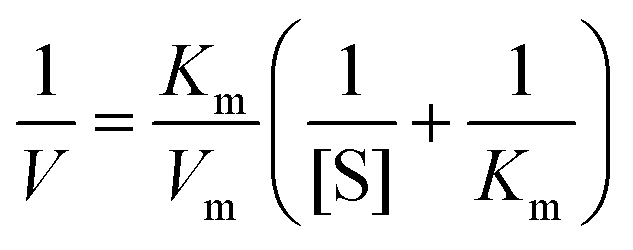Fabrication of noble metal nanoparticles decorated on one dimensional hierarchical polypyrrole@MoS2 microtubes†
Received
1st June 2020
, Accepted 19th July 2020
First published on 20th July 2020
Abstract
MoS2-based hybrids have aroused great interest for their outstanding performance in the application fields of biochemical sensing, catalysis and energy storage. Herein, we present a facile strategy to fabricate hierarchical microtubes by cultivating a MoS2 sheet-like nanostructure on polypyrrole microtubes (designated as PPy@MoS2 microtubes) using MoO3@PPy micro-cables as self-sacrificial templates. Such a dissolution–regrowth mechanism is demonstrated for the formation of hierarchical PPy@MoS2 microtubes by studying the morphology of the intermediate products in the process of the sulfidation reaction. The PPy microtubes are able to effectively improve the electrical conductivity of the hybrid architecture and greatly alleviate the agglomeration of the MoS2 nanosheets. Notably, the sheet-like MoS2 nanostructure can load more noble metal nanoparticles (NPs) owing to MoS2 released photogenerated electrons irradiated by light. Then, metal (Ag, Au, and Pd) NPs are reduced and in situ decorated on PPy@MoS2 microtubes, thus forming ternary PPy@MoS2@Ag, Au, and Pd nanohybrids, respectively. This decoration method also expands the wide range of application fields of PPy@MoS2. As a proof of application, the ternary PPy@MoS2@Au hybrids reveal excellent enzyme-like catalytic performance. Owing to the high coverage of Au NPs as well as one dimensional hierarchical MoS2-based ternary unique structures, the resultant PPy@MoS2@Au hybrid composites exhibited synergistically enhanced peroxidase-like catalytic activity relative to MoS2, MoS2@Au, and PPy@MoS2 alone, demonstrating the remarkable prospects of MoS2-based hybrids in chemical/biological molecule sensing application.
Introduction
Noble metal (Ag, Au, Pd, Pt, etc.) nanoparticles (NPs) have aroused enormous interest in catalysis,1 biosensing2 and even cancer therapy.3 However, owing to their high surface energy, the individual noble metal NPs can be easily aggregated together after being used, greatly limiting their further application. As an alternative, loading noble metal NPs on a solid support is considered as an effective route to resolve the aggregation. Moreover, the resultant support@noble metal NP composites exhibit more intriguing properties than individual noble metal NPs or solid supports with their synergistic effect between noble metal NPs and solid supports.4,5 In addition, a strong interaction between noble metal NPs and supports was proved to adjust the coverage size, dispersion state, stability and the resulting catalytic performance.6,7 Grounded on the above advantages, much effort has been made to fabricate noble metal NPs for the decoration of a variety of supports. For example, Guo et al. have developed a facile and auniversal approach to prepare graphene-based nanocomposites by in situ nucleation and growth of diverse noble metals (Au, Ag, and Pd) on the surface of RGO with the assistance of a PDA coating layer.8 Nanocelluloses can serve as potential supports for metal NPs to form CNC–metal (Ag, Au, and Pd) hybrid NPs without the addition of an external reducing agent due to their reductive surface functional groups and good water dispersibility.9,10 Although a variety of substrates have been developed to fabricate noble metal NP based nanocomposites, the development of facile and universal substrates to prepare noble metal NP based nanocomposites by in situ nucleation and growth of diverse noble metals on the surfaces of the substrates still remains a considerable challenge.
Molybdenum disulfide (MoS2), as a typical two dimensional (2D) material, has demonstrated great potential in various application fields, including sensing,11 electrocatalysis,12 and energy storage.13 To improve the poor electronic conductivity and provide other functionalities of MoS2, decoration of MoS2 materials with noble metal NPs has aroused great interest in the application of catalysis, biosensing etc. Notably, the noble metal NPs can be in situ generated on the surface of MoS2 nanosheets without using an extra reducing agent, greatly facilitating the preparation process. However, the individual MoS2 nanosheets suffered from fewer accessible active sites, and the high surface energy and interlayer van der Waals interaction caused serious aggregation of MoS2 layers. These greatly impede their decoration on noble metal NPs. To overcome these problems, many strategies have been developed to stabilize the individual MoS2 nanosheet with more exposed active sites. The most commonly used method is to decorate the 2D MoS2 nanosheets with carbonaceous materials,14,15 conductive polymers16 and metal oxides/sulfides,17–19 which could serve as supports to prevent the aggregation of MoS2. Among these backbones, the conducting polymers (CPs) are widely employed to fabricate functional CPs@MoS2 owing to their high electrical and thermal conductivity, low cost and good mechanical strength. Moreover, CPs offer a desirable matrix toward fast electron transport in the faradaic redox reaction. Therefore, hybridizing MoS2 nanosheets with CPs, especially polypyrrole (PPy), so as to improve the charge mobility and long-term stability, would be expected to obtain satisfactory overall electrochemical performance.20 For instance, Sun et al. have fabricated ultrathin MoS2/N-doped carbon nanowires with a largely expanded (002) plane of MoS2 using PPy@MoS2 as a precursor, which exhibited excellent rate capabilities and long-term durability as an anode for LIBs. However, the formation mechanism of PPy@MoS2 is not clearly explored.21 Chang et al. have deposited MoS2 nanosheets on the PPy nanotubes by a facile hydrothermal method, which showed enhanced electrochemical performance as supercapacitor electrodes, but the experimental procedures are complex and time-consuming due to the preparation of PPy nanotubes first.22,23 Thus, it is highly desired but remains challenging to develop a facile strategy for the fabrication of one dimensional hierarchical architectures with highly dispersed MoS2 immobilized on conducting polymers.
On the basis of those mentioned above, a creative idea was proposed to fabricate3D hierarchical PPy@MoS2 microtubes which can act as both a support and a reductant for the in situ loading of noble metal (Ag, Au, and Pd) NPs, so as to improve the catalytic behaviour. The resultant noble metal NP-based PPy@MoS2 microtubes would offer the following advantages: (i) the 2D hierarchical MoS2 nanosheet decorated 1D PPy microtubes would provide confined spaces for the in situ formation of uniform noble metal NPs with high coverage, owing to the good reduction ability of the MoS2 layers; (ii) the resulting ternary PPy@MoS2@noble metal NP heterostructure provides specific metal–support interactions at the interface which would facilitate the reactivity of the noble metal NPs, further improving the mass/electron transport during the electrocatalytic reaction. Therefore, the synergy of noble metal NPs and PPy@MoS2 nanostructures can greatly influence the heterogeneous electron transfer reactions and allow rapid detection of analytes with high performances. Taking PPy@MoS2@Au as an example, the application of PPy@MoS2@Au nanocomposites toward enzyme-like catalysis is well demonstrated.
Experimental
Materials
Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) and concentrated nitric acid were purchased from Sinopharm Chemical Reagent Co. Ltd. Thiourea was obtained from the Alfa Aesar Chemical Company. All reagents were used directly without further purification.
Synthesis of MoO3
The MoO3 microrods were synthesized through hydrothermal treatment according to the literature.24–26 In a typical procedure, 1 g of ammonium heptamolybdate tetrahydrate (H24Mo7N6O24·4H2O) was dissolved in deionized water (20 mL), and 5 mL 65% HNO3 was added. Then, the reaction solution was transferred into a Teflon-lined stainless steel autoclave and heated at 180 °C for 20 h. After cooling, white solution was filtrated and then dried at 60 °C to obtain MoO3.
Synthesis of MoO3@PPy
The as-prepared MoO3 (0.1 g) microrods were dissolved into a mixed solution of 40 mL deionized water and 5 mL ethanol. Then, 0.1 mL of pyrrole was added during the stirring process. Subsequently, 0.329 g of (NH4)2S2O8 (dissolved in 5 mL of deionized water) was slowly dropped into the above solution, and the polymerization was performed for 4.5 h. Finally, the black solution was filtrated and then dried at 60 °C to obtain MoO3@PPy.
Synthesis of PPy@MoS2
342.5 mg thiourea was homogenously dispersed into 30 mL deionized water. Then, 50 mg of the as-obtained MoO3@PPy was added to the above dispersion. After being fully dissolved, the mixture was transferred to a 50 mL Teflon-lined stainless steel autoclave and treated in an oven at 200 °C for 24 h. The resulting precipitate was collected and washed with deionized water and ethanol, and then dried at 50 °C for 12 h.
Synthesis of PPy@MoS2@Au
Typically, 10 mg PPy@MoS2 was dissolved in 20 mL deionized water. Then 1 mg of HAuCl4 (dissolved in 10 mL of deionized water) was slowly dropped into the above solution, and the obtained mixture was sonicated for 1 h. The products were harvested by centrifugation, washed with deionized water as well as ethanol and dried at 50 °C overnight to attain PPy@MoS2@Au.
Peroxidase-like catalytic activities of PPy@MoS2@Au
In order to evaluate the peroxidase-like activity of PPy@MoS2@Au microtubes, a model experiment for the oxidation of the TMB substrate in the presence of H2O2 was performed. In the experiment, 10 μL of TMB solution (10 mM in ethanol) and 10 μL of H2O2 (10 mM) were subsequently added into 370 μL of acetate buffer solution. Then, 10 μL of PPy@MoS2@Au microtube suspension (1.0 mg mL−1) was added into the above solution. After the mixture was incubated at 50 °C for 12 minutes, PPy@MoS2@Au was centrifuged quickly. Finally, the UV-vis spectra were recorded in a wavelength-scan mode under the optimal conditions, unless otherwise stated. Furthermore, the steady-state kinetics analysis was measured by keeping the concentration of H2O2 fixed when using TMB as the substrate, while the concentration of TMB was constant when using H2O2 as the substrate.
Colorimetric detection of H2O2 and tannic acid
For the detection of H2O2, 10 μL of TMB (10 mM) and 10 μL of PPy@MoS2@Au microtubes (1.0 mg mL−1) along with varied concentrations of H2O2 were added into 370 μL of acetate buffer solution (pH 3.0). Then, the absorbance change at 652 nm was used to produce a dose of H2O2 concentration–dependent responsive curves. The detection of tannic acid (TA) is similar to that of H2O2. In the experiment, varied concentrations of TA were added into 370 μL of acetate buffer solution (pH 3.0), which contained 10 μL of PPy@MoS2@Au microtube suspension (1.0 mg mL−1), 10 μL of TMB solution (10 mM in ethanol) and 10 μL of H2O2 (10 mM). Then, the solution was centrifuged rapidly, and the absorbance change at 652 nm was measured by UV-vis absorption spectroscopy. In order to evaluate the selectivity of the proposed method, 10.0 mM of uric acid (UA), urea, oxalic acid (OA), citric acid (CA), ascorbic acid (AA), cholesterol, L-homocysteine, Cu2+, Zn2+ and K+ were added into the above reaction system instead of TA.
Preparation of black and green tea samples
The purchased dry black tea and green tea were accurately weighed, placed in a beaker containing 100 mL of deionized water, heated in a water bath at 100 °C for 30 minutes, and cooled to room temperature, and the supernatant obtained by filtration through a filter was diluted twice to obtain a sample to be tested.
Characterization
The crystal structures of the sample at different stages were characterized by using X-ray diffraction (XRD, BRUKER D2 PHASER with Cu K radiation). The morphologies of the products were captured using a scanning electron microscope (SEM, JEOL-4800) and a transmission electron microscope (TEM, JEOL-1011). The specific surface area of PPy@MoS2 was analysed by the Brunauer–Emmett–Teller (BET) method. The chemical composition of PPy@MoS2 and PPy@MoS2@Au were analyzed by X-ray photoelectron spectroscopy (XPS) measurements that were performed on a VGES-CALAB MKII X-ray photoelectron spectrometer.
Results and discussion
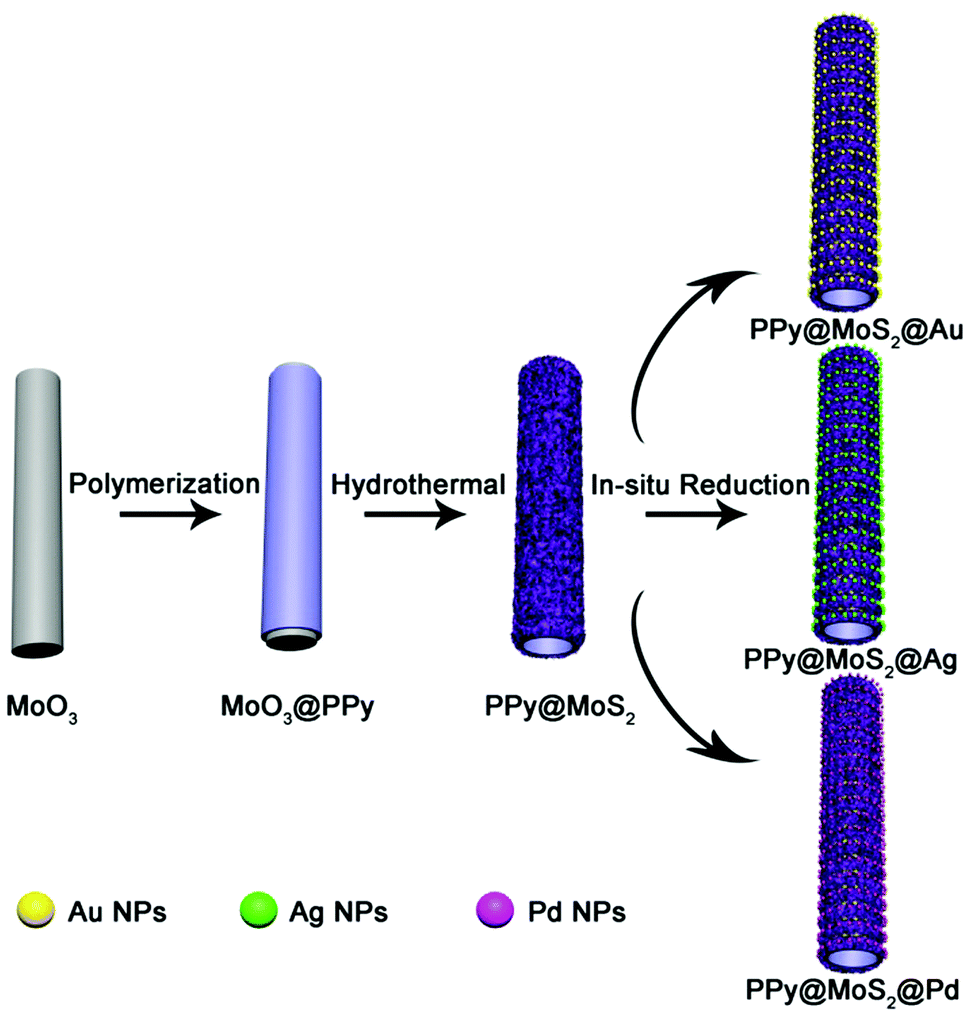
The illustration of the preparation of PPy@MoS2@Au/Ag/Pd is described in Scheme 1. MoO3 microrods were synthesized with a facile hydrothermal reaction. Afterwards, the resultant MoO3 microrods were employed as a hard template for the synthesis of MoO3@PPy microcables through a facile oxidation polymerization process. After sulfurization under hydrothermal conditions with thiourea as a source of sulfur, the hierarchical PPy@MoS2 was successfully synthesized by the self-template method. The construction of PPy layers surrounding the MoO3 microrods plays a critical role in preserving the unique 1D configuration. Without the coating of the polypyrrole layer, the MoO3 microrods are completely collapsed, resulting in the formation of aggregated MoS2 flowers. Owing to MoS2 released photogenerated electrons under light irradiation,27 the Au, Ag, and Pd NPs are in situ formed without adding any additional reducing agents, leading to the formation of PPy@MoS2@Au, Ag, and Pd composites. The in situ reduction strategy does not need a foreign reductant, and only the precursors of metal salts are involved. This is greatly beneficial for the synthesis procedure.
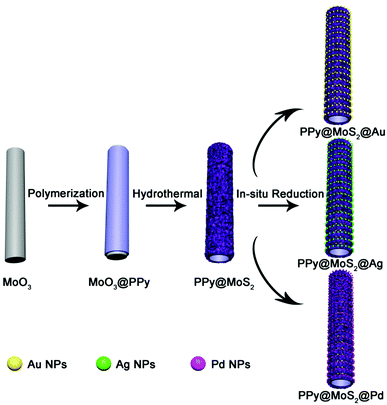 |
| | Scheme 1 Schematic synthesis of PPy@MoS2@Au/Ag/Pd composites. | |
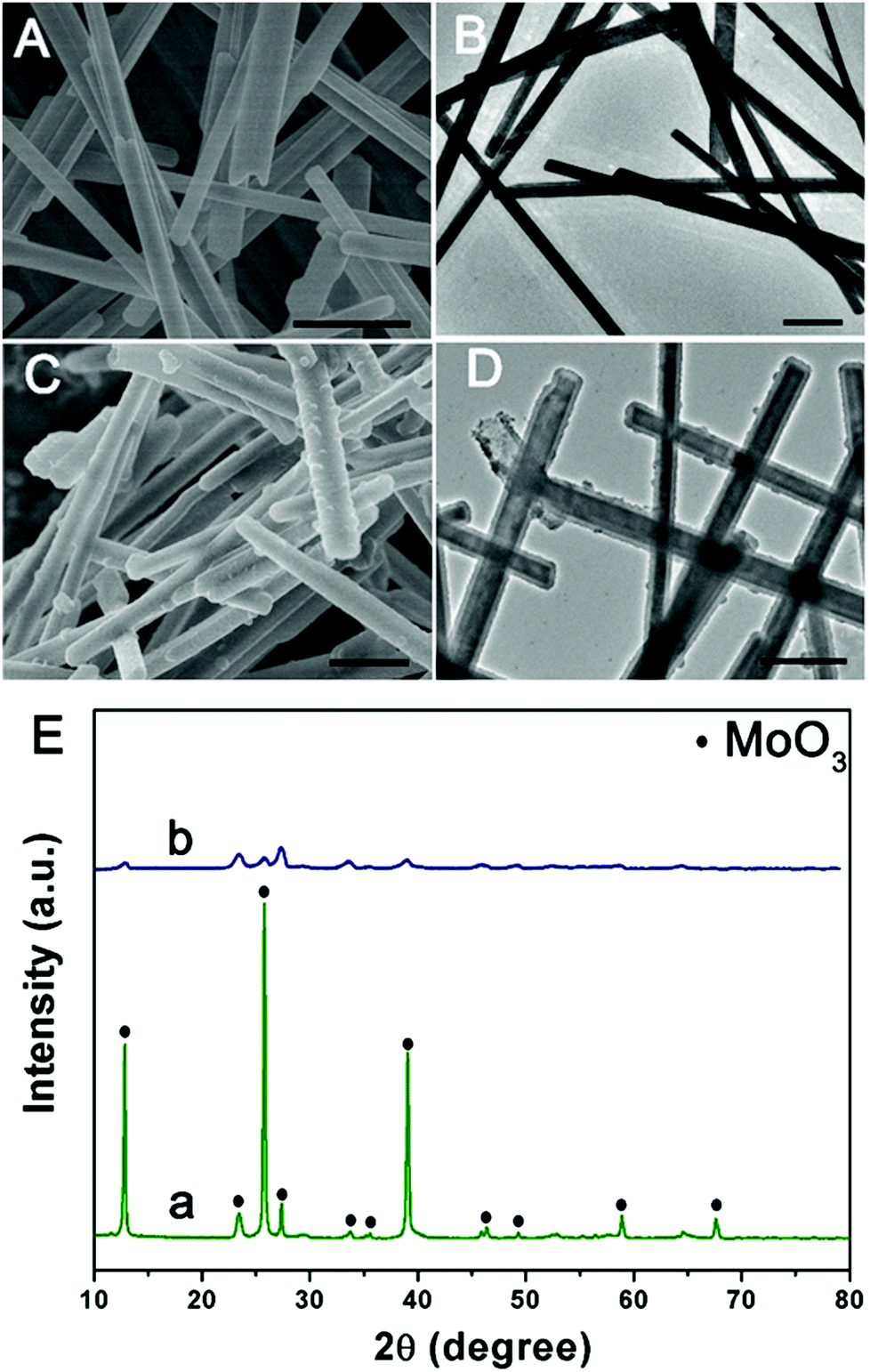
The morphology and structure of the product at each stage were characterized by SEM and TEM. Initially, the MoO3 microrods are obtained by a simple hydrothermal method, and exhibit a rod-like morphology with a smooth surface. As shown in Fig. 1(A and B), the MoO3 microrods are ∼300 nm in diameter and ∼10 μm in length. The XRD patterns of MoO3 were indexed to the orthorhombic MoO3 phase (JCPDS card no. 05-0508) as shown in Fig. 1E(a).28 Polypyrrole (PPy) has been considered as a kind of CP with π-conjugated polymeric backbones as well as a promising carbonaceous precursor, which could increase the conductivity as well as the ionic diffusion rate. Then, the MoO3 microrods were successfully coated with the PPy shell without the need for any surfactant by mixing pyrrole and ammonium persulfate in the ethanol/water suspension of MoO3 microrods at room temperature. As shown in Fig. 1(C and D), the MoO3@PPy microcables inherit a uniform microrod-like shape of the MoO3 templates, and the surface is much rougher compared to that of MoO3. The XRD pattern (Fig. 1E(b)) also showed that the peak intensity of the MoO3@PPy composite is significantly weaker than that of the virgin MoO3. This is mainly due to the semi-crystalline nature of the PPy coating.29,30
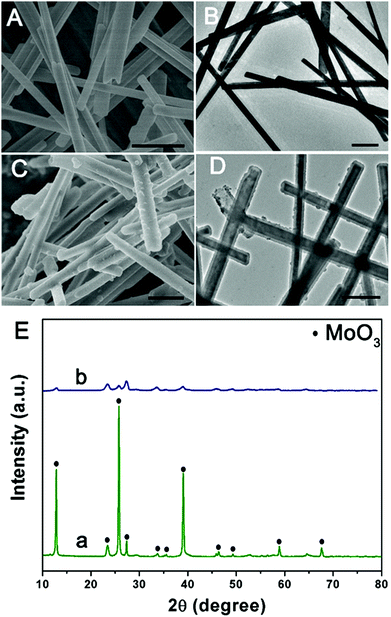 |
| | Fig. 1 SEM and TEM images of MoO3 (A and B) and MoO3@PPy (C and D), and (E) XRD patterns of MoO3 (a) and MoO3@PPy (b). Scale bars: 1 μm in (A–D). | |
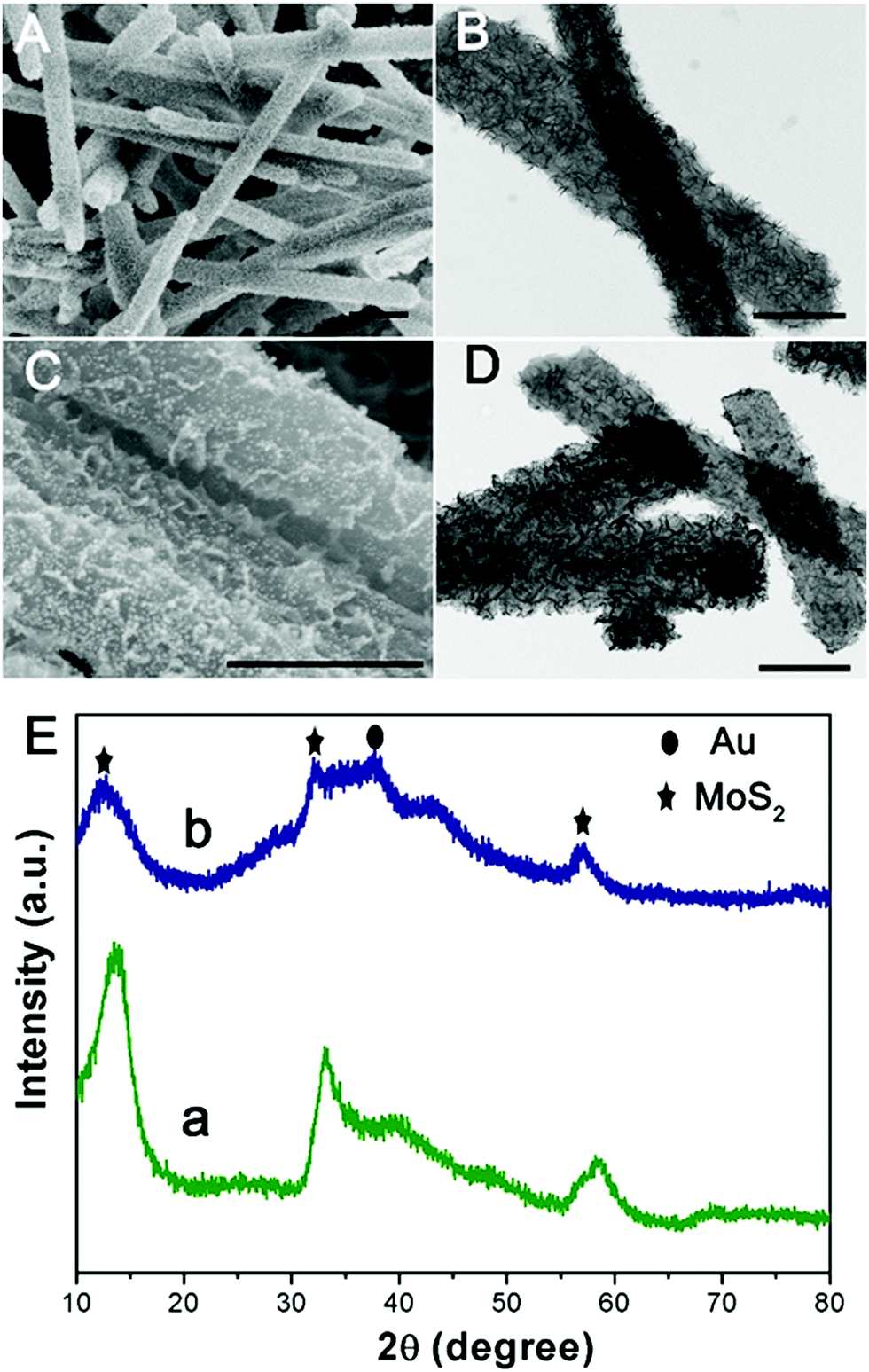
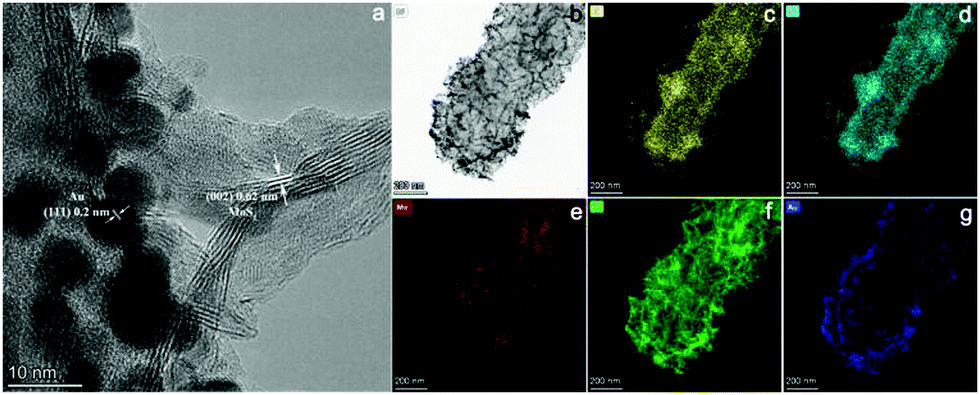
Subsequently, one dimensional hierarchical PPy@MoS2 composites are formed by a one-step hydrothermal process, in which MoO3@PPy as the precursor and thiourea provides the sulfur source. Owing to strong interactions between PPy and MoS2, the MoS2 nanosheets are closely decorated on the surface of PPy microtubes as a support. As shown in Fig. 2(A and B), the composite also inherits a uniform microrod-like shape from the MoO3@PPy precursor, which has a rough surface with vertically aligned layered MoS2 nanosheets. It is worth noting that the 2D MoS2 nanosheets are assembled into integrative 1D microrods, indicating the combined features of both 1D and 2D nanostructures. Indeed, XRD investigation (Fig. 2E(a)) also confirmed that the newly formed phase was MoS2 because all diffraction peaks clearly pointed to MoS2 (JCPDS number 37-1492).16 The high-resolution TEM (HRTEM) image shown in Fig. 3a provides further insight into the interior structure. The visible lattice fringes with a layer distance of 0.62 nm are consistent with the d-spacing of the (002) planes of 2H-MoS2.31 And the lattice fringes with a d-spacing of 0.2 nm correspond to the (111) plane of Au.32 The elemental mapping images in Fig. 3b–f show that the C, N, Mo, S and Au five elements were evenly distributed in the PPy@MoS2@Au microtubes.
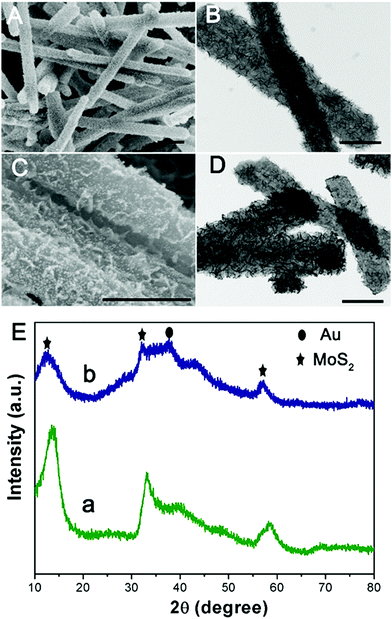 |
| | Fig. 2 SEM and TEM images of PPy@MoS2 (A and B) and PPy@MoS2@Au and (C and D) and (E) XRD patterns of PPy@MoS2 (a) and PPy@MoS2@Au (b). Scale bars: 1 μm in (A) and 500 nm in (B–D). | |
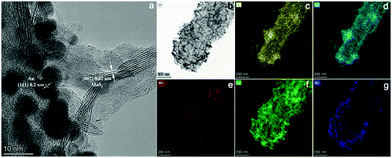 |
| | Fig. 3 The HRTEM image of PPy@MoS2@Au (a and b), and the corresponding elemental mapping images of C (c), N (d), Mo (e), S (f) and Au (g). | |
To demonstrate this vital role of polymer coating's strategy, polyaniline (PANI) is used as another coating polymer for the synthesis of hierarchical PANI@MoS2 microtubes. Previous work reported that PANI showed a similar property to PPy as a conducting polymer. Thus, it is expected that this strategy can be easily extended to the synthesis of the hierarchical polymer@MoS2 composites by simply alternating PPy to PANI polymers. Using the same experimental procedures, MoO3@PANI and PANI@MoS2 were easily fabricated (Fig. S1, ESI†), further proving that the organic polymer coating plays a vital role in the self-template formation of hierarchical polymer@MoS2 composites. The decoration of noble metal NPs on hierarchical PPy@MoS2 microtubes enables the formation of abundant interfaces, which generate an energy filtration effect that introduces energetic barriers to limit the transport of lower energy charge-carriers, which is greatly beneficial for the improvement of the Seebeck coefficient.23 A few methods for realizing functional decorative layered two-dimensional transition metal dichalcogenide (TMDC) materials have been reported. Unfortunately, modification processes are complicated, and the current research on decoration with noble metal NPs for PPy@MoS2 is insufficient. Herein, the PPy@MoS2@Au composites are obtained by direct reduction of HAuCl4 by PPy@MoS2. The driving force for the immobilization of the Au nanoparticles could be ascribed to MoS2 released photogenerated electrons under light irradiation, which contribute to the reduction of Au3+ to Au0. As shown in Fig. 2(C and D), high coverage of tiny Au NPs is well dispersed on the surfaces of PPy@MoS2. For the Au decorated PPy@MoS2 composites, due to the tiny Au NPs, only one characteristic peak related to Au (111) (JCPDS 04-0784) is observed in the diffraction pattern (Fig. 2E, curve b),6 and all other diffraction peaks could be indexed to PPy cores and hierarchical MoS2, revealing the coexistence of Au, PPy, and MoS2. Moreover, the density of Au NPs can be facilely adjusted by changing the amount of chloroauric acid. When the added weights of chloroauric acid were increased from 1 mg to 2 mg and 4 mg, respectively, the density of Au NPs is significantly increased while the size of Au NPs was almost kept constant (Fig. S2, ESI†).
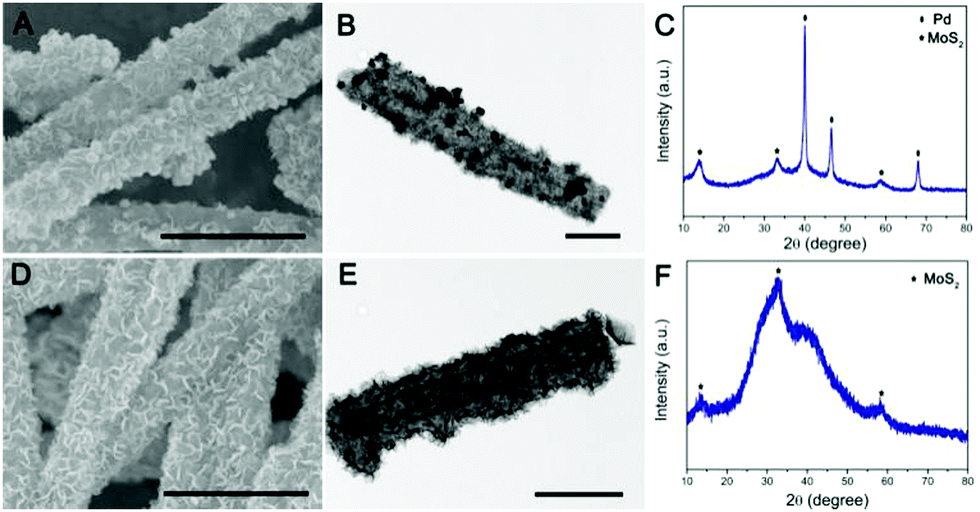
To further validate the versatile applicability of this strategy, we have also decorated other noble metals, such as Pd and Ag, on the surface of PPy@MoS2 with a similar procedure. Fig. 4 shows the SEM and TEM images of PPy@MoS2@Pd and PPy@MoS2@Ag composites. As shown in Fig. 4(A, B, D and E), all the metal NPs are well dispersed on the PPy@MoS2 surface. The formation of a noble metal–PPy@MoS2 composite crystal structure was also verified by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). As shown in Fig. 4C, besides the diffraction peaks for MoS2, several diffraction peaks for Pd (JCPDS number 46-1043) are also found in the XRD pattern of PPy@MoS2@Pd, which proves that PPy@MoS2@Pd composites were successfully fabricated. Meanwhile, the XRD patterns of PPy@MoS2@Ag composites are almost the same as those of the PPy@MoS2 sample, indicating that the crystal structure of MoS2 does not change after surface modification (Fig. 4F). However, the XRD peaks of Ag are not observed, which were due to the tiny size of Ag NPs decorated on PPy@MoS2, which is also consistent with Sun's work.33 X-ray photoelectron spectroscopy (XPS) measurements are performed to further confirm the chemical compositions of PPy@MoS2@Pd and PPy@MoS2@Ag (Fig. S3, ESI†). The high-resolution spectra are shown in Fig. S3b (ESI†), as for the Pd 3d spectrum of PPy@MoS2@Pd nanocages, the peaks located at 341.4 and 336.3 eV are indexed to Pd 3d3/2 and Pd 3d5/2, respectively, indicative of Pd0.34 As presented in the high resolution Ag 3d spectrum (Fig. S3d, ESI†), the two main peaks at around 368.8 and 374.8 eV should be assigned to Ag0.35 To observe the distribution patterns of these supported noble metals, STEM-EDS was used. As seen from Fig. S4 and S5 (ESI†), energy-dispersive spectroscopy (EDS) mapping revealed that C, N, O, Mo, S, and Ag were doped into the PPy@MoS2@Ag composite. Similarly, elemental C, N, O, Mo, S and Pd were uniformly dispersed in the PPy@MoS2@Pd composite. The above results further illustrate that noble metals have been successfully decorated on the surface of PPy@MoS2 microtubes, consistent with SEM and TEM observations. Therefore, this strategy is a facile, efficient and general method for controlling the tailored formation of noble metal NPs decorated on one-dimensional hierarchical PPy@MoS2 nanostructures.
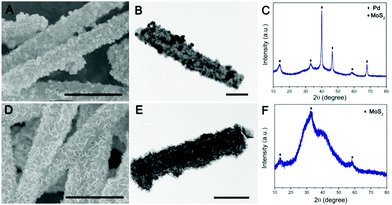 |
| | Fig. 4 SEM and TEM images of PPy@MoS2@Pd (A and B) and PPy@MoS2@Ag (D and E), and (C) XRD patterns of PPy@MoS2@Pd (C) and PPy@MoS2@Ag (F). Scale bars: 1 μm in (A and D), and 500 nm in (B and E). | |
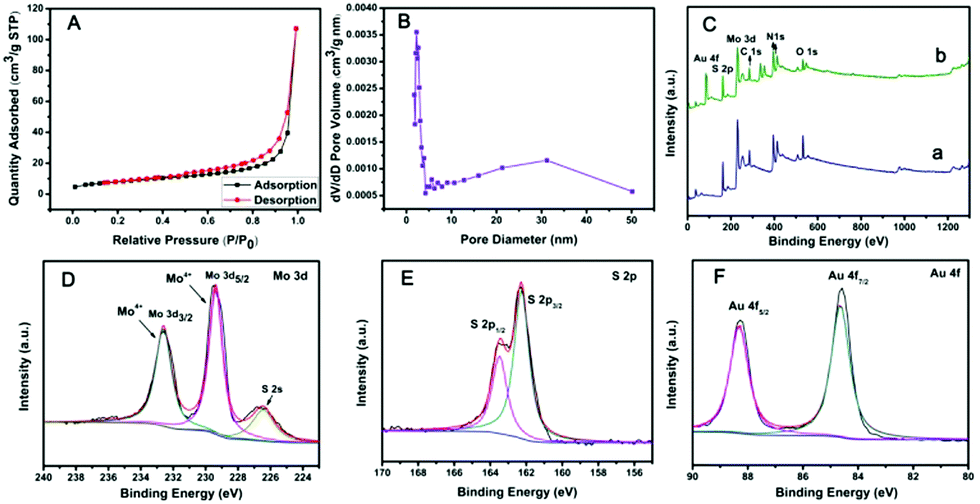
The specific surface area of PPy@MoS2 was characterized by N2 adsorption/desorption experiments, as shown in Fig. 5(A and B). PPy@MoS2 shows a high Brunauer–Emmett–Teller (BET) specific surface area of 28.57 m2 g−1 and the BJH adsorption average pore diameter was about 24.81 nm. In addition, the N2 adsorption/desorption isotherm is in well accord with the IUPAC IV type, revealing the mesoporous feature of the PPy@MoS2 composites.36–41 The high specific surface area and mesoporous architecture of PPy@MoS2 could afford sufficient accessible active sites during the catalytic reaction. The chemical compositions and valence states of PPy@MoS2 and PPy@MoS2@Au were further examined by X-ray photoelectron spectroscopy (XPS). Both of them indicated similar element peaks, except for Au in PPy@MoS2@Au in Fig. 5C. The high resolution XPS spectra of Mo 3d, S 2p and Au 4f regions of PPy@MoS2@Au are shown in Fig. 5D–F. The high-resolution Mo 3d XPS spectrum (Fig. 5D) indicated that the oxidation state of the Mo ion is 4+. Meanwhile, the low binding energy peak at around 226.29 eV is ascribed to S 2s in MoS2.42Fig. 5E shows a high-resolution spectrum for S 2p, the main doublet located at 161.9 eV and 163.1 eV corresponds to S 2p3/2 and S 2p1/2, respectively. As can be seen from the Au XPS spectrum (Fig. 5F), the peaks at 87.5 and 83.8 eV are attributed to the 4f5/2 and 4f7/2 binding energies of the zero valence state of metallic Au, respectively.43 All of the above results indicate that we have successfully prepared PPy@MoS2 and PPy@MoS2@Au.
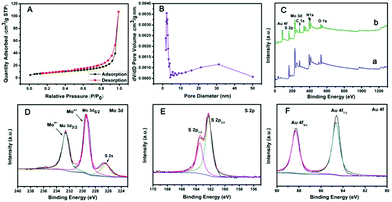 |
| | Fig. 5 (A) N2 adsorption–desorption isotherms and (B) the pore size distribution curve of PPy@MoS2, (C) XPS full spectrum of PPy@MoS2 (a, blue line) and PPy@MoS2@Au (b, green line), (D) XPS spectra of Mo 3d of PPy@MoS2@Au, (E) XPS spectra of S 2p of PPy@MoS2@Au and (F) XPS spectra of Au 4f of PPy@MoS2@Au. | |
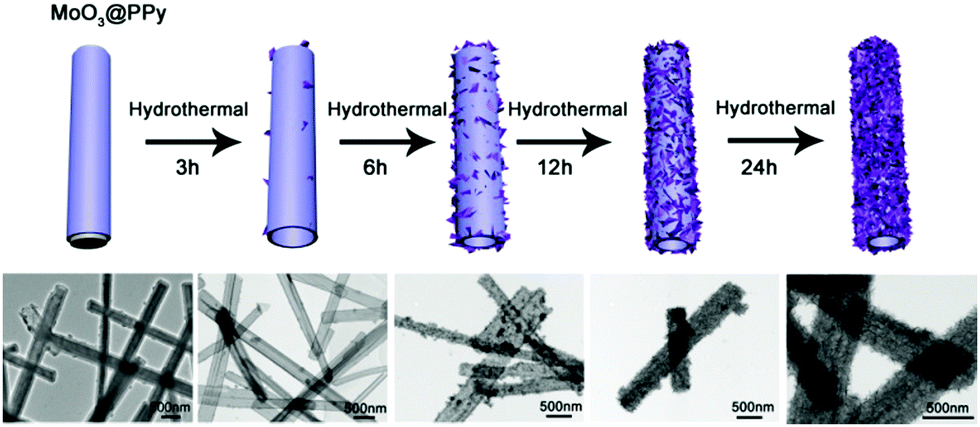
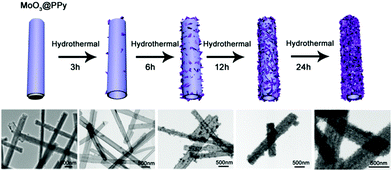
To investigate the possible formation process of MoO3@PPy converting to PPy@MoS2, we carried out a series of control experiments by varying the solvothermal time. As shown in Fig. 6a and b, almost no MoS2 nanosheets were found on the final products, and the PPy hollow microtubes with smooth surfaces were obtained when the reaction time was 3 h. This indicates that MoO3 microrod cores start to transform to MoO42− when the reaction time increases to 3 h. As the solvothermal time reaches 6 h, some MoS2 nanosheets were covered on the PPy microtubes (Fig. 6(c and d)). When the solvothermal time was further prolonged to 12 hours, the density of the MoS2 nanosheet on the PPy surface was further increased (Fig. 6(e and f)). After reacting for 24 h, the morphology of PPy@MoS2 did not obviously change (Fig. 6(g and h)). Based on the all above results, the formation of the one dimensional hierarchical structures of PPy@MoS2 probably involved an interesting dissolution–regrowth process in which MoO3@PPy and thiourea were involved (Fig. 7). At first, thiourea was decomposed to CS2 and NH3 in solution, which can dissolve the MoO3 cores to form (NH4)2MoO4, and the hollow PPy microtubes were easily obtained. This was confirmed by the SEM and TEM results of intermediate products after 3 hours. Subsequently, MoS2 began to form gradually resulting from the reaction between MoO42− and CS2, which can slowly cover the surface of PPy microtubes. When the reaction time reached up to 12 and 24 hours, there is no obvious difference in the morphology of PPy@MoS2 synthesized at 12 h and 24 h, indicating that the inner MoO3 cores were totally converted to MoS2 covering the surface of PPy microtubes. To gain further insight into the formation process of MoO3@PPy converting to PPy@MoS2, X-ray photoelectron spectroscopy (XPS) measurements of the samples at different reaction times were performed and Mo 3d spectra were deconvoluted. The summary of XPS analysis of the valence states, peak position and relative content in these composites are listed in Table S1 (ESI†). As can been seen in Fig. S6 (ESI†), when the reaction time reached 3 h, the Mo 3d XPS spectrum is fitted to the doublet peaks at 232.6 and 235.8 eV, corresponding to Mo6+ peaks with ca. 3.2 eV spin–orbit splitting, which reveals that only Mo6+ ions are present in the composites (shown in Fig. S6 and Table S1, ESI†).44 When the reaction time increased to 6 h, there are five peaks at 226.1, 229.0, 232.2, 233.3 and 236.1 eV of Mo 3d, which belong to S 2s, Mo4+ 3d5/2, Mo4+ 3d3/2, Mo6+ 3d5/2, and Mo6+ 3d3/2, revealing that there are two molybdenum oxidation states, 67.01% Mo4+ and 32.99% Mo6+ in the composites (shown in Fig. S6 and Table S1, ESI†).44 With the reaction time increasing from 6 h to 12 h, the content of MoS2 increases from 67.01 to 72.98%. With a further increase in reaction time to 24 h, as shown in Fig. S6d (ESI†), only Mo4+ is present in the composites, which indicates that the inner MoO3 cores were totally converted to MoS2. The XPS results verify the conversion of MoO3 into (NH4)2MoO4, and then formation of MoS2 nanosheets, which is also in accordance with the SEM and TEM results. In fact, similar growth processes have also been exemplified in many other materials with the development of many unique structures.45–47 Notably, the in situ interfacial construction of PPy nanolayers plays a vital role in maintaining the one-dimensional (1D) hybrid structure. As a comparison, we tried to prepare MoS2 composites without the participation of PPy. As seen from Fig. S7 (ESI†), without the coating of an organic polypyrrole layer, the MoO3 microrods completely collapsed, resulting in the formation of aggregated MoS2 flowers.
 |
| | Fig. 6 SEM and TEM images of PPy@MoS2, 3 h (a and b), 6 h (c and d), 12 h (e and f) and 24 h (g and h). Scale bars: 1 μm in (a, c, e and g), and 500 nm in (b, d, f and h). | |
 |
| | Fig. 7 Schematic illustration of the evolution of the formation of PPy@MoS2. | |
In particular, Zhu's group reported a similar route to synthesize a composite with ultrathin MoS2 nanosheets on Mo2C-embedded N-doped carbon nanotubes.42 In their report, the growth of MoS2 nanosheets on Mo2C-embedded NCNTs is based on in situ solids and subsequent hydrothermal reactions. The difference in them is that the synthetic strategy we employed does not require a high temperature carbonization process. As a comparison, we also applied their method to synthesize one dimensional hierarchical C@MoS2 (MoO2@C was obtained by calcination of MoO3@PPy at 500 °C under an N2 atmosphere). Initially, the S2− ions decomposed from thiourea react with Mo4+ of MoO2@C microcables, forming thin MoS2 nanosheets on the surface of MoO2@C. These MoS2 nanosheets act as physical barriers, which will hinder the further inward diffusion of S2− ions. With the increase of reaction time, more Mo4+ ions diffuse to the surface and then react with S2− ions, leading to the sustained growth of MoS2 nanosheets.48 Interestingly, we found that this synthesis method is affected by the thickness of PPy. When we also used the same MoO3@PPy-100 μL as the precursor, followed by high-temperature calcination, and then the hydrothermal reaction to synthesize MoS2-based composites. The test results indicate that the MoS2 nanosheets were almost not found on the surface of MoO2@C (Fig. S8(a and b), ESI†). We suspect that the coated carbon layer is too thick, so that Mo4+ cannot diffuse out and react with S2− to form MoS2 nanosheets. In order to prove this assumption, we reduced the pyrrole to 50 μL while keeping other conditions unchanged. The SEM and XRD results of C@MoO2@MoS2-50 showed that the MoS2 nanosheets grew on the surface of the one-dimensional structure (Fig. S8(c, d) and Fig. S9, ESI†). It is worth noting that our method is not affected by the thickness of PPy. We reduced the amount of pyrrole to 50 μL and increased the amount to 150 μL respectively, to obtain MoO3@PPy-50 and MoO3@PPy-150. Under the same experimental conditions, PPy@MoS2 with one dimensional hierarchical morphology could also be obtained (Fig. S10A, ESI†). The corresponding XRD results also proved that the hierarchical PPy@MoS2 can be easily obtained, which is not related to the thickness of PPy (Fig. S10B, ESI†). The reason may be attributed to the different formation mechanisms of MoS2. In Zhu's work, the mechanism of forming MoS2 is mainly that during the heating process, O2− ions in MoO2/Mo2C–NCNTs were replaced with S2− ions, leading to the transformation of MoO2 to MoS2. However, in our work, the formation of PPy@MoS2 is demonstrated to involve the dissolution–regrowth process.
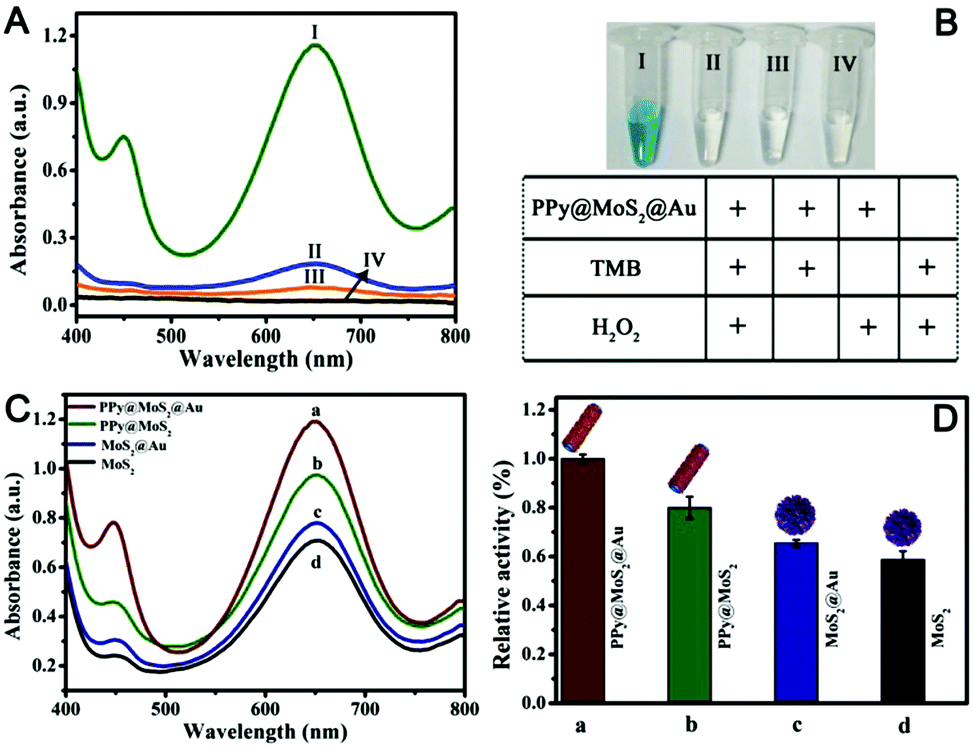
Recently, MoS2 nanosheets have also been demonstrated to exhibit inherent enzyme-like activity, which have been used for sensing and antibacterial applications.49,50 Nevertheless, owing to the aggregated MoS2, the peroxidase-like activity of individual MoS2 is not high, which greatly limits its practical applications. To further enhance the peroxidase-like activity of MoS2 nanosheets, coupling MoS2 nanosheets with one or two kinds of nanomaterials to produce binary or ternary components of MoS2 based nanozymes have been designed and fabricated for synergistic enhanced enzyme-like catalysis.51,52 As a proof of application, the as-prepared PPy@MoS2@Au (the mass ratio of PPy@MoS2 to HAuCl4 is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was employed to evaluate the enzyme-like activity using the TMB–H2O2 reaction as a model reaction system because the colourless TMB can be oxidized to a blue product in the presence of a catalyst. As shown in Fig. 8A(I), there is an obvious absorption peak at 652 nm originating from the oxidation of TMB in the presence of H2O2. Compared with no PPy@MoS2@Au, the response rate is 595%, indicating that the nanocomposites do have excellent catalytic properties. Meanwhile, the TMB–H2O2 reaction systems in the presence of PPy@MoS2@Au produced a deep blue colour, while no obvious reactions occurred in the control systems (Fig. 8B). This again proves the important role of the empty tube and layered structure PPy@MoS2@Au nanozymes as the case with horseradish peroxidase (HRP). To investigate the ternary compound advantages of PPy@MoS2@Au, the catalytic activities with MoS2, MoS2@Au and PPy@MoS2 were tested for comparison. Fig. 8A indicates that the UV-Vis spectrum of PPy@MoS2@Au has the largest absorption peak at 652 nm. Meanwhile, the absorption peaks of PPy@MoS2, MoS2@Au, and MoS2 nanoflowers are all inferior to that of PPy@MoS2@Au (the corresponding SEM images of MoS2 and MoS2@Au are shown in Fig. S7 and S11, ESI†). As shown in Fig. 8C and D, it was found that PPy@MoS2@Au composite showed remarkably higher catalytic activity than the controlled samples and other nanomaterials, which could be owing to the synergistic effect from the strong interfacial interactions between MoS2 nanosheets and PPy microtubes, as well as Au nanoparticles. In addition, the TMB–H2O2 reaction solutions catalyzed by different nanomaterials can be visually observed through the inset photograph, which are in accordance with the foregoing results. First, the substrate can access the inner surface of the PPy microtubes through the holes and form a local concentration effect, speeding up the catalytic reaction rate. Secondly, the MoS2 nanosheets dispersed on PPy microtubes can provide more exposed active sites. After being decorated with high coverage of gold NPs, the catalytic effect on the oxidation of TMB was greatly enhanced owing to multiple synergistic catalytic effects among PPy, MoS2 and Au.
:2) was employed to evaluate the enzyme-like activity using the TMB–H2O2 reaction as a model reaction system because the colourless TMB can be oxidized to a blue product in the presence of a catalyst. As shown in Fig. 8A(I), there is an obvious absorption peak at 652 nm originating from the oxidation of TMB in the presence of H2O2. Compared with no PPy@MoS2@Au, the response rate is 595%, indicating that the nanocomposites do have excellent catalytic properties. Meanwhile, the TMB–H2O2 reaction systems in the presence of PPy@MoS2@Au produced a deep blue colour, while no obvious reactions occurred in the control systems (Fig. 8B). This again proves the important role of the empty tube and layered structure PPy@MoS2@Au nanozymes as the case with horseradish peroxidase (HRP). To investigate the ternary compound advantages of PPy@MoS2@Au, the catalytic activities with MoS2, MoS2@Au and PPy@MoS2 were tested for comparison. Fig. 8A indicates that the UV-Vis spectrum of PPy@MoS2@Au has the largest absorption peak at 652 nm. Meanwhile, the absorption peaks of PPy@MoS2, MoS2@Au, and MoS2 nanoflowers are all inferior to that of PPy@MoS2@Au (the corresponding SEM images of MoS2 and MoS2@Au are shown in Fig. S7 and S11, ESI†). As shown in Fig. 8C and D, it was found that PPy@MoS2@Au composite showed remarkably higher catalytic activity than the controlled samples and other nanomaterials, which could be owing to the synergistic effect from the strong interfacial interactions between MoS2 nanosheets and PPy microtubes, as well as Au nanoparticles. In addition, the TMB–H2O2 reaction solutions catalyzed by different nanomaterials can be visually observed through the inset photograph, which are in accordance with the foregoing results. First, the substrate can access the inner surface of the PPy microtubes through the holes and form a local concentration effect, speeding up the catalytic reaction rate. Secondly, the MoS2 nanosheets dispersed on PPy microtubes can provide more exposed active sites. After being decorated with high coverage of gold NPs, the catalytic effect on the oxidation of TMB was greatly enhanced owing to multiple synergistic catalytic effects among PPy, MoS2 and Au.
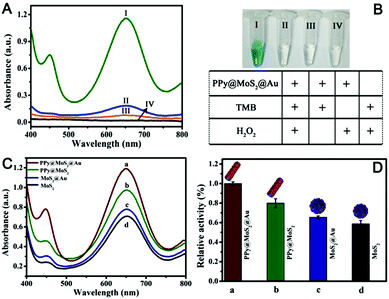 |
| | Fig. 8 (A and B) Typical UV-Vis spectra of different reaction systems. Concentration of different substances in the reaction system: [TMB] = 0.25 mM; [H2O2] = 2.5 mM; [PPy@MoS2@Au] = 0.025 mg mL−1; the reaction in 0.2 M NaAc–HAc buffer solution (pH 3.0) at 50 °C for 12 min. (C and D) UV-visible absorption curves of different materials were compared, (a–d) (PPy@MoS2@Au, PPy@MoS2, and MoS2@Au, MoS2), respectively. The reaction in 0.2 M NaAc–HAc buffer solution (pH 3.0) at 50 °C; [TMB] = 0.25 mM; [H2O2] = 2.5 mM. | |
Optimization of experimental conditions
In order to further study the catalytic performance of the material, the experimental conditions were optimized. The catalytic activity of PPy@MoS2@Au is closely dependent on the pH of the reaction, temperature, time and concentration of the catalyst. The peroxidase-like activity of PPy@MoS2@Au was measured by varying the reaction pH from 1.0 to 7.0. As can be seen from Fig. S12A (ESI†), with increasing pH from 1.0 to 3.0, the catalytic activity of PPy@MoS2@Au increased, and then decreased with a further increase to 7.0. This phenomenon may be due to the fact that the PPy@MoS2@Au framework structure exhibits a more efficient catalytic site at pH 3.0 and achieves significant activity. This result is similar to previous reports on nanostructured peroxidase mimics.53 Also, the peroxidase-like activity of the nanocomposite was measured by changing reaction temperatures from 30 to 60 °C. As can be observed from Fig. S12B (ESI†), the catalytic activity gradually increased with the increase of the reaction temperature up to 50 °C. This may be because the kinetic energy and diffusion rate of H2O2 molecules increase with the increase of reaction temperature, which leads to the increase of molecular formula between H2O2 and PPy@MoS2@Au catalytic centers. Nevertheless, when the temperature exceeded 50 °C, the catalytic efficiency significantly decreased. This is probably owing to the fact that with increasing temperature, H2O2 molecules are converted to other products before a collision with the catalytic centers of PPy@MoS2@Au could occur. In addition, we further examined the effects of reaction time and catalyst concentration on the catalytic activity of PPy@MoS2@Au nanocomposites toward the oxidation of TMB by H2O2 (Fig. S12C and D, ESI†). To understand the response rate of the catalyst to absorbance, the time-dependent evolution of the absorption spectra was examined. Fig. S12C (ESI†) shows that the catalytic activity gradually increases with the reaction time and reaches a saturation value after about 12 minutes. The absorbance monotonically increases with the amount of PPy@MoS2@Au; when the concentration reached 1.0 mg mL−1, the absorbance was nearly saturated, and there was no obvious increase trend, therefore 1.0 mg mL−1 was chosen as the optimal concentration (Fig. S12D, ESI†). Therefore, to obtain more reliable results, subsequent experiments will maintain the reaction under optimal conditions.
Steady-state kinetic analysis
To further examine the peroxidase-like catalytic activity of PPy@MoS2@Au microtubes, the apparent kinetic data of the peroxidase-like reaction were obtained by changing one substrate concentration of TMB or H2O2 while keeping the other substrate concentration constant in this system. The initial reaction rates of the reaction were obtained and fitted with the Lineweaver–Burk double reciprocal plot:54
where V is the initial velocity, Km is the Michaelis–Menten constant, Vm is the maximal reaction velocity and [S] is the concentration of the substrate.
Fig. S13A and B (ESI†) depict typical Michaelis–Menten curves by fixing concentrations of H2O2 (10 mM) and TMB (10 mM), respectively. A reciprocal relationship between the initial velocity and the substrate concentration was obtained (Fig. S13C and D, ESI†). Furthermore, the Michaelis–Menten curves can be fitted to Lineweaver–Burk plots, which are shown in Fig. S13C and D (ESI†). From the Lineweaver–Burk diagram, the key enzyme kinetic parameters such as the maximum initial velocity (Vm) and the Michaelis–Menten constant (Km) were obtained by the Lineweaver–Burk equation (Table S2, ESI†).55 It is well known that Km is generally considered to be an indicator of the affinity of an enzyme for a substrate. The lower the Km, the higher the affinity of the enzyme and vice versa. The results indicate that PPy@MoS2@Au composites possess a much higher binding affinity of nanocomposites towards H2O2, suggesting that H2O2 could be detected at a lower concentration. It can be seen from Table S2 (ESI†) that the Km value of PPy@MoS2@Au to the substrate H2O2 is 247 times smaller than that of horseradish peroxidase (HRP). These data indicate that the prepared PPy@MoS2@Au microtubes are efficient peroxidase mimics, suggesting that H2O2 can be detected at lower concentrations.
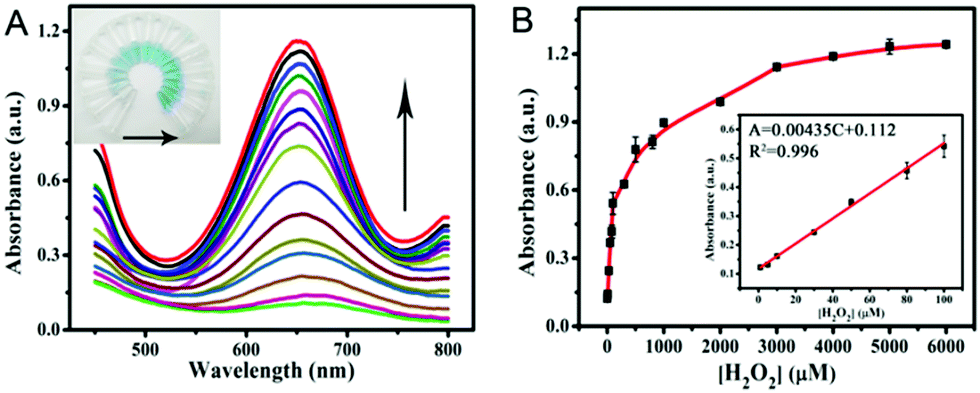
Colorimetric detection of H2O2
According to the intrinsic peroxidase-like properties of PPy@MoS2@Au microtubes, a colorimetric method for the detection of H2O2 is realized. Since the catalytic activity of the PPy@MoS2@Au nanocomposite is H2O2 concentration-dependent, this can be applied to detect H2O2 by monitoring the absorbance changes. Fig. 9A depicts a typical H2O2 concentration–response curve, indicating that the intensity of the absorbance at 652 nm increases with the increased concentration of H2O2. The inset of Fig. 9A shows evident color variation, revealing that H2O2 can be visually measured by the naked eye. From the corresponding calibration plot in the inset of Fig. 9B, it has been found that H2O2 could be well detected in the linear range from 1.0 μM to 100 μM (R2 = 0.996), and the detection limit was estimated to be about 0.57 μM, its sensitivity is better than some other reported H2O2 sensors.56–58 The affinity of PPy@MoS2@Au for H2O2 is relatively high, which is consistent with previous studies.
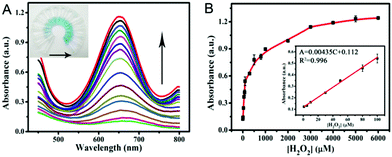 |
| | Fig. 9 (A) The dose–response curve and (B) calibration curves of the analytical approach for the colorimetric detection of H2O2. The inset shows the linear response plot of absorbance to the concentration of H2O2. The absorbance changes of the system consisting of NaAc–HAc (0.2 M, pH 3.0), TMB (0.25 mM), and PPy@MoS2@Au catalyst suspension (0.025 mg mL−1) in the presence of various concentrations of H2O2 (1.0–6000 μM) (Fig. 9A). Error bars show the standard deviations of three repeated measurements. | |
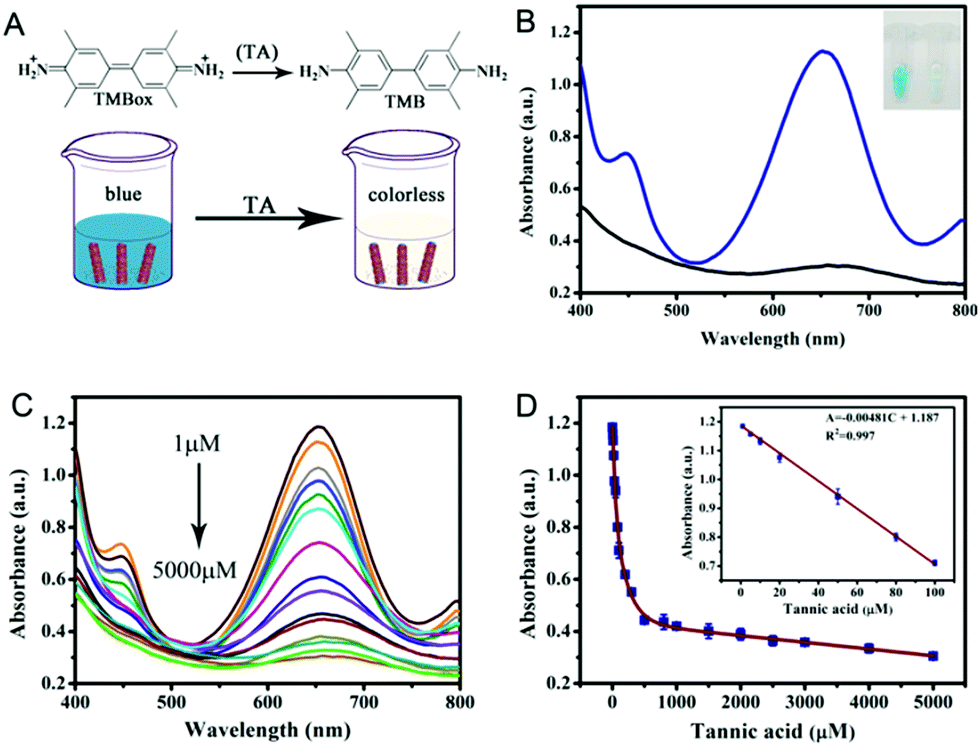
Colorimetric biosensing of TA
Based on the above findings, we have developed a simple colorimetric approach for the detection of TA, and the method does not require the participation of any natural enzymes. The biosensor works by reducing TMBox by TA in the presence of PPy@MoS2@Au peroxidase mimics and H2O2 (Fig. 10A). The reaction system changes from blue to colorless. As shown in Fig. 10C, the absorbance of the reaction system decreases as the concentration of TA increase. In Fig. 10D, a linear calibration curve was obtained with an equation A = −0.00481C + 1.187 (R2 = 0.997) and a linear range from 1.0 to 100 μM. And the detection limit obtained is 0.87 μM (S/N = 3) on the basis of the linear calibration curve. This detection limit is much better than those of most of the previously reported nanomaterial-based peroxidase colorimetric detection and other analytical methods of TA (Table 1).59,60 This result suggests that the PPy@MoS2@Au microtube-based peroxidase-like system possesses potential practical applications.
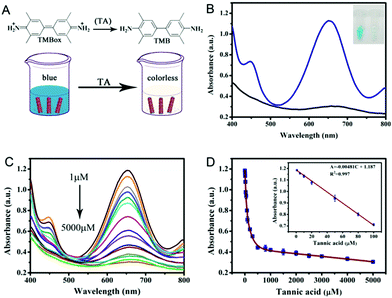 |
| | Fig. 10 (A) TA detection schematic and (B) UV-vis absorption principle diagram of TA detection. (C) The UV-vis absorption spectra of the colorimetric sensor in the presence of different concentrations of TA (5000, 4000, 3000, 2500, 2000, 1000, 300, 200, 100, 80, 50, 30, 20, 10, and 1 μM). (D) A dose–response curve for TA detection in the range of 1 to 5000 μM using PPy@MoS2@Au as a peroxidase mimic. Inset: The linear calibration plot for TA. The experiments were performed in the reaction system in 0.2 M NaAc–HAc buffer solution (pH 3.0) at 50 °C; [TMB] = 0.25 mM; [H2O2] = 0.25 mM, [PPy@MoS2@Au] = 0.119 mg mL−1. Error bars show the standard deviation derived from three repeated measurements. | |
Table 1 Comparison of the linear range and LOD with other composite based colorimetric methods of this work with PPy@MoS2@Au composites as peroxidase mimics for the colorimetric detection of TA
| Analytical method (material) |
Linear range (μM) |
Detection limit (μM) |
Ref. |
|
PrTu (1-benzoyl-3-(pyrrolidine) thiourea).
|
| Spectrophotometry |
1.0–100.0 |
1.0 |
61
|
| Electrochemistry (PrTu)a |
10–500 |
3.0 |
62
|
| Spectrophotometry |
40–147 |
10 |
63
|
| Fluorescence |
0.8–4.8 |
0.24 |
64
|
| Chemiluminescence |
0.07–10.0 |
0.6 |
65
|
| Colorimetric method |
0.1–10.0 |
0.048 |
58
|
| Colorimetry (Mn3O4) |
0.05–1.4 |
0.019 |
66
|
| Colorimetric detection |
1.0–100.0 |
0.87 |
This work |
Interference analysis
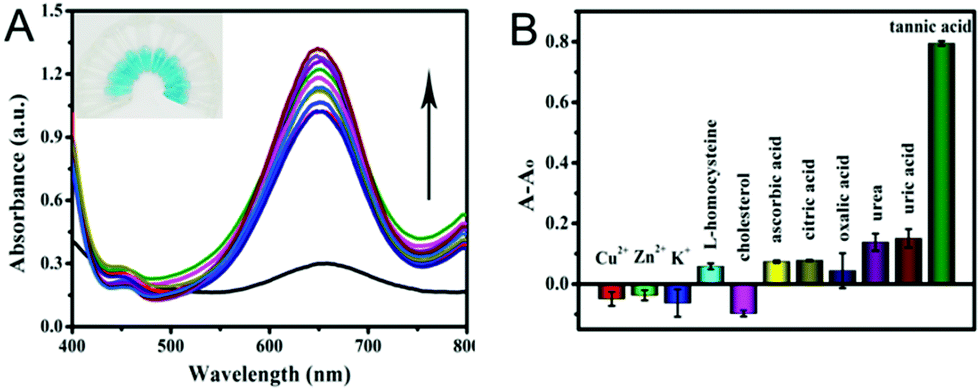
Furthermore, the selectivity based on PPy@MoS2@Au nanotubes for TA detection was investigated. The influence of some typical common interfering components has been investigated and the results are displayed in Fig. 11. The UV-vis absorption spectrum of PPy@MoS2@Au solution after addition of different analytes 10 times is shown in Fig. 11A. It is observed that the reaction system containing TA exhibits tremendous colorimetric contrast with those systems containing other interferences including Cu2+, Zn2+, K+, L-homocysteine, cholesterol, AA, CA, OA, urea and UA. In detail, the colorimetric change in the presence of TA is much more sensitive than that in the presence of other interferential substances (Fig. 11B). This result indicates that our proposed detection system shows good selectivity for the detection of TA.
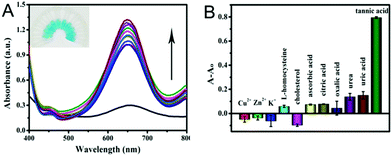 |
| | Fig. 11 (A) UV-visible absorption spectrum of TA selective detection, the photo of the illustration corresponds to the UV-visible absorption spectrum from bottom to top, and (B) the corresponding histogram. Selectivity analysis for TA determination by relative changes in absorbance. The concentration of TA was 0.02439 mM, Cu2+, Zn2+, K+, L-homocysteine, cholesterol, AA, CA, OA, urea and UA applied in the experiments was 0.2439 mM. The different values of absorbance at 652 nm in 0.2 M NaAc–HAc buffer solution (pH 3.0) systems containing fixed concentrations of TMB (0.25 mM), H2O2 (0.25 mM), and catalyst dispersion (0.119 mg mL−1). Error bars show the standard deviation derived from three repeated measurements. | |
Real sample analysis
According to the highly selective and sensitive detection of TA, the proposed method could be directly applied to measure the TA concentration in different kinds of tea. Table 2 shows the results of TA obtained in black tea and green tea samples. The results showed that the TA concentrations in black tea and green tea were 28.93 μM and 44.08 μM. In order to further verify the accuracy of the method, a recovery experiment was carried out and the results showed that the recoveries of TA in black tea ranged from 89.26 to 95.83%, while that in green tea ranged from 90.34 to 102.82%. The relative standard deviations (RSDs) are all less than 3.2%. It is obvious that satisfactory recoveries were obtained for the spiked samples, and the results demonstrated that it provided the potential for the quantitative determination of TA in real samples.
Table 2 Determination of TA in tea samples
| Samples |
Detected (μM) |
Added (μM) |
Found (μM) |
Recovery (%) |
RSD (n = 3, %) |
| Black tea |
28.93 |
25.0 |
51.25 |
89.26 |
1.8 |
| 30.0 |
56.64 |
92.04 |
2.5 |
| 35.0 |
62.47 |
95.83 |
3.0 |
| 35.0 |
76.49 |
92.60 |
2.1 |
| Green tea |
44.08 |
45.0 |
84.73 |
90.34 |
1.4 |
| 50.0 |
95.49 |
102.82 |
3.2 |
Conclusions
Herein, hierarchical PPy@MoS2 microtubes were prepared by a self-template process, which involves the coating of the PPy layer on MoO3 microrods followed by the hydrothermal treatment with thiourea. This unique architecture shows a hierarchical microtube configuration, which is constructed using edge-oriented MoS2 nanosheets and PPy microtubes. The existence of the PPy layer not only enhances the electron conductivity of MoS2, but also improves the dispersion of MoS2 and the exposure of more edge sites. SEM and TEM tests of intermediate products confirmed the dissolution–regrowth mechanism of the hierarchical PPy@MoS2 microtubes in the sulfurization reaction process. Profiting from the photogenerated electrons under light irradiation of MoS2 in PPy@MoS2, high coverages of noble metal NPs such as Ag, Au, and Pd NPs were in situ decorated on the surface of the as-prepared hierarchical PPy@MoS2 microtubes, giving rise to the formation of ternary PPy@MoS2@Ag, Au, and Pd microtubes. As a proof of application for PPy@MoS2@Au, the prepared ternary PPy@MoS2@Au nanotubes exhibit an obviously higher peroxidase-like catalytic activity than independent MoS2, MoS2@Au, and PPy@MoS2 owing to the synergistic catalytic effect among the three components. The present strategy may also pave a new way for developing high performance electrode materials for energy storage application.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support provided by the Natural Science Foundation of Shanghai City (18ZR1416400) and the National Key R&D Program of China (Grant 2018YFC1801503).
Notes and references
- T. S. Rodrigues, A. G. M. D. Silva and P. H. C. Camargo, J. Mater. Chem. A, 2019, 7, 5857–5874 RSC.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- S. Kim, G. H. Jeong, D. Choi, S. Yoon, H. B. Jeon, S. M. Lee and S. Kim, J. Colloid Interface Sci., 2013, 389, 85–90 CrossRef CAS PubMed.
- X. Zhou, M. Dorn, J. Vogt, D. Spemann, W. Yu, Z. Mao, I. Estrelalopis, E. Donath and C. Gao, Nanoscale, 2014, 6, 8535–8542 RSC.
- Q. Fang, J. Zhang, L. Bai, J. Duan, H. Xu, K. C. Leung and S. Xuan, J. Hazard. Mater., 2019, 367, 15–25 CrossRef CAS PubMed.
- J. Duan, L. Bai, K. Xu, Q. Fang, Y. Sun, H. Xu, K. C.-F. Leung and S. Xuan, J. Hazard. Mater., 2020, 384, 121276 CrossRef CAS PubMed.
- L. Guo, Q. Liu, G. Li, J. Shi, J. Liu, T. Wang and G. Jiang, Nanoscale, 2012, 4, 5864–5867 RSC.
- B. R. Evans, H. Oneill, V. P. Malyvanh, I. Lee and J. Woodward, Biosens. Bioelectron., 2003, 18, 917–923 CrossRef CAS PubMed.
- M. Kaushik and A. Moores, Green Chem., 2016, 18, 622–637 RSC.
- Y. Hu, Y. Huang, C. Tan, X. Zhang, Q. Lu, M. Sindoro, X. Huang, W. Huang, L. Wang and H. Zhang, Mater. Chem. Front., 2017, 1, 24–36 RSC.
- G. Zhang, H. Liu, J. Qu and J. Li, Energy Environ. Sci., 2016, 9, 1190–1209 RSC.
- W.-J. Zhang and K.-J. Huang, Inorg. Chem. Front., 2017, 4, 1602–1620 RSC.
- F. Tu, Y. Han, Y. Du, X. Ge, W. Weng, X. Zhou and J. Bao, ACS Appl. Mater. Interfaces, 2019, 11, 2112–2119 CrossRef CAS PubMed.
- W. Liu, M. Zhu, J. Liu, W. Hu, X. Li and J. Liu, Inorg. Chem. Front., 2018, 5, 2198–2204 RSC.
- L. Yang, S. Wang, J. Mao, J. Deng, Q. Gao, Y. Tang and O. G. Schmidt, Adv. Mater., 2013, 25, 1180–1184 CrossRef CAS PubMed.
- H. He, J. Lin, W. Fu, X. Wang, H. Wang, Q. Zeng, Q. Gu, Y. Li, C. Yan and B. K. Tay, Adv. Energy Mater., 2016, 6, 1600464 CrossRef.
- Y. Zhang, W. Sun, X. Rui, B. Li, H. T. Tan, G. Guo, S. Madhavi, Y. Zong and Q. Yan, Small, 2015, 11, 3694–3702 CrossRef CAS PubMed.
- Y. Zhao, W. Wang, M. Chen, R. Wang and Z. Fang, CrystEngComm, 2018, 20, 7266–7274 RSC.
- J. Zhu, Y. Ren, B. Yang, W. Chen and J. Ding, Nanoscale Res. Lett., 2017, 12, 627 CrossRef PubMed.
- H. Sun, J.-G. Wang, Y. Zhang, W. Hua, Y. Li and H. Liu, J. Mater. Chem. A, 2018, 6, 13419–13427 RSC.
- C. Chang, X. Yang, S. Xiang, H. Que and M. Li, J. Mater. Sci.: Mater. Electron., 2017, 28, 1777–1784 CrossRef CAS.
- X.-Q. Qiao, Z.-W. Zhang, F.-Y. Tian, D.-F. Hou, Z.-F. Tian, D.-S. Li and Q. Zhang, Cryst. Growth Des., 2017, 17, 3538–3547 CrossRef CAS.
- M. Zhang, J. Zheng, J. Wang, J. Xu, T. Hayat and N. S. Alharbi, Sens. Actuators, B, 2019, 282, 85–95 CrossRef CAS.
- M. Zhang, L. Chen, J. Zheng, W. Li, T. Hayat, N. S. Alharbi, W. Gan and J. Xu, Dalton Trans., 2017, 46, 9172–9179 RSC.
- L. Chen, M. Zhang, X. Yang, W. Li, J. Zheng, W. Gan and J. Xu, J. Alloys Compd., 2017, 695, 3339–3347 CrossRef CAS.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- J. Wang, M. Zhang, T. Miao, Y. Ling, Q. Wen, J. Zheng, J. Xu, T. Hayat and N. S. Alharbi, Inorg. Chem. Front., 2018, 5, 844–852 RSC.
- Y. Liu, B. Zhang, Y. Yang, Z. Chang, Z. Wen and Y. Wu, J. Mater. Chem. A, 2013, 1, 13582–13587 RSC.
- W. Tang, X. Gao, Y. Zhu, Y. Yue, Y. Shi, Y. Wu and K. Zhu, J. Mater. Chem., 2012, 22, 20143–20145 RSC.
- S. Xia, Y. Wang, Y. Liu, C. Wu, M. Wu and H. Zhang, Chem. Eng. J., 2018, 332, 431–439 CrossRef CAS.
- X. Ma, M. Liu, L. Gan, P. K. Tripathi, Y. Zhao, D. Zhu, Z. Xu and L. Chen, Phys. Chem. Chem. Phys., 2014, 16, 4135–4142 RSC.
- Y. Sun, Y. Wang, J. Y. Chen, K. Fujisawa, C. F. Holder, J. T. Miller, V. H. Crespi, M. Terrones and R. E. Schaak, Nat. Chem., 2020, 12, 284–293 CrossRef CAS PubMed.
- J. Sheng, J. Kang, Z. Hu, Y. Yu, X. Fu, R. Sun and C. Wong, J. Mater. Chem., 2018, 6, 15789–15796 RSC.
- Y. Zhu, R. Zhu, L. Yan, H. Fu, Y. Xi, H. Zhou, G. Zhu, J. Zhu and H. He, Appl. Catal., B, 2018, 239, 280–289 CrossRef CAS.
- W. He, X. Guo, J. Zheng, J. Xu, T. Hayat, N. S. Alharbi and M. Zhang, Inorg. Chem., 2019, 58, 7255–7266 CrossRef CAS PubMed.
- M. Zhang, X. Liu, Q. Luo, Q. Wang, L. Zhao, G. Deng, R. Ge, L. Zhang, J. Hu and J. Lu, Chem. Eng. J., 2020, 389, 124450 CrossRef CAS.
- G. Zhai, J. Wang, Z. Chen, W. An and Y. Men, Chem. Eng. J., 2018, 337, 488–498 CrossRef CAS.
- J. Zheng, M. Zhang, T. Miao, J. Yang, J. Xu, N. S. Alharbi and M. Wakeel, Mater. Chem. Front., 2019, 3, 224–232 RSC.
- J. Hu, M. Zhang, L. Liu, J. Zheng, H. Alsulami, M. A. Kutbi and J. Xu, Inorg. Chem., 2020, 59, 9356–9363 CrossRef CAS PubMed.
- M. Zhang, Y. Ling, L. Liu, J. Xu, J. Li and Q. Fang, Inorg. Chem. Front., 2020 10.1039/D0QI00596G.
- K. Zhang, Y. Zhao, S. Zhang, H. Yu, Y. Chen, P. Gao and C. Zhu, J. Mater. Chem., 2014, 2, 18715–18719 RSC.
- S. Zhang, H. Li, Z. Wang, J. Liu, H. Zhang, B. Wang and Z. Yang, Nanoscale, 2015, 7, 8495–8502 RSC.
- P. Qin, G. Fang, W. Ke, F. Cheng, Q. Zheng, J. Wan, H. Lei and X. Zhao, J. Mater. Chem. A, 2014, 2, 2742–2756 RSC.
- F. Lyu, Y. Bai, Z. Li, W. Xu, Q. Wang, J. Mao, L. Wang, X. Zhang and Y. Yin, Adv. Funct. Mater., 2017, 27, 1702324 CrossRef.
- M. Liu, Y. Zheng, L. Zhang, L. Guo and Y. Xia, J. Am. Chem. Soc., 2013, 135, 11752–11755 CrossRef CAS PubMed.
- T. Zhang, J. Ge, Y. Hu, Q. Zhang, S. Aloni and Y. Yin, Angew. Chem., 2008, 47, 5806–5811 CrossRef CAS.
- L. Chen, H. Jiang, Y. Hu, H. Wang and C. Li, Sci. China Mater., 2018, 61, 1049–1056 CrossRef CAS.
- T. I. Kim, B. Kwon, J. Yoon, I. J. Park, G. S. Bang, Y. Park, Y. Seo and S. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 7908–7917 CrossRef CAS PubMed.
- V. Yadav, S. Roy, P. Singh, Z. Khan and A. Jaiswal, Small, 2019, 15, 1803706 CrossRef PubMed.
- M. Chi, Y. Zhu, L. Jing, C. Wang and X. Lu, Anal. Chim. Acta, 2018, 1035, 146–153 CrossRef CAS PubMed.
- W. Zhu, M. Chi, M. Gao, C. Wang and X. Lu, J. Colloid Interface Sci., 2018, 528, 410–418 CrossRef CAS PubMed.
- J. Zheng, D. Song, H. Chen, J. Xu, N. S. Alharbi, T. Hayat and M. Zhang, Chin. Chem. Lett., 2020, 31, 1109–1113 CrossRef CAS.
- Y. Dong, H. Zhang, Z. Rahman, L. Su, X. Chen, J. Hu and X. Chen, Nanoscale, 2012, 4, 3969–3976 RSC.
- R. Zhang, N. Lu, J. Zhang, R. Yan, J. Li, L. Wang, N. Wang, M. Lv and M. Zhang, Biosens. Bioelectron., 2020, 150, 111881 CrossRef CAS PubMed.
- H. Wei and E. Wang, Anal. Chem., 2008, 80, 2250–2254 CrossRef CAS PubMed.
- Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097–1105 CrossRef CAS.
- C. Cui, Q. Wang, Q. Liu, X. Deng, T. Liu, D. Li and X. Zhang, Sens. Actuators, B, 2018, 277, 86–94 CrossRef CAS.
- N. Song, Y. Zhu, F. Ma, C. Wang and X. Lu, Mater. Chem. Front., 2018, 2, 768–774 RSC.
- N. Lu, M. Zhang, L. Ding, J. Zheng, C. Zeng, Y. Wen, G. Liu, A. Aldalbahi, J. Shi and S. Song, Nanoscale, 2017, 9, 4508–4515 RSC.
- C. Zhaohui, Z. Xiaodan, C. Haiyan and H. Yuming, Analyst, 2013, 138, 2343–2349 RSC.
- P. A. Kilmartin, H. Zou and A. L. Waterhouse, J. Agric. Food Chem., 2001, 49, 1957–1965 CrossRef CAS PubMed.
- T. J. Cheng, H. Y. Hsiao, C. Y. Chung, P. C. Chen and R. L. C. Chen, Microchim. Acta, 2010, 169, 117–122 CrossRef CAS.
- H. Yang, L. He, S. Pan, H. Liu and X. Hu, Spectrochim. Acta, Part A, 2019, 210, 111–119 CrossRef CAS PubMed.
- H. Cui, Q. Li, R. Meng, H. Zhao and C. He, Anal. Chim. Acta, 1998, 362, 151–155 CrossRef CAS.
- X. Zhang and Y. Huang, Anal. Methods, 2015, 7, 8640–8646 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 showing SEM and TEM images of MoO3@PAIN (a and b) and PAIN@MoS2 (c and d). Scale bars: 1 μm in (a and b) and 500 nm in (c and d); Fig. S2 showing SEM and TEM images of PPy@MoS2@Au, 2 mg (a and b) and 4 mg (c and d). Scale bars: 1 μm in (a and c) and 200 nm in (b and d); Fig. S3 showing the XPS full spectrum of PPy@MoS2@Pd (a) and PPy@MoS2@Ag (c), (b) XPS spectra of Pd 3d of PPy@MoS2@Pd and (d) XPS spectra of Ag 3d of PPy@MoS2@Ag; etc. See DOI: 10.1039/d0tb01387k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Jingli
Xu
a,
Jing
Zheng
c,
Jingli
Xu
a,
Jing
Zheng