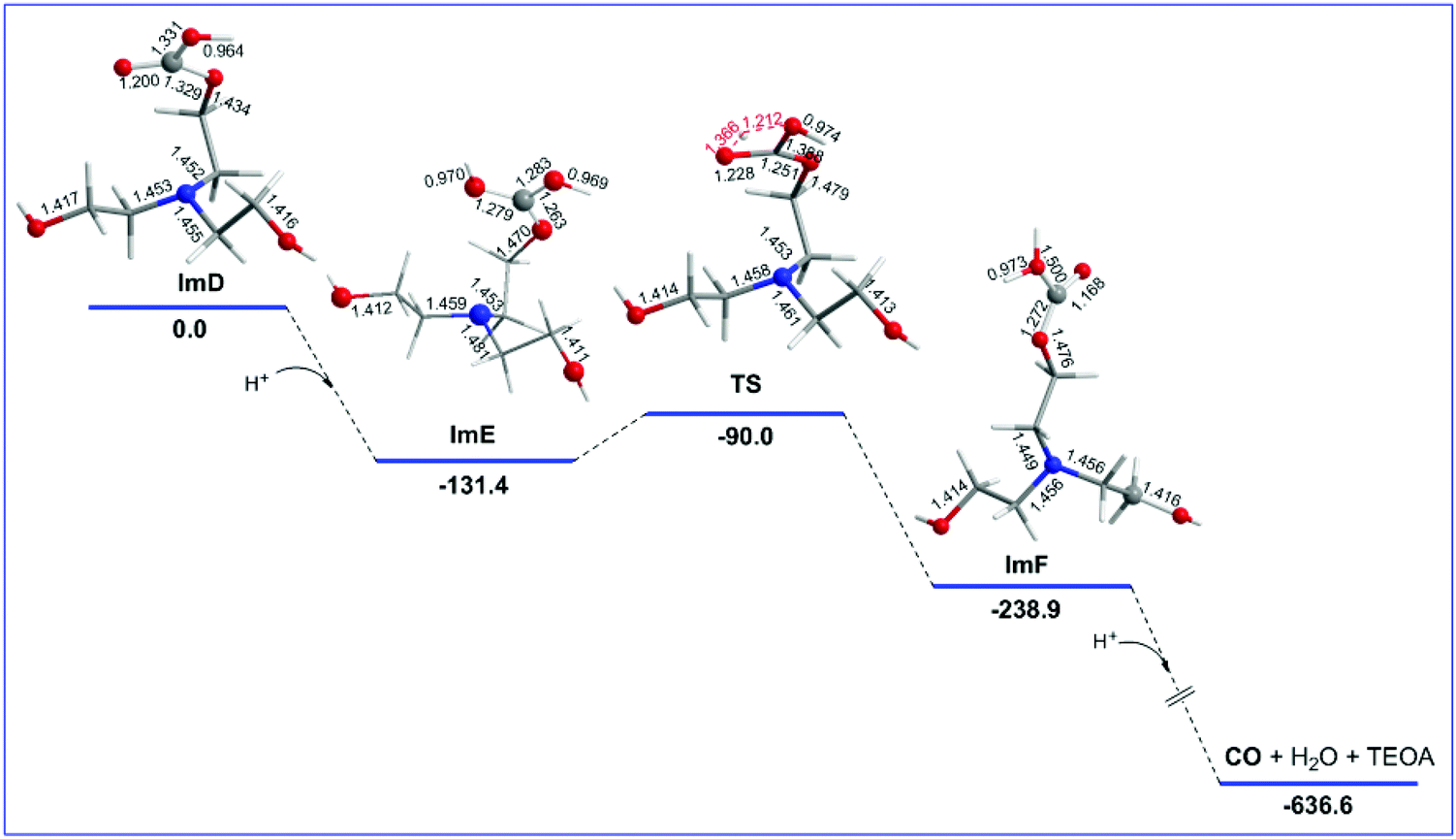 | ||
| Fig. 3 Geometric and free energy, ΔG (in kcal mol−1), reaction profiles for step C of the reduction of CO2 to CO catalyzed by the [(en)(CO)3ReCl] catalyst calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
Reaction steps B1′ and B1′′: CO2 capture
Alternative reaction pathways (steps B1′ and B1′′) involving direct capture of CO2 by the active ImA1′ catalytic species (step B1′) or its one electron reduced ImA1′′ species (step B1′′) yielding the [Re(en)(CO)3(CO2)] and [Re(en)(CO)3(CO2)]− intermediates, respectively have also been explored by DFT methods. The proposed catalytic cycles for steps B1′ and B1′′ are shown in Fig. 4. The geometric and free energy reaction profiles calculated for steps B1′ and B1′′ are given in Fig. 5 and 6 respectively.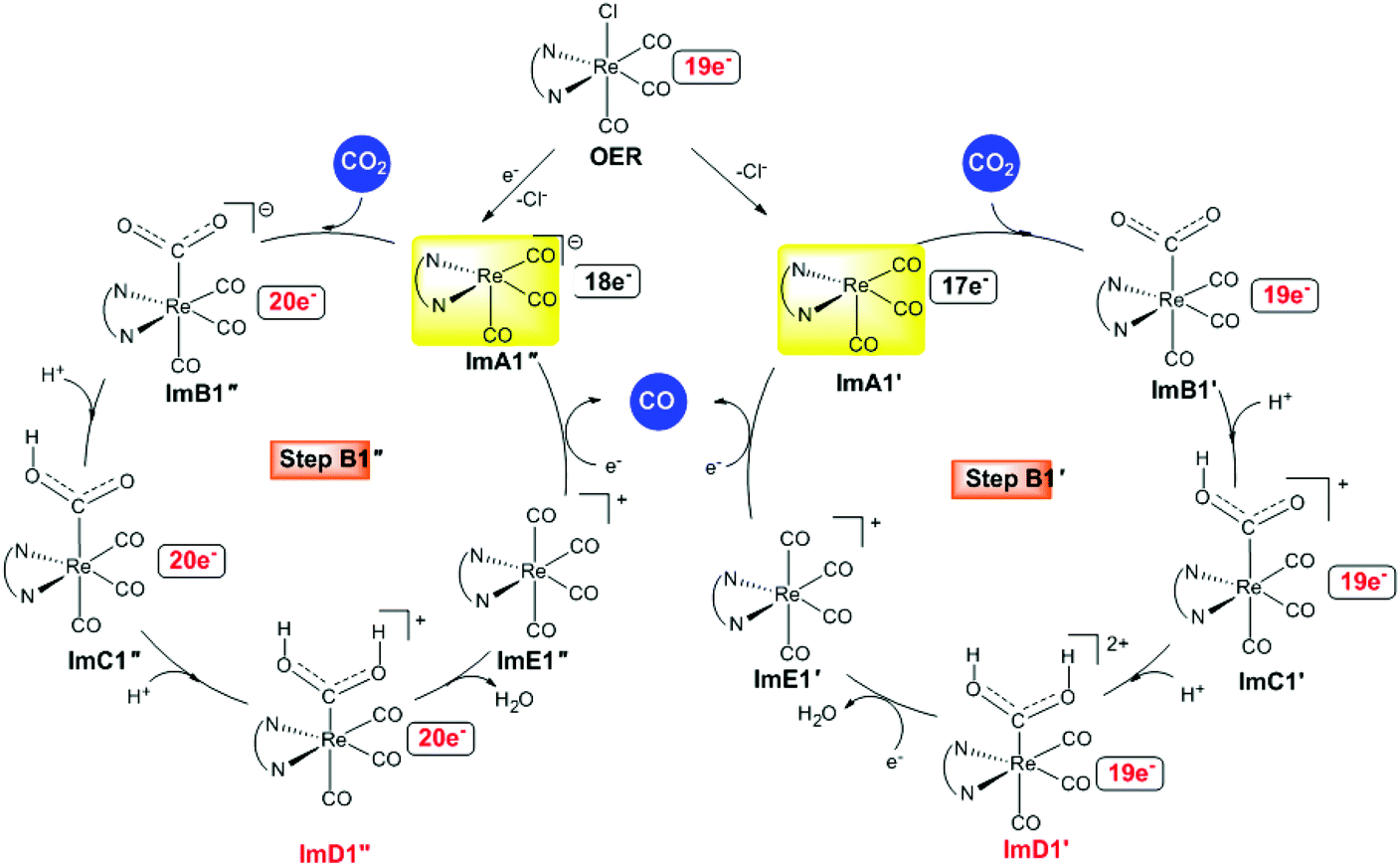 | ||
| Fig. 4 Proposed catalytic cycles for steps B1′ and B1′′ calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
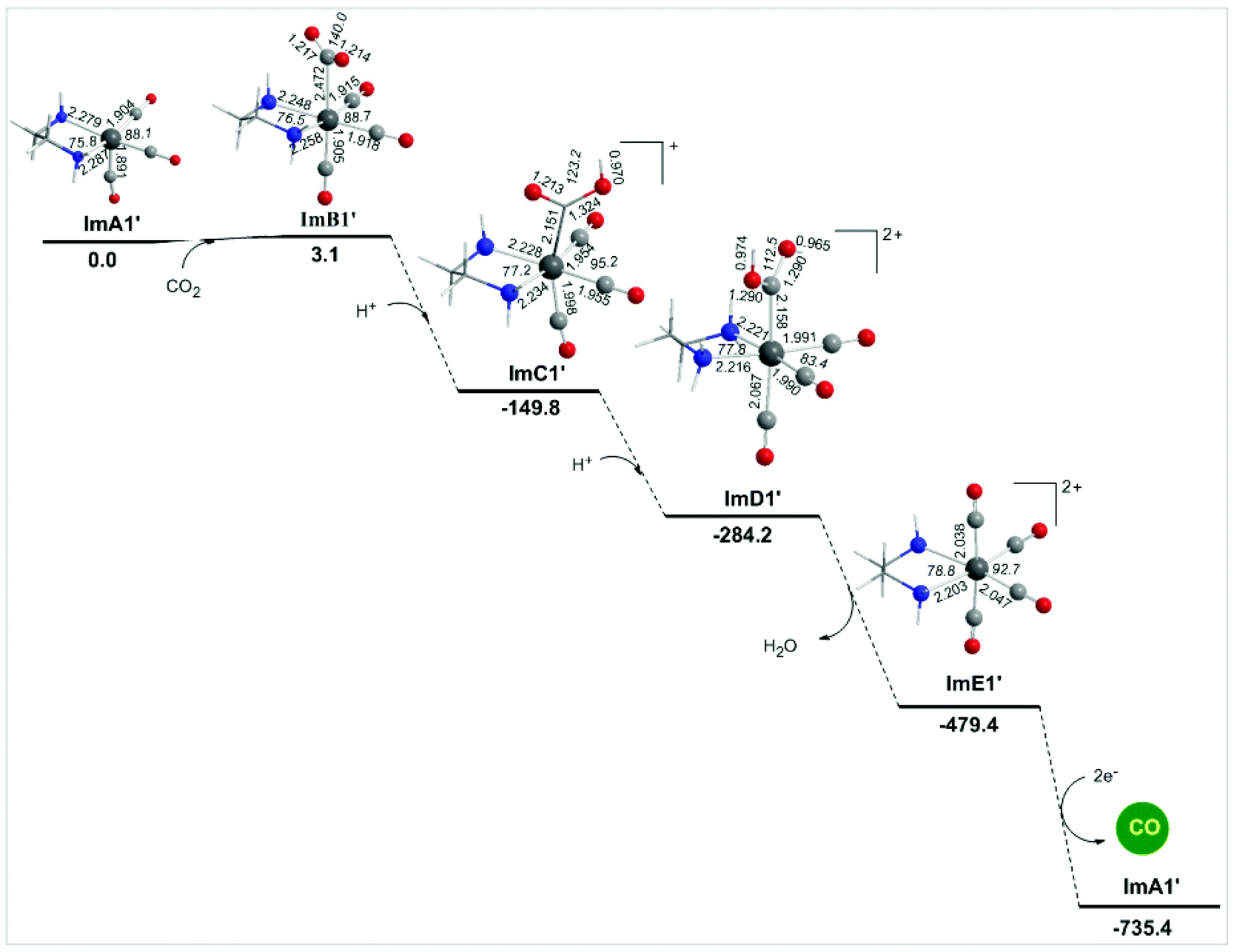 | ||
| Fig. 5 Geometric and free energy, ΔG (in kcal mol−1), reaction profiles for step B1′ of the reduction of CO2 to CO catalyzed by the [(en)(CO)3ReCl] catalyst calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
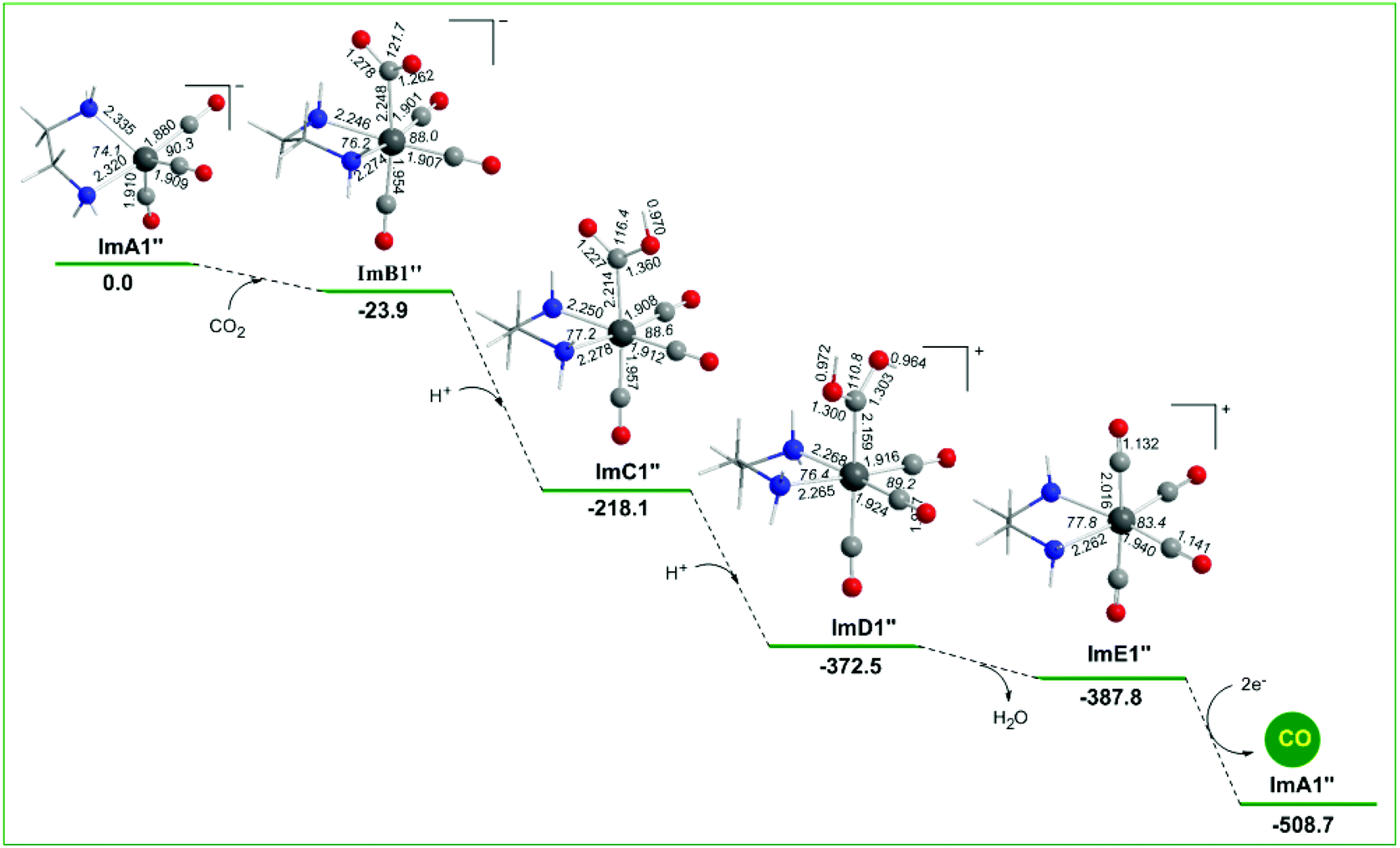 | ||
| Fig. 6 Geometric and free energy, ΔG (in kcal mol−1), reaction profiles for step B1′′ of the reduction of CO2 to CO catalyzed by the [(en)(CO)3ReCl] catalyst calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
After excitation of the S0 ground state of the [Re(en)(CO)3Cl] complex to the triplet T1 state, the latter could undergo one and two electron reductions by TEOA accompanied by Cl− dissociation affording the catalytically active 17e−ImA1′ and the anionic 18e−ImA1′′ intermediates which initiate reaction steps B1′ and B1′′ respectively (Fig. 4).
According to NBO population analysis, the Re metal centers in ImA1′ and ImA1′′ intermediates acquire negative natural atomic charges of −0.752 and −1.174|e| respectively. Noteworthily the Re metal center in ImA1′ and ImA1′′ is a nucleophilic center and therefore could not be attacked by a nucleophile. In this regard, direct coordination of CO2 (CO2 capture) to the Re metal center of ImA1′ and ImA1′′ intermediates seems to be highly unlikely in line with the experimental observations reported by Ishitani's group28 that no reaction occurs when CO2 is added to a solution of the fac-[ReI(bpy)(CO)3(DMF)]+ complex, while the capture of CO2 was observed when bubbling CO2 into a equilibrium mixture of fac-[ReI(bpy)(CO)3(DMF)]+/fac-[ReI(bpy)(CO)3(OCH2CH2NR2)] (bpy = 2,2′-bipyridine) complexes in solution by 1H and 13C NMR spectroscopy yielding the fac-[ReI(bpy)(CO)3{R2N–CH2CH2O–COO}] complex which was isolated and fully characterized.
Scheme 3 shows the 3D plots of the frontier molecular orbitals (FMOs) and spin density distributions of ImA1′ and ImA1′′ catalytic species.
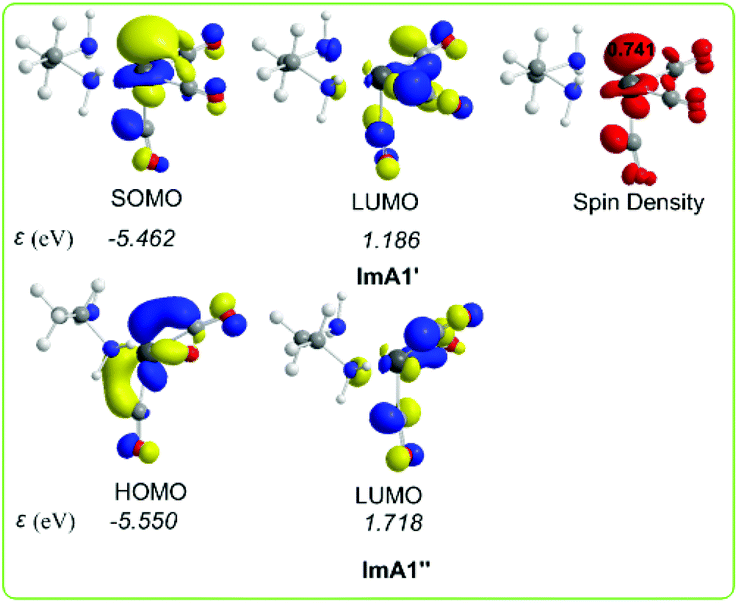 | ||
| Scheme 3 3D plots of the frontier molecular orbitals (FMOs) and spin density distribution of ImA1′ and ImA1′′ catalytic species calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
The capture of CO2 by ImA1′ in the reaction step B1′ yielding ImB1′ (Fig. 6) is slightly endergonic (3.1 kcal mol−1), while the estimated interaction energy is 7.0 kcal mol−1. According to the NBO population analysis, the CO2 molecule is loosely associated with the Re metal centre of the ImA1′ catalytic species, with the Re–CO2 bond having a WBI(Re–CO2) of only 0.348. Note that the Re metal centre acquires a negative natural atomic charge of −0.808|e| and the C atom of the coordinated CO2 a positive natural atomic charge of 0.755|e|. The side-on orientation of the CO2 molecule in the ImB1′ intermediate indicates that non-covalent (dipole–dipole and electrostatic) interactions predominate in the coordination of CO2 to the ImA1′ catalytic species. The weak covalent interactions are mirrored on the bonding σ(Re–CO2) NBO which is constructed from the interaction of the sp3.52d2.74 hybrid orbitals (48.4% p and 37.7% d character) of Re with an sp2.30 hybrid (69.6% p-character) on the carbon donor atom of the CO2 ligand and described as σ(Re–CO2) = 0.605hRe + 0.797hC. The occupancy or the σ(Re–CO2) NBO is 0.947|e|.
Next the successive protonations of the coordinated CO2 yielding ImC1′ and ImD1′ intermediates are exergonic processes, and the estimated ΔG values are −152.9 and −134.6 kcal mol−1 respectively. ImD1′ releasing H2O affords the dicationic [Re(en)(CO)4]2+ complex ImE1′ which upon 2e− reduction releases the desired CO product and regenerates the catalytically active ImA1′ species.
In reaction step B1′′ (Fig. 6), the capture of CO2 by ImA1′′ yielding ImB1′′ is exergonic (−23.9 kcal mol−1), indicative of the stronger non-covalent and covalent interactions between ImA1′′ and CO2 (with an interaction energy of 23.9 kcal mol−1). The NBO population analysis revealed that the CO2 molecule is coordinated to the Re metal centre of the ImA1′′ catalytic species, forming a stronger nearly single Re–CO2 bond with a WBI(Re–CO2) of 0.755. The Re metal centre acquires a negative natural atomic charge of −1.016|e| and the C atom of the coordinated CO2 a positive natural atomic charge of 0.631|e|. The covalent component of the Re–CO2 bond is presented by the bonding σ(Re–CO2) NBO which is constructed from the interaction of sp2.83d2.52 hybrid orbitals (44.5% p and 39.7% d character) of Re with an sp1.62 hybrid (61.8% p-character) on the carbon donor atom of the CO2 ligand and described as σ(Re–O) = 0.614hRe + 0.789hC. The occupancy or the σ(Re–O) NBO is 0.947|e|.
Successive protonations of the coordinated CO2 yielding ImC1′′ and ImD1′′ are exergonic processes with estimated ΔG values of −194.2 and −154.4 kcal mol−1 respectively. ImD1′′ releasing H2O in an exergonic process (ΔG = −15.3 kcal mol−1) affords the cationic [Re(en)(CO)4]+ complex ImE′′ which upon 2e− reduction releases the desired CO product and regenerates the catalytically active ImA1′′ species.
In summary reaction steps B1′ and B1′′ are less favoured than reaction step B1 due to higher energy consumption to achieve the release of the desired CO product from the cationic [Re(en)(CO)4]+ complex by 2e− reductions. Furthermore, the CO2 insertion is more feasible than CO2 capture illustrating the crucial role of the sacrificial electron donor TEOA in the photocatalytic reduction of CO2 to CO catalyzed by the Re(en)(CO)3Cl complex.
Reaction step B2
Reaction step B2 starts with the five-coordinated [(en)(CO)2ReCl]− intermediate that resulted from the dissociation of one of the equatorial CO ligands of the excited T1 triplet state (step A2). It is important to note that Jean-Marie Lehn and co-workers19 showed, by a set of experiments concerning the carbonyl ligand exchange in [fac-Re(bpy)(CO)3Cl], that reductive quenching of the excited state of the [Re(bpy)(CO)3Cl] complex by tertiary amines gives an unstable five-coordinated [Re(bpy)(CO)2Cl] species upon labilization of a CO ligand.Two possible catalytic cycles following reaction steps B2′ and B2′′ have been thoroughly investigated. The reaction steps for the photocatalytic reduction of CO2 to CO catalyzed by the five-coordinated [(en)(CO)2ReCl]−, ImA2 intermediate are depicted schematically in Fig. 7.
 | ||
| Fig. 7 Proposed catalytic cycles for steps B2′ and B2′′ calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
Scheme 4 shows the 3D plots of the frontier molecular orbitals (FMOs) and spin density distribution of ImA2 and TEOA˙+ species.
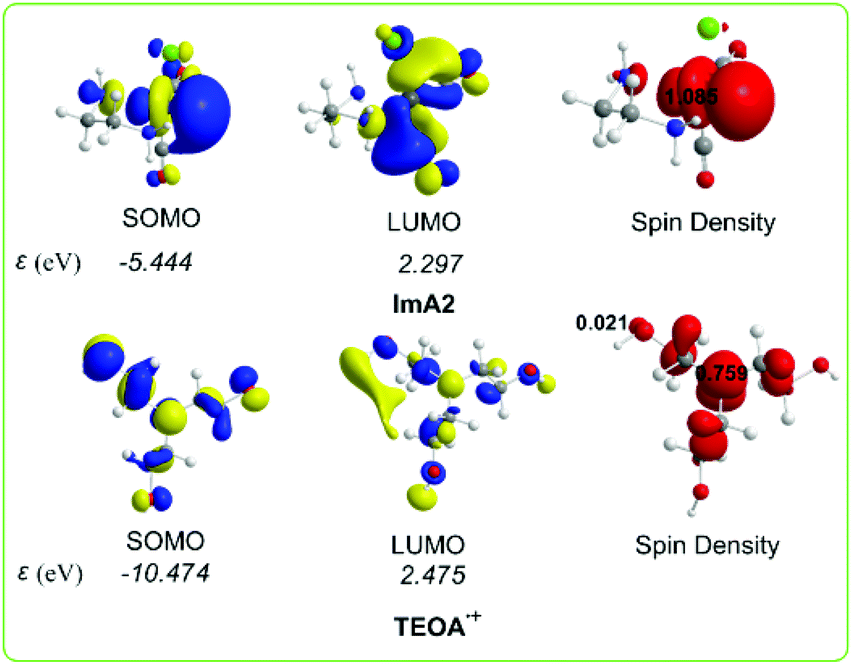 | ||
| Scheme 4 3D plots of the frontier molecular orbitals (FMOs) and spin density distribution of ImA2 and TEOA˙+ species calculated at the wB97XD/Def2-TZVP level of theory. | ||
The SOMO of ImA2 is composed mainly of 84.8% Re spzdz2 hybrid orbitals, while the LUMO is composed of 72.0% Re pd hybrid orbitals and 8.3% 2p AOs of the carbon atom of the CO ligand in trans position to the chloride ligand. A spin density of 1.085|e| is localized on the Re central atom. According to the NBO population analysis, the Re metal centre acquires a negative natural atomic charge of −0.552|e|. The SOMO of TEOA˙+ is mainly localized on one of the CH2CH2OH alkyl groups (50.0%) mainly on the O atom (32.5%) and on the nitrogen atom (16.5%) of TEOA. Both the nature of the FMOs and the negative natural atomic charge on the Re metal centre could not support covalent interactions with the CO2 ligand, thereby the capture of CO2 by ImA2 might be supported mainly by non-covalent interactions. Indeed the interaction energy for the ImA2⋯CO2 association is only 4.0 kcal mol−1 calculated according to the wB97XD/Def2-TZVP/PCM computational protocol. However the nature of the FMOs of ImA2 and TEOA˙+ allows SOMOImA2–SOMOTEOA˙+ interactions giving rise to the covalent component of the ImA2–TEOA bonding upon the formation of a bonding σ(Re–OCH2CH2NR2) NBO.
Reaction step B2′: CO2 insertion
The geometric and free energy reaction profiles calculated for step B2′ are shown in Fig. 8.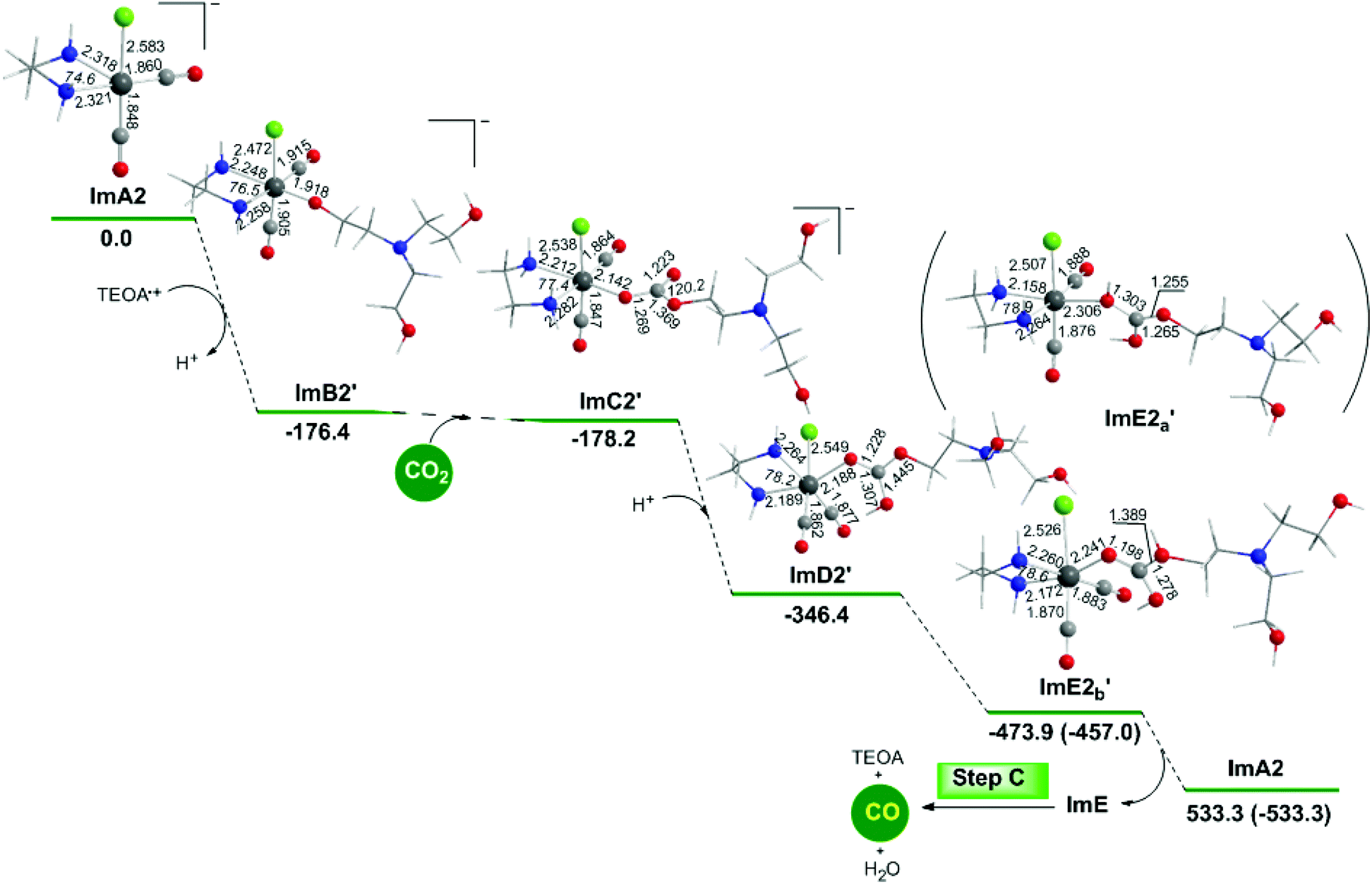 | ||
| Fig. 8 Geometric and free energy, ΔG (in kcal mol−1), reaction profiles for step B2′ of the reduction of CO2 to CO catalyzed by the active [(en)(CO)3Re]− catalytic species, ImA2, calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
Perusal of Fig. 8 shows that an R2N–CH2CH2O˙ radical is coordinated to the Re metal centre of the catalytically active ImA2 species which disposes of a vacant coordination site yielding ImB2′. The interaction of R2N–CH2CH2O˙ with ImA2 is exergonic (ΔG = −176.4 kcal mol−1). The NBO population analysis of ImB2′ revealed that the R2N–CH2CH2O˙ species is coordinated to the Re metal centre of ImA2 forming a Re–OCCH2CH2NR2 bond with a WBI(Re–O) of 0.604. The covalent component of the Re–O bond is described by the bonding σ(Re–O) NBO, with an occupancy of 1.929|e|. The σ(Re–O) NBO results from the interaction of the sp2.89d4.15 hybrid orbitals (35.8% p and 51.4% d character) of Re with an sp2.16 hybrid (68.3% p-character) of the O donor atom of the R2N–CH2CH2O˙ ligand and is described as σ(Re–O) = 0.399hRe + 0.917hO. The O atom of the coordinated R2N–CH2CH2O ligand acquires a negative natural atomic charge of −0.759|e| indicative of an anionic R2N–CH2CH2O− ligand. The Re metal center in ImB2′ acquires also a negative natural atomic charge of −0.249|e|.
Next insertion of a CO2 molecule into the Re–OCH2CH2NR2 transforms the OCH2CH2NR2 ligand to OC(O)OCH2CH2NR2 yielding ImC2′. The insertion process is slightly exergonic (ΔG = −1.8 kcal mol−1). The Re–OC(O)OCH2CH2NR2 coordination bond is a relatively weak Re–O dative bond with a WBI(Re–O) of 0.485 described by the bonding σ(Re–O) NBO as shown in Scheme 5 with an occupancy of 1.934|e|.
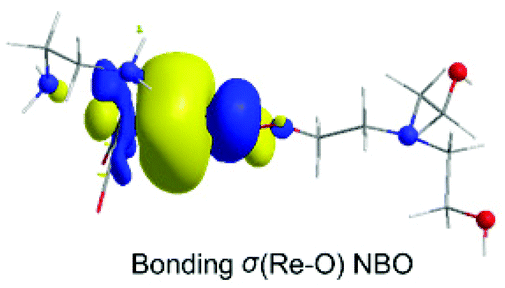 | ||
| Scheme 5 3D plot of the bonding σ(Re–O) NBO of ImC2′ calculated at the wB97XD/Def2-TZVP level of theory. | ||
The σ(Re–O) NBO is constructed from the interaction of the sp3.00d3.39 hybrid orbitals (40.5% p and 45.7% d character) of Re with an sp1.73 hybrid (63.3% p-character) of the O donor atom of the R2N–CH2CH2OC(O)O ligand and is described as σ(Re–O) = 0.364hRe + 0.931hO. The carbonate O atoms of the coordinated R2N–CH2CH2OC(O)O ligand acquiring negative natural atomic charges of −0.744, −0.666 and −0.526|e| are susceptible to protonation, particularly the carboxylate O atoms yielding the mono-ImD2′ and di-protonated ImE2a′ or ImE2b′ intermediates. ImE2a′ and ImE2b′ differ in the protonation sites of the coordinated R2N–CH2CH2OC(O)O ligand. The successive protonations are exergonic, and the estimated ΔG values are −168.2 kcal mol−1 and −127.5 (−110.6) kcal mol−1. Both ImE2a′ and ImE2b′ intermediates are unstable transient species directly releasing the cationic di-protonated [R2N–CH2CH2OC(OH)2], ImE, species and regenerating the catalytically active ImA2 species. The ImE species participates in reaction step C (Fig. 1) that delivers the desired CO product.
Reaction step B2′′: CO2 capture
The geometric and free energy reaction profiles calculated for step B2′′ are shown in Fig. 9. The capture of CO2 by ImA2 in reaction step B2′′ yielding ImB2′′ is slightly exergonic (−4.0 kcal mol−1) and it is achieved by the nucleophilic attack of the electrophilic carbon atom of CO2 since the Re metal centre acquires a negative natural atomic charge of −0.507|e| and the C atom of the coordinated CO2 a positive natural atomic charge of 0.737|e|. The NBO population analysis of ImB2′′ revealed that the CO2 molecule is coordinated to the Re metal centre of the ImA2 catalytic species, forming a Re–CO2 bond with a WBI(Re–CO2) of 0.634. The covalent component of the Re–CO2 bond is described by the bonding σα(Re–CO2) and σβ(Re–CO2) NBOs. The σα(Re–CO2) NBO, with an occupancy of 0.920|e|, results from the interaction of sp2.47d2.27 hybrid orbitals (42.9% p and 39.4% d character) of Re with an sp1.79 hybrid (64.1% p-character) on the carbon donor atom of the CO2 ligand and is described as σ(Re–CO2) = 0.610hRe + 0.792hC. The σβ(Re–CO2) NBO, with an occupancy of 0.767|e|, results from the interaction of the sp4.25d3.94 hybrid orbitals (46.2% p and 42.8% d character) of Re with an sp1.96 hybrid (66.2% p-character) on the carbon donor atom of the CO2 ligand and is described as σβ(Re–CO2) = 0.679hRe + 0.735hC. The O atoms of the coordinated C(O)O ligand acquiring negative natural atomic charges of −0.646 and −0.746|e| are susceptible to protonation. The first and second protonations transforming the coordinated C(O)O ligand to C(O)OH and C(OH)OH ligands and yielding ImC2′′ and ImD2′′ intermediates respectively are strongly exergonic, with the estimated ΔG values being −184.6 and −149.5 kcal mol−1. ImD2′′ is a transient species directly decomposed to give H2O and ImE2′′ formulated as the [Re(en)(CO)3Cl]+ complex which by 2e− reduction releases the desired CO product and regenerates the starting ImA2 complex. The two electrons are provided by the TEOA sacrificial electron donor. Note that the proposed catalytic cycle for step B2′′ is exactly the same as a possible catalytic cycle for the photocatalytic reduction of CO2 to CO catalyzed by the [Re(bpy)(CO)2Cl] catalytic species generated from the loss of a CO ligand from the reduced form of the [Re(bpy)(CO)3Cl] complex proposed by Lehn and co-workers.19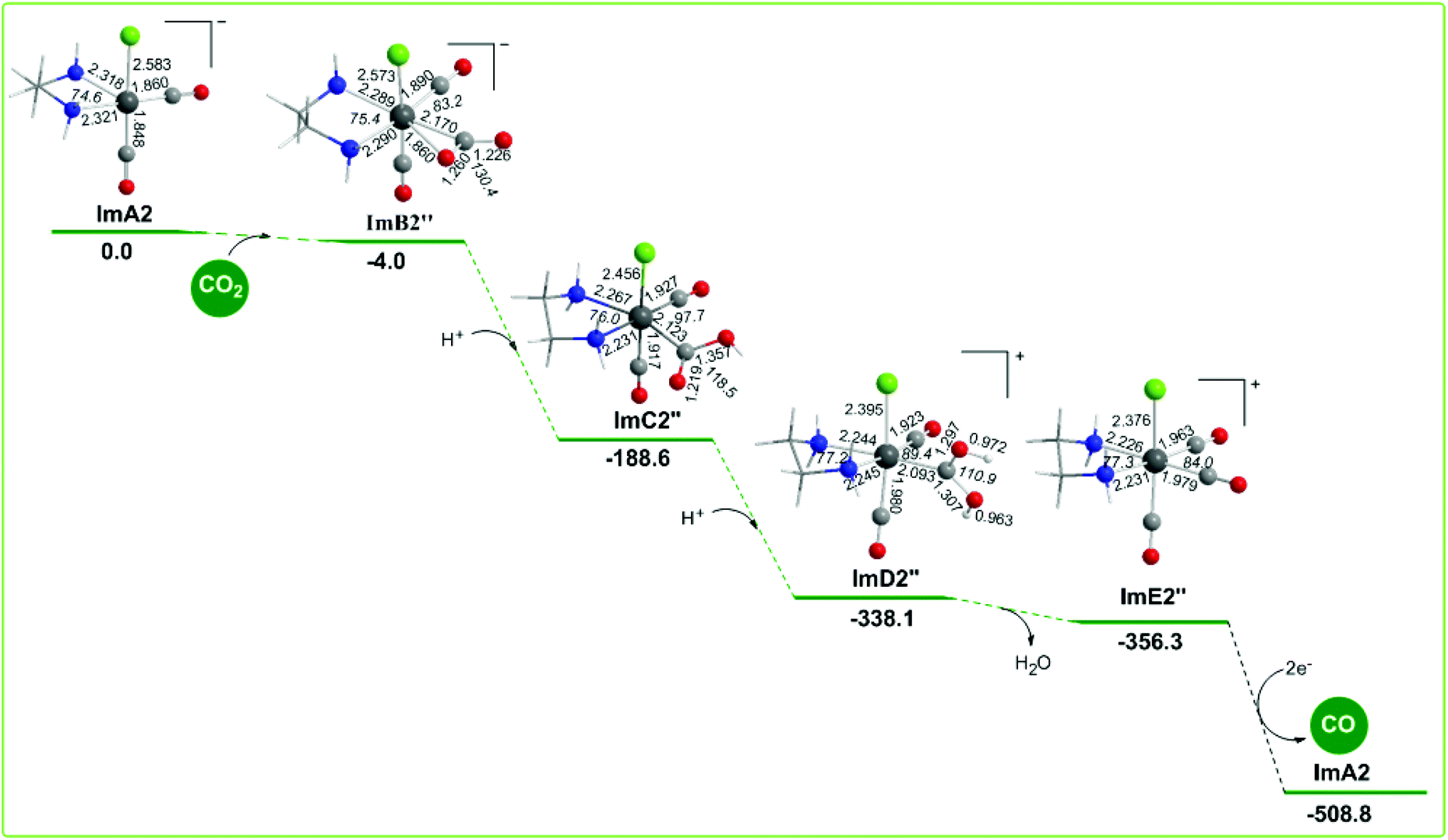 | ||
| Fig. 9 Geometric and free energy, ΔG (in kcal mol−1), reaction profiles for step B2′′ of the reduction of CO2 to CO catalyzed by the active [(en)(CO)3Re]− catalytic species, ImA2, calculated at the wB97XD/Def2-TZVP level in the DMF solution. | ||
In summary the free energy reaction profiles for reaction steps B2′ and B2′′ suggest that both the reaction pathways would proceed in parallel, but the CO2 insertion pathway should be most favorable than the CO2 capture pathway, since in the latter the CO2 capture by the ImA2 catalytic species is difficult and the release of CO from the cationic [Re(en)(CO)3Cl]+, ImE2′′ intermediate should be subject to 2e− reduction, that is the energy consuming process. The CO2 capture pathway is expected to be the most favorable reaction pathway in the electrochemical reduction of CO2 to CO mediated by the [Re(en)(CO)3Cl] catalyst in line with the catalytic cycle for the reduction of CO2 to CO mediated by the [Re(bpy)(CO)3Cl] catalyst proposed by Lehn and co-workers.19
Conclusions
This study reveals the crucial role of the TEOA sacrificial electron donor in the photocatalytic reduction of CO2 → CO catalyzed by the [Re(en)(CO)3Cl] complex. Despite its electron and proton donor abilities, TEOA can capture CO2 upon coordination to the Re metal centre of the “real” catalytic species [Re(en)(CO)3], [Re(en)(CO)3]− and/or [Re(en)(CO)2Cl]− generated from the T1 state upon single and double reductive quenching by TEOA or photodissociation of a CO ligand. The oxidized R2NCH2CH2O˙ radical of TEOA upon coordination with the Re metal centre of the catalytic species promotes the CO2 insertion in the Re–OCH2CH2NR2 bond forming stable intermediates that upon successive protonations yield unstable transient species that release CO and regenerate the catalytic species. In particular, in the CO2 insertion pathways, CO is delivered by an unstable diprotonated [R2NCH2CH2OC(OH)(OH)]+ species regenerating TEOA and the catalyst. The CO2 insertion reaction pathway is the favourable pathway for the photocatalytic reduction of CO2 → CO catalysed by the [Re(en)(CO)3Cl] complex in the presence of TEOA manifesting its crucial role as an electron and proton donor, capturing CO2 and releasing CO. The CO2 capture reaction pathways involving direct capture of CO2 by the catalytic species is less favorable because the direct capture of CO2 is a difficult task and the release of CO from the [Re(en)(CO)4]+ and [Re(en)(CO)2Cl]+ intermediates formed upon successive protonation of the captured CO2 ligand corresponds to energy consuming processes. This is why the CO2 capture reaction pathways are favourable pathways in the electrochemical reduction of CO2 to CO mediated by the [Re(en)(CO)3Cl] catalyst in line with the catalytic cycle proposed by Lehn and co-workers.19 We believe that the present in-depth mechanistic investigations would obviously inspire the chemists towards new and more efficient, selective and stable catalytic systems for the CO2 insertion step and easily releasing the reduction product of CO.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been performed under the Project HPC-EUROPA3 (INFRAIA-2016-1-730897), with the support of the EC Research Innovation Action under the H2020 Programme; in particular, the author gratefully acknowledges the support of Prof. M. Bickelhaupt, Department of Chemistry, Vrije Universiteit Amsterdam and the computer resources and technical support provided by the SURFsara supercomputing centre.Notes and references
- M. Aresta and D. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- M. Cokoja, C. Bruckmeier, B. Rieger, F. Kühnand and W. A. Hermann, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- J. A. Keith, K. A. Grice, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2013, 135, 15823–15829 CrossRef CAS PubMed.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- K. S. Rawat, A. Mahata, I. Choudhuri and B. Pathak, J. Phys. Chem. C, 2016, 120(16), 8821–8831 CrossRef CAS.
- D. H. Apaydin, S. Schlager, E. Portenkirchner, S. Niyaziand and N. S. Sariciftci, ChemPhysChem, 2017, 18, 3094–3116 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamur, K. Nozaki and O. Ishitani, Chem. Sci., 2019, 10, 3080–3088 RSC.
- Y. Asai, H. Katsuragi, K. Kita, T. Tsubomura and Y. Yamazaki, Dalton Trans., 2020, 49, 4277–4292 RSC.
- I. Choudhuri and D. G. Truhlar, J. Phys. Chem. C, 2020, 124(16), 8504–8513 CrossRef CAS.
- S. Gong, J. Fan, V. Cecenc, C. Huang, Y. Min, Q. Xu and H. Li, Chem. Eng. J., 2021, 405, 126913–112627 CrossRef CAS.
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Nat. Commun., 2021, 12, 1835–1844 CrossRef CAS PubMed.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- C. Kutal, M. A. Weber, G. Ferraudi and D. A. Geiger, Organometallics, 1985, 4, 2161–2166 CrossRef CAS.
- C. Kutal, M. A. Weber, G. Ferraudi and D. A. Geiger, Organometallics, 1987, 6, 553–557 CrossRef CAS.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283 CrossRef CAS PubMed.
- J. M. Smieja, E. E. Bensona, B. Kumara, K. A. Gricea, C. S. Seua, J. M. Miller, J. M. Mayer, P. Clifford and C. P. Kubiaka, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15646–15650 CrossRef CAS PubMed.
- K. A. Grice, N. X. Gu, M. D. Sampson and C. P. Kubiak, Dalton Trans., 2013, 42, 8498–8503 RSC.
- E. E. Benson, K. A. Grice, J. M. Smieja, P. Clifford and C. P. Kubiak, Polyhedron, 2013, 58, 229–234 CrossRef CAS.
- J. Agarwal, E. Fujita, H. F. Schaefer III and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS PubMed.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825 CrossRef CAS PubMed.
- P. Kurz, B. Probst, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2006, 2966–2974 CrossRef CAS.
- Y. Kuramochi, K. Fukaya, M. Yoshida and H. Ishida, Chem. – Eur. J., 2015, 21, 10049–10060 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef.
- D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Q. Wu and W. T. Yang, J. Chem. Phys., 2002, 116, 515–524 CrossRef CAS.
- Y. Minenkov, A. Singstad, O. Occhipinti and V. Jensen, Dalton Trans., 2012, 41, 5526–5541 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold, in The Encyclopedia of Computational Chemistry, ed. P. V. R. Schleyer, John Wiley & Sons, Chichester, U.K., 1998 Search PubMed.
- S. J. A. van Gisbergen, F. Kootstra, P. R. T. Schipper, O. V. Gritsenko, J. G. Snijders and E. J. J. Baerends, Phys. Rev. A, 1998, 57, 2556–2571 CrossRef CAS.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Chem. Phys., 1996, 104, 5134–5147 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction steps for the photocatalytic reduction of CO2 to CO catalyzed by the [(en)(CO)3ReCl] catalyst starting with the [(en)(CO)3Re] intermediate resulted upon one electron reduction of the T1 state by TEOA (Fig. S1). Cartesian coordinates of the reactants, intermediates and products involved in the catalytic cycles (Table S1). See DOI: 10.1039/d1dt02188e |
| This journal is © The Royal Society of Chemistry 2021 |