
N-Heterocyclic carbene complexes enabling the α-arylation of carbonyl compounds†
Sylwia
Ostrowska
 a,
Thomas
Scattolin
b and
Steven P.
Nolan
*b
a,
Thomas
Scattolin
b and
Steven P.
Nolan
*b
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
bDepartment of Chemistry and Center for Sustainable Chemistry, Ghent University, Krijgslaan 281 (S-3), 9000, Ghent, Belgium. E-mail: steven.nolan@ugent.be
First published on 26th March 2021
Abstract
The considerable importance of α-arylated carbonyl compounds, which are widely used as final products or as key intermediates in the pharmaceutical industry, has prompted numerous research groups to develop efficient synthetic strategies for their preparation in recent decades. In this context, the α-arylation of carbonyl compounds catalyzed by transition-metal complexes have been particularly helpful in constructing this motif. As illustrated in this contribution, tremendous advances have taken place using palladium– and nickel–NHC (NHC = N-heterocyclic carbene) complexes as pre-catalysts for the arylation of a wide range of ketones, aldehydes, esters and amides with electron-rich, electron-neutral, electron-poor, and sterically hindered aryl halides or pseudo-halides. Despite significant progress, especially in asymmetric α-arylations promoted by chiral NHC ligands, there are numerous challenges which have and continue to encourage further studies on this topic. Some of these are presented in this report.
 Sylwia Ostrowska | Sylwia Ostrowska conducted her MS degree in 2011 at Faculty of Chemistry, Lodz University of Technology under the supervision of Prof. Krzysztof Strzelec. She has defended her PhD thesis entitled “New N-heterocyclic palladium complexes-synthesis, structure, reactivity and catalytic activity” at Adam Mickiewicz University in Poznań under supervision Prof. Cezary Pietraszuk. In the years 2019–2020 she completed a postdoctoral fellowship at the Institute of Organic Chemistry of the Polish Academy of Sciences, Warsaw. Laboratory of Organic Synthesis in the group of Prof. Rafał Loska working in the project “Transition metal catalyzed C–H activation of aldonitrons.” Her research is primarily focused on the synthesis, reactivity, and separation of Late Transition metal N-heterocyclic carbenes homogenous catalysts. |
 Thomas Scattolin | Thomas Scattolin was born in Padua (Italy) in 1990 and studied chemistry at the Ca’ Foscari University of Venice (BSc 2012 and MSc 2014, summa cum laude). He completed his PhD in 2019 under the supervision of Prof. Fabiano Visentin in the same university (inter-university programme with University of Trieste). In 2018 he was a visiting scientist in the laboratories of Prof. Antonio Togni at the ETH in Zurich, Switzerland. In 2020, he joined the laboratory of Prof. Steven P. Nolan at the Ghent University. His research is primarily focused on the synthesis and reactivity of late transition metal complexes with applications in homogeneous catalysis and medicinal chemistry. |
 Steven P. Nolan | Steven P. Nolan received his BSc in Chemistry from the University of West Florida and his PhD from the University of Miami where he worked under the supervision of Professor Carl D. Hoff. After a postdoctoral stay with Professor Tobin J. Marks at Northwestern University, he joined the Department of Chemistry of the University of New Orleans in 1990. In 2006 he joined the Institute of Chemical Research of Catalonia (ICIQ) as Group leader and ICREA Research Professor. In early 2009, he joined the School of Chemistry at the University of St Andrews and in 2017 joined the Department of Chemistry of Ghent University as Senior Full Professor. Professor Nolan's research interest revolve around the design and synthesis of catalytic complexes enabling organic transformations. |
1. Introduction
One of the challenges in organic synthesis is the formation of a C–C bond between an aryl group and the α-carbon of carbonyl compounds. Aryl carbonyl derivatives are present in many natural products such as lucuminic acid, welwistatin and vancomycin (Fig. 1), as well as crucial building blocks in the pharmaceutical industry. In this respect, the well-known and important class of nonsteroidal anti-inflammatory drugs (i.e. Naproxen, Flurbiprofen, Diclofenac and Indomethacin) are particularly worthy of mention (Fig. 1).1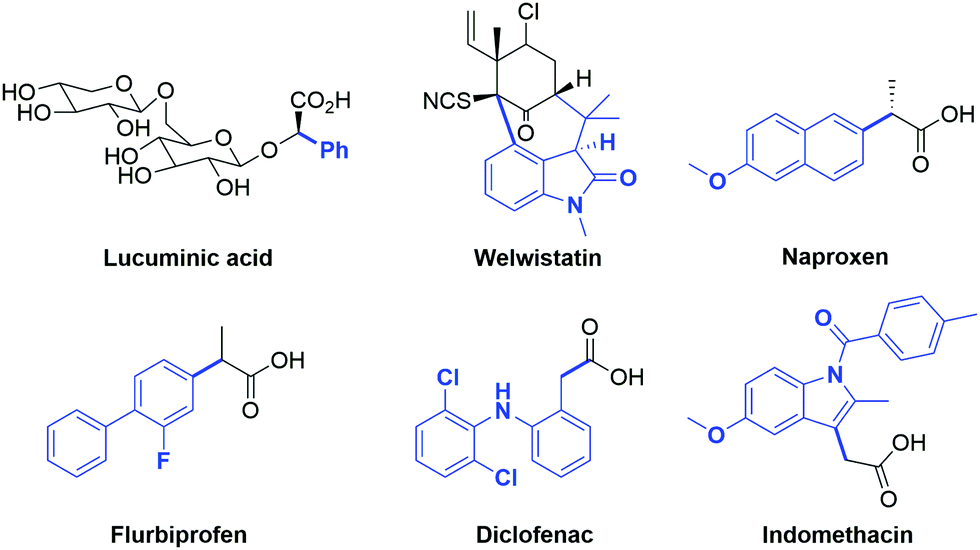 | ||
| Fig. 1 Examples of biologically active molecules bearing α-arylated carbonyl moieties. | ||
Until the early 2000s, the arylation process of carbonyl substrates involved classical organic reactions such as nucleophilic aromatic substitution, which is suitable only when highly activated aryl halides are employed,2 or the use of stoichiometric amounts of metal complexes with preformed enolates.3 However, these transformations suffer from many practical drawbacks such as functional group incompatibility, air and moisture sensitivity, and toxicity of reagents. A much more appealing strategy is represented by the use of transition metal complexes able to catalyze this transformation.3d,e In this context, the use of in situ generated versus well-defined metal catalysts, the nature and stoichiometry of the reagents (aryl halide, carbonyl compound and base), the solvent and the reaction conditions (i.e. temperature and time) have proven to be decisive in the outcome of arylation reactions. Moreover, as in all cross-coupling reactions, the choice of the ancillary ligands present on the metal catalyst is fundamental as their steric and electronic features can heavily influence the conversion of substrates into the desired products. For example, in palladium-catalyzed α-arylations the presence of electron-rich ligands tends to stabilize the palladium(II) intermediate, thereby facilitating the oxidative addition of the aryl halide.4
The last decade has witnessed a tremendous progress in catalyst design by the fine-tuning of the supporting ligands used in many catalytic transformations including a wide range of metal-catalyzed α-arylation of carbonyl compounds. Although catalysts based on simple transition metal precursors with the addition of phosphine ligands are often used in α-arylation reactions,5 in recent times also N-heterocyclic carbene ligands (NHC) have been applied successfully to this end. This is due to the unique properties of these ligands, such as: (i) the ability of NHCs to enable difficult oxidative additions through strong σ-donation to various metal centers,6 (ii) the weak π-acceptor character of most NHC ligands enable this high electron donation to metal centers,7 (iii) the possibility to modulate the steric environment around the metal by N-wingtip substitution and backbone modifications are attractive design/control elements,6,8 (iv) the high stability of metal–NHC derivatives both in organic solvents and aqueous media in view of strong M–NHC bonds.6,8
In this contribution, we propose an overview of the most important α-arylation reactions of carbonyl compounds promoted by transition metal–NHC complexes, covering the literature since 2002 (the year of the first report on metal–NHC catalyzed α-arylation) through 2020. The reactions discussed are limited to the use of aryl halides or pseudo-halides with enolates generated in situ from various carbonyl compounds such as ketones, amides, esters and aldehydes.
2. Metal–NHC catalyzed α-arylation of ketones: general aspects
The synthesis of α-aryl substituted ketones has been the subject of intense research over the past 20 years or so. In the early years of research one of the most common methods involved the reaction of an enolate with a benzyne derivative affording an Ar–C bond at the α-position of a carbonyl compound.9 Several metal and main group aryl reagents have been reported for the synthesis of α-arylated ketones. Among them were included aryl-bismuth,10 aryl-cadmium,11 aryl-copper,12 aryl-lead,13 aryl-boron,14 (π-halogeno-benzene) chromium tricarbonyls,15 diaryl iodonium salts,16 aryl-magnesium halides,17 and arylazo tert-butyl sulfides.18 This protocol is limited by the cost and time needed to prepare stoichiometric amounts of the arylating reagents.Later, the success of phosphine ligands in the metal-catalyzed α-arylation of ketones has partially solved some problems associated with the transition metal and main group arylating reagents. However, issues such as high costs, nontrivial synthesis, and possible formation of unwanted by-products in the reaction encouraged researchers to evaluate other category of ligands such as the N-heterocyclic carbenes (NHCs).5,19 NHCs are the best tertiary phosphine surrogates available (and are much more, indeed) and their use as ligands in organometallic complexes has been applied in benchmark reactions as well as for the synthesis of promising anticancer agents.20 Their strong σ-electron-donating ability enables the formation of stable metal species and facilitates oxidative addition even of challenging substrates, while their important steric occupation is responsible for rapid reductive elimination.6
2.1 Palladium–NHC catalyzed α-arylation of ketones
In 2002, Nolan and co-workers reported on the use of well-defined palladium–NHC complexes as homogeneous catalysts for the α-arylation of ketones.21 Initially, they investigated a panel of palladium complexes (Fig. 2) in the arylation of propiophenone with chlorobenzene.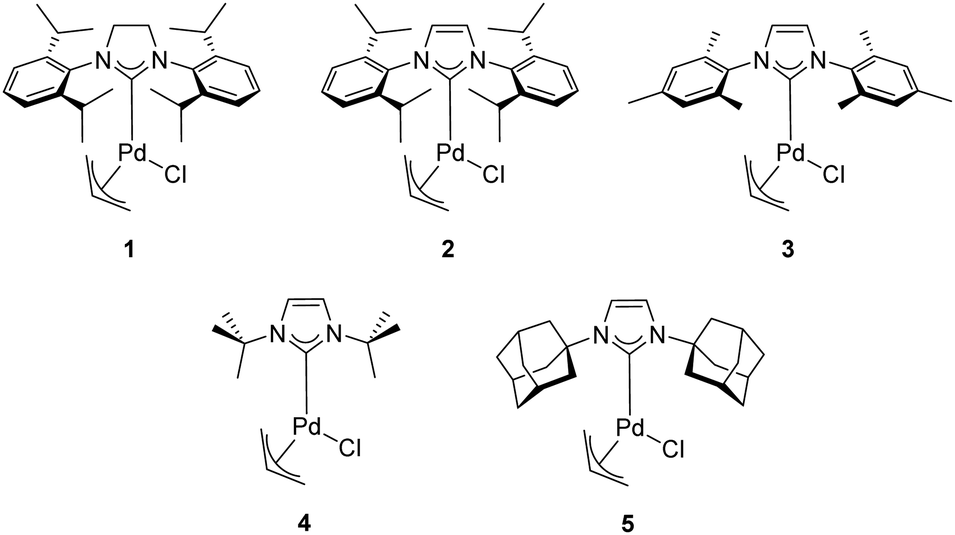 | ||
| Fig. 2 Pd-Allyl complexes used by Nolan and co-workers in the α-arylation of ketones. | ||
In particular, the air stable palladium-allyl complex 1 (allyl[1,3-bis(2,6-diisopropylphenyl)-2-imidazolidinylidene]chloro palladium(II), [Pd(SIPr)(η3-allyl)Cl]) afforded the highest yield of α-arylation product, using NaOtBu as the base in THF at 50–70 °C, with a short reaction time and low catalyst loading (1 mol%) (Scheme 1).
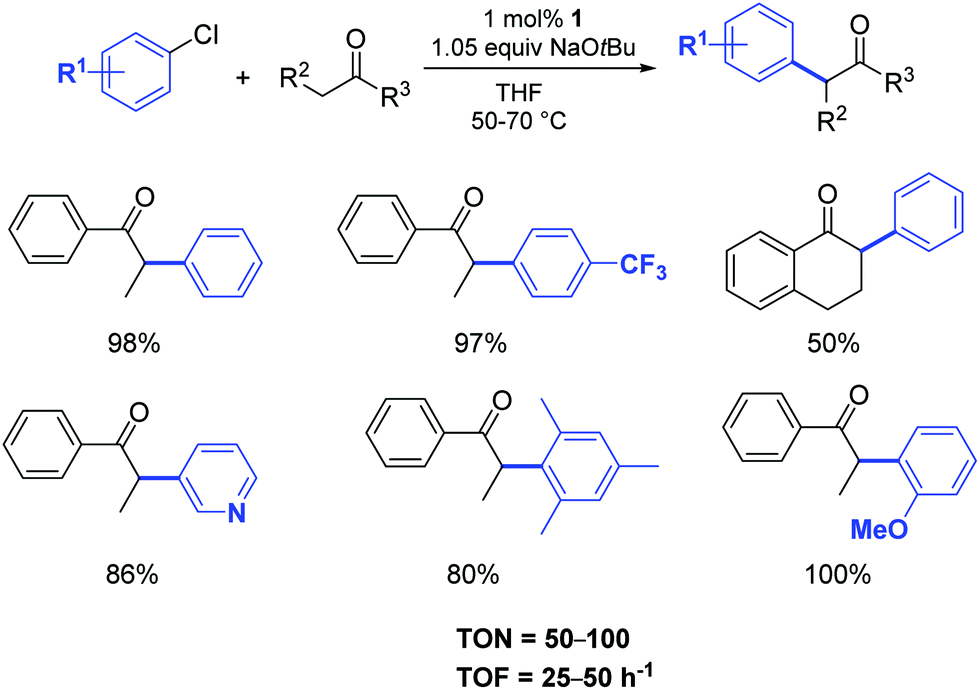 | ||
| Scheme 1 α-Arylation of ketones using aryl chlorides according to Nolan et al.21 | ||
The same pre-catalyst proved to be equally efficient when aryl triflates were used as starting materials (Scheme 2).21a
 | ||
| Scheme 2 α-Arylation of ketones using aryl triflates according to Nolan.21 | ||
Nolan and co-workers proposed that the activation step of these catalysts involves the inter- or intramolecular nucleophilic attack of a base such as NaOtBu or KOtBu on the allyl moiety (Scheme 3). Additionally, they showcased the importance of the steric hindrance of the NHC ligand on the outcome of the reaction. For example, the difference in catalytic activity, at 50 °C in 1 h, between [Pd(SIPr)(η3-allyl)Cl] (1) and its unsaturated and sterically less demanding congener [Pd(IPr)(η3-allyl)Cl] (2) is noteworthy (yield: 97% vs. 51%, respectively).
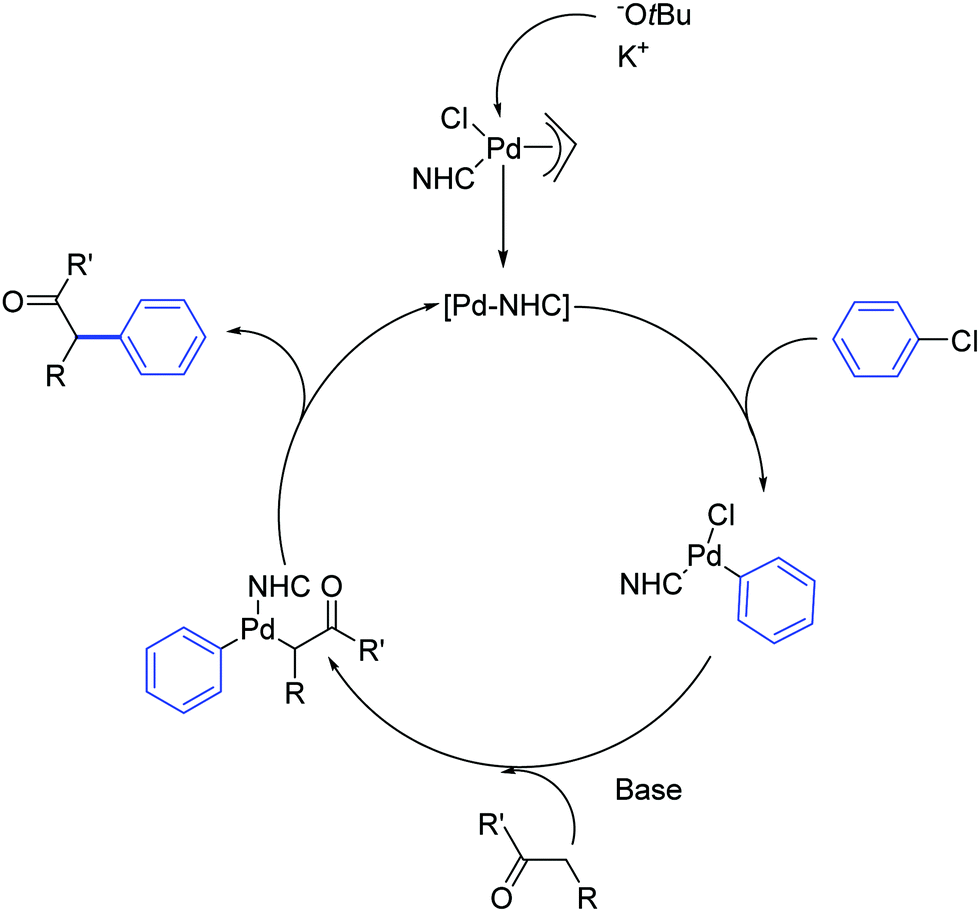 | ||
| Scheme 3 Proposed catalytic cycle for the α-arylation of ketones promoted by Pd-allyl complexes. | ||
One year later, Nolan and co-workers investigated the catalytic activity of a second family of palladium–NHC complexes that combined the beneficial properties of the carbene ligand with the robustness of the palladacyclic structure.22 In particular, the palladacyclic species 6 showed a similar catalytic activity to that of the allyl complex 1 (Scheme 4). In this case, the catalyst activation could occur by an attack of the alkoxide on the palladium center, thereby generating a palladium alkoxide species. Subsequent reductive elimination of this aryl palladium alkoxide intermediate would generate the active catalytic species. Alternatively, activation could occur through the formation of a palladacycle–NHC-aryl complex, which would undergo reductive elimination to form an arylated amino-aryl fragment and a [Pd(NHC)] species. The exact route employed is still unclear.
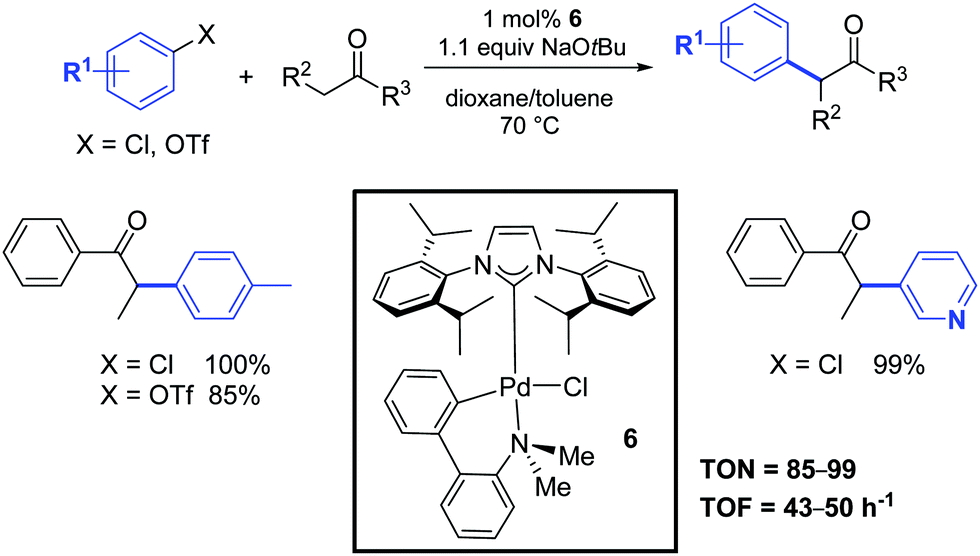 | ||
| Scheme 4 α-Arylation of ketones according to Nolan and co-workers.22 | ||
The authors demonstrated that it is possible to reduce the amount of catalyst 6 from 1 to 0.25 mol% by operating under microwave irradiation (Scheme 5).23 Furthermore, they highlighted that a variety of functional groups was tolerated on the aryl moiety (with the exception of aldehydes and nitriles) and that substituents in the α-position to the carbonyl group had a detrimental effect on the arylation reaction. In this respect, unsymmetrically substituted ketones were preferentially arylated at the least sterically hindered α-carbon atom position.
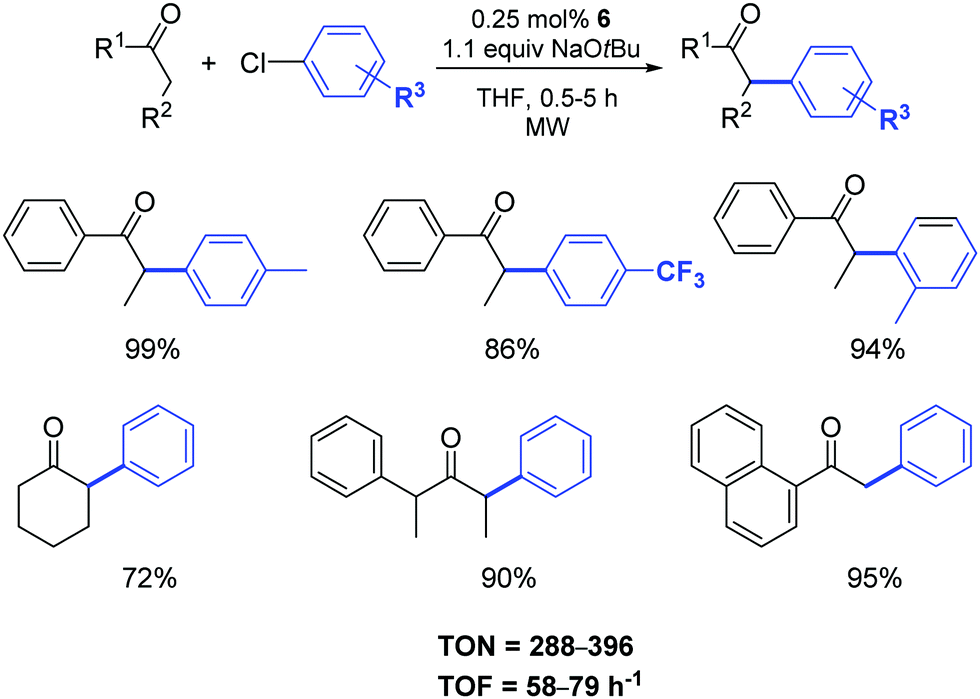 | ||
| Scheme 5 α-Arylation of ketones under microwave irradiation.23 | ||
When the temperature was raised to 130 °C, the reactions reached completion within 2 min with no decrease in the yields. As expected, aryl bromides were suitable substrates for reactions under these conditions and a variety of aryl and alkyl ketones can be easily arylated using unactivated and sterically demanding aryl bromides in very good yields and, in general, shorter reaction times compared to their chloride analogues.
A few years later, Singh and Nolan described a convenient protocol for the α-arylation of ketones catalyzed by the palladium–NHC acetate complex 7 (Scheme 6).24 A wide selection of functionalized halides and ketones was investigated in order to better understand the influence of electronic and steric characteristics on the reaction.
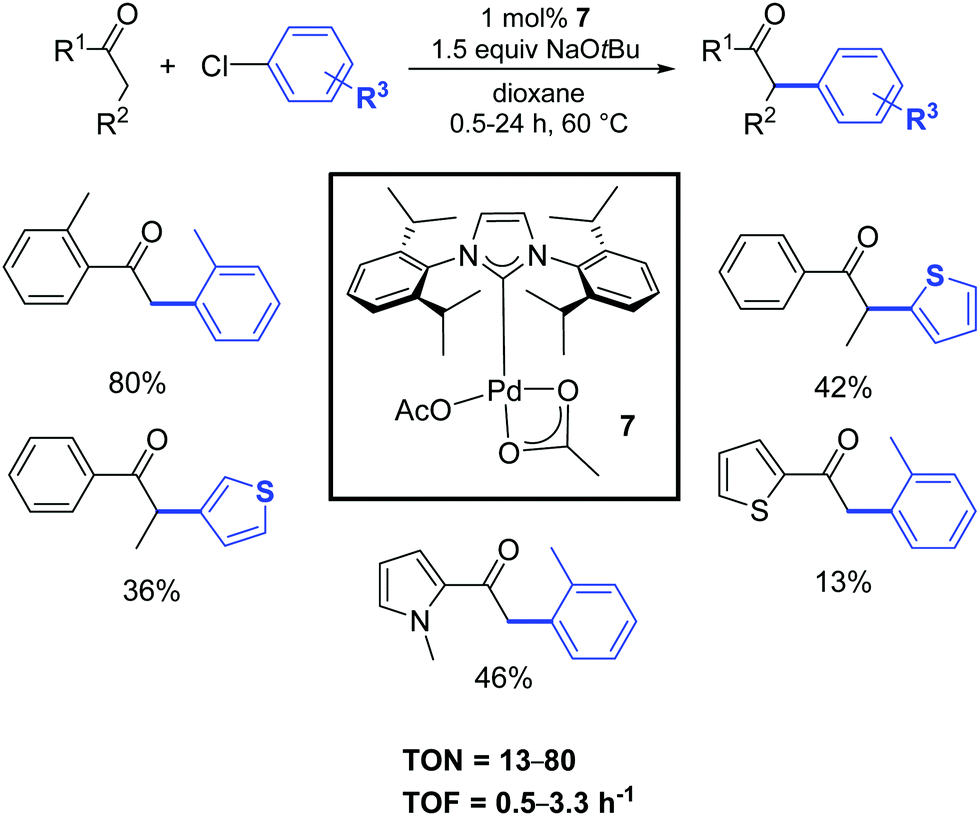 | ||
| Scheme 6 α-Arylation of various substrates including heterocyclic ketones and chlorides, according to Nolan and coworkers.24 | ||
In 2005, Nolan and co-workers synthesized, with a simple and easily scalable protocol, the air- and moisture-stable complex [Pd(IPr)(acac)Cl] (8), that displayed high catalytic activity for the α-arylation of ketones (Scheme 7).25 All reactions proceeded under extremely mild conditions and short reaction times.25,26
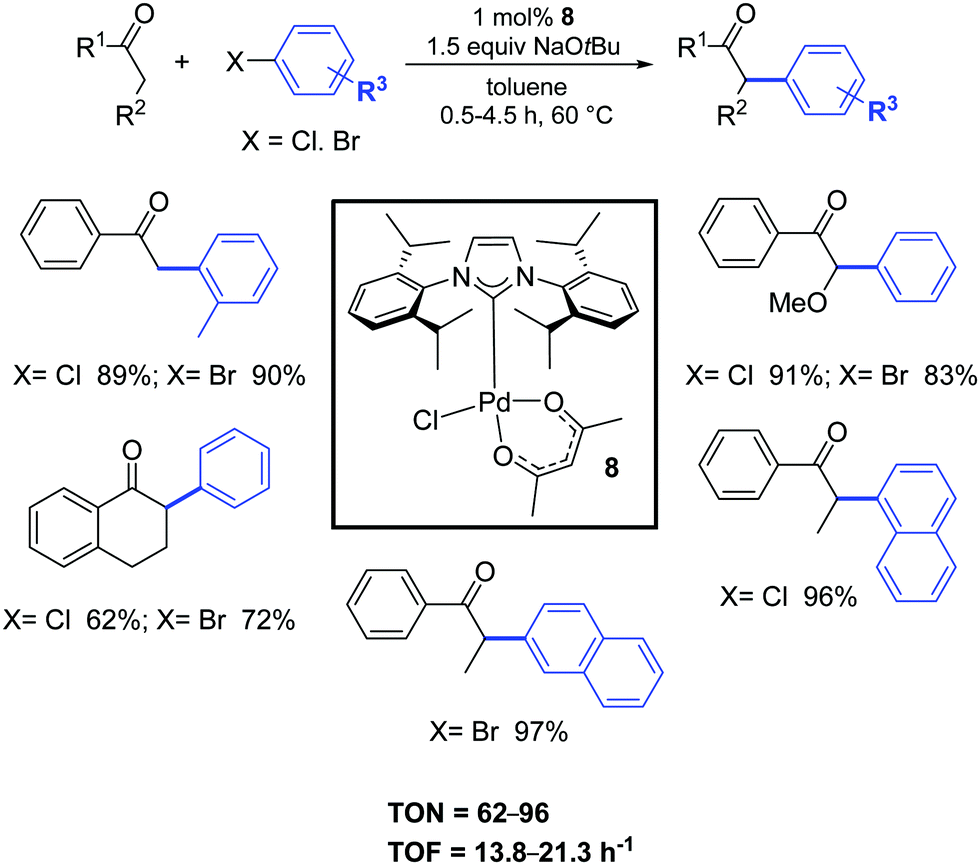 | ||
| Scheme 7 α-Ketones arylation reactions of aryl chlorides and bromides.26b | ||
Nine years later, the use of the ligand IHept instead of IPr, allowed to significantly decrease the catalyst loading (100–500 ppm) for a wide range of ketones.26c
In 2006, Matsubara and co-workers carried out a polycondensation of haloaryl ketones catalyzed by palladium complexes bearing NHC ligands.27 They demonstrated that [Pd(IPr)(OAc)2] (7) and other palladium–NHC derivatives are capable of efficiently promoting this type of transformation (Scheme 8).
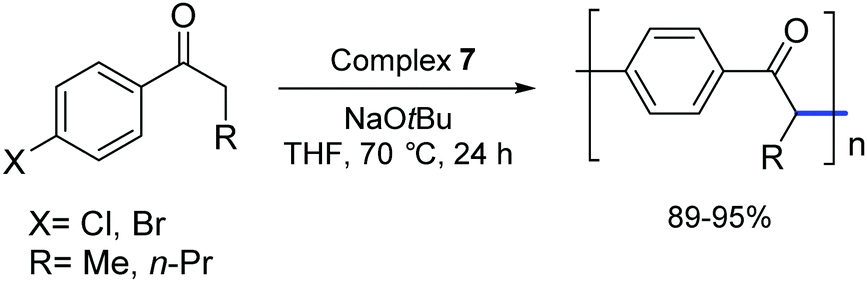 | ||
| Scheme 8 Polymerization of 4-haloaryl alkyl ketones catalyzed by 7 as described by Matsubara and co-workers.27 | ||
Five years later, Wu and colleagues described a cyclopalladated ferrocenyl-imine complex (9) that exhibited high catalytic activity for the α-arylation of ketones with aryl halides using a very low catalyst loading (0.01 mol%) as illustrated in Scheme 9.28 This protocol was successfully applied to various ketones and a broad range of aryl chlorides and bromides. The corresponding products were obtained in moderate to excellent yields.
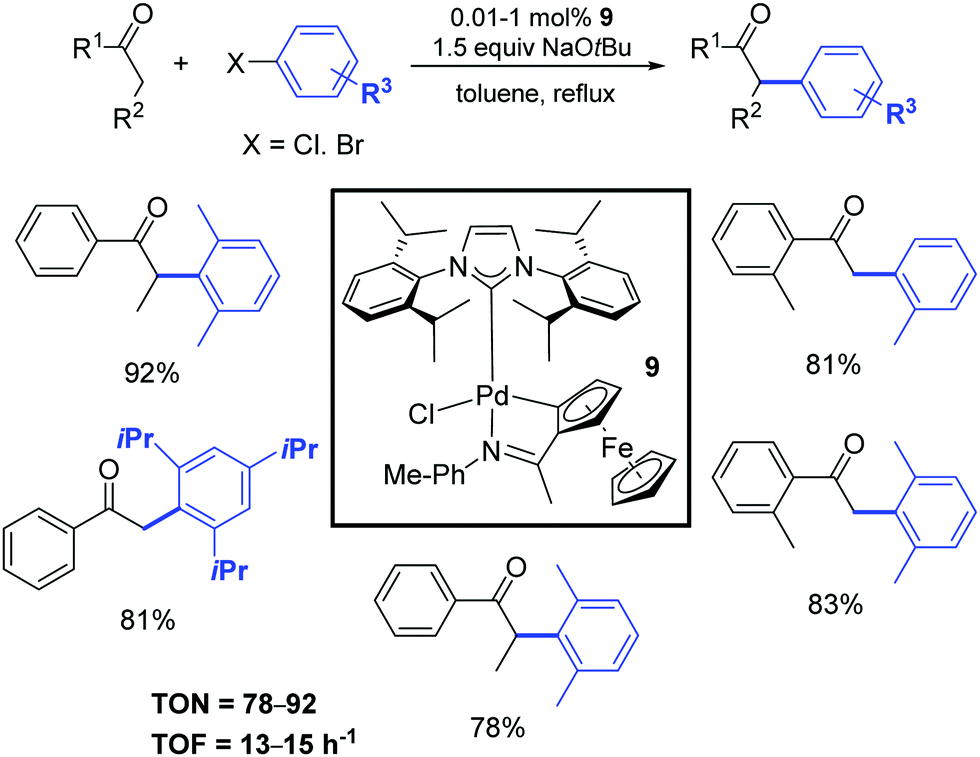 | ||
| Scheme 9 α-Arylation of ketones with aryl halides, according to Wu and colleagues.28 | ||
In the same year, Shi et al. investigated the use of the easily prepared and air-stable PEPPSI (pyridine-enhanced pre-catalyst preparation stabilization and initiation) palladium complexes 10–13 as pre-catalysts in the α-ketone arylation with unactivated aryl chlorides under mild conditions (Scheme 10).29 Among the complexes tested, and maybe unsurprisingly, [Pd(SIPr)(Py)Cl2] (12) displayed high activity for a variety of activated, unactivated and sterically hindered aryl halides. With this approach, both aryl and alkyl ketones can be arylated. Interestingly, the α-arylation of some alkyl ketones can even be carried out at room temperature.30
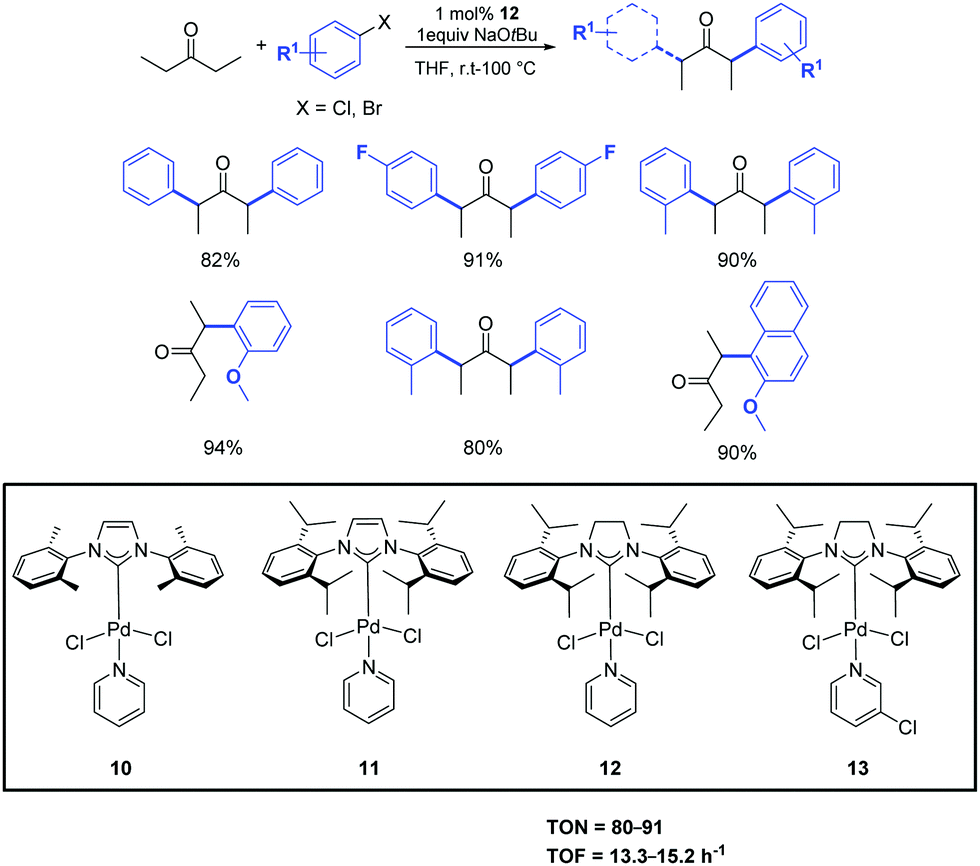 | ||
| Scheme 10 α-Arylation of pentanone with aryl halide catalyzed by complex 12.30 | ||
The mechanism proposed by the authors for this Pd-catalyzed α-arylation process is shown in Scheme 11. The catalytically active Pd(0) species is generated from the Pd(II) pre-catalyst by transmetalation and reductive elimination of the organometallic reaction partner.30
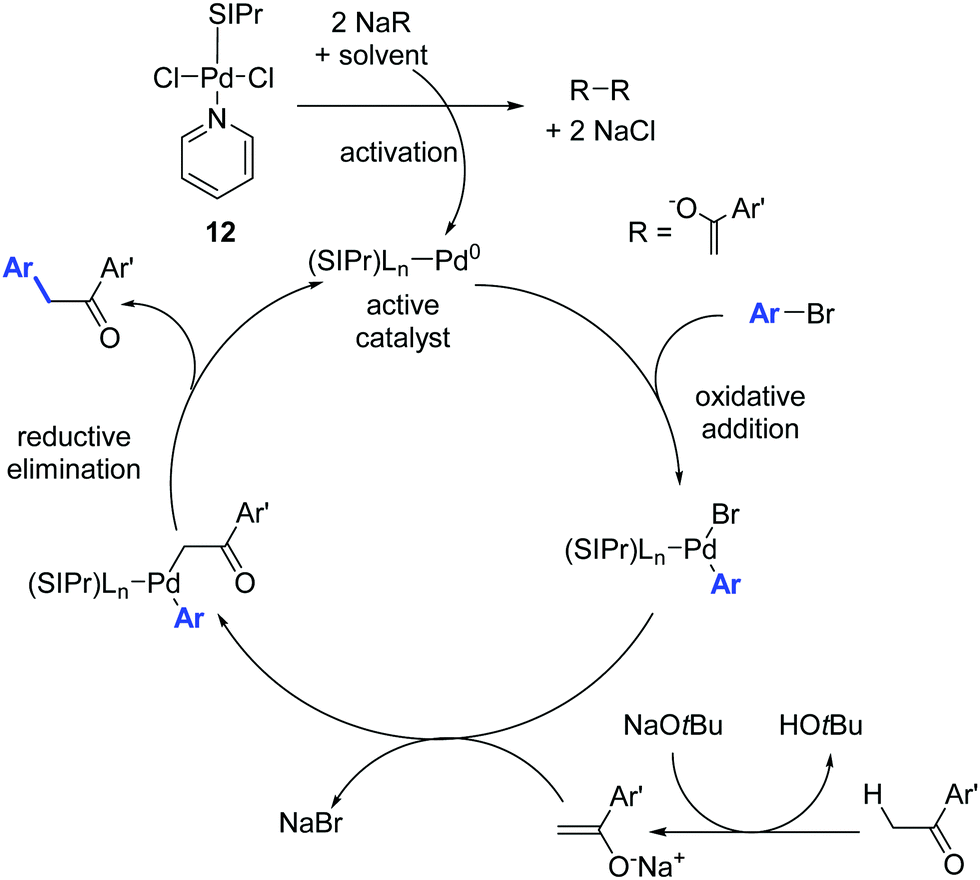 | ||
| Scheme 11 Proposed mechanism for the Pd catalyzed α-arylation reaction described by Shi and co-workers.30 | ||
Shao and coworkers described the application of [Pd(IPr)Cl2(Me-imidazole)] (14) in the α-arylation reaction between ketones and aryl chlorides under mild conditions (Scheme 12).31 No mention of the activity or synthesis is mentioned in this contribution of the SIPr analogue.
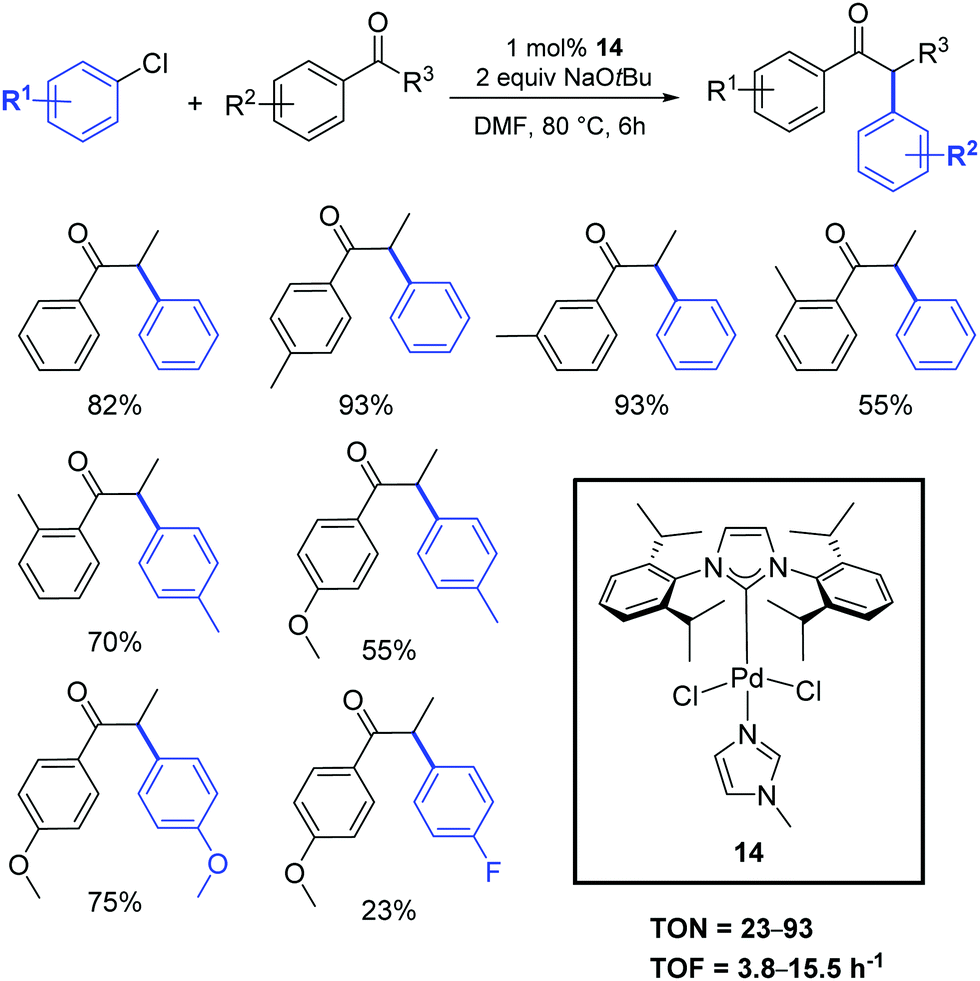 | ||
| Scheme 12 α-Arylation of ketones with aryl chlorides described by Shao et al.31 | ||
Complex 14 was also successfully used in the two-component, three-molecule reaction between 2,3-dihydroinden-1-ones and aryl chlorides (Scheme 13).32 It appears that three steps: a Pd-catalyzed α-arylation, subsequent aldol condensation and final isomerization are involved in the reactions. The authors examined a broad scope of substrates (with electron-rich, electron-poor and sterically hindered substituents), achieving the corresponding products in acceptable to high yields.
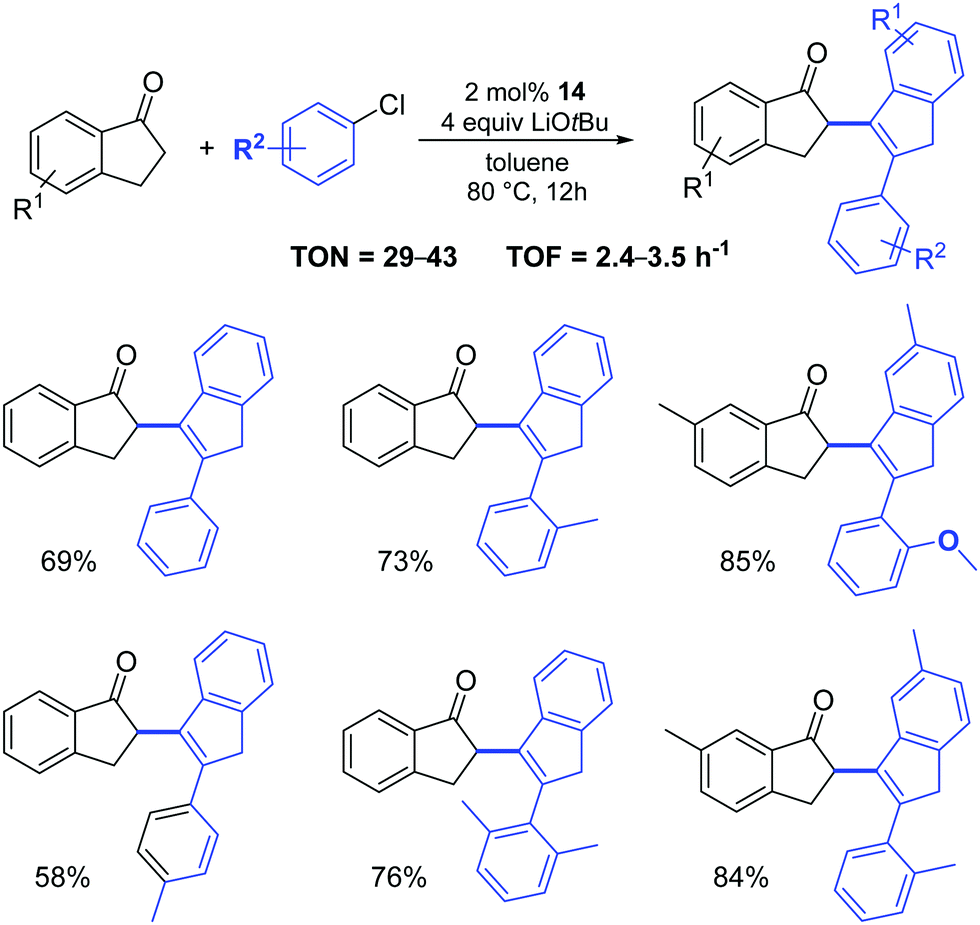 | ||
| Scheme 13 Reactions of 2,3-dihydroinden-1-ones with aryl chlorides.32 | ||
In 2012, Glorius and co-workers explored the effects of a quinine additive in the asymmetric α-arylation of 2-methyl-1-tetralone catalyzed by Pd(dba)2 or [Pd(SIPr)(η3-allyl)Cl] (1).33 They showed that without any addition of quinine, the reaction proceeded quickly within 1.5 h (yield = 48%). In the presence of quinine full conversion was observed after 4 h. The slower reaction rate could be caused by the binding of quinine to Pd, therefore blocking or hindering access to active sites around the metal complex. Interestingly, the yield increased to 92% and an ee of 80% was observed, which clearly showed that the addition of quinine forms a more chemo- and enantio-selective catalytically active species, one where quinine acts as a labile chiral auxiliary. The lower ee obtained with complex 1 compared to Pd(dba)2 could be explained by a faster racemic background reaction of small amounts of the free NHC–Pd(0) species, as the reaction rate without quinine is much faster for the NHC–Pd complex than for Pd(dba)2 (Table 1). Obviously the number and labile nature of ligands around the Pd(0) complex play a key role here.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Quinine [mol%] | Aryl halide | Time [h] | Yieldb [%] | eec [%] |
a Ketone (0.3 mmol), aryl halide (0.6 mmol), cat. (1.4 mol%), quinine (7.5 mol%), NaOtBu (2.0 equiv.), toluene (2.0 mL), 100 °C, reaction time: aryl iodides 3–8 h, aryl bromides 16–30 h.
b Isolated yields.
c The ee value was measured using a chiral AD-H column (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 hexane:i-PrOH).
d NMR yield (CH2Br2 as internal standard). :2 hexane:i-PrOH).
d NMR yield (CH2Br2 as internal standard).
|
||||||
| 1 | Pd(dba)2 | 0 | PhI | 24 | 44 | 0 |
| 2 | Pd(dba)2 | 7.5 | PhI | 3 | 85 | 92 |
| 3 | 1 | 0 | PhI | 1.5 | 48 | 0 |
| 4 | 1 | 7.5 | PhI | 4 | 92 | 80 |
| 5 | 1 | 0 | PhCl | 72 | 33d | 0 |
| 6 | 1 | 7.5 | PhCl | 72 | 28d | 5 |
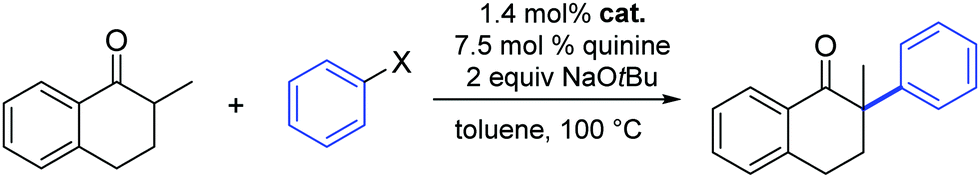
Finally, Glorius also investigated the use of aryl chlorides as substrates. Unfortunately, the reactivity with and without quinine was quite low and no full conversion was observed even after reaction times of three days and with exhibiting minimal product enantioselectivity (Table 1).33
In 2014, a series of PEPPSI-type complexes bearing imidazolylidene, benzimidazolylidene or pyrene–imidazolylidene ligands (15–17) were prepared and studied by Peris and co-workers (Fig. 3).34
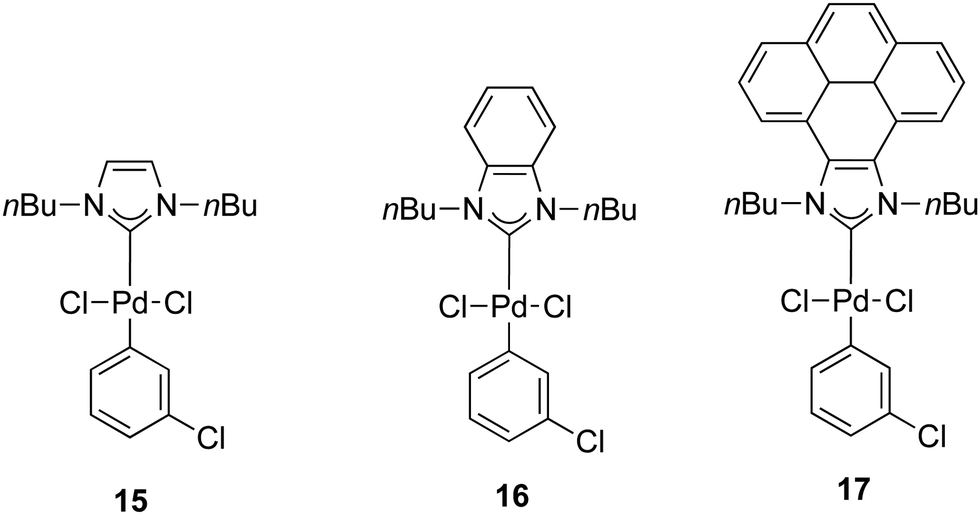 | ||
| Fig. 3 N-Heterocyclic palladium complexes described by Peris and co-workers.34 | ||
The α-arylation of different ketones and aryl halides was carried out at 80 °C in 1,4-dioxane with a catalyst loading of 1 mol%. The benzimidazolylidene-containing pre-catalyst (16) offered the best activity and the pyrene derivative 17 proved to be more active than complex 15.34 Moreover, they showed that the addition of pyrene to the catalytic α-arylation of ketones results in a decrease in the activity of the reactions catalyzed by the pyrene–imidazolylidene palladium complex 16, whereas the other two pre-catalysts did not modify their activity in the presence of this π-stacking additive. Interestingly, when 16 was used for the α-arylation of aliphatic ketones, the addition of pyrene did not modify the activity of the catalyst (entries 15 and 16, Table 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | R1 | R2 | R3 | Additivea | Yieldb [%] |
| a Reactions were carried out with aryl halide (0.6 mmol), ketone (0.5 mmol), NaOtBu (0.7 mmol), and cat. (1 mol%) in 1,4-dioxane (2 mL) at 80 °C for 6 h. b Yields obtained by GC analysis using anisole as internal reference. c 0.05 mmol of pyrene was added. | ||||||
| 1 | 15 | H | C6H5 | CH3 | — | 7 |
| 2 | 15 | H | C6H5 | CH3 | Pyrenec | 9 |
| 3 | 15 | CH3 | C6H5 | CH3 | — | 23 |
| 4 | 15 | CH3 | C6H5 | CH3 | Pyrenec | 20 |
| 5 | 15 | H | C(CH3)3 | H | — | 50 |
| 6 | 15 | H | C(CH3)3 | H | Pyrenec | 46 |
| 7 | 16 | H | C6H5 | CH3 | — | 36 |
| 8 | 16 | H | C6H5 | CH3 | Pyrenec | 31 |
| 9 | 16 | CH3 | C6H5 | CH3 | — | 79 |
| 10 | 16 | CH3 | C6H5 | CH3 | Pyrenec | 81 |
| 11 | 17 | H | C6H5 | CH3 | — | 31 |
| 12 | 17 | H | C6H5 | CH3 | Pyrenec | 16 |
| 13 | 17 | CH3 | C6H5 | CH3 | — | 36 |
| 14 | 17 | CH3 | C6H5 | CH3 | Pyrenec | 27 |
| 15 | 17 | H | C(CH3)3 | H | — | 64 |
| 16 | 17 | H | C(CH3)3 | H | Pyrenec | 66 |
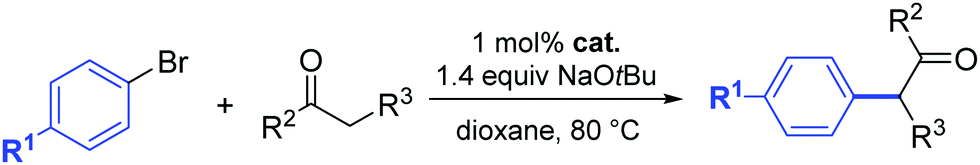
In 2015, Shao and colleagues reused [Pd(IPr)Cl2(Me-imidazole)] (14) as an efficient pre-catalyst in the reaction between tetralone and aryl chlorides, affording the desired α-arylated tetralones in good to high yields under the conditions illustrated in Scheme 14.35
 | ||
| Scheme 14 α-Arylation of tetralone as described by Shao and co-workers.35 | ||
In 2018, an interesting palladacyclic complex (18) was obtained using a one-pot procedure and under mild conditions by Lu and co-workers (Scheme 15).36 Detailed studies demonstrated the high catalytic activity of 18 toward α-arylation of ketones using aryl chlorides (Scheme 15).
 | ||
| Scheme 15 Complex (18) catalyzed α-arylation of ketones as described by Lu et al. | ||
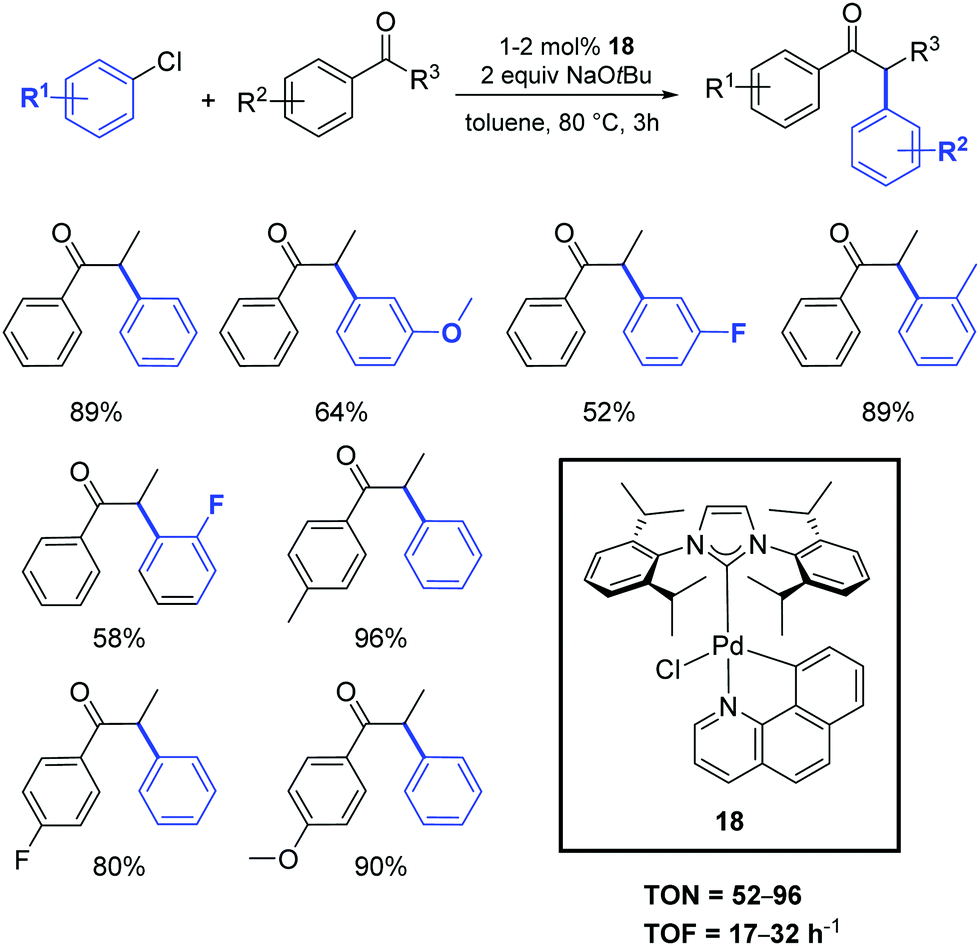
Recently, Cazin and Nolan reported a family of air- and moisture-stable palladate complexes (19–24), which are considered key intermediates in the synthesis of classical [Pd(NHC)(η3-allyl)Cl] or [Pd(NHC)(η3-cinnamyl)Cl])] derivatives via the weak-base route (Fig. 4).37 These complexes can be easily synthesized by the simple addition of an azolium salt to the dimeric palladium precursor of interest ([Pd(μ-Cl)(η3-allyl)]2 or [Pd(μ-Cl)(η3-cinnamyl)]2).
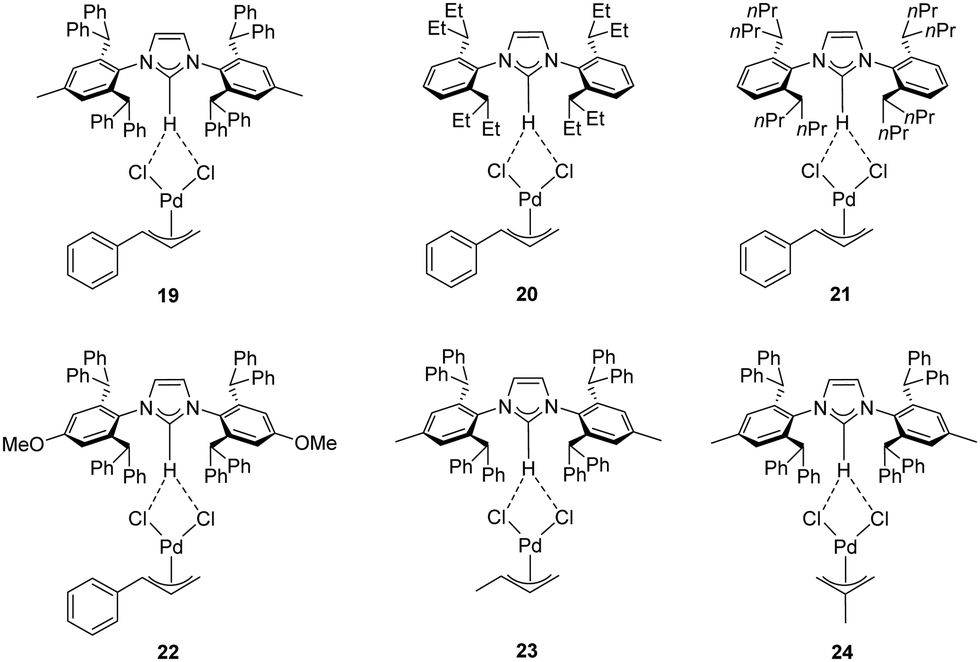 | ||
| Fig. 4 Palladates described by Cazin and Nolan.37 | ||
All tested complexes 19–24 showed high catalytic activity (conversion: 94–99%) in the reaction of propiophenone with 4-chlorotoluene (Scheme 16).37 The arylation was carried out under air, using non-anhydrous solvents and at low catalyst loading (0.2 mol%).
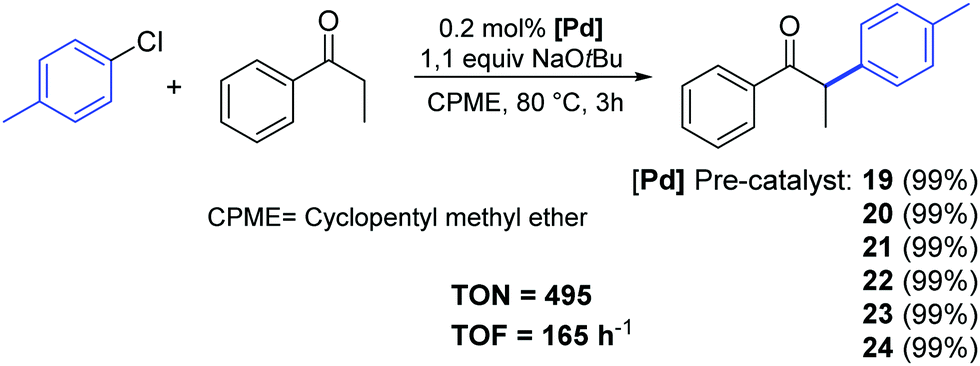 | ||
| Scheme 16 α-Arylation of 4-chlorotoluene with propiophenone catalyzed by palladates. | ||
Gratifyingly, the sterically hindered aryl chlorides were successfully coupled with the more challenging α-tetralone and acetophenone (Scheme 17).
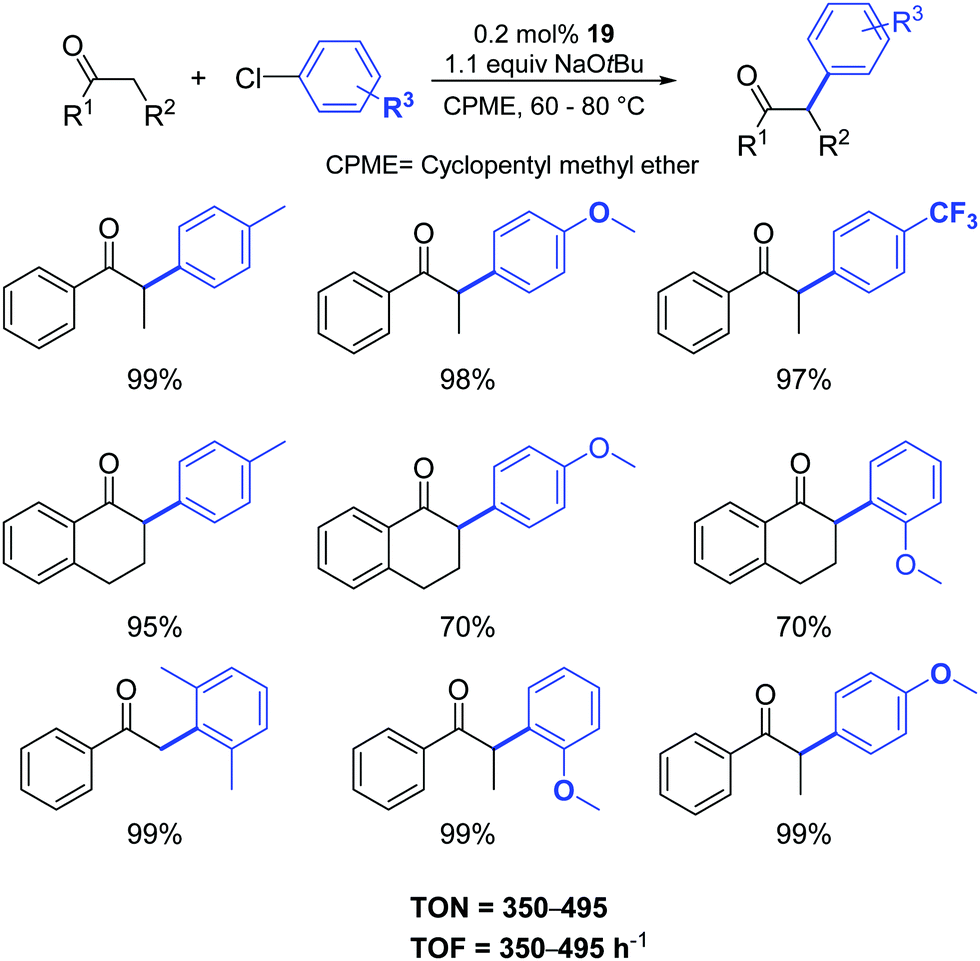 | ||
| Scheme 17 α-Arylation of ketones catalyzed by 19 as described by Cazin and Nolan.37 | ||
In 2020, the palladium-imine complex 25 was successfully synthesized and used as a pre-catalyst for the α-arylation of ketones by Shen and colleagues.38 The transformation of the substrates into products occurs with similar performances by operating under inert atmosphere or in the air (Scheme 18). Unactivated aryl and heteroaryl chlorides were effectively deployed in the reaction with only 0.5 mol% of catalyst loading.
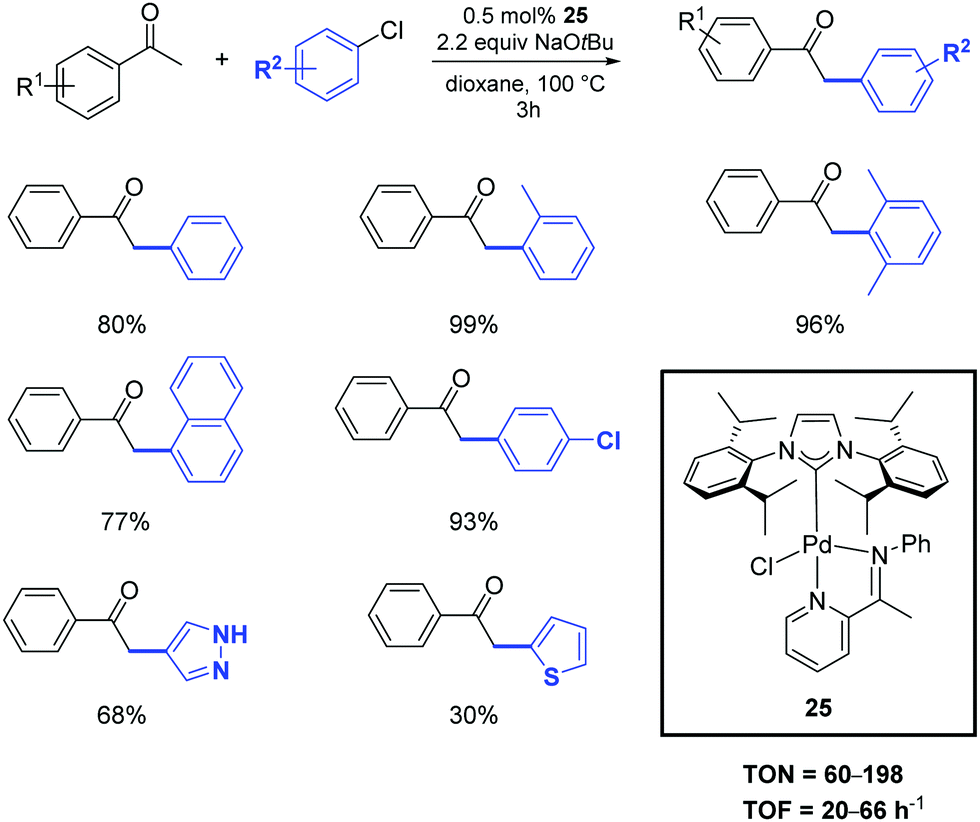 | ||
| Scheme 18 α-Arylation of acetophenone with aryl and heteroaryl chlorides catalyzed by 25. | ||
In addition to the classical bulky electron-rich cyclic diaminocarbenes, another fascinating family of NHC ligands was developed by Bertrand and co-workers in 2005.39 These ligands are known as cyclic alkylaminocarbenes (CAACs), in which one of the amino substituents has been replaced by an alkyl group. This structural modification makes the CAACs significantly more σ-donors than classical imidazolylidenes. Furthermore, the presence of the tertiary carbon atom provides a “flexible steric bulk” that differs from the steric bulk of cyclic diaminocarbenes.40,41 The strong σ-donating properties combined with the flexible steric bulk should, in theory, further accelerate the oxidative addition, transmetalation and reductive elimination in the catalytic cycle of the α-arylation reaction compared to imidazolylidenes. Therefore, Bertrand and co-workers explored the effect of air-stable CAAC ligands in the α-arylation reaction of propiophenone with aryl chlorides at room temperature (Table 3).39 The experimental results highlighted a dramatic difference in activity between the tested catalysts 26–28 (Fig. 5). While 27 proved to be the most effective catalyst for the coupling of less hindered aryl chlorides, complex 28 was more active for bulky aryl chlorides. This difference can be rationalized on the basis of the “steric and electronic match/mismatch” between substrate and catalyst.
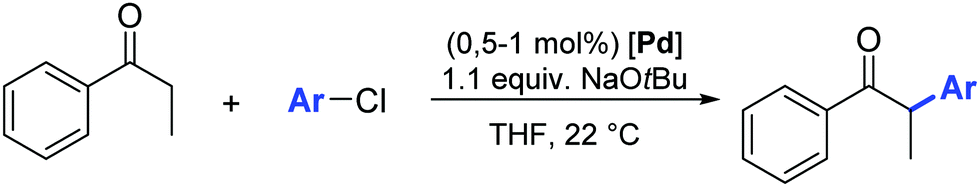
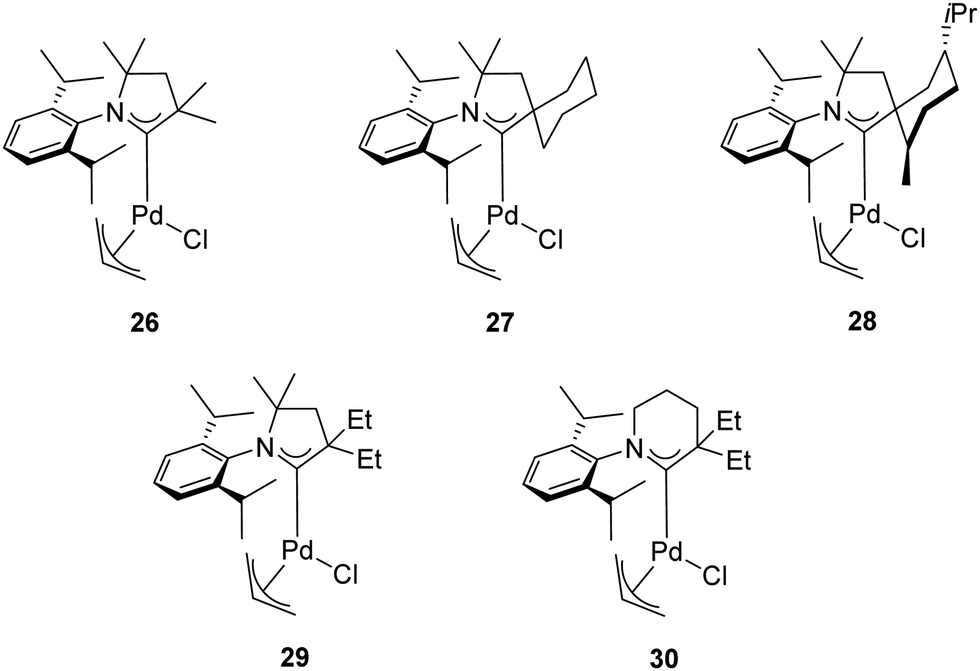 | ||
| Fig. 5 Pd(CAAC)(η3-allyl)Cl] complexes used in α-arylation of ketones. | ||
In 2018, the same research group disclosed new palladium complexes bearing five-membered (29) and six-membered (30) CAAC ligands (Fig. 5).42 Complex 30 showed higher activity compared to the palladium complexes with five-membered CAACs in the α-arylation of propiophenone with aryl chlorides at room temperature (Table 3). This is probably due to the stronger donor and acceptor properties of 30. The high ambiphilic character of 30 allows the intramolecular carbene insertion into an unactivated C(sp3)–H bond.
A third broad category of NHCs are the 1,2,3-triazol-5-ylidenes, which were discovered and even isolated in their free form in 2010 year.43 This new generation of NHCs is commonly known as “mesoionic carbenes” (MICs) since no uncharged resonance forms can be obtained from these systems.43 Due to the combination of their strong donor character (higher than classical imidazolylidenes) and the interesting synthetic flexibility of their ligand precursors via click chemistry, their use in the area of catalysis has attracted a great deal of interest in both academia and industry. Selected complexes with Late Transition Metals (Pd, Au, Ru, Ir) have been reported and exploited as catalysts for a wide range of organic transformations.44 In this context, Espinosa and colleagues prepared a series of 1,2,3-triazolium salts constructed form “clicked” hydroxybenzene derivatives. They showed that deprotonation with potassium bis(trimethylsilyl)amide (KHMDS), followed by in situ metalation, allowed for the synthesis of a series of mono-, di- and trinuclear triazol-5-ylidene palladium complexes 31–33 (Fig. 6).45
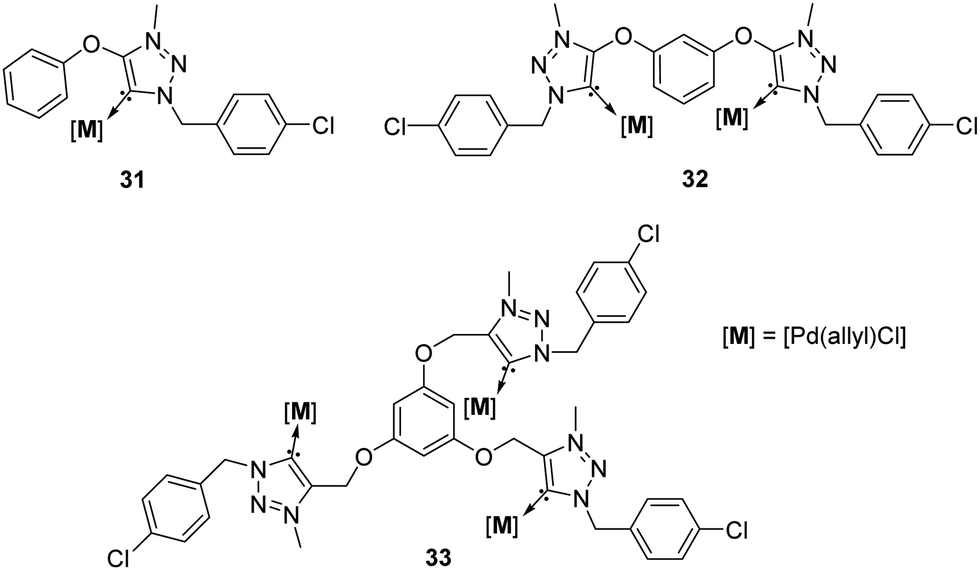 | ||
| Fig. 6 Mono-, di- and trinuclear MIC complexes, described by Mendoza-Espinosa.45 | ||
These complexes were tested in the α-arylation of propiophenone with aryl bromides using 2 mol% of catalyst (Table 4).45 The catalytic experiments established the enhanced performance of di- and trimetallic palladium complexes 32–33 in this reaction.
2.2 Nickel–NHC catalyzed α-arylation of ketones
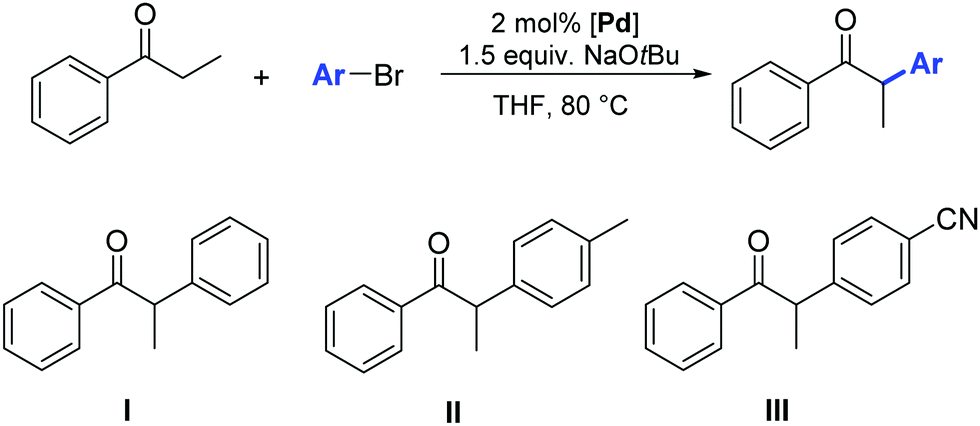
Among the other transition metals studied in the α-arylation of ketones, the results obtained with nickel–NHC pre-catalysts are particularly worthy of mention. Such compounds are generally less expensive and more sustainable than their palladium congeners. However, the disadvantage of nickel-based systems is often the toxicity of Ni(0) intermediates (or residues) and their high sensitivity to oxygen and moisture.The α-arylation of acyclic ketones was achieved for the first time using a new, simple, stable, and easy-to-access nickel(II)–halide complex bearing mixed PPh3/N-heterocyclic carbene ligands (34).46 The activity of this pre-catalyst in the α-arylation of propiophenones with bromo- and chloro-aryl derivatives was investigated using 10 mol% of 34 in the presence of NaOtBu. The reactions proceed in toluene at 100 °C, affording the products in moderate to good yields (Scheme 19). Early results showed the poorer reactivity of Ni vs. Pd counterparts.
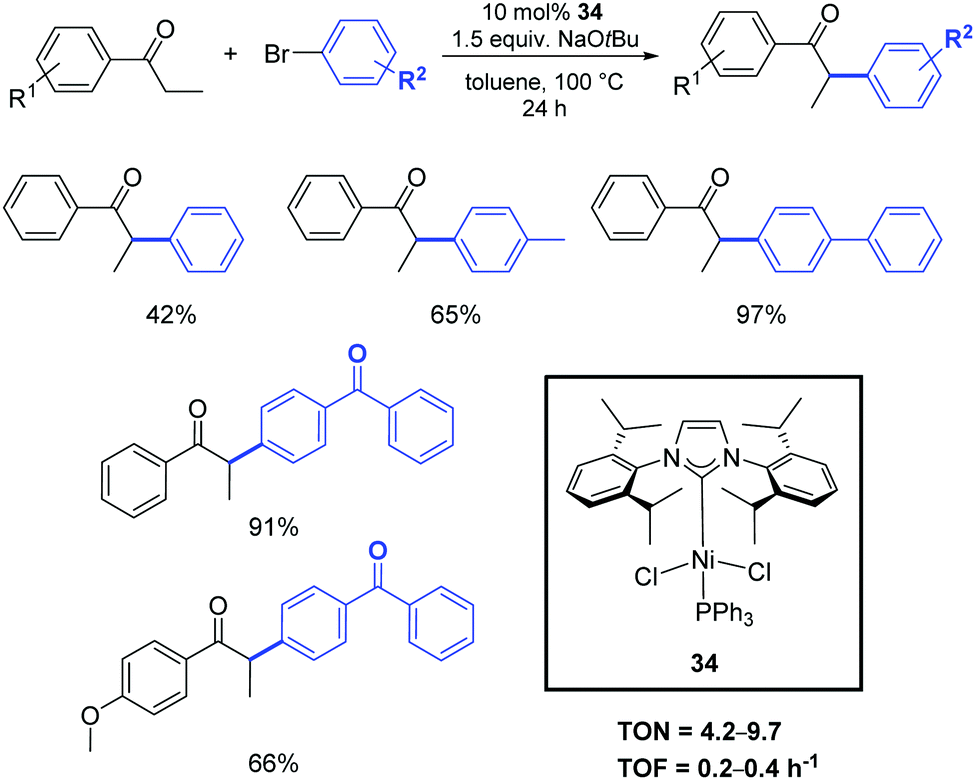 | ||
| Scheme 19 α-Arylation of ketones catalyzed by 34, described by Matsubara et al.46 | ||
The catalytic activity of 34 is attributed to its ability to eliminate triphenylphosphine, thereby creating a vacant coordination site around the nickel center (Scheme 20).
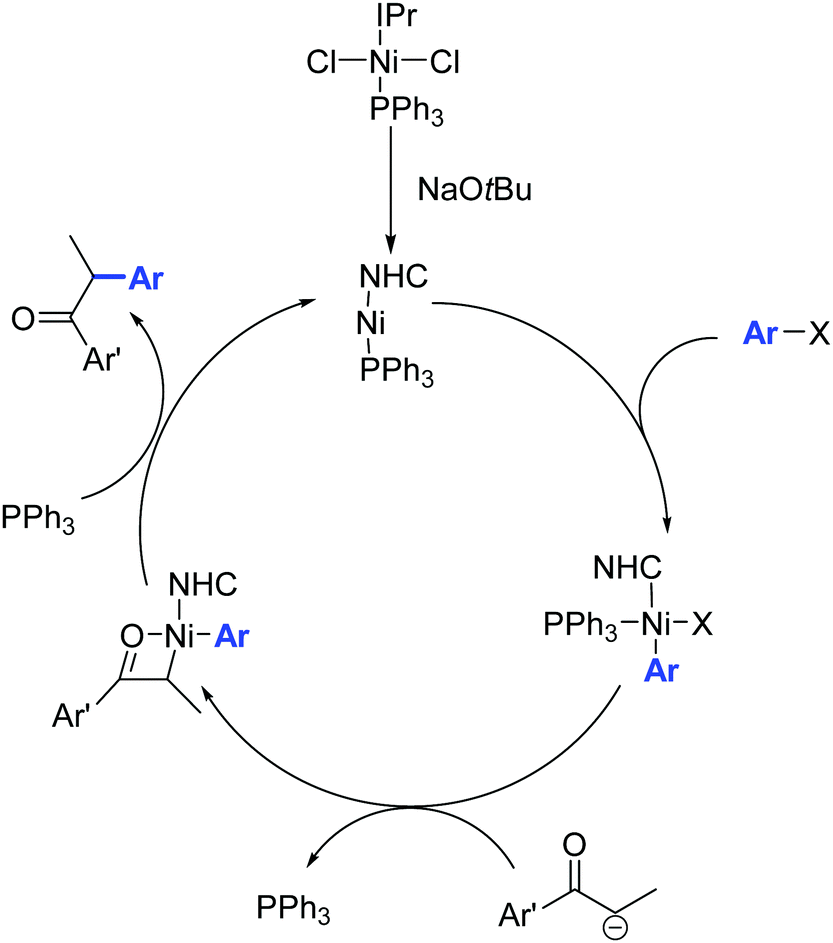 | ||
| Scheme 20 Proposed mechanism for the α-arylation of ketones in the presence of the Ni complex 34, described by Matsubara and co-workers.46 | ||
In 2014, Peris and colleagues prepared a series of Ni–NHC complexes with three different types of NHC ligands: imidazolylidenes, benzimidazolylidenes and pyrene–imidazolylidenes (35–37). They evaluated the catalytic activity of the synthesized nickel complexes (Fig. 7) in the α-arylation of ketones with aryl halides, but these did not show any activity. The reactions were carried out at 80 °C in 1,4-dioxane with a catalyst loading of 1 mol%.34
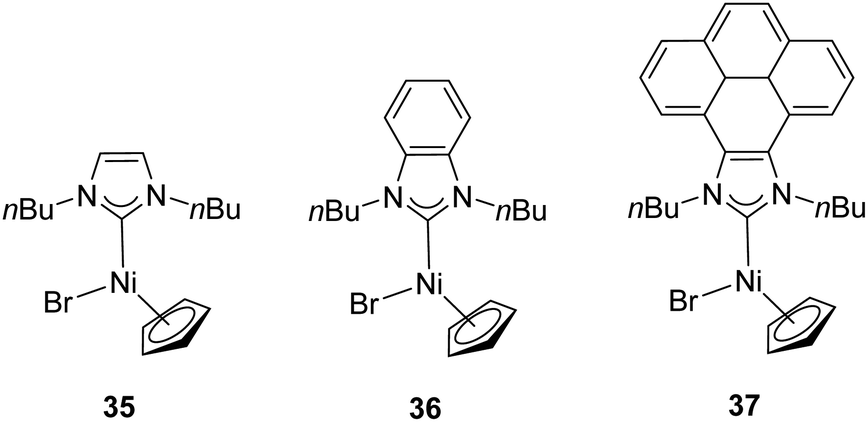 | ||
| Fig. 7 Ni–NHC complexes reported by Peris and co-workers. | ||
In the same year, Ritleng and co-workers described the air-stable nickel–NHC complexes 38–43, that exhibited good catalytic activity for the α-arylation of acyclic ketones (Scheme 21).47
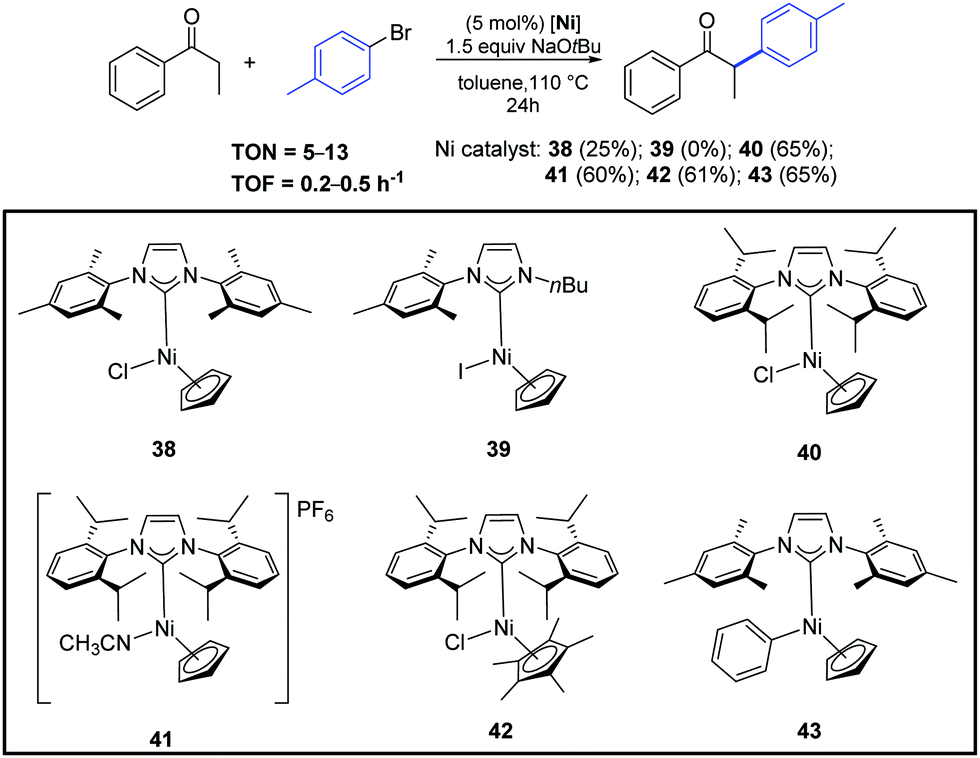 | ||
| Scheme 21 α-Arylation of propiophenone with 4-bromotoluene catalyzed by complexes 38–43, described by Ritleng and co-workers.47 | ||
Among the complexes tested, 40 enabled efficient α-arylation of acyclic ketones with aryl bromides already at 3 mol% as catalyst loading (Scheme 22).47 The authors suggested that the main mechanism in this nickel-catalyzed α-arylation was of a radical nature and that a C-bound ketone enolate derivative of 40, if involved, had only a minor role.
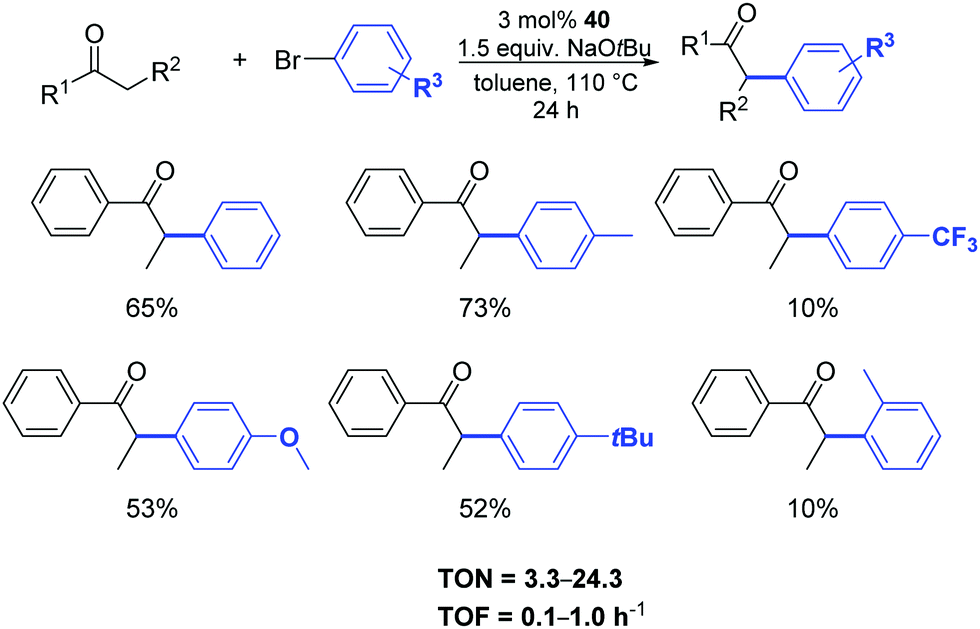 | ||
| Scheme 22 α-Arylation of ketones with aryl bromides catalyzed by 40, described by Ritleng and co-workers.47 | ||
In 2015 Nolan and colleagues reported a general methodology for the α-arylation of ketones promoted by the nickel complexes 44–47. In particular, the new well-defined [Ni(IPr*)(η3-cinnamyl)Cl] (46) pre-catalyst showed high performance, allowing the coupling of a wide range of ketones, including acetophenone derivatives, with various functionalized aryl chlorides (Scheme 23).48
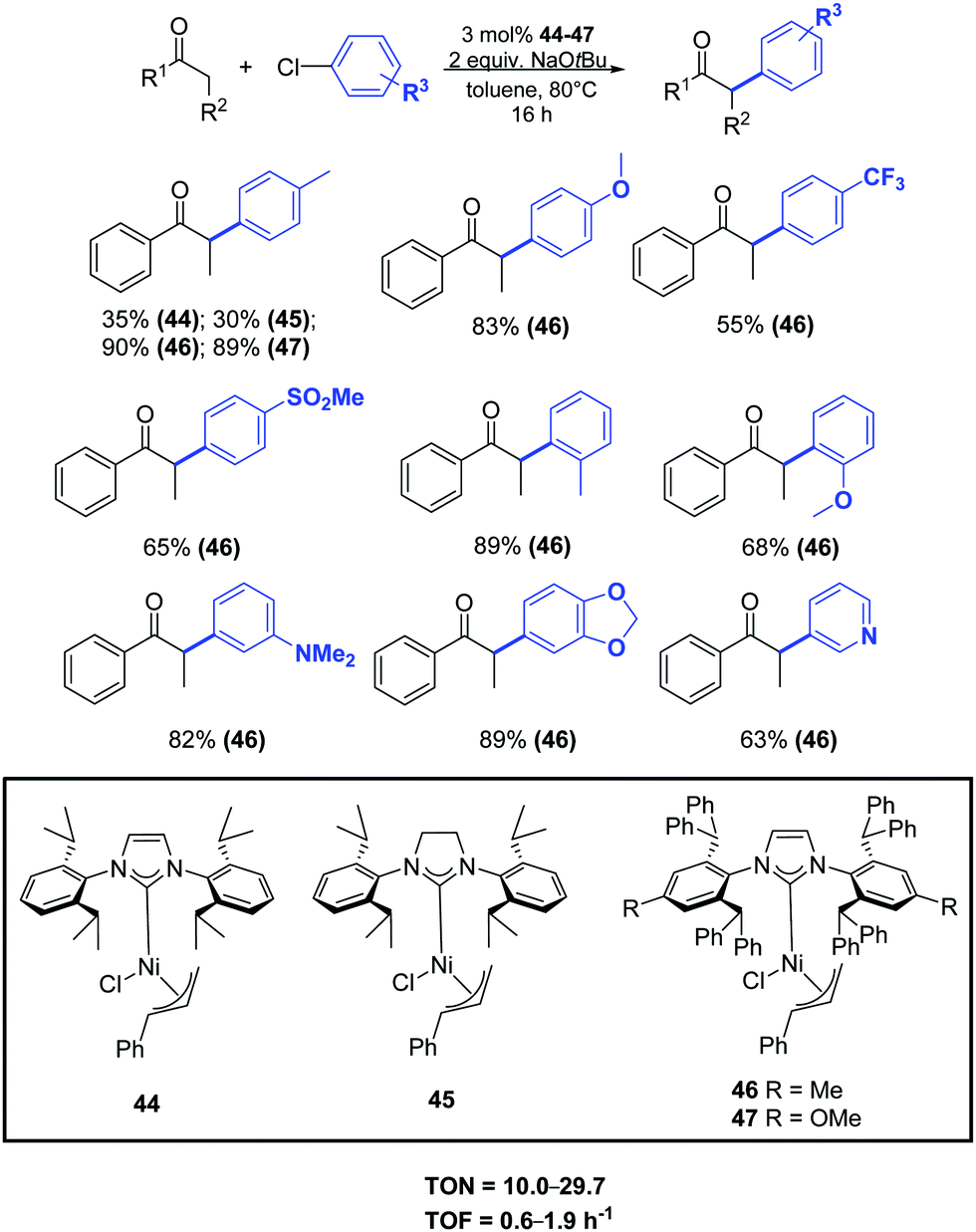 | ||
| Scheme 23 α-Arylation of ketones catalyzed by [Ni(NHC)(η3-cinnamyl)Cl] complexes (46) as described by Nolan and coworkers.48 | ||
The catalytic cycle proposed by Nolan and colleagues is illustrated in Scheme 24.48
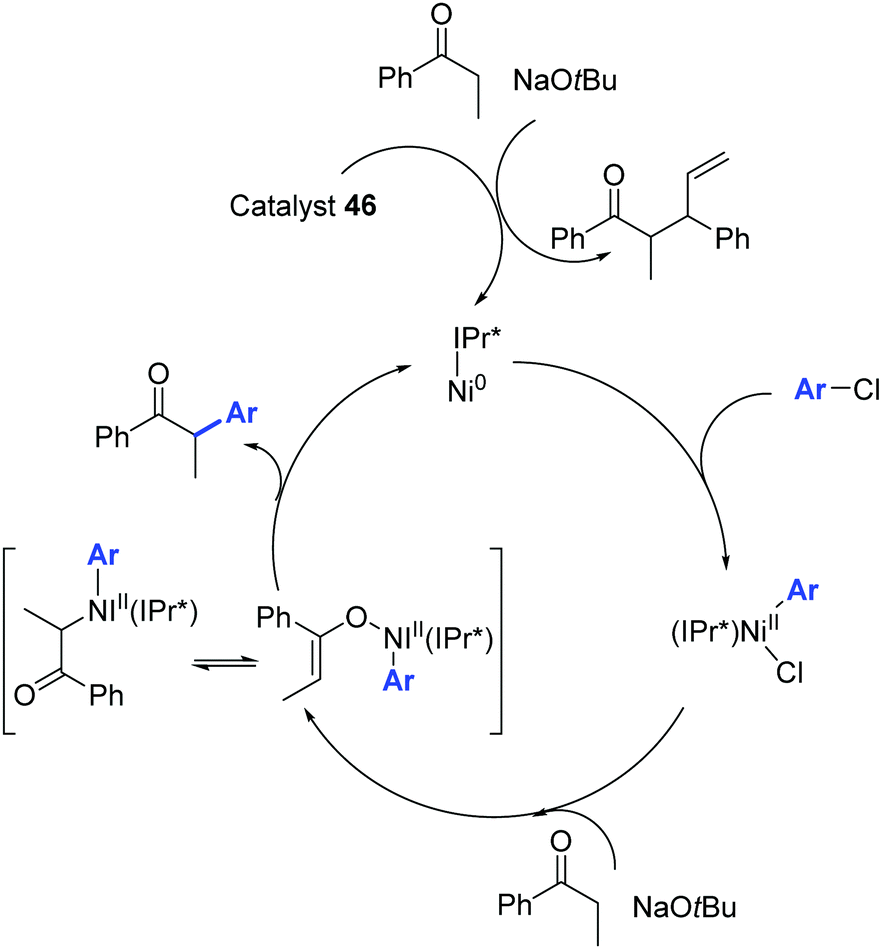 | ||
| Scheme 24 Ni0/NiII catalytic cycle described by Nolan and co-workers.48 | ||
Nickel-allyl complexes 44–47 were then used for the synthesis of 6-methoxy-2-phenyl-1-tetralone (Scheme 25).49 The arylation of the starting tetralone (1.1 equiv.) proceed in toluene at 80 °C for 16 h in the presence of a catalytic amount (2 mol%) of complex 47, which bear the IPr*OMe ligand.
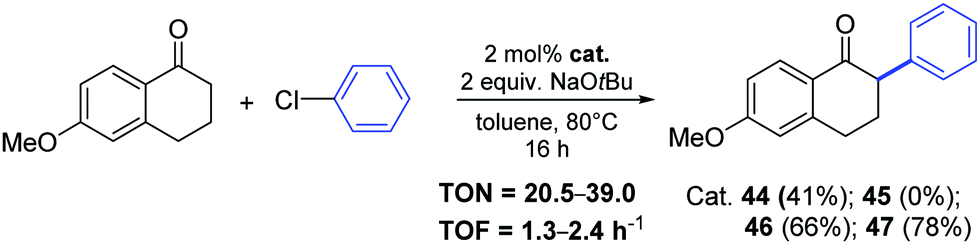 | ||
| Scheme 25 Synthesis of 6-methoxy-2-phenyl-1-tetralone via ketone arylation towards nafoxidine and lasofoxifene, described by Nolan and co-workers.49 | ||
In 2015, Helquist et al. described a procedure for Ni-catalyzed cross-coupling of ketone enolates with alkenyl halides in the presence of the N-heterocyclic carbene precursors L1–L6 (Scheme 26).50
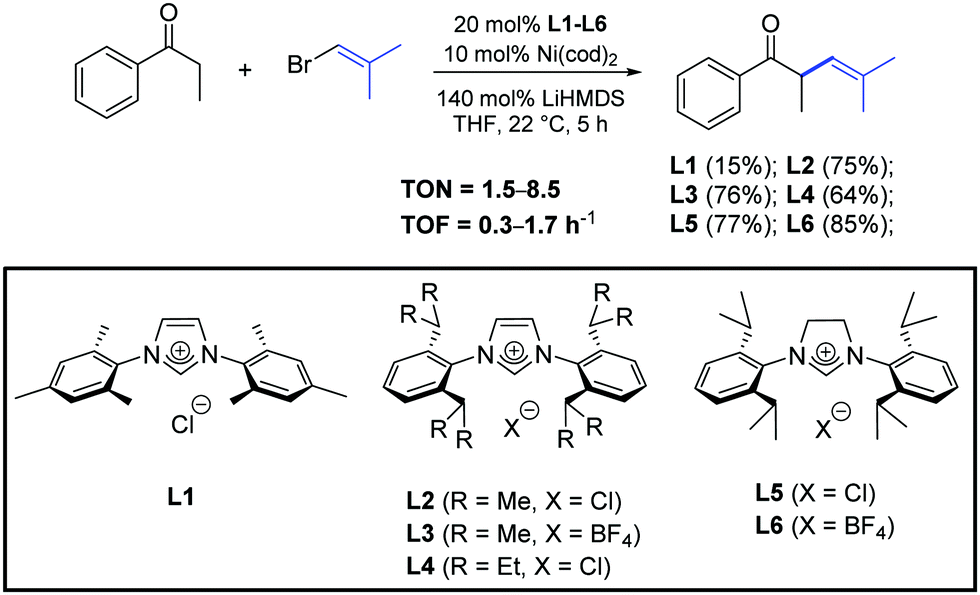 | ||
| Scheme 26 N-Heterocyclic carbene precursors used in the α-arylation of ketones, described by Helquist and co-workers.50 | ||
The examination of the scope of aromatic and aliphatic ketone lithium enolates with a variety of alkenyl bromides was carried out in the presence of the azolium salt L2 and Ni(cod)2 (5 mol%), as illustrated in Scheme 27.50
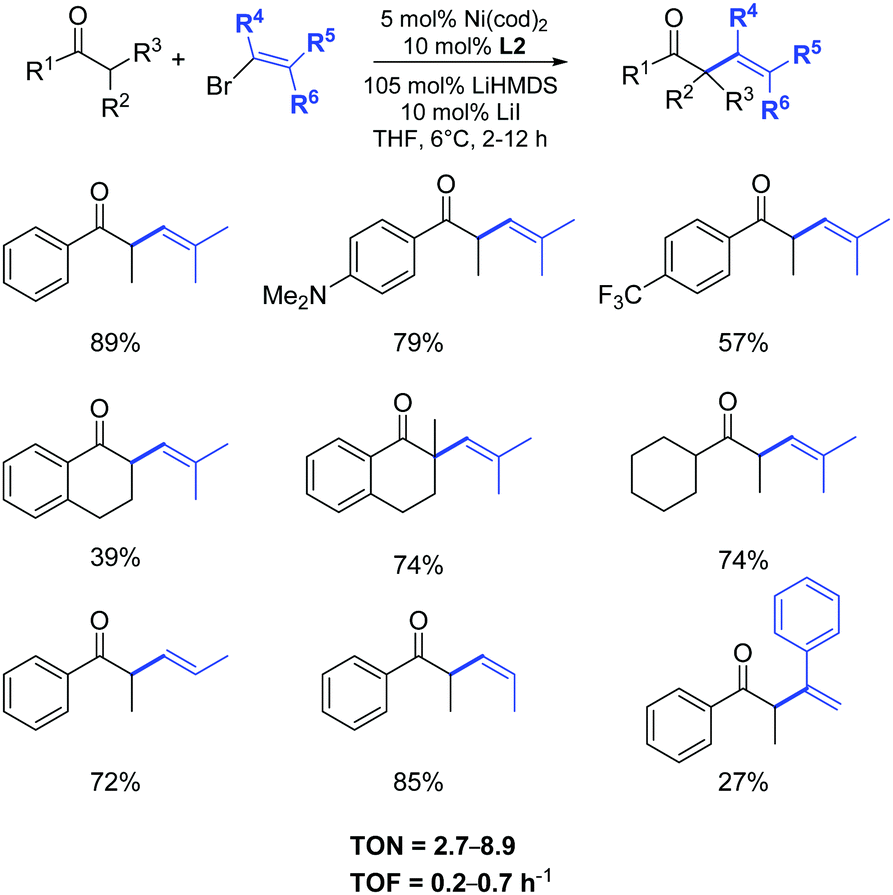 | ||
| Scheme 27 α-Arylation of ketones in the presence of Ni(cod)2/L2. | ||
Using the same approach, Wang and Li developed an efficient catalytic system (Ni(cod)2, L2 and LiOtBu) for the α-arylation of ketones with aryl trimethylammonium triflates. Activated, non-activated and deactivated aryl trimethylammonium triflates proved suitable for the coupling. The ketones used in this transformation included propiophenone, butyrophenone and acetophenone derivatives as well as a cyclohexanone derivative (Scheme 28).51
 | ||
| Scheme 28 Nickel catalyzed α-arylation of 1-arylpropan-1-ones with aryl trimethyl-ammonium triflates as described by Wang and Li.51 | ||
3. Metal–NHC catalyzed α-arylation of aldehydes
Metal-catalyzed α-arylation of aldehydes is a notoriously difficult transformation because of their propensity to readily undergo aldol condensation under basic conditions. For this reason, only a limited number of reports on α-arylations of aldehydes are present in the literature. Most of them require the use of air-sensitive and rather expensive tertiary phosphine ligands.5,52 However, although this area is not yet fully developed, significant progresses have recently been achieved. For instance, Bertrand and co-workers reported the α-arylation of isobutanal with 2-chlorotoluene catalyzed by the efficient palladium–CAAC complex 28 (Scheme 29).39 This reaction was performed in THF at room temperature using 1 mol% of the catalyst. The desired product was obtained after 16 h in 98% yield.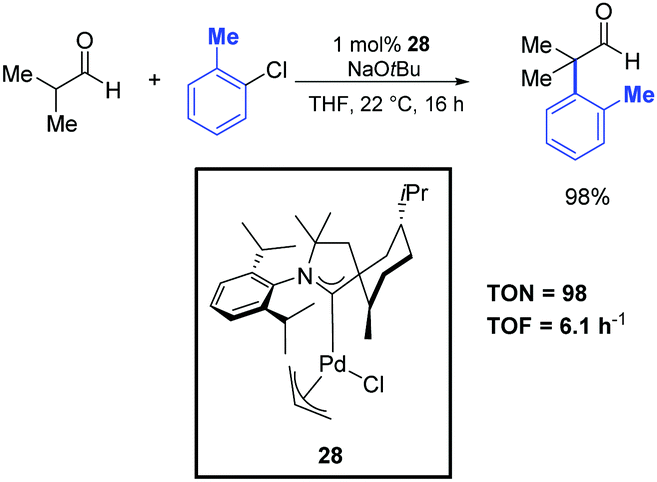 | ||
| Scheme 29 α-Arylation of isobutanal with 2-chlorotoluene, described by Bertrand and co-workers.39 | ||
In 2013, Nareddy and Mazet developed a protocol that relied on the use of the air-stable palladium–NHC complex 1.53 As illustrated in Scheme 30, they successfully obtained the α-arylation of linear and α-branched aldehydes in dioxane at 100 °C in the presence of 5 mol% of the catalyst.
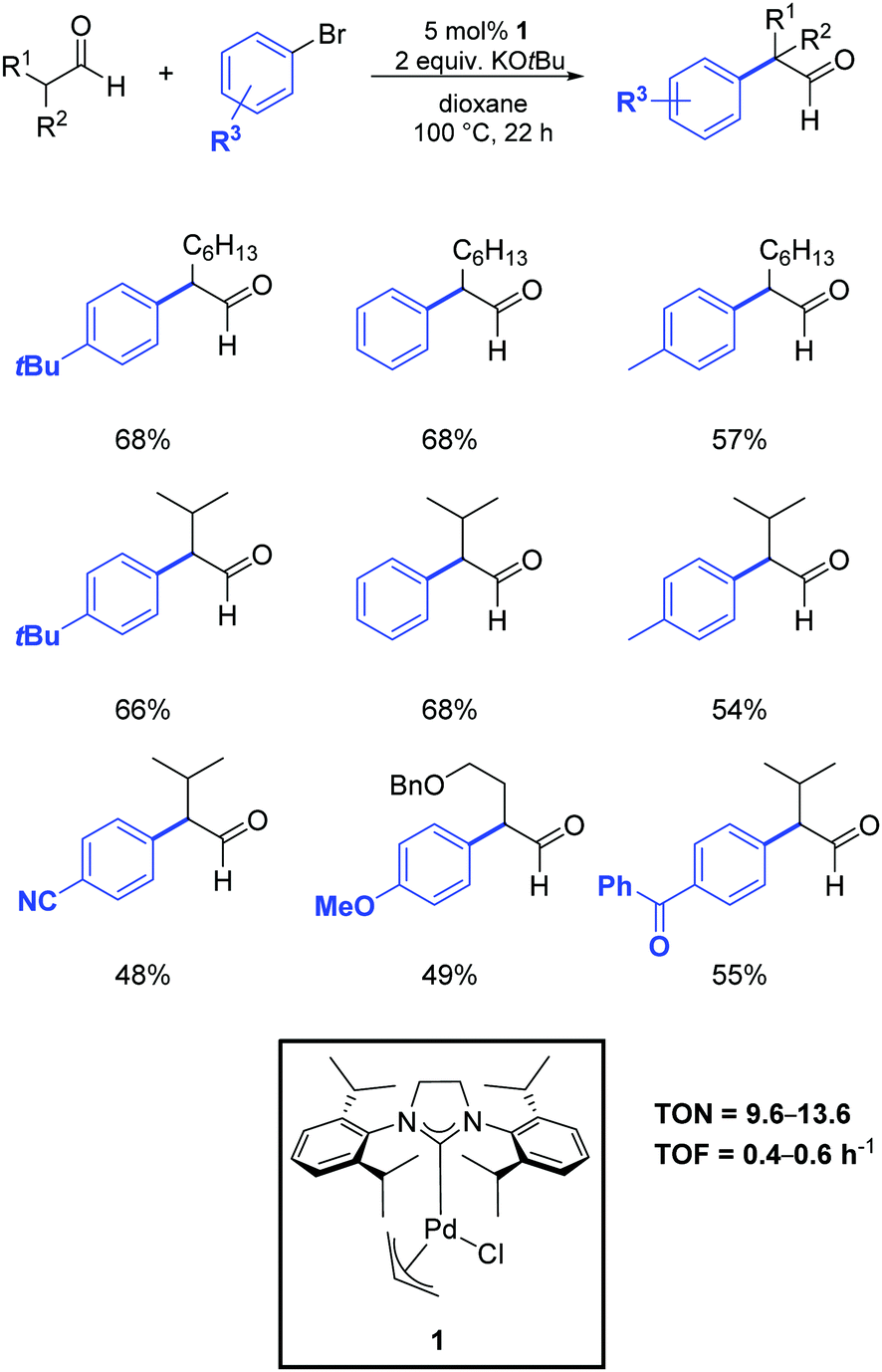 | ||
| Scheme 30 α-Arylation of linear and branched aldehydes with aryl bromides, as described by Nareddy and Mazet.53 | ||
4. Metal–NHC catalyzed α-arylation of esters
As previously mentioned, α-aryl esters and their derivatives are of great synthetic importance in organic chemistry by virtue of their presence in many pharmaceuticals and biologically active compounds (i.e. naproxen, flurbiprofen and dichlofenac). Besides these target molecules, α-aryl carboxylic acid and esters are valuable building blocks, as they can act as precursors for the synthesis of aryl alcohols, amines, and nitriles.During the last decades, very few catalytic and economically viable routes to α-aryl esters have been described.54 While there are no regioselectivity issues associated with the α-arylation of esters, their lower reactivity and relative sensitivity to basic conditions makes this transformation intrinsically challenging. Moreover, the possibility for ester enolates to undergo Claisen condensation also complicates the synthetic assembly protocols. Although the most frequently used ligands in metal catalyzed α-arylation of esters are tertiary phosphine ligands,5,55 recent developments in the area have demonstrated the ability of palladium–NHC complexes to promote this type of reaction very effectively.5,56 For example, Hartwig and co-workers observed that α-arylation of esters can be carried out at room temperature by using 2 mol% of Pd(dba)2 in the presence of the N-heterocyclic carbene precursor L6 (Scheme 31).57 An evaluation of several simple and inexpensive ligands showed that a combination of Pd(dba)2 and tri-tert-butylphosphine or the hindered carbene precursor L6 generated efficient catalysts. Although tert-butoxides were strong enough bases to couple ketones with aryl halides, low conversions were observed from reactions of esters. Thus, lithium hexamethyldisilazane (LiHMDS) was tested and proved to be more suitable. To the best of our knowledge, this represents the only reported example of the use of N-heterocyclic carbene ligands in the α-arylation of esters.
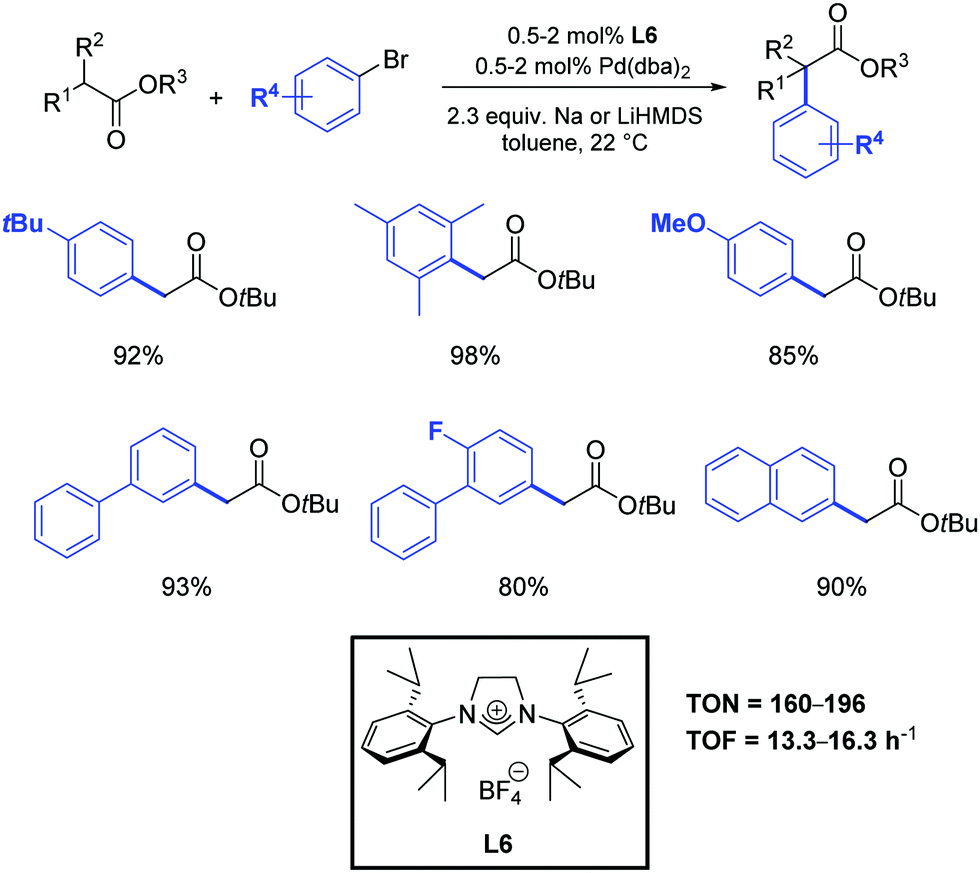 | ||
| Scheme 31 α-Arylation of esters catalyzed by Pd(dba)2 and L6, as described by Hartwig and co-workers.57 | ||
5. Metal–NHC catalyzed α-arylation of amides
The lower acidity of amides compared to ketones requires the use of strong bases for their functionalization. Consequently, problems associated with intolerance of other functional groups, decomposition and diarylation processes as well as dehydrohalogenation from β-hydride elimination pathways might be expected, especially for intermolecular reactions.Many examples have shown the applicability of the α-arylation of amides to the total synthesis of biologically active compounds.58 As for the previously described categories of carbonyl compounds, also in the α-arylation of amides, the use of metal complexes supported by tertiary phosphine ligands has been extensively developed.5,59 However, these ligands are increasingly being replaced by the more stable and cheaper N-heterocyclic carbene ligands.
In 2001, Lee and Hartwig demonstrated the first α-arylation of amides with NHC-based complexes, although their work was mainly focused on the study of a series of phosphine ligands.60 Interestingly, they observed similar performances between PCy3 and NHC precursors (L5 and L6) in the palladium-catalyzed synthesis of oxindoles by intramolecular α-arylation of ortho-bromoanilides (Scheme 32).
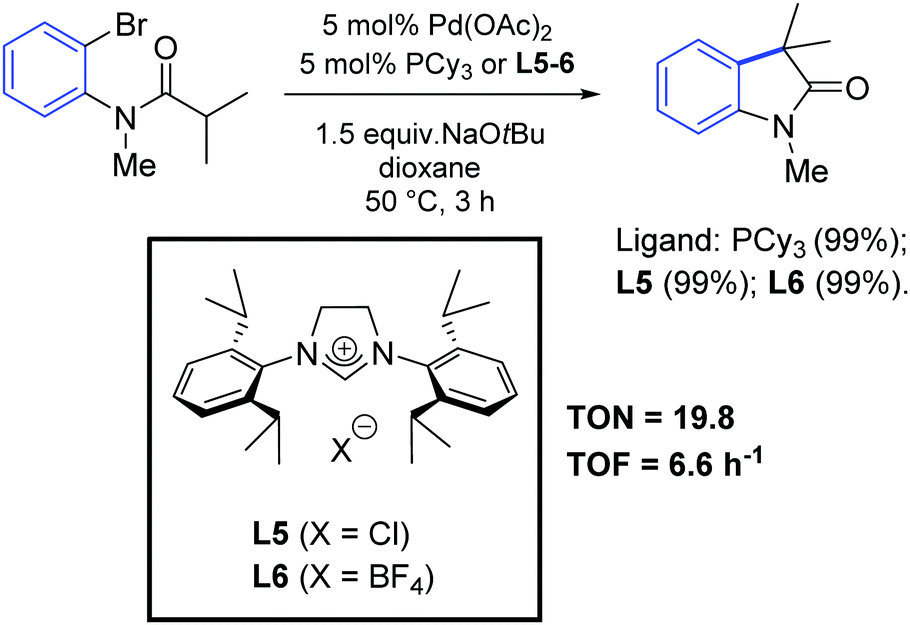 | ||
| Scheme 32 Synthesis of oxindoles via intramolecular arylation of 2-bromoanilides, as described by Lee and Hartwig.60 | ||
Furthermore, an asymmetric version of this reaction was reported, using the optically active NHC precursors L7 and L8. It was found that the catalysts generated in situ from [Pd(dba)2] and the above-mentioned azolium salts provided the oxindole products in up to 77% of enantiomeric excess (Scheme 33). Surprisingly, the two ligand precursors (L7 and L8) gave the products as opposite enantiomers.
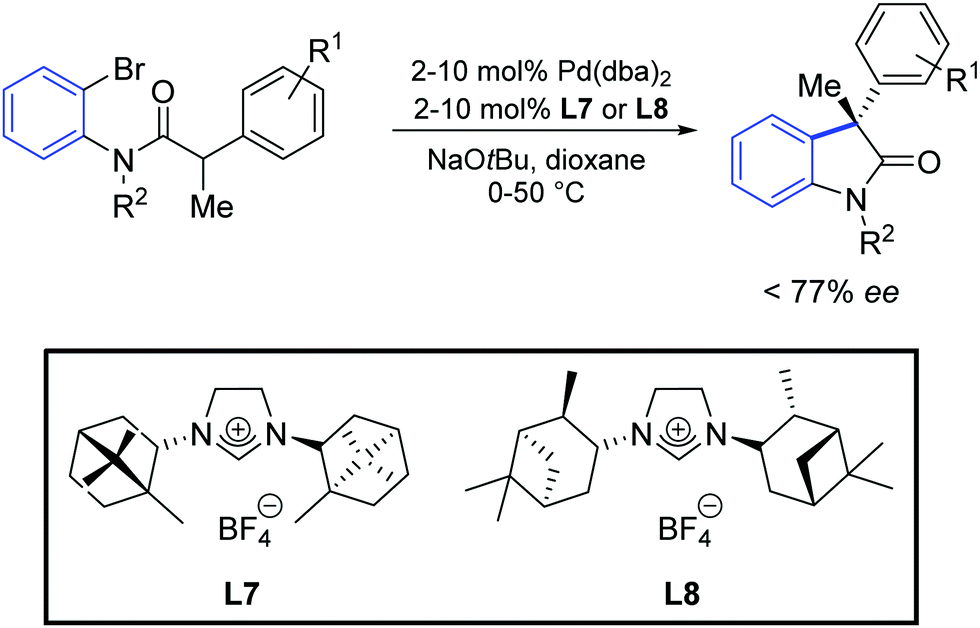 | ||
| Scheme 33 Enantioselective synthesis of oxindole described by Lee and Hartwig.60 | ||
One year later, Zhang et al. developed a [Pd2(dba)3]/ligand L9 catalytic system for the synthesis of oxindoles that operated at lower catalyst loadings (1.5–3 mol%) as is illustrated in Scheme 34.61
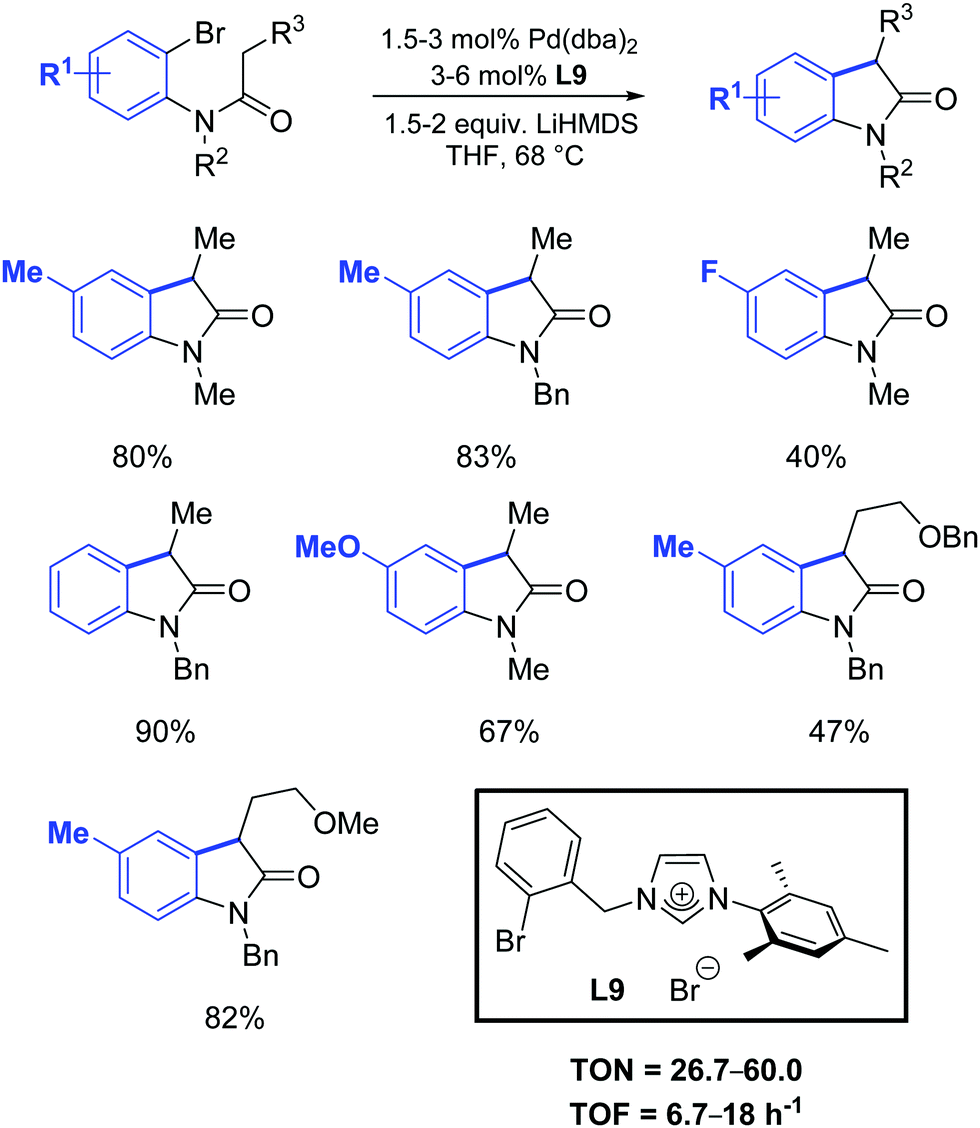 | ||
| Scheme 34 Oxindole formation according to Zhang et al.61 | ||
In analogy with observations made by Nolan and co-workers,62 it was suggested that two close coordination sites on the metal were necessary for the formation of the C–C bond. Instead of having two carbene ligands bound to palladium, the oxidative addition of ligand L9 to the palladium(0) center provide a less bulky pre-catalyst, that can promote the formation of an oxindole from several substrates. In addition, they proposed that the initial step of the catalytic cycle may involve the formation of a palladacyclic species through oxidative insertion of palladium(0) into the aryl-bromide bond (Scheme 35).
 | ||
| Scheme 35 The mechanism proposed by Zhang for the synthesis of oxindoles. | ||
Subsequent to the seminal work of Lee and Hartwig, other research groups have investigated the enantioselective formation of oxindoles through the use of different N-heterocyclic carbene precursors. Only modest enantioselectivities were independently obtained by Glorius,63 Kondo and Aoyama64 in asymmetric cyclizations of 2-halo-anilides to oxindoles, using catalytic systems composed of either Pd(0) or Pd(II) and a panel of chiral azolium salts (L10–L18 in Fig. 8).
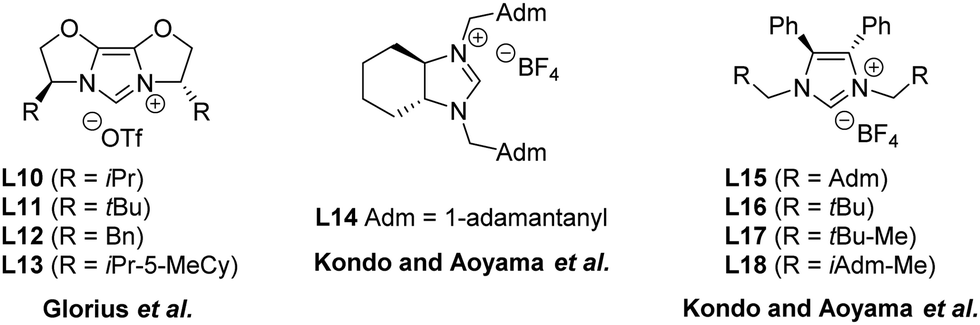 | ||
| Fig. 8 Chemical structures of chiral NHC precursors.63,64 | ||
In 2007, Kündig and co-workers reported a high enantioselectivity in the intramolecular arylation of N-substituted ortho-bromoanilides by using the imidazolium iodides L19–L21 (Scheme 36).65 The reaction yields were generally very high and combined with ee values up to 90%.
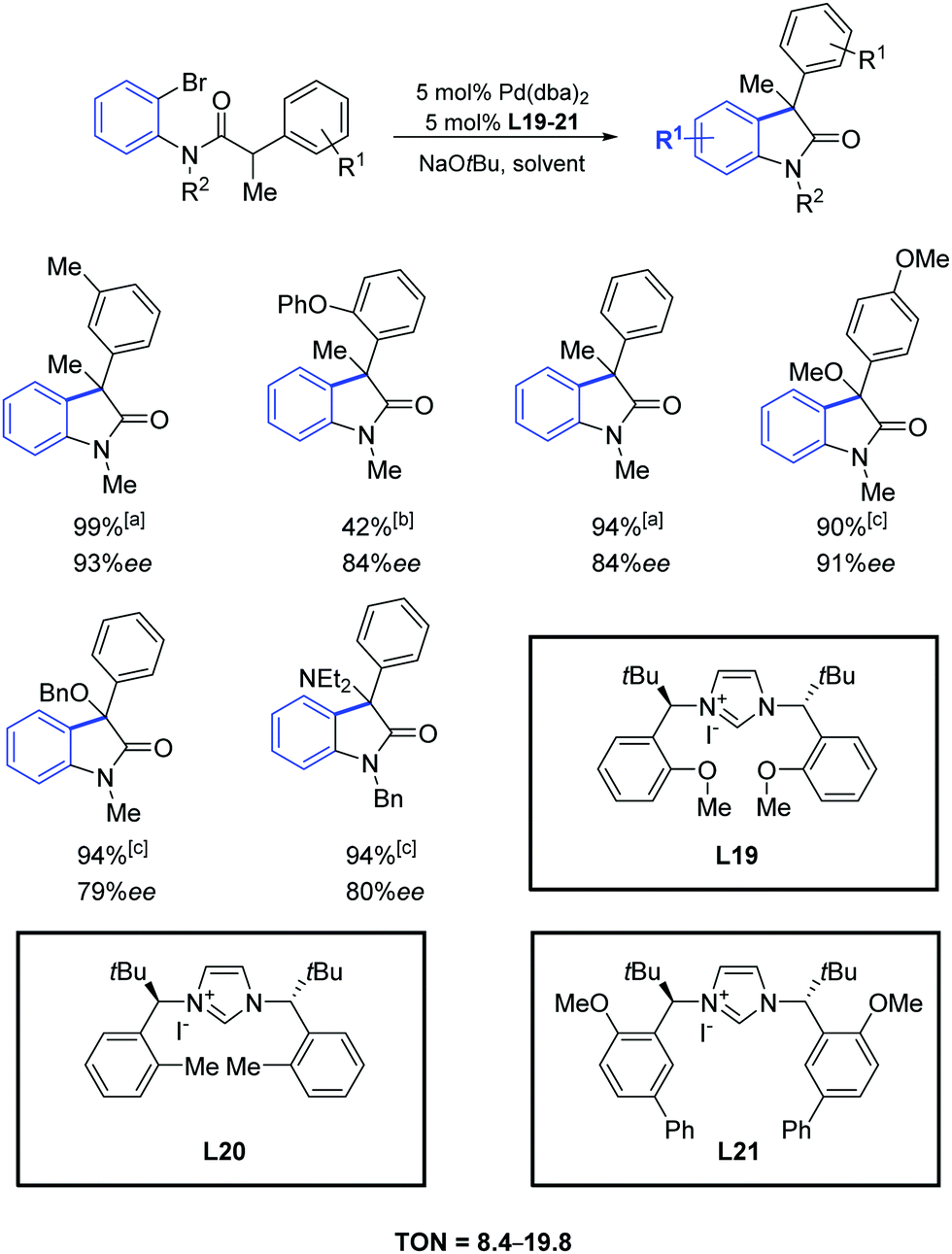 | ||
| Scheme 36 Chiral azolium salts in the palladium catalyzed intramolecular cyclization of amides to oxindoles, as described by Küding and coworkers. aLigand L19, DME, 23 °C; bLigand L20, DME, 23 °C; cLigand L21, toluene, 50 °C. | ||
More recently, Glorius and colleagues described a new sterically demanding chiral IBiox ligand L22.66 Specifically, this ligand has one of the largest percent buried volume among NHCs, thus making it attractive for its use in cross-coupling processes with challenging substrate combinations. Glorius demonstrated that the intramolecular α-arylation of amides could be conducted using aryl chlorides as substrates with very high enantioselectivity, even for ortho substituted aryl moieties (Scheme 37).
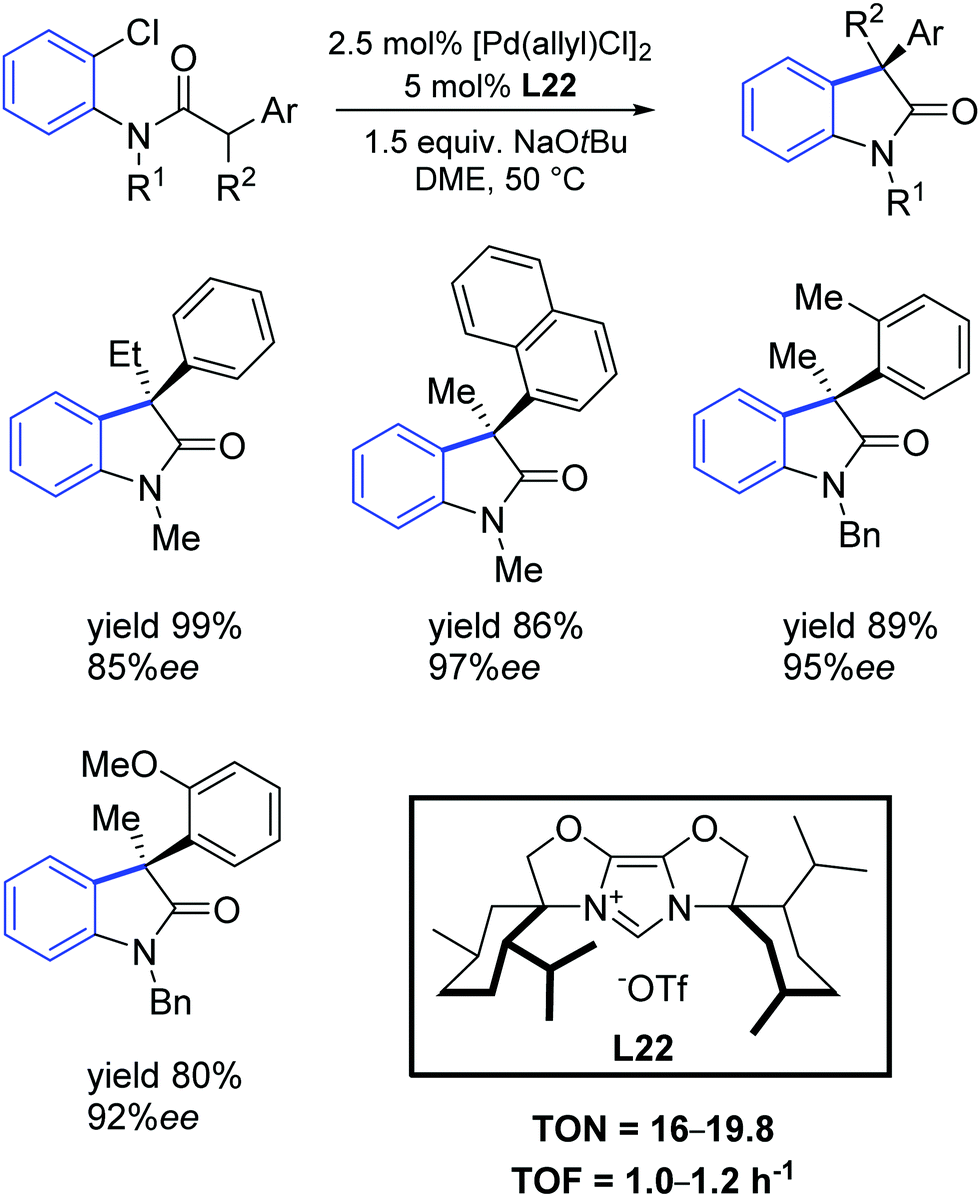 | ||
| Scheme 37 Intramolecular α-arylation described by Glorius and co-workers.66 | ||
In 2008, Dorta and coworkers described the preparation of [Pd(NHC)(η3-cinnamyl)Cl] complexes (48–50) derived from C2-symmetric diamines with naphthyl side-chains. These compounds exist as a mixture of diastereomers and the palladium complexes can be successfully separated, and their absolute configuration assigned (Fig. 9).67
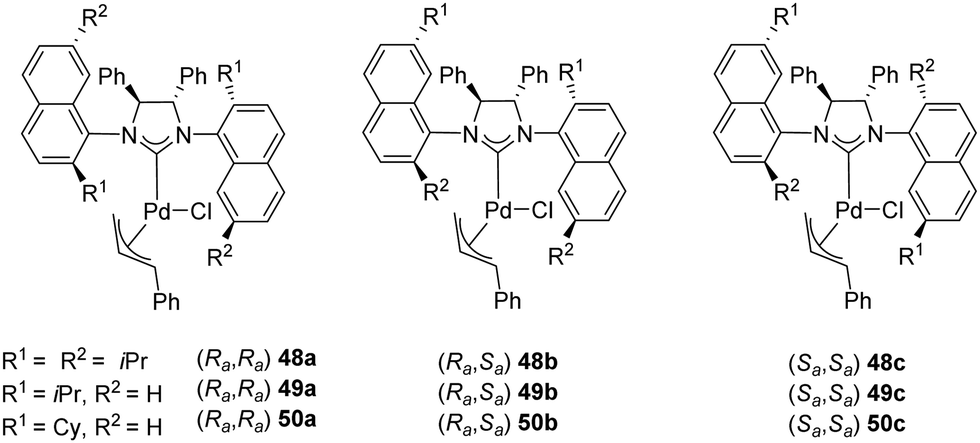 | ||
| Fig. 9 Palladium complexes reported by Dorta and co-workers. | ||
Complexes 48–50 were successfully employed as pre-catalysts in the asymmetric intramolecular α-arylation of amides, as attested by the high yields and enantioselectivity summarized in Table 5.
|
|
||||||
|---|---|---|---|---|---|---|
| Ent. | R1 | R2 | [Pd] | R a,Ra | % yielda and (eebc) | |
| R a,Sa | S a,Sa | |||||
| a Isolated yields. b Determined by chiral HPLC. c Absolute stereochemistry determined as (R)-configuration. d Reaction run at 50 °C. | ||||||
| 1 | Me | Ph | 48 | 98 (86) | 98 (67) | 98 (20) |
| 2d | Me | Ph | 49 | 94 (67) | 95 (52) | 95 (34) |
| 3d | Me | Ph | 50 | 98 (51) | 98 (46) | |
| 4 | Bn | Ph | 48 | 98 (88) | 98 (45) | 97 (50) |
| 5 | Bn | Ph | 49 | 89 (76) | 96 (61) | |
| 6 | Me | p-Tol | 48 | 99 (87) | 98 (79) | 97 (44) |
| 7 | Me | p-Tol | 49 | 98 (84) | 98 (74) | |
| 8 | Me | p-Tol | 50 | 98 (62) | 98 (70) | |
| 9 | Me | o-Tol | 48 | 99 (85) | 97 (89) | 98 (44) |
| 10 | Me | m-Tol | 48 | 99 (80) | 96 (65) | 98 (24) |
| 11d | Me | m-Tol | 49 | 98 (71) | 98 (49) | |
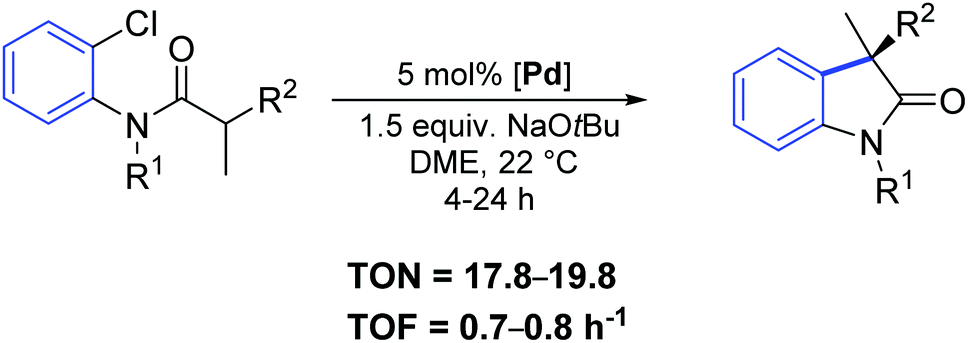
Two years later, the same group reported a new synthetic strategy to 3-allyl oxindoles bearing a chiral quaternary carbon stereocenter via a direct palladium-catalyzed α-arylation reaction.68 In particular, the chiral palladium allyl complex 51 offered excellent activity and chemo- and enantio-selectivity (Scheme 38).
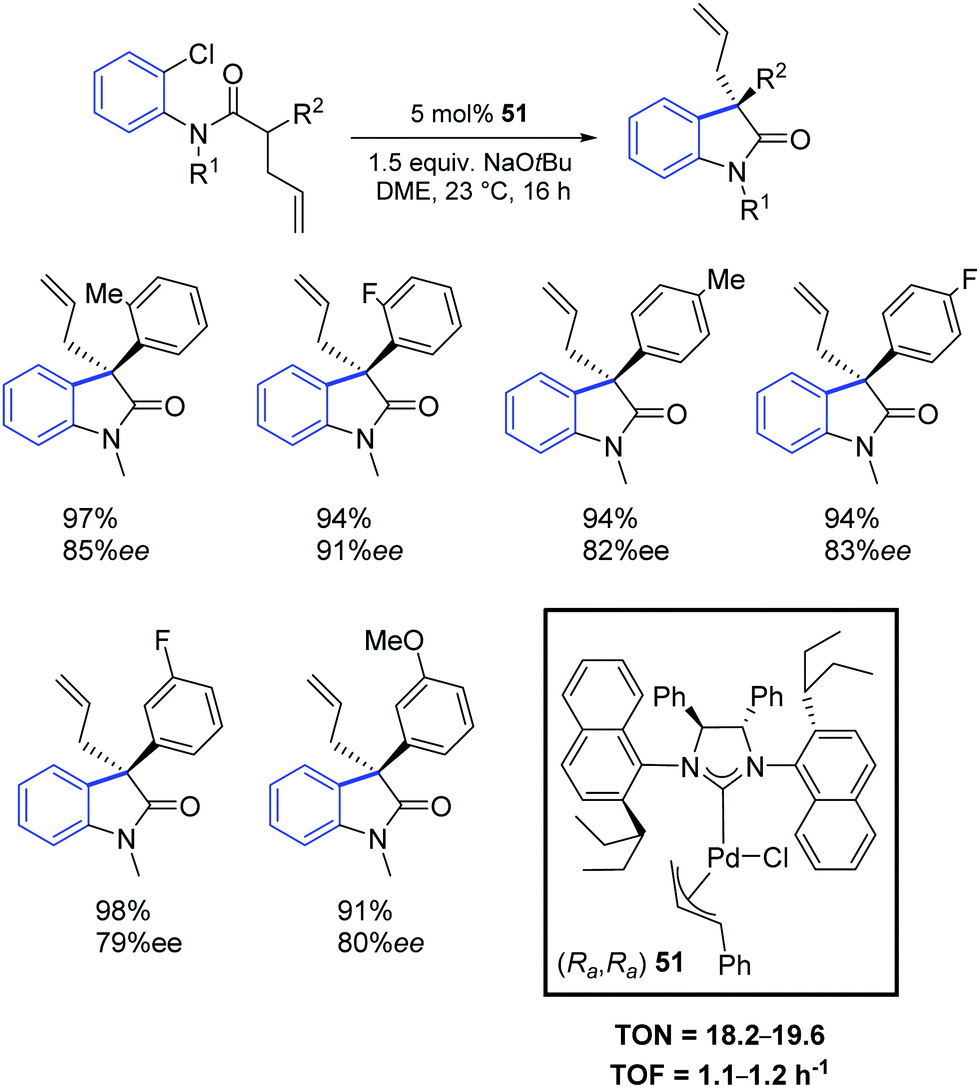 | ||
| Scheme 38 Asymmetric oxindoles synthesis with (Ra,Ra)-51 as described by Dorta. | ||
In a subsequent report, Dorta and colleagues reported a chiral Pd–NHC complex (52) with naphthyl side groups, which enabled the synthesis of the interesting family of 3-fluoro-3-aryl oxindoles (Scheme 39).69
 | ||
| Scheme 39 Asymmetric Pd–NHC catalyzed arylation of amides, as described by Dorta and co-workers.69 | ||
In 2011, Murakami and co-workers reported that chiral NHC ligands having a 2,2′-bisquinoline-based C2 symmetric skeleton (L23) were efficient ligands in the palladium-catalyzed synthesis of 3,3-disubstituted oxindoles with good yields and enantioselectivities (Scheme 40).70
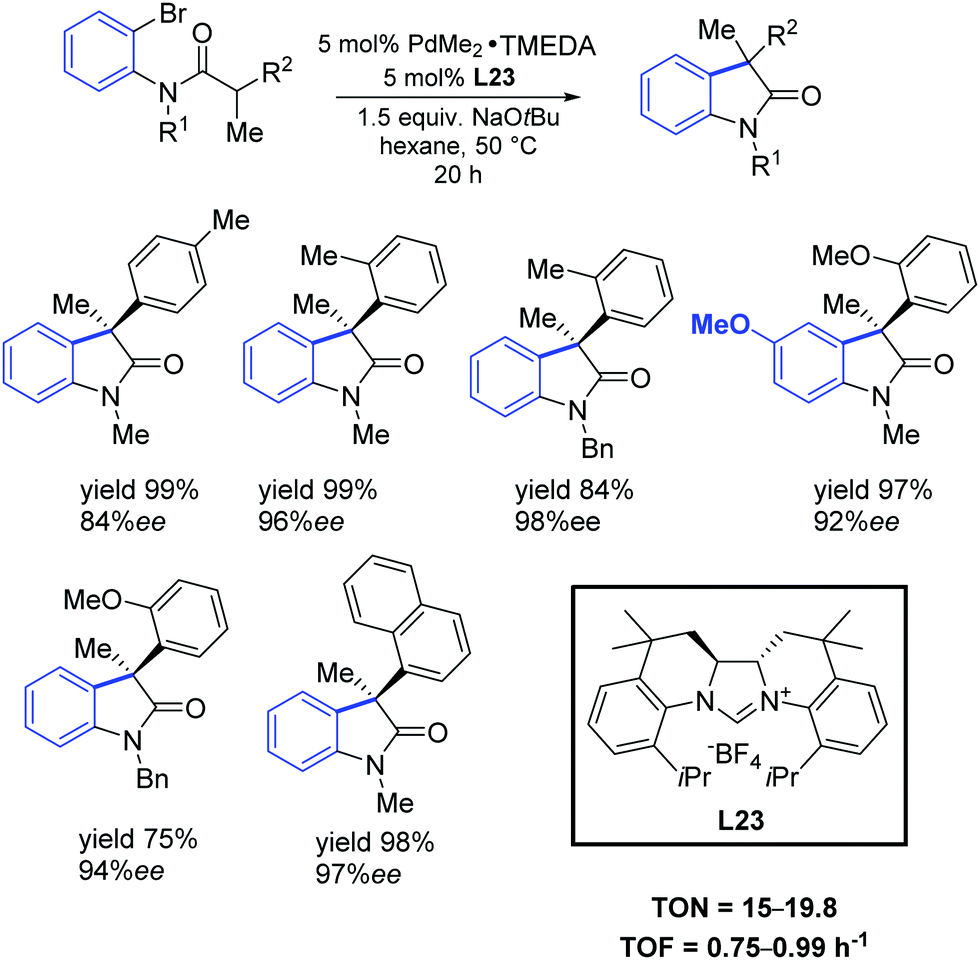 | ||
| Scheme 40 Asymmetric intramolecular α-arylation of amides, as described by Murakami and co-workers.70 | ||
In 2012, Trapp and co-workers prepared a series of three palladium complexes (53–55) bearing ring-expanded (hexahydropyrimidine core) NHC ligands with natural D-(+)-camphor chiral groups.71 These pre-catalysts showed different reaction profiles in the asymmetric intramolecular α-arylation of amides. In particular, all palladium complexes provided good conversions of bromo as well as chloro substrates with 2.5 mol% catalyst loading (Scheme 41). With the sterically demanding catalyst 53, a reaction profile leading either to an arylation or dehalogenation product depending on the used substrate was observed. Higher enantioselectivities compared to the five-membered bornylamine-derived congeners were observed even at higher temperatures.
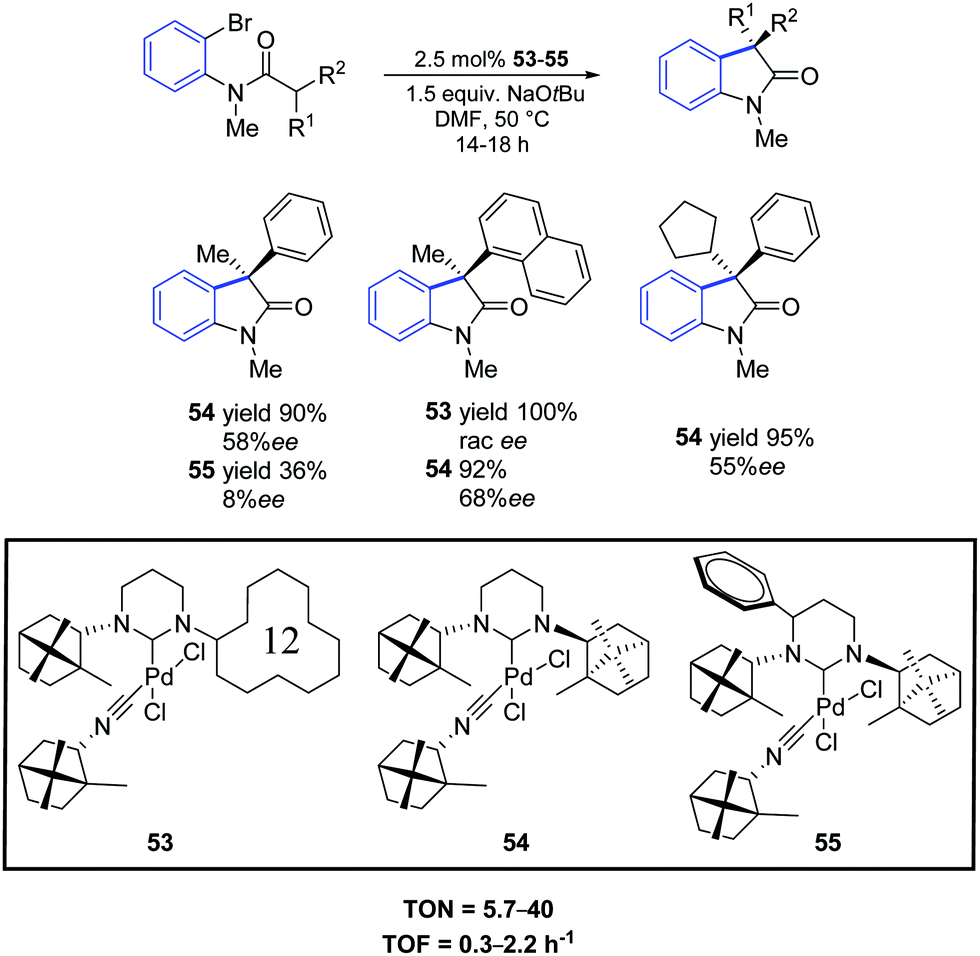 | ||
| Scheme 41 Asymmetric oxindole synthesis using NHC–Pd–isocyanide complexes 53–55, as described by Trapp and co-workers.71 | ||
In 2013, Kündig et al. extended their previous discovery [66] to include the synthesis of new bulky NHC precursors that were used in the asymmetric intramolecular α-arylation of amides producing 3,3-disubstituted oxindoles.72 One of them, L24, containing tert-butyl and 1-naphthyl substituents at the stereogenic centre, proved to efficiently promote the asymmetric synthesis of new spiro- and azaspiro-oxindoles in high yields (up to 99%) and enantioselectivities (up to 97% ee) (Scheme 42).
 | ||
| Scheme 42 Asymmetric synthesis of spirooxindoles as described by Kündig et al.72 | ||
The authors also performed detailed DFT calculations addressing the reaction mechanism. DFT data suggested that the oxidative insertion of palladium–NHC into Caryl–Br bond initially generates a palladacycle, which upon deprotonation yields an O-enolate. The subsequent conversion of O-enolate to C-enolate followed by reductive elimination explains the asymmetric induction to give highly enantioenriched oxindoles (Scheme 43).72
 | ||
| Scheme 43 Energetically most preferred mechanistic pathway for Pd–NHC catalyzed synthesis of oxindole (S) as described by Kündig and co-workers.72 | ||
In 2015, Wilhelm and colleagues synthesized new sterically hindered chiral imidazolinium salts via a simple route from low-cost amino alcohols incorporating different sterically demanding silyl groups. These new derivatives were analyzed and investigated as carbene precursors in a palladium-catalyzed asymmetric intramolecular α-arylation of an amide. The easily accessible carbene precursor L25 gave enantiomeric excesses of up to 72% with nearly quantitative yields (Scheme 44).73
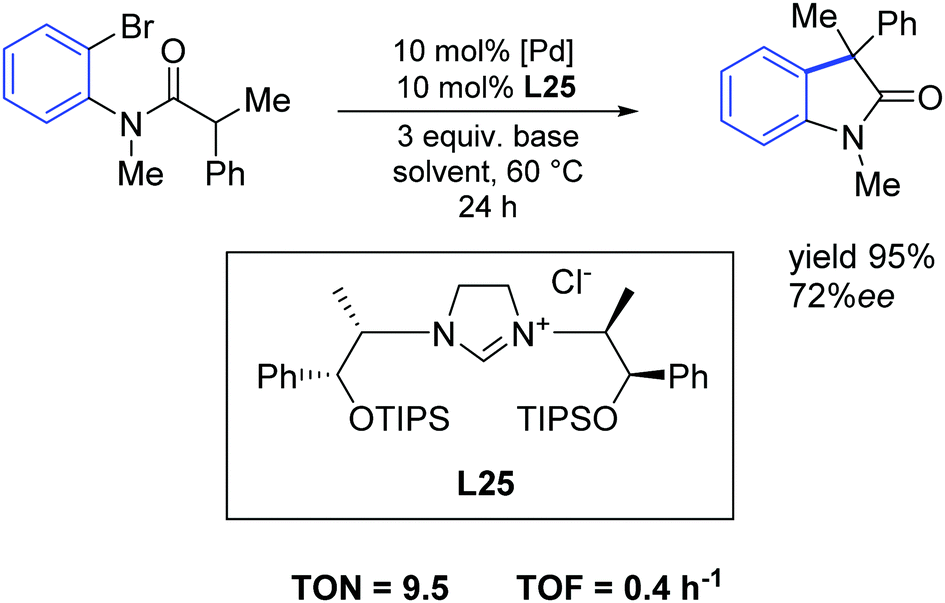 | ||
| Scheme 44 Palladium catalyzed α-arylation described by Wilhelm and co-workers.73 | ||
One year later, Shi and Li found that their synthesized C2-symmetric chiral benzimidazolium salts L26–L29, in conjunction with [Pd(allyl)Cl]2, efficiently catalyzed the synthesis of chiral diarylmethanol derivatives with high yields and moderate enantioselectivities (Scheme 45).74
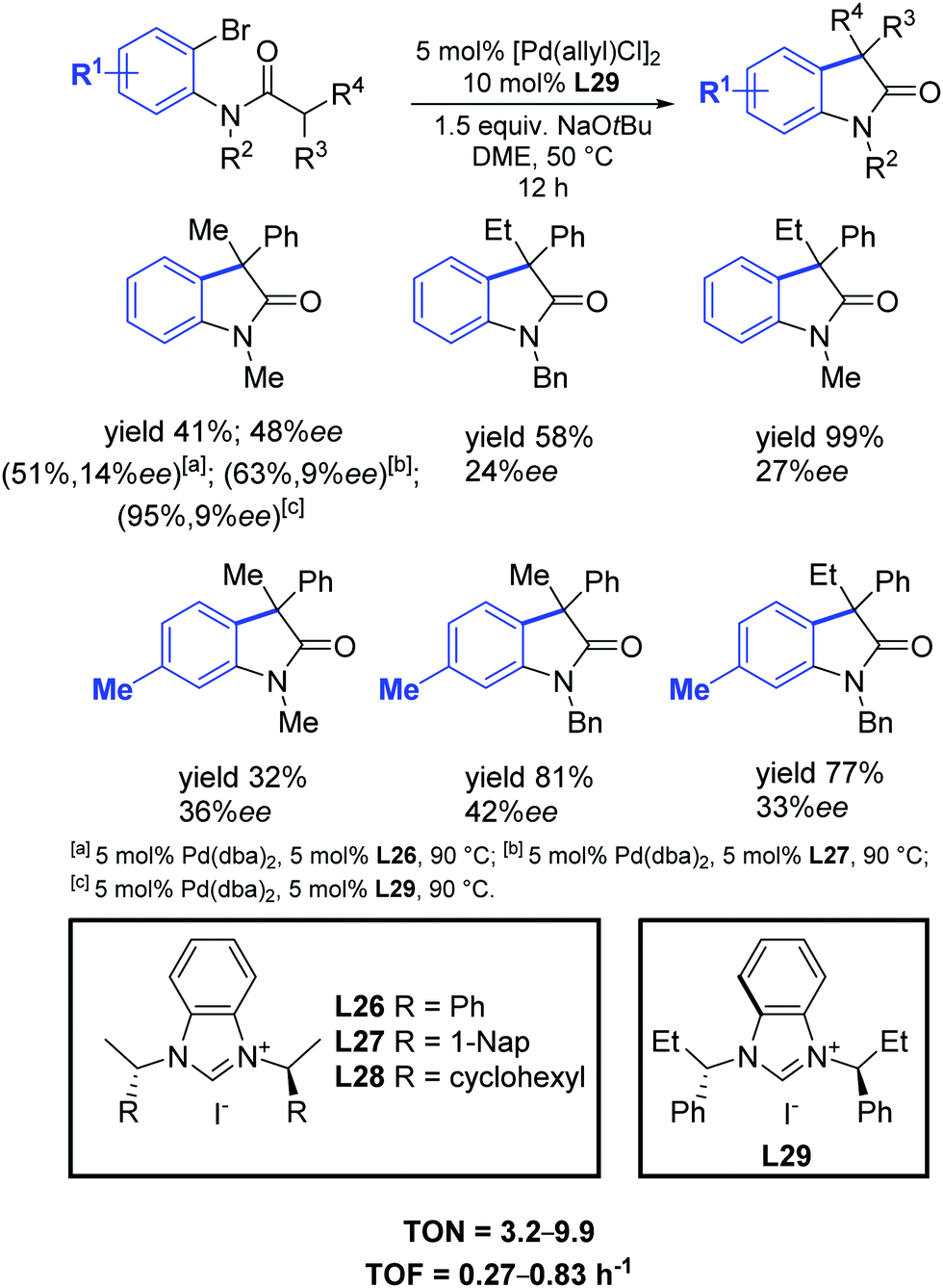 | ||
| Scheme 45 Chiral NHC precursors in the palladium catalyzed intramolecular cyclization of amides into oxindoles, as described by Shi and Li.74 | ||
In 2018, Gandhi and coworkers presented the synthesis of fluorene-based mono- and bimetallic Pd–PEPPSI complexes (56–61) which were demonstrated to be effective for the one-pot sequential α-arylation/alkylation of oxindoles (Fig. 10).75
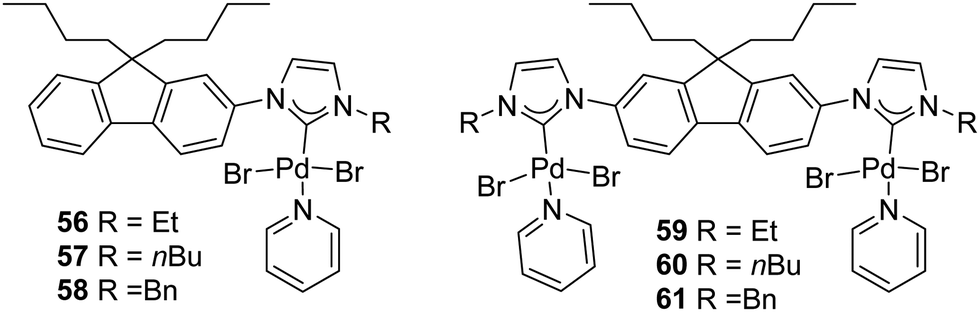 | ||
| Fig. 10 Mono- and bis(palladium)–PEPPSI complexes described by Gandhi et al.75 | ||
This streamlined approach provided functionalized 3,3-disubstituted oxindoles in excellent yields under mild reaction conditions (Scheme 46). Complex 61 showed higher catalytic activity towards various aryl halides and N-substituted oxindoles.75
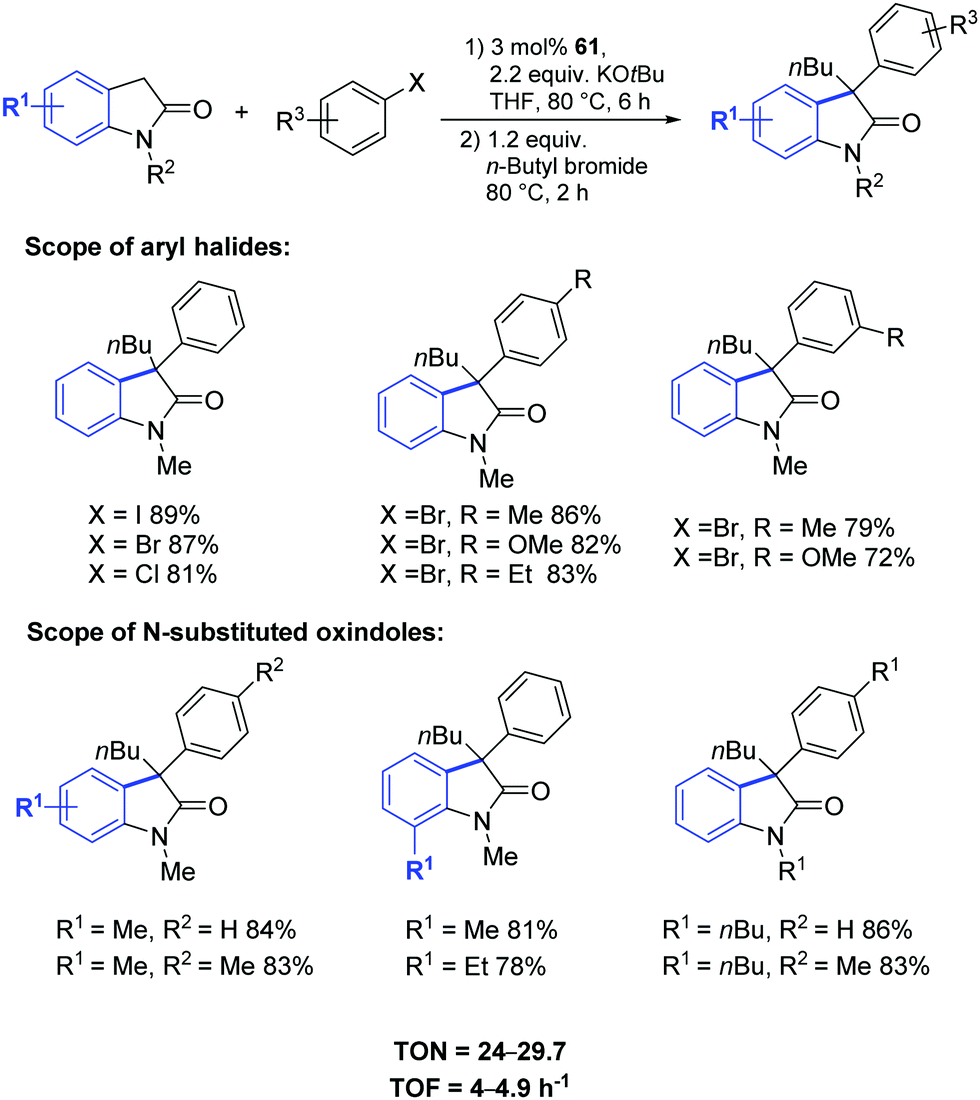 | ||
| Scheme 46 Coupling between aryl halides and N-substituted oxindoles. | ||
Moreover, the authors proposed a mechanism based on previous literature reports (Scheme 47).76 The first step is the oxidative addition between the palladium(0) derivative and the aryl bromide leading to intermediate A which then undergoes bromide substitution by potassium enolate of the oxindole moiety, leading to the formation of intermediate B. Reductive elimination from the intermediate B leads to the formation of C which then undergoes alkylation in the presence of n-butyl bromide affording the desired 3,3-disubstituted oxindole.
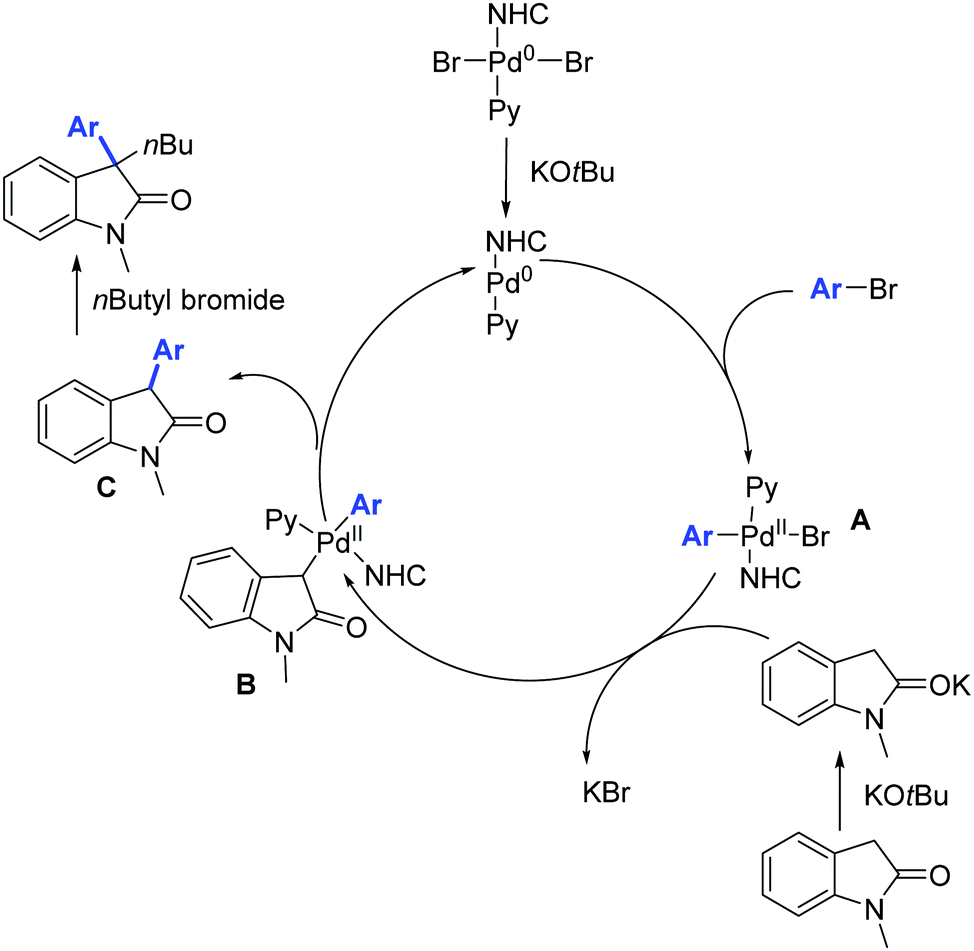 | ||
| Scheme 47 Mechanism proposed by Gandhi and co-workers. | ||
In 2019, Maity and co-workers synthesized a family of dinuclear PEPPSI-type complexes (62–64) which proved to be active pre-catalysts for the α-arylation of oxindole (Scheme 48).77
 | ||
| Scheme 48 Catalytic α-arylation of oxindole, as described by Maity and co-workers.77 | ||
Finally, in 2019, Babu and Shukla investigated the diastereoselective Pd-catalyzed intramolecular amide α-arylation of tertiary C(sp3)–H bond using mono spiro-oxindole-based carboxamide substrates and the synthesis of various sterically hindered 1,2-bisspirooxindolopyrrolidines possessing contiguous vicinal quaternary stereocenters with high diastereoselectivity.78 Among the tested catalytic systems, the most effective was the PEPPSI complex 13 in the presence of NaOtBu (Scheme 49).
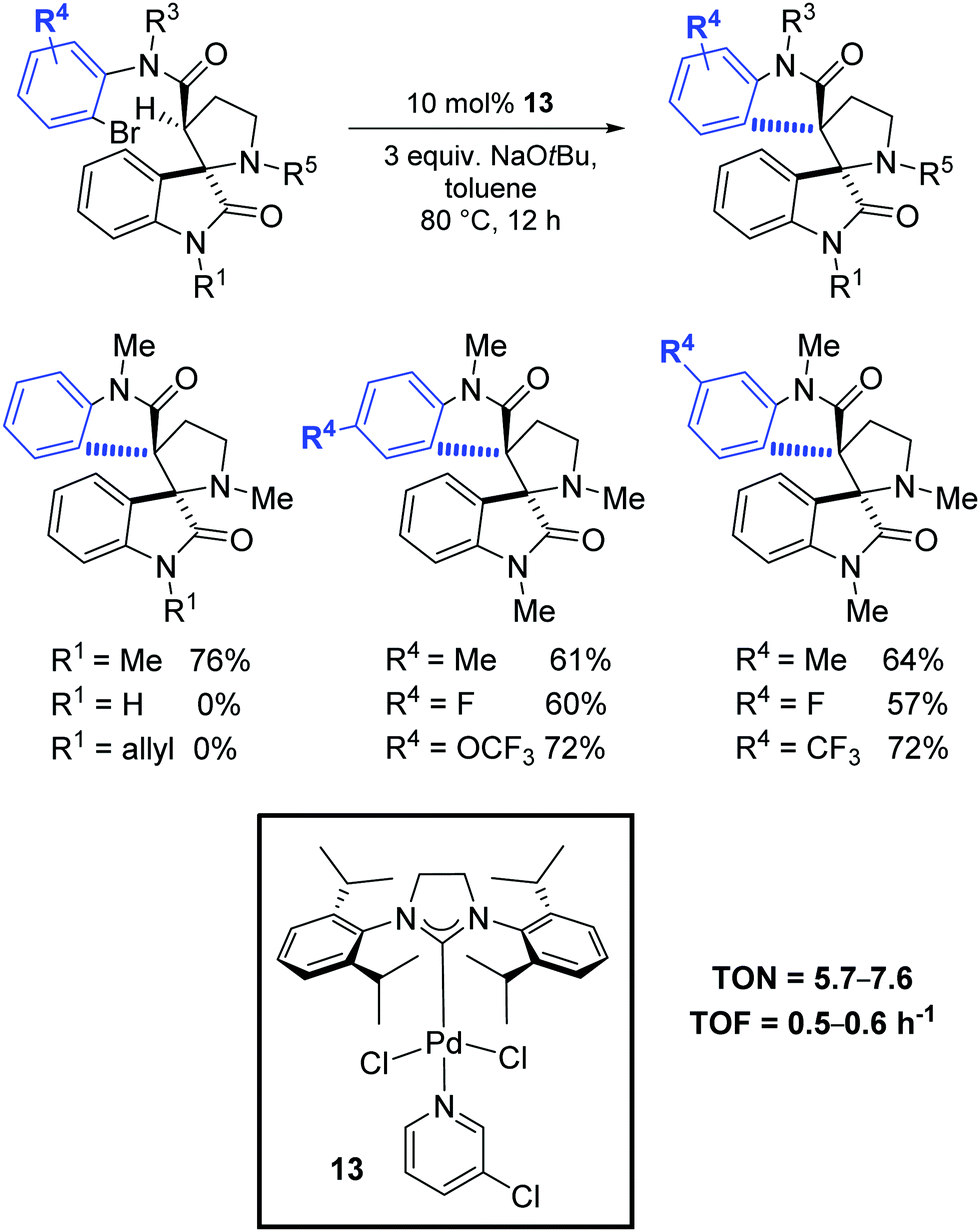 | ||
| Scheme 49 Diastereoselective Pd-catalyzed intramolecular amide α-C–H arylation, as described by Babu and Shukla.78 | ||
6. α-Arylation of carbonyl compounds with pseudo-halide substrates
In light of the importance of α-arylation of carbonyl compounds, many researchers have focused on modification of this reaction in pursuit of high efficiency, high selectivity, and a wider scope. Compared to aryl halides and phenol derivatives, aryl sulfides have been scarcely explored as aryl electrophiles in catalytic transformations as they are thought to poison transition-metal catalysts (and because of their unpleasant smells).79Known catalytic C–S bond-cleavage reactions required large amounts of catalysts (over 2 mol%, but generally 5–20 mol%)80 and/or stoichiometric amounts of transition metal salts (typically copper salts). Since C–S moieties are commonly found in synthetic intermediates, it is important to develop efficient transformations of these bonds with the minimum amount of catalyst.
In this respect, Osuka and co-workers presented a new efficient palladium-catalyzed α-arylation reaction of ketimines with aryl sulfides. Low catalyst loadings (ca. 0.5 mol%) of palladium–NHC complexes 65–66 were sufficient for efficient arylation (Scheme 50).81 Noteworthy, α-arylated ketimine products are useful for the synthesis of various azaarenes, including 2,3-diarylpyrroles, indoles and pyrrole-diones.
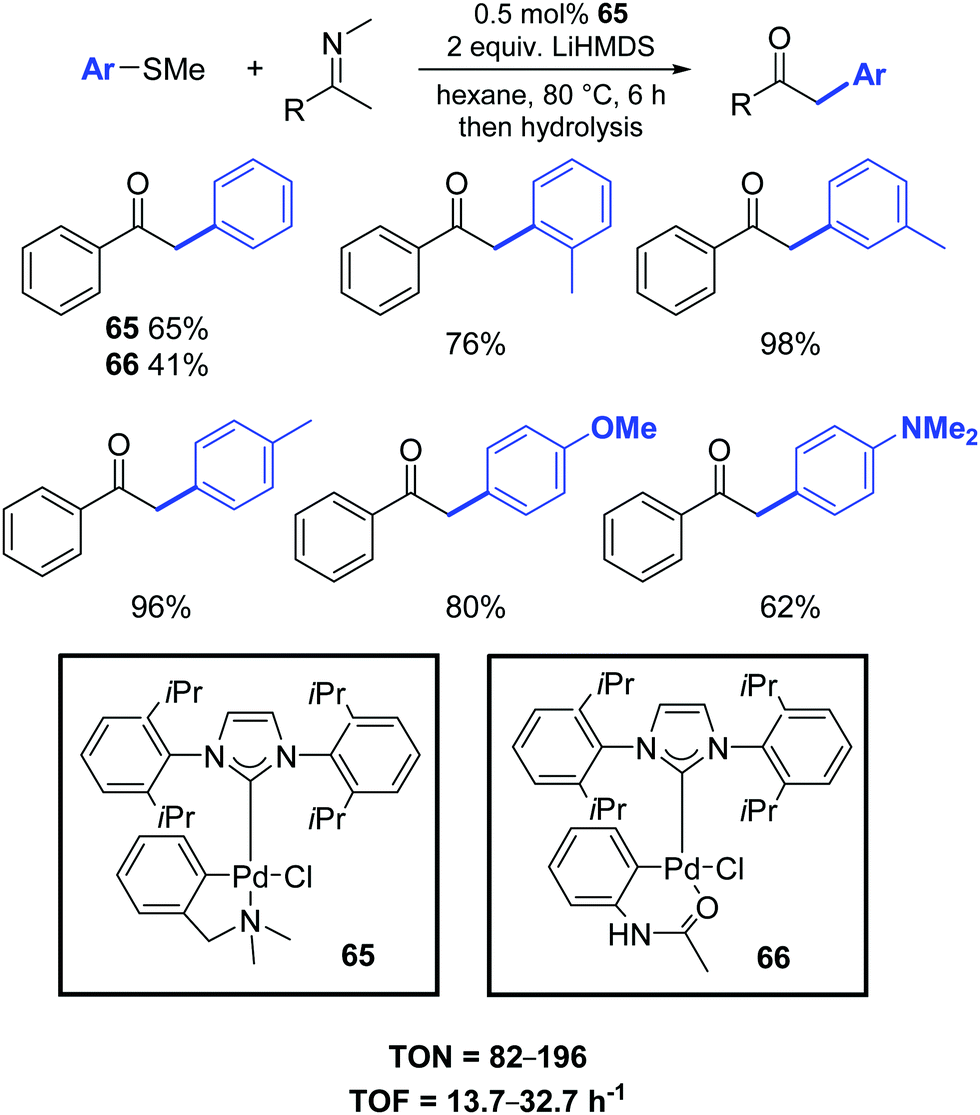 | ||
| Scheme 50 Palladium catalyzed α-arylation of ketoimines, as described by Osuka et al. | ||
7. Conclusion and perspectives
Over the past twenty years, great advances have been made in the field of α-arylation of carbonyl compounds catalyzed by transition-metal complexes. As illustrated throughout this contribution, tremendous advances have taken place using palladium– and nickel–NHC complexes. The unique electronic and steric properties of NHCs, in particular, their greater σ-donor character compared to tertiary phosphine ligands and capacity to form well-defined complexes with diverse transition metals, combined with their remarkable stability to oxidative conditions have enabled significant improvements of the existing α-arylation methods.Today, a large number of carbonyl compounds-including ketones, esters, amides, aldehydes, can be coupled with electron-rich, electron-neutral, electron-poor, and sterically hindered aryl halides or pseudo-halides. An especially important direction has been the development of asymmetric α-arylation promoted by chiral NHC ligands. Despite significant progress, there are numerous challenges that still require solutions.
The studies carried out in the last decades have in fact highlighted a good selectivity towards the products of interest but often low TON and TOF values, which represent a crucial issue in industry. It should be remembered that high catalyst loadings heavily influence the cost and sustainability of any given process.
Moreover, compared to phosphines, much fewer NHC ligands are commercially available. This needs to be addressed so that the beneficial impact of NHC ligands will be available to all researchers and therefore facilitate screening of reaction conditions in a rapid and modular fashion. For this simple reason of availability, only a small number of NHC ligands have been tested in α-arylation reactions. The discovery of new NHC ligands can facilitate further progress in the field and contribute to the full study of the influence of steric and electronic effects on the course of the reaction. The method of generation of the active catalyst needs to be explored in more detail. The most desirable approach, we feel, is the use of well-defined NHC–metal complexes because this eliminates the ligand precursor metalation step and reduces the formation of side-products. There is a need to extend research to the α-arylation reactions catalyzed by NHC complexes with metals other than palladium and nickel, understanding that nickel has been little studied thus far. Studies focusing catalytic functionalization via α-arylation reactions of various other (unsaturated) compounds such as nitriles, 1,3-dicarbonyl compounds, aldehydes, nitroalkanes, sulfoximines, and sulfones that have been successfully tested in the presence of phosphine ligands should be extended to well-defined NHC-bearing systems.
In summary, it is clear that the use of transition metal NHCs has significantly expanded the portfolio of α-arylation methods and has the potential to address future challenges in this field by capitalizing on novel design. Such approaches using NHC ligands should be routinely leveraged by researchers because the catalytic α-arylation reaction represents a powerful tool for the efficient, straightforward, regioselective and chemoselective preparation of fine chemicals including pharmacologically active compounds, their synthetic building blocks, and interesting naturally occurring substances.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ghent University Special Research Fund (BOF) starting and advanced grants to SPN as well as the SBO (D2M) and the iBOF C3 project for financial support.Notes and references
- (a) H. Venkatesan, M. C. Davis, Y. Altas, J. P. Snyder and D. C. Liotta, J. Org. Chem., 2001, 66, 3653 CrossRef CAS PubMed; (b) T. Y. Shen, Angew. Chem., Int. Ed. Engl., 1972, 11, 460 CrossRef CAS PubMed; (c) W. B. Wright, J. B. Press, P. S. Chan, J. W. Marsico, M. F. Haug, J. Lucas, J. Tauber and A. S. Tomcufcik, J. Med. Chem., 1986, 29, 523 CrossRef CAS PubMed; (d) R. R. Goehring, Y. P. Sachdeva, J. S. Pisipati, M. C. Sleevi and J. F. Wolfe, J. Am. Chem. Soc., 1985, 107, 435 CrossRef CAS; (e) S. Edmondson, S. J. Danishefsky, L. Sepp-Lorenzino and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147 CrossRef CAS; (f) H. R. Sonawane, N. S. Bellur, J. R. Ahuja and D. G. Kulkarni, Tetrahedron: Asymmetry, 1992, 3, 163 CrossRef CAS; (g) T. Takeda, R. Gonda and K. Hatano, Chem. Pharm. Bull., 1997, 45, 697 CrossRef CAS; (h) T. Stratmann, R. E. Moore, R. Bonjouklian, J. B. Deeter, G. M. L. Patterson, S. Shaffer, C. D. Smith and T. A. Smitka, J. Am. Chem. Soc., 1994, 116, 9935 CrossRef; (i) J. P. Rieu, A. Boucherle, H. Cousse and G. Mouzin, Tetrahedron, 1986, 42, 4095 CrossRef CAS.
- R. K. Norris, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and M. F. Semmelhack, Pergamon, New York, 1991, ch. 2.2, vol. 4, and references therein Search PubMed.
- (a) J. Morgan, J. T. Pinhey and B. A. Rowe, J. Chem. Soc., Perkin Trans., 1997, 1, 1005 RSC; (b) J. H. Ryan and P. J. Stang, Tetrahedron Lett., 1997, 38, 5061 CrossRef CAS; (c) T. Mino, T. Matsuda, K. Maruhashi and M. Yamashita, Organometallics, 1997, 16, 3241 CrossRef CAS; (d) M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108 CrossRef CAS; (e) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed.
- (a) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676 CrossRef CAS PubMed; (b) P. Novák and R. Martin, Curr. Org. Chem., 2011, 15, 3233 CrossRef; (c) Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955 CrossRef CAS.
- (a) T. Scattolin and S. P. Nolan, Trends Chem., 2020, 2, 721–736 CrossRef CAS; (b) H. V. Huynh, Chem. Rev., 2018, 118, 9457 CrossRef CAS PubMed; (c) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC; (d) S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gomez-Suarez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895 RSC; (e) A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269 CrossRef CAS; (f) S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487 CrossRef CAS; (g) G. Meng, L. Kakalis, S. P. Nolan and M. Szostak, Tetrahedron Lett., 2019, 60, 378 CrossRef CAS; (h) A. Comas-Vives and J. N. Harvey, Eur. J. Inorg. Chem., 2011, 5025 CrossRef CAS; (i) E. L. Rosen, C. D. Varnado, A. G. Tennyson, D. M. Khramov, J. W. Kamplain, D. H. Sung, P. T. Cresswell, V. M. Lynch and C. W. Bielawski, Organometallics, 2009, 28, 6695 CrossRef CAS; (j) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687 CrossRef CAS; (k) D. G. Gusev, Organometallics, 2009, 28, 763 CrossRef CAS; (l) R. A. Kelly, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202 CrossRef CAS; (m) S. Diez-Gonzalez and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef CAS.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- (a) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407 CrossRef CAS; (b) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650 RSC; (c) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440 CrossRef CAS PubMed; (d) S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589 CrossRef CAS PubMed; (e) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC; (f) O. Kühl, Coord. Chem. Rev., 2009, 253, 2481 CrossRef; (g) S. Wuertz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523 CrossRef CAS PubMed; (h) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef; (i) T. Scattolin, L. Canovese, F. Visentin, S. Paganelli, P. Canton and N. Demitri, Appl. Organomet. Chem., 2018, 32, e4034 CrossRef; (j) L. Canovese, F. Visentin, T. Scattolin, C. Santo and V. Bertolasi, Polyhedron, 2016, 119, 377 CrossRef CAS; (k) T. Scattolin, I. Caligiuri, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, F. Rizzolio, C. Santo and F. Visentin, Dalton Trans., 2018, 47, 13616 RSC; (l) A. Simoens, T. Scattolin, T. Cauwenbergh, G. Pisanò, C. S. J. Cazin, C. V. Stevens and S. P. Nolan, Chem. – Eur. J., 2021, 27, 5653–5657 CrossRef CAS PubMed.
- T. Kametani, S. Noguchi, I. Agata, T. Aono, K. Kigasawa, M. Hiiragi, T. Hayasaka and O. Kusama, J. Chem. Soc. C, 1971, 1047 RSC.
- J.-P. Finet, Chem. Rev., 1989, 89, 1487 CrossRef CAS.
- P. R. Jones and J. R. Young, J. Org. Chem., 1968, 33, 1675 CrossRef CAS.
- M. W. Rathke and D. Vogiazoglou, J. Org. Chem., 1987, 52, 3697 CrossRef CAS.
- J. Morgan, J. T. Pinhey and B. A. Rowe, J. Chem. Soc., Perkin Trans., 1997, 1, 1005 RSC.
- C. Liu, C. He, W. Shi, M. Chen and A. Lei, Org. Lett., 2007, 9, 5601 CrossRef CAS PubMed.
- T. Mino, T. Matsuda, K. Maruhashi and M. Yamashita, Organometallics, 1997, 16, 3241 CrossRef CAS.
- V. K. Aggarwal and B. Olofsson, Angew. Chem., Int. Ed., 2005, 44, 5516 CrossRef CAS PubMed.
- M. S. Newman and M. D. Farbman, J. Am. Chem. Soc., 1944, 66, 1550 CrossRef CAS.
- C. Dell’Erba, M. Novi, G. Petrillo and C. Tavani, Tetrahedron, 1993, 49, 235 CrossRef.
- (a) H. Muratake, A. Hayakawa and M. Nataume, Tetrahedron Lett., 1997, 38, 7577 CrossRef CAS; (b) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740 CrossRef CAS; (c) G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2002, 41, 953 CrossRef CAS; (d) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473 CrossRef CAS; (e) A. Ehrentraut, A. Zapf and M. Beller, Adv. Synth. Catal., 2002, 344, 209 CrossRef CAS; (f) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360 CrossRef CAS; (g) Y. Terao, T. Satoh, M. Miura and M. Nomura, Tetrahedron, 2000, 56, 1315 CrossRef CAS; (h) T. Satoh, Y. Kametani, Y. Terao, M. Miura and M. Nomura, Tetrahedron Lett., 1999, 40, 5345 CrossRef CAS; (i) Y. Terao, Y. Kametani, H. Wakui, T. Satoh, M. Miura and M. Nomura, Tetrahedron, 2001, 57, 5967 CrossRef CAS; (j) T. Sperger and F. Schoenebeck, Synthesis, 2018, 4471 CAS; (k) M. Orlandi and G. Licini, J. Org. Chem., 2020, 85, 11511 CrossRef CAS PubMed.
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (b) T. Scattolin, I. Caligiuri, N. Mouawad, M. El Boustani, N. Demitri, F. Rizzolio and F. Visentin, Eur. J. Med. Chem., 2019, 179, 325 CrossRef CAS PubMed; (c) T. Scattolin, N. Pangerc, I. Lampronti, C. Tupini, R. Gambari, L. Marvelli, F. Rizzolio, N. Demitri, L. Canovese and F. Visentin, J. Organomet. Chem., 2019, 899, 120857 CrossRef CAS; (d) T. Scattolin, E. Bortolamiol, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio, L. Cavallo, B. Dereli, M. V. Mane, S. P. Nolan and F. Visentin, Chem. Commun., 2020, 56, 12238 RSC; (e) T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 11868 CrossRef CAS PubMed; (f) T. Scattolin, S. Giust, P. Bergamini, I. Caligiuri, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, F. Rizzolio and F. Visentin, Appl. Organomet. Chem., 2019, 33, e4902 CrossRef.
- (a) M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2020, 4, 4053 CrossRef PubMed; (b) M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470 CrossRef CAS.
- M. S. Viciu, R. A. Kelly III, E. D. Stevens, F. Naud, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479 CrossRef CAS PubMed.
- O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly III and S. P. Nolan, J. Org. Chem., 2006, 71, 685 CrossRef CAS PubMed.
- R. Singh and S. P. Nolan, J. Organomet. Chem., 2005, 690, 5832 CrossRef CAS.
- O. Navarro, N. Marion, N. M. Scott, J. González, D. Amoroso, A. Bell and S. P. Nolan, Tetrahedron, 2005, 61, 9716 CrossRef CAS.
- (a) N. Marion, P. D. Frémont, I. M. Puijk, E. C. Ecarnot, D. Amoroso, A. Bell and S. P. Nolan, Adv. Synth. Catal., 2007, 349, 2380 CrossRef CAS; (b) N. Marion, E. C. Ecarnot, O. Navarro, D. Amoroso, A. Bell and S. P. Nolan, J. Org. Chem., 2006, 71, 3816 CrossRef CAS PubMed; (c) E. Marelli, M. Corpet, S. R. Davies and S. P. Nolan, Chem. – Eur. J., 2014, 1 Search PubMed.
- K. Matsubara, H. Okazaki and M. Senju, J. Organomet. Chem., 2006, 691, 3693 CrossRef CAS.
- J. Zhang, X. Yang, X. Cui and Y. Wu, Tetrahedron, 2011, 67, 8800 CrossRef CAS.
- C. J. O’Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2006, 12, 4743 CrossRef PubMed.
- C. Cao, L. Wang, Z. Cai, L. Zhang, J. Guo, G. Pang and Y. Shi, Eur. J. Org. Chem., 2011, 1570 CrossRef CAS.
- Z.-K. Xiao and L.-X. Shao, Synthesis, 2012, 8800 Search PubMed.
- H.-Y. Yin, M.-Y. Liu and L.-X. Shao, Org. Lett., 2013, 15, 6042 CrossRef CAS PubMed.
- C. Richter, K. V. S. Ranganath and F. Glorius, Adv. Synth. Catal., 2012, 354, 377 CrossRef CAS.
- H. Valdes, M. Poyatos, G. Ujaque and E. Peris, Chem. – Eur. J., 2014, 20, 1 CrossRef.
- H.-Y. Yin, X.-L. Lin, S.-W. Li and L.-X. Shao, Org. Biomol. Chem., 2015, 13, 9012 RSC.
- F. Liu, Y.-Y. Hu, D. Li, Q. Zhou and J.-M. Lu, Tetrahedron, 2018, 74, 5683 CrossRef CAS.
- C. M. Zinser, K. G. Warren, F. Nahra, A. Al-Majid, A. Barakat, M. S. Islam, S. P. Nolan and C. S. J. Cazin, Organometallics, 2019, 38, 2812 CrossRef CAS.
- H.-Y. Lu, A. Shen, Y.-Q. Li, Y.-C. Hu, C. Ni and Y.-C. Cao, Tetrahedron Lett., 2020, 61, 152124 CrossRef CAS.
- V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 117, 5851 CrossRef.
- (a) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195 CrossRef CAS PubMed; (b) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690 CrossRef CAS PubMed.
- (a) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed; (b) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 55, 10046 CrossRef PubMed; (c) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed.
- C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255 CrossRef CAS PubMed.
- (a) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed; (b) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (c) J. D. Crowley, A. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118 CrossRef CAS; (d) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC.
- (a) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304 CrossRef CAS PubMed; (b) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS.
- D. Mendoza-Espinosa, R. Gonzalez-Olvera, G. E. Negron-Silva, D. Angeles-Beltran, O. R. Suarez-Castillo, A. Alvarez-Hernandez and R. Santillan, Organometallics, 2015, 34, 4529 CrossRef CAS.
- K. Matsubara, K. Ueno, Y. Koga and K. Hara, J. Org. Chem., 2007, 72, 5069 CrossRef CAS PubMed.
- (a) M. Henrion, M. J. Chetcuti and V. Ritleng, Chem. Commun., 2014, 50, 4624 RSC; (b) M. Henrion, B. de P. Cardoso, V. César, M. J. Chetcuti and V. Ritleng, Organometallics, 2017, 36, 1113 CrossRef CAS.
- J. A. Fernández-Salas, E. Marelli, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2015, 21, 1 CrossRef PubMed.
- E. Marelli, J. A. Fernández-Salas and S. P. Nolan, Synthesis, 2015, 2032 CAS.
- M. Grigalunas, T. Ankner, P.-O. Norrby, O. Wiest and P. Helquist, J. Am. Chem. Soc., 2015, 137, 7019 CrossRef CAS PubMed.
- J. Li and Z.-X. Wang, Org. Biomol. Chem., 2016, 14, 7579 RSC.
- (a) Y. Terao, T. Satoh, M. Miura and M. Nomura, Tetrahedron Lett., 1998, 39, 6203 CrossRef CAS; (b) H. Muratake and H. Nakai, Tetrahedron Lett., 1999, 40, 2355 CrossRef CAS; (c) Y. Terao, Y. Fukuoka, T. Satoh, M. Miura and M. Nomura, Tetrahedron Lett., 2002, 43, 101 CrossRef CAS; (d) R. Martín and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7236 CrossRef PubMed; (e) G. D. Vo and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 2127 CrossRef CAS PubMed; (f) J. Aleman, S. Cabrera, E. Maerten, J. Overgaard and K. A. Jřrgensen, Angew. Chem., Int. Ed., 2007, 46, 5520 CrossRef CAS PubMed; (g) C. Mazet, Chimia, 2013, 67, 658 CrossRef CAS PubMed; (h) I. Franzoni, L. Guenee and C. Mazet, Tetrahedron, 2014, 70, 4181 CrossRef CAS; (i) N. Humbert, E. Larionov, L. Mantilli, P. Nareddy, C. Besnard, L. Guenee and C. Mazet, Chem. – Eur. J., 2014, 20, 745 CrossRef CAS PubMed; (j) S. Plunkett, L. G. DeRatt, S. D. Kuduk and J. Balsells, Org. Lett., 2020, 22, 7662 CrossRef CAS.
- P. Nareddy and C. Mazet, Chem. – Asian J., 2013, 8, 2579 CrossRef CAS PubMed.
- J. March, Advanced Organic Chemistry, John Wiley and Sons, New York, 4th edn, 2001, ch. 10 Search PubMed.
- (a) W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996 CrossRef CAS PubMed; (b) T. Hama and J. F. Hartwig, Org. Lett., 2008, 10, 1545 CrossRef CAS PubMed; (c) T. Hama and J. F. Hartwig, Org. Lett., 2008, 10, 1549 CrossRef CAS PubMed; (d) X. Liu and J. F. Hartwig, Org. Lett., 2003, 5, 1915 CrossRef CAS PubMed; (e) B. M. Trost and X. Ariza, J. Am. Chem. Soc., 1999, 121, 10727 CrossRef CAS; (f) O. Gaertzen and S. L. Buchwald, J. Org. Chem., 2002, 67, 465 CrossRef CAS PubMed; (g) M. R. Biscoe and S. L. Buchwald, Org. Lett., 2009, 11, 1773 CrossRef CAS PubMed; (h) D. Solé and O. Serrano, J. Org. Chem., 2008, 73, 2476 CrossRef PubMed; (i) T. Hama, X. Liu, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 11176 CrossRef CAS PubMed; (j) T. Hama, S. Ge and J. F. Hartwig, J. Org. Chem., 2013, 78, 8250 CrossRef CAS PubMed; (k) E. Koch, R. Takise, A. Studer, J. Yamaguchi and K. Itami, Chem. Commun., 2015, 51, 855 RSC; (l) T. Sperger and F. Schoenebeck, Synthesis, 2018, 4471 CAS.
- F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS PubMed.
- (a) M. Jorgensen, S. Lee, X. Liu, J. P. Wolkowski and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 12557 CrossRef CAS PubMed; (b) S. Lee, N. A. Beare and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 8410 CrossRef CAS.
- (a) R. Freund and W. W. K. R. Mederski, Helv. Chim. Acta, 2000, 83, 1247 CrossRef CAS; (b) B. Evans, J. L. Leighton, K. E. Rittle, K. F. Gilbert, G. F. Lundell, N. P. Gould, D. W. Hobbs, R. M. DiPardo, D. F. Veber, D. J. Pettibone, B. V. Clineschmidt, P. S. Anderson and R. M. Freidinger, J. Med. Chem., 1992, 35, 3919 CrossRef CAS PubMed; (c) S. M. N. Efange, A. P. Kamath, A. B. Khare, M.-P. Kung, R. H. Mach and S. M. Parsons, J. Med. Chem., 1997, 40, 3905 CrossRef CAS PubMed; (d) T. Honda, H. Namiki and F. Satoh, Org. Lett., 2001, 3, 631 CrossRef CAS PubMed; (e) S. T. Sivanandan, A. Shaji, I. Ibnusaud, C. C. C. Johansson Seechurn and T. J. Colacot, Eur. J. Org. Chem., 2015, 38 CrossRef CAS.
- (a) K. H. Shaughnessy, B. C. Hamann and J. F. Hartwig, J. Org. Chem., 1998, 63, 6546 CrossRef CAS; (b) S. Rivara, A. Lodola, M. Mor, A. Bedini, G. Spadoni, V. Lucini, M. Pannacci, F. Fraschini, F. Scaglione, R. O. Sanchez, G. Gobbi and G. Tarzia, J. Med. Chem., 2007, 50, 6618 CrossRef CAS PubMed; (c) C. Botuha, C. M. S. Galley and T. Gallagher, Org. Biomol. Chem., 2004, 2, 1825 RSC; (d) T. Honda, H. Namiki and F. Satoh, Org. Lett., 2001, 3, 631 CrossRef CAS PubMed; (e) T. Y. Zhang and H. Zhang, Tetrahedron Lett., 2002, 43, 1363 CrossRef CAS; (f) T. Y. Zhang and H. Zhang, Tetrahedron Lett., 2002, 43, 193 CrossRef CAS; (g) G. C. Bignan, K. Battista, P. J. Connolly, M. J. Orsini, J. Liu, S. A. Middletona and A. B. Reitz, Bioorg. Med. Chem. Lett., 2005, 15, 5022 CrossRef CAS PubMed; (h) S. P. Marsden, E. L. Watson and S. A. Raw, Org. Lett., 2008, 10, 2905 CrossRef CAS PubMed; (i) B. M. Trost and M. U. Frederiksen, Angew. Chem., Int. Ed., 2005, 44, 308 CrossRef CAS PubMed; (j) M. J. Durbin and M. C. Willis, Org. Lett., 2008, 10, 1413 CrossRef CAS PubMed; (k) R. A. Altman, A. M. Hyde, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 9613 CrossRef CAS PubMed; (l) L. Ackermann, R. Vicente and N. Hofmann, Org. Lett., 2009, 11, 4274 CrossRef CAS PubMed; (m) T. Hama, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 4976 CrossRef CAS PubMed; (n) B. Zheng, T. Jia and P. J. Walsh, Org. Lett., 2013, 15, 4190 CrossRef CAS PubMed; (o) B. Zheng, T. Jia and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 165 CrossRef CAS PubMed; (p) E. Barde, A. Guerinot and J. Cossy, Synthesis, 2019, 178 CAS.
- S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402 CrossRef CAS PubMed.
- T. Y. Zhang and H. Zhang, Tetrahedron Lett., 2002, 43, 193 CrossRef CAS.
- C. Zhang, J. Huang, M. L. Trudell and S. P. Nolan, J. Org. Chem., 1999, 64, 3804 CrossRef CAS.
- F. Glorius, G. Altenhoff, R. Goddard and C. Lehmann, Chem. Commun., 2002, 2704 RSC.
- (a) T. Arao, K. Sato, K. Kondo and T. Aoyama, Chem. Pharm. Bull., 2006, 54, 1576 CrossRef CAS PubMed; (b) T. Arao, K. Kondo and T. Aoyama, Tetrahedron Lett., 2006, 47, 1417 CrossRef CAS; (c) T. Arao, K. Kondo and T. Aoyama, Chem. Pharm. Bull., 2006, 54, 1743 CrossRef CAS PubMed.
- (a) E. P. Kündig, T. M. Seidel, Y.-X. Jia and G. Bernardinelli, Angew. Chem., Int. Ed., 2007, 46, 8484 CrossRef PubMed; (b) Y.-X. Jia, J. M. Hillgren, E. L. Watson, S. P. Marsden and E. P. Kündig, Chem. Commun., 2008, 4040 RSC; (c) Y.-X. Jia, D. Katayev, G. Bernardinelli, T. M. Seidel and E. P. Kündig, Chem. – Eur. J., 2010, 16, 6300 CrossRef CAS PubMed; (d) D. Katayev and E. P. Kündig, Helv. Chim. Acta, 2012, 95, 2287 CrossRef CAS.
- S. Wurtz, C. Lohre, R. Frohlich, K. Bergander and F. Glorius, J. Am. Chem. Soc., 2009, 131, 8344 CrossRef PubMed.
- X. Luan, R. Mariz, C. Robert, M. Gatti, S. Blumentritt, A. Linden and R. Dorta, Org. Lett., 2008, 10, 5569 CrossRef CAS PubMed.
- X. Luan, L. Wu, E. Drinkel, R. Mariz, M. Gatti and R. Dorta, Org. Lett., 2010, 12, 1912 CrossRef CAS PubMed.
- L. Wu, L. Falivene, E. Drinkel, S. Grant, A. Linden, L. Cavallo and R. Dorta, Angew. Chem., Int. Ed., 2012, 51, 2870 CrossRef CAS PubMed.
- L. Liu, N. Ishida, S. Ashida and M. Murakami, Org. Lett., 2011, 13, 1666 CrossRef CAS PubMed.
- M. J. Spallek, D. Riedel, F. Rominger, A. S. K. Hashmi and O. Trapp, Organometallics, 2012, 31, 1127 CrossRef CAS.
- D. Katayev, Y.-X. Jia, A. K. Sharma, D. Banerjee, C. Besnard, R. B. Sunoj and E. P. Kündig, Chem. – Eur. J., 2013, 19, 11916 CrossRef CAS PubMed.
- M. A. Gilani, E. Rais and R. Wilhelm, Synlett, 2015, 1638 CAS.
- W. He, W. Zhao, B. Zhou, H. Liu, X. Li, L. Li, J. Li and J. Shi, Molecules, 2016, 21, 742 CrossRef.
- P. Kumar, R. Panyam, B. Ugale and T. Gandhi, J. Org. Chem., 2018, 83, 7622 CrossRef.
- (a) M. J. Durbin and M. C. Willis, Org. Lett., 2008, 10, 1413 CrossRef CAS PubMed; (b) J. Cossy, A. de Filippis and D. G. Pardo, Org. Lett., 2003, 5, 3037 CrossRef CAS PubMed.
- A. Majumder, R. Naskar, P. Roy and R. Maity, Eur. J. Inorg. Chem., 2019, 1810 CrossRef CAS.
- D. Shukla and S. A. Babu, Adv. Synth. Catal., 2019, 361, 2075 CrossRef CAS.
- (a) I. P. Beletskaya and V. P. Ananikov, Eur. J. Org. Chem., 2007, 3431 CrossRef CAS; (b) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (d) C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590 CrossRef CAS PubMed; (e) H. Liu and X. Feng, Chem. – Asian J., 2013, 8, 2546 CrossRef CAS PubMed; (f) C.-F. Lee, Y.-C. Liu and S. S. Badsara, Chem. – Asian J., 2014, 9, 706 CrossRef CAS.
- (a) Y. Ookubo, A. Wakamiya, H. Yorimitsu and A. Osuka, Chem. – Eur. J., 2012, 18, 12690 CrossRef CAS PubMed; (b) K. Murakami, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 7510 CrossRef CAS; (c) T. Sugahara, K. Murakami, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 9329 CrossRef CAS PubMed; (d) S. Otsuka, D. Fujino, K. Murakami, H. Yorimitsu and A. Osuka, Chem. – Eur. J., 2014, 20, 13146 CrossRef CAS PubMed; (e) K. Kunchithapatham, C. C. Eichman and J. P. Stambuli, Chem. Commun., 2011, 47, 12679 RSC; (f) J. Torres-Nieto, A. Arvalo, P. Garcia-Guterrez, A. Acosta-Ramirez and J. J. Garcia, Organometallics, 2004, 23, 4534 CrossRef CAS; (g) J. Torres-Nieto, A. Arevalo and J. J. Garcia, Organometallics, 2007, 26, 2228 CrossRef CAS.
- K. Gao, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2016, 128, 4649 CrossRef.
Footnote |
| † Dedicated to the memory of Professor Victor Snieckus. |
| This journal is © The Royal Society of Chemistry 2021 |