
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging heterogeneous catalysts for biomass conversion: studies of the reaction mechanism
Longfei
Lin
 ab,
Xue
Han
b,
Buxing
Han
*a and
Sihai
Yang
*b
ab,
Xue
Han
b,
Buxing
Han
*a and
Sihai
Yang
*b
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: hanbx@iccas.ac.cn
bDepartment of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: Sihai.Yang@manchester.ac.uk
First published on 18th August 2021
Abstract
The development of efficient catalysts to break down and convert woody biomass will be a paradigm shift in delivering the global target of sustainable economy and environment via the use of cheap, highly abundant, and renewable carbon resources. However, such development is extremely challenging due to the complexity of lignocellulose, and today most biomass is treated simply as waste. The solution lies in the design of multifunctional catalysts that can place effective control on substrate activation and product selectivity. This is, however, severely hindered by the lack of fundamental understanding of (i) the precise role of active sites, and (ii) the catalyst–substrate chemistry that underpins the catalytic activity. Moreover, active sites alone often cannot deliver the desired selectivity of products, and full understanding of the microenvironment of the active sites is urgently needed. Here, we review key recent advances in the study of reaction mechanisms of biomass conversion over emerging heterogeneous catalysts. These insights will inform the design of future catalytic systems showing improved activity and selectivity.
 Longfei Lin | Dr Longfei Lin received his PhD in Chemistry (2017) from Université Pierre et Marie Curie under the supervision of Professor Hélène Lauron-Pernot. He worked as a postdoctoral researcher at the Department of Chemistry, University of Manchester (2017–2020). In 2021, Dr Lin joined the Institute of Chemistry, Chinese Academy of Sciences, as an academic member of staff. His research interest lies in biomass conversion over new solid catalysts, particularly in understanding the essential relationship between the reaction performance and active sites/structure of the catalysts by combined experimental and computational techniques. |
 Xue Han | Dr Xue Han received her BSc from Peking University in 2007 and PhD in Chemistry from the University of Nottingham in 2010 under the supervision of Sir Professor Martyn Poliakoff. She is currently a Research Fellow at the Department of Chemistry, University of Manchester. Her research interest lies in the development of functional porous materials for the capture and conversion of gaseous air pollutants, such as NH3, NOx and SO2, as well as separation and catalysis of valuable organic compounds. The research will promote the development of a clean-air future. |
 Buxing Han | Professor Buxing Han received his PhD (1988) from the Institute of Chemistry, Chinese Academy of Sciences (CAS), and did postdoctoral research (1989–1991) at the University of Saskatchewan. He has been a Professor at the Institute of Chemistry, CAS, since 1993 and is the Chairman of Green Chemistry Committee, Chinese Chemical Society. He works on green chemistry and sustainable chemistry, especially the catalytic transformation of biomass and CO2 into valuable chemicals. He is an Academician of Chinese Academy of Sciences, a Fellow of the Academy of Sciences for the Developing World (TWAS), and a Fellow of the Royal Society of Chemistry. |
 Sihai Yang | Dr Sihai Yang received his BSc and PhD in Chemistry from Peking University (2007) and the University of Nottingham (2010), respectively. He is currently a Reader in Inorganic Chemistry at the University of Manchester. Yang's group develops porous materials based upon stable metal–organic frameworks and zeolites for applications in clean-air technology, heterogeneous catalysis, separation and conductivity. He has made pioneering contributions for the application of cutting-edge techniques, such as synchrotron X-ray diffraction, spectroscopy, neutron diffraction and inelastic/quasi-elastic neutron scattering, to in situ and operando studies of many dynamic and complex chemical processes. |
1. Introduction
The increasing concerns on the longevity of fossil fuels and current emissions of an astronomical amount of greenhouse gases are powerful drivers to develop new sustainable and environmentally-friendly chemical processes to produce fuels and feedstocks.1,2 Biomass, which is produced from carbon dioxide (CO2) and water through photosynthesis by plants or microbes, is a renewable resource of carbon. Lignocellulose is the most abundant biomass resource with an annual production of ca. >170 billion metric tonnes, but currently only <5% of the produced lignocellulose is utilised for conversion into value-added products and fuels while the majority is treated as waste owing to the lack of efficient catalysts to break down and convert lignocellulose.3Lignocellulosic biomass mainly contains cellulose, hemicellulose and lignin (Fig. 1).4 Cellulose is a homopolymer consisting of β-D-glucopyranose units linked by β-glycosidic bonds, and accounts for 40–50% of lignocellulosic biomass. Hemicellulose is a branched polymer with low degrees of polymerisation (ca. 200). Hemicellulose is composed of pentose, hexose and uronic acids, and accounts for 15–30% of lignocellulose. Lignin, rich in aromatic functionalities, is a heavily cross-linked, complex polymer with coumaryl, coniferyl and sinapyl alcohols, accounting for 15–30% of lignocellulosic biomass.4 Due to the complex and highly robust structure of lignocellulosic biomass, energy-efficient and cost-effective production of chemicals and fuels from lignocellulosic biomass remains an extremely challenging target for both academia and industry.
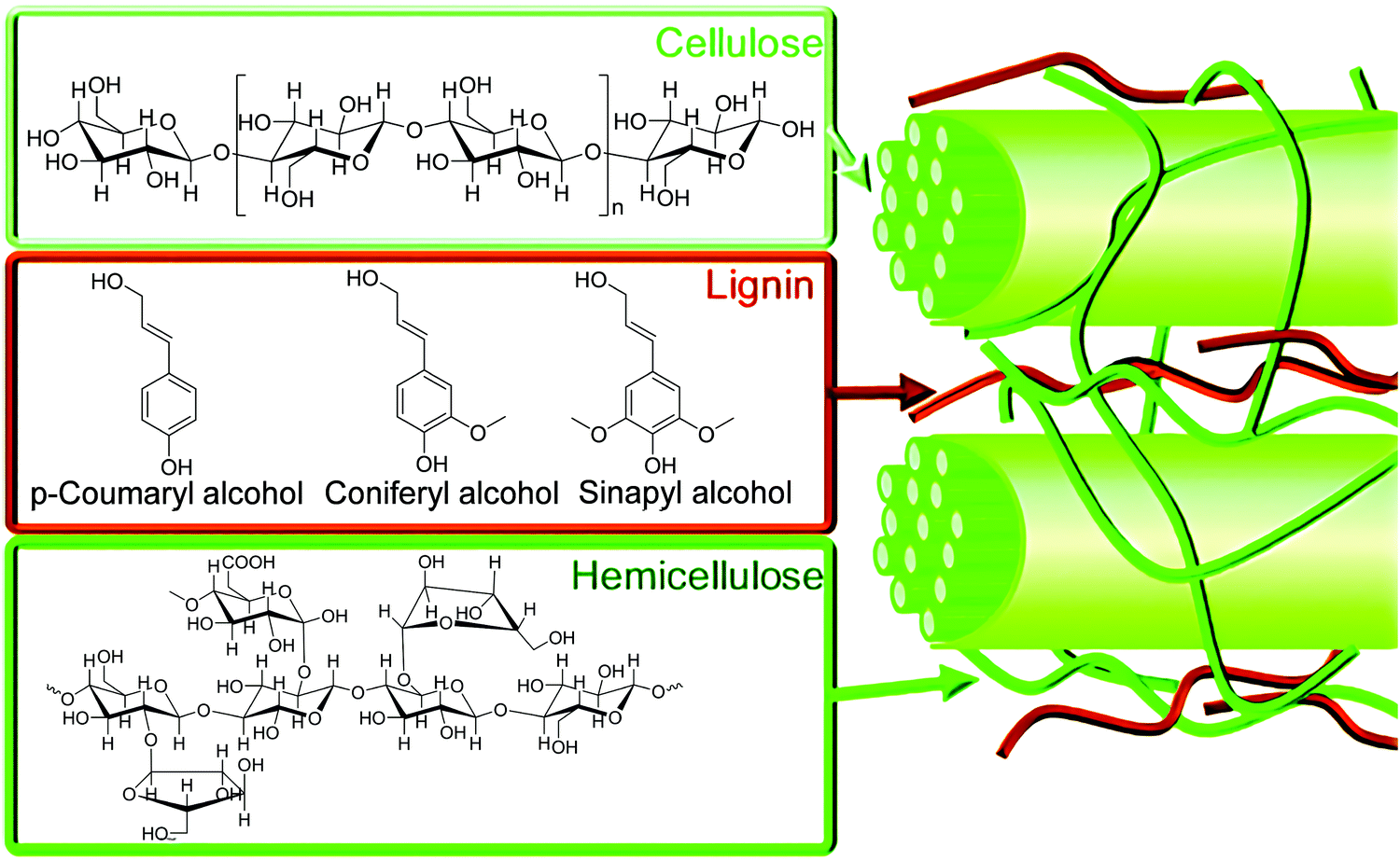 | ||
| Fig. 1 Structures of cellulose, hemicellulose, and lignin components of lignocellulosic biomass and associated molecular building blocks. Reproduced with permission from ref. 4, Copyright 2012 Royal Society of Chemistry. | ||
One state-of-the-art strategy relies on the thermochemical treatment of lignocellulose to produce bio-oils or syngas by pyrolysis or gasification, respectively, coupled with subsequent catalytic upgrading to produce hydrocarbons.5,6 These processes require high reaction temperature, and are generally non-selective and highly energy-intensive. Research efforts have been focused on developing new active catalysts to directly convert lignocellulose to liquid alkanes as transportation fuels under mild conditions.7 However, due to the complex composition of lignocellulose, it is almost impossible to obtain a single component product from a given one-pot process.8 One promising solution is to pre-treat lignocellulose by supercritical CO2,9 ionic-liquid-assisted deconstruction,10,11 or utilisation of deep eutectic solvents12 to isolate cellulose, hemicellulose and lignin. According to their different properties, designed processes can then be applied to promote the selective conversion of each component into target chemicals and fuels for direct use.
An enormous amount of efforts have been devoted to converting biomass and derived materials via (hydro)pyrolysis, solvolysis, hydrogenolysis, hydrodeoxygenation (HDO), dehydration, oxidation and reduction of lignin, and “lignin-first” approach-based depolymerisation. Various homogeneous and heterogeneous catalysts have been employed, such as polyoxometalates, ionic liquids, zeolites, mesoporous silica, transition metal complexes, carbon materials, and supported metal particles.3,10,13–18 Heterogeneous catalysts attract greater attention due to their advantages in separation, catalyst recycling and environmental impact.19 However, there are difficulties and challenges with these processes due to limited catalyst stability, low yields of the product and poor selectivity, and thus the formation of complex mixtures of products. Downstream separation is costly and often even impossible;20 indeed, the separation of chemical mixtures is often the bottleneck of modern chemical industries.21 As a result, products derived from biomass are subject to high cost, thus restricting their widespread application and implementation within renewable energy cycles and supplies. Therefore, powerful drivers exist to design highly selective and robust production processes of liquid fuels and value-added chemicals for direct use from biomass and its derivatives. Such design requires unambiguous understanding of the catalytic mechanism of active sites and their microenvironment within the emerging heterogeneous catalysts. Although several excellent reviews are published on the upgrading of biomass to fuels and chemicals from various aspects,1,3,4,15,16,22–28 a review focused on the analysis of the catalytic mechanism for biomass conversion is not yet available in the literature to date.
The isotope-labelling technique,29 nuclear magnetic resonance (NMR) spectroscopy,30 synchrotron X-ray powder diffraction (SXPD),31 X-ray absorption spectroscopy (XAS),32 density functional theory (DFT) calculations and,33 more recently, inelastic neutron scattering (INS)7,34–36 have been employed to investigate the reaction mechanism of biomass conversion. INS has several advantages compared with the optical spectroscopies, such as infrared and Raman spectroscopy.37–43 For example, INS is not subject to any optical selection rule, and all vibrations (0–4000 cm−1) are allowed and, in principle, measurable in INS. INS spectra can be readily and accurately modelled by DFT: the intensities are proportional to the concentration of elements in the sample and the features in the spectra can be fully assigned. Last but not least, neutrons penetrate deeply into materials and pass readily through the walls of metal containers, making cell design straightforward. Thus, neutrons are ideally positioned to measure the bulk properties of materials, which is particularly beneficial to the study of species residing on the inner surface of porous catalysts.
This review assesses the reaction mechanism studied by the isotope-labelling technique, NMR, SXPD, XAS, INS and DFT, and particularly focuses on the binding interactions between substrates and catalysts during the conversion of biomass (Fig. 2). The reaction mechanism is closely related to (i) the structure, active sites and support of the catalysts; (ii) the properties, nature of the chemical bonds, and intermediates of the substrates; and (iii) the reaction conditions, such as the solvent, pressure, additives and temperature, all of which will be discussed in detail (Fig. 2). The selected reactions in this review include hydrolysis, dehydration, isomerisation, aldol condensation, retro-aldol condensation, hydrogenation, hydrogenolysis, HDO, hydride shift, ring-opening, decarboxylation, Diels–Alder cycloaddition and oxidation (Scheme 1). The main body of this review is divided into four sections (Section 2–5) based on the end products of conversion of biomass. In Section 2, we discuss the reaction mechanism over porous solid catalysts for the production of aromatics and light olefins from biomass, because light olefins (ethylene, propylene and butene) and aromatics (benzene, toluene and xylene) are two families of the most important bulk chemicals with an annual global production of ca. 400 million44 and 160 million tonnes,45–47 respectively. They are basic building blocks for petrochemicals and polymers such as fibres, rubbers and plastics. In Section 3, we discuss the reaction mechanism for the synthesis of bio-plastics, for example the conversion of biomass to bio-plastic precursors, such as lactic acid and 2,5-furandicarboxylic acid (FDCA). In Section 4, we review the mechanism for the synthesis of liquid alkanes as fuels, which includes three strategies, (i) conversion of carbohydrate to 2,5-dimethylfuran (DMF), (ii) direct conversion of lignocellulose to liquid alkanes, and (iii) tandem production of long chain alkanes. Finally in Section 5, we summarise reactions involving C–O and C–C bonds in biomass conversion. Instead of summarising the catalytic performance of various catalysts, this review attempts to analyse the nature of the active sites and mechanism of the reaction over selected heterogeneous catalysts to provide key insights into the design of future efficient systems for biomass conversion.
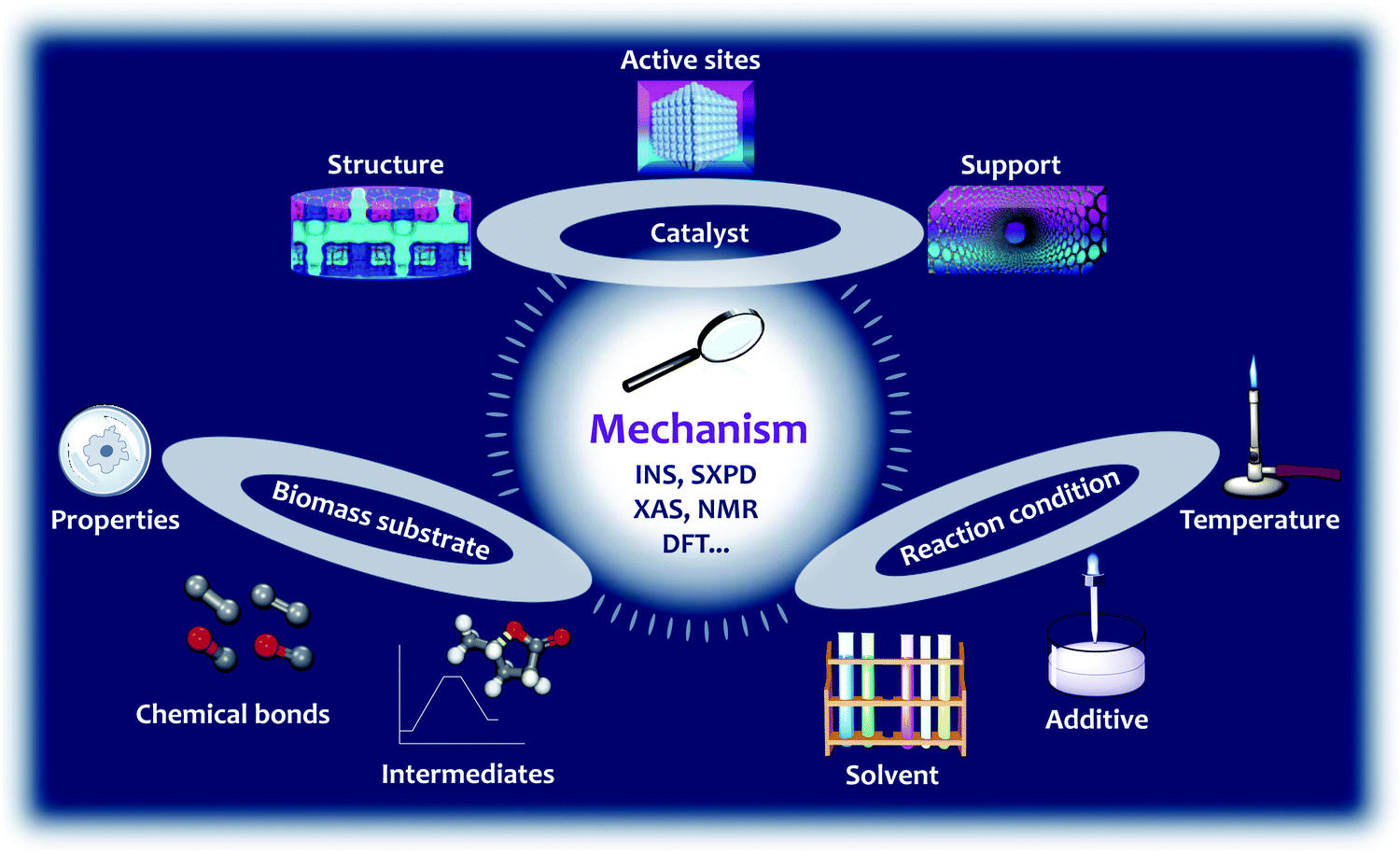 | ||
| Fig. 2 Central theme of this review: analysis of the reaction mechanism of biomass conversion over emerging heterogeneous catalysts studied by the INS, SXPD, XAS, NMR, and DFT techniques. The structure, active site and support of the catalyst, properties, chemical bonds, intermediates of the substrate, and reaction conditions such as the solvent, additives and temperature are systematically reviewed. | ||
 | ||
| Scheme 1 Routes of the catalytic conversion of lignocellulose to the representative chemicals and liquid fuels. | ||
2. Reaction mechanism for the production of bulk chemicals
2.1. Production of aromatic compounds
Lignin is constructed from monomers of coumaryl, coniferyl and sinapyl alcohols, and represents the most abundant renewable source of aromatic compounds on the earth.8,48 Because of its highly irregular polymeric structure and recalcitrant nature, efficient production of arenes from lignin is extremely challenging and often requires energy-intensive processes. For example, hydropyrolysis of lignin to aromatic compounds over the zeolite catalyst H-USY (yield: ∼30 wt%)49 or Pd/HZSM-5 (yield: ∼44 wt%)50 must operate at temperatures as high as 923 K.Recently, an HDO reaction based on a porous Ru/Nb2O5 catalyst has been developed to convert lignin under milder conditions (523 K) to enable the almost quantitative cleavage of Caromatic–O bonds and the complete removal of oxygen content from lignin, resulting in a total mass yield of 35.5 wt% and an exceptional arene selectivity of 71 wt%.34 The HDO process over Ru/Nb2O5 involves the cleavage of Caromatic–O bonds but preserving the aromatic rings, which is difficult because the C–O bond in aryl ethers is strong, and the hydrogenation of aromatic rings is thermodynamically favoured. To reveal the catalytic mechanism behind the selective cleavage of Caromatic–O bonds over Ru/Nb2O5, operando INS and DFT calculations were carried out. Phenol was selected as a model compound and several common catalyst supports such as ZrO2, Al2O3, and TiO2 were also studied for comparison to reveal the uniqueness of the Nb2O5 support. The INS study of adsorbed phenol on Nb2O5 showed that the intensity of the Caromatic–OH bending mode reduced significantly, indicating that the Caromatic–OH group of phenol interacts with Nb(V) sites and is deprotonated to form phenoxide bound to the vacant surface Nb(V) sites in an end-on model, resulting in the selective activation of C–O bonds. DFT calculations show distinct disassociation energies for the C–O bond of adsorbed phenoxide species on different catalysts. The energy required to cleave the C–O bonds in pristine phenol is 556 kJ mol−1, which is reduced to 425, 500, 491 and 510 kJ mol−1 upon adsorption on Nb2O5, ZrO2, Al2O3 and TiO2, respectively (Fig. 3a and b). The largest reduction of energy (Δ = 131 kJ mol−1) over Nb2O5 promotes the selective cleavage of C–O bonds, while it hinders the hydrogenation of the benzene ring owing to the confined adsorption geometry of phenoxide (end-on), leading to the observed high selectivity of arenes.
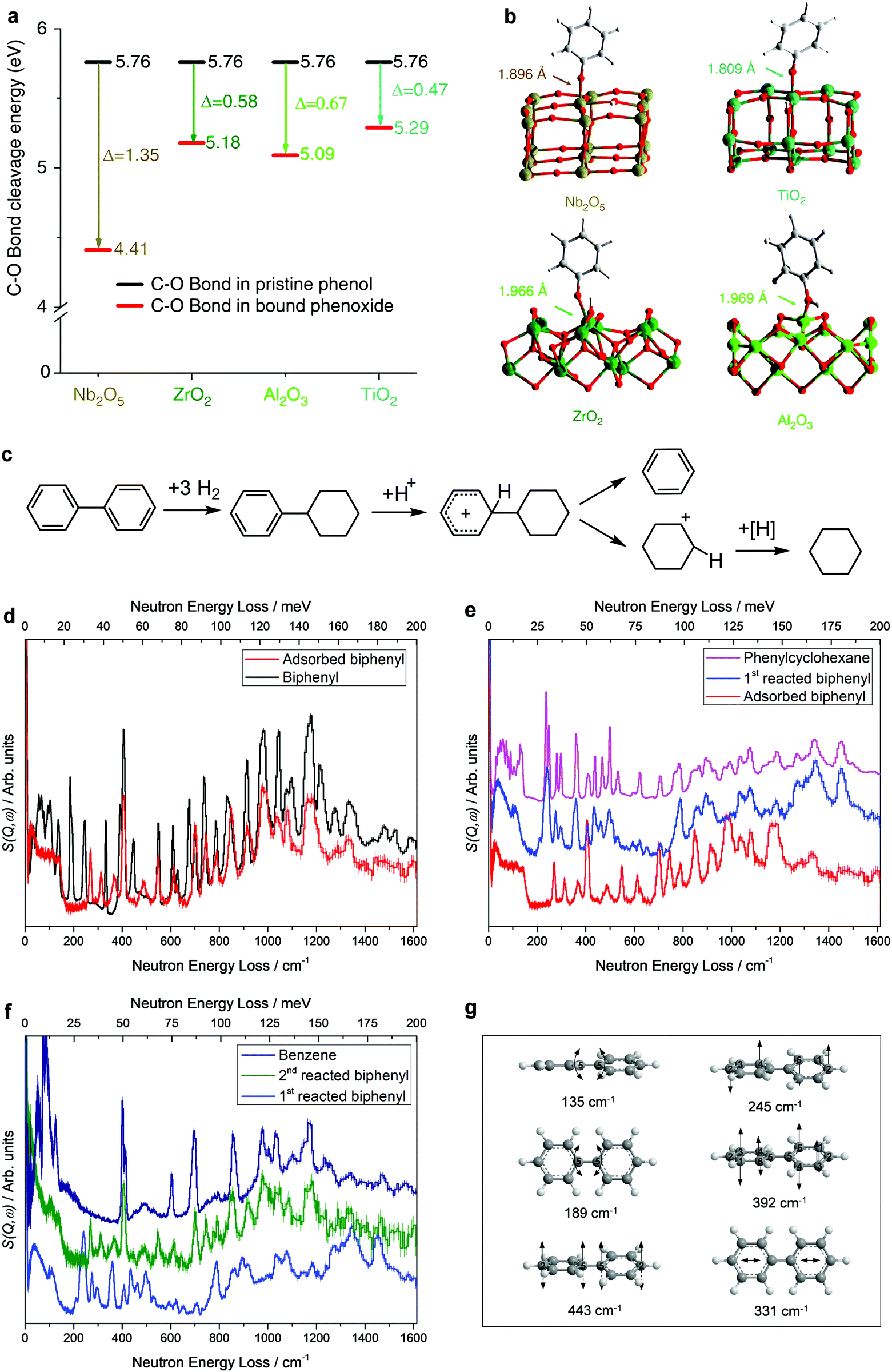 | ||
| Fig. 3 Conversion of lignin to aromatics. (a) Calculated energies for disassociation of the C–O bonds of phenol upon adsorption over Nb2O5, ZrO2, Al2O3 and TiO2. (b) Views of the corresponding DFT-optimised structural models for phenoxide bound on the Nb2O5(001), ZrO2(010), Al2O3(110) and TiO2(101) surfaces. Adapted from ref. 34 published by Springer Nature, licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/40/legalcode). (c) Possible reaction mechanism in the conversion of biphenyl over the Ru/NbOPO4 catalyst. Inelastic neutron scattering (INS) spectra for Ru/NbOPO4 in the adsorption and catalytic conversion of biphenyl. All spectra shown here are difference spectra. (d) Comparison of INS spectra for solid and adsorbed biphenyl on Ru/NbOPO4. INS spectra for Ru/NbOPO4 during the first (e) and second (f) catalytic conversion of biphenyl. (g) Views of selected C–C vibration modes of biphenyl. Reproduced with permission from ref. 35, Copyright© 2019 Elsevier. | ||
Compared with the cleavage of C–O bonds, the selective cleavage of interunit C–C bonds in lignin while preserving the aromatic ring is even more challenging, as the C–C bonds have notably higher dissociation energy (226–494 kJ mol−1) than that of C–O bonds (209–348 kJ mol−1). This naturally restricts the yield of aromatic monomers from the depolymerisation of lignin. A mesoporous multifunctional catalyst, Ru/NbOPO4, combining NbOx species and phosphates that contain strong Brønsted acid sites, has successfully broken down both interunit C–O and C–C bonds in lignin under mild conditions (583 K for 40 h).35 To investigate the catalytic mechanism, biphenyl, which contains the C5–C5 linkage (dissociation energy of 481 kJ mol−1), was selected as the model compound to study the cleavage of C–C bonds over Ru/NbOPO4 by operando INS/DFT. It revealed four steps involved in the catalytic process (Fig. 3c–g). (i) The biphenyl molecule adsorbs onto the catalyst with a flat orientation. This results in the disappearance of the modes of out-of-plane and in-plane bending (135 and 189 cm−1, respectively) of the C5–C5 bond. Meanwhile, the intensities of the deformational modes of the benzene rings at 628, 736, and 986 cm−1 reduced dramatically, indicating that both benzene rings of biphenyl adsorb on the catalyst (Fig. 3d). (ii) The adsorbed biphenyl molecule is partially protonated by the acid sites residing on the catalyst surface and an intermediate carbocation is formed. This is reflected by the shifts of the INS features at 245 and 392 cm−1, corresponding to the twisting mode of the C–C bond within the benzene rings, to 269 and 366 cm−1, respectively, and the shift of the peak at 331 cm−1 (assigned to the intra-ring stretching mode of benzene) to lower energy at 313 cm−1 (Δ = 18 cm−1) (Fig. 3d). (iii) One benzene ring of biphenyl is hydrogenated rapidly in the presence of H2 to give phenylcyclohexane adsorbed on the catalyst solely via its phenyl group. This is observed directly by INS as upon the reaction in H2 the peaks at 736 and 986 cm−1 (ring deformational modes of the phenyl group) have disappeared, and the peaks at 269, 313 and 366 cm−1 related to the intermediate carbocation shift to 275, 295 and 358 cm−1, respectively, and several new features appeared which are all consistent with the INS spectrum of phenylcyclohexane (Fig. 3e). (iv) C5–C5 bonds are efficiently cleaved over Ru/NbOPO4. In this step, the INS peaks for phenylcyclohexane significantly reduced or completely disappeared and features related to benzene (609, 405, 702, 857, 980, and 1185 cm−1) appeared and increased in intensity as the reaction proceeds, demonstrating the cleavage of C5–C5 bonds of adsorbed phenylcyclohexane on Ru/NbOPO4 (Fig. 3f). Besides the NbOx species for the cleavage of C–O bonds, Ru/NbOPO4 integrates Brønsted acid sites and Ru centres with moderate hydrogenation ability, which enables the cleavage of robust interunit C–C bonds to maximise the production of lignin monocyclic hydrocarbons.
In addition to lignin, aromatics can also be produced from derivatives of cellulose and hemicellulose.51–56 Bio-derived furans can be converted into aromatics by a Diels–Alder (DA) cycloaddition with ethene or propene on Lewis acid sites, followed by dehydration on Brønsted acid sites.51,52 Side reactions, such as the hydrolysis of DMF to 2,5-hexanedione and ethene oligomerisation, can occur on acid sites as well (Scheme 2).52 DFT calculations demonstrate that the hydrolysis of DMF occurs more readily over beta zeolite with strong acid sites than over t-ZrO2 with weak acid sites with an activation energy of 166 and 275 kJ mol−1, respectively (Scheme 2). Ethene oligomerisation was also strongly favoured over the beta zeolite (Scheme 2).52 Thus, the highly selective and efficient production of aromatics from furans requires weak acid sites and cooperation between Lewis and Brønsted acid sites. Moreover, the structure of the catalyst is one of the key factors for the catalytic performance.53 A mimic of the reaction intermediate, oxanorbornene, was used as the organic structure directing agent (OSDA) to synthesise the DS-ITQ-2 zeolite (Fig. 4a and b).53,54 The resultant zeolite has the MWW structure with hemi-cavities, which can stabilise the reaction intermediate, oxanorbornene, due to the pore confinement effect, and thus the DA cyclodimerization is greatly enhanced. This was further demonstrated by DFT calculations which show that the stabilisation energies of the reaction intermediates over DS-ITQ-2 are higher than those over the BEA and FAU zeolites (−82 vs. −66 and −50 kJ mol−1, respectively, Fig. 4b–d).53
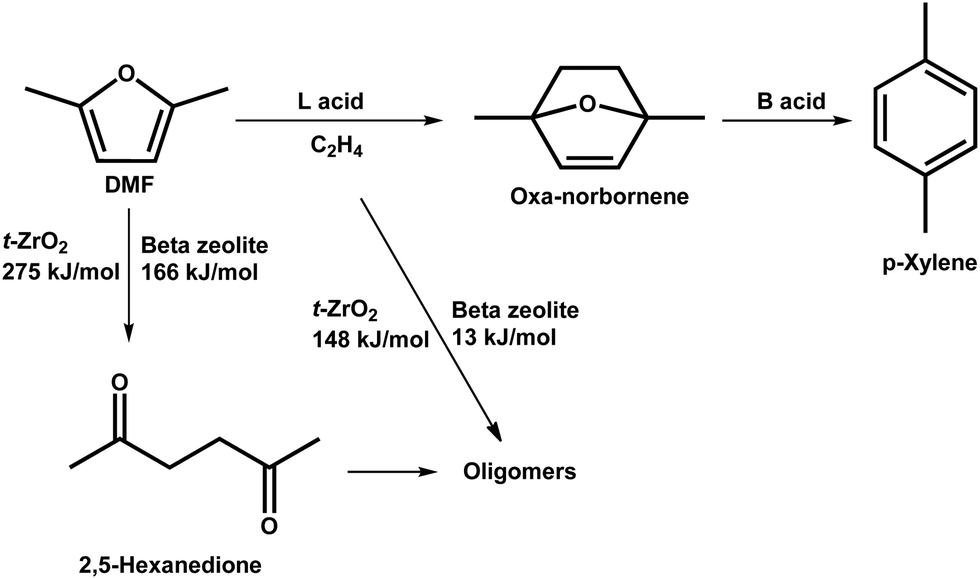 | ||
| Scheme 2 Reaction network of dehydrative aromatization of DMF with ethylene. Reproduced with permission from ref. 52, Copyright© 2019 Elsevier. | ||
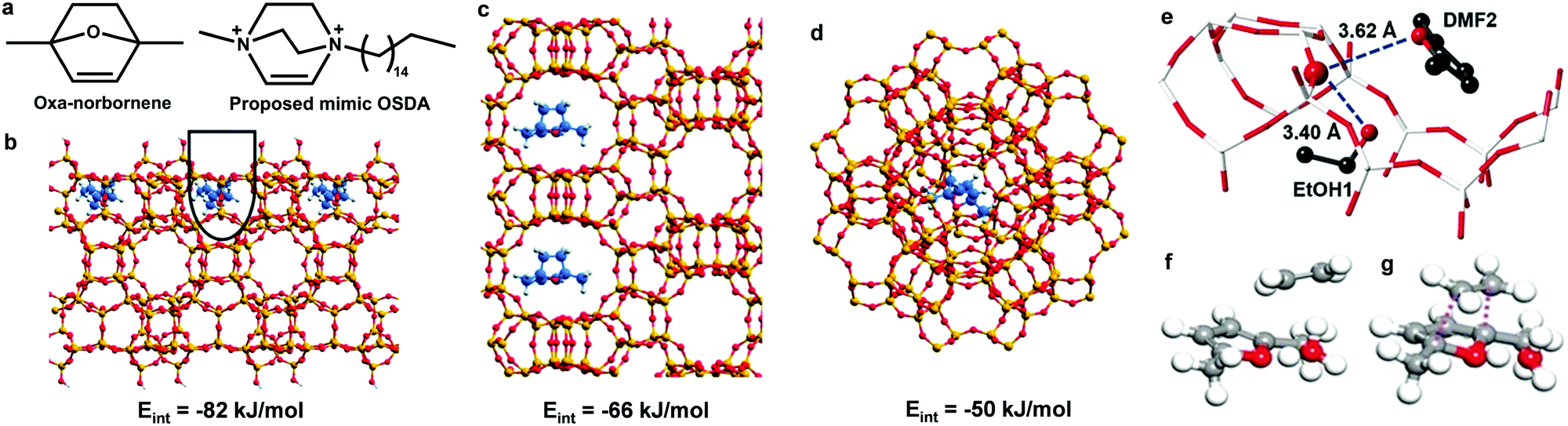 | ||
| Fig. 4 Conversion of cellulose/heicellulose derivatives to aromatics. (a) Comparison of oxanorbornene and the proposed mimic organic structure directing agent. Optimised structures of the oxanorbornene intermediate in the cavities of external hemi-cavities of MWW (b) and pores of BEA (c) and FAU (d). Adapted with permission from ref. 53, published by the Royal Society of Chemistry, licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported Licence (https://creativecommons.org/licenses/by-nc/3.0/). (e) Views of the crystal structure of EtOH + DMF@HZSM-5, obtained from Rietveld refinements based upon synchrotron X-ray powder diffraction data. Atoms are represented as balls/sticks: Si/Al, white; O, red (large dark red for acidic T6-OH); C, black. No hydrogen atoms are plotted. A configuration of DMF–H3O+–C2H4 (f) and the Diels–Alder transition state (g) on HZSM-5 based on DFT calculations. Grey, C; red, O; white, H. Adapted with permission from ref. 57, Copyright© 2016 John Wiley & Sons. | ||
Bio-derived ethanol has been used to generate ethene in situ over zeolite catalysts and this renders the production of renewable aromatics notably straightforward.57 In the ethene/DMF system, DMF is firstly protonated by Brønsted acid sites, followed by DA cycloaddition on Lewis acid sites with ethene. In the ethanol/DMF system, it was observed by SXPD that ethanol can be protonated by Brønsted acid sites of the zeolite more easily than DMF (Fig. 4e). The protonated ethanol undergoes dehydration to release ethene as a dienophile and a protonated water molecule (hydronium ion, H3O+). The latter interacts with ethene and DMF through hydrogen bonds to form a particular configuration (Fig. 4f) that is close to the Diels–Alder transition state (Fig. 4g), resulting in a decreased activation barrier of the ethanol path by 11.2 kJ mol−1 as compared to the ethene route. Thus, the promoted protonation of ethanol is a key step to enhance the production of aromatics from bio-derived ethanol and DMF.
In summary, the limiting step in the production of aromatics from DMF is DA cycloaddition, which can be promoted in suitable cavities of porous catalysts due to the stabilisation of the reaction intermediates. The hydronium ion, H3O+, is also helpful for the stabilisation of the reaction intermediate. Both Lewis and Brønsted acid sites are involved during the transformation and their cooperation is essential to achieve high catalytic performance. Catalysts incorporating weak acid sites show clear advantages of preventing the formation of by-products.
2.2. Production of lower olefins
Olefins can be produced from biomass through pyrolysis of lignocellulose to bio-oils which include ketones, aldehydes, alcohols, esters, ethers, sugars, carboxylic acids, phenols and furans, followed by catalytic cracking processes at elevated temperatures (typically >873 K).58 This route, however, is highly energy-intensive and non-selective. An alternative route is to convert cellulose and hemicellulose from agricultural waste to gamma-valerolactone (GVL) via low-cost and high-yield commercialised processes.59 GVL then undergoes ring-opening and decarboxylation to produce olefins under mild conditions.60 GVL obtained from biorefineries is usually in 20–40 wt% aqueous solution. The conversion of GVL to olefins in water is the key step to bridge biomass and petroleum processing61 and thus has attracted much attention. A series of solid acids including SiO2/Al2O3,60 γ-Al2O3,62 La/ZSM-5,63 Zn-AlPO-5,64 and Pd/Nb2O565 were reported as catalysts for the conversion of GVL to butenes. However, most catalysts partially or completely lose activity in water owing to the reaction between water and Lewis acid sites. Recently, quantitative production of butenes from GVL in aqueous solution was achieved over a new heteroatomic zeolite, NbAlS-1,36,66 which was synthesised by incorporating both Nb(V) and Al(III) centres into the MFI-type framework.A combination of XAS, SXPD, INS and DFT has successfully revealed the reaction mechanism of the ring-opening and decarboxylation of GVL over the NbAlS-1 zeolite.36 On HZSM-5 (an isostructural analogue to NbAlS-1 but without Nb sites), GVL adsorbs on the Brønsted acid sites via its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group67 (Fig. 5a), while on NbS-1 (an isostructural analogue to NbAlS-1 but without Al sites), GVL adsorbs on the Nb(V) site through its intra-ring O-centre (Fig. 5b), which is confirmed by XAS. On the multifunctional NbAlS-1 zeolites, GVL interacts with a bridging O(H)-centre on the framework through its CO group [C
O group67 (Fig. 5a), while on NbS-1 (an isostructural analogue to NbAlS-1 but without Al sites), GVL adsorbs on the Nb(V) site through its intra-ring O-centre (Fig. 5b), which is confirmed by XAS. On the multifunctional NbAlS-1 zeolites, GVL interacts with a bridging O(H)-centre on the framework through its CO group [C![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) ⋯ = 3.574(141) Å] via a weak hydrogen bond and with framework T-sites (NbO4 moieties) via its intra-ring O-centre [Nb⋯C4 = 3.789(75) Å] (Fig. 5c), which was demonstrated by the Rietveld refinement of the SXPD data. The adsorption of GVL on NbAlS-1 in the unique “chelating” mode (Fig. 5c) significantly enhances its reactivity. Then selective cleavage of the C4–O1 bond and ring-opening of GVL were observed by INS due to the strong activation by Nb(V) sites. Meanwhile, C1O2 is protonated via proton transfer from the Brønsted acid sites. On addition of the H-shift in the C5-skeleton, a decarboxylation reaction via the cleavage of the C1–C2 bond occurs, yielding butene and equimolar CO2 (Fig. 5d).
⋯ = 3.574(141) Å] via a weak hydrogen bond and with framework T-sites (NbO4 moieties) via its intra-ring O-centre [Nb⋯C4 = 3.789(75) Å] (Fig. 5c), which was demonstrated by the Rietveld refinement of the SXPD data. The adsorption of GVL on NbAlS-1 in the unique “chelating” mode (Fig. 5c) significantly enhances its reactivity. Then selective cleavage of the C4–O1 bond and ring-opening of GVL were observed by INS due to the strong activation by Nb(V) sites. Meanwhile, C1O2 is protonated via proton transfer from the Brønsted acid sites. On addition of the H-shift in the C5-skeleton, a decarboxylation reaction via the cleavage of the C1–C2 bond occurs, yielding butene and equimolar CO2 (Fig. 5d).
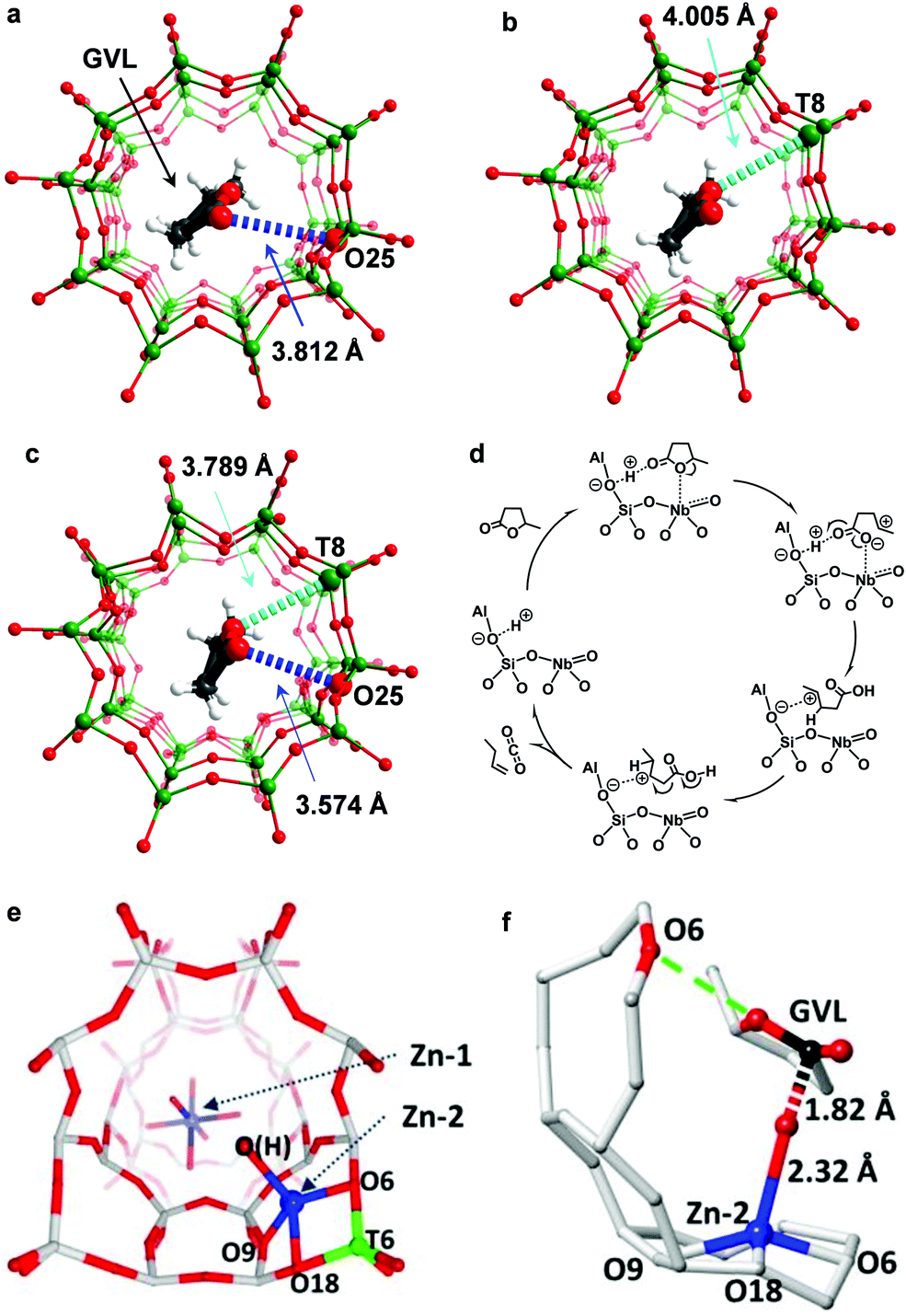 | ||
| Fig. 5 Production of butenes. Views of the crystal structures of GVL loaded on HZSM-5 (a), NbS-1 (b) and NbAlS-1 (c), obtained from Rietveld refinements based upon in situ synchrotron X-ray powder diffraction data. GVL molecules and the functional sites involved in the cooperative binding are highlighted by the use of an amplified ball-and-stick model (Nb/Al/Si, green; C, grey; O, red; H, white). The O1⋯Nb interactions and O2⋯O(H) interactions are highlighted in cyan and blue, respectively. Owing to the uncertainty in the locations of protons, all hydrogen bonds in this report are described as the distance between the OGVL and the Ozeolite centres. (d) Proposed reaction mechanisms for the conversion of GVL over NbAlS-1 based on INS/SXPD experiments. Reproduced with permission from ref. 36, Copyright© 2019 Springer Nature. Views of the crystal structures of Zn/ZSM-5 (e) and GVL loaded-Zn/ZSM-5 (f). Atoms are represented as balls/sticks: Si, grey; Al, green; Zn, blue; O, red; C, black. Adapted with permission from ref. 63, Copyright© 2017 John Wiley & Sons. | ||
ZnO/HZSM-5 has also shown excellent catalytic performance for the ring-opening and decarboxylation of GVL.63 SXPD, XAS and 1H NMR identified that the active sites were the Brønsted acid site located at the framework T6 site68 and a tetrahedrally coordinated Zn (Zn–OH) centre attached to the 5-membered ring of ZSM-5 through the wall-oxygens next to Al(T6)63 (Fig. 5e). These two sites cooperate to interact with the intra-ring O-centre [O⋯C4 = 3.34(8) Å] and CO [Zn–⋯![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O = 1.82(4) Å] group of the adsorbed GVL molecule (Fig. 5f). The activated GVL molecule then undergoes ring opening and hydrolytic decarboxylation with a water molecule to form CO2 and 1-butene. The latter can further undergo aromatisation to produce aromatics over the ZnO/ZSM-5 catalyst.
O = 1.82(4) Å] group of the adsorbed GVL molecule (Fig. 5f). The activated GVL molecule then undergoes ring opening and hydrolytic decarboxylation with a water molecule to form CO2 and 1-butene. The latter can further undergo aromatisation to produce aromatics over the ZnO/ZSM-5 catalyst.
The production of aromatics and olefins from biomass derivatives including lignin, cellulose and hemicellulose is attracting increasing attention given the demonstrated success. Based on the studies above, the key for the efficient and selective production of aromatics and olefins is to design task-specific active sites (e.g., Nb site for cleavage of C–O bonds) for the activation of target chemical bonds in the substrates, and the cooperativity of different types of active sites within the microenvironment also plays an important role in achieving the unique geometry of adsorbed substrates, promoting their selective conversion.
3. Reaction mechanism for the production of bio-plastic precursors
Plastic is ubiquitous and plays an indispensable role in most synthetic products sold today.69 At present, the global production of plastics is approximately 350 million tonnes per year,26 which is mainly derived from non-renewable petrochemical resources. The production process of plastics causes an enormous amount of CO2 emission. In addition, over 80 million tonnes of used plastic materials are burned, buried, or disposed of in the ocean, leading to serious problems for the environment and ecological systems owing to their high resistance to biological degradation processes.69 It is a matter of great urgency to develop new recycling concepts,70 clean incineration methods, and rapidly degradable plastics71–81 to resolve these problems. The production of biodegradable plastics from renewable biomass resources can alleviate the dual pressures of shortage of petrochemical resources and environmental pollution and is therefore of great significance to the development of a sustainable society.3.1. Production of lactic acid
Polylactic acid is a polymer synthesised from lactic acid and a new type of biodegradable material based on biomass.74 It can be used as packaging materials, fibres and textiles, and thus has a wide range of applications in the fields of clothing and various industries (e.g., construction, agriculture, forestry, and papermaking). The global production of the monomer of lactic acid is ca. 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes every year via both fossil and biomass resources.82
000 tonnes every year via both fossil and biomass resources.82
Owing to the abundant functional groups in biomass components, it is promising to transform them directly to target chemicals, rather than to fully decompose them, followed by the re-introduction of functional groups. A functionality index (F/C) has been introduced to guide the production of chemicals from biomass, where C is the number of carbon atoms in the molecule and F is the functionality parameter [F(CC) = 1, F(–OH) = 1, F(CO) = 2].83 Besides, the atom economy is an important index in green chemistry. The change of O/C means input or output of O to the molecule (O and C are the number of oxygen and carbon atoms in the molecule), which is related to the atom economy. The plot of the F/C vs. the O/C ratio (Fig. 6) compares the production of lactic acid from biomass and fossil resources. The F/C and O/C values for lactic acid are 1.33 and 1.00, respectively. If lactic acid is obtained from fossil resources, such as C3H6, which has an F/C of only 0.33, functional groups with high F/C, such as –COOH and –OH, must be introduced. In contrast, biomass saccharides are intrinsically functionalised, such as glucose (F/C = 1.17, close to the F/C of lactic acid), which are ideal candidates to be transformed to lactic acid. Moreover, the transformation of saccharides with C6H12O6 units to lactic acid has an atom economy of 100%, demonstrating the great promise of the development of biomass-based routes to produce lactic acid from saccharides.
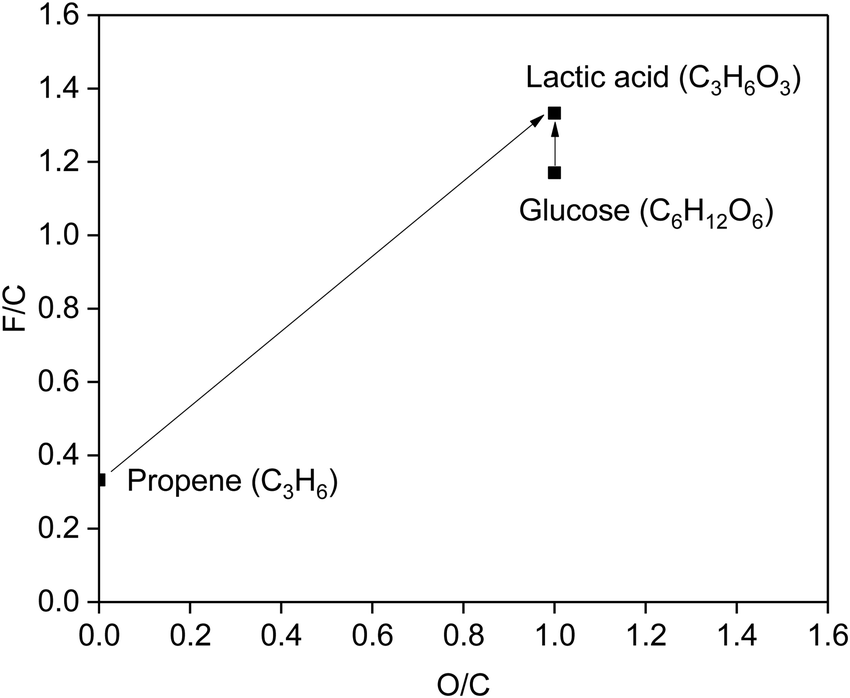 | ||
| Fig. 6 F/C vs. O/C ratio for propene, lactic acid and glucose. F/C refers to the functionality index, where C is the number of carbons in the molecule and F is the functionality parameter. A change of O/C indicates input or output of O to the molecule, where O and C are the number of oxygens and carbons in the molecule. | ||
Fermentation of edible biomass to lactic acid has been commercialised for decades.84,85 However, biological fermentation methods suffer from long cycles and are labour-intensive. Moreover, pH buffering with an alkali during the fermentation results in producing dilute Ca-lactate salt in water. As a result, large quantities of solid waste of CaSO4 (1.0 tonne per tonne of lactic acid) are produced to release lactic acid from this salt by acidification with H2SO4. Moreover, complex separation and purification steps are required to obtain pure lactic acid from the fermentation living broth. These drawbacks result in high production cost and low efficiency, which hamper the megatonne-scale production of polylactic acid as a commodity plastic. Thus, the development of cheap, efficient and clean catalytic technology to produce lactic acid in large quantities from inedible biomass attracts increasing attention.
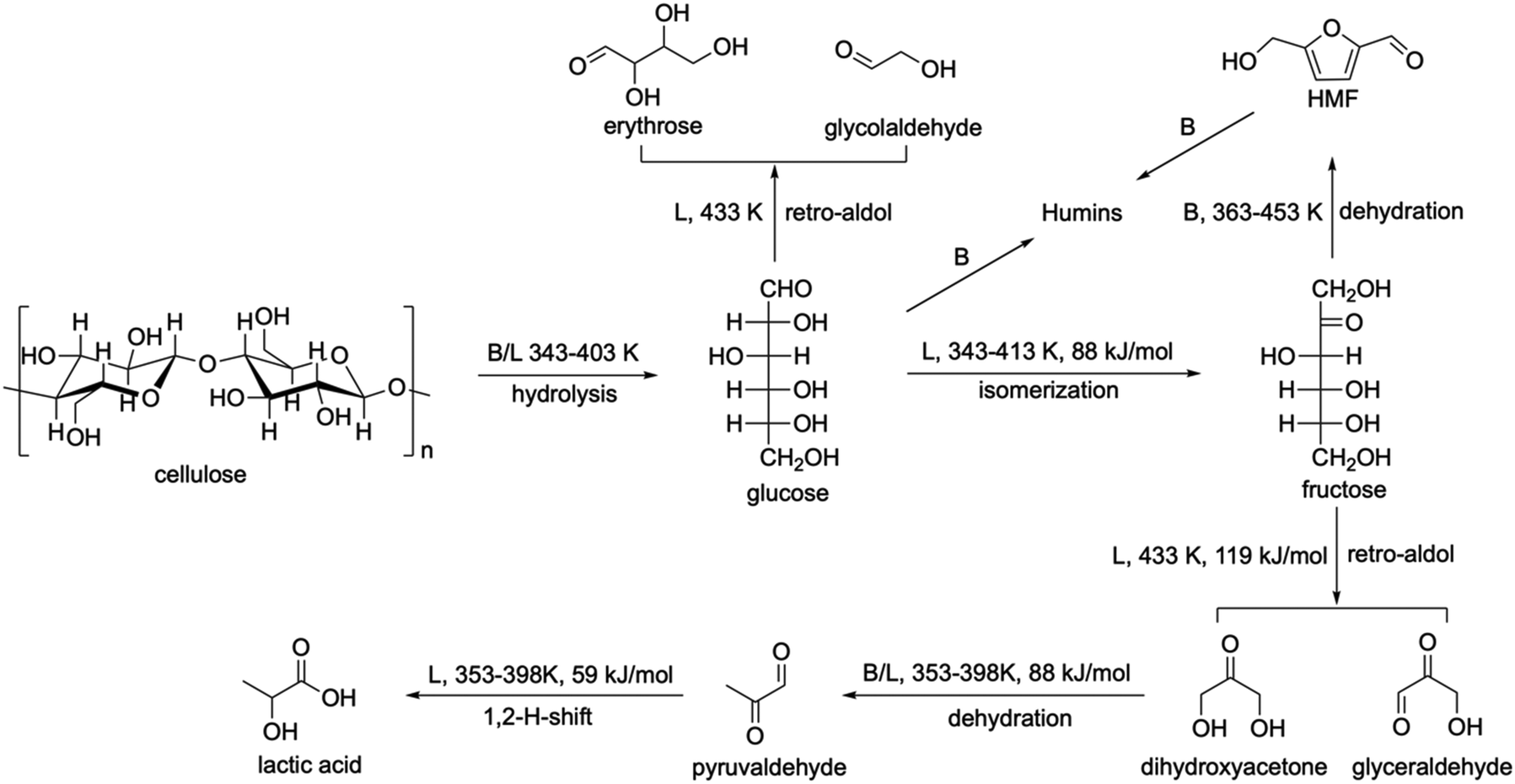
The chemical conversion of saccharides to lactic acid involves cascade reactions, including hydrolysis of polysaccharides to monosaccharides, isomerisation of monosaccharides to fructose, retro-aldol condensation, dehydration, and hydride shift (Scheme 3a). Homogeneous catalysts, such as alkalis,86 Pb(NO3)2,87 and Er(OTf)3,88 exhibit high efficiency (up to 96% yield) to produce lactic acid (Table 1). However, it has the same drawbacks as the fermentation process due to the complex separation and purification steps. Heterogeneous catalysts such as AlZrOx89 and AlWOx90 were investigated but showed low yield (<35%) of lactic acid (Table 1).
 | ||
| Scheme 3 Production of lactic acid. (a) Reaction pathway of cellulose to lactic acid. (b) Proposed reaction mechanism for the hydrolysis of cellulose over a Brønsted acid. Reproduced with permission from ref. 95, Copyright© 2011 John Wiley & Sons. (c) Proposed reaction mechanism for the hydrolysis of cellulose over a Lewis acid. Adapted with permission from ref. 96, Copyright© 2007 Elsevier. (d) Glucose isomerization mechanisms by way of intramolecular hydride shift over Sn-beta zeolite. Adapted with permission from ref. 97. Copyright© 2011 John Wiley & Sons. (e) Plausible mechanisms for retro-aldol condensation of fructose to dihydroxyacetone and glyceraldehyde, catalysed by open tin sites. Adapted with permission from ref. 26 and 100, Copyright© 2018 John Wiley & Sons. | ||
| Catalysts | Substrate | Reaction conditions | Conv (%) | LA/methyl lactate yield (%) | Ref. |
|---|---|---|---|---|---|
| Ba(OH)2 | Glucose | 298 K, 48 h, H2O | 99.5 | 95.4 (LA) | 86 |
| Ba(OH)2 | Fructose | 298 K, 48 h, H2O | 98.7 | 83.5 (LA) | 86 |
| Ba(OH)2 | Cellulose | 298 K, 48 h, H2O | — | 42.2 (LA) | 86 |
| Pb(NO3)2 | Glucose | 463 K, 4 h, H2O | 100 | 62 (LA) | 87 |
| Pb(NO3)2 | Fructose | 463 K, 4 h, H2O | 100 | 74 (LA) | 87 |
| Pb(NO3)2 | Cellulose | 463 K, 4 h, H2O | 100 | 68 (LA) | 87 |
| Er(OTf)3 | Glucose | 513 K, 0.5 h, H2O | 100 | 47 (LA) | 88 |
| Er(OTf)3 | Fructose | 513 K, 0.5 h, H2O | 100 | 55 (LA) | 88 |
| Er(OTf)3 | Cellulose | 513 K, 0.5 h, H2O | 100 | 61 (LA) | 88 |
| AlZrOx | Glucose | 453 K, 2 h, H2O | 92 | 34 (LA) | 89 |
| AlWOx | Cellulose | 463 K, 24 h, H2O | 47 | 28 (LA) | 90 |
| Sn-Beta | Glucose | 433, 20 h, MeOH | 99 | 43 | 72 |
| Sn-Beta | Fructose | 433, 20 h, MeOH | 99 | 44 | 72 |
| Sn-Beta | Sucrose | 433, 20 h, MeOH | 100 | 64 (methyl lactate) | 72 |
| H–Al-Beta | Sucrose | 433, 20 h, MeOH | 99 | 0 | 72 |
| Ti-Beta | Sucrose | 433, 20 h, MeOH | 98 | 44 (methyl lactate) | 72 |
| Zr-Beta | Sucrose | 433, 20 h, MeOH | 99 | 40 (methyl lactate) | 72 |
| Sn-MWW | Glucose | 433, 20 h, MeOH | 99 | 44 (methyl lactate) | 91 |
| Sn-MWW | Fructose | 433, 20 h, MeOH | 99 | 46 (methyl lactate) | 91 |
| Sn-MWW | Sucrose | 433, 20 h, MeOH | 99 | 50 (methyl lactate) | 91 |
A major breakthrough in the conversion of saccharides to lactic acid over heterogeneous catalysts was achieved by using Sn-beta zeolite, which is remarkably efficient to convert sucrose to methyl lactate (Table 1, yield 64%) at 433 K in methanol.72 For comparison, H–Al-beta (Brønsted acidic), Ti- and Zr-beta (Lewis acidic) and Si-beta (non-acidic) were also investigated.72 H–Al-Beta converts sugars in methanol to hydroxymethylfurfural (derivatives, methyl levulinate and methyl-D-pyranoside through dehydration. Zr-Beta and Ti-beta with weak Lewis acidity produce only moderate yields (31–44%) of methyl lactate. The non-acidic Si-beta has limited ability to produce methyl lactate (<10%). Other Sn-based zeolites such as Sn-MWW also exhibit similar catalytic performance (Table 1).91 These results indicate that the Sn sites on the zeolite play a key role in the production of methyl lactate. A large amount of efforts were devoted to studying the reaction mechanism over Sn-zeolites to understand their high catalytic performance. Isotopic labelling, NMR spectroscopy and DFT calculations were employed to investigate the catalytic processes including hydrolysis, isomerisation, retro-aldol condensation, hydride shift and dehydration, which are discussed below.
The two retro-aldol products, dihydroxyacetone (DHA) and glyceraldehyde (GLY), are interconvertible through intramolecular hydride shift.104 They can be converted easily to lactic acid (activation energies of 58–88 kJ mol−1) through dehydration over Lewis or Brønsted acid sites, hydride shift over Lewis acid sites and rehydration over Brønsted acid sites under mild reaction conditions (353–398 K).104–108
Compared with fermentation processes, the selectivity on the Sn-beta system is lower (Table 2). A series of Sn-based zeolites have been prepared and investigated, such as Zn–Sn-beta and Pb–Sn-beta (LA 48% and 52%),109 hierarchical Sn-beta (methyl LA 58%),110 nanosize Sn-beta (methyl LA 57%),111 Sn-beta + K2CO3 (methyl LA 72%),112 Sn-MCM-41 COOH (methyl LA 45%),113 and Sn-MFI + MoO3 (ethyl LA 66%),114 as well as other catalysts, such as Zr-SBA-15 (ethyl LA 33%),115,116 Ga-doped Zn/H-nanozeolite Y catalysts (methyl LA 58%),117 and Er-exchanged montmorillonite K10 (LA 68%).118 The yield of lactic acid or methyl lactate was improved to 72% in the system of Sn-beta + K2CO3,112 but it is still lower than that achieved by the fermentation process (95%, Table 2). Therefore, we summarise the reaction network including the details of active sites, reaction temperatures and activation energies (Scheme 4) to inform the design of new efficient catalysts. Among the conversion of cellulose to lactic acid, hydrolysis and dehydration can be catalysed by both Brønsted and Lewis acid sites, and the former has higher reactivity. Isomerisation, retro-aldol, and 1,2-H-shift can readily occur on Lewis acid sites. The formation of by-products such as 5-hydroxymethylfurfural (5-HMF) and humins mainly occurs on Brønsted acid sites. Thus, the decrease of Brønsted acid sites would significantly hinder the formation of by-products. This was confirmed by a study which shows that the selectivity of lactic acid can be improved by decreasing the ratio of Brønsted/Lewis acid sites.119 Although the hydrolysis of cellulose and dehydration of GLY can be affected in the meantime, it would have little effect on the overall reaction, because the rate-determining step is the retro-aldol reaction (the activation energy on Sn-beta is 119 kJ mol−1). Moreover, the accumulation of glucose from cellulose via hydrolysis can promote the formation of by-products, such as erythrose and glycolaldehyde. Thus, a slight reduction of the hydrolysis in turn is beneficial for improving the yield of lactic acid. Although the retro-aldol reaction of saccharides on Sn-beta zeolites occurs predominantly from fructose rather than glucose,100 the latter accounts for 10%, which needs further strategies to reduce the retro-aldol reaction of glucose. In conclusion, four aspects need to be considered to design an efficient catalyst: (i) controlling precisely the Brønsted acid sites in order to adjust the hydrolysis reaction and to selectively reduce the formation of 5-HMF and humins; (ii) developing new strategies to promote the retro-aldol cleavage between C3 and C4 centres and to hinder other retro-aldol reactions; (iii) discovering suitable Lewis acid sites to operate under mild reaction conditions and hence to reduce side reactions; and (iv) designing porous structures of the catalyst to improve the mass transfer of the reactants, intermediates and products.
| Criterion | Heterogeneous72 | Fermentation84,85 |
|---|---|---|
| Media | Alcohol | Aqueous broth |
| Feedstock | Glucose, sucrose | Glucose, sucrose |
| Concentration feed | Up to 10 wt% | Up to 15 wt% |
| Catalyst | Sn-Beta zeolite | Bacteria (yeast) |
| Co-reagents | None | Alkali + nutrients |
| Major product | Alkyl lactates | Ca-lactate salt |
| Stereopurity product | Racemic | L |
| Selectivity (mol%) | Up to 64% | Up to 95% |
| Productivity | Up to 3.3 g Lreactor−1 h−1 | 0.3–5 g Lreactor−1 h−1 |
| Gypsum co-product | No | 1 kg per kg of LA |
| Work-up | Easy: filtration, distillation, alcohol reuse | Complex: acidification, two filtrations, purification, esterification, distillation |
| Catalyst reuse | Yes | No (in a complex broth) |
 | ||
| Scheme 4 Summary of the main and side reactions during the conversion of cellulose to lactic acid. B and L represent Brønsted and Lewis acid sites, respectively. | ||
3.2. Production of 2,5-furandicarboxylic acid (FDCA)
FDCA is a precursor to produce sustainable polymers, such as polyethylene 2,5-furandicarboxylate, which can replace the petroleum-derived polyethylene terephthalate.120 The conversion of fructose to FDCA undergoes two steps: dehydration of fructose to 5-HMF and oxidation of 5-HMF to FDCA. Heterogeneous catalysts based upon noble metals, such as Pt, Au, Ru and Pd nanoparticles, combined with homogeneous base catalysts have been studied for the oxidation of 5-HMF to FDCA.121 These catalytic systems show excellent catalytic performance with a >99% yield of FDCA under mild conditions. However, homogeneous catalysts cause serious corrosion of the equipment and can hardly be separated or recycled. A homogeneous base-free route for the oxidation of 5-HMF to FDCA by using Mg–Al–CO3 hydrotalcite-supported Pd nanoparticles as the catalyst has been developed, achieving yields of FDCA of >99% at 373 K under ambient pressure.122 The excellent catalytic performance of Mg–Al–CO3 hydrotalcite-supported Pd nanoparticles under homogeneous base free-conditions is attributed to their suitable basicity and abundant –OH groups on the surface.The reaction mechanism of aerobic oxidation of 5-HMF to FDCA over Mg–Al–CO3 hydrotalcite-support Pd catalysts is proposed (Fig. 7).122 In the first step, the aldehyde group of 5-HMF adsorbs onto the surface of Pd nanoparticles, followed by nucleophilic addition of a hydroxyl group from hydrotalcite to form a new hydroxyl group on active Pd sites. The formed hydroxyl group and C–H group are then dehydrogenated to form a carboxylic group, leaving two hydrogen atoms on the surface of the Pd nanoparticles. Then, 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) is produced through a hydrolysis reaction, accompanied by regeneration of the hydroxyl group on hydrotalcite. The H atoms on the Pd nanoparticles react with O2 to form peroxide and water. In the second step, the hydroxymethyl group in HMFCA adsorbs onto the surface of the Pd nanoparticles, and O–H and C–H bonds are activated, followed by an elimination reaction of β-hydride to generate 5-formyl-2-furancarboxylic acid (FFCA). In the same way as the first step, the carbonyl group of FFCA is oxidised to FDCA.
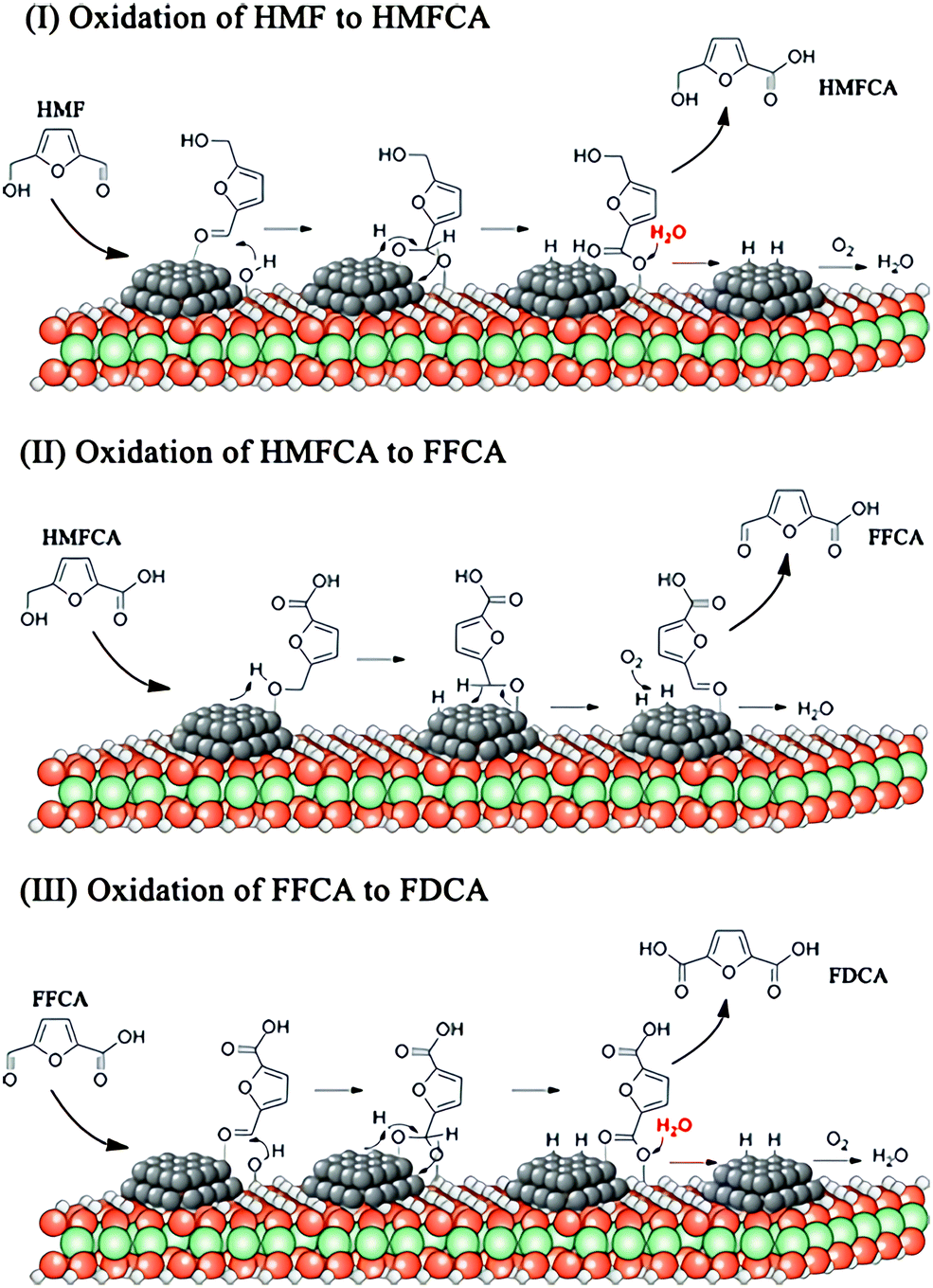 | ||
| Fig. 7 View of the plausible reaction mechanism of oxidation of 5-HMF to FDCA over Pd/HT catalysts. Reprinted with permission from ref. 122, Copyright© 2016 American Chemical Society. | ||
The proposed reaction mechanism reveals that the cooperativity between noble metals and basic sites of the support is of critical importance for the oxidation of 5-HMF to FDCA. Future improved catalysts need to be ideally based upon earth-abundant metals, which can retain synergistic effects with basic supports. The low yield of 5-HMF from the conversion of fructose is another major barrier to the production of FDCA from biomass, and this is discussed below.
4. Reaction mechanism for the production of liquid fuels
Conversion of biomass to liquid fuels is considered as less appealing than to bulk chemicals in terms of added value per tonne of biomass ($200–400 vs. ∼$1000 for liquid fuels and chemicals, respectively).123 However, the sum of demand for liquid fuels is significantly higher than for bulk chemicals. For example, ∼90% of crude oil is consumed as fuels while only the remaining 10% is upgraded to chemicals. Thus, the synthesis of sustainable fuels represents an enormous market in the conversion of biomass. First-generation biodiesel and bioethanol are manufactured from edible plant crops, which competes with food production in terms of land use.1 Therefore, the conversion of inedible lignocellulosic biomass to fuels represents a target sustainable approach.7,35,124–127 The strategy that breaks down lignocellulosic biomass to syngas (CO + H2) through gasification, followed by the Fischer–Tropsch process to upgrade the syngas to alkanes, is effective but energy-intensive. Alternatively, through liquefaction, hydrolysis, or dehydroxygenation, lignocellulosic biomass can be converted to liquid fuels, such as alkanes, aromatic hydrocarbons or other small molecules.128–1304.1. Conversion of fructose to DMF
DMF has a suitable boiling point (366 K), low water solubility (2.3 g L−1), high research octane number (119) and high energy density (30 kJ cm−1), which make it an ideal candidate as liquid fuel for transportation.126 A two-step route to convert fructose to DMF has been reported.126 In the first step, fructose is transformed to 5-HMF through dehydration in an HCl/NaCl/solvent/water system at 180 °C with a 5-HMF yield of 70%. After separation and purification, in the second step, 5-HMF is converted to DMF (yield 76%) on a CuRu/C catalyst via hydrogenation and HDO at 220 °C. The reaction mechanisms of dehydration, hydrogenation and HDO over these solid catalysts were studied by the isotope-labelling technique and NMR spectroscopy.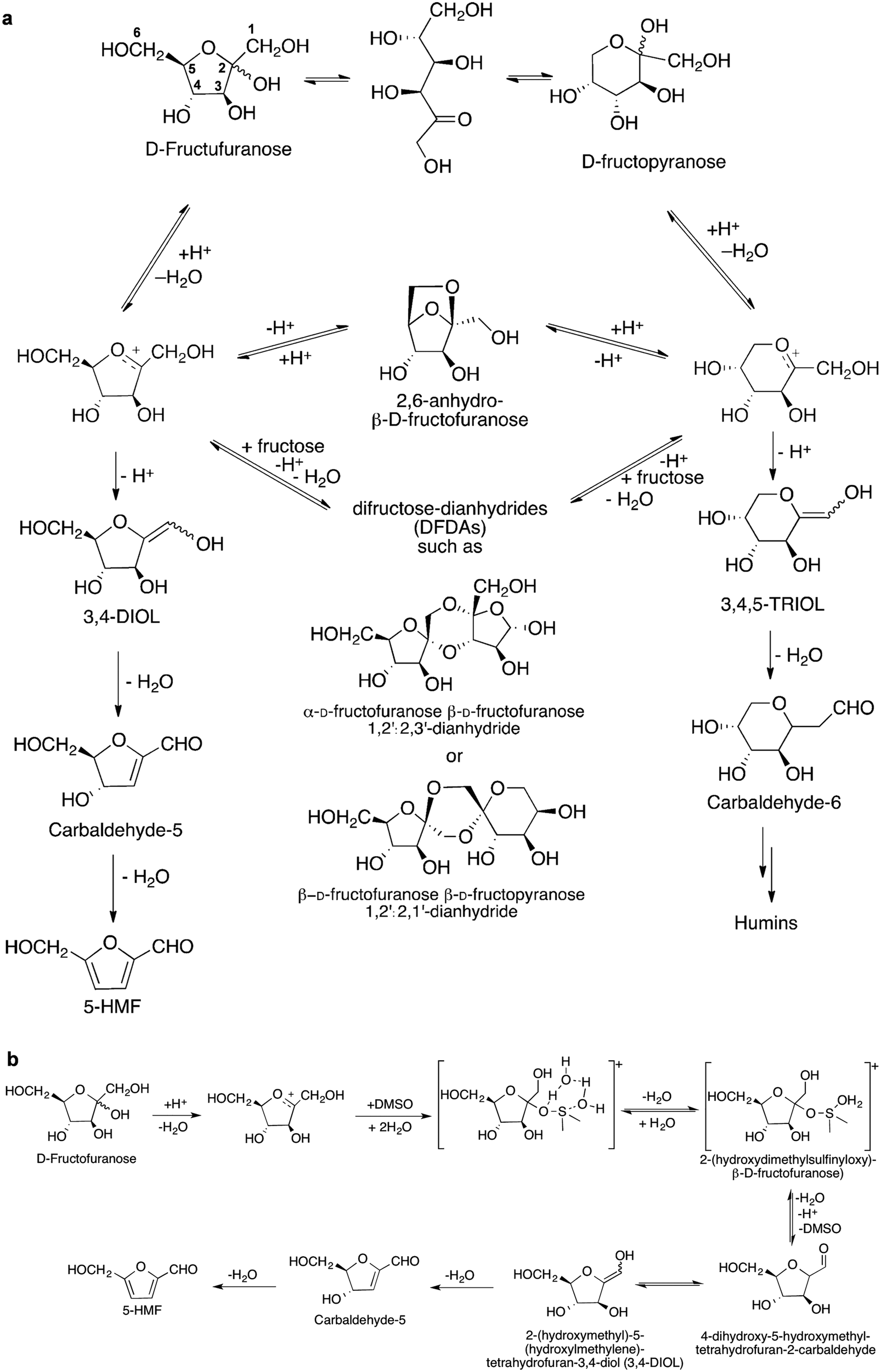 | ||
| Scheme 5 Dehydration of fructose to 5-HMF. (a) Proposed reaction mechanism of acid-catalysed dehydration of fructose to 5-HMF via the cyclic pathway. Reproduced with permission from ref. 29, Copyright© 2012 Royal Society of Chemistry. (b) Proposed role of DMSO in conversion of fructose to 5-HMF. Adapted with permission from ref. 134, Copyright© 2016 Elsevier. | ||
Dimethyl sulfoxide (DMSO) has been identified as a suitable solvent for the synthesis of 5-HMF catalysed by Amberlyst 70 and it can limit side reactions. Among fructose isomers, the concentration of D-fructofuranose is significantly higher in DMSO than in other solvents such as H2O and MeOH. The nucleophilic attack of DMSO to the fructopyranosil oxocarbenium ions forms a key intermediate [2-(hydroxydimethylsulfinyloxy)-β-D-fructofuranose] (Scheme 5b).134 This intermediate can be readily converted to 5-HMF rather than to oligomers and humins. In addition, 5-HMF has high reactivity and can transform to humins, and, in order to avoid any further reaction of 5-HMF, a bi-phase system to extract 5-HMF from the reaction mixtures was developed.135
Recently, porous solid solvents (PSSs), which are comprised of solvent moieties (i.e., DMSO and ionic liquids) and solid acid sites (i.e. sulphonic groups), have been designed for enhancing the conversion of fructose to 5-HMF.136 The solvent moieties in PSSs as hydrogen-bond acceptors mimic liquid-phase solvents to break the hydrogen bonding network of fructose and to stabilise the intermediate (Fig. 8). Moreover, the solvent moieties surrounding the acid sites can enhance their cooperation, resulting in higher catalytic performance compared with that in liquid solvent systems. This new approach significantly improves the yield of 5-HFM in a green manner by avoiding costly separation processes.
 | ||
| Fig. 8 Schematic illustration of fructose in various systems. (a) Solvent DMSO containing hydrogen bond acceptors interacting with fructose through hydrogen bonds. (b) Solvent moieties (purple) anchored on a porous solid material interacting with fructose through hydrogen bonds. (c) The interruption of the hydrogen network of fructose by solvent moieties (X−). C in DMSO, yellow; C in fructose, grey; H, white; S, light magenta; O in DMSO, dark magenta; O in fructose, red; hydrogen bond, dash line. Reproduced from ref. 136, published by Springer Nature, licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/legalcode). | ||
The reaction mechanism of the conversion of 5-HMF to DMF over PtCo nanocrystals was investigated by XRD, XAS and DFT calculations.137 XRD and XAS analysis suggests that the Pt3Co2 catalyst consists of a Pt-rich core (88% Pt and 12% Co) and a Co3O2 surface monolayer (Fig. 9a), which are responsible for the dissociation of H2 and cleavage of C–O bonds, respectively. The first step is the dissociation of H2, which can occur via homolytic and/or heterolytic dissociation paths. The homolytic splitting of H2 molecules over Co and Pt atoms is energetically favourable, resulting in a Co–H–Pt bridging configuration. Then, the two dissociated H atoms are added to the carbonyl group of weakly adsorbed 5-HMF, yielding 2,5-bis(hydroxymethyl)furan. Subsequently, the HDO of 2,5-bis(hydroxymethyl)furan occurs on the Co3O2 coating (Fig. 9b). The two Co atoms break the C–O bonds to yield a loosely bound radical and an –OH group. Then, the radical moiety drags a hydrogen atom from the –OH group, yielding 2-hydroxylmethyl-5-methyl furan and a chemisorbed oxygen atom. Similarly, C–O scission on 2-hydroxylmethyl-5-methyl furan results in the formation of DMF. Finally, the chemisorbed O atom reacts with H2 to form water and complete the catalytic cycle. The scission of the C–O bond can be catalysed over the surface layer of Co3O2, whereas the chemisorption of the furan ring is difficult on Co3O2. Thus, the ring-opening and decarboxylation of the formed DMF is hindered, leading to the selective production of DMF.
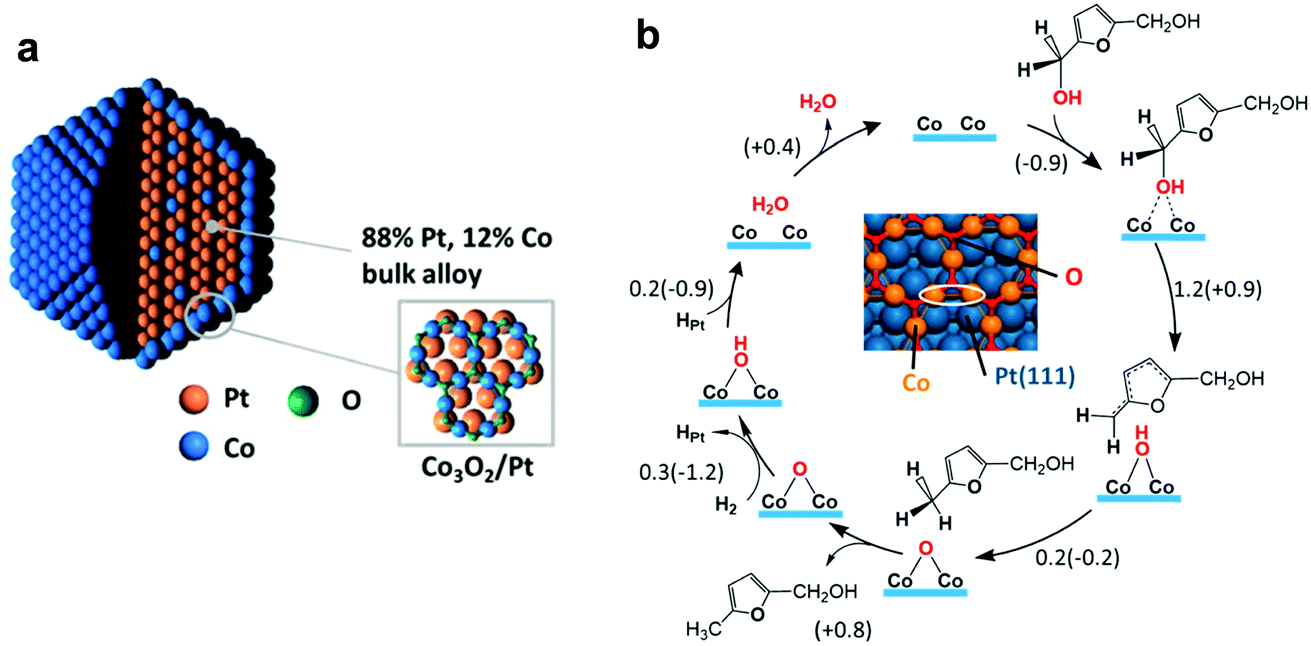 | ||
| Fig. 9 Mechanism of HDO of 5-HMF. (a) Pt3Co2 nanocrystal model involving an alloy core (88% Pt, 12% Co) covered with a Co3O2 surface oxide monolayer with a honeycomb structure. (b) Reaction mechanism of 2,5-bis(hydroxymethyl)furan hydrodeoxygenation to 2-hydroxylmethyl-5-methyl furan on the Co3O2/Pt(111) surface. DFT reaction barriers (energies) are given in eV. The inset depicts a portion of a Co3O2/Pt(111) surface. Two Co atoms participating in C–O bond activation are encircled with a white ellipsoid. Reproduced with permission from ref. 137, Copyright© 2016 American Chemical Society. | ||
The reaction pathway described above can be altered over Pd-based N-doped mesoporous carbon (Pd/NMC) materials with the assistance of formic acid, and a high yield of DMF can be achieved (>97%, Fig. 10a).145 In the Pd/NMC material, two pyridinic N atoms interact with one Pd atom, leading to a change in the oxidation state of Pd0 to Pd2+ (Fig. 10b and c). Formic acid is activated by Pd2+ centres and decomposes to a formate anion and H+ (Fig. 10d). The H+ from formic acid protonates the C–OH group of HMF that is bound to the Pd2+ species, thus driving the cleavage of the C–OH bond during the hydrogenolysis of HMF to 5-methylfurfural (5-MF) with H− from heterolytic dissociation of H2 on Pd0 nanoparticles. Then the –CHO group of 5-MF is hydrogenated on Pd0, followed by a similar hydrogenolysis process on Pd2+ to produce DMF.145 In this system, the synergistical cooperation of Pd2+ species and formic acid in the hydrogenolysis of the C–OH bond leads to the improvement of the activity,145 and formic acid also acts as a mild hydrogen source and suppresses the ring-hydrogenation to improve the selectivity of DMF.146–148
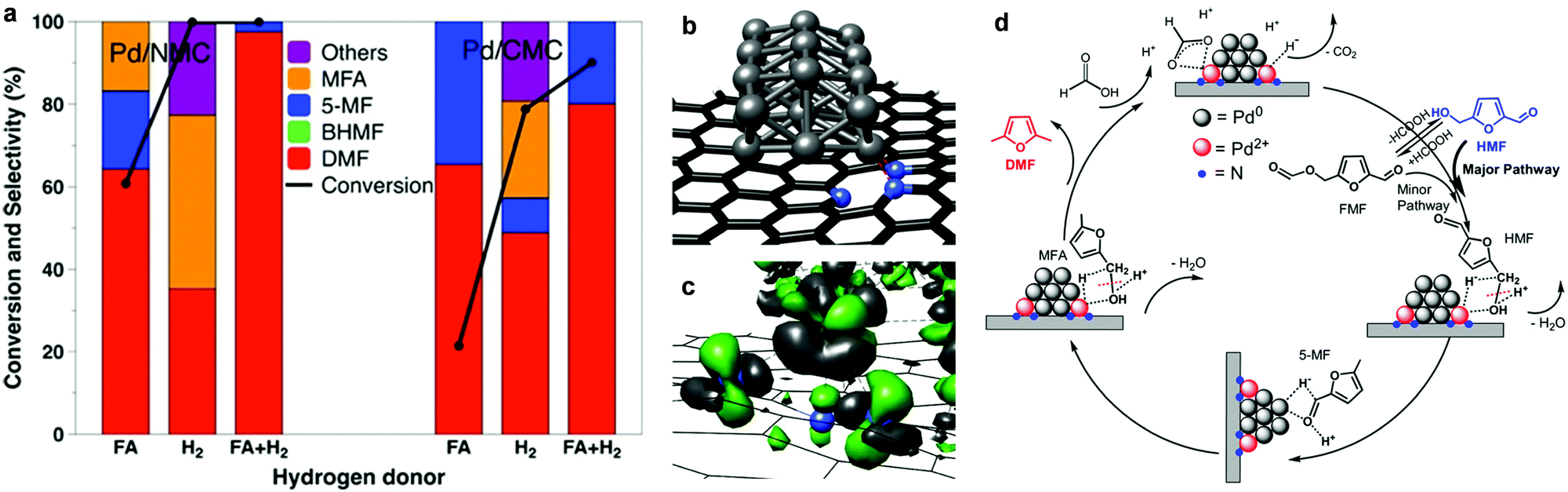 | ||
| Fig. 10 Conversion of HFM to DMF using different hydrogen sources over Pd-based catalysts. (a) Most stable structure of Pd21 on a pyridinic carbon support (two pyridinic N connected to Pd21) calculated for the Pd/NMC model. (b) Change in the electron density induced by the interaction between the Pd21 cluster and the pyridinic N atoms (green: density gain; grey: density loss). (c) Comparison of Pd/NMC and Pd/CMC with respect to the conversion of HMF to DMF using different hydrogen sources after 3 h. (d) Proposed reaction pathway for the conversion of HMF to DMF over Pd/NMC in FA. Reproduced from ref. 145, published by John Wiley and Sons, licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/legalcode). | ||
Mechanism studies provide insights into the design of catalysts for the selective production of DMF from fructose, and inspire a series of studies on one-pot production of DMF from fructose.149 A carbon-based solid functionalised with p-toluenesulfonic acid (TsOH) and an encapsulated CuCo multifunctional catalyst (CuCo@C-TsOH) can directly convert fructose to DMF with a yield of 71%.150 Recently, a multifunctional catalyst Cu–Pd/UiO-66(NH2)@SGO was developed for direct conversion of saccharides to DMF.151 The Cu–Pd/UiO-66(NH2)@SGO (SGO = sulfonated graphene oxide) catalyst, which has Brønsted acid sites, Lewis acid sites and HDO active sites, shows excellent catalytic performance for direct conversion of fructose to DMF with a yield of 85%.
4.2. Production of C5/C6 alkanes
Production of liquid alkanes from cellulose/hemicellulose has attracted tremendous attention because C5/C6 alkanes are components of liquid fuels.152–154 It has been demonstrated that sorbitol can be transformed to C5/C6 alkanes under mild conditions in an aqueous-phase reforming process.155 This discovery is the cornerstone to developing new processes for production of liquid fuels from biomass. By using Ir–ReOx/SiO2 combined with HZSM-5 as a catalyst, cellulose has been successfully converted to n-hexane.156 A multifunctional Pt/NbOPO4 catalyst has shown excellent catalytic performance for the direct conversion of raw woody biomass into liquid alkanes,7 where the major barrier is the ring-opening and the sequential HDO of tetrahydrofuran (THF) derivatives (e.g., DMF). The reaction mechanism of key steps, i.e. ring-opening and HDO of THF on Pt/Nb2O5, were studied by INS and computational methods. Firstly, THF is adsorbed on the surface via interactions between its ring O(δ−) centre and the open Nb(δ+) site (O⋯Nb = 2.33 Å) (Fig. 11a). The adsorbed THF molecule reacts on strong Lewis acid sites [Nb(V)], and forms an intermediate bound to two adjacent Nb(V) centres simultaneously (O⋯Nb = 1.98 Å) via ring-opening (Fig. 11b). This intermediate is unstable, and quickly transforms to 1-butanoxide bound to the surface Nb(V) sites under H2 (dissociation over Pt sites), which was observed experimentally by INS (Fig. 11c). Further cleavage of the C–O bond of 1-butanoxide on NbOPO4 was investigated by first-principles calculations and the widely used ReOx catalyst in biomass conversion was also studied for comparison. Catalysed by surface Nb5c, the C–O bond can be cleaved with only a barrier of 76.2 kJ mol−1, whereas it has to overcome a larger barrier of 124 kJ mol−1 on Re2O7 (Fig. 11d). The dissociation barrier of the C–O bond was found to be related to their corresponding adsorption energies and the stronger the bond strength of M5c–O (M = Nb, Re), the easier the C–O bond cleavage. An analysis of the electronic structure shows electron accumulation between O (or OH) and Nb5c (or Re5c) (Fig. 11e), indicating a typical covalent bond character, of which the strength is determined by the size and energy level of the d-orbital of M5c cations. The d-orbitals of Nb5c near the Fermi level are more delocalised, and the energy level of the highest occupied d-bands is higher for Nb5c compared to Re5c (Fig. 11f). Thus, NbOPO4 has strong binding ability and can effectively catalyse the deoxygenation reaction.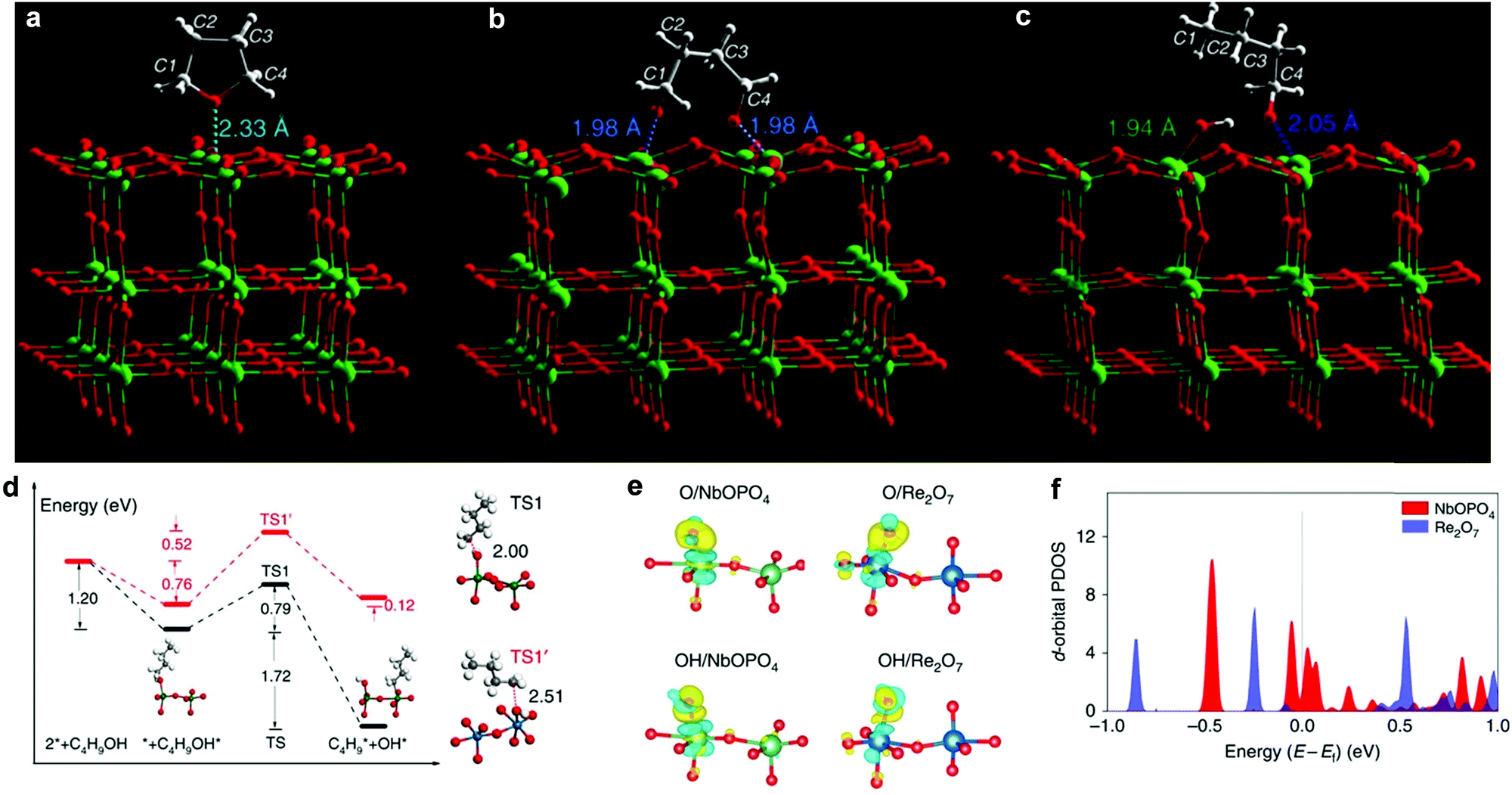 | ||
| Fig. 11 View of the optimised structural models of adsorbed THF (a), reacted (ring-opened) THF (b) and 1-butanol after adsorption to generate 1-butanoxide and surface hydroxyl (c) on the (001) plane of the catalyst Pt/Nb2O5 (Nb: green; O: red; C; grey; H: white). (d) Calculated energy profiles of C–O bond cleavage of C4H9OH. Black and red lines indicate NbOPO4(100) and Re2O7(010), respectively. The structures of the initial and final states on NbOPO4(100) are shown, whereas the transition state involving C–O bond cleavage of C4H9OH is depicted as TS1 and TS1′, with the elongated C–O bond lengths shown. Emerald balls represent Nb atoms, dark blue for Re, white for H, grey for C and red for O. (e) The isosurfaces of the charge density difference for O and OH adsorption on the NbOPO4(100) and Re2O7(010) surfaces. For clarity, only the local active sites are shown. The regions depicted in yellow indicate charge accumulation and light blue for charge depletion. Emerald balls represent Nb atoms, dark blue for Re, white for H and red for O. (f) The d-orbital projected density of states for the surface Nb5c and Re5c atoms, demonstrating their relative energies, in which the energy is aligned to the Fermi level. Reproduced from ref. 7, published by Springer Nature, licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/legalcode). | ||
4.3. Production of long-chain alkanes
As the boiling points of C5/C6 alkanes are low (309–342 K), production of long-chain alkanes from biomass is more desirable for direct use as gasoline. A stepwise route has been proposed to produce C7–C15 liquid alkanes.89 It proceeds through dehydration of biomass-derived carbohydrates on acid catalysts, aldol condensation of furanic compounds with ketones to form large organic compounds on base catalysts, and HDO to form long-chain alkanes on acid/metal sites. Among these steps, the aldol condensation is the step that determines the overall number of carbon atoms and, hence, impacts the quality of the obtained fuel.The reaction mechanism of aldol condensation of furfural-acetone on acid–base pairs is proposed in Scheme 6 based on kinetic studies.157 Acetone firstly adsorbs on acid–base pairs and its α-proton is abstracted by base sites, resulting in the formation of a carbanion intermediate, which is stabilised by weak acid sites. The adsorbed furfural molecule on a weak acid site is then attacked by the carbanion via the interaction between the carbanion and carbonyl group. This step leads to the formation of an α-hydroxyl ketone which is unstable and rapidly dehydrates to C8 compounds and water, accompanied by the regeneration of active sites. Similarly, further aldol condensation of C8 compounds with furfurals produces C13 compounds. Kinetic studies indicate that the rate-determining step is the abstraction of α-protons by base sites. Thus, the basicity of the active site and the acidity of that proton are key factors affecting the aldol condensation between furanic compounds and ketones. The roles of weak acid sites are to adsorb reagents via carbonyl groups and to stabilise the reaction intermediates.
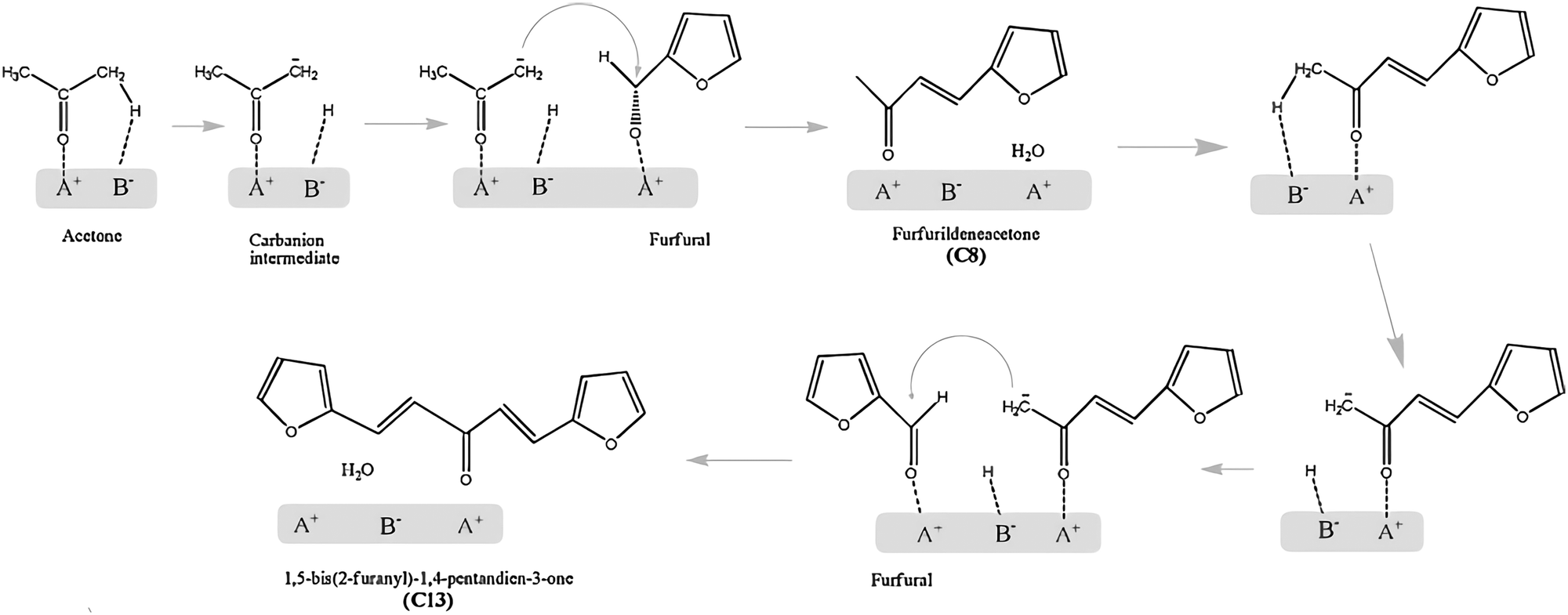 | ||
| Scheme 6 Proposed reaction mechanism of furfural-acetone aldol condensation on acid–base pairs. Reproduced with permission from ref. 157, Copyright© 2011 Elsevier. | ||
5. Reactions of C–O/C–C bonds in biomass
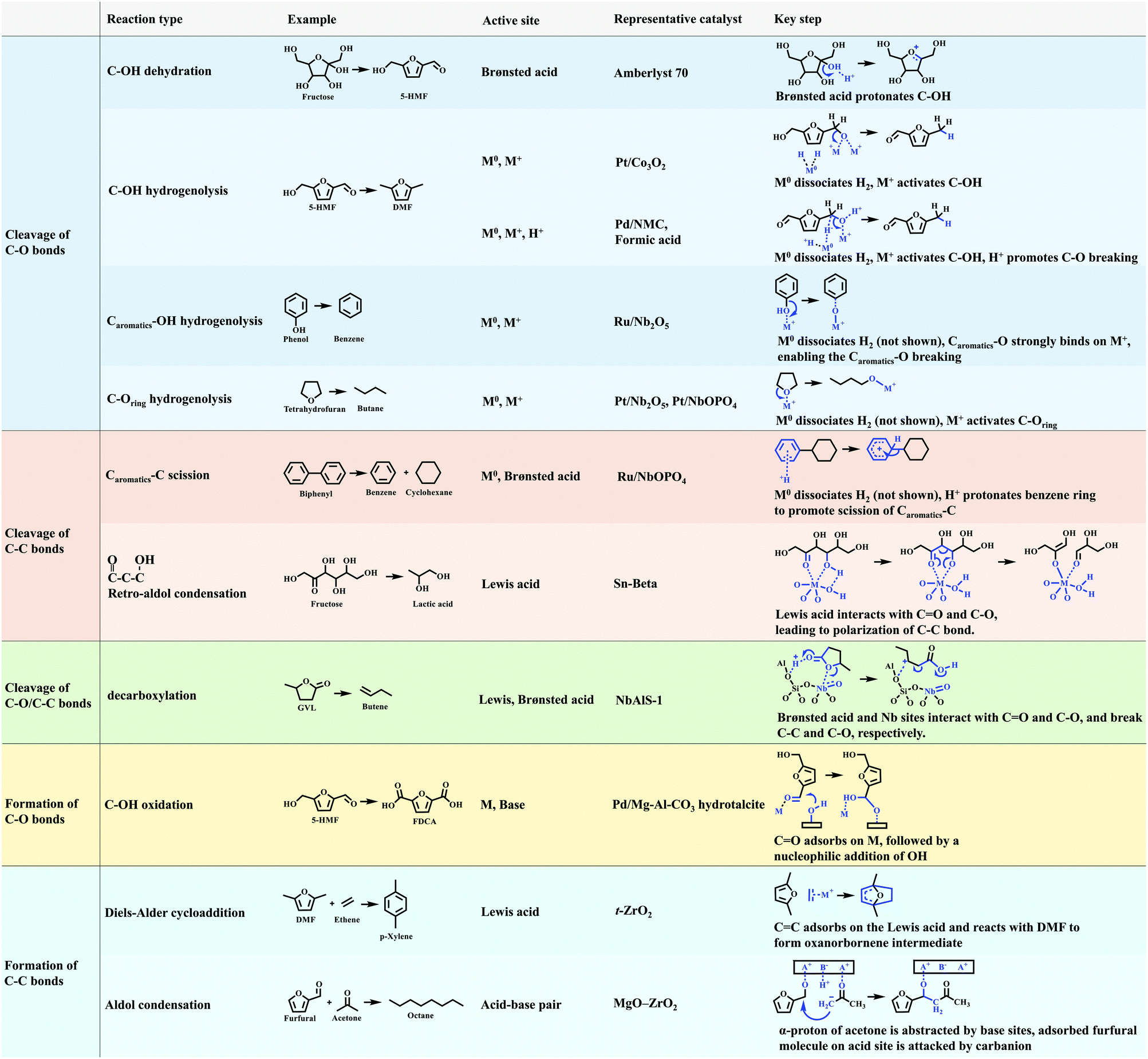
Lignocellulosic biomass is mainly constructed from C–O and C–C bonds. The conversion of lignocellulosic biomass involves numerous reactions, which can be generally classified as: cleavage of C–O bonds, cleavage of C–C bonds, cleavage of C–O/C–C bonds, and formation of C–O and C–C bonds (Fig. 12). The heterogeneous catalysts for catalysing these reactions are summarised in Fig. 13. C–O bonds can be cleaved via either dehydration or hydrogenolysis. In the former, –OH is protonated by Brønsted acids (such as Amberlyst 70), resulting in the cleavage of the C–O bond. The hydrogenolysis of C–OH usually occurs on M0/M+ sites (M0, metal in the reduced state; M+, metal in the oxidised state). The M0 site dissociates H2 and the M+ site activates C–OH, enabling the cleavage of C–O bonds. Where the CC bond needs to be protected during hydrogenolysis, such as 5-HMF to DMF, and phenol to benzene, efforts need to be made to slow down the H2 dissociation and/or to enhance the activation of the C–O bond. Several strategies have been reported. (i) A Pt/Co3O2 core/shell structure is designed, in which Pt is enriched in the core but lean in the shell. Thus, the dissociation of H2 is reduced and hydrogenation of the CC bond is hindered. (ii) A Pd/NMC/formic acid system is proposed. Pd sites partially exist in the divalent state due to interaction with pyridinic N centres, so the dissociation of H2 is hindered by reducing the amount of Pd0 sites. In the meantime, Pd2+ and formic acid both reduce the activation energy of C–O bonds. (iii) A Ru/Nb2O5 catalyst is designed, which consists of metal sites that dissociate H2 mildly and Lewis acid sites that strongly bind to the C–O group. The strong Lewis acid and metal with a strong ability of H2 dissociation (Pt/Nb2O5) are favourable for the cleavage of C–Oring. The cleavage of the C–C bond is challenging and needs very strong acid sites. The functional groups nearby which polarise the C–C bond determine the pathway of its cleavage. Because the aromatic group can be protonated by the Brønsted acid, the Caromatic–C bond is cleaved by β-scission. The –CHO and –OH groups can be activated by a strong Lewis acid (e.g., Sn-beta), and the cleavage of the C–C bond of fructose undergoes retro-aldol condensation on Lewis acid sites. In terms of the cleavage of intramolecular C–O/C–C bonds (such as in GVL), the precise locations of the Brønsted and Lewis acid sites (NbAlS-1) and their close cooperation play a key role. The formation of C–O and C–C bonds is only involved in a few reactions for biomass conversion, such as the oxidation of –OH to –COOH, Diels–Alder cycloaddition to produce p-xylene, and aldol condensation for increasing the length of the carbon chain. In summary, the cleavage of C–O and C–C bonds is catalysed by acid sites and metal sites, while the formation of C–O and C–C bonds usually requires the participation of a base. Overall, reactions involving C–O or C–C bonds vary in different systems, and detailed studies of the reaction mechanism are required for the design of optimal catalysts for a given system.
 | ||
| Fig. 12 Comparison of the reaction mechanisms of the cleavage of C–O, C–C, and C–O/C–C bonds, and the formation of C–O and C–C bonds. M0 is the metal in the reduced state, and M+ is the metal in the oxidised state. A+B− is the acid–base pair. | ||
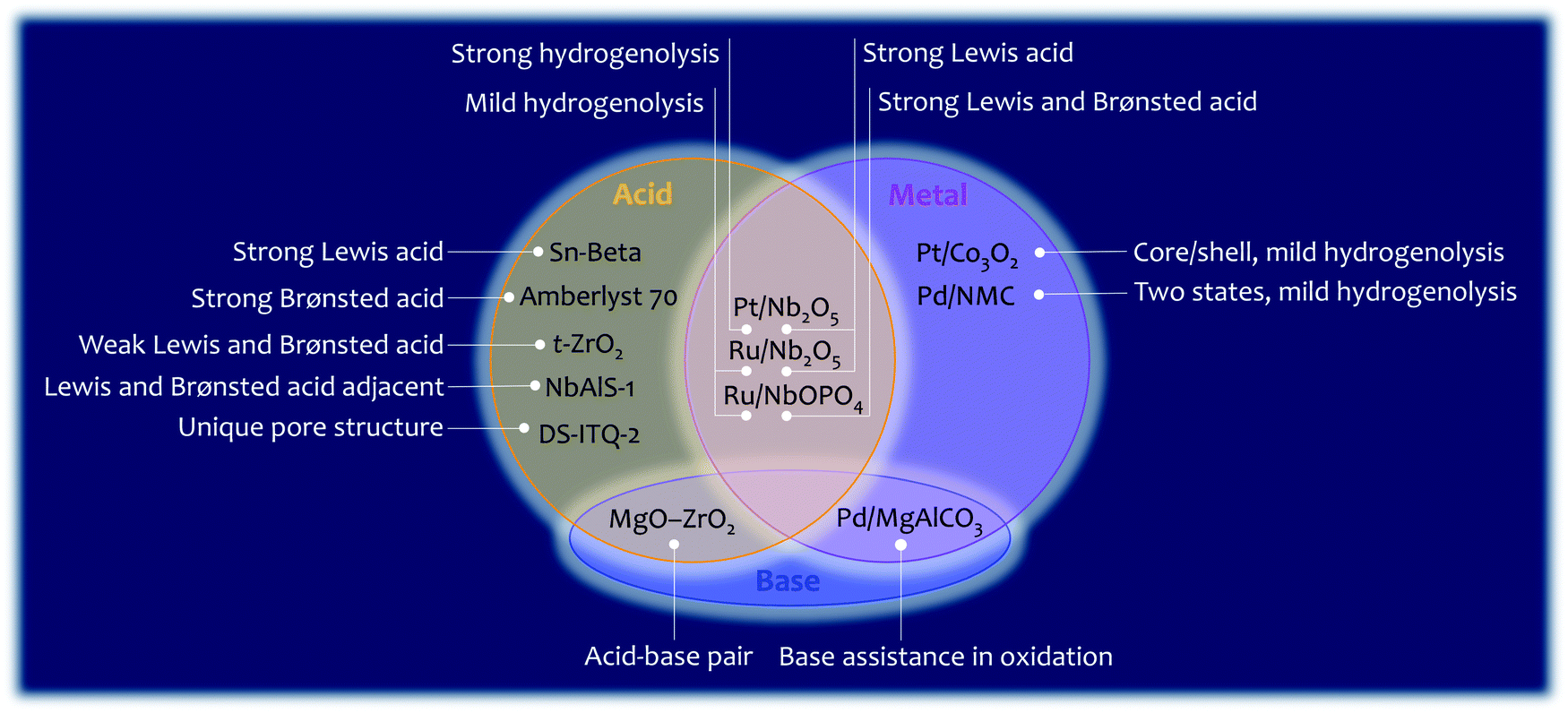 | ||
| Fig. 13 An overview of the activity of selected heterogeneous catalysts discussed in this review. | ||
6. Summary and outlook
The conversion of lignocellulosic biomass to useful carbon feedstocks is of great importance for the development of a sustainable and carbon-neutral economy. A number of research efforts have been devoted to developing efficient catalysts and catalytic systems for the conversion of lignin, cellulose and hemicellulose to value-added chemicals and liquid fuels. Lignin consists of aromatic units, linked by C–O and C–C bonds. To produce aromatics, the key step is to cleave C–O and C–C bonds selectively and to preserve aromatic functionalities in the meantime. Emerging niobium-based porous solids incorporating both NbOx and Brønsted acid sites can catalyse the scission of both C–O and C–C bonds under an H2 atmosphere, while largely preserving the aromatic functionalities with an arene selectivity up to ∼70%. These promising systems give key insights into the design of new catalysts: (i) construction of cooperative catalytic sites (e.g., early transition metals and strong Brønsted acid sites) to promote the cleavage of C–O and C–C bonds; (ii) weakening the adsorption of benzene rings on the catalyst supports to hinder their hydrogenation; and (iii) optimising the active sites to promote the dissociation of hydrogen such as by using bimetallic nanoparticles.The conversion of cellulose and hemicellulose to useful chemicals often involves cascade reactions. Dehydration and decarboxylation often occur on Brønsted acid sites. Isomerisation, retro-aldol condensation, hydride shift, and ring-opening prefer Lewis acid sites. Aldol condensation can be catalysed by either an acid or base. Subtle changes in the strength of the acid/base can greatly affect the performance of various catalysts. Hydrogenation, hydrogenolysis, hydrodeoxygenation and oxidation often need active metal sites, and the key to catalyst design is to precisely control the activity of these sites and to tune the microenvironments around these active sites, which all requires fundamental understanding of the reaction mechanism. It is therefore of critical importance to link catalyst design and catalysis testing to state-of-the-art structural and dynamic characterisation of the catalytic chemistries that underpin the product selectivity. Fundamental understanding of how catalysts function (or not) at a molecular level will enable us to ‘see’ and thus precisely manipulate the microenvironment to deliver the desired catalytic performance. A number of operando and in situ characterisation techniques have been employed to gain deep understanding, such as high-resolution synchrotron X-ray powder diffraction, inelastic neutron scattering, X-ray absorption spectroscopy and NMR spectroscopy. Vibrational spectroscopy is one of the most important techniques to capture the reaction intermediates and de-convolute the reaction network. While INS is usually collected at temperatures below 50 K to minimise the Debye–Waller effect, IR/Raman spectra can be collected at reaction temperatures, although their interpretation is often challenging and subject to uncertainties. The development of new measurement facilities to enable the simultaneous measurements of INS and optical spectroscopies would significantly promote the studies of reaction mechanisms by allowing the unambiguous assignment of vibrational modes with the aid of computational methods. The reaction mechanism discussed in this review will provide insights into further design and optimisation of future targets and identification of industrial applications in bio-refineries.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank the Royal Society, the University of Manchester, the National Key Research and Development Program of China (2017YFA0403103, 2017YFA0403003 and 2017YFA0403101, 2017YFA0403102), and the National Natural Science Foundation of China (21890761, 21733011) for funding.References
- X. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS PubMed.
- D. Yang, Q. Zhu, X. Sun, C. Chen, W. Guo, G. Yang and B. Han, Angew. Chem., Int. Ed., 2020, 59, 2354–2359 CrossRef CAS PubMed.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162 CrossRef PubMed.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- A. R. C. Morais, A. M. da Costa Lopes and R. Bogel-Łukasik, Chem. Rev., 2015, 115, 3–27 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- S. Xia, G. A. Baker, H. Li, S. Ravula and H. Zhao, RSC Adv., 2014, 4, 10586–10596 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. V. den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- P. Sudarsanam, R. Zhong, S. V. den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- T. W. Walker, A. H. Motagamwala, J. A. Dumesic and G. W. Huber, J. Catal., 2019, 369, 518–525 CrossRef CAS.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. M. Alonso, R. Mariscal and M. L. Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- H. Li, A. Bunrit, N. Li and F. Wang, Chem. Soc. Rev., 2020, 49, 3748–3763 RSC.
- C. Mondelli, G. Gözaydın, N. Yan and J. Pérez-Ramírez, Chem. Soc. Rev., 2020, 49, 3764–3782 RSC.
- G. R. Akien, L. Qi and I. T. Horváth, Chem. Commun., 2012, 48, 5850–5852 RSC.
- T. Blasco, Chem. Soc. Rev., 2010, 39, 4685 RSC.
- B. T. W. Lo, L. Ye and S. C. E. Tsang, Chem, 2018, 4, 1778–1808 CAS.
- M. A. Newton and W. van Beek, Chem. Soc. Rev., 2010, 39, 4845 RSC.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8, 16104 CrossRef PubMed.
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudić, S. F. Parker, S. Yang and Y. Wang, Chem, 2019, 5, 1521–1536 CAS.
- L. Lin, A. M. Sheveleva, I. da Silva, C. M. A. Parlett, Z. Tang, Y. Liu, M. Fan, X. Han, J. H. Carter, F. Tuna, E. J. L. McInnes, Y. Cheng, L. L. Daemen, S. Rudić, A. J. Ramirez-Cuesta, C. C. Tang and S. Yang, Nat. Mater., 2020, 19, 86–93 CrossRef CAS PubMed.
- A. J. O’Malley, S. F. Parker and C. R. A. Catlow, Chem. Commun., 2017, 53, 12164–12176 RSC.
- D. Lennon and S. F. Parker, Acc. Chem. Res., 2014, 47, 1220–1227 CrossRef CAS PubMed.
- P. W. Albers and S. F. Parker, Adv. Catal., 2007, 51, 99–132 CAS.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS PubMed.
- L. Lin, Q. Mei, X. Han, S. F. Parker and S. Yang, Top. Catal., 2021, 64, 593–602 CrossRef CAS.
- M. Bender, ChemBioEng Rev., 2014, 1, 136–147 CrossRef CAS.
- M. Garside, Global capacity of benzene 2017 & 2022, Statista Inc., New York, 2020 Search PubMed.
- M. Garside, Global production capacity of toluene 2018 & 2023, Statista Inc., New York, 2019 Search PubMed.
- M. Garside, Global xylene capacity projection 2019-2021, Statista Inc., New York, p. 2021 Search PubMed.
- X. Shen, Q. Meng, Q. Mei, H. Liu, J. Yan, J. Song, D. Tan, B. Chen, Z. Zhang, G. Yang and B. Han, Chem. Sci., 2020, 11, 1347–1352 RSC.
- Z. Ma, E. Troussard and J. A. van Bokhoven, Appl. Catal., A, 2012, 423–424, 130–136 CrossRef CAS.
- O. Jan, R. Marchand, L. C. A. Anjos, G. V. S. Seufitelli, E. Nikolla and F. L. P. Resende, Energy Fuels, 2015, 29, 1793–1800 CrossRef CAS.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- L. Tao, T.-H. Yan, W. Li, Y. Zhao, Q. Zhang, Y.-M. Liu, M. M. Wright, Z.-H. Li, H.-Y. He and Y. Cao, Chem, 2018, 4, 2212–2227 CAS.
- V. J. Margarit, E. M. Gallego, C. Paris, M. Boronat, M. Moliner and A. Corma, Green Chem., 2020, 22, 5123–5131 RSC.
- V. J. Margarit, M. E. Martínez-Armero, M. T. Navarro, C. Martínez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 13724–13728 CrossRef CAS PubMed.
- E. A. Uslamin, N. Kosinov, G. A. Filonenko, B. Mezari, E. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9, 8547–8554 CrossRef CAS.
- F. Wang, X. Chu, P. Zhao, F. Zhu, Q. Li, F. Wu and G. Xiao, Fuel, 2020, 262, 116538 CrossRef CAS.
- I. F. Teixeira, B. T. W. Lo, P. Kostetskyy, M. Stamatakis, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. E. Tsang, Angew. Chem., Int. Ed., 2016, 55, 13061–13066 CrossRef CAS PubMed.
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, Appl. Catal., A, 2014, 469, 490–511 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- J. J. Bozell, Science, 2010, 329, 522–523 CrossRef CAS PubMed.
- D. Wang, S. H. Hakim, D. M. Alonso and J. A. Dumesic, Chem. Commun., 2013, 49, 7040–7042 RSC.
- L. Ye, Q. Song, B. T. W. Lo, J. Zheng, D. Kong, C. A. Murray, C. C. Tang and S. C. E. Tsang, Angew. Chem., Int. Ed., 2017, 56, 10711–10716 CrossRef CAS PubMed.
- W. Lin, L. Ye, S. Wu, B. Lo, Y.-K. Peng, P. Zhao, I. McPherson and S. C. E. Tsang, ChemSusChem, 2018, 11, 4214–4218 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- B. Donoeva and K. P. de Jong, Nat. Mater., 2020, 19, 5–6 CrossRef CAS PubMed.
- J. Q. Bond, C. S. Jungong and A. Chatzidimitriou, J. Catal., 2016, 344, 640–656 CrossRef CAS.
- B. T. W. Lo, L. Ye, J. Qu, J. Sun, J. Zheng, D. Kong, C. A. Murray, C. C. Tang and S. C. E. Tsang, Angew. Chem., Int. Ed., 2016, 55, 5981–5984 CrossRef CAS PubMed.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- A.-C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- R. F. Service, Science, 2006, 312, 1861 CrossRef CAS PubMed.
- M. Dusselier, P. V. Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 3210–3215 CrossRef CAS PubMed.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357–8362 CrossRef CAS PubMed.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- R. D. Clercq, M. Dusselier and B. F. Sels, Green Chem., 2017, 19, 5012–5040 RSC.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- San Jose, Global Lactic Acid Market, PRWEB, California, 2012.
- M. Dusselier, M. Mascal and B. F. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, Cham, 2014, pp. 1–40 Search PubMed.
- Y. J. Wee, J. N. Kim and H. W. Ryu, Food Technol. Biotechnol., 2006, 44, 163–172 CAS.
- W. Groot, J. van Krieken, O. Sliekersl and S. de Vos, Poly(Lactic Acid), John Wiley & Sons, Ltd, 2010, pp. 1–18 Search PubMed.
- L. Li, F. Shen, R. L. Smith and X. Qi, Green Chem., 2017, 19, 76–81 RSC.
- Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4, 2141 CrossRef PubMed.
- F.-F. Wang, C.-L. Liu and W.-S. Dong, Green Chem., 2013, 15, 2091–2095 RSC.
- W. Zeng, D. Cheng, F. Chen and X. Zhan, Catal. Lett., 2009, 133, 221 CrossRef CAS.
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, Appl. Catal., B, 2011, 105, 171–181 CrossRef CAS.
- Q. Guo, F. Fan, E. A. Pidko, W. N. P. van der Graaff, Z. Feng, C. Li and E. J. M. Hensen, ChemSusChem, 2013, 6, 1352–1356 CrossRef CAS PubMed.
- K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya and A. Satsuma, Green Chem., 2009, 11, 1627–1632 RSC.
- C. C. Chang, H. Je Cho, Z. Wang, X. Wang and W. Fan, Green Chem., 2015, 17, 2943–2951 RSC.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS PubMed.
- N. Shimada, H. Kawamoto and S. Saka, Carbohydr. Res., 2007, 342, 1373–1377 CrossRef CAS PubMed.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- S. G. Elliot, E. Taarning, R. Madsen and S. Meier, ChemCatChem, 2018, 10, 1414–1419 CrossRef CAS.
- X. Yang, B. Lv, T. Lu, Y. Su and L. Zhou, Catal. Sci. Technol., 2020, 10, 700–709 RSC.
- S. Yamaguchi, M. Yabushita, M. Kim, J. Hirayama, K. Motokura, A. Fukuoka and K. Nakajima, ACS Sustainable Chem. Eng., 2018, 6, 8113–8117 CrossRef CAS.
- M. Kim, S. Ronchetti, B. Onida, N. Ichikuni, A. Fukuoka, H. Kato and K. Nakajima, ChemCatChem, 2020, 12, 350–359 CrossRef CAS.
- Y. Sun, L. Shi, H. Wang, G. Miao, L. Kong, S. Li and Y. Sun, Sustainable Energy Fuels, 2019, 3, 1163–1171 RSC.
- M. Dusselier and B. F. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, Cham, 2014, pp. 85–126 Search PubMed.
- E. Taarning, S. Saravanamurugan, M. Spangsberg Holm, J. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef CAS PubMed.
- K. Nakajima, J. Hirata, M. Kim, N. K. Gupta, T. Murayama, A. Yoshida, N. Hiyoshi, A. Fukuoka and W. Ueda, ACS Catal., 2018, 8, 283–290 CrossRef CAS.
- X. Wang, Y. Song, L. Huang, H. Wang, C. Huang and C. Li, Catal. Sci. Technol., 2019, 9, 1669–1679 RSC.
- M. Xia, W. Dong, M. Gu, C. Chang, Z. Shen and Y. Zhang, RSC Adv., 2018, 8, 8965–8975 RSC.
- X. Yang, J. Bian, J. Huang, W. Xin, T. Lu, C. Chen, Y. Su, L. Zhou, F. Wang and J. Xu, Green Chem., 2017, 19, 692–701 RSC.
- X. Yang, Y. Liu, X. Li, J. Ren, L. Zhou, T. Lu and Y. Su, ACS Sustainable Chem. Eng., 2018, 6, 8256–8265 CrossRef CAS.
- S. Tolborg, I. Sádaba, C. M. Osmundsen, P. Fristrup, M. S. Holm and E. Taarning, ChemSusChem, 2015, 8, 613–617 CrossRef CAS PubMed.
- F. de Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef CAS PubMed.
- M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11777–11782 CrossRef CAS PubMed.
- L. Yang, X. Yang, E. Tian, V. Vattipalli, W. Fan and H. Lin, J. Catal., 2016, 333, 207–216 CrossRef CAS.
- L. Yang, X. Yang, E. Tian and H. Lin, ChemSusChem, 2016, 9, 36–41 CrossRef CAS PubMed.
- D. Verma, R. Insyani, Y.-W. Suh, S. M. Kim, S. K. Kim and J. Kim, Green Chem., 2017, 19, 1969–1982 RSC.
- F. F. Wang, J. Liu, H. Li, C.-L. Liu, R. Z. Yang and W. S. Dong, Green Chem., 2015, 17, 2455–2463 RSC.
- A. A. Marianou, C. C. Michailof, D. Ipsakis, K. Triantafyllidis and A. A. Lappas, Green Chem., 2019, 21, 6161–6178 RSC.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4, eaap9722 CrossRef PubMed.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- Y. Wang, K. Yu, D. Lei, W. Si, Y. Feng, L.-L. Lou and S. Liu, ACS Sustainable Chem. Eng., 2016, 4, 4752–4761 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- X. Han, Y. Guo, X. Liu, Q. Xia and Y. Wang, Catal. Today, 2019, 319, 2–13 CrossRef CAS.
- C. Wang, X. Zhang, Q. Liu, Q. Zhang, L. Chen and L. Ma, Fuel Process. Technol., 2020, 208, 106485 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2019, 193, 404–422 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- T. Chhabra, A. Bahuguna, S. S. Dhankhar, C. M. Nagaraja and V. Krishnan, Green Chem., 2019, 21, 6012–6026 RSC.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS.
- J. Zhang, A. Das, R. S. Assary, L. A. Curtiss and E. Weitz, Appl. Catal., B, 2016, 181, 874–887 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- Q. Sun, S. Wang, B. Aguila, X. Meng, S. Ma and F. S. Xiao, Nat. Commun., 2018, 9, 3236 CrossRef PubMed.
- J. Luo, H. Yun, A. V. Mironenko, K. Goulas, J. D. Lee, M. Monai, C. Wang, V. Vorotnikov, C. B. Murray, D. G. Vlachos, P. Fornasiero and R. J. Gorte, ACS Catal., 2016, 6, 4095–4104 CrossRef CAS.
- R. Gunawan, H. S. Cahyadi, R. Insyani, S. K. Kwak and J. Kim, J. Phys. Chem. C, 2021, 125, 10295–10317 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- J. Luo, J. D. Lee, H. Yun, C. Wang, M. Monai, C. B. Murray, P. Fornasiero and R. J. Gorte, Appl. Catal., B, 2016, 199, 439–446 CrossRef CAS.
- G. H. Wang, J. Hilgert, F. H. Richter, F. Wang, H. J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- B. Chen, F. Li, Z. Huang and G. Yuan, Appl. Catal., B, 2017, 200, 192–199 CrossRef CAS.
- C. Zhu, H. Wang, H. Li, B. Cai, W. Lv, C. Cai, C. Wang, L. Yan, Q. Liu and L. Ma, ACS Sustainable Chem. Eng., 2019, 7, 19556–19569 CrossRef CAS.
- G. Meng, K. Ji, W. Zhang, Y. Kang, Y. Wang, P. Zhang, Y. G. Wang, J. Li, T. Cui, X. Sun, T. Tan, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4139–4146 RSC.
- B. Hu, L. Warczinski, X. Li, M. Lu, J. Bitzer, M. Heidelmann, T. Eckhard, Q. Fu, J. Schulwitz, M. Merko, M. Li, W. Kleist, C. Hättig, M. Muhler and B. Peng, Angew. Chem., Int. Ed., 2021, 60, 6807–6815 CrossRef CAS PubMed.
- J. Zhao, M. Liu, G. Fan, L. Yang and F. Li, Ind. Eng. Chem. Res., 2021, 60, 5826–5837 CrossRef CAS.
- Y. Sun, C. Xiong, Q. Liu, J. Zhang, X. Tang, X. Zeng, S. Liu and L. Lin, Ind. Eng. Chem. Res., 2019, 58, 5414–5422 CrossRef CAS.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2010, 49, 6616–6618 CrossRef CAS PubMed.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- J. Li, Z. Song, Y. Hou, Z. Li, C. Xu, C.-L. Liu and W.-S. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 12481–12491 CrossRef CAS PubMed.
- R. Insyani, D. Verma, H. S. Cahyadi, S. M. Kim, S. K. Kim, N. Karanwal and J. Kim, Appl. Catal., B, 2019, 243, 337–354 CrossRef CAS.
- L. Jin, W. Li, Q. Liu, L. Ma, C. Hu, A. T. Ogunbiyi, M. Wu and Q. Zhang, Bioresour. Technol., 2020, 297, 122492 CrossRef CAS PubMed.
- L. Jin, W. Li, Q. Liu, L. Ma, S. Li, Y. Liu, B. Zhang and Q. Zhang, Fuel Process. Technol., 2019, 196, 106161 CrossRef CAS.
- A. Romero, A. Nieto-Márquez, N. Essayem, E. Alonso and C. Pinel, Microporous Mesoporous Mater., 2019, 286, 25–35 CrossRef CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS PubMed.
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- L. Faba, E. Díaz and S. Ordóñez, Appl. Catal., B, 2012, 113–114, 201–211 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |