
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an antibacterial nanocomposite hydrogel for human dental pulp engineering†
M.
Bekhouche
 *ab,
M.
Bolon
a,
F.
Charriaud
a,
M.
Lamrayah
a,
D.
Da Costa
ac,
C.
Primard
c,
A.
Costantini
a,
M.
Pasdeloup
a,
S.
Gobert
a,
F.
Mallein-Gerin
a,
B.
Verrier
a,
M.
Ducret
abd and
J.-C.
Farges
abd
*ab,
M.
Bolon
a,
F.
Charriaud
a,
M.
Lamrayah
a,
D.
Da Costa
ac,
C.
Primard
c,
A.
Costantini
a,
M.
Pasdeloup
a,
S.
Gobert
a,
F.
Mallein-Gerin
a,
B.
Verrier
a,
M.
Ducret
abd and
J.-C.
Farges
abd
aLaboratoire de Biologie Tissulaire et Ingénierie Thérapeutique, UMR5305 CNRS/Université Lyon 1, Lyon, France
bFaculté d'Odontologie, Université de Lyon, Université Lyon 1, Lyon, France
cAdjuvatis®, Lyon, France
dHospices Civils de Lyon, Service de Consultations et Traitements Dentaires, Lyon, France
First published on 3rd August 2020
Abstract
Hydrogel-based regenerative endodontic procedures (REPs) are considered to be very promising therapeutic strategies to reconstruct the dental pulp (DP) tissue in devitalized human teeth. However, the success of the regeneration process is limited by residual bacteria that may persist in the endodontic space after the disinfection step and contaminate the biomaterial. The aim of this work was to develop an innovative fibrin hydrogel incorporating clindamycin (CLIN)-loaded Poly (D,L) Lactic Acid (PLA) nanoparticles (NPs) to provide the hydrogel with antibacterial properties. CLIN-PLA-NPs were synthesized by a surfactant-free nanoprecipitation method and their microphysical properties were assessed by dynamic light scattering, electrophoretic mobility and scanning electron microscopy. Their antimicrobial efficacy was evaluated on Enteroccocus fæcalis by the determination of the minimal inhibitory concentration (MIC) and the minimal biofilm inhibition and eradication concentrations (MBIC and MBEC). Antibacterial properties of the nanocomposite hydrogel were verified by agar diffusion assays. NP distribution into the hydrogel and release from it were evaluated using fluorescent PLA-NPs. NP cytotoxicity was assessed on DP mesenchymal stem cells (DP-MSCs) incorporated into the hydrogel. Type I collagen synthesis was investigated after 7 days of culture by immunohistochemistry. We found that CLIN-PLA-NPs displayed a drug loading of 10 ± 2 μg per mg of PLA polymer and an entrapment efficiency of 43 ± 7%. Antibiotic loading did not affect NP size, polydispersity index and zeta potential. The MIC for Enterococcus fæcalis was 32 μg mL−1. MBIC50 and MBEC50 were 4 and 16 μg mL−1, respectively. CLIN-PLA-NPs appeared homogenously distributed throughout the hydrogel. CLIN-PLA-NP-loaded hydrogels clearly inhibited E. faecalis growth. DP-MSC viability and type I collagen synthesis within the fibrin hydrogel were not affected by CLIN-PLA-NPs. In conclusion, CLIN-PLA-NP incorporation into the fibrin hydrogel gave the latter antibacterial and antibiofilm properties without affecting cell viability and function. This formulation could help establish an aseptic environment supporting DP reconstruction and, accordingly, might be a valuable tool for REPs.
1. Introduction
Regenerative endodontic procedures (REPs) aim at reconstructing a dental pulp (DP) tissue in the endodontic space of devitalized human teeth, in order to restore major functionalities involved in injury detection such as immune surveillance and sensitivity.1–5 Over the last decade, many tissue engineering approaches have been developed to achieve this goal. Most are based on the injection, into the cleaned endodontic space, of a three-dimensional (3D) scaffold incorporating or not DP mesenchymal stem cells (DP-MSCs).4 Numerous kinds of scaffolds were tested, such as natural and synthetic polymers/co-polymers, hydroxyapatite/tricalcium phosphate powders, self-assembling peptide systems, platelet-rich plasma and decellularized extracellular matrices (ECM).6–11 However, all of them only partially reach the materiobiological criteria necessary for DP regeneration which include bioinspired structural design, low cytotoxicity, physiological degradation by host and/or incorporated cells, and rapid replacement with an ECM characteristic of the tissue.8,11,12 In addition, authors rarely took into consideration one major limit of REPs which is the persistence of residual bacteria in the endodontic space after the disinfection step. Indeed, bacteria are hardly eliminated by the disinfectants classically used such as sodium hypochlorite, owing to their organization into biofilms on the dentin surface and their deep penetration within dentin tubules. They clearly hamper DP regeneration13–15 by promoting host immune/inflammatory events instead of regenerative ones. To overcome this difficulty, polydioxanone-based nanofibrous hydrogels containing metronidazole, ciprofloxacin, and/or clindamycin16–19 have been proposed. Although these scaffolds displayed effective antibacterial properties against species present into the endodontic space such as Enterococcus fæcalis and Porphyromonas gingivalis, they strongly affected DP-MSCs viability (up to 50%). Therefore, designing new hydrogel formulations with antibacterial and antibiofilm properties, capable of preserving cell viability and function, is clearly required.8,11,20Fibrin was recently reported as a promising scaffold material for REPs since it promotes DP-like tissue formation both in vitro and in vivo.11,21 We recently developed an injectable fibrin hydrogel formulation highly suitable to dental practice since it clearly supported DP-MSCs viability and DP-like collagenous ECM deposition.20 However, neither fibrin nor its degradation products display any antimicrobial activities.22 We decided in the present study to give our newly developed fibrin scaffold antibacterial properties by adding antibiotic-loaded nanoparticles (NPs).
NPs used as carriers have demonstrated high relevance in antibiotic delivery since they preserve the native structure of the drug, enhance its bioavailability and biodistribution in infected tissues and ensure its high concentration locally. By slowly releasing the molecule, they increase the exposure time of the bacterial population to the antibiotic whereas, by restraining the drug localization within the infected site, they minimize putative systemic side effects.23 Antibiotic-loaded NPs appear well adapted to biofilm eradication owing to their ability to penetrate deep into biofilm matrices.24 This penetration enables the antibiotic to reach the deepest and most persistent bacteria which are difficult to eliminate.25 Over the last years, Poly (D,L) Lactic Acid NPs (PLA-NPs) received considerable attention, because of their biodegradable nature, biocompatibility, and low immunogenicity.26 PLA is a biopolymer with high safety profile27 which has been approved by the US Food and Drug Administration for various applications.26 PLA-NPs constitute a versatile carrier of drugs and vaccines26–28 and have been used to deliver antifungal and antibacterial drugs such as amphotericin B,29 minocycline30 and pipemidic acid.31 Our hypothesis was that PLA-NPs could constitute a suitable antibiotic carrier that, once added to our fibrin hydrogel formulation, would provide the hydrogel with antibacterial and antibiofilm properties. The antibiotic clindamycin (CLIN) was chosen because of its broad spectrum activity against endodontic bacteria and its routine use in oral medicine in patients allergic to β-lactams.32 Also, CLIN can be easily incorporated into PLA-NPs owing to its hydrophobicity.33–35
This study aimed at designing a nanocomposite scaffold made of a fibrin hydrogel supplemented with CLIN-loaded PLA-NPs in order to provide the aseptic environment necessary for DP regeneration without affecting DP-MSC viability and function (Fig. 1).
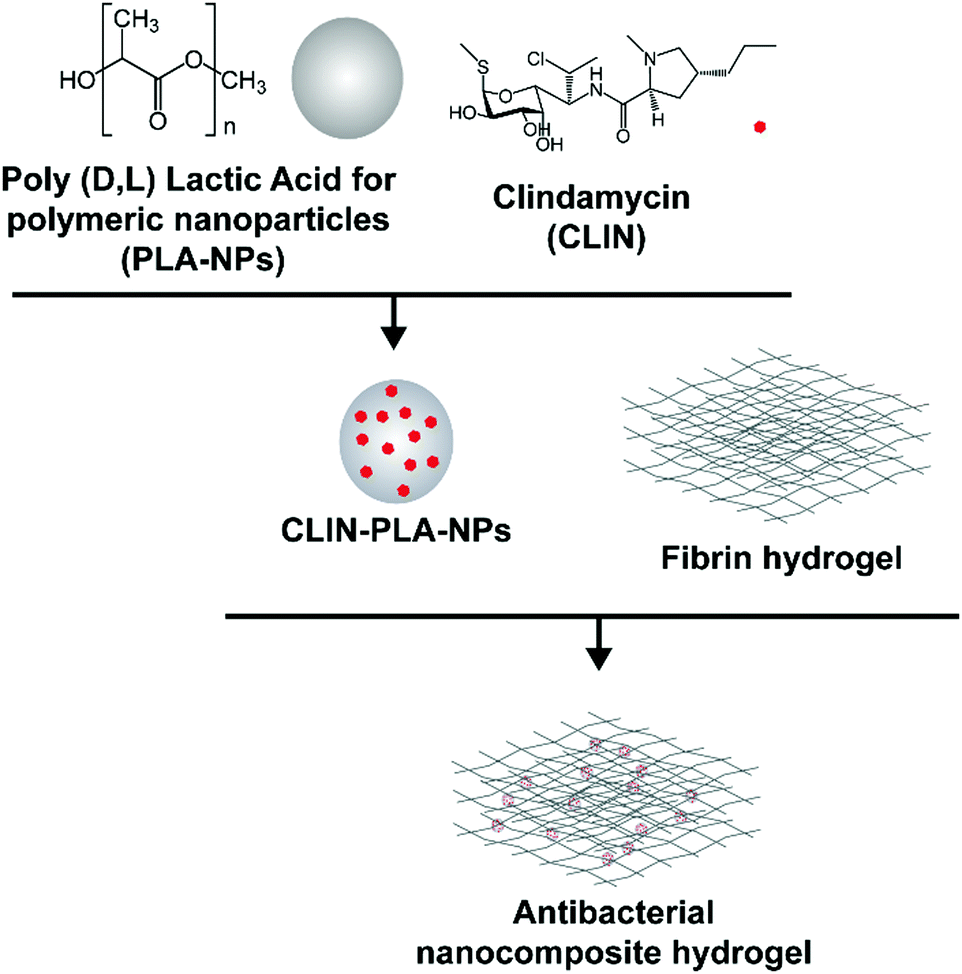 | ||
| Fig. 1 Schematic representation of the two-step synthesis of the nanocomposite hydrogel. | ||
2. Experimental
2.1 NP synthesis
PLA (MW 45 g mol−1, pKa 3.8) was produced according to the patent FR2745005A1 of Phusis (France). CLIN (MW 424.98 g mol−1, pKa 7.6, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P 2.16) was purchased from Sellekchem (USA). CLIN-PLA-NPs were synthesized as previously described using a surfactant-free solvent diffusion method.36 Briefly, CLIN and PLA were dissolved in acetone (2.5% w/w) and this solution was slowly added to an aqueous phase composed of ethanol and 4 mM carbonate buffer (pH 10) under moderate stirring. Pyrogen free water (Otec, Aguettant, France) was added at the end of pouring to favour solvents evaporation under reduced pressure and controlled temperature using a Rotavapor coupled to a heating bath (Buchi, France). The final volume of particles was precisely measured as well as the final pH of the solution (pH 8.5, storage buffer). NPs without CLIN were synthesized in parallel using the same method.
P 2.16) was purchased from Sellekchem (USA). CLIN-PLA-NPs were synthesized as previously described using a surfactant-free solvent diffusion method.36 Briefly, CLIN and PLA were dissolved in acetone (2.5% w/w) and this solution was slowly added to an aqueous phase composed of ethanol and 4 mM carbonate buffer (pH 10) under moderate stirring. Pyrogen free water (Otec, Aguettant, France) was added at the end of pouring to favour solvents evaporation under reduced pressure and controlled temperature using a Rotavapor coupled to a heating bath (Buchi, France). The final volume of particles was precisely measured as well as the final pH of the solution (pH 8.5, storage buffer). NPs without CLIN were synthesized in parallel using the same method.
2.2 NP morphophysical properties
NP hydrodynamic diameter, polydispersity index (PDI) and zeta potential were measured for the different CLIN-PLA-NP and PLA-NP batches after dilution (1/150) of the NP suspension in 1 mM NaCl using a Zetasizer Nano ZS Plus (Malvern Instruments, UK). Diameter and PDI were measured by dynamic light scattering (DLS) at 25 °C at a 173 degree angle. The zeta potential was determined by measuring electrophoretic mobilities by laser Doppler velocimetry using the same device, at a scattering angle of 12.5 degree. Each value was the average of 4 measurements.NP morphology was visualized with a scanning electron microscope equipped with a field emission gun (FEI Quanta 250 SEM, Thermo Fischer Scientific, USA). For sample preparation, the NP solution was diluted 10 times and a drop was deposited on a glass coverslip then air-dried. The coverslip was then copper sputtered (BAL-TEC, MCS 010, UK) prior to observation.
2.3 CLIN loading into PLA-NPs
The amount of CLIN incorporated into the PLA-NPs was determined using the high performance liquid chromatography (HPLC) with UV/Visible detection method developed by Na-Bangchang et al.37 The HPLC system was composed of a Degasser 1260 (Agilent, USA), a Quaternary Pump G1311A, an Autosampler G1329A, a Column Heater G1316A and a Diode Array Detector G1315A, all four from Hewlett Packard (USA). Chromatograms were obtained using the Chem Station for Liquid Chromatography 3D System software (Hewlett Packard, USA). The stationary phase was a CLIPEUS C18 column (5 μm, 100 × 4 mm, Higgins Analytical, USA). The mobile phase was composed of 0.02 M disodium hydrogenophosphate (Sigma Aldrich, France) (pH 2.8) and acetonitrile (ACN, Carbo Elba, Italy) in proportion 76:24 (v/v). The solvent used was HPLC grade. The chromatographic analysis was operated at 25 °C. A 100 μL volume was injected at a flow rate of 0.75 mL min−1. The wavelength was set at 200 nm. This method showed linearity (R2 = 0.997) in the CLIN concentration range 1–60 μg mL−1 (ESI† 1).
The CLIN-PLA-NP suspension was centrifuged at 11000 rpm for 45 min and the supernatant was transferred in a new tube for quantification. CLIN-PLA-NPs present in the pellet were solubilized using ACN to release loaded CLIN. PLA was precipitated by adding pure methanol (Carbo Elba, Italy) with a total solvent volume of 50 μL per g of PLA. The ratio ACN:methanol was adjusted to 1:14 (v/v).
Entrapment efficiency (EE) was calculated using the formula:
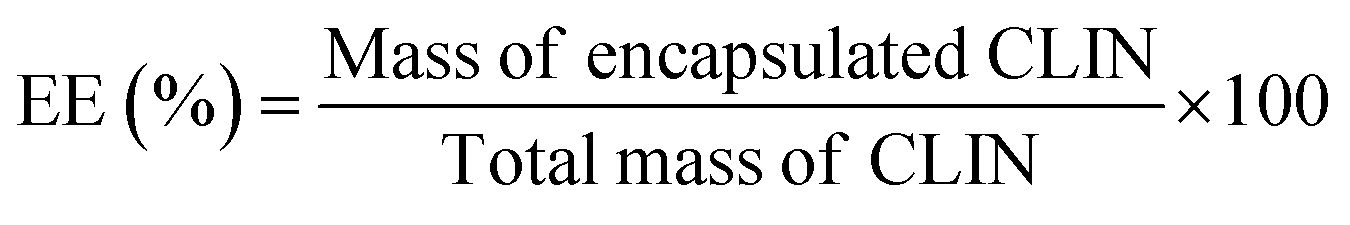
Drug loading (DL) was calculated using the formula:
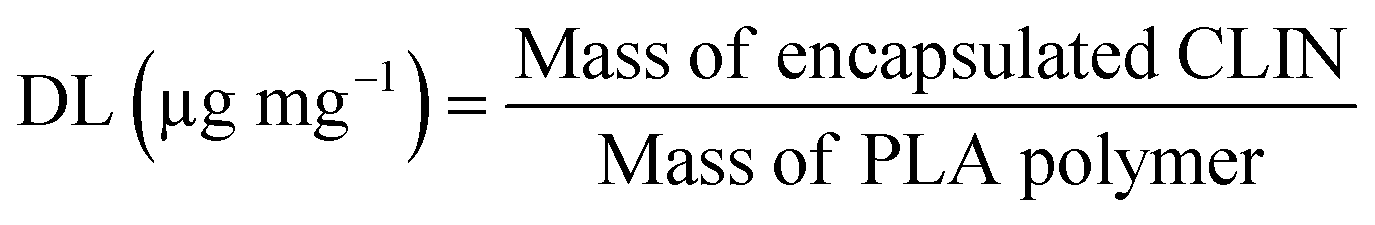
To assess CLIN release from the NPs, CLIN-PLA-NPs were diluted in PBS buffer (1:1, v:v) and incubated in triplicate at 37 °C for 9 days. At different time points (5, 22, 46, 90, 162, 214 h), the NPs were collected by centrifugation at 11000 rpm for 45 min and the CLIN content was determined as described above. The clindamycin release from the nanoparticles at each time was reported as a percentage of the starting mass of CLIN. Stability of clindamycin in PBS at 37 °C was assessed by HPLC for 21 days (ESI† 1).
2.4 Antibacterial and antibiofilm properties
The E. fæcalis (ATCC® 47077) strain was used to evaluate the antibacterial properties of CLIN-PLA-NPs. The minimal inhibitory concentration (MIC), defined as the lowest antibiotic concentration that inhibits a visible planktonic bacterial growth after an overnight incubation at 37 °C, was determined using the broth microdilution method. An E. fæcalis suspension was prepared to obtain an optical density (OD) of 0.004 at 600 nm, which corresponds to 106 colony forming units (CFU) per mL. The suspension was cultured in a 96-well plate in brain-heart infusion (BHI) broth (Becton Dickinson, USA), in the presence of serial dilutions of free CLIN (0.004 to 256 μg mL−1) or of the corresponding CLIN-PLA-NPs or PLA-NPs dilutions (see Result section 3.1). The plate was incubated for 24 h at 37 °C and OD was measured at 600 nm with a Multiskan GO spectrophotometer (Thermo Fischer Scientific). The lowest CLIN concentration that yields an OD similar to the negative control was determined as the MIC. The MIC value was confirmed by seeding the bacterial suspensions used for MIC determination on BHI-agar plates (Sigma Aldrich, France).The minimal biofilm inhibitory concentration (MBIC), defined as the lowest antibiotic concentration that inhibits the growth of a visible biofilm, was determined by culturing E. fæcalis in BHI supplemented with 1% glucose (Sigma Aldrich), then by plating bacteria at an initial OD of 0.004 during 24 h at 37 °C, in the presence of the same CLIN, CLIN-PLA-NP or PLA-NP serial dilutions as previously described in this section. Biofilms formed after 24 h38 were washed three times with PBS (Gibco, France) and stained with 0.1% crystal violet (Sigma Aldrich). After a 15 min incubation, biofilms were washed again three times with water and dried at room temperature (RT). Glacial acetic acid was added to solubilize crystal violet and biofilm absorbance was measured at OD 590 nm.
The minimal biofilm eradication concentration (MBEC), defined as the lowest antibiotic concentration that eradicates a 24 hour old biofilm38 was determined in the same experimental conditions as for MBIC measurement, except that free CLIN, CLIN-PLA-NPs or PLA-NPs were added on a 24 hour old E. fæcalis biofilm.
Antimicrobial properties of the nanocomposite fibrin hydrogel (see Section 2.5 for hydrogel preparation) were assessed using an agar diffusion method based on the plating of hydrogels containing 50, 100 or 200 μg mL−1 of CLIN, CLIN-PLA-NPs or equivalent 0.2, 0.4 or 0.8% solid content PLA-NPs, on BHI broth-agar previously seeded with E. fæcalis at 1.5 OD. Each experiment was performed three times in triplicate. After 24 h at 37 °C, inhibition areas were photographed and measured using the Fiji software.
2.5 PLA-NP distribution and release from the nanocomposite hydrogel
The fibrin hydrogel formulation was prepared as previously described by Ducret et al.20 to obtain a final fibrin hydrogel composed of 10 mg mL−1 fibrinogen (MW ∼ 340 kDa, pI 5.8) (EMD Millipore Corp., USA) which had been polymerized in the presence of 4 U mL−1 thrombin (Sigma Aldrich, France). CLIN-PLA-NPs or free CLIN were added to obtain a final concentration of 50 μg mL−1 CLIN into the hydrogel.To characterize the microstructure of the fibrin hydrogel and NP localization, fibrin-alone and CLIN-PLA-NP-loaded fibrin hydrogels were treated for scanning electron microscopy (SEM). Briefly, samples were rinsed with water, frozen in pasty nitrogen, sublimated at −90 °C to unveil the fibrin network and sputtered with gold/palladium. Samples were then observed with a FEI Quanta 250 SEM (Thermo Fisher Scientific, USA).
To assess CLIN release, hydrogels were prepared as above in the presence of 50 μg mL−1 clindamycin in free (CLIN) or formulated (CLIN-PLA-NPs) forms, or of the equivalent 0.2% solid content empty particles (PLA-NPs). After polymerization, 1 volume of PBS was added on the top of the hydrogel and incubated in a humidified chamber at 37 °C for 24 h. At different time points (0.25, 0.5, 2, 3, 6, 24 h), the solution was collected and replaced with a fresh one. Samples were analysed by HPLC-UV and the cumulative CLIN release was determined. The assay was realized in triplicate using three different batches of nanoparticles.
General distribution of NPs in the hydrogel was assessed using PLA-NPs coupled with a green-Bodipy fluorophore (PLA-fluoNPs) (Adjuvatis®, France). PLA-fluoNPs had a diameter of 171 nm, a PDI of 0.03 and a zeta potential of −57 ± 1 mV, all values comparable to non-fluorescent PLA-NPs and CLIN-PLA-NPs (see Result section 3.1). PLA-fluoNPs displayed an EE of 95–98%, which indicated that the measured fluorescence intensity reflected directly the NP amount present in the hydrogel. Nanocomposite hydrogels were formulated as previously described, using the same amount of PLA-fluoNPs as for CLIN-PLA-NPs. They were observed with an Eclipse TE300 fluorescence microscope (Nikon, Japan) using an excitation wavelength of 505 nm and an emission wavelength of 515 nm. Images were analysed using the Fiji software.
To assess NP release over time, hydrogels containing PLA-fluoNPs were incubated in PBS at 37 °C for 4 days. The incubation medium was collected at different time points (24, 48, 72 and 96 h) and the fluorescence intensity was determined using a multimode flagship microplate reader (Infinite M1000, Tecan, Switzerland). The percentage of released PLA-fluoNPs was calculated by comparing the released fluorescence to the total fluorescence incorporated in the hydrogel, because fluorescence intensity is directly proportional to PLA-fluoNPs concentration in these conditions (R2 > 0.99) (ESI† 2).
2.6 DP-MSC isolation and expansion
Isolation and expansion of DP-MSCs were performed with a good manufacturing practice approach as described previously.39,40 Briefly, healthy impacted human third molars were collected from donors aged 13–17 years with informed consent of the patients and their parents, in accordance with the World Medical Association's Declaration of Helsinki and the French Public Health Code (Article R1211-49), and following a protocol approved by the French Ministry of Higher Education and Research (CODECOH: DC-2014-2325). DPs from teeth between Nolla developmental stages 5 (crown almost completed) and 7 (one third root completed) were gently extirpated from pulp cavities and cut into fragments of about 0.5–2 mm3. Pulp fragments were seeded as explants on dishes pre-coated with a mixture of human placental collagens I and III (0.5 μg cm−2) and cultured in Optistem® serum-free medium (Univercell Biosolutions, France) supplemented with 100 IU mL−1 penicillin and 100 mg mL−1 streptomycin (Thermo Fischer Scientific, USA). After 2 weeks, cells outgrowing from the explants were harvested using TrypLE® (Thermo Fischer Scientific, USA) and checked for stemness by flow cytometry as previously described.38 DP-MSCs were passaged 2–3 times before being incorporated into the fibrin hydrogel formulation (1667 cells per μL). Cellularized formulations were then poured into 96-well plates for polymerization and cultured in 120 μL Optistem® medium.2.7 Cell viability determination
Cytotoxicity was determined after 48 h of culture in NP-free fibrin hydrogels and in fibrin hydrogels containing 0.2% solid content PLA-NPs, CLIN-PLA-NPs, or the equivalent 50 μg mL−1 free CLIN, using a Live/Dead Cell Double Staining® kit (Sigma Aldrich, France). Experiments were performed in triplicate using three different PLA-CLIN-NPs and PLA-NPs batches and DP-MSCs from two female patients (13 and 14 year old). DP-MSCs viability is defined as the percentage of living cells compared to total cells. Briefly, hydrogels were quickly rinsed three times with PBS and incubated for 15 min at 37 °C in a PBS solution containing 0.2 μM acetoxymethyl ester of calcein and 0.2 μM propidium iodide. They were then observed using an Eclipse TE300 fluorescence microscope (Nikon Instruments Europe B.V., Netherlands) and cells were counted using the Fiji software. Negative controls were realized by replacing DP-MSCs with PBS to confirm signal specificity.2.8 Type I collagen immunohistochemistry
Type I collagen synthesis by DP-MSCs was determined using immunohistochemistry as described previously20 with slight modifications. Briefly, DP-MSCs were cultured for 7 days in fibrin-alone, fibrin/CLIN-PLA-NP or fibrin/PLA-NP hydrogels as described in Section 2.7. Hydrogels were fixed in acidified formal alcohol (AFA; Microm Microtech, France) at 7 °C for 24 h and classically embedded in paraffin. Five μm serial sections were cut, deparaffinized and rehydrated. For antigen retrieval, sections were incubated at 37 °C first in 0.5% hyaluronidase diluted in PBS for 1 h then in Pepsin Reagent (ref R2283, Sigma Aldrich, France) for 15 min. Endogenous peroxidase activity was blocked by incubating the sections in 0.5% hydrogen peroxide for 10 min at RT. To reduce non-specific binding of antibodies, sections were treated with BlockAidTM (Thermo Fisher Scientific, USA) at RT for 1 h. Sections were then incubated with the anti-type I collagen rabbit polyclonal antibody (1/2000) (ref. 20111[380k], Novotec, Bron, France) overnight at 4 °C. Antibody detection was performed with an ImmPRESS® HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection kit (Vector Laboratories, USA). Sections were finally counterstained with hematoxylin, mounted with DEPEX medium (ref 06522, Sigma Aldrich, France) and observed with an Eclipse TE300 microscope.2.9 Statistical analyses
Data are presented as the mean values ± standard deviation. Statistical analyses were performed on GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla, CA, USA) using ANOVA followed by multiple comparison using statistical hypothesis testing and Bonferroni correction. The number of samples (N) is indicated in figure captions. A difference among means with a P-value < 0.05 was considered significant.3. Results
3.1 Characterization of the nanoparticles with or without clindamycin
The CLIN-PLA-NPs were synthesized by the surfactant-free nanoprecipitation method36 which includes three stages: an initial nucleation stage, a growth phase, and the formation of stable particles when an equilibrium is reached. A small size distribution results from a high nucleation rate and a low growth of particles.41 At high pH, the carboxylic acid groups of the PLA polymer are highly ionized, leading to a rapid stabilization of the nanoparticles driven by electrostatic repulsions between them. This method is applicable for the encapsulation of lipophilic or amphiphilic compounds, driven by hydrophobic interactions with the polymer,42 as previously characterized by molecular modelling.36The CLIN-PLA-NPs exhibited a Z-average size of 159 ± 2 nm and a polydispersity index of 0.09 ± 0.01 which indicated a relatively narrow particle size distribution (Table 1). PLA-NPs and CLIN-PLA-NPs exhibited a regular spherical morphology, were fairly uniform in size and did not show any form of aggregation (Fig. 2A–D). The zeta potential of −60 ± 2 mV was negative according to the net charge of PLA in the storage buffer (pH 8.5). CLIN incorporation into PLA-NPs did not significantly modify their diameter, PDI, zeta potential, or morphology. Entrapment efficiency was 43 ± 7%, a value corresponding to a drug loading of 10 ± 2 μg CLIN per mg PLA (Table 1). The profile of clindamycin release from PLA nanoparticles was evaluated for 9 days in PBS at 37 °C. It was characterized by a fast burst release of more than 60% of the clindamycin loaded onto the nanoparticles (Fig. 2E).
| PLA-NPs | CLIN-PLA-NPs | |
|---|---|---|
| Hydrodynamic diameter (nm) | 155 ± 2 | 159 ± 2 |
| Polydispersity index | 0.09 ± 0.01 | 0.09 ± 0.01 |
| Zeta potential (mV) | −66 ± 3 | −60 ± 2 |
| Entrapment efficiency (%) | — | 43 ± 7 |
| Drug loading (μg mg−1) | — | 10 ± 2 |
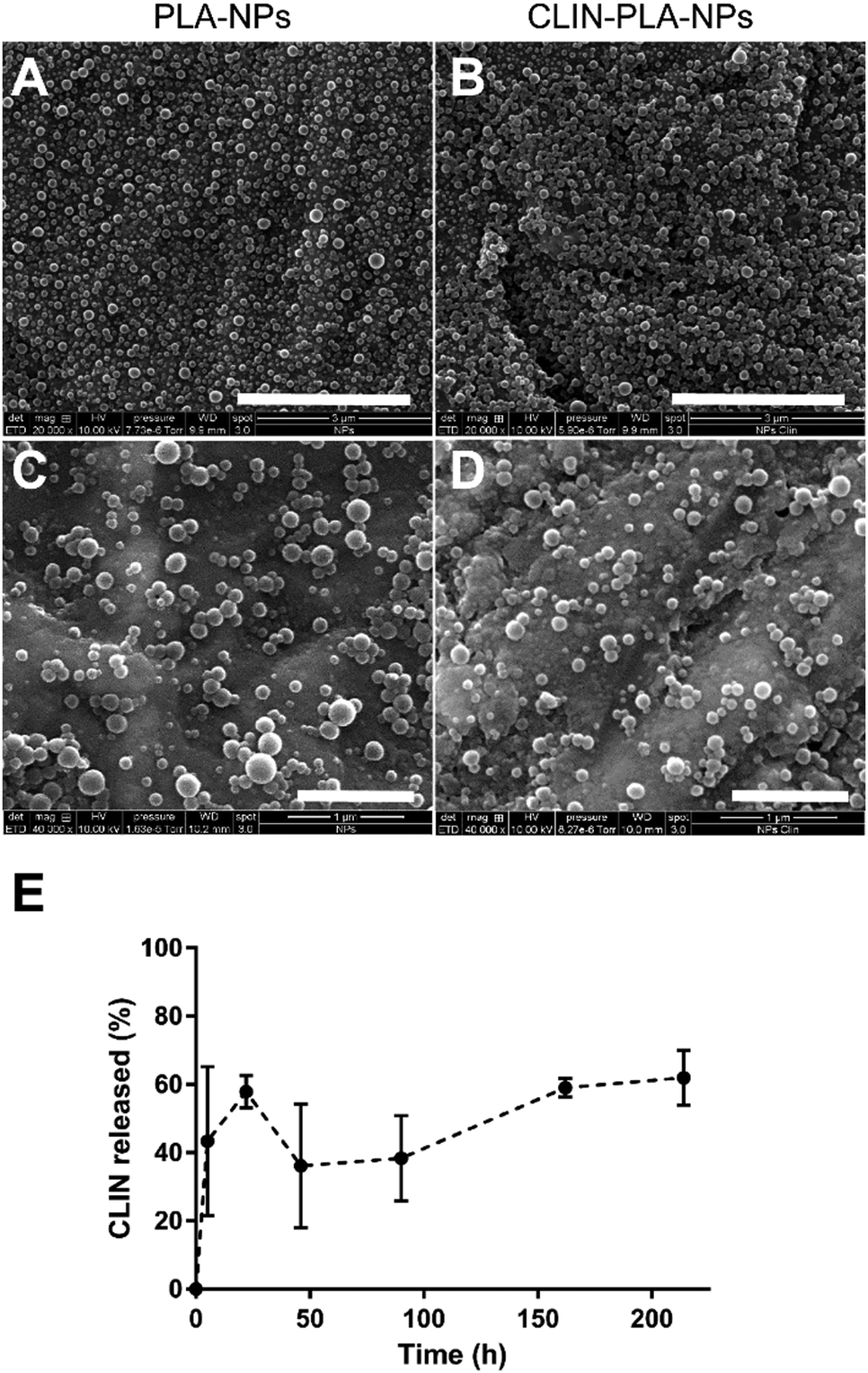 | ||
| Fig. 2 Representative SEM images of PLA-NPs (A and C) and CLIN-PLA-NPs (B and D) at low and high magnification. Scale bars: 3 μm (top) and 1 μm (bottom). Profile of clindamycin release from the nanoparticles in PBS at 37 °C (N = 3) (E). | ||
3.2 Antibacterial and antibiofilm properties of CLIN-PLA-NPs
To assess the antimicrobial properties of CLIN-PLA-NPs, the Minimum Inhibitory Concentration (MIC) against E. fæcalis was determined according to the broth microdilution method, in the presence of serial concentrations of clindamycin (0.004 to 256 μg mL−1), free or loaded onto nanoparticles (respectively CLIN and CLIN-PLA-NPs), or equivalent concentrations of empty PLA-NPs (0.016 to 1.02% solid content). A similar value of 32 μg mL−1 was obtained for free CLIN and CLIN-PLA-NPs (Table 2), whereas PLA-NPs were unable to inhibit E. fæcalis growth. To evaluate the ability of CLIN-PLA-NPs to prevent biofilm formation or degrade a 24 hour old biofilm, MBIC50 and MBEC50 were estimated by measuring E. fæcalis biofilm density after crystal violet staining. The MBIC50, defined as the lowest CLIN concentration that results in a 50% inhibition of biofilm formation, was found to be 4 μg mL−1 for CLIN-PLA-NPs and free CLIN. PLA-NPs did not inhibit E. fæcalis biofilm formation. The MBEC50, defined as the lowest CLIN concentration that eradicates 50% of a 24 hour old biofilm, was determined following the addition of free CLIN, CLIN-PLA-NPs or PLA-NPs on a 24 hour old biofilm. It was found to be 16 μg mL−1 for CLIN-PLA-NPs and free CLIN, whereas PLA-NPs did not affect the biofilm.| MIC (μg mL−1) | MBIC50 (μg mL−1) | MBEC50 (μg mL−1) | |
|---|---|---|---|
| CLIN | 32 | 4 | 16 |
| CLIN-PLA-NPs | 32 | 4 | 16 |
| PLA-NPs | >256 | >256 | >256 |
3.3 Antimicrobial properties of the nanocomposite hydrogel
To assess the antimicrobial properties of the nanocomposite hydrogel, agar diffusion assays were designed by plating hydrogel formulations containing fibrin alone (control) or fibrin supplemented with 50–200 μg mL−1 of free (CLIN) or clindamycin-loaded nanoparticles (CLIN-PLA-NPs), or with equivalent concentrations of free PLA-NPs (0.2–0.8% solid content) on E. fæcalis-seeded agar dishes (Fig. 3A). Fibrin hydrogels containing clindamycin or clindamycin-loaded nanoparticles clearly inhibited E. fæcalis growth in a dose-dependent manner, whereas the fibrin hydrogel loaded with PLA-NPs did not exhibit any antimicrobial activity (Fig. 3B).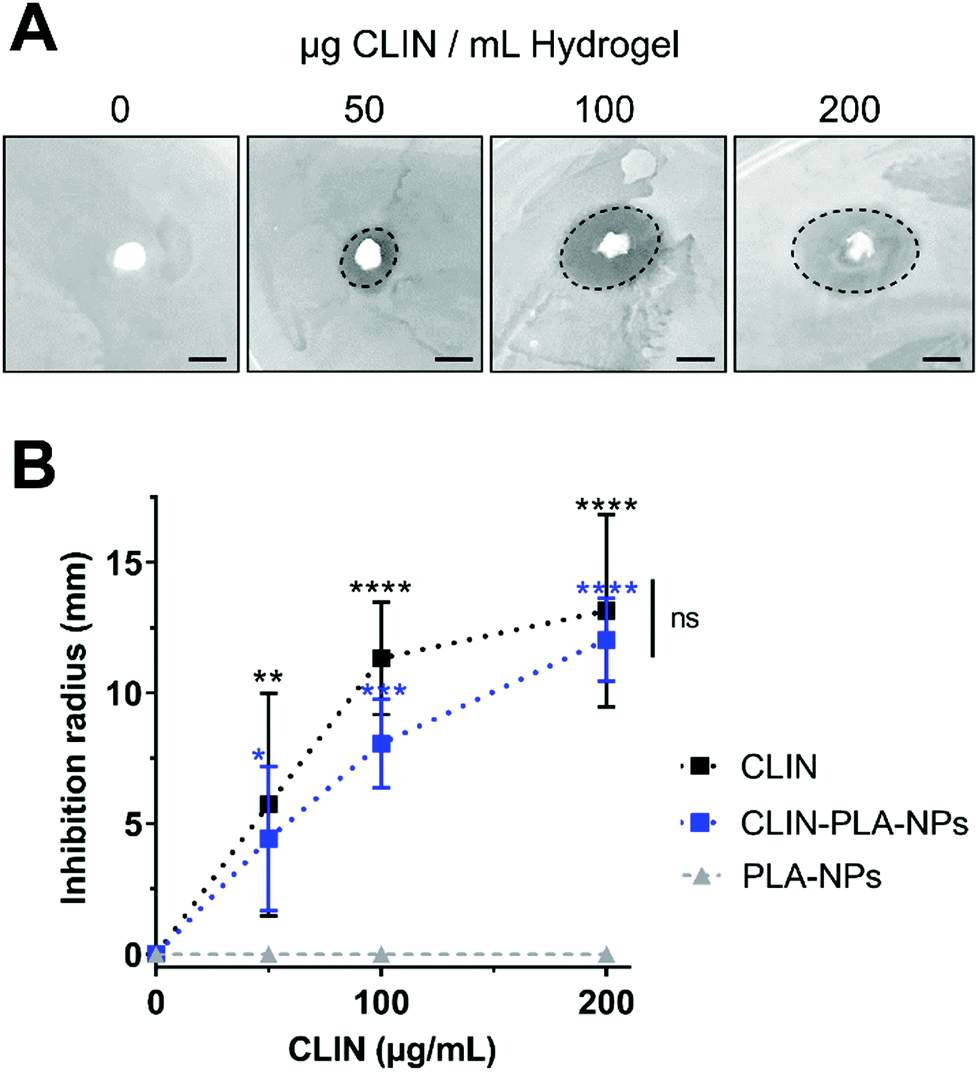 | ||
| Fig. 3 (A) Representative pictures of agar diffusion assays in the presence of the fibrin-alone hydrogel (far left) or fibrin hydrogels loaded with CLIN-PLA-NPs to get final concentrations of 50, 100 or 200 μg mL−1 CLIN. Scale bar is 10 mm. (B) Graphical representation reporting the inhibition radius in the presence of 50, 100 or 200 μg mL−1 free CLIN (black squares) or 2, 4 or 8 mg mL−1 CLIN-PLA-NPs (blue squares) or PLA-NPs (grey triangles). N = 3. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. PLA-NPs as control. Differences between free CLIN and CLIN-PLA-NPs are not significant (ns). | ||
3.4 Nanocomposite hydrogel formulation
A nanocomposite hydrogel containing 50 μg mL−1 clindamycin in free form or loaded onto nanoparticles, which corresponds to 0.2% solid content of PLA, was formulated to provide the hydrogel with efficient antibacterial and antibiofilm properties. Fibrin hydrogel microstructure was then analysed at high magnification using SEM. Fibrin formed a loose meshwork (Fig. 4A). Clindamycin-loaded nanoparticles appeared localized within the holes of the meshwork and/or in close contact with fibrin (Fig. 4B). Cumulative clindamycin release from fibrin hydrogels was assessed to determine the impact of the formulation on the release of the antibiotic. After 24 h, 54.0 ± 0.9% and 43.5 ± 1.9% of the clindamycin was released from the fibrin hydrogel when it was added under the free form or in addition to empty nanoparticles respectively, while only 24.8 ± 3.0% of the initially introduced clindamycin was released from the nanocomposite hydrogel formulated with clindamycin-loaded nanoparticles (Fig. 4C). This suggests that clindamycin-loaded nanoparticles restrain CLIN release from the hydrogel.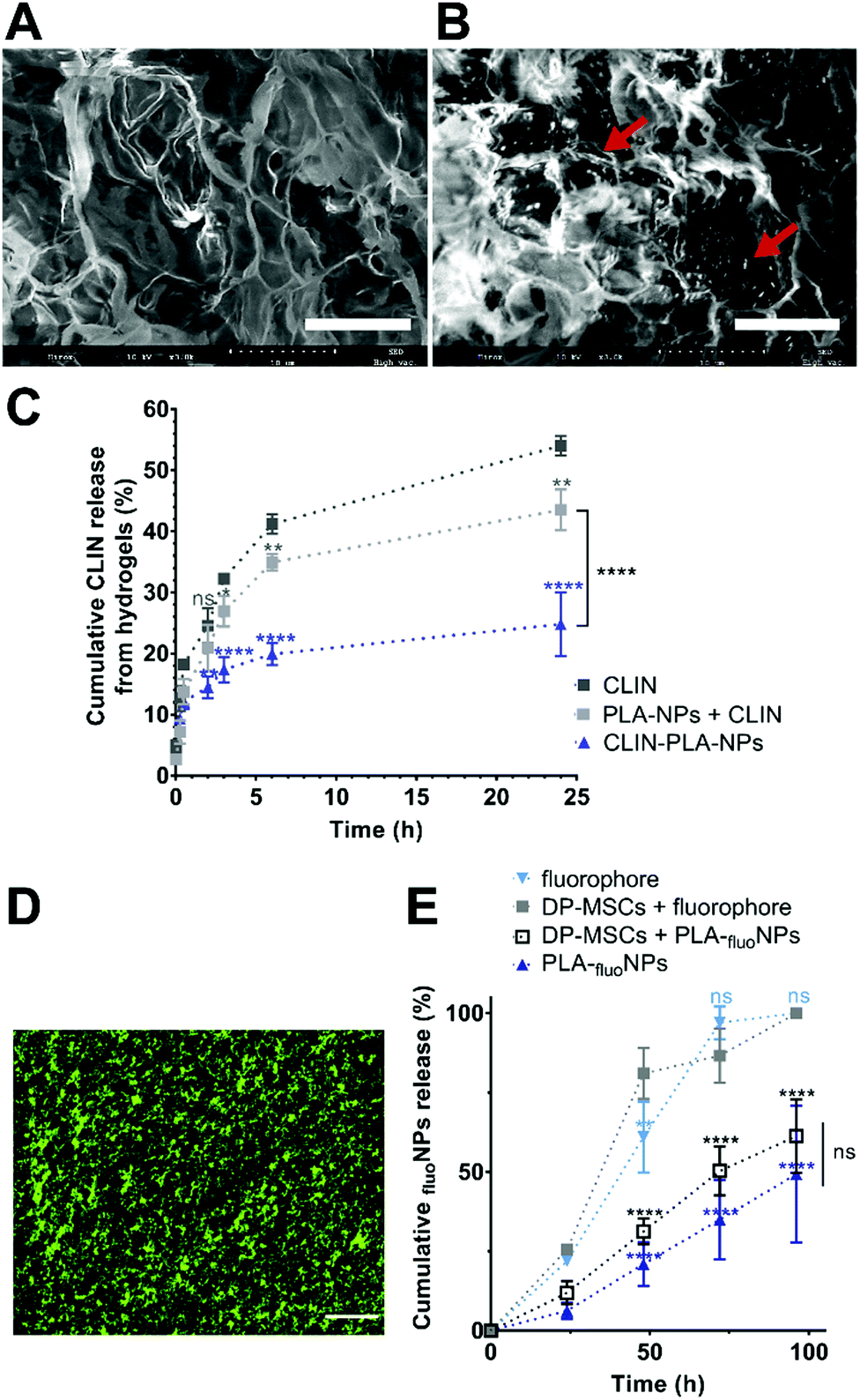 | ||
| Fig. 4 Representative SEM pictures of the microstructure of a fibrin-alone hydrogel (A) and of a fibrin hydrogel containing CLIN-PLA-NPs (B). NPs are indicated by red arrows for illustration. Scale bar: 10 μm. (C) Cumulative release of CLIN from fibrin hydrogels formulated with free CLIN, free CLIN + empty PLA-NPs, or CLIN encapsulated in PLA-NPs (N = 3). *P < 0.05, **P < 0.01, ****P < 0.0001 vs. free CLIN control. The statistical difference between PLA-NPs + CLIN and CLIN-PLA-NPs at 24 h is indicated by a bracket. No significant difference was observed below 2 h. (D) Distribution of PLA-fluoNPs within the fibrin hydrogel. Scale bar: 25 μm. (E) Graphical representation showing the cumulative release of PLA-fluoNPs from non-cellularized or DP-MSC-containing hydrogels (dark blue triangles and empty black squares, respectively), and of the fluorophore from non-cellularized or DP-MSC-containing hydrogels (light blue triangles and gray squares, respectively). The fluorophore alone was used as a control. Released fluorescence is expressed as the percentage of the starting one incorporated into the hydrogel. N = 3. ****P < 0.0001 vs. free fluorophore as control. No significant difference was observed below 48 h. Differences between DP-MSCs + PLA-fluoNPs and PLA-fluoNPs are not significant (ns). | ||
To investigate the release of nanoparticles from the hydrogel and their distribution within the hydrogel, green-fluorescent NPs (PLA-fluoNPs) were used. No release of the fluorophore from the particles into the aqueous medium (PBS) was observed within the four days of the experiment (ESI† 2). NPs appeared homogeneously distributed in the fibrin hydrogel and did not form agglomerates or patchwork (Fig. 4D). Time-course analyses of PLA-fluoNP release were then performed from hydrogels incorporated with or without DP-MSCs. Cumulative release profiles highlighted a complete release of the free fluorophore from the fibrin hydrogel after 4 days in the presence or absence of DP-MSCs, demonstrating that the fluorophore was not trapped by the fibrin hydrogel (Fig. 4E). After the same time, 61.3 ± 6.7% and 49.3 ± 12.4% of PLA-fluoNPs were released from cellularized and non-cellularized hydrogels, the difference being not significant.
3.5 Nanocomposite hydrogel cytotoxicity
CLIN-PLA-NP toxicity on DP-MSCs was investigated using a Live/Dead® assay and fluorescence imaging (Fig. 5A–D). After 48 h of culture, similar percentages of living DP-MSCs were observed in the presence of empty or clindamycin-loaded nanoparticles (77 ± 4% and 75 ± 3%, respectively) (Fig. 5E). Values were not statistically different from control cultures (in absence of NPs) or in the presence of free CLIN which showed 78 ± 7% and 73 ± 3% of living cells, respectively (Fig. 5E).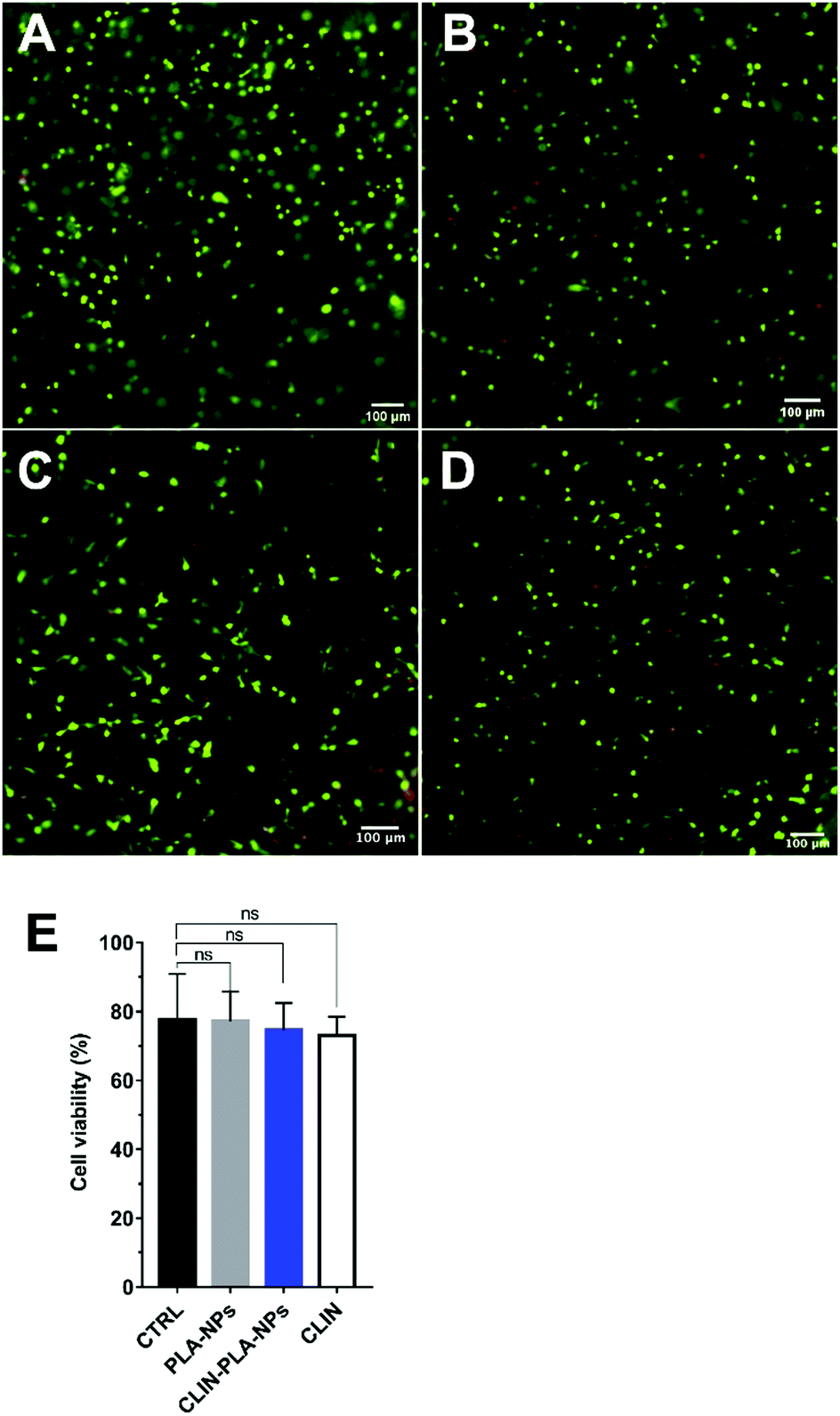 | ||
| Fig. 5 Cell viability assessment with the Live/Dead® assay. (A–D) Representative pictures of DP-MSCs stained after culture for 48 h in a fibrin-alone hydrogel (A) or in a fibrin hydrogel containing 0.2% solid content PLA-NPs (B) or 50 μg mL−1 clindamycin-loaded nanoparticles (0.2% solid content) (C), or 50 μg mL−1 free CLIN (D). Scale bar: 100 μm. (E) Graphical representation of DP-MSC viability in the fibrin-alone hydrogel (CTRL) or in the presence of 0.2% solid content PLA-NPs or 50 μg mL−1 clindamycin loaded onto nanoparticles (CLIN-PLA-NPs), or free (CLIN) (N = 6; ns: not significant). | ||
To confirm that NPs did not alter DP-MSC functionality, the synthesis of type I collagen, the main component of DP ECM, was assessed by immunohistochemistry after 7 days of culture (Fig. 6). While no staining was detected in the controls performed without primary antibody (Fig. 6A and B), we observed that DP-MSCs produced type I collagen both in the fibrin-alone hydrogel (Fig. 6C) and in the fibrin hydrogel containing CLIN-PLA-NPs (Fig. 6D).
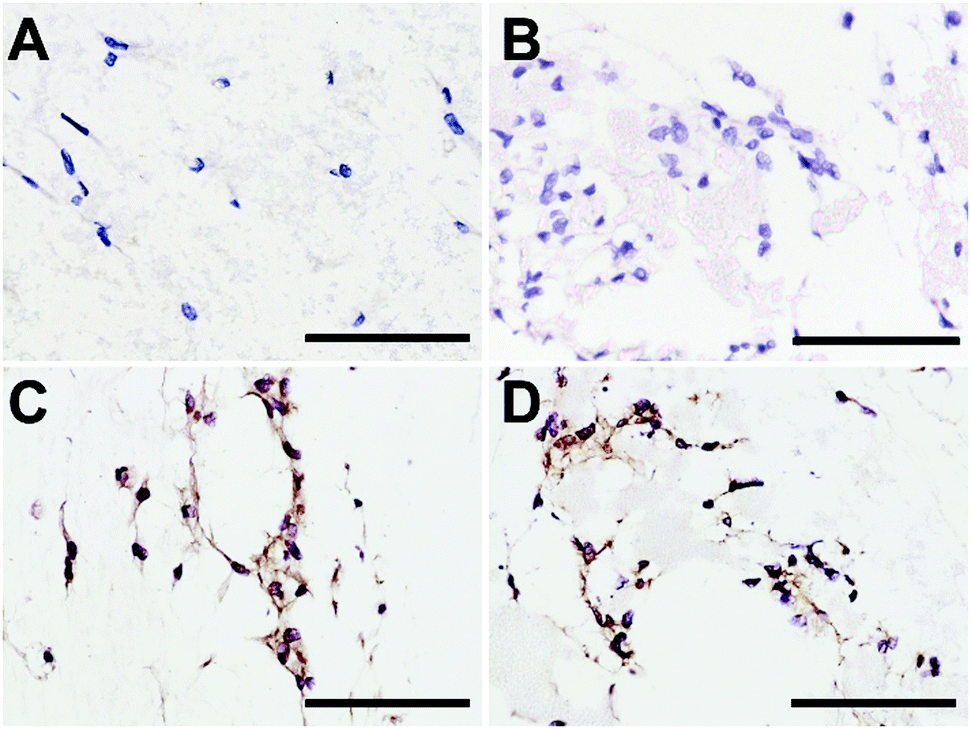 | ||
| Fig. 6 Representative pictures of type I collagen immunostaining after 7 days of culture of DP-MSCs in a fibrin-alone hydrogel (A and C) or in a fibrin hydrogel containing 2 mg mL−1 CLIN-PLA-NPs (B and D). Controls without the primary antibody (A and B). Scale bar: 20 μm. | ||
4. Discussion
This paper reports the development of an innovative fibrin-based hydrogel supplemented with antibiotic-loaded PLA-NPs to establish an aseptic environment favourable to tissue regeneration. Antimicrobial properties were provided to the hydrogel via clindamycin-loaded nanoparticles. NPs have been proposed in dentistry for various clinical applications including endodontic space (root canal) disinfection.23,43,44 Most studies relied on the use of metal-based NPs (silver, gold, copper oxide, iron oxide or zinc oxide) which are known to have intrinsic antimicrobial properties.45 However, the eco and cytotoxicity of these NPs is a major drawback that still remains to be solved.23 For this reason, non-metal polymeric 160–230 nm NPs made of chitosan or poly-lactic-co-glycolic acid (PLGA) were incorporated into toothpastes to deliver the antiseptic chlorhexidine in the dental biofilm. However, they displayed an in vitro toxicity between 20 and 60% on human gingival fibroblasts,46 which precluded their clinical use. Therefore, designing novel NP formulations that are both antimicrobial and non-cytotoxic is required.NP formation and drug entrapment efficiency (EE) depend on several parameters including the chemical synthesis method, the nature of the selected solvent, and the physicochemical properties of the drug.47 By using a solvent diffusion method previously reported,36 we obtained PLA-NPS and CLIN-PLA-NPs with similar hydrodynamic diameters (about 155–160 nm), polydispersity index (0.09) and zeta potential values (about −65 mV). This suggests that the CLIN incorporation did not significantly modify the physical parameters of PLA-NPs. Particle size influences many properties and it is therefore important to characterize precisely this physicochemical parameter. DLS data are not sufficient to assess particles’ size and size distribution because this method is intensity-based and therefore more sensitive to large particles. We accordingly used SEM which allows a number-based particles’ size measurement. SEM results highlighted a large number of small-sized particles. In addition, particles observed by SEM were dehydrated, which may lead to their artificial compaction compared to DLS measurements.
Nanoparticle synthesis by nanoprecipitation was conducted in carbonate buffer at pH 10. At this pH, clindamycin, which exhibits a pKa of 7.6, is to a very small extend ionized and therefore less soluble than in water. This reduced solubility results in a decreased diffusion of clindamycin in the aqueous phase during the nanoprecipitation process, favouring its coprecipitation with the hydrophobic polymer. The entrapment rate into PLA particles is thus enhanced in carbonate buffer compared to nanoprecipitation in water for example. Such a phenomenon was previously described by Govender et al. with procaine hydrochloride as a drug and PLGA as a polymer.48 We thus obtained a drug loading of 10 ± 2 μg CLIN per mg PLA and an entrapment efficiency of 43 ± 7%. These values were higher than those obtained during PLGA-nanosphere vectorization of antibiotics such as minocycline which was reported to give the highest EE (29.9%).49 Higher antibiotic EE could only be obtained with the use of surfactants,29,31 but the latter are known to be deleterious for many host cells and tissues. On the contrary, one major benefit of our CLIN-PLA-NPs is that they were prepared following a protocol that did not need any surfactant or stabilizer that may be pro-inflammatory or toxic for cells incorporated into or close to the fibrin hydrogel.
The lincosamide antibiotic CLIN inhibits bacterial protein synthesis by acting at the level of the 50S ribosome.32 It was chosen in this study to provide the hydrogel with antimicrobial properties for various reasons, first owing to its broad spectrum activity and its routine use in oral medicine in patients allergic to β-lactams such as amoxicillin.32 CLIN is for these patients the antibiotic of choice for treating oral infections or to prevent endocarditis.50 Interestingly, CLIN has the original ability to stimulate the host immune system by promoting the mobilization and chemotaxis of polymorphonuclear leukocytes to the infected sites.50 This property may be highly valuable to attract, into the fibrin scaffold, periodontal/blood immune cells in order to potentiate the CLIN antibacterial effect. CLIN was also shown to promote angiogenesis, another interesting property in the context of DP regeneration where the development of new vessels into the endodontic space is needed.51 Finally, CLIN was selected as an example of antibiotic since it can be easily incorporated into PLA-NPs owing to its positive charge and hydrophobicity.33–35
Antibacterial properties of CLIN-PLA-NPs were assessed on E. fæcalis, a microorganism frequently found in persistent endodontic infections, hard to eradicate and responsible for many endodontic treatment failures.52 The MIC of 32 μg mL−1 reported in this work for the E. fæcalis ATCC® 47077TM strain in the presence of CLIN-PLA-NPs was the same as for free CLIN, suggesting that CLIN incorporation into PLA-NPs did not decrease its antimicrobial properties. It was similar to the MIC reported by the European Committee on Antimicrobial Susceptibility Testing for E. fæcalis growth inhibition by CLIN. CLIN was found to be efficient at relatively high concentration, but its incorporation into PLA-NPs could strongly limit its putative systemic effects and adverse reactions.53 Experiments in animal models are on-going to investigate in vivo the safety of CLIN-PLA-NPs and the ability of this innovative medical device to promote DP regeneration. Interestingly, CLIN-PLA-NPs were able to inhibit the formation of an E. fæcalis biofilm. This results contrast with the reported inability of CLIN-containing electrospun nanofibers to inhibit the formation of E. fæcalis and Actinomyces naeslundii biofilms in the absence of ciprofloxacin and metronidazole.18 We suggest that this difference is due to the presence of PLA-NPs which are known to protect the structure of the drug, enhance its bioavailability and biodistribution, and ensure a high local antibiotic concentration.23 It may also be a result of the microstructure of the fibrin hydrogel which could allow for a better diffusion of the antibiotic through the meshwork.54 Indeed, the agar diffusion assay clearly displayed that CLIN-PLA-NPs progressively released from the hydrogel efficiently inhibited E. fæcalis growth, in a similar way to 50 μg mL−1 free CLIN incorporated into the hydrogel. This result demonstrates that CLIN-PLA-NPs maintain their activity upon incorporation within the nanocomposite hydrogel. Other works have demonstrated that CLIN inhibits Streptococcus aureus biofilm formation,55 and the MBIC50 results reported here extend the CLIN antibiofilm property to E. fæcalis. In addition, our MBEC50 data indicates that CLIN-PLA-NPs can induce E. fæcalis biofilm dispersal. This property may be highly valuable in the context of DP regeneration since the eradication of the endodontic bacterial biofilm present on the dentin wall or deeply entrapped in dentin tubules remains a major challenge to REPs.14,15,56
Investigation of the nanocomposite hydrogel microstructure using SEM showed that the fibrin formed a loose meshwork with CLIN-PLA-NPs that seemed to be free and/or in contact with the fibrin fibrils. Time-course analysis of CLIN release from the hydrogel revealed that, after 24 h, CLIN was released approximately two times slower from the hydrogel when it was loaded onto PLA nanoparticles compared to free CLIN. Clindamycin release from nanoparticles in a physiological buffer (PBS at 37 °C) was rapid, which suggests the dissolution of the drug which is close to the surface and, accordingly, rapidly available. The small size of the particles, characterized by a large surface to volume ratio, may enhance this effect. These results suggest that the CLIN-PLA-NPs allows for the maintenance of the CLIN within the nanocomposite hydrogels in comparison to free CLIN. Since HPLC equipped with UV/Visible detection at 200 nm is prone to background-limited sensitivity, our study of clindamycin release from the hydrogel was restricted to 24 h. Further studies are necessary to precisely characterize the time-course release of clindamycin from the various fractions of this three component system, using for example a more accurate detection method such as HPLC-MS/MS.57 The use of fluorescent PLA-based nanoparticles demonstrated that the NPs were homogeneously distributed into the hydrogel and did not form aggregates. Time-course analyses further revealed that PLA-fluoNPs were progressively released from the hydrogel into the medium without any impact of the DP-MSCs on the NP release profile. This result indicates that the nanocomposite hydrogel could be used for a cellularized approach of REPs. Altogether, these results suggest that the hydrogel may constitute a reservoir capable of bringing NP close to the dentin wall after injection into the endodontic space. This property is extremely valuable to destroy bacterial biofilms present on the dentin surface and in dentin tubules.58 This finding suggests that the use of PLA-NPs as a CLIN carrier and their progressive release from the hydrogel could allow for a prolonged effect of the antibiotic by maintaining high drug concentration within the hydrogel and at its periphery compared to the drug alone. This could provide a suitable environment lasting long enough to allow the formation of a novel immunocompetent DP-like tissue. It is possible that the hydrophobic nature of both fibrin and PLA could be involved in the progressive release of the NPs from the hydrogel. However, the precise mechanisms by which NPs are maintained into the hydrogel and released from it remain to be solved. Deciphering the close relationships that we observed between NPs and the fibrin network will help answer this question.
The biocompatibility of the nanocomposite hydrogel was assessed on DP-MSCs. Cell viability was found to be about 75% after 48 h of culture in the presence of CLIN-PLA-NPs, which corresponds to the ISO 10993-5 standards that require at least 70% cell viability.59 These results are similar to those reported for various types of mesenchymal stem cells, including DP-MSCs, seeded in a fibrin scaffold.11,60 They are higher than the DP-MSC viability of 50–60% measured in clindamycin, ciprofloxacin and metronidazole-loaded electrospun nanofibers.19 This difference may be due to the high biocompatibility reported for fibrin scaffolds compared to synthetic ones.11 At the functional level, our immunostaining experiments showed that DP-MSCs cultured within fibrin-alone and CLIN-PLA-NP-containing hydrogels synthesized type I collagen, which confirms the previously reported safety of fibrin-based scaffolds for DP-MSCs.20 Our results are consistent with a recent study reporting that CLIN was safe for DP-MSCs in vitro in the same range of concentrations (i.e. below 500 μg mL−1).51 However, the mechanism of action of CLIN on DP-MSCs within the nanocomposite hydrogel deserves further characterization to determine the real impact of the antibiotic on cell differentiation and hydrogel degradation in favour of dental pulp matrix neo-synthesis.
5. Conclusions
In the present study, we successfully developed an innovative, antibacterial and biocompatible hydrogel that may be valuable for DP regeneration in a previously infected endodontic space. CLIN-loaded PLA-NPs released from the hydrogel displayed antibiotic and antibiofilm properties that could overcome a major challenge of REPs which is the persistence, in the endodontic space, of bacteria that hamper the DP regeneration process. Work is on-going to determine what proportion of NPs accumulate along the dentin wall and penetrate into the micrometer-sized dentin tubules. Investigations of the antibacterial properties of these NPs loaded with CLIN or other antibiotic molecules against multispecies endodontic biofilms are also in progress. We believe that the presented technology could be transferable to other tissue engineering and regenerative medicine strategies where asepsis is difficult to reach by conventional disinfection means.Author contributions
BV, FMG and JCF conceived the original idea. JCF and MD designed the study, supervised the work and wrote the article. MBe analysed the results and drafted the article. MBo, FC and ML synthesized and characterized the CLIN-PLA-NPs. MBo performed the release analyses and assessed the antimicrobial properties. MBe prepared the samples for SEM analyses and immunohistochemistry. SG extracted and characterized the DP-MSCs. MP and AC performed the Live/Dead analyses. CP synthesized the PLA-fluoNPs. DD performed the biofilm analyses. CP and DD gave technical advices regarding NPs synthesis and characterizations.Conflicts of interest
BV, DD and CP work for the Adjuvatis® company which manufactured PLA-NPs and have accordingly a commercial interest. However, these authors only supplied the material and were not involved in data analyses or interpretation.Acknowledgements
This work was supported by the French National Centre for Scientific Research (CNRS) and the French Institute for Odontological Research (IFRO). DDC is a recipient of a CIFRE PhD grant from the French National Association of Research and Technology (ANRT). ML was supported by a PhD studentship of the Ligue contre le Cancer (France) for three years. The authors acknowledge the contribution of Adjuvatis® for providing PLA-NPs and its expertise in the NP domain, and the Centre Technologique des Microstructures (Université Lyon 1, Villeurbanne, France) for help with electron microscopy. Immunohistochemistry experiments were performed by N. El Kholti in the PRIMATISS facility housed in the laboratory (UMR 5305 CNRS/Université Lyon 1).References
- J. J. Mao, S. G. Kim, J. Zhou, L. Ye, S. Cho, T. Suzuki, S. Y. Fu, R. Yang and X. Zhou, Dent. Clin. North Am., 2012, 56(3), 639–649 CrossRef PubMed.
- A. Diogenes, N. B. Ruparel, Y. Shiloah and K. M. Hargreaves, J. Am. Dent. Assoc., JADA, 2016, 147(5), 372–380 CrossRef PubMed.
- T. Gong, B. C. Heng, E. C. M. Lo and C. Zhang, Stem Cells Int., 2016, 2016, 9204574 Search PubMed.
- M. Ducret, H. Fabre, A. Celle, F. Mallein-Gerin, E. Perrier-Groult, B. Alliot-Licht and J. C. Farges, Biomed. Mater. Eng., 2017, 28(s1), S159–S168 Search PubMed.
- T. Morotomi, A. Washio and C. Kitamura, Jpn Dent. Sci. Rev., 2019, 55(1), 5–11 CrossRef PubMed.
- K. M. Galler, R. N. D’Souza, J. D. Hartgerink and G. Schmalz, Adv. Dent. Res., 2011, 23(3), 333–339 CrossRef CAS PubMed.
- V. Rosa, A. Della Bona, B. N. Cavalcanti and J. E. Nör, Dent. Mater., 2012, 28(4), 341–348 CrossRef CAS PubMed.
- M. T. Albuquerque, M. C. Valera, M. Nakashima, J. E. Nör and M. C. Bottino, J. Dent. Res., 2014, 93(12), 1222–1231 CrossRef CAS PubMed.
- P. J. Palma, J. C. Ramos, J. B. Martins, A. Diogenes, M. H. Figueiredo, P. Ferreira, C. Viegas and J. M. Santos, J. Endod., 2017, 43(8), 1279–1287 CrossRef PubMed.
- C. C. Huang, R. Narayanan, N. Warshawsky and S. Ravindran, Front. Physiol., 2018, 9, 495 CrossRef PubMed.
- K. M. Galler, F. P. Brandl, S. Kirchhof, M. Widbiller, A. Eidt, W. Buchalla, A. Göpferich and G. Schmalz, Tissue Eng., Part A, 2018, 24(3–4), 234–244 CrossRef CAS PubMed.
- Y. Li, H. Meng, Y. Liu and B. P. Lee, Sci. World J., 2015, 2015, 685690 Search PubMed.
- A. F. Fouad, Adv. Dent. Res., 2011, 23(3), 285–289 CrossRef CAS PubMed.
- L. Vishwanat, R. Duong, K. Takimoto, L. Phillips, C. O. Espitia, A. Diogenes, S. B. Ruparel, D. Kolodrubetz and N. B. Ruparel, J. Endod., 2017, 43(6), 916–922 CrossRef PubMed.
- P. Verma, A. Nosrat, J. R. Kim, J. B. Price, P. Wang, E. Bair, H. H. Xu and A. F. Fouad, J. Dent. Res., 2017, 96(1), 100–106 CrossRef CAS PubMed.
- M. C. Bottino, K. Kamocki, G. H. Yassen, J. A. Platt, M. M. Vail, Y. Ehrlich, K. J. Spolnik and R. L. Gregory, J. Dent. Res., 2013, 92(11), 963–969 CrossRef CAS PubMed.
- J. Palasuk, K. Kamocki, L. Hippenmeyer, J. A. Platt, K. J. Spolnik, R. L. Gregory and M. C. Bottino, J. Endod., 2014, 40(11), 1879–1884 CrossRef PubMed.
- K. Kamocki, J. E. Nör and M. C. Bottino, Int. Endod. J., 2015, 48(12), 1147–1156 CrossRef CAS PubMed.
- A. Karczewski, S. A. Feitosa, E. I. Hamer, D. Pankajakshan, R. L. Gregory, K. J. Spolnik and M. C. Bottino, J. Endod., 2018, 44(1), 155–162 CrossRef PubMed.
- M. Ducret, A. Montembault, J. Josse, M. Pasdeloup, A. Celle, R. Benchrih, F. Mallein-Gerin, B. Alliot-Licht, L. David and J. C. Farges, Dent. Mater., 2019, 35(4), 523–533 CrossRef CAS PubMed.
- N. Ruangsawasdi, M. Zehnder and F. E. Weber, J. Endod., 2014, 40(2), 246–250 CrossRef PubMed.
- H. Redl, G. Schlag, G. Stanek, A. Hirschl and T. Seelich, Biomaterials, 1983, 4(1), 29–32 CrossRef CAS PubMed.
- L. Wang, C. Hu and L. Shao, Int. J. Nanomed., 2017, 12, 1227–1249 CrossRef CAS PubMed.
- K. Forier, K. Raemdonck, S. C. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, J. Controlled Release, 2014, 190, 607–623 CrossRef CAS PubMed.
- W. S. Cheow and K. Hadinoto, Methods Mol. Biol., 2014, 1147, 227–238 CrossRef CAS PubMed.
- B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS PubMed.
- A. Mahapatro and D. K. Singh, J. Nanobiotechnol., 2011, 9, 55 CrossRef CAS PubMed.
- V. Pavot, M. Berthet, J. Rességuier, S. Legaz, N. Handké, S. C. Gilbert, S. Paul and B. Verrier, Nanomedicine, 2014, 9(17), 2703–2718 CrossRef CAS PubMed.
- D. M. Casa, T. C. Carraro, L. E. de Camargo, L. F. Dalmolin, N. M. Khalil and R. M. Mainardes, J. Nanosci. Nanotechnol., 2015, 15(1), 848–854 CrossRef CAS PubMed.
- W. Yao, P. Xu, Z. Pang, J. Zhao, Z. Chai, X. Li, H. Li, M. Jiang, H. Cheng, B. Zhang and N. Cheng, Int. J. Nanomed., 2014, 9, 3963–3970 Search PubMed.
- E. Pancani, M. Menendez-Miranda, A. Pastor, F. Brisset, M. F. Bernet-Camard, D. Desmaële and R. Gref, Acta Pharm. Sin. B, 2018, 8(3), 420–431 CrossRef PubMed.
- M. Smieja, Can. J. Infect. Dis., 1998, 9(1), 22–28 CrossRef CAS PubMed.
- S. J. Siegel, J. B. Kahn, K. Metzger, K. I. Winey, K. Werner and N. Dan, Eur. J. Pharm. Biopharm., 2006, 64(3), 287–293 CrossRef CAS PubMed.
- P. Gao, X. Nie, M. Zou, Y. Shi and G. Cheng, J. Antibiot., 2011, 64(9), 625–634 CrossRef CAS PubMed.
- S. R. Shah, A. M. Henslee, P. P. Spicer, S. Yokota, S. Petrichenko, S. Allahabadi, G. N. Bennett, M. E. Wong, F. K. Kasper and A. G. Mikos, Pharm. Res., 2014, 31(12), 3379–3389 CrossRef CAS PubMed.
- M. Lamrayah, F. Charriaud, S. Hu, S. Megy, R. Terreux and B. Verrier, Int. J. Pharm., 2019, 568, 118569 CrossRef CAS PubMed.
- K. Na-Bangchang, V. Banmairuroi, B. Kamanikom and D. Kiod, Southeast Asian J. Trop. Med. Public Health, 2006, 37(1), 177–184 CAS.
- L. Thieme, M. Klinger-Strobel, A. Hartung, C. Stein, O. Makarewicz and M. W. Pletz, J. Antimicrob. Chemother., 2018, 73(6), 1553–1561 CrossRef CAS PubMed.
- H. Fabre, M. Ducret, O. Degoul, J. Rodriguez, E. Perrier-Groult, E. Aubert-Foucher, M. Pasdeloup, C. Auxenfans, C. McGuckin, N. Forraz and F. Mallein-Gerin, Stem Cells Int., 2019, 2019, 2186728 Search PubMed.
- M. Ducret, H. Fabre, J. C. Farges, O. Degoul, G. Atzeni, C. McGuckin, N. Forraz, F. Mallein-Gerin and E. Perrier-Groult, J. Endod., 2015, 41(9), 1492–1499 CrossRef PubMed.
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B, 2019, 9(1), 4–18 CrossRef PubMed.
- M. Chorny, I. Fishbein, H. D. Danenberg and G. Golomb, J. Controlled Release, 2002, 83(3), 389–400 CrossRef CAS PubMed.
- R. A. Bapat, C. P. Joshi, P. Bapat, T. V. Chaubal, R. Pandurangappa, N. Jnanendrappa, B. Gorain, S. Khurana and P. Kesharwani, Drug Discovery Today, 2019, 24(1), 85–98 CrossRef CAS PubMed.
- M. Samiei, A. Farjami, S. M. Dizaj and F. Lotfipour, Mater. Sci. Eng., C, 2016, 58, 1269–1278 CrossRef CAS PubMed.
- M. Polívková, T. Hubáček, M. Staszek, V. Švorčík and J. Siegel, Int. J. Mol. Sci., 2017, 18(2), 419 CrossRef PubMed.
- L. Chronopoulou, G. Nocca, M. Castagnola, G. Paludetti, G. Ortaggi, F. Sciubba, M. Bevilacqua, A. Lupi, G. Gambarini and C. Palocci, New Biotechnol., 2016, 33(1), 23–31 CrossRef CAS PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42(3), 1147–1235 RSC.
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57(2), 171–185 CrossRef CAS.
- T. S. Kashi, S. Eskandarion, M. Esfandyari-Manesh, S. M. Marashi, N. Samadi, S. M. Fatemi, F. Atyabi, S. Eshraghi and R. Dinarvand, Int. J. Nanomed., 2012, 7, 221–234 Search PubMed.
- I. Brook, M. A. Lewis, G. K. Sándor, M. Jeffcoat, L. P. Samaranayake and J. Vera Rojas, Oral Surg., Oral Med., Oral Pathol., Oral Radiol. Endod., 2005, 100(5), 550–558 CrossRef PubMed.
- N. Dubey, J. Xu, Z. Zhang, J. E. Nör and M. C. Bottino, J. Endod., 2019, 45(7), 882–889 CrossRef PubMed.
- A. A. Hoelscher, J. K. Bahcall and J. S. Maki, J. Endod., 2006, 32(2), 145–147 CrossRef PubMed.
- M. H. Thornhill, M. J. Dayer, M. J. Durkin, P. B. Lockhart and L. M. Baddour, J. Dent. Res., 2019, 98(10), 1081–1087 CrossRef CAS PubMed.
- J. Chotitumnavee, T. Parakaw, R. L. Srisatjaluk, C. Pruksaniyom, S. Pisitpipattana, C. Thanathipanont, T. Amarasingh, N. Tiankhum, N. Chimchawee and N. Ruangsawasdi, J. Dent. Sci., 2019, 14(1), 7–14 CrossRef PubMed.
- K. Schilcher, F. Andreoni, V. Dengler Haunreiter, K. Seidl, B. Hasse and A. S. Zinkernagel, Antimicrob. Agents Chemother., 2016, 60(10), 5957–5967 CrossRef CAS PubMed.
- S. N. Kaushik, B. Kim, A. M. Walma, S. C. Choi, H. Wu, J. J. Mao, H. W. Jun and K. Cheon, Biomater. Res., 2016, 20, 14 CrossRef PubMed.
- M. Seifrtová, L. Nováková, C. Lino, A. Pena and P. Solich, Anal. Chim. Acta, 2009, 649(2), 158–179 CrossRef PubMed.
- P. Neelakantan, M. Romero, J. Vera, U. Daood, A. U. Khan, A. Yan and G. S. P. Cheung, Int. J. Mol. Sci., 2017, 18(8), 1748 CrossRef PubMed.
- A. L. B. Maçon, S. Li, J. J. Chung, A. Nommeots-Nomm, A. K. Solanki, M. M. Stevens and J. R. Jones, J. Mater. Chem. B, 2016, 4(36), 6032–6042 RSC.
- W. Ho, B. Tawil, J. C. Dunn and B. M. Wu, Tissue Eng., 2006, 12(6), 1587–1595 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Clindamycin detection by HPLC and stability (SI1) – calibration curve of PLA-fluoNPs and fluorescence release (SI2). See DOI: 10.1039/d0tb00989j |
| This journal is © The Royal Society of Chemistry 2020 |