
First global analysis of the GSK database of small molecule crystal structures†
Leen N.
Kalash
 a,
Jason C.
Cole
b,
Royston C. B.
Copley
a,
Colin M.
Edge
a,
Alexandru A.
Moldovan
b,
Ghazala
Sadiq
b and
Cheryl L.
Doherty
*a
a,
Jason C.
Cole
b,
Royston C. B.
Copley
a,
Colin M.
Edge
a,
Alexandru A.
Moldovan
b,
Ghazala
Sadiq
b and
Cheryl L.
Doherty
*a
aMedicinal Science & Technology, GlaxoSmithKline, GSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK. E-mail: Cheryl.x.Doherty@GSK.com
bThe Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK
First published on 14th July 2021
Abstract
Information gleaned from crystal structure databases has previously been reported on several pharmaceutically relevant compounds to make knowledge-based predictions of polymorphism. Access to a large dataset that is highly relevant to the molecules under study is considered to be essential for these studies. We present a survey of the GlaxoSmithKline (GSK) database of small molecule crystal structures containing X-ray diffraction results from GSK and heritage companies from the past 40 years for this purpose. These structures were collected at different stages of the pharmaceutical pipeline and are not limited to marketed products. We found that the GSK database matches the CSD Drug Subset in terms of crystal descriptors, but not in the diversity of solid form space. Applying the hydrogen bond propensity model to GSK polymorphs has demonstrated the increased value in using combined published and proprietary data sources to build the training data sets. Within GSK, we have also shown the value of applying knowledge-based predictions in the de-risking of active pharmaceutical ingredient forms of development candidates. The work described here illustrates the importance of database curation to improve the accuracy of the results obtained.
Introduction
Pharmaceutical materials scientists choose from the different accessible solid states for each active pharmaceutical ingredient (API) to develop a solid form, which exhibits the properties and behaviour most suitable to produce a successful drug product. The selection of this final solid form, and the crystallisation route to produce it, are key milestones in the drug development process. Solid state properties critical to the success of the final dosage form are locked in at this stage and these features contribute to determining the safety, manufacturability and bioavailability of the drug.1 Single component free drugs, salts, hydrates, co-crystals and occasionally even solvates are potentially viable options and may be considered for selection if those solid forms have the solid state properties required.2 It is also well known that many APIs spontaneously crystallise in multiple arrangements to form polymorphs.3 In polymorphs the molecular components are the same but the overall arrangement is not, differing in some combination of the molecular conformation, hydrogen bonding and overall packing. These changes can be subtle, significant or fall somewhere in between. As a result, polymorphs of the same API will have different physical properties, including features key to the successful development of that API such as solubility, dissolution rate, chemical and physical stability, melting point and habit.3–5 Indeed, this is of such importance that solid form screening is a regulatory requirement for new drugs, to provide confidence in the safety and efficacy of the product.6Typically, experimental solid form screening to identify the thermodynamically stable and relevant metastable polymorphs are performed initially during the pre-formulation phase of drug development, with more comprehensive screens being carried out later on. The solid form which is most stable under conditions relevant to the manufacturing and storage of the drug product is typically preferred for development, so a wide range of crystallisation conditions and storage protocols are commonly explored to identify the most suitable stable solid form for each candidate.2,7 Despite these efforts, unexpected new polymorphs can still appear, even in well-screened systems. Notable examples such as ritonavir8 and rotigotine9,10 show that the late discovery of a new stable polymorph can result in significant challenges to providing a safe and efficacious drug product.2,11,12 Similar bioavailability issues were also encountered with other polymorphic drugs such as chloramphenicol palmitate, oxytetracycline, carbamazepine, atorvastatin calcium, axitinib, phenylbutazone, and rifaximin.13
It is clear that the best chance of finding the most stable solid form comes from designing the widest experimental screen possible, but it is not feasible to explore every possible experimental condition in a reasonable timescale. It is therefore crucial to understand the potential solid form landscape for each candidate and to use that information to do the right subset of experiments that will allow the isolation of the stable form for every case.
One route for the investigation of solid form landscapes is the application of structural chemistry knowledge derived from the Cambridge Structural Database (CSD), which is a collection of every organic, organometallic and metal–organic crystal structure published and now totals over one million entries. Data mining these structures using bespoke solid form informatics tools14–16 developed by the Cambridge Crystallographic Data Centre (CCDC) allows a deeper understanding of the solid form, helps identify weaknesses that may relate to the risk of alternate forms, reveals opportunities for intervention and provides validation and reassurance. Solid form informatics is not a replacement for experimental work but a complementary tool which allows a more informed experimental design to probe risks and opportunities. Solid form informatics is now well established and widely used within the pharmaceutical industry in drug development.17–20
By evaluating a structure in the context of existing knowledge in the CSD, it is relatively straightforward to identify both common and unusual structural features. As examples, an unusual conformation of a molecule, ring or functional group, a geometrically unusual hydrogen bonded interaction, or an unusual donor–acceptor combination may suggest that alternative crystal forms without these compromises could potentially exist.14–16 Statistics and comparative CSD analysis can give answers easily and quickly which can influence and advance the decision-making process with respect to risk mitigation in solid form selection.21
To draw useful conclusions from structural informatics analyses the available training data must be relevant to the compound of interest. Bryant et al.,22 as part of the advanced manufacturing supply chain initiative (AMSCI) funded advanced digital design of pharmaceutical therapeutics (ADDoPT) project created the CSD drug subset (CSD-DS) consisting of every published small molecule crystal structure containing an approved drug molecule (8632 entries). A strong overlap in molecular features including size and flexibility between this drug subset and in-house crystal structure data23 for AstraZeneca and Pfizer was demonstrated providing support for the use of statistical informatics models.
As the application of the models described here increases it is valuable to demonstrate the relevance of the publicly available data to modern drug candidates. This allows the accuracy of these models to be assessed for pharmaceutical solid form development. We report herein the first global analysis of the GSK database of small molecule crystal structures. Unlike the CSD-DS, the structures have been obtained across the various stages of the pharmaceutical pipeline and were originally collected to answer structural problems. As such, the database is not limited to marketed products but also includes medicinal chemistry leads, candidate molecules, intermediates and impurities. As of November 2019, there were 2473 entries in the database. It is worth noting that as the structures were collected to answer specific problems, there is no guarantee (indeed it is highly unlikely) that all solid-state forms of a particular material are contained within the database. In addition, the morphology of many of the crystals studied was accurately recorded in the GSK database, with many samples having been routinely face-indexed.
Our analysis described here: offers insights into the difference between proprietary and public domain data; illustrates the relevance of including both CSD-DS and proprietary structural data to build models for GSK candidates; and demonstrates the structural informatics methodology to explore the polymorph landscape of real pharmaceutical candidates.
Results and discussion
Some definitions
In the remainder of this paper we use several terms to describe different classes of crystal structure. The names of different classes of structure have been a subject of debate24 and so we feel it is useful to clarify the exact definitions of these terms used in this publication (Table 1). As we wish to compare our results to those of Bryant et al.,22 our definitions are broadly based on those in the previous paper.| Term | Definition |
|---|---|
| Free drug | A structure containing a single type of component. Note that this classification can include structures with multiple symmetry independent copies of a given molecule in the asymmetric unit; vis-à-vis any value of Z′. It includes zwitterionic systems. This term has been applied whether the material is formally a proven drug in its own right or not i.e. includes all organics, such as intermediates etc. |
| Salt | A structure where any of the components are charged (not including zwitterions) |
| Hydrate | A structure containing more than one type of component where at least one of the components is a water molecule |
| Solvate | A structure containing more than one type of component where at least one of the components is a solvent molecule other than water. A solvent was defined as being a liquid whose role is to dissolve the drug in any stage of the synthetic process |
| Co-Crystal | A structure containing more than one type of component where at least one of these components is charge-neutral and not a solvate molecule, a water molecule or the free drug |
Note that, other than free drugs, no class of crystal structure is mutually exclusive; indeed, it is possible to have a crystal structure that is in all the other classes at once. For completeness whilst discussing definitions, we use the term API in this paper to describe the biologically active substance of a structure.25 This term is not necessarily interchangeable with the free drug, since the API will include any salt counter ions or co-crystal co-formers.
Identifying crystallisation families and assessing polymorphism in the GSK database
Detailed manual analysis of the GSK database of 2473 crystal structures identified eight structures that were deemed either irrelevant (e.g. octasulfur) or to have sufficient errors as to warrant their exclusion. In addition, eight structures were found to be exact duplicates of other entries (i.e. the same cell constants and R1 value) and a further 40 were adjudged to be less preferred determinations of the same structure. All of these were omitted, leaving 2417 structures for further analysis. These criteria are consistent with those used in the preparation of the CSD-DS.22In this phase of the analysis, we built crystallisation families from the database. Any solid forms that might reasonably be produced from a solvent crystallisation of an API were regarded as being in such a family. To illustrate this, an API in isolation or a hydrate and/or solvate of this would be in a crystallisation family but a different salt for instance would not; that salt would have its own family. These groupings of solid state forms and their interconversion either by crystallisation or desolvation are important to understand in a pharmaceutical context since the hydrated and solvated forms are often not desirable; a better understanding of these families could help improve the risk profile of APIs under development.
Crystallisation families in the GSK database were identified by sorting and grouping the chemical formulas and then manually inspecting the entries to ensure the same API was present in each case. The initial intention was to do this in an automated fashion by pulling together entries with the same canonicalised SMILES for the highest molecular weight component. This was not a successful approach owing to a number of factors, the most notable being the inability to discern the stereochemistry present. Recognising different enantiomers and diastereomers proved difficult without manual intervention, as did separating structures that were single enantiomers versus racemates. Although care was taken to ensure stereochemical differences were taken into consideration, potential atropisomers were considered to be the same entity. This was on the basis that information on the conversion rate was not available within the systems being investigated. SMILES were helpful to identify molecules with the same heaviest component formula and different atomic connectivity but even this was hampered as it is dependent on the correct and identical assignment of bond orders in the database. The discovery that some of the entries within the GSK database were in error or inconsistent was a disappointment but perhaps one of the greatest learnings from this exercise. As a result of this work, a detailed list of database corrections has been drawn up.
Based on the above methodology, 137 crystallisation families were identified in the GSK database. Fig. 1A shows the count of families in the database based on the type of members present in each, where an API could be a free drug or salt (there are no co-crystal examples). We explain the content of this Venn diagram in more detail by means of an example. The intersection of all four sets contains the number 5. This means there are five crystallisation families in the database that contain at least: one structure of the API alone, one hydrated form, one solvated form and one hydrate/solvate. Note that an individual hydrate/solvate does not occur at the intersection of hydrates and solvates since this diagram describes the whole family, not individual structures. By far the largest subset are the families that are composed of only API polymorphs (43 families), followed by the API and solvates (30 families) and the API and hydrates (17 families). These proportions are not representative of family compositions generally, since the search for polymorphic structures and their differences is a primary deliverable for X-ray diffraction studies in GSK. Since the largest fraction of the crystallisation families corresponded to polymorphic systems, these were reviewed separately in more detail. The polymorphs were identified using the procedure outlined in the Experimental section. Based on this analysis, 141 structures or 5.83% of the GSK database were polymorphs, which is notably smaller than the proportion reported in the CSD-DS (approximately 25%).22 The difference is likely to be a result of the different primary deliverables behind the two databases.
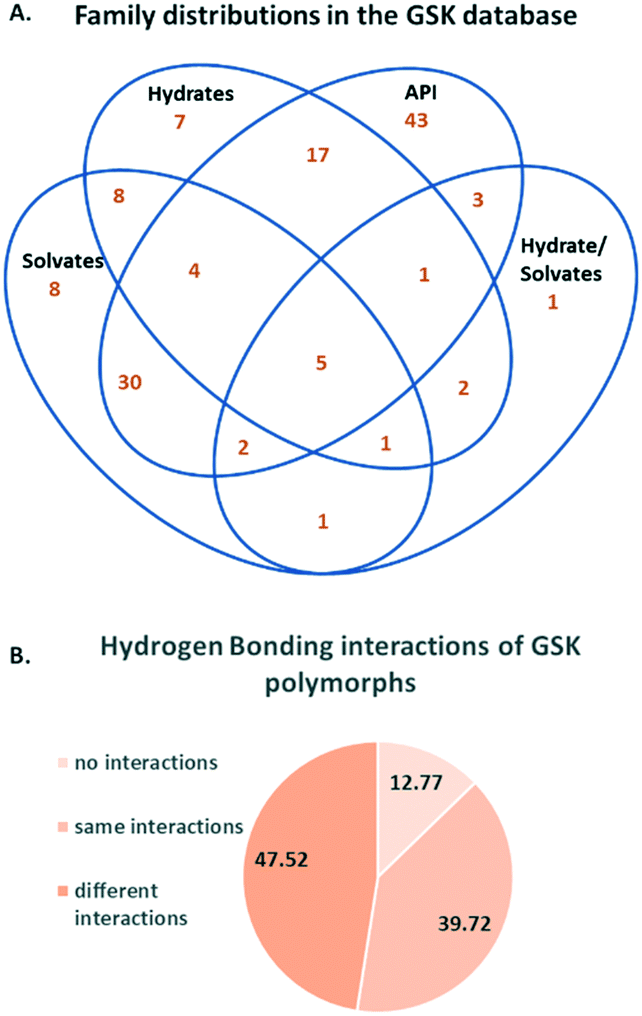 | ||
| Fig. 1 A. Distribution of crystallisation families in the GSK database B. Percentage distribution of categories of hydrogen bonding interactions in polymorphs. | ||
The hydrogen bonding interactions in polymorphic structures were obtained using a python script (see Experimental section for details). Fig. 1B contains a breakdown of these interactions. The percentage of compounds with hydrogen bonding interactions that are identical in the polymorphs (in terms of the identity of donors and acceptors) is 39.7%. The hydrogen bonding arrangement for polymorphs is different in 47.5% of cases. The difference in hydrogen bonding across polymorphs supports the use of the CCDC's hydrogen bond propensity tool,26–28 in which the identification of possible polymorphs is based on the likelihood of obtaining different hydrogen bonding arrangements. The limitation of the HBP methodology is that polymorphs with the same hydrogen bonding cannot be distinguished from one another. The finding presented here based on the GSK database is that polymorphs with different hydrogen bonding are sufficiently common to support the use of the HBP tool in the pharmaceutical industry. 12.8% of the polymorphs were found to have no hydrogen bonding interactions. Comparison of the percentage of polymorphs from the GSK database that exhibit hydrogen bonding (87.2%) with the percentage of the total number of GSK structures (all 2417 structures) that are involved in hydrogen bonding (77.3%) indicates agreement with the analysis done by Cruz-Cabeza et al.,29 where it was found that compounds that are able to hydrogen bond have higher tendency to form polymorphs than those which do not. Despite the fact that the group of polymorphs that do not exhibit hydrogen bonding are not as frequently observed as those with hydrogen bonding, these should not be overlooked and developments in the CCDC's solid form tools (such as the aromatic analyser and the full interaction maps) are beginning to address these cases.30,31
Another factor relating to polymorphism is chirality. It was found by Cruz-Cabeza et al.29 that chiral molecules are less prone to polymorphism than their achiral counterparts. The sum of chiral arrangements were computed for molecules of polymorphs (71 molecules). It turned out that only 49.3% of the polymorphs have chiral arrangements, as compared to all unique molecules of the GSK database (2009 molecules) where 59% have chiral arrangements. Hence, these percentages agree with the work of Cruz-Cabeza et al. suggesting that chiral molecules are less prone to polymorphism; however, this percentage might not be representative of the actual polymorphs obtained in experimental screens.
Differences in the solid form distribution between the GSK database and CSD-DS
The solid forms present in the whole GSK database were analysed (see Experimental section for details). The Venn diagram in Fig. 2 shows considerable differences between the solid form distribution of the GSK database (2416 crystal structures are shown as one structure is a clathrate that could not be classified into the Venn diagram) and the CSD-DS (8632 crystal structures).5 58.1% of crystal structures in the GSK database are free drugs compared to 19.6% in the CSD-DS. The percentage of forms that might be considered as salts in the GSK database (23.1%) is approximately half of the percentage in the CSD-DS as a whole (45.5%). Looking more closely at the GSK salts, these could be sub-divided into three categories, where the drug-like component was cationic (19.5%), anionic (3.1%) or neutral (0.5%). The first two categories are easy to understand and exemplified by hydrochloride or sodium salts respectively. The final category is less intuitive and results when the drug-like molecule is neutral but other components present are charged. In many of these cases there is chelation of the neutral species around a metal cation. It should be noted that in some of the salt cases, the largest organic component of a structure was not the drug. Where possible, the classification into the above three categories was based on a knowledge of the drug component from internal registry databases. If this information was not available, it was assumed that the largest organic component was the drug.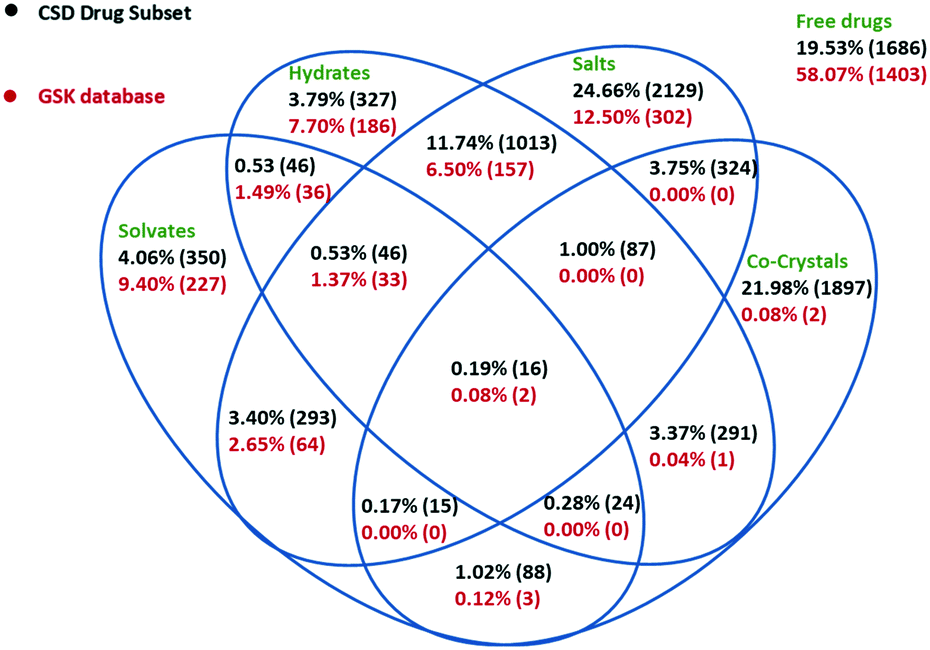 | ||
| Fig. 2 Venn diagram detailing the solid form distribution of the GSK database in comparison to the CSD-DS. Forms could be either free drugs, salts, co-crystals, hydrates, solvates, or a combination of these. Each of these forms is displayed by their percentage of occurrence in each database. | ||
The different balance between the free drug and salt structures in the two databases is highly likely to be a result of the GSK collection including pharmaceutically relevant molecules from all parts of the pipeline, not just marketed products: medicinal chemistry samples generated as part of lead optimisation studies would rarely be prepared as salts for instance. Another marked difference between the GSK database and the CSD-DS is the number of co-crystal structures (0.3% vs. 31.0% respectively). Historically, GSK has not actively prioritised the development of co-crystal APIs and this is reflected in the numbers. The above co-crystal definition removes a number of GSK structures from this category, in the case of salts where the drug-like molecule is present as both a charged and a neutral molecule.
The total number of hydrated structures is found to be 17.2% in GSK and 20.4% in the CSD-DS. Despite the overall differences in the composition already discussed, these numbers for the hydrates are reasonably similar. This might suggest that hydration as a phenomenon is not unduly influenced by whether the pharmaceutical material is charged and/or multicomponent. This seems a little counterintuitive at first, as one might expect the salts more prevalent in the CSD-DS to be more hydrated since they are charged.7 There is a slight bias in that direction in the figures, but it is not as pronounced as was expected and more work may be needed to rationalise this finding further. By comparison, there is a bigger difference in the number of solvates found in the GSK and CSD-DS, 15.1% and 10.0% respectively. This difference again probably comes down to the composition of the two databases, mainly due to GSK strategy, where questions (particularly from Discovery) could often be answered using solvates whereas their existence in marketed drugs would be seen as a clear disadvantage.
The crystal descriptor space of the GSK database and the CSD-DS
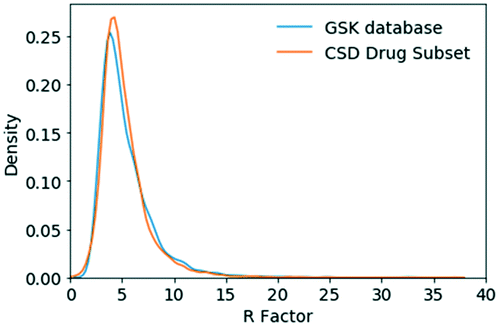 | ||
| Fig. 3 R-Factor distribution for small molecule crystal structures in the GSK database and the CSD-DS. | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (23.3%) and third ranking is P21 (12.4%). Clearly GSK's database exhibits more examples of the Sohncke space groups, which can support the packing of chiral molecules, whereas the CSD-DS has more of the centrosymmetric space groups composed of achiral molecules and racemates.
(23.3%) and third ranking is P21 (12.4%). Clearly GSK's database exhibits more examples of the Sohncke space groups, which can support the packing of chiral molecules, whereas the CSD-DS has more of the centrosymmetric space groups composed of achiral molecules and racemates.
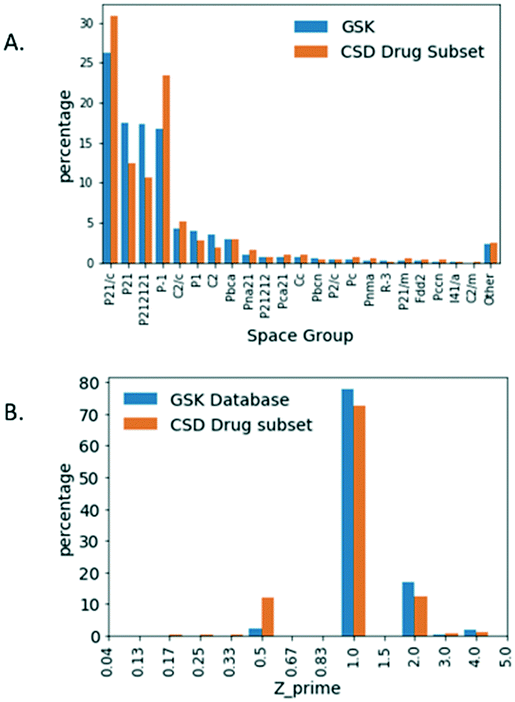 | ||
| Fig. 4 A. Percentage distribution of space groups. B. Z′ for small molecule crystal structures in the GSK database and the CSD-DS. | ||
The relative Z′ distribution profiles obtained for both databases are shown in Fig. 4B. By far the most frequent structures are those with Z′ = 1 in both the GSK database (77.8%) and for the CSD-DS (72.5%). There are far more structures containing symmetry (Z′ < 1) in the CSD-DS by comparison to the GSK database. In particular, Z′ = 0.5 represents 12.06% for the CSD-DS but just 2.40% for the GSK database. Closer inspection of Z′ = 0.5 structures in the CSD-DS is illuminating. 98% of structures that are Z′ = 0.5 in the CSD-DS have more than one distinct component in the asymmetric unit, so the high observation in the CSD-DS of Z′ = 0.5 structures compared to GSK is an artefact of the high number of co-formers that often straddle centres of symmetry in the CSD set, and how Z′ is defined in these cases. The Z′ = 2 structures for the GSK database (17.05%) are slightly greater than the CSD-DS (12.53%). Both these findings fit with the larger number of Sohncke space groups in GSK, since inversion symmetry in these are impossible and chiral molecules tend to mimic a centrosymmetric arrangement33 and this requires two independent molecules to achieve. Otherwise the broad Z′ profiles are similar for both databases.
The packing distributions of these two databases are very similar as shown in Fig. 5A and also agree well with those values originally reported for aromatic molecules by Kitaigorodskii (0.6–0.8).35 The difference in the median values for these packing coefficients were investigated with a Mann–Whitney test and shown to be significant (Table S2†). This analysis uses highly relevant datasets to confirm the typical range of packing coefficients for drug-like crystal systems. This allows future low density materials to be identified clearly as a risk early in development, further allowing mitigating development activities to be targeted.
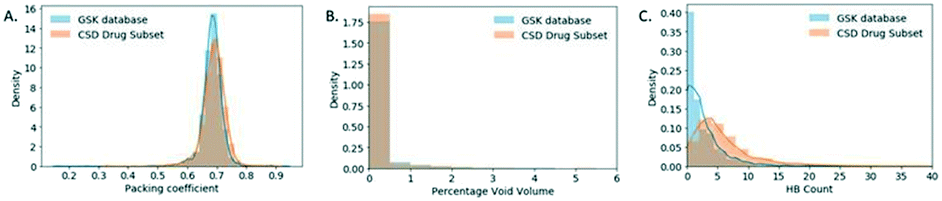 | ||
| Fig. 5 (A). Packing coefficient distribution. (B) Percentage void volume density distribution (C) number of hydrogen bonding pairs (HBP) distribution. For small molecule crystal structures in the GSK database and the CSD-DS. | ||
As part of the packing coefficient analysis, an attempt was made to understand structures at the extremes (in the tails) of the GSK distribution. The most obvious reason for structures with low packing coefficients was the use of SQUEEZE procedures.36 These have not generally been used for GSK structures (with a preference for modelling disordered solvent whenever possible) but in some cases there was no alternative. A ConQuest search was performed on the GSK database in an attempt to identify structures that used SQUEEZE and to see the effect of this on the packing coefficient. Fourteen structures were identified as using SQUEEZE, with nine of these have packing coefficients between 0.46 and 0.59, which is within the tail of the distribution.
To gain a better understanding of GSK structures with low packing coefficients the percentage void volume density distributions (relative ratio) were obtained for both databases as illustrated in Fig. 5B. 9.40% of the CSD-DS has a non-zero percentage void volume (see Experimental section for the definition used to calculate void space) whereas the GSK database has a higher percentage (15.39%) of structures with a non-zero percentage void volume, which could be attributed to the CSD-DS containing a larger proportion of marketed products in which the drug substances have optimised solid state properties.
The number of hydrogen bond pairs (HBP) in each structure was computed with a script written in Python (refer to Experimental section for details). The HBP distributions of the two databases are very different as shown in Fig. 5C, where the distribution of the CSD-DS is shifted towards higher HBP values. The mean value for the CSD-DS is 6, compared to just 3 in the GSK database. This could be explained by the predominance of free drugs in the GSK database (58% of the total solid form distribution) in comparison to the CSD-DS that has fewer of these forms and more salts, which are generally capable of exhibiting more hydrogen bonding interactions. The lack of co-crystals in the GSK entries may also have an impact here. Another interesting finding is that 22.7% of the structures in the GSK database exhibit no H-bonding, which is a much higher percentage than the CSD-DS structures where only 4.8% of the structures do not display any H-bonding.
For the molecular descriptor space analysis, unique molecules in the GSK database (2099 molecules) and the CSD-DS (778 molecules) were considered in an attempt to draw out relations to observed crystal descriptors. The increased percentage of GSK structures with no H-bonding interactions in comparison to the CSD-DS comes in line as well with the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P distribution (Fig. 6A), where for the GSK database it is shifted towards higher values, with a mean value of 3.17 ± 2.24, indicating that the molecules are more hydrophobic in nature in comparison to the CSD-DS with a mean value of 1.79 ± 2.65.
P distribution (Fig. 6A), where for the GSK database it is shifted towards higher values, with a mean value of 3.17 ± 2.24, indicating that the molecules are more hydrophobic in nature in comparison to the CSD-DS with a mean value of 1.79 ± 2.65.
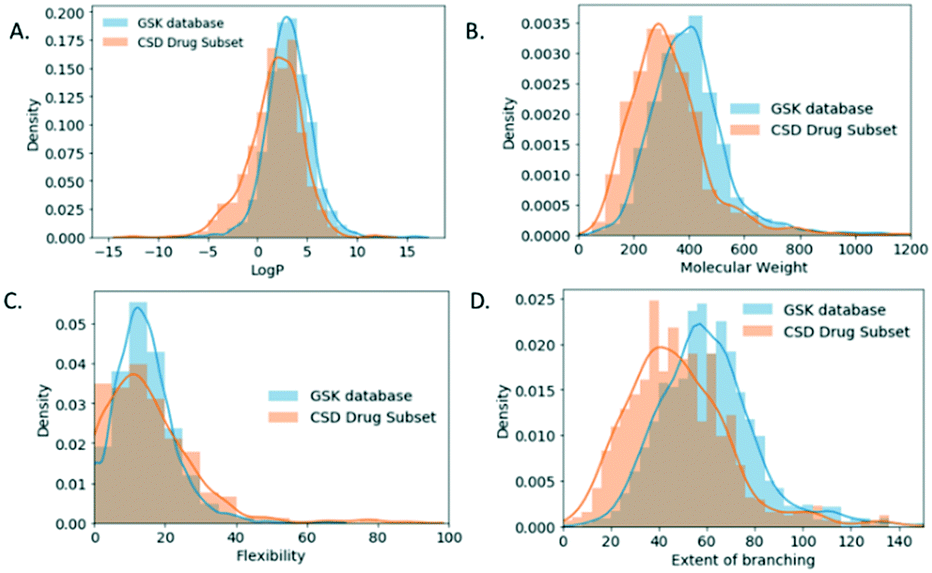 | ||
| Fig. 6 (A). LogP (B) molecular weight and (C) flexibility (flexibility = int(100*rotatable bonds/total bonds) (D) extent of branching is walk count, which is the return of the number of walks of order 2 that start and end at the same atom, density (relative ratio) distribution for small molecules crystal structures in the GSK database and the CSD drug subset. | ||
Furthermore, the molecular weight, flexibility, and branching density distribution profiles were obtained for both databases (Fig. 6B–D respectively). Flexibility is defined as integer * (100*rotatable bonds/total bonds), where the rotatable bonds percentage distribution were obtained for each database. Branching37 is the return of the number of walks of order 2 that start and end at the same atom which is an indication of the branching within a molecule. The mean MW value for the GSK molecules is 399 g mol−1 as compared to the CSD-DS molecules, which is 324 g mol−1. The mean value of the flexibility for GSK molecules is 14 ± 8 as compared to the CSD DS molecules 15 ± 13. Finally, the mean branching value for GSK molecules is 61 as compared to CSD DS molecules, which is 41. It is noted that Mann–Whitney tests were performed on each of the GSK and CSD-DS distributions in Fig. 6 showing no significant difference in flexibility but a significant difference in each of the others (see Table S2 in ESI†). The molecular weight and extent of branching distribution profiles for the GSK dataset are shifted towards higher values, which might contribute to the decreased values of the packing coefficient distribution.38 Flexibility does not correlate with these observed trends since the medians were not shown to differ significantly. However, it has been previously reported that crystalline molecules, tend to have low molecular weight and generally are simpler structures with a lower number of rotatable bonds. On the contrary, uncrystallisable molecules tend to be more structurally complex and flexible molecules which makes them harder to crystallise.39 It is noted as well that the walk count/branching descriptors are used in machine learning methods for the prediction of crystallisability of small organic molecules.40,41
The benefits of combining datasets when building knowledge-based models of hydrogen bond propensity
The hydrogen bond propensity (HBP) model approach is used for predicting the likelihood of hydrogen bonding between donors and acceptors in a crystal structure, helping in rationalising stable and metastable crystalline forms.26,42 The model implements logistic regression and uses training data obtained with a survey method based on the CSD system that works by categorising the hydrogen bonds and extracting model parameter values employing descriptive structural and chemical properties from three-dimensional organic crystal structures. Predictions are then formed based on two-dimensional features of the input test structure.In this section, we show the importance of having a good coverage of functional groups when using HBP to build a reliable model. An interesting example that illustrates this is the molecule, GW825964X. We investigate whether the statistical analysis of hydrogen bonds in the publicly available CSD-DS alone is sufficient to predict the likelihood of the hydrogen bonding for GW825964X, or if there is value in incorporating the proprietary data in the GSK database. The GSK database contains a much smaller number of structures but they are highly relevant, and it often includes close analogues from the same chemical series. Training datasets were built from GSK, CSD-DS, and combined GSK/CSD-DS data to build three comparative models, which were used to evaluate the performance and predictive power of these models.
The HBP model performance was evaluated using different training data sets of GSK, CSD-DS, and a combination of the two by comparing the area under the curve (AUC) (Table 2). The GSK database exhibits a larger dataset than the CSD-DS training set when mined for relevant structures. However, two functional groups were not well represented using either the GSK training set or the CSD-DS alone, whereas a combination of GSK and CSD-DS ensured that all functional groups are well represented. This indicates there is value in collecting more structures for compounds containing certain functional groups that are poorly represented in the CSD-DS, in order to build better models for compounds containing these features in the future.
| Polymorphs of GW825964X | |||
|---|---|---|---|
| Training set | AUC | Number of well represented functional groups | Data size |
| GSK | 0.93 | Two not well represented | 729 |
| CSD-DS | 0.90 | Two not well represented | 450 |
| GSK + CSD-DS | 0.92 | All well represented | 1179 |
Plots of the mean hydrogen bond (H-bond) co-ordination versus mean H-bond propensity were generated by the HBP models using the three different types of training sets (GSK, CSD-DS, and GSK + CSD-DS) for two polymorphs of GW825964X and are shown in Fig. 7A–C respectively. Using two of the training sets (GSK and GSK + CSD-DS) as shown in Fig. 7A and C respectively, the more stable form exhibits higher mean H-bond co-ordination and mean H-bond propensity values. With the CSD-DS training set alone (Fig. 7B), the more stable form exhibits a lower H-bond propensity value.
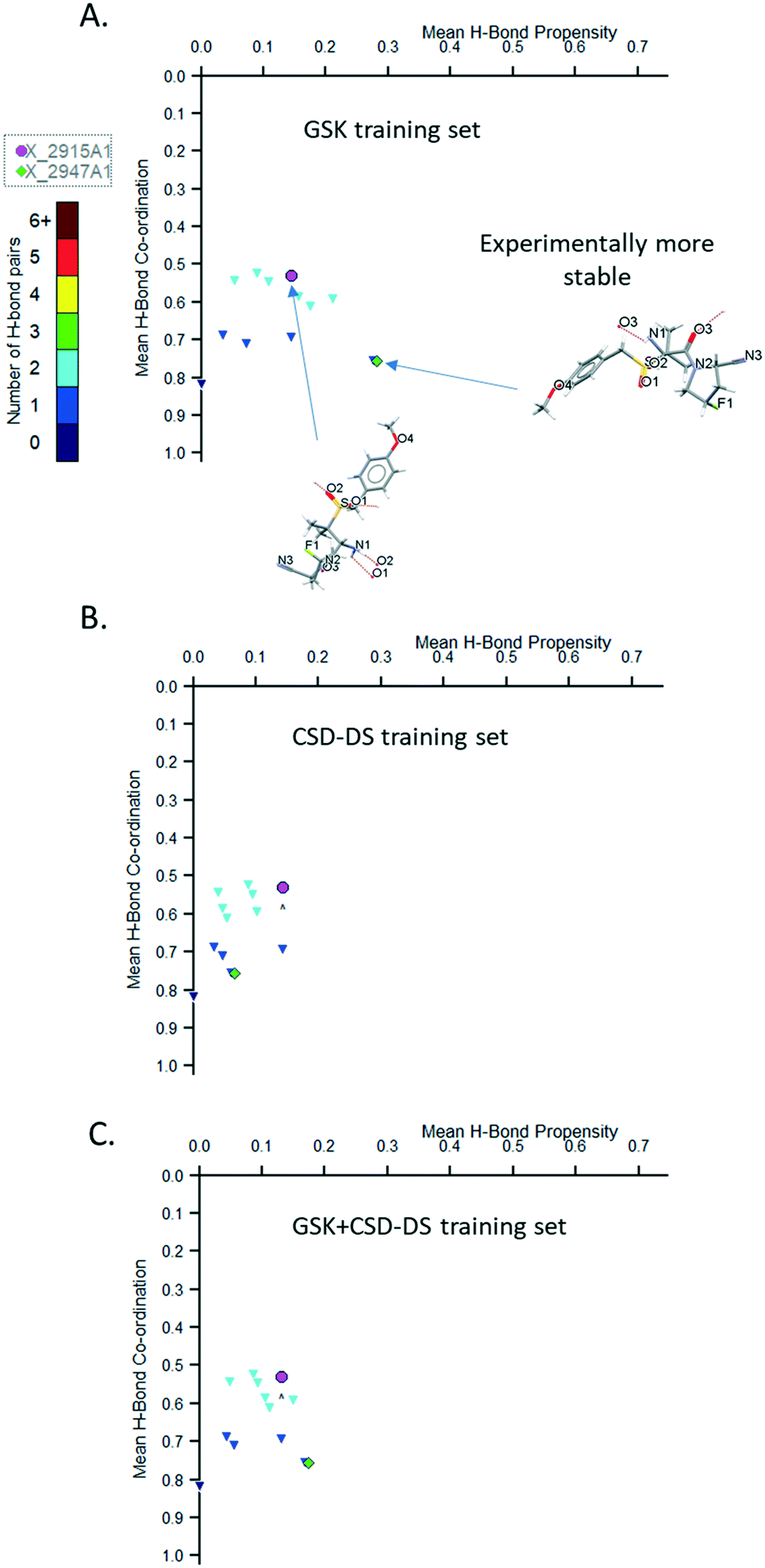 | ||
| Fig. 7 Plot of mean H-bond co-ordination versus mean H-bond propensity for the models using (A) GSK training set, (B) CSD-DS training set, and (C) GSK+CSD-DS training set for GW825964X (structure X_2947A1 is experimentally more stable than structure X_2915A1), min. donor co-ordination P(n) value set to 0.05 and min. acceptor co-ordination P(n) value set to 0.05. | ||
The cause for the lower propensity in the CSD-DS data set is the low representation of the amide carbonyl acceptor compared to the sulfonyl acceptor. The more stable structure contains a hydrogen bond to this carbonyl, whereas the less stable structure contains a hydrogen bond to the sulfonyl. The model underestimates the likelihood of the hydrogen bond to the carbonyl as compared to the sulfonyl (see Tables S3–S8†).
In the GSK training set there are also two underrepresented groups, but neither form hydrogen bonds in the two structures, and so the relative impact of these being under-represented in the model is small (see Tables S9–S11 ESI†).
The coordination score does not change from model to model as these are based on pre-calculated information from the CSD. The model only considers the donors or acceptors in isolation, and so are less affected by a lack of data in the CSD for the more chemically specific functional groups used to build the training dataset. The scores differ here (see Coordination_Score Table S12 in ESI†) because the amide carbonyl is generally more likely to accept the observed number of hydrogen bonds than the sulfonyl carbonyl in the two structures.
This analysis highlights the benefit of combining our in-house database with the CSD-DS to build more statistically certain HBP models. This shows the value of incorporating as much relevant data as possible into knowledge based modeling methods.
Assessing crystal morphology in the GSK database
Morphology is a key quality attribute for processing and manufacturing unit operations (e.g. filtration, flow and agglomeration) in pharmaceutical manufacturing.43,44 Delivering consistent particles with acceptable mechanical properties is therefore also an essential consideration in the form selection process. Database driven prediction that could lead to purposeful modification of morphologies would be a valuable tool, so the morphology data available in the GSK database was assessed in some detail. The morphologies of the small molecule crystals captured in the GSK database were first categorised into 1D (e.g. needle, rod), 2D (e.g. plate, lath) and 3D (e.g. prism, block). Further details of how this classification was achieved can be found in the ESI.† As shown in Fig. 8A, the morphology has not been captured for just over half of the GSK database, in many cases for structures that were collected before the introduction of crystallographic information files. Whilst the number missing morphology information was high, a study of the remaining 45% was felt valuable since in many cases the information available was very reliable, including full face-indexing. Returning to the morphology categories defined above, there is a higher percentage proportion of 2D (38.3%) and 3D (39.9%) crystal structures in the GSK database compared to 1D (21.8%) (Fig. 8B). It is worth noting that this distribution is representative of the crystal structures that have been collected and the lower percentage of the 1D crystals could be partly attributed to the fact that, from an experimental point of view, good diffraction data are generally harder to collect from these.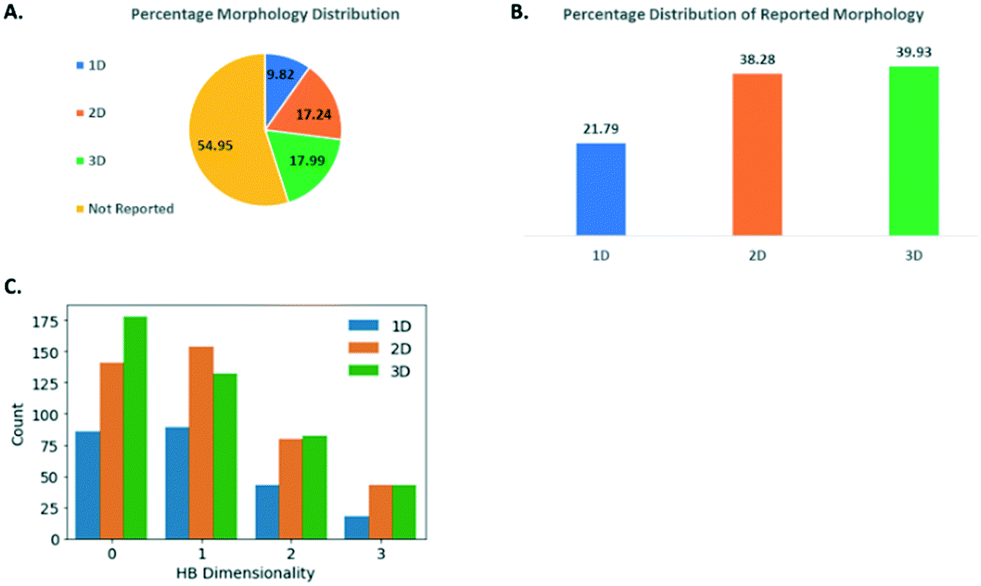 | ||
| Fig. 8 (A) Percentage distribution of morphology for small molecule crystal structures in the GSK database. (B) Count of hydrogen bond dimensionality of small molecule crystal structures with 1D, 2D, and 3D morphology. (C) Relationship between the HDB (discrete, chains, sheets and networks) of the structure with the external crystal morphology. | ||
The hydrogen bond dimensionality (HBD) (see Experimental section: protocol for generating molecular descriptor distributions) is a useful descriptor to assess the networks formed, and its distribution has been investigated in relation to the morphology of the small molecule crystals studied as part of the GSK database. This analysis aims to seek a correlation between the dimensionality of hydrogen bonding networks and the morphology of the crystals. HBD could be categorised as: discrete (when molecules do not possess hydrogen bonds or hydrogen bonds are formed in discrete units without any long-range extension; chains (when there are chains of hydrogen bonded molecules); sheets (when there are hydrogen bonded sheets); and networks (with extensive interconnected hydrogen bonding in all directions). As shown in Fig. 8C, for the 1D and 2D morphologies, the HBD has a similar distribution, with the chains HBD being the most frequent, followed by discrete, sheets and finally, networks. This does not indicate any clear correlation existing between HBD and morphology. For the 3D morphology crystal structures however, the discrete HBD is clearly the most frequent followed by chains, sheets, then networks. This is clearly different and warrants further consideration. The count of 3D structures with discrete HBD was shown to be different from the count of non-3D structures with discrete HBD and we used a chi-squared test to show that this was significant. This is not only due to a lack of any hydrogen bonding because for discrete HBD structures hydrogen bonds are found in 51.7% of 3D structures and in similar proportion of 45.3% of all structures.
One hypothesis is that the lack of long range hydrogen bonding in these discrete structures leads to the greater influence of van der Waals' interactions and more even growth in all directions, subject to BFDH considerations. Work to investigate this further might include a closer inspection of the unit cell dimensions and indeed the difference between observed and BFDH calculated dimensions. Having accurately face-indexed crystals in GSK would help in this regard.
Conclusions
In this work the first global analysis of the GSK database of small molecule crystal structures has been reported. The survey has demonstrated some notable differences between this proprietary dataset and the public Cambridge Structural Database-drug subset (CSD-DS). Both databases are of a historic nature and may no longer represent typical molecules under active consideration today and neither dataset is comprehensive due to the different reasons for data collection and publication. Given this the subsets are as complete as possible, bearing in mind quality considerations. In the GSK database there is a predominance of crystal structures of free drugs, with a reduced number of salts and almost no co-crystals. Despite this, the number of hydrates is similar and solvates are more prevalent in the GSK structures.Crystallisation families of structures containing the same API have been identified in the GSK database and structures for related polymorphs identified. The number of the latter are generally quite low however since most GSK structures have arisen from individual requests where a single structure was used to answer the problem in hand. The resulting study of the hydrogen bonding in these polymorphs was very informative. A large number of polymorph pairs in the GSK dataset have been shown to have different hydrogen bonding arrangements. This is highly encouraging for the use of the CCDC's HBP tools for the prediction of alternative polymorphs.
The R-factor distribution for the GSK database and the CSD-DS were comparable. On the basis of this broad metric, the structures in the GSK database and the CSD-DS are regarded as being of a similar quality. A greater difference was observed when considering the space groups and Z′ values in the two databases. The GSK database favors Sohncke space groups that can contain chiral molecules and consequently, there are fewer Z′ = 1/2 and more Z′ = 2 structures.
A similar distribution of packing coefficients is found in the two databases. Structures treated with the SQUEEZE procedure occupy the lower coefficient GSK tail. The number of hydrogen bonded pairs in the GSK database are much lower than the CSD-DS whereas percentage void volume, molecular weight, and flexibility density distributions are higher for the GSK database, which are factors that could contribute to the shift of the packing coefficient distribution towards lower values for the GSK database.
A study of the morphology of GSK's crystals involved segregating these into 1D, 2D and 3D categories. The 3D morphology crystals, which are most suited for the pharmaceutical industry due to their enhanced handling and processing characteristics, were the most abundant in the GSK database. However some caution is required when considering this as they are also the easiest to measure experimentally. Perhaps more interestingly, an analysis of the hydrogen bond dimensionality shows that structures with no directionality (hence where van der Waals' forces play a larger role) clearly favor the 3D morphologies.
The work described in this paper greatly increases the understanding of the contents of the GSK database and demonstrates the value of this small number of chemically relevant additional structures in models assessing polymorph stability. It has highlighted the need for large amounts of accurate and relevant crystal structure data to build more reliable hydrogen bond propensity (HBP) models, and it demonstrates the value of using them in de-risking API forms of pharmaceutical candidates. In order to extract useful information in this way, particularly using automated scripts, the contents of the database need to be carefully curated to ensure consistency. The work also emphasises the advantages of the database such as the accurate recording of the morphology of many of the crystals that have been routinely face-indexed. This would allow for the successful analysis of the factors impacting the formation of crystal structures with different morphologies, which would also be invaluable to the development of modelling and prediction algorithms for crystal habits and strategies for habit modification. Overall, the data analysed could be used for building more reliable models for the prediction of materials properties and improvement of the performance of GSK materials, aiding crystal engineering and de-risking materials selection earlier in the drug development process to the benefit of patients.
Experimental
Initial attempts to inspect the internal GSK database using python scripts produced some unexpected results and manual inspection of these revealed some logical but computationally ‘hard to fix’ issues. The level of crystallographic interpretation needed to get robust results was such that a decision was made to fully review the GSK database contents before carrying out the data mining activities described herein. Although the work related to duplicates, polymorphs and crystallisation families outlined below yielded the final lists of these, the analysis of the initial script-produced output speeded up the crystallographic decision making in many cases, as the necessary structural comparisons had already been carried out.Building a master spreadsheet
To document the review and extract some of the information presented in this paper, a master spreadsheet was prepared using Microsoft Excel for Office 365 (evolving version numbers). The starting point for this spreadsheet was to output tab delimited relevant information for all the GSK database entries using CCDC ConQuest [all references to CCDC ConQuest and CCDC Mercury relate to version 2020.1 (Build 280197)]. Additional columns were added to record the solid form, crystallisation family and polymorph information. A final column was used to log future corrections needed for the internal database. A line number based on the order of the entries at the start of the work was also included on each row. This was summed at the bottom of the file to flag if any lines in the spreadsheet were accidentally deleted during the manual editing processes that were to follow. In the next step of the spreadsheet preparation, Excel was used to sort the file contents based on the chemical formula column. Once sorted automatically in this way, the spreadsheet had to be manually sorted again since the component containing the highest number of carbon atoms was not always the first listed in the formula. With the sorting completed on the component containing the highest number of carbon atoms, the spreadsheet was inspected line by line. If the sorted component formula was unique then clearly this structure could never be considered as a duplicate, a polymorph or part of a crystallisation family. In such cases, the solid form information (see below) was completed in the spreadsheet and the result was briefly viewed in CCDC Mercury to sanity check the entry before continuing to the next row. On the basis of the sanity check, eight structures in the database were considered not worthy of further consideration, either in terms of their non-pharmaceutical content or their incomplete or inappropriate modelling. These eight structures were removed from further analysis and ultimately will be removed from the GSK database.
Identifying duplicates and polymorphs
If the inspection of the spreadsheet showed two or more entries having the same formula with respect to all the components present, work was carried out to establish the relationship between the structures. A generally trivial step was checking whether the chemical connectivity was the same. Clearly if not then the structures represent different materials and the entries are unrelated. If the connectivities were the same, the next thing considered were any chiral centres present. To be duplicates or polymorphs, the materials would have to display the same stereochemistry: careful separation of diastereoisomers, single enantiomers and racemates (with due consideration to the space group) significantly reduced the number of related structures. If the entries were still judged to be the same entity (and now also considering the centrosymmetric structures as well) then a decision was taken on whether the entries represented duplicate structures or polymorphs. Eight exact duplicates (i.e. the same structure entered into the database twice) were easily identified by observing identical cell constants, R-values, molecule overlays and packing similarities. One of each pair of these structures was marked for removal from the database and not used for data mining. Separating redeterminations of the same structure and polymorphs was harder and was carried out by considering the following in CCDC Mercury: the space group and unit cell constants; a visual and a scripted (compare_loaded_crystals_by_pattern.py) comparison of the simulated XRPD patterns; the molecule overlay; and the packing similarity. If the decision was that the structures were duplicates (redeterminations effectively) they were kept in the database for completeness but only one of each was considered for the data mining described below. This led to the removal of a further 40 structures from the analysis. Generally speaking (unless there was a compelling reason otherwise) the structure with the lowest R-value was the one kept for further study. All polymorph structures were kept for analysis.Identifying crystallisation families
In a crystallisation family, the API has to be the same but the overall formula could differ in terms of the amount of solvent and/or water that is present. Checking that APIs were the same was done analogously to that just described for duplicates and polymorphs. Once all the structures containing a particular API were identified, the make-up of the family was assessed. If a family contained one or more API only structures then the family was considered part of the API set of Fig. 1A. If it also contained one or more hydrate structures, it was also in the Hydrates set of Fig. 1A and so on. Note that this broad classification does not distinguish between the amount of water in a hydrate or the amount (or number of types) of solvent in a solvate.Identifying hydrogen bonding interactions in polymorphs
The hydrogen bonding interactions in polymorphic structures were obtained using a python script (hbp_dims_calculation.py) where a mol2 file of the polymorphs was used as input. The interactions were classified as the same, different or no interactions according to the method described in ESI.†Assessing crystal morphology in the GSK database
The morphology distribution was obtained by the habit description of each GSK entry in the CIF file extracted from the in-house database using the crystal_habit.py script. The morphology of the small molecule crystal structures was categorised as 1D, 2D, and 3D according to the criteria described in the ESI.† The hydrogen bond dimensionality and hydrogen bonding interactions were computed from a mol2 file of the GSK structures using hbp_dims_calculation.py.Preparation of solid form distributions
Our work described here was done in comparison to the work done by Bryant et al. assessing the solid form space of all the CSD-DS including duplicate values, stereoisomers and families etc. (8632 structures; currently only 8614 are retrieved due to changes in the identifiers of the CSD_Drug_Subset_cleanup.gcd file). All the statistics for the CSD-DS are reported as published by Bryant et al.22 The GSK entries were manually categorised (using the definitions of free drugs, salts, co-crystals, hydrates and solvates outlined at the start of the Results and discussion section) in the above spreadsheet based on the chemical formula and where necessary, viewing the database entry using CCDC Mercury. The salts were subdivided in the spreadsheet depending on whether the drug-like component was cationic, anionic or neutral. In salts with two organic fragments, if it was unclear which represented the drug component, GSK registration information was taken into consideration. If such information was not available, the largest organic fragment was considered the drug-like component. The resulting solid form distribution was extracted from the master spreadsheet.Crystal descriptor space for the GSK database
The final list of 2417 structures was used for this analysis. Crystal descriptors were computed for the GSK database (without the duplicates and identical structures as already described) and were compared to the CSD-DS without duplicates (best_representative_full_subset_updated.gcd file). The R-factor to assess the quality of the two databases was obtained using the crystal_r_factor.py script. The older structures in the GSK database have been assigned an arbitrary R-value of 10 are not considered in the analysis. The space group and Z′ distributions of the GSK database and the CSD drug subset were obtained using the Space_group_GSK.py and crystal_z_prime.py scripts. Packing coefficients and temperature (K) were extracted for the GSK database (using crystal_packing_coefficient.py and entry_temperature.py) in order to compute the mean, median, and standard deviation values at different temperatures to investigate the temperature effect and its significance in relation to the natural variation according to the structure's packing. The packing coefficient distribution was also obtained for the CSD-DS. SQUEEZEd GSK structures were found using a CCDC Conquest search with the word SQUEEZE with the all text option. Void volume density (using default settings: grid spacing 0.7 and probe radius 1.2 Å) and hydrogen bonding pair distributions for the GSK database and CSD-DS were obtained using the crystal_void_analysis.py and hbp_dims_calculation.py scripts. Mann–Whitney statistical tests which included statistical analysis on each of the GSK and CSD-DS distributions were performed using the Mann_Whitney.py script. The chi2 statistical test was performed for the morphology data using the chi2_statistics.py script. Histogram and KDE plots were generated for the CSD-DS and GSK structures using the matplotlib 3.1.0 (ref. 45) and seaborn 0.9.0 packages.46Protocol for generating molecular descriptor distributions
For the molecular descriptor space analysis, we considered 2099 unique molecules in the GSK database and 778 molecules in the CSD-DS. SMILES were generated for all structures and are matched with the SMILES of APIs/Free drugs obtained with Helium for Excel 4.0.29.0 (an in-house plug-in powered by ChemAxon)47 based on the GSK Registry Number. The SMILES of all GSK compounds were canonicalised using Helium for Excel then MW, flexibility, logp distributions, and the sum of chiral atoms and bonds were generated using Helium for Excel. The extent of branching/walk counts for the heaviest components in each structure, were generated from a mol2 file using the walk_count.py script. The same protocol was followed for the CSD-DS molecules (best_representative_molecule_updated.gcd). KDE plots were generated for CSD-DS and GSK molecules using matplotlib and seaborn packages. A Mann–Whitney test, which included statistical analysis on each of the GSK and CSD-DS distributions, was performed to ensure that the differences in the medians of distributions were statistically significant.Hydrogen bond propensity analysis on GW825964X polymorphs
The hydrogen bond propensity (HBP) tool in CCDC Mercury was applied on two GW825964X crystal structures with the identifiers, X_2915A1 and X_2947A1. The model output can be found in the ESI.† As part of selecting suitable fragments, the default fragments were output from the HBP tool as CCDC ConQuest template (cqt) files. Then each fragment file (Table S13†) was used in a CCDC search for related structures containing relevant and similar fragments. The results of these searches were used to build training sets for each of the polymorphs from GSK data, CSD-DS data, and a combination of GSK and CSD-DS data (Table 2). Logit models prepared from each training set (Tables S9–S11†) were built in the HBP tool and, along with coordination scores generated for the functional groups present (Table S12†), were used to prepare a propensity prediction tables and charts (Tables S3–S8,†Fig. 7).Python scripts
The python scripts listed in this section can be found in the ESI.†Author contributions
The last author named is corresponding author, the first named author is primary author. All other authors are listed in alphabetical order. We describe contributions to the paper using the CRediT taxonomy in the same order as above, contributions in each category are equal unless stated otherwise: conceptualization and methodology, L. N. K., R. C. B. C., C. L. D.; software, L. N. K. (lead), J. C. C., C. M. E., A. A. M.; data curation, L. N. K., R. C. B. C., C. L. D.; validation, L. N. K., J. C. C., R. C. B. C., C. L. D.; investigation, L. N. K. (lead), R. C. B. C., C. M. E.; writing – original draft, L. N. K. (lead), R. C. B. C., G. S., C. L. D.; writing – review & editing, L. N. K., J. C. C., R. C. B. C., C. M. E., A. A. M., G. S., C. L. D. (lead); supervision, J. C. C., R. C. B. C. (lead), C. M. E., C. L. D. (lead); project administration, R. C. B. C., C. L. D.; funding acquisition, R. C. B. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Andrew Maloney (CCDC, Cambridge) for helpful discussions around the previous Bryant et al. paper and Drs. Robert Willacy and Susan Reutzel-Edens (CCDC, Cambridge) for their valuable feedback on this paper. Dr. Luca Russo is thanked for helpful discussions. The GSK Fellows are thanked for the post-doctoral funding (LNK) that allowed this work.Notes and references
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, 2nd edn, 2016 Search PubMed.
- A. Newman, Org. Process Res. Dev., 2013, 17, 457–471 CrossRef CAS.
- J. Bernstein, Polymorphism in Molecular Crystals, 2020 Search PubMed.
- A. Nangia, J. Indian Inst. Sci., 2007, 87, 133–147 CAS.
- S. Mirza, I. Miroshnyk, J. Heinamaki, O. Antikainen, J. Rantanen, P. Vuorela, H. Vuorela and J. Yliruusi, AAPS PharmSciTech, 2009, 10, 113–119 CrossRef CAS PubMed.
- ICH, Specifications: Test Procedures and Acceptable Criteria for New Drug Substances and New Drug Products: Chemical Substances Q6A, https://database.ich.org/sites/default/files/Q6A%20Guideline.pdf, (accessed 8th October 2020, 2020) Search PubMed.
- G. P. Stahly, Cryst. Growth Des., 2007, 7, 1001–1026 CrossRef.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS.
- I. B. Rietveld and R. Ceolin, J. Pharm. Sci., 2015, 104, 4117–4122 CrossRef CAS.
- K. R. Chaudhuri, Expert Opin. Drug Delivery, 2008, 5, 1169–1171 CrossRef CAS PubMed.
- A. Newman and R. Wenslow, AAPS Open, 2016, 2, 2 CrossRef.
- D.-K. Bucar, R. W. Lancaster and J. Bernstein, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef CAS PubMed.
- R. Censi and P. Di Martino, Molecules, 2015, 20, 18759–18776 CrossRef CAS PubMed.
- J. Chrisholm, E. Pidcock, J. van de Streek, L. Infantes, S. Motherwell and F. H. Allen, CrystEngComm, 2006, 8, 11–28 CAS.
- E. Pidcock, J. A. Chisholm, P. A. Wood, P. T. A. Galek, L. Fabian, O. Korb, A. J. Cruz-Cabeza, J. W. Liebeschuetz, C. R. Groom and F. H. Allen, Supramolecular Chemistry: From Molecules to Nanomaterials, 2012, pp. 2927–2946 Search PubMed.
- P. T. A. Galek, E. Pidcock, P. A. Wood, I. J. Bruno and C. R. Groom, CrystEngComm, 2012, 14, 2391–2403 RSC.
- N. Feeder, E. Pidcock, A. M. Reilly, G. Sadiq, C. L. Doherty, K. R. Back, P. Meenan and R. Docherty, J. Pharm. Pharmacol., 2015, 67, 857–868 CrossRef CAS PubMed.
- T. Grecu, H. Adams, C. A. Hunter, J. F. McCabe, A. Portell and R. Prohens, Cryst. Growth Des., 2014, 14, 1749–1755 CrossRef CAS.
- D. E. Braun, J. A. McMahon, L. H. Koztecki, S. L. Price and S. M. Reutzel-Edens, Cryst. Growth Des., 2014, 14, 2056–2072 CrossRef CAS.
- S. N. Black, H. P. Wheatcroft, R. Roberts, M. F. Jones, I. McFarlane and A. Pettersen, J. Pharm. Sci., 2020, 109, 1509–1518 CrossRef CAS PubMed.
- P. T. A. Galek, E. Pidcock, P. A. Wood, N. Feeder and F. H. Allen, Computational Approaches in Pharmaceutical Solid State Chemistry, 2016, pp. 15–35 Search PubMed.
- M. J. Bryant, S. N. Black, H. Blade, R. Docherty, A. G. P. Maloney and S. C. Taylor, J. Pharm. Sci., 2019, 108, 1655–1662 CrossRef CAS PubMed.
- C. J. Tilbury, J. Chen, A. Mattei, S. Chen and A. Y. Sheikh, Cryst. Growth Des., 2018, 18, 57–67 CrossRef CAS.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. Roy Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 4290–4291 CrossRef CAS.
- ICH, Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, https://www.fda.gov/media/71518/download, (accessed 8th June 2021) Search PubMed.
- P. T. A. Galek, F. H. Allen, L. Fabian and N. Feeder, CrystEngComm, 2009, 11, 2634–2639 RSC.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- P. A. Wood, T. S. G. Olsson, J. C. Cole, S. J. Cottrell, N. Feeder, P. T. A. Galek, C. R. Groom and E. Pidcock, CrystEngComm, 2013, 15, 65–72 RSC.
- R. Montis, R. J. Davey, S. E. Wright, A. J. Cruz-Cabeza and G. R. Woollam, Angew. Chem., Int. Ed., 2020, 59(46), 20357–20360 CrossRef CAS PubMed.
- A. J. C. Wilson, Acta Crystallogr., Sect. A: Found. Crystallogr., 1989, 45, 210 CrossRef.
- G. Mueller and M. Lutz, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 871–880 CrossRef CAS.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultants Bur., 1964 Search PubMed.
- A. I. Kitaigorodskii, Physical Chemistry, Molecular Crystals and Molecules, Academic, 1973, vol. 29 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- O. Ivanciuc and A. T. Balaban, Croat. Chem. Acta, 1996, 69, 63–74 CAS.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- J. G. P. Wicker and R. I. Cooper, CrystEngComm, 2015, 17, 1927–1934 RSC.
- B. C. Hancock, J. Pharm. Sci., 2017, 106, 28–30 CrossRef CAS PubMed.
- F. Pereira, CrystEngComm, 2020, 22, 2817–2826 RSC.
- P. T. A. Galek, L. Fabian, W. D. S. Motherwell, F. H. Allen and N. Feeder, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 768–782 CrossRef CAS PubMed.
- M. D. Ticehurst and I. Marziano, J. Pharm. Pharmacol., 2015, 67, 782–802 CrossRef CAS PubMed.
- R. Docherty, G. O'Connor, R. Y. Penchev, J. Pickering and V. Ramachandran, From Molecules to Crystals to Functional Form: Science of Scale, Engineering Crystallography: From Molecule to Crystal to Functional Form, ed. K. Roberts, R. Docherty and R. Tamura, Springer, Dortrecht, 2017 Search PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- M. L. Waskom, J. Open Source Softw., 2021, 6(60), 3021 CrossRef.
- ChemAxon, https://chemaxon.com/, (accessed 10th June 2021, 2021) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Hydrogen bond propensity model tables, protocols for hydrogen bonding and morphology analyses, python scripts and CSD-DS gcd files. See DOI: 10.1039/d1ce00665g |
| This journal is © The Royal Society of Chemistry 2021 |