
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New frontiers in enzyme immobilisation: robust biocatalysts for a circular bio-based economy†
Roger A.
Sheldon
 *ab,
Alessandra
Basso
c and
Dean
Brady
a
*ab,
Alessandra
Basso
c and
Dean
Brady
a
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits 2050, Johannesburg, South Africa. E-mail: roger.sheldon@wits.ac.za
bDepartment of Biotechnology, Section BOC, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands
cPurolite, Unit D, Llantrisant Business Park, Llantrisant, CF72 8LF, UK
First published on 30th March 2021
Abstract
This tutorial review focuses on recent advances in technologies for enzyme immobilisation, enabling their cost-effective use in the bio-based economy and continuous processing in general. The application of enzymes, particularly in aqueous media, is generally on a single use, throw-away basis which is neither cost-effective nor compatible with a circular economy concept. This shortcoming can be overcome by immobilising the enzyme as an insoluble recyclable solid, that is as a heterogeneous catalyst.
 Roger A. Sheldon | Roger Sheldon, a recognised authority on Green Chemistry, is widely known for developing the E factor for assessing environmental impacts of chemical processes. He is currently Distinguished Professor of Biocatalysis Engineering at the University of the Witwatersrand (SA). He received the RSC Green Chemistry Award and the Biocat Lifetime Achievement Award for important and lasting contributions to biocatalysis and was elected a Fellow of the Royal Society in 2015 and a honorary Fellow of the RSC in 2019. He has a PhD from Leicester University (UK) and was at Shell Research Amsterdam (1969–1980), DSM-Andeno (1980–1990), Delft University of Technology (1991–2007) and CEO of CLEA Technologies (2006–2015). |
 Alessandra Basso | Alessandra Basso received her PhD in Trieste, Italy in Pharmaceutical Sciences in 2001. Her interest in immobilized enzymes started earlier in 1998 and in 2007 she founded SPRIN, a company involved in the development, manufacture and sales of resins for enzyme immobilization and ready to use immobilized enzymes for the industry. In 2012 she joined Purolite and the Life Sciences product line started. From then more than 600 products have been successfully launched in the market for the industrial biocatalysis and downstream purification. Author of 70 scientific publication. |
 Dean Brady | Dean Brady obtained his PhD from Rhodes University under the supervision of Professor John Duncan. He then joined AECI (African Explosives and Chemical Industries) in 1994, working on industrial biotechnology R&D. From 1999 he worked at the CSIR (South Africa) as a Chief Research Scientist and Biocatalysis Group Leader, including a period as a Visiting Research Fellow in Professor Roger Sheldon's group at the Delft University of Technology (Netherlands). In 2013 he became Professor and the Head of the School of Chemistry at the University of the Witwatersrand. |
Key learning points(1) The advantages and limitations of immobilised enzymes in industrial applications, in particular in continuous processing.(2) The different technical and regulatory requirements dictated by the end use of the immobilised enzymes in e.g. commodity chemicals vs pharma and food applications. (3) The different immobilisation methods – on carriers (supports) or carrier-free – and the nature of the carriers used, including new nanomaterials, such as graphene oxide, metal organic frameworks and magnetically recoverable enzymes. (4) The different reactor technologies used and the emergence of continuous flow biocatalysis in combination with multi-enzyme cascades, including microreactors for more efficient and cost-effective processing. (5) Recent advances in enzyme immobilisation, including the integration of protein engineering and enzyme immobilisation and in vivo immobilization, for better design and more cost effective production. |
1. Introduction
With the looming prospect of anthropogenic climate change, widespread environmental degradation and mass extinctions, the transition to a greener, more sustainable manufacture of commodity chemicals and liquid fuels is an imperative. Catalysis is the most powerful tool in the green chemistry toolbox, reducing waste, improving atom economy, minimising energy requirements and intensifying reactions. Riding on the wave of advances in genomics, bioinformatics and protein engineering, catalysis with enzymes, i.e. biocatalysis, has become a mature green and sustainable technology that is widely applied in industry.1–4Enzymes are biocompatible, biodegradable and are manufactured from inexpensive, renewable resources. In contrast with precious metal catalysts, enzyme prices are reasonably stable over time and expensive product purification steps to remove traces of catalyst are not required. Enzymatic reactions are performed at close to ambient temperatures and pressures in water at physiological pH, affording high rates and selectivities. Furthermore, there is no need for functional group protection or activation and processes are more atom and step economic, generating less waste and consuming less energy than conventional processes. In short, biocatalytic processes are more cost-effective, have a smaller environmental footprint and are more sustainable than traditional processes for chemicals manufacture.
Enzymes are now used in a wide range of applications, from detergents, through production of paper and pulp, textiles, leather and food and beverages,5 to commodity chemicals, agrochemicals and pharmaceuticals manufacture. Thanks to advances in protein engineering, enzymes can be optimised to exhibit exquisite selectivities, in particular stereoselectivities, and high activities with non-natural substrates under the challenging conditions of high substrate concentrations and elevated temperatures of industrial processes. Nowhere are their capabilities more evident than in the application of biocatalysis in the industrial synthesis of a broad range of important pharmaceuticals.1,5,6
1.1 Types of biocatalytic processes
Biocatalytic processes can be divided into two types: (i) involving suspensions of growing microbial whole cells, i.e. fermentations, and (ii) involving dead cells which contain the particular enzyme or the isolated enzyme. The former have the advantage that cofactors and fresh enzyme are automatically regenerated with the machinery of the cell when necessary but the disadvantage that product needs to be efficiently secreted in the medium and not be toxic for the living cells. In this review we shall be mainly concerned with the use of isolated enzymes in their immobilised form. The isolated (free) enzyme has the advantage that there is no competition with other enzymes which may be present in whole microbial cells but the added costs of isolation and eventual purification make it more expensive.Notwithstanding the numerous advantages, industrial applications of enzymes are often hampered by a lack of long term operational stability owing to loss of the tertiary structure (denaturation) under the extreme conditions extant in industrial synthesis. Moreover, free enzymes are soluble, homogeneous catalysts in water, which makes them difficult to recover unless expensive membrane systems are used and this limits their use in continuous processing. A free enzyme may also aggregate in hydrophobic media or in aqueous solutions at a pH near its isoelectric point, resulting in diffusion limitations and decreased enzyme activity.7
1.2 Enzyme immobilisation: advantages and limitations
The above mentioned problems can be remedied by immobilising the enzyme as a solid heterogeneous catalyst which is insoluble in water and, hence, is readily recovered by filtration or centrifugation, thus facilitating its reuse and product separation in downstream processing. Since an immobilised enzyme cannot easily penetrate biological membranes it has low or no allergenicity. Furthermore, immobilisation suppresses unfolding of the enzyme's tertiary structure, thus affording a more stable biocatalyst which can be used under a wider range of reaction conditions, including water immiscible organic solvents. Another advantage is that immobilised enzymes are suitable for use in continuous flow operation, e.g. in a packed bed or a plug flow reactor. A limitation of immobilisation is that it generally leads to some loss of activity during immobilisation, often due to interaction of the wrong conformation with the carrier and, of course, the additional cost of the carrier has to be considered. Because it is a heterogeneous catalyst activity loss can also result from diffusion limitations, especially with macromolecular substrates, and dependent on the particle size (see later). Loss of activity can also result from abrasion of the solid particles, e.g. by a mechanical stirrer in the bioreactor. All these factors are compensated by the capacity to recycle the immobilised enzyme and usually an industrial evaluation of feasibility requires an initial calculation of the minimum number of cycles needed to reach the financial break-even point. It is worth noting, however, that immobilisation is not always the answer on an industrial scale. Novozymes has recently developed a liquid formulation of Thermomyces lanuginosus lipase (TLL), instead of the commonly used immobilised lipases, for the production of fatty acid methyl esters (FAMEs) for use as biodiesel. The liquid formulation, Callera™ L, performed admirably with less expensive non-degummed oils, such as crude soybean oil containing 3–5% water.8Historically, immobilisation of enzymes has been essential to the commercial viability of many large scale biocatalytic processes.9 Examples involving immobilised enzymes or immobilised whole cells are shown in Table 1. They include glucose isomerase for the production of high fructose corn syrup, penicillin G amidase for the production of semisynthetic antibiotics, the use of lipases for the production of cocoa butter analogues and the production of chiral amines in organic solvents, all at multi-thousand to multi-hundred thousand tonnes per annum scale. The glucose isomerase process involves the conversion of 107 tonnes of glucose per annum. It is worth noting, however, that glucose production involves conversion of 109 tonnes of corn starch per annum using soluble amylases.
| Enzyme | Industry | Type | Process |
|---|---|---|---|
| IWC = Immobilised Whole Cells; IME = Immobilised Enzyme. | |||
| Nitrile hydratase | Chemical | IWC | Acrylonitrile hydrolysis to acrylamide |
| Penicillin amidase | Pharma | IME | Manufacture of β-lactam antibiotics |
| Glucose isomerase | Food | IWC/IME | High-fructose corn syrup |
| Aminoacylase | Chemical | IME | D- and L-amino acids |
| Lipase | Food | IME | Cocoa butter analogues |
| Lipase | Food | IME | Omega-3 ethyl esters |
| Epimerase | Food | IME | Allulose sweetener |
| Lipase | Chemical | IME | Enantiomerically pure amines |
| Lipase | Chemical | IME | Enantiomerically pure herbicides, e.g. outlook |
| Transaminase | Pharma | IME | Sitagliptin |
1.3 The bio-based economy
The ongoing transition from chemicals manufacture from fossil resources to a more circular production from renewable biomass in a so-called bio(mass)-based economy10,11 has provided an important boost to the widespread application of biocatalysis. A truly bio-based economy is by definition circular12 since it uses only renewable resources and is totally unreliant on fossil feedstocks. Nonetheless, multiple recycling of the catalyst, i.e. the enzyme, is obviously an additional benefit. The goal is to achieve carbon neutrality, i.e. no net green-house gas (GHG) emissions consisting mainly of carbon dioxide. This is essential in connection with climate change mitigation and the preservation of natural resources and biodiversity. It is worth noting that the 450 million tonnes of carbon per annum used for the global chemical industry represents ca. 0.1% of the global production of biomass.A switch from oil and natural gas to a renewable biomass feedstock means a switch from hydrocarbons to carbohydrates as base chemicals13 and the latter are more amenable to biocatalytic conversion in aqueous media. Bio-based chemicals are not new. Fermentation has long provided a variety of chemicals but the availability of cheap crude oil stunted the development of bio-based chemicals. Furthermore, until recently the lack of viable expertise to manipulate cellular metabolism limited the range of bio-based chemicals which could be produced on an industrial scale. However, commercialisation of fermentation processes for industrial monomers such as lactic acid, succinic acid, ethylene, 1,3-propanediol and 1,4-butanediol has paved the way for increasing substitution of fossil fuel derived commodity chemicals.
Bio-refineries are expected to increasingly provide product and by-product streams useful as chemical feedstocks. However, first generation (1G) renewable biomass such as cane and beet sugar and corn starch are not viewed as being sustainable in the long term owing to competition with food production. In contrast, second generation (2G) feedstocks, in particular polysaccharides in agricultural and forestry residues and food supply chain waste (FSCW), are seen as eminently sustainable.
Biocatalysis has two major contributions in a biorefinery: (i) the selective deconstruction of polysaccharide biomass to C6 and C5 sugars using cocktails of isolated enzymes and (ii) conversion of the sugars to biofuels and commodity chemicals by fermentation. In addition the sugars can be converted to commodity chemicals and polymers using combinations of chemocatalysis and biocatalysis with free enzymes.13
2. Immobilisation methods
There are basically three methods to immobilise either whole microbial cells or isolated enzymes (Fig. 1): (i) on an insoluble organic or inorganic carrier (support), (ii) entrapment in a carrier such as hydrogel or polymer matrix which is generated in the presence of the cells or free enzyme, (iii) carrier-free self-immobilisation by cross-linking of enzymes or whole cells. In all three methods a free enzyme is converted from a water soluble homogeneous catalyst into a solid heterogeneous catalyst. In a fourth method, encapsulation, the free enzyme remains a homogeneous water-soluble catalyst but is, in emulation of the intracellular environment, confined behind a membrane which is permeable to substrate and product.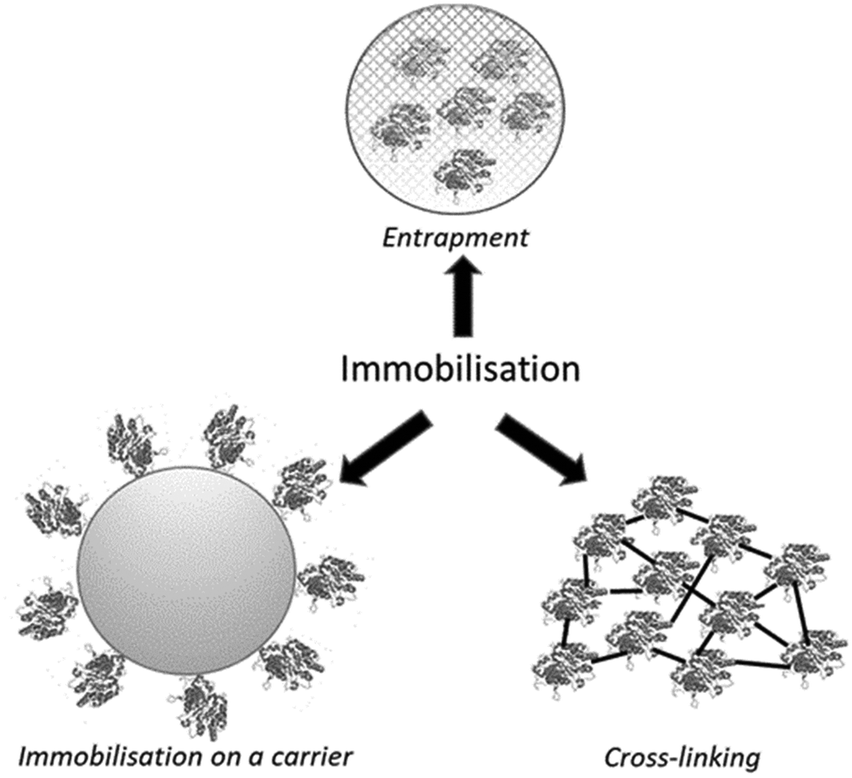 | ||
| Fig. 1 Immobilisation of enzymes or whole microbial cells. | ||
2.1 Immobilisation of whole cells
The primary method for whole-cell immobilisation is entrapment in hydrogels, for example by ionotropic gelation of water-soluble polyelectrolytes consisting of charged functional group-containing polysaccharides (alginate, pectate, carrageenan, chitosan). In particular, entrapment of whole cells in calcium alginate beads is widely used for the immobilisation of both dead and viable whole cells, for example in the fermentative production of ethanol.14 It involves dropwise addition of a solution of calcium chloride to a solution of sodium alginate and the whole cells in water (Fig. 2). Alginate is a natural polysaccharide, extracted from seaweed. It is stable at high temperatures, biocompatible and biodegradable and approved for use in food, cosmetic and pharmaceutical applications. Moreover, alginate beads have good mechanical stability and low-cost.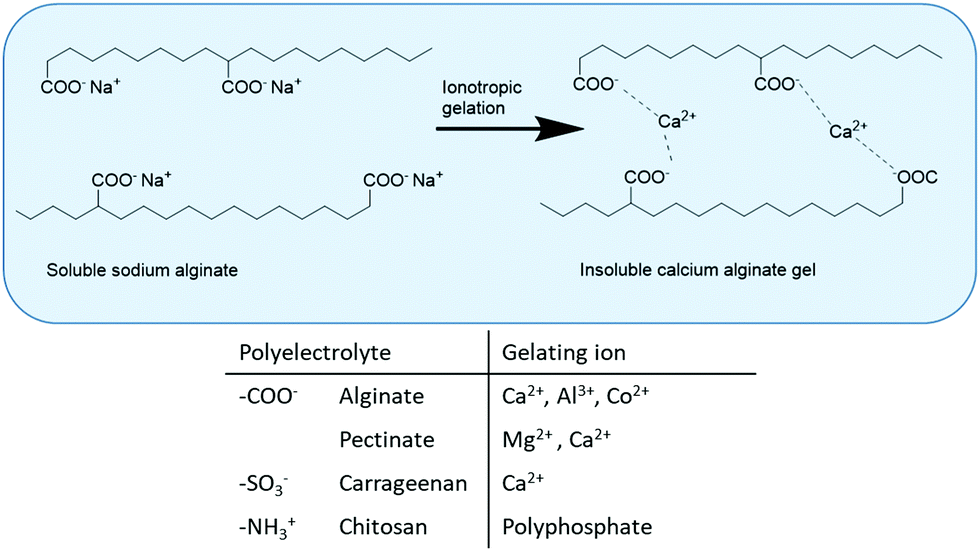 | ||
| Fig. 2 Immobilisation of whole cells by ionotropic gelation. | ||
Another method for whole cell immobilisation is entrapment in hydrogel polymer matrices, in particular polyacrylamide. An interesting industrial example is the immobilisation of whole cells of a Rhodococcus rhodochrous J1 containing a nitrile hydratase in a polyacrylamide hydrogel for use as a highly active and selective catalyst for production of the corresponding monomer, acrylamide by hydration of acrylonitrile.15
Entrapment is not used as much for the immobilisation of isolated enzymes because enzyme molecules are much smaller than whole cells and are, therefore, more easily subject to activity loss by leaching of enzyme. The primary method for immobilisation of isolated enzymes involves binding to a prefabricated carrier (support).16 The latter can be a synthetic polymer, a biopolymer or an inorganic solid such as alumina or (mesoporous) silica. Immobilisation comprises (i) simple physical adsorption (via van der Waals and hydrophobic interactions) on the surface, (ii) ionic bonding or (iii) covalent attachment or (iv) metal affinity binding.
2.2 Immobilisation of isolated enzymes on insoluble carriers
Simple adsorption is the easiest and least invasive procedure. It is the method of choice for applications in water free media as organic solvents and oils. The binding is sufficiently strong and lipases immobilised on hydrophobic resins, in particular poly methyl methacrylates, are widely used. Indeed, the quintessential example, Novozyme 435, comprising C. antarctica lipase (CaLB) immobilised on a macroporous resin consisting of polymethyl methacrylate cross-linked with divinyl benzene, is probably the most widely used immobilised enzyme in industry and academia.17 Lipases immobilised on hydrophobic resins are used in packed columns for, e.g. the production of edible oils as cocoa butter analogues, fats used in infant formula or omega-3 fish oil derivatives, for multiple cycles over many months of activity.18 We also note that Evonik has been using CaLB for the synthesis of emollient esters (e.g. myristyl myristate or coconut oil esters) on a large scale for many years. It replaced an environmentally unfavourable chemical synthesis under harsh conditions.19Lipases typically possess a hydrophobic face with a lid that covers the active site, presumably to prevent it hydrolysing the non-lipid esters in a cell. This lid opens in a hydrophobic environment, such as the surface of a lipid droplet, activating the enzyme. By using a hydrophobic support the lipase can be bound while simultaneously locking the lid into the open position.
Another pertinent example involving an organic solvent is the immobilised (R)-selective transaminase (TA) on a very hydrophobic resin used in the key step for the synthesis of sitagliptin, the active ingredient of the drug Januvia (Fig. 3).20
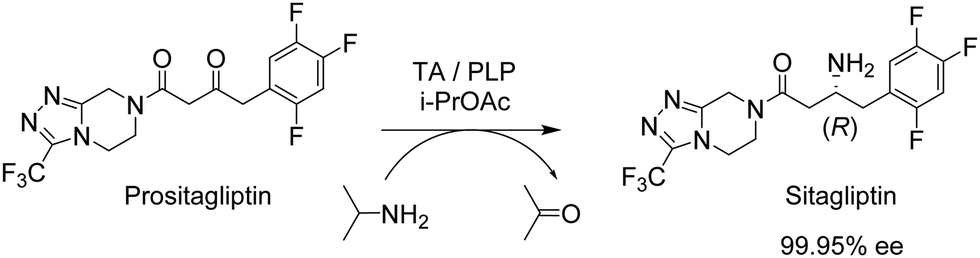 | ||
| Fig. 3 Synthesis of sitagliptin using an R-transaminase. | ||
The TA had already been optimised by directed evolution but an undesirable constraint of the developed protocol was the need for a DMSO/water solvent system. Immobilisation of the TA at 4% loading on a hydrophobic octadecyl functionalised polymethacrylate resin, afforded an immobilisate which permitted a 91% product yield and >99% ee at 200 g L−1 ketone substrate concentration in isopropyl acetate saturated with water at 50 °C. No detectable loss of activity was observed during 10 consecutive recycles over a 200 h period. This enabled >90% reduction in the amount of TA used (despite a 45% loss of activity on immobilisation) compared with the soluble enzyme process. Not surprisingly, the lyophilised free enzyme was completely denatured in the organic solvent immediately after addition and, consequently, exhibited zero activity. Finally, the generality of the method was demonstrated with another nine prochiral ketone substrates.
Ionic binding of enzymes can involve the use of an ion-exchange resin. Selection of a commercial resin (either cationic or anionic) can be made to complement the overall surface charge on the enzyme which depends on the protein isoelectric point and the pH of the solution. The first full scale industrial use of an immobilised enzyme was reportedly the synthesis of an L-amino acid using an L-amino acid hydrolase by Tanabe Seiyaku in 1969. It involved ionic binding of the enzyme to a DEAE-Sephadex resin for use in a packed bed reactor (Fig. 4).
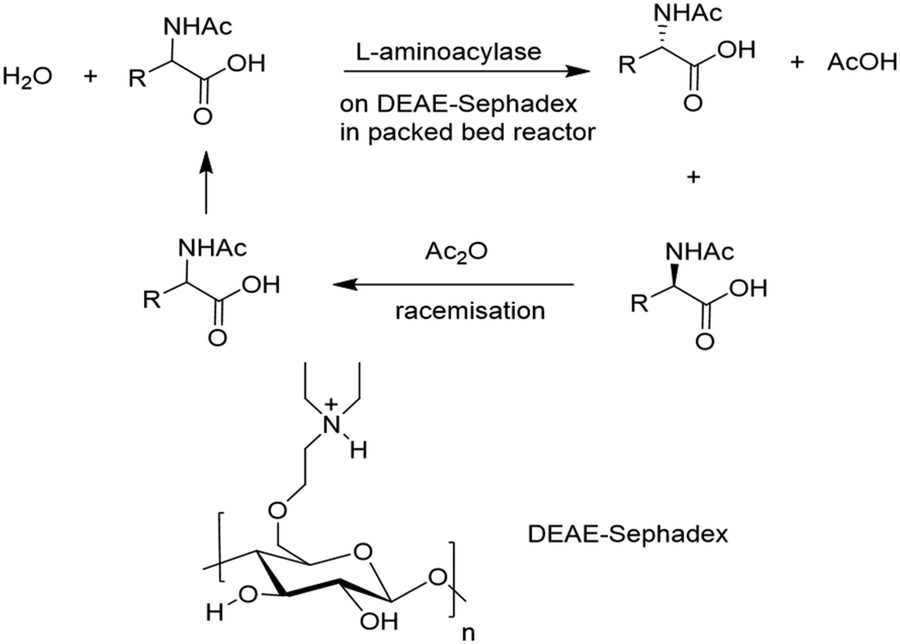 | ||
| Fig. 4 L-Amino acid catalysed synthesis of L-amino acids. | ||
Immobilisation by simple adsorption or ionic binding has the disadvantage that the enzyme is susceptible to leaching in aqueous media depending on the pH and ionic strength and, hence, limits its application to water free systems. This shortcoming can be overcome by employing covalent bonding through reaction of free amino groups in, for example, lysine residues on the surface of most enzymes, with epoxide groups on the carrier surface. A widely used class of carriers are, for example, epoxy methacrylates (Fig. 5).21 A major advantage of the latter is that they are chemically and physically very stable, they can be produced by suspension polymerization in particle sizes suitable for either batch or column applications and conform to the regulatory requirements for applications in pharma and food.
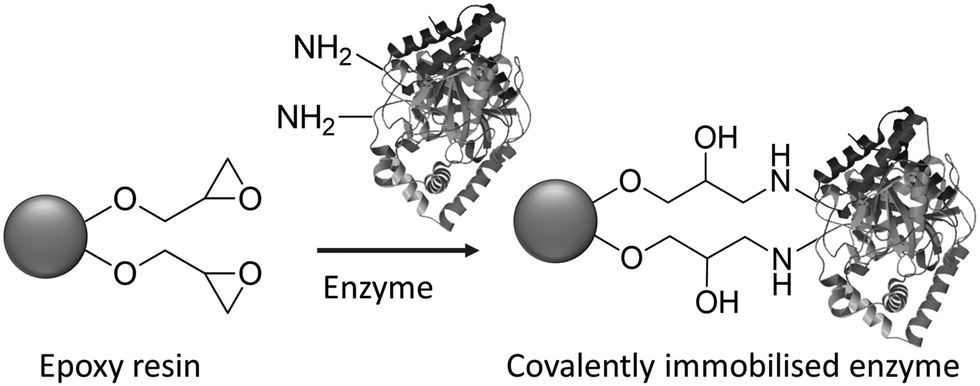 | ||
| Fig. 5 Covalent attachment of enzymes to epoxy methacrylate resins. | ||
Recombinant proteins are generally produced with a string of six to nine histidine residues, a so-called His-tag, attached to the N- or C-terminus. This enables enzyme purification by binding of the histidine imidazole groups in the His-tag to immobilised transition metal ions under specific buffer conditions. This technique forms the basis for affinity immobilisation that exploits chelating groups on the surface of polymers such as iminodiacetic acid (IDA) with preloaded metals such as Ni2+, Zn2+, Fe2+, Co2+ and Cu2+. These metals have great affinity for the histidine group on the surface of the enzyme and this provides the advantage of high immobilisation yield and increased enzyme activity compared to other immobilisation methods through such non-invasive binding.22 Moreover the possibility exists for highly selective simultaneous isolation of the expressed enzyme and its immobilisation in a single efficient step. A proper design of polymers and ligands ensures minimisation of metal leaching, which can cause regulatory issues in industrial scale-up.
A shortcoming of carrier immobilised enzymes in general is the huge dilution of activity, ranging from 90% to >99%, with proportional diminishing space-time yields and productivities, caused by the large volume of the catalytically inert carrier. This problem can be circumvented by using carrier-free, self-immobilisation techniques whereby enzyme molecules are covalently linked together, forming microscopic particles.
2.3 Carrier-free self-immobilisation of isolated enzymes: CLEAs
The most convenient and popular method for self-immobilisation of enzymes is as cross-linked enzyme aggregates (CLEAs).23 Addition of, for example, a saturated solution of ammonium sulfate to a solution of the enzyme at optimum pH results in precipitation of the enzyme as physical aggregates held together by non-covalent bonding without perturbation of the tertiary structure, that is without denaturation. Subsequent cross-linking by addition of glutaraldehyde renders the aggregates permanently insoluble while maintaining their pre-organised tertiary structure and, hence, their activity. Cross-linking involves reaction of the aldehyde functionalities with free amino groups, usually contained in lysine residues, on the surface of neighbouring enzyme molecules. Other di-aldehydes derived from polysaccharides, such as pectin dialdehyde, can be used as cross-linkers but glutaraldehyde is by far the most cost-effective.The CLEA technology has many advantages. CLEAs are cost-effective as they avoid the often substantial costs of a carrier and, since precipitation with ammonium sulfate is typically used to purify enzymes, CLEAs can be produced directly from crude enzyme preparations, for example directly from fermentation broth. As they are carrier-free, they exhibit high catalyst productivities (kg product per kg catalyst) and space time yields (STYs). They exhibit improved storage and operational stability to denaturation by heat, organic solvents and autolysis and are eminently stable towards leaching in aqueous media and high ionic strength. They can be used in aqueous media or organic solvents and are easily recovered for recycling by filtration, centrifugation or decantation, thus facilitating downstream processing. The CLEA technology has broad scope and has been successfully applied to countless enzymes from the five classes of enzymes typically used in industrial synthesis: hydrolases, oxidoreductases, lyases, transferases and isomerases.23,24 CLEAs are highly porous materials and diffusional limitations are not observed with bioconversions commonly used in organic synthesis.
Immobilisation of an enzyme as a CLEA can lead to dramatic enhancement of storage and operational stability and is particularly effective with multimeric enzymes which often have limited thermal stability outside the cell owing to facile dissociation and loss of activity. For example, the industrially very important nitrile hydratases (NHases) are multimers and notoriously unstable outside the cell. Hence, processes involving NHases, e.g. the earlier mentioned process for acrylamide manufacture, are generally performed as whole cell processes. However, a NHase-CLEA showed a dramatic increase in storage stability compared to the free enzyme and could be recycled 36 times with only minor loss of activity.23 The industrially important enzyme halohydrin dehalogenase (HHDH) is a tetramer and immobilisation as a CLEA afforded 90% activity recovery, a strong tolerance to organic solvents and 70% activity retention after 10 recycles.23
Recently, increasing emphasis on renewable feedstocks has precipitated a growing attention for enzymatic conversion of macromolecular substrates, such as polysaccharides and proteins, which cannot easily access the pores of standard CLEAs. Consequently, so-called porous CLEAs with better internal mass transfer, were developed by adding starch to the enzyme solution prior to precipitation and cross-linking.23 Following cross-linking, α-amylase is added to catalyse hydrolysis of the starch and produce a highly porous CLEA with improved mass transfer of macromolecular substrates. Alternatively, macromolecular cross-linkers can be used to produce porous CLEAs.
2.4 Size matters: activity and the particle size dilemma
The success, or failure, of an immobilisation procedure is largely determined by the activity recovery which is simply the activity of the total amount of immobilisate produced divided by the total activity of the free enzyme offered, expressed as a percentage. The masses of the free enzyme offered and the resulting immobilisate may, of course, be totally different. The often quoted immobilisation yield, in contrast, corresponds to the percentage of the free enzyme offered that has been immobilised and says nothing about the recovered activity which could be zero.The activity of heterogeneous catalysts is directly related to their surface area and porosity. The smaller the particles the higher the relative surface area, the better the diffusion of substrates and products in the catalysts. Hence, from the point of view of activity the smaller the better. With large particles activity loss may be due to diffusional limitations and it is important, therefore, to determine if an observed activity loss is intrinsic or owing to diffusion limitations. This can be achieved by ascertaining if grinding the immobilised enzyme particles leads to an increase in activity. Whether or not diffusion limitations are observed is dependent on the absolute rate of the free enzyme catalysed reaction. If the reaction rate is very high, then the observed rate will often be diffusion controlled; but most of the reactions that we are concerned with in organic synthesis are not very fast reactions and, hence, not easily subject to diffusion control.
The problem with small particles is that they are difficult to recover by filtration or centrifugation and for batch processing a compromise has to be found. This is the dilemma of particle size. For the processing of large volumes of substrate, as in oil refining and petrochemicals, continuous processing over highly active catalysts with small particle sizes is required. This is achieved by conducting the processes with a bed of catalyst which is fluidised by the rapid upwards flow of the substrate. The particles must be sufficiently dense, non-compressible and of a uniform size that they are not swept out of the reactor by the substrate.
The limitations of CLEAs are more related to their physical properties. They have a relatively small, non-uniform particle size (5–50 μm and typically below 10 μm). This is generally sufficient for separation by filtration or centrifugation in a batch operation but the small particle size together with limited rigidity is a handicap for application in fixed-bed reactors due to a high pressure drop. This can be counteracted by blending with non-compressible particles such as controlled-pore glass or perlite (volcanic glass) but the size and fragility of enzyme particles remains an issue for large scale applications.
An effective way to tackle the particle size and uniformity problem of CLEAs involves self-immobilisation by addition of a cross-linker to a suspension of an enzyme in a water-in-oil emulsion. This affords uniform enzyme microspheres, aptly named ‘spherezymes’, with a particle diameter of 50 μm and a size distribution within 3% and more controlled structural features.25
When using polymers in industrial processes, they should preferably be in the form of spherical beads as uniform as possible in terms of particle size distribution. Generally, industrial processes use particles larger than 100 micron that can fit either batch reactors or column reactors. Packed columns usually utilise polymers with larger particle size to minimise pressure drop issues.
3. Regulatory requirements
Regulatory issues can shape the direction of technical innovation in enzyme immobilisation. When designing immobilised enzymes for commercial applications, such as in the food or pharmaceutical industry, or for medical applications, a key aspect to consider is that the chemicals used are not hazardous, can be used safely, are easily removed and do not pose risks to human or animal health. Consequently adherence to regulatory codes (such as ISO and CFR) supersedes attaining maximum biocatalyst performance when deciding the materials involved. Extractability, leaching, cytotoxicity and toxicological considerations all relate to the material selection. Some materials are generally acceptable, such as cellulose triacetate for immobilisation of lactase in dairy applications. Heavy metals and toxic chemicals should be avoided but glutaraldehyde is allowed as a cross-linker by CFR regulations.Enzymes from animals are of particular concern owing to the possibility of transmissible diseases, including transmissible spongiform encephalopathy (TSE) diseases such as bovine spongiform encephalopathy (BSE), which is associated with contaminating proteins, as well as transgression of various religious codes. In food applications it may also be required that the immobilised enzyme does not contain any residual DNA if originating from a genetically modified microorganism.
For pharmaceutical applications it is necessary to consult the Pharmacopoeia for the suitability of materials. In the case of unlisted materials a toxicological analysis is required by the pharmaceutical user on each extractable and leachable used in the manufacture of the immobilised enzyme, including the carrier and the enzyme. The maximum level allowed for each chemical used is based on the number of cycles, application, human daily dosage, and other factors. Such information is subsequently included in the Drug Master File submitted to the national regulatory body – such as the FDA.
It is also self-evident that standard immobilisation techniques with broad industrial applicability should be available so that it is not necessary to go through the regulatory process with each application.
4. Novel carrier materials and separation technologies
The ideal carrier for enzyme immobilisation should have:Minimum cost-contribution, i.e. enzyme costs are <5% of the total process costs, sufficient availability, as well as environmental compatibility.
Good physical and mechanical stability to be used in different solvents and reactor/configurations.
A large surface area and good porosity for efficient mass transfer and high activity.
Optimum affinity for the enzyme in terms of hydrophilicity or hydrophobicity, depending on the application.
A readily functionalised surface to attach the enzyme.
Facile recovery and multiple recycling.
As discussed in the preceding section, activity is dependent on the size of particles and nanomaterials are, therefore, potentially attractive carriers for enzyme immobilisation in special applications such as medicine, diagnostics, micro-electronics and environmental monitoring.26 They are mainly classified into carbonaceous, metallic, mesoporous silica-, cellulose- and chitosan-based and metal organic frameworks (MOFs).
4.1 Graphene oxide
Graphene oxide (GO), the precursor of graphene in its large scale synthesis from graphite, is an ideal candidate for immobilising enzymes and has attracted much attention recently.27 It combines high surface area and exceptional thermal and mechanical properties with an abundance of surface functional groups (carbonyl, carboxyl, hydroxyl and epoxy) that confer it with good dispersibility in water.Crude naringinase, comprising α-rhamnosidase and β-glucosidase, was immobilised by covalent attachment to GO. The resulting immobilisate catalysed the two-step hydrolysis of naringin to naringenin via the intermediacy of prunin (Fig. 6). Prunin was produced in high yield by immobilising a purified fraction containing only the α-rhamnosidase or by selective inactivation of the β-glucosidase fraction.
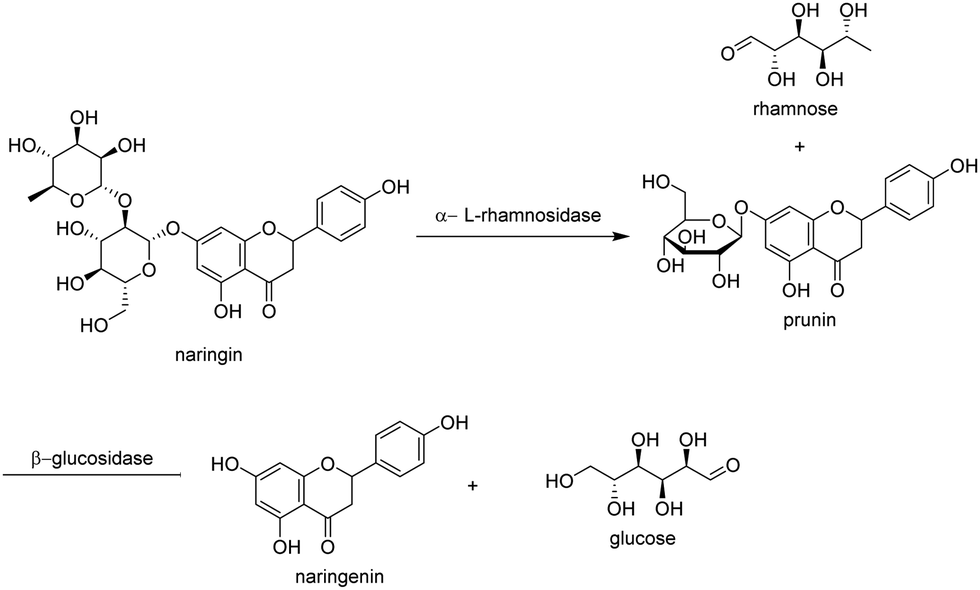 | ||
| Fig. 6 Hydrolysis of naringin catalysed by naringinase on graphene oxide. | ||
The pure and crude GO supported naringinase were obtained in high activity recoveries, exhibiting high productivities and long term stabilities. They clearly have excellent potential for enabling a green and more sustainable large-scale production of the citrus flavonoids by enzymatic hydrolysis of naringin.
4.2 Metal–organic frameworks (MOFs)
Metal–organic frameworks (MOFs) are highly ordered microporous crystalline hybrid materials that are constructed from metal ions and organic linkers. Based on their extremely high specific surface areas and excellent porosity coupled with extraordinary multi-functionality and relatively high stability, MOFs have attracted much attention as novel carriers for enzyme immobilisation.28 The highly ordered structures of MOFs provides a uniform microenvironment for enzymes and many MOF–enzyme composites exhibit unprecedented catalytic performances compared with the corresponding free enzymes, including improved activity, stability, selectivity, and recyclability.Physical adsorption of cellulose in UiO-66-NH2, a MOF derived from ZrCL4 and 2-amino terephthalic acid, afforded an immobilisate that catalysed the hydrolysis of water soluble carboxymethyl cellulose at 30–80 °C retaining 70% activity after 10 recycles.29 Incorporation of magnetic nanomaterials within MOFs such as UiO-66-NH2 permits their recovery through the application of magnetic fields. For example, magnetic cellulase CLEAs with high activity, enhanced pH and thermal stability, were prepared from a magnetic core–shell MOF, Fe3O4–UiO-66-NH2. The system could be recovered magnetically and reused. Notably it was less inhibited by formic acid and vanillin, typically present in lignocellulosic prehydrolysates, than the free enzyme.30
It is evident that MOFs present an exciting opportunity for enzyme immobilisation, but considerable research and development is required to turn these into commercial biocatalysts. An important question to be answered is: what is the commercial availability and price of MOFs such as UiO-66 and UiO-66-NH2?
4.3 Magnetisable carriers and CLEAs for magnetic separation
As noted earlier, it is also important that the particles can be easily separated from reaction mixtures and one way to enable this is to use magnetic (nano)particles as carriers. Facile separation and recovery of small, highly active particles of immobilised enzymes is enabled by incorporating nanoparticles of magnetite (Fe3O4) or maghemite (γ-Fe2O3) to create enzyme–ferromagnetic particle hybrids.31 These can be easily separated magnetically on an industrial scale using standard commercial equipment, enabling a novel combination of biocatalysis and downstream processing. Furthermore, magnetic recovery enables facile separation of the solid biocatalyst from other suspended solids in the reaction mixture, as is the case in the processing of polysaccharides, in particular lignocellulosic feedstocks in biorefineries.32 Other uses include slurry processes and polymer synthesis.Enzyme–magnetic particle hybrids can involve attaching enzymes to a magnetic carrier or formation of carrier-free magnetisable CLEAs by conducting cross-linking in the presence of amino functionalised Fe3O4 particles produced by reaction of surface hydroxyl groups with 3-aminopropyltriethoxysilane (APTES).23 It was subsequently found that stable magnetic CLEAs are produced more cost-effectively using non-functionalised magnetite particles. However, these m-CLEAs undergo leaching of iron at acidic pH which is accelerated in the presence of free carboxylic acids, thus posing a serious problem in the processing of lignocellulose, for example.
This shortcoming was overcome by using commercially available zerovalent carbonyl iron particles (CIP), produced by thermal decomposition of iron pentacarbonyl. The particles were coated with a protective nanometer layer of silica to afford m-CLEAs with a particle size in the range 1–15 μm that combined stability towards leaching with a surprisingly high magnetic susceptibility, which facilitates their magnetic separation. An m-CLEA of glucoamylase produced in this way was shown to be effective in, for example, the conversion of starch to glucose.23
m-CLEAs have been successfully used with a variety of enzymes: for example, carbohydrases, lipases, proteases and laccases involved in biomass conversion processes.23,33 In a recent example, a m-CLEA of a recombinant arabinose isomerase prepared from Fe3O4 was used for the isomerisation of D-galactose to D-tagatose, a functional sweetener (Fig. 7). It exhibited increased storage and operational stability and could be recycled several times with little loss of activity.34 Higher losses observed at long reaction times could be due to dissolution of Fe3O4.
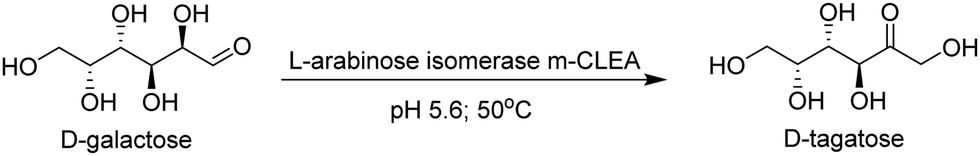 | ||
| Fig. 7 L-Arabinose isomerase m-CLEA catalysed isomerisation of D-galactose to D-tagatose. | ||
4.4 Biocatalyst separation and reactor design
Industrial organic syntheses, in particular in the pharmaceutical industry, are mostly conducted in stirred tank reactors (STRs) where the immobilised enzyme is maintained in suspension by physical mixing with a propeller stirrer. The immobilised enzyme is separated usually by filtration with stainless steel filters at the bottom of the reactor. However, a compromise has to be made between the high activity of small particles and the facile separation of large particles (see Section 2.4). Moreover, mechanical attrition of the immobilised enzyme particles, resulting from shear forces caused by the propeller stirrer, is a frequently encountered problem with STRs. In STRs the preferred particle size is >100 micron, with a very tight control on fines, since these can block the filters. Materials used in STRs are required to be highly robust and survive in some processes up to 1000 cycles. Processes involving larger volumes, e.g. in fine chemicals and food processing, are often conducted in packed bed column reactors, which require relatively large particles (usually larger than 300 micron) to avoid pressure drop over the column, or fluidised bed reactors which require relatively dense particles (see Fig. 8).23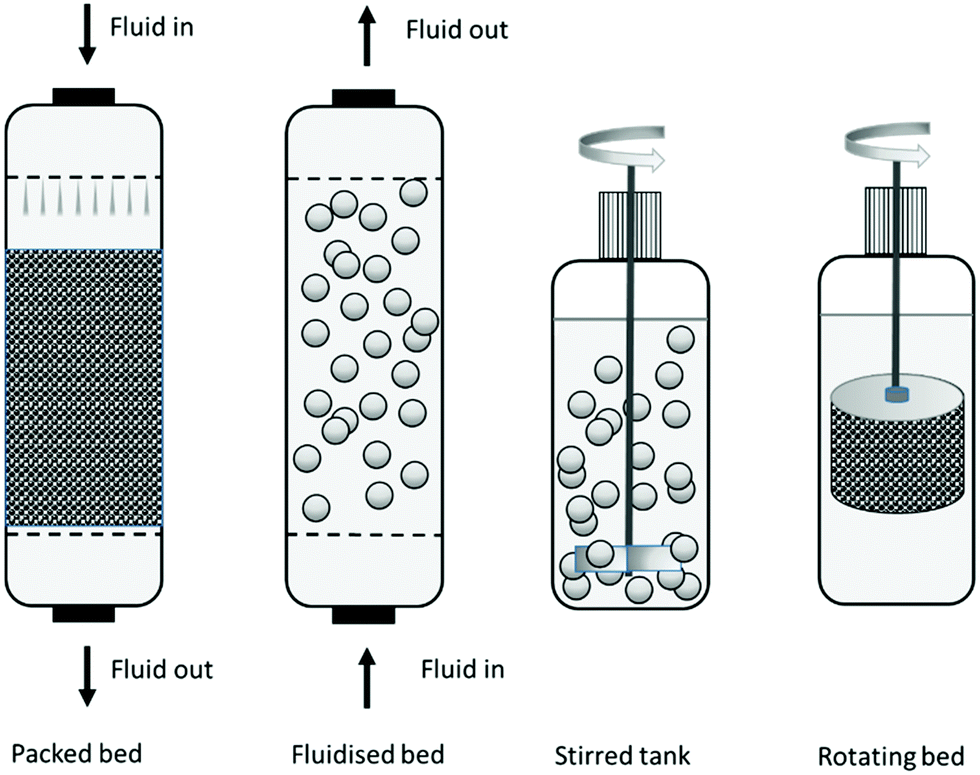 | ||
| Fig. 8 Reactor design. | ||
Fluidised beds are ideal when viscous media are involved, such as in the manufacture of edible oils. Fluidised beds are obtained either by counter flow with a liquid or by bubbling inert gasses such as nitrogen. In fluidised bed or packed bed columns more porous materials can be used, since the mechanical stress is much lower compared to an STR and, hence, diffusion can be improved.
A perfusion basket reactor is an ideal solution when the immobilised enzyme needs to be maintained separated from the bulk medium due to mechanical instability. This is a variation on the tea bag concept which involves confining the catalyst in a filtration membrane-like module that is suspended in the STR in order to avoid contact with the stirrer. A further refinement, the rotating bed reactor developed by SpinChem (http://www.spinchem.com), involves using a catalyst-containing compartment attached to the propeller stirrer. This technology combines the benefits of an STR and a packed bed and has been scaled up successfully to more than 100 litres scale.35
In the context of biocatalyst separation it should also be mentioned that an alternative to enzyme immobilisation is to use enzyme membrane reactors (EMRs). Degussa already commercialised the use of, for example, aminoacylases and amino acid dehydrogenases (with cofactor recycling) in EMRs for the production of D-amino acids in the 1980s.36
An interesting variation on this theme is the use of immobilised enzymes in membrane slurry reactors (MSRs) whereby immobilised enzymes are retained inside the reactor because they are too large to pass through the pores of a membrane patch in the reactor wall. This enables the use of a broad range of catalyst particle sizes including the relatively small particles of CLEAs. The reaction and biocatalyst separation are combined into a single operation. High catalyst loadings, longer catalyst life-times owing to reduced mechanical attrition, and higher volumetric and catalyst productivities are some of the many advantages of an MSR. Its practical utility was demonstrated in the industrially important penicillin G amidase-CLEA catalysed hydrolysis of penicillin G to 6-aminopenicillanic acid (6-APA).23
5. Continuous processing: biocatalysis in flow
The use of both biocatalysis and continuous processing in the manufacture of fine chemicals and pharmaceuticals has increased exponentially in recent years. Hence, much attention has recently shifted to merging these two technological trends into the use of continuous flow biocatalysis37,38 in fine chemicals and pharmaceuticals production. This has, in turn, provided an important stimulus for the wide-spread application of immobilised enzymes in, for example, packed bed flow reactors.The merging of flow chemistry with biocatalysis provides solutions to various practical problems observed in batch processing. For example, feedback inhibition and intermediate degradation are no longer observed in continuous processing. The future for flow-biocatalysis is bright and the field will continue to grow.
It can also be used in conjunction with the co-immobilisation of multi-enzyme systems and the design of cofactor regeneration methods to enable cost effective redox biocatalysis. For example, the co-immobilisation of a secondary alcohol dehydrogenase and an amine dehydrogenase for the continuous conversion of an alcohol to the corresponding amine in a biocatalytic hydrogen-borrowing cascade (Fig. 9).39
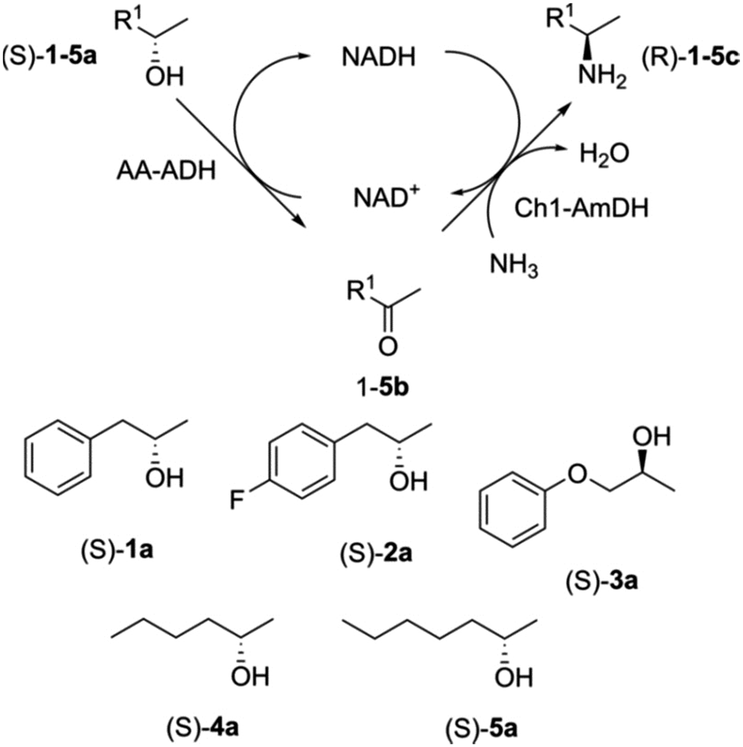 | ||
| Fig. 9 Enzymatic hydrogen-borrowing cascade in continuous flow. | ||
Microfluidic immobilised enzyme reactors (μ-IMERs) are the focus of much current interest.40 They offer several practical advantages for conducting continuous processes with biocatalysts: (i) smaller dimensions, (ii) lower equipment costs and energy consumption, (iii) high surface area to volume ratio resulting in rapid heat exchange and mass transfer and high activities. The enzymes can be immobilised onto inner-walls of the microtubes or as particles in packed-beds or as monoliths.
Microreactors allow for better control of reaction parameters – temperature, pressure and pH, residence time – and minimal attrition and denaturation of the immobilised enzyme is observed compared to the high shear experienced in stirred tank reactors. Furthermore, μ-IMERs are robust and easy to scale up to industrial scale production. This can involve scaling up or scaling out (see Fig. 10) or, for commercial scale production, a combination of both.
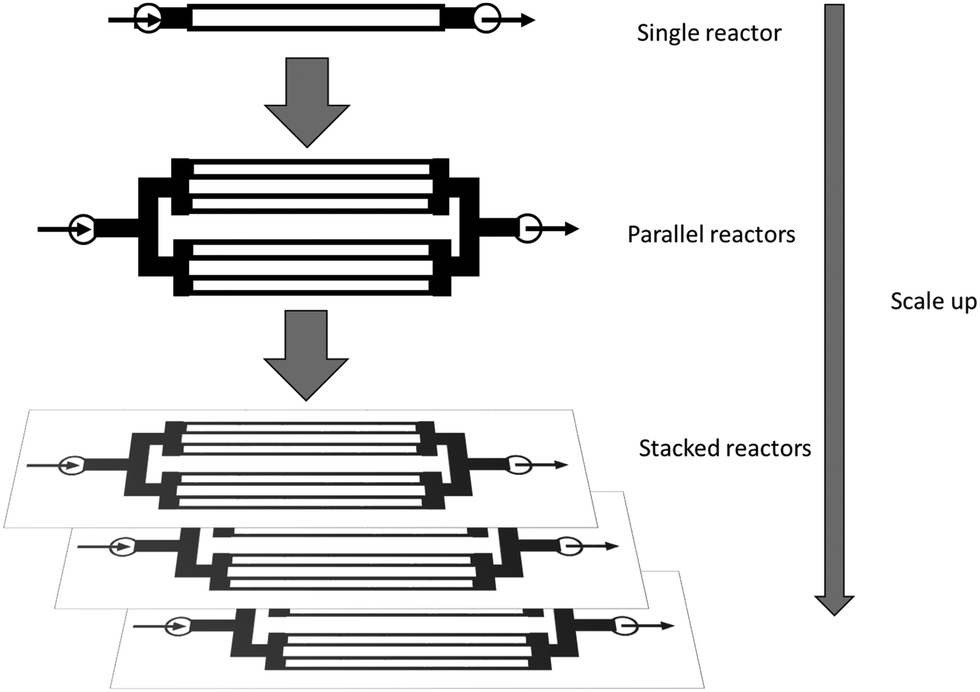 | ||
| Fig. 10 Scaling up and scaling out of μ-IMERs. | ||
6. New frontiers in enzyme immobilisation: pushing the envelope
6.1 Integrating protein engineering and enzyme immobilisation protocols
As noted in the Introduction, two important drivers of the growing application of biocatalysis in, inter alia, chemicals manufacture, pharmaceuticals and food technology, are protein engineering and enzyme immobilisation. The earlier mentioned production of sitagliptin is a prime example of what is possible by improving the performance of an enzyme by using both technologies in sequence.In some cases, however, immobilisation of enzymes using standard techniques can lead to substantial loss of activity resulting from reactions of reactive functional groups in amino acid residues with reagents used in the immobilisation procedure. This can be alleviated by using protein engineering to evolve the enzyme to be more compatible with the immobilisation procedure. In an early example of this approach two cycles of error-prone PCR were used to optimise a formate dehydrogenase (FDH) for immobilisation in a polyacrylamide (PA) gel.41 This afforded a variant with 4.4 fold higher activity compared with the wild-type enzyme when immobilised in PA. The increased activity resulted from an exchange of lysine, glutamic acid and cysteine residues remote from the active site.
Covalent attachment of molecules of enzymes to carriers, or to other enzyme molecules by cross-linking, generally involves reaction with lysine residues on the enzyme surface. This can be problematic with some enzymes having a paucity of lysine residues on the surface. In this case protein engineering can be used to replace surface amino acids with lysine to provide multiple attachment points. This approach can provide additional benefits by allowing for optimum orientation of the enzyme on the solid surface. Alternatively, genetic engineering can enable site-specific incorporation of nonstandard amino acids (NSAAs) containing functional groups which enable cross-linking of enzyme aggregates in a highly controlled manner, thus avoiding undesirable reactions of glutaraldehyde which lead to loss of enzyme activity.42
The complementarity of PE and EI is readily apparent from a comparison of their main features (Table 2). For example, EI focuses on enzyme reuse and controlling the microenvironment whereas PE focuses on genetic variability but both techniques contribute to improving stability. Advances in PE using directed evolution techniques enabled the development of isolated enzymes with predefined properties.43 However, an optimised variant may not necessarily have a high performance when immobilised since that was not a selection criterion. Conversely, variants with unremarkable stability as free enzymes may be exceptionally stable when immobilised. The logical next step towards optimum performance of enzymes is to integrate the immobilisation step into the screening process of directed evolution.44 Hence, immobilised biocatalyst engineering (IBE) combines the strengths of both PE and EI to produce immobilised enzymes conforming to preset process parameters.
| Main feature | Enzyme immobilisation | Protein engineering |
|---|---|---|
| Reusability | Yes | No |
| Genetic variability | No | Yes |
| Improved selectivity | No | Yes |
| Improved stability | Yes | Yes |
| Performance in non-conventional media | Yes | Yes |
| Non-natural reactions | No | Yes |
| Substrate promiscuity | Yes | Yes |
| Generate large numbers of variants | No | Yes |
| Control of the microenvironment | Yes | No |
6.2 In vivo vs. in vitro immobilisation
Even when EI forms an integral part of the screening of variants produced by directed evolution it is still a separate step in the production of the immobilised enzyme. The production of the free enzyme is in vivo while the immobilisation is conducted in vitro. It would have obvious advantages, however, to combine the production of the enzyme with its in vivo immobilisation, thus providing a one-step production of an already immobilised enzyme. This can be achieved by engineering enzymes to self-assemble into nano- or micro-sized insoluble aggregates inside the cell without the need for attachment to a prefabricated carrier.Genetic engineering can be used to fuse a self-assembly promoting partner protein to the target enzyme. For example, various microorganisms produce, as a means of energy storage, inclusions of polyhydroxyalkanoates (PHAs). Genetic fusion of the polyester synthase (PhaC), the central enzyme of PHA biosynthesis, to the target enzyme in vivo results in the formation of insoluble PHA beads displaying the target enzyme in a highly functional mode and covalently linked to the bead. This is truly an elegant example of a biocatalyst immobilised on a bio-based plastic and produced in one step from a renewable feedstock.45
A variation on this theme involves the use of Cry3Aa, a member of a family of Cry proteins which are produced and directly crystallised within the bacterial cells of Bacillus thuringiensis (Bt). They are well-known as biological insecticides. The crystals are stable and insoluble in water at neutral pH. Hence, one-step immobilisation can be achieved by genetically fusing the target enzyme to Cry3Aa and isolating the Cry-fusion crystals (CFCs).46 The CFC approach constitutes a novel, cost effective and biocompatible approach to immobilisation of enzymes.
For example, a CFC of Bacillus subtilis lipase A was produced and used in the production of fatty acid methyl esters for biodiesel by methanolysis of triglycerides.46 The crystal framework significantly stabilised the lipase with regard to denaturation at elevated temperatures and in organic solvents. Recycling was tested with a low (2.5%) catalyst loading and 93% conversion was maintained over 9 recycles, corresponding to a total turnover of 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 713.
713.
Yet another variation on the theme of in vivo immobilisation is to use protein engineering to enable the production of catalytic inclusion bodies (CatIBs).47 Bacterial inclusion bodies were long considered as unfolded waste material produced by heterologous over-expression of recombinant genes. Recently this opinion has changed dramatically with the application of protein engineering techniques. CatIB formation is enabled by the fusion of short peptide tags or aggregation-inducing protein domains to the target protein, resulting in the in vivo formation of CatIBs as carrier-free immobilisates. The technique has already been shown to have broad scope, suggesting that on-demand, cost-effective production of CatIBs, as biologically produced immobilisates from target enzymes, is within reach. Smart magnetisation of CatIBs to facilitate isolation could be a further improvement for large scale applications.48
7. Biocatalytic cascades: co-immobilisation of multiple enzymes
In stark contrast with in vivo cellular metabolism, where a vast array of enzymes work in concert, classical multi-step organic syntheses are conducted in a step-by-step fashion where intermediates are isolated and often purified after every step. This leads to low volumetric productivities and copious waste generation. Telescoping of multi-step syntheses into one-pot procedures automatically reduces the number and amounts of solvents used and the amount of waste generated. In addition, it affords processes with higher productivities using fewer unit operations, smaller reactor volumes and in shorter cycle times.49Conducting biocatalytic conversions as multi-step enzymatic cascade processes is greatly facilitated by the fact that a multitude of enzymes perform admirably under roughly the same conditions, that is in water at ambient temperature and pressure and defined pH. Coupling of enzymatic steps together provides the possibility of driving equilibria towards product formation by removing the product and, because of the superb specificity of enzymes, reaction steps involving protection and deprotection of functional groups can be dispensed with. A further advantage is that it is possible to co-immobilise enzymes which would not be compatible in homogeneous solution. A good example is the successful co-immobilisation of a lipase and a protease in a combi-CLEA.50
In order to facilitate their removal and reuse, multiple enzymes can be co-immobilised on, or in, a carrier or by cross-linking. This affords the additional benefit of enzyme proximity which enables substrate channelling, leading to reductions in diffusion times and resulting in higher overall activities and less accumulation of inhibitory intermediates. This effect is manifest in nature in the multi-enzyme cellulosomes which contain mainly carbohydrases, bound together by non-catalytic scaffoldins, to catalyse the hydrolysis of cellulose and hemicellulose.51 Co-immobilisation also facilitates the orchestration of enzymatic cascade processes in continuous operation and we have already given an example of such a system in Section 5, Fig. 9.
The cellulosome can be emulated by immobilising the different glycosides in a combi-CLEA, e.g. a reusable magnetic combi-CLEA produced from ‘ultra clear’ enzyme preparation, containing pectinase, cellulase and xylanase, was used for the hydrolysis of a mixture of cellulose and hemicellulose.52
Of particular interest is redox cofactor recycling by co-immobilisation of an oxidative and reductive enzyme into a cascade. For example, a combi-CLEA of glycerol dehydrogenase (GDH) and NADH oxidase was developed for highly efficient in situ NAD+ regeneration (Fig. 11).53
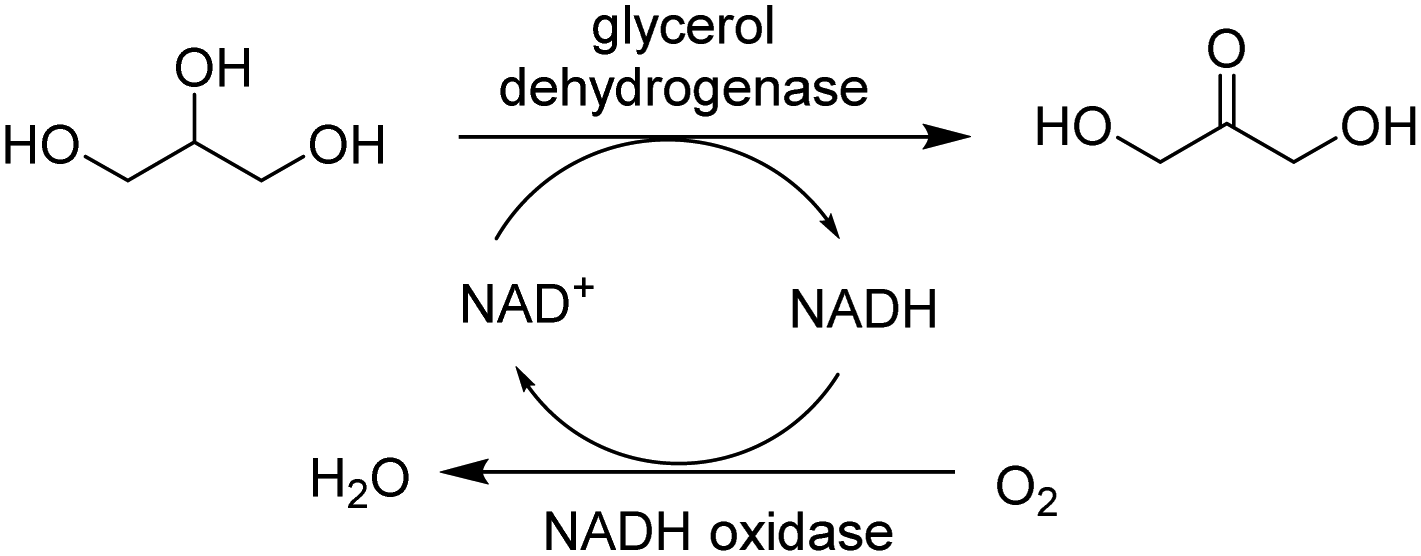 | ||
| Fig. 11 Oxidoreductases in cascade reactions. | ||
Another variation on this theme is the combination of biocatalysis and chemocatalysis in chemoenzymatic cascade processes. A striking example is the combination of a ligand-free Pd catalysed Suzuki–Myaura cross-coupling reaction in aqueous dimethylformamide (DMF) with a transaminase (TA) catalysed amination (Fig. 12).54 Enzymatic and chemoenzymatic cascade processes go hand-in-hand with flow chemistry using immobilised biocatalysts. It is not surprising, therefore, that further improvements were obtained by immobilising the engineered TA on an EziG His-Tag affinity resin and conducting the one-pot reaction in continuous flow, thus combining the features of one-pot cascade processes with an immobilised combination of enzymes in continuous flow operation.
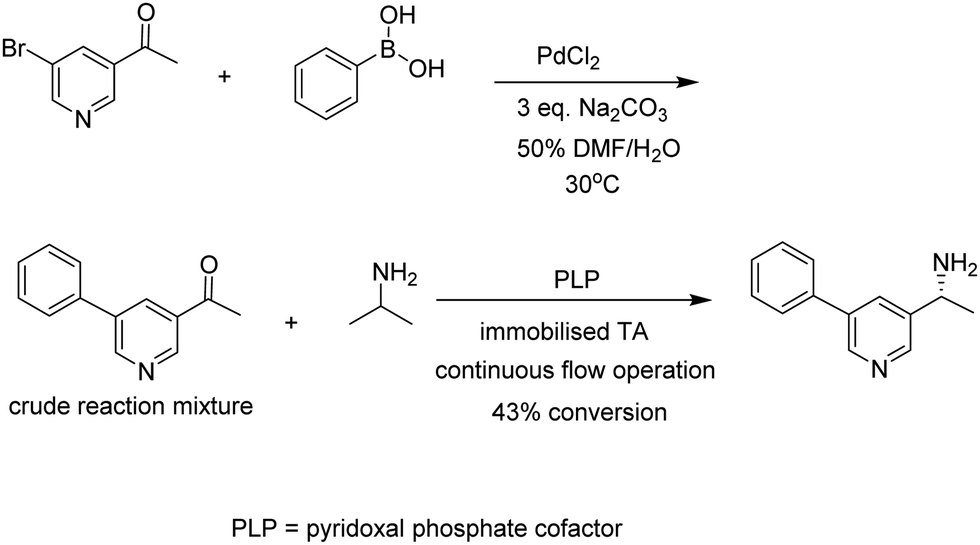 | ||
| Fig. 12 Chemoenzymatic cascade process with an immobilised transaminase in continuous flow. | ||
Microfluidic immobilised enzyme reactors (μ-IMERs) are also suitable for performing multi-enzyme cascade reactions. For example, a microfluidic packed-bed reactor containing a His-tagged ketoreductase (KRED) and His-tagged GDH attached to functionalised magnetic beads for enantioselective reduction of prochiral ketones (Fig. 13).55
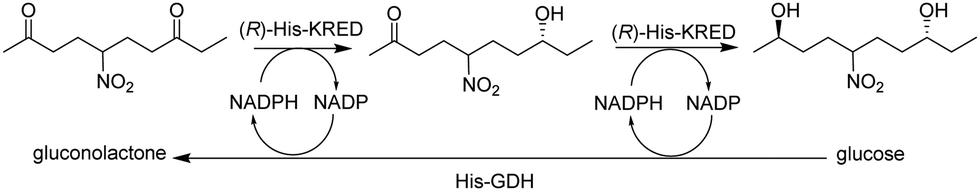 | ||
| Fig. 13 Cascade process with immobilised KRED and GDH. | ||
8. Conclusions and outlook
Biocatalysis has undergone a phenomenal growth in the last two decades to become a mature technology with enormous commercial potential. This growth was largely enabled by advances in genomics, bioinformatics and protein engineering.However, notwithstanding numerous important advantages as catalysts, the water solubility of enzymes seriously hampers their reuse and limits their broad industrial application. Consequently, immobilisation as a recyclable insoluble solid is generally needed in order to make their industrial use cost-effective. Moreover, the increasing use of continuous biocatalytic processing in industrial scenarios constitutes a further stimulus for the use of immobilised enzymes.
Enzymes can be immobilised on a wide variety of natural and synthetic carriers (supports) via simple adsorption, ionic or covalent bonding and affinity binding or as carrier-free cross-linked enzymes. Recent advances involve the use of novel carriers, e.g. various highly porous nanomaterials, in combination with novel separation and reactor technologies such as magnetic separation. However, different applications have different technical requirements, e.g. in terms of particle size, method and materials of immobilisation, and specific regulatory demands. The requirements for an immobilised enzyme used in diagnostic or biosensor applications is obviously completely different from the requirements of an immobilised enzyme used in the production of food such as a sweetener.
Advances in protein engineering and enzyme immobilisation were major enablers of the biocatalysis revolution but they were originally applied as separate technologies. Hence, further performance evolution is envisaged as a result of integration of the two techniques in immobilised biocatalyst engineering. Similarly, there are great expectations from the integration of enzyme production with genetically engineered in vivo immobilisation, particularly in combination with novel separation techniques. In short, the future is bright for applications of immobilised enzymes as the cornerstone of a circular bio-based economy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dean Brady would like to thank Dr Robin Drennan (of Wits) and Dr Konanani Rashamuse (of the DSI) for their continued support of biocatalysis research.References
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60(1), 88–119 CrossRef CAS.
- R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- B. Hauer, ACS Catal., 2020, 10, 8418–8427 CrossRef CAS.
- V. V. Vinogradov, N. N. Skvortsova and E. F. Krivoshapkina, J. Agric. Food Chem., 2019, 67, 11553–11567 CrossRef PubMed.
- H. Sun, H. Zhang, E. L. Ang and H. Zhao, Bioorg. Med. Chem., 2018, 26, 1275–1284 CrossRef CAS PubMed.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- S. Cesarini, P. Diaz and P. M. Nielsen, Process Biochem., 2013, 48, 484–487 CrossRef CAS.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- M. C. R. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. M. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- A. Pellis, S. Cantone, C. Ebert and L. Gardossi, New Biotechnol., 2018, 40, 154–169 CrossRef CAS PubMed.
- R. A. Sheldon, Philos. Trans. R. Soc., A, 2020, 378, 20190274 CrossRef CAS PubMed.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 4464–4480 CrossRef CAS.
- J. C. Duarte, J. A. R. Rodrigues, P. J. S. Moran, G. P. Valença and J. R. Nunhez, AMB Express, 2013, 3, 31 CrossRef PubMed.
- B.-Y. Kim and H.-H. Hyun, Biotechnol. Bioprocess Eng., 2002, 7, 194–200 CrossRef CAS.
- For an excellent recent review see: C. L. Borges Reis, E. Yvay, A. de Sousa, J. de França Serpa, R. C. Oliviera and J. C. Sousa dos Santos, Quim. Nova, 2019, 42, 768–783 Search PubMed.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. Dos Santos, R. C. Rodriguez, A. Berenguer, L. E. Brand and R. Fernandez-Lafuenta, Catal. Sci. Technol., 2019, 9, 2380–24520 RSC.
- R. C. Rodriguez, J. J. Virgen-Ortiz, J. C. S. dos Santos, A. Berenguer-Murcia, A. R. Alcantara, O. Barbosa, C. Ortiz and R. Fernandez-Lafuente, Biotechnol. Adv., 2019, 37, 746–770 CrossRef PubMed.
- M. B. Ansorge-Schumacher and O. Thum, Chem. Soc. Rev., 2013, 42, 6475–6490 RSC and references cited therein.
- M. D. Truppo, H. Strotman and G. Hughes, ChemCatChem, 2012, 4, 1071–1074 CrossRef CAS.
- B. Chen, E. M. Miller, W. Xie, M. Cai and R. A. Gross, Biomacromolecules, 2008, 9, 463–471 CrossRef CAS PubMed.
- P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef.
- R. A. Sheldon, Catalysts, 2019, 9, 261 CrossRef CAS.
- H. Yamaguchi, Y. Kityota and M. Miyazaki, Catalysts, 2018, 8, 174 CrossRef.
- M. B. Mbanjwa, K. J. Land, T. Windvoel, J. G. Korvink, D. Visser and D. Brady, Proc. Biochem., 2018, 69, 75–81 CrossRef CAS.
- D.-M. Liu and C. Dong, Process Biochem., 2020, 92, 464–475 CrossRef.
- J. M. Carceller, J. P. Martinez Galan, R. Monti, J. C. Bassan, M. Filice, S. Iborra, J. Yu and A. Corma, Green Chem., 2019, 21, 839–849 RSC.
- S. Liang, X.-L. Wua, J. Xiong, M.-H. Zong and W.-Y. Lou, Coord. Chem. Rev., 2020, 406, 213149 CrossRef CAS.
- I. N. Ahmed, X.-L. Yang, A. A. Dubale, R.-F. Li, Y.-M. Ma, L.-M. Wang, G.-H. Hou, R.-F. Guan and M.-H. Xie, Bioresour. Technol., 2018, 270, 377–382 CrossRef CAS PubMed.
- B. Qi, J. Luo and Y. Wan, Bioresour. Technol., 2018, 268, 577–582 CrossRef CAS PubMed.
- K. Khoshnevisan, E. Poorakbar, H. Baharifar and M. Barkh, Magnetochemistry, 2019, 5, 36 CrossRef CAS.
- M. Asgher, M. Shahid, S. Kamal and H. M. F. Iqbal, J. Mol. Catal. B: Enzym., 2014, 101, 56–66 CrossRef CAS.
- G. N. Lucena, J. Solid State Chem., 2019, 270, 58–70 CrossRef CAS.
- M. de Sousa, B. Silva Gurgel, B. C. Pessela and L. R. B. Gonçalvez, Enzyme Microb. Technol., 2020, 138, 109566 CrossRef CAS PubMed.
- H. Mallin, J. Muschiol, E. Byström and U. T. Bornscheuer, ChemCatChem, 2013, 5, 3529–3532 CrossRef CAS.
- J. Wöltinger, K. H. Drauz and A. S. Bommarius, Appl. Catal., A, 2001, 221, 171–185 CrossRef and references cited therein.
- M. Romero-Fernández and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef PubMed.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- W. Böhmer, T. Knaus and F. G. Mutti, ChemCatChem, 2018, 10, 731–735 CrossRef PubMed.
- Y. Zhu, Q. Chen, L. Shao, Y. Jia and X. Zhang, React. Chem. Eng., 2020, 5, 9–32 RSC.
- M. B. Ansorge-Schumacher, H. Slusarczyk, J. Schümers and D. Hirtz, FEBS J., 2006, 273, 3938–3945 CrossRef CAS.
- H. Li, R. Wang, A. Wang, J. Zhang, Y. Yin, X. Pei and P. Zhang, ACS Sustainable Chem. Eng., 2020, 8(16), 6466–6478 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- C. Bernal, K. Rodriguez and R. Martinez, Biotechnol. Adv., 2018, 36, 1470–1480 CrossRef CAS.
- F. B. H. Rehm, S. Chen and B. H. A. Rehm, Bioengineered, 2018, 9, 6–11 CrossRef PubMed.
- B. S. Heater, M. M. Lee and M. K. Chan, Sci. Rep., 2018, 8, 12783 CrossRef PubMed.
- V. D. Jäger, R. Lamm, K. Küsters, G. Ölçücü, M. Oldiges, K.-E. Jaeger, J. Büchs and U. Krauss, Appl. Microbiol. Biotechnol., 2020, 104, 7313–7329 CrossRef PubMed.
- R. Koszagova, T. Krajcovic, K. Palencarova-Talafova, V. Patoprsty, A. Vikartovska, K. Pospiskova, I. Safarik and J. Nahalka, Microb. Cell Fact., 2018, 17, 139 CrossRef.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- S. S. Mahmod, F. Yusof, M. S. Jami, S. Khanahmahdi and H. Shah, Process Biochem., 2015, 12, 2144–2157 CrossRef.
- C. You and Y.-H. P. Zhang, ACS Synth. Biol., 2013, 2, 102–110 CrossRef CAS PubMed.
- M. Perwez, J. A. Mazumder and M. Sardar, Enzyme Microb. Technol., 2019, 131, 109389 CrossRef CAS PubMed.
- M.-Q. Xu, F.-L. Li, W.-Q. Yu, R.-F. Li and Y.-W. Zhang, Int. J. Biol. Macromol., 2020, 144, 1013–1021 CrossRef CAS PubMed.
- A. W. H. Dawood, J. Bassut, R. O. M. A. de Souza and U. T. Bornscheuer, Chem. – Eur. J., 2018, 24, 16009–16013 CrossRef CAS PubMed.
- T. Peschke, P. Bitterwolf, K. S. Rabe and C. M. Niemeyer, Chem. Eng. Technol., 2019, 42(10), 2009–2017 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cs00015b |
| This journal is © The Royal Society of Chemistry 2021 |