
An artificial protein-probe hybrid as a responsive probe for ratiometric detection and imaging of hydrogen peroxide in cells†
Xing
Zhang
ab,
Youxin
Fu
 b,
Guangren
Qian
*a,
Run
Zhang
b and
Zhi Ping
Xu
*b
b,
Guangren
Qian
*a,
Run
Zhang
b and
Zhi Ping
Xu
*b
aSchool of Environmental Science and Chemical Engineering, Shanghai University, Shanghai 200444, China. E-mail: grqian@shu.edu.cn
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, QLD 4072, Australia. E-mail: gordonxu@uq.edu.au
First published on 21st May 2020
Abstract
An artificial protein-probe hybrid, Cm–Np-B@BSA, was prepared via host–guest interactions between hydrogen peroxide (H2O2)-responsive Cm–Np-B molecule and bovine serum albumin (BSA). The Cm–Np-B@BSA probe exhibited high sensitivity and selectivity towards H2O2 under physiological conditions and had excellent biocompatibility, allowing for sensitive ratiometric detection and imaging of endogenous H2O2 in live cells.
Fluorescent protein has emerged as an attractive platform in designing responsive probes for real-time detection and monitoring of specific biomarkers in biological systems.1–3 In the past few decades, many efforts have been made to produce genetically encoded fluorescent protein probes to respond to biomarkers and investigate their applications in biology and biomedicine.4–7 Despite the advantages of fluorescent protein probes in biosensing and bioimaging (such as excellent biocompatibility), only a limited number of responsive fluorescent protein probes have been reported.4,8,9 This is mainly because the tedious procedure for fluorescent protein production restricts their expression on a large scale.
In order to overcome this limitation, several artificial protein-probe hybrids were made by integrating a fluorophore (e.g. organic fluorescent dye, luminescent metal complex, or quantum dot) with a suitable protein via supermolecular and host–guest interactions, or chemical conjugations.10–13 This kind of hybrid provides an alternative scaffold for designing responsive fluorescent protein-probe hybrids for biological applications.10,12 Incorporating various fluorophores, especially fluorescent organic dyes, into proteins to yield artificial protein-probe hybrids enhances the biocompatibility and bioavailability of fluorescent dyes. More importantly, the artificial hybrid enables biomarker detection in aqueous solutions and physiological conditions without fluorescence quenching caused by dye molecule aggregation.14,15 For example, coumarin was loaded into the cavities of bovine serum albumin (BSA),16–18 and emitted characteristic fluorescence that was quenched in its aggregation state in water. With interactions between the fluorophore and protein, several fluorescent dyes, including coumarin, have been reported for protein detection in aqueous solutions.12,13,19–21 In this work, we expanded this idea and developed an artificial protein-probe hybrid specific for hydrogen peroxide (H2O2) detection, and also highlighted the strategy of developing artificial protein-probe hybrids beyond the aforementioned protein detection.
H2O2, produced during mitochondrial respiration, is one of the most important reactive oxygen species (ROS) in biological systems.22–28 Endogenous H2O2 is the most stable ROS, having the lowest reactivity and the highest intracellular concentration (∼10−8–10−4 M).29 In living organisms, this biomolecule serves as an essential biomarker in regulating a wide variety of physiological events, such as activation of diverse signalling pathways to stimulate cell proliferation, differentiation, migration, and apoptosis.29–32 The role of H2O2 in biological systems is concentration-dependent.33 It is, therefore, highly demanded to develop suitable responsive fluorescence probes for H2O2 detection in live cells for a better understanding of H2O2-mediated biofunctions.34
Different from previously reported molecule probes for H2O2 detection, this communication presents a biocompatible responsive protein-probe hybrid for ratiometric detection of H2O2 in aqueous solutions and living systems through a simple one-step self-assembly of organic dye Cm–Np-B with BSA. As shown in Scheme 1, the dye (Cm–Np-B) was designed based on the elegant modulation of the intramolecular charge transfer (ICT) and the Förster resonance energy transfer (FRET).35–37 The emission of energy transfer (ET) donor, i.e. coumarin (Cm, blue channel) can be activated after loading into the cavity of BSA through host–guest interactions in PBS buffer. Within the BSA system (Cm–Np-B@BSA), the emission of the ET acceptor, i.e. naphthalimide (Np, green channel), is quenched due to the inability of the ICT process. Through an H2O2-triggered reaction to cleave boronate group,38 the “donor-π-acceptor (D-π-A)” hydroxyl-Np derivative (Cm–Np@BSA) is generated and thus the emission of Np is switched “ON”. Moreover, the formation of Cm–Np@BSA also enables FRET from Cm to Np, allowing ratiometric fluorescence detection and bioimaging of H2O2 with a blue-to-green fluorescence colour change.
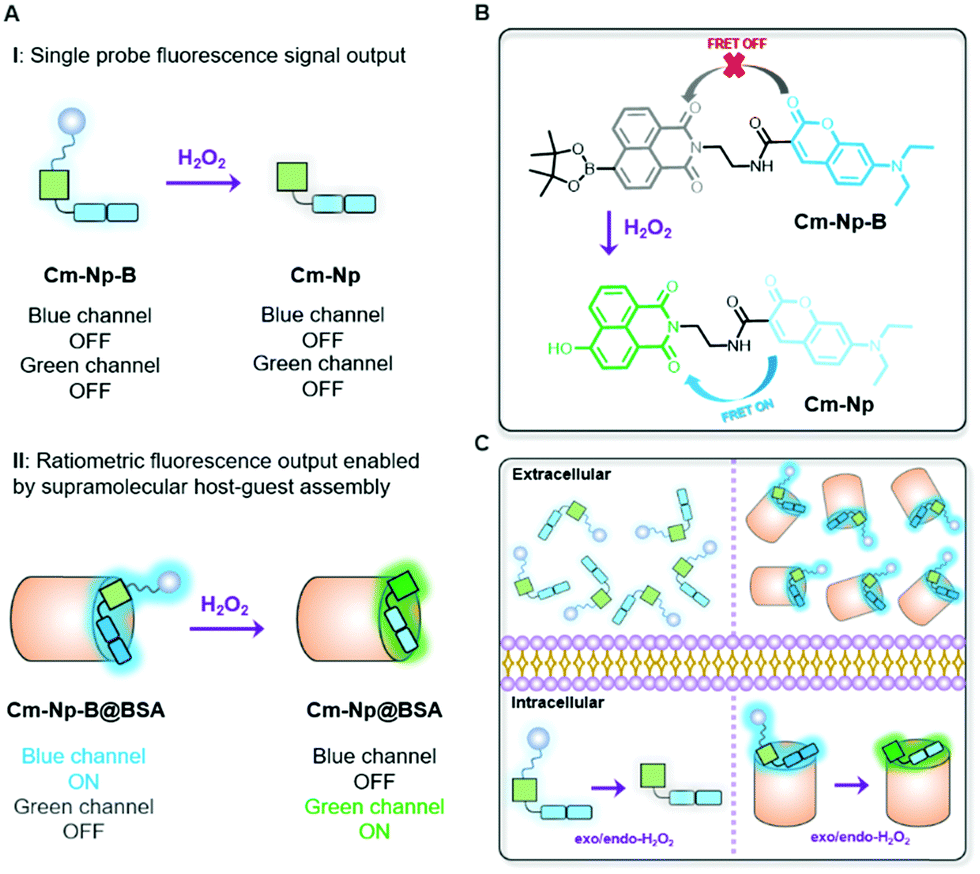 | ||
| Scheme 1 Design of an artificial protein-probe hybrid (Cm–Np-B@BSA) for responsive H2O2 detection. (A) ON–OFF non-response of molecular probe (i) and response of BSA-probe (ii); (B) ON–OFF responsive mechanism; (C) cellular responses for H2O2 detection. | ||
The fluorescent dye, Cm–Np-B, was synthesized in a four-step procedure with a reasonable yield (Scheme S1, ESI†), and the chemical structures of Cm–Np-B and the intermediates were identified with NMR and ESI-MS (Fig. S1–S5, ESI†). Then the protein-probe hybrid (Cm–Np-B@BSA) was made through simply mixing the probe and BSA in PBS buffer (Page S6, ESI†).
The blue channel activation of Cm–Np-B@BSA was first evaluated by UV-vis absorption and fluorescence spectra. As shown in Fig. 1A, Cm–Np-B was essentially non-fluorescent in both the blue and green channels in PBS buffer (pH 7.4, black dashed curve), largely attributed to the aggregation-caused quenching (ACQ).39 In sharp contrast, the fluorescence intensity was gradually increased upon mixing with more BSA (from 0 to 50 μM) and maximum enhancement of emission intensity (>145-fold) at 460 nm was achieved at 50 μM of BSA. Simultaneously, a new peak in the UV-vis absorption spectrum at 280 nm was observed, which is attributed to the characteristic absorbance of BSA (Fig. S6, ESI†). Similarly, Cm–Np-B is also responsive to human serum albumin (HSA). As shown in Fig. S7 (ESI†), the fluorescence intensity gradually increased upon mixing with more HSA (from 0 to 50 μM), presumably due to the similar 3D structure, chemical composition, and biological functions of these two albumins.
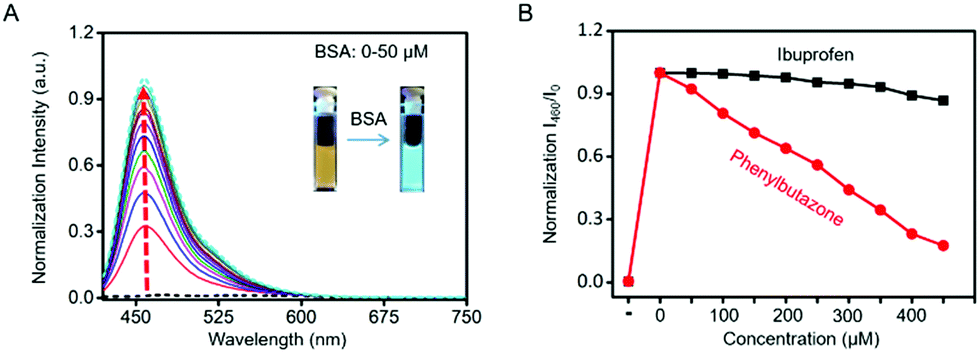 | ||
| Fig. 1 Formation of Cm–Np-B@BSA and its fluorescence. (A) fluorescence spectra of Cm–Np-B (5 μM) at different concentrations of BSA (0–50 μM); (B) fluorescence intensity changes in Cm–Np-B@BSA (5/50 μM, means 5 μM probe with 50 μM protein) with the concentration of ibuprofen (black solid line) and phenylbutazone (red solid line) increasing from 0 to 450 μM in PBS buffer (0.5% DMSO (v/v), pH 7.4; excitation wavelength: 410 nm). | ||
Dynamic light scattering (DLS) analysis showed a large size of molecule probe Cm–Np-B (600–700 nm) in PBS buffer (Fig. S8, ESI†), due to aggregation of Cm–Np-B molecules in PBS. Upon assembling Cm–Np-B with BSA, the hydrodynamic diameter of the Cm–Np-B@BSA hybrids was 17.8 ± 1.3 nm, slightly larger than that of BSA (13.0 ± 0.3 nm). The size increase and disappearance of Cm–Np-B aggregates together confirm formation of the guest–host complexes (Cm–Np-B@BSA)via binding Cm–Np-B into the hydrophobic pocket of BSA with a slightly big volume.
As shown in Fig. 1A, the emission intensity of Cm–Np-B dye was BSA-concentration dependent. Fluorescence Job's plot analysis showed a maximum intensity at the molecular fraction of 0.5 (Fig. S9, ESI†), indicating the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry between Cm–Np-B and BSA. Cm–Np-B is thus located only at one binding site of BSA. Previous reports have shown that most small hydrophobic molecules are wrapped into the hydrophobic pockets of BSA, i.e. site I and site II.40–42 Phenylbutazone and ibuprofen are known to bind site I and site II of BSA, respectively. In order to determine the binding site, a competitive assay was carried out to displace Cm–Np-B from BSA using these two albumin-binding molecules. As shown in Fig. 1B, the intensity of blue emission at 460 nm was decreased upon adding phenylbutazone, while adding ibuprofen caused a negligible change in the fluorescence intensity. The selective replacement of Cm–Np-B by phenylbutazone suggests that the preferred binding site of Cm–Np-B is site I of BSA. In addition, the blue channel emission of the probe largely decreased upon adding trypsin, suggesting the release of Cm–Np-B into PBS buffer and subsequent aggregation after BSA degradation (Fig. S10, ESI†).
:1 binding stoichiometry between Cm–Np-B and BSA. Cm–Np-B is thus located only at one binding site of BSA. Previous reports have shown that most small hydrophobic molecules are wrapped into the hydrophobic pockets of BSA, i.e. site I and site II.40–42 Phenylbutazone and ibuprofen are known to bind site I and site II of BSA, respectively. In order to determine the binding site, a competitive assay was carried out to displace Cm–Np-B from BSA using these two albumin-binding molecules. As shown in Fig. 1B, the intensity of blue emission at 460 nm was decreased upon adding phenylbutazone, while adding ibuprofen caused a negligible change in the fluorescence intensity. The selective replacement of Cm–Np-B by phenylbutazone suggests that the preferred binding site of Cm–Np-B is site I of BSA. In addition, the blue channel emission of the probe largely decreased upon adding trypsin, suggesting the release of Cm–Np-B into PBS buffer and subsequent aggregation after BSA degradation (Fig. S10, ESI†).
To test the fluorescence response of Cm–Np-B@BSA to H2O2, Cm–Np-B@BSA in PBS buffer was treated with H2O2 at different concentrations and the changes in UV-vis absorbance and fluorescence spectra were recorded. As shown in Fig. S11A (ESI†), the absorbance at λabs = 350 nm was gradually decreased and the maximum absorbance band at λabs = 430 nm was gradually intensified upon the addition of H2O2 (0–200 μM). The change of UV-vis absorption spectra is ascribed to the ICT feature in Cm–Np dye due to the transformation of Cm–Np-B@BSA to Cm–Np@BSA (Scheme 1). Clearly, the UV-vis absorption spectral change in Cm–Np-B@BSA towards H2O2 was more sensitive than that of Cm–Np-B (Fig. S11B, ESI†), suggesting the higher sensitivity of the artificially devised fluorescent protein-probe hybrid.
As shown in Fig. 2A, Cm–Np-B@BSA exhibited a blue emission band from the coumarin donor (λex = 410 nm, λem = 460 nm) in the absence of H2O2. Upon the addition of H2O2 to test solution, the green emission band from the Np acceptor at λem = 525 nm was gradually intensified, accompanied by blue emission weakening. The green emission enhancement is contributed by the ICT-dominated fluorescence of Cm–Np and the FRET process from Cm to Np, while the blue emission decrease is ascribed to the FRET process. Similar to the unchanged UV-vis absorption spectrum with H2O2, Cm–Np-B showed no fluorescence response to H2O2 (Fig. S12, ESI†), corroborating the superior capability of the artificial protein-probe hybrid in H2O2 detection. Besides these, the time-dependent fluorescence response of probe-protein hybrid Cm–Np-B@BSA and Cm–Np-B toward H2O2 in PBS buffer was also measured (Fig. S13, ESI†), and Cm–Np-B@BSA showed a faster response than Cm–Np-B.
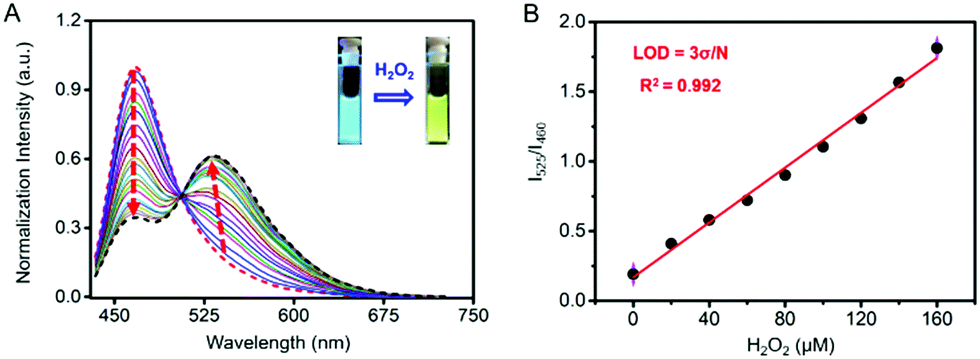 | ||
| Fig. 2 Sensitivity and selectivity of Cm–Np-B@BSA (5/50 μM) for H2O2 detection. (A) Fluorescence spectral response of Cm–Np-B@BSA (5/50 μM) to H2O2 at the concentration of 0–200 μM; (B) linear detection range and limit of detection (LOD) of Cm–Np-B@BSA (5/50 μM). All measurements were carried out in PBS buffer, pH 7.4, excitation wavelength: 410 nm. | ||
The blue-to-green fluorescence emission change in Cm–Np-B@BSA enables ratiometric fluorescence detection of H2O2 in PBS buffer. As shown in Fig. 2B, the dose-dependent fluorescence response (F525/F460) of Cm–Np-B@BSA exhibited a good linearity against the concentration of H2O2 in the range of 0–160 μM. The detection limit (3σ/k) was calculated to be 1 μM. The low detection limit and the broad detection range indicate the high sensitivity of artificial protein-probe hybrid Cm–Np-B@BSA for H2O2 detection.
The selectivity of Cm–Np-B@BSA towards H2O2 over other interfering species, including some other ROS, anions, and biomolecules, was also confirmed. Cm–Np-B@BSA in PBS buffer was treated with 200 μM of H2O2 and 500 μM of other species, respectively. After 10 min incubation, remarkable changes of the fluorescence of Cm–Np-B@BSA were noticed in the presence of H2O2, but not in the presence of other species (F525/460, Fig. S14, ESI†). Furthermore, the competitive selectivity of Cm–Np-B@BSA to H2O2 over other species was also tested. As shown in Fig. S15 (ESI†), the fluorescence (F525/460) was increased exclusively to H2O2 and the enhancement of fluorescence intensity was not affected in the presence of all competitive species. These data indicate the high selectivity of protein-probe hybrid Cm–Np-B@BSA for a ratiometric fluorescence response to H2O2 in PBS buffer.
Then, the effect of pH on the ratiometric fluorescence response of Cm–Np-B@BSA to H2O2 was examined in PBS buffer with pH 5–8. As shown in Fig. S16E (ESI†), no obvious difference in the emission spectra was observed in solutions with pH 6–8, suggesting that the Cm–Np-B@BSA probe is able to detect H2O2 in an environment with pH 6–8, while the peak at 525 nm in pH 5 solution was relatively weaker, probably due to the acidity slowing down the H2O2 detection reaction.
Encouraged by the excellent sensitivity and selectivity of the Cm–Np-B@BSA response to H2O2, we further demonstrated the application of this artificial protein-probe hybrid for imaging H2O2 in living cells. MTT assay showed that Cm–Np-B@BSA had low cytotoxicity towards RAW 264.7 macrophage cells (Fig. S17, ESI†). For example, the cell viability remained over 85% after incubation of RAW 264.7 macrophage cells with Cm–Np-B@BSA (60 μM/50 μM) for 24 h. For safety, 5 μM/50 μM of Cm–Np-B@BSA was used in most subsequent experiments.
The capability of Cm–Np-B@BSA in visualizing intracellular H2O2 in RAW 264.7 cells was then examined. As shown in Fig. 3A, clear blue fluorescence was noticed after incubating RAW 264.7 cells with Cm–Np-B@BSA for 2 h, which was remarkably increased in comparison with the control group (RAW 264.7 macrophage cells only) (Fig. S18, ESI†). The flow cytometric data showed a similar change. Both observations confirm the effective cell internalization of Cm–Np-B@BSA (Fig. S19, ESI†). Interestingly, supplying Cm–Np-B@BSA-internalized cells with H2O2 (200 μM), the intracellular blue channel fluorescence was largely quenched, while intense green channel fluorescence emerged (Fig. 3A). This remarkable change was also confirmed by flow cytometric analysis (Fig. 3B), in agreement with spectrometric analysis (Fig. 2A) and in sharp contrast to that using Cm–Np-B for the same fluorescence imaging and flow cytometric analysis, where only small alternation in the fluorescence blue and green channels was observed (Fig. S20, ESI†). Thus the sharp contrast corroborates the high sensitivity of the fluorescent protein-probe hybrid (Cm–Np-B@BSA) for intracellular H2O2 detection.
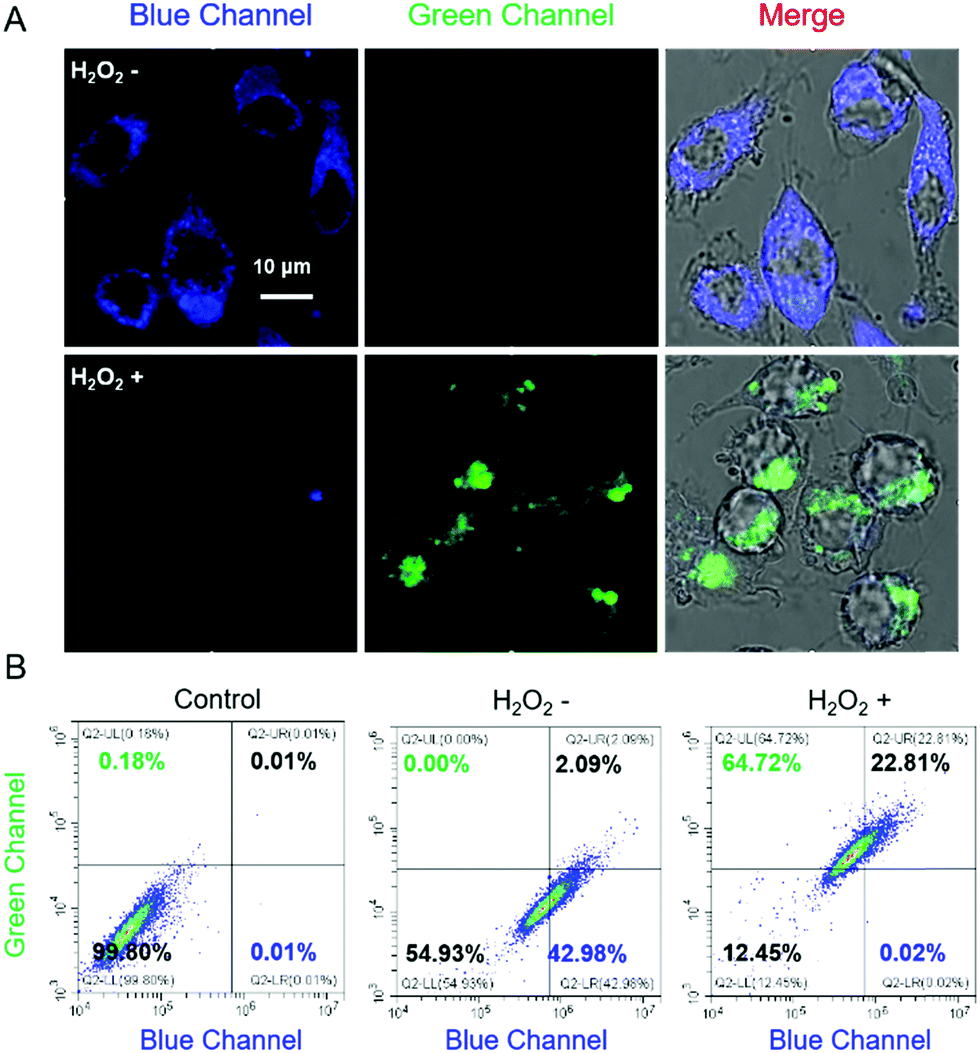 | ||
| Fig. 3 Fluorescence imaging and flow cytometric data of Raw 264.7 cells using Cm–Np-B@BSA (5/50 μM) as the probe. (A) Fluorescence imaging of living cells with (H2O2+) or without (H2O2−) exogenous H2O2 (200 μM); (B) flow cytometric data of cells with (H2O2+) or without (H2O2−) exogenous H2O2 (200 μM). λex = 405 nm, λem = 460–480 and 520–560 nm for blue and green channels, respectively. The images were recorded by confocal laser-scanning microscopy (scale bar = 10 μm). | ||
The capability of Cm–Np-B@BSA for ratiometric fluorescence visualization of intracellular H2O2 generation was further evaluated in LPS-stimulated RAW 264.7 macrophage cells. As shown in Fig. 4, obvious fluorescence emissions in both the blue and green channels were observed, suggesting that Cm–Np-B@BSA is able to detect endogenous H2O2 in live cells.
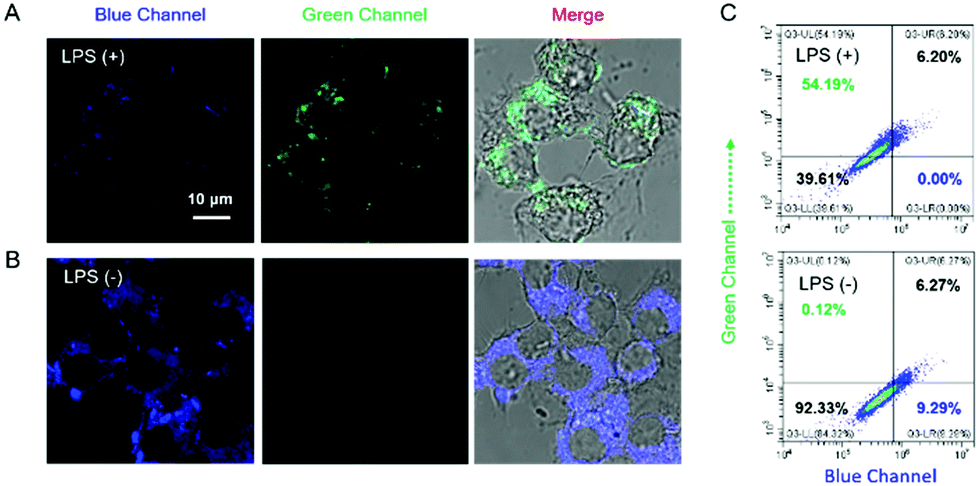 | ||
| Fig. 4 Fluorescence imaging of Raw 264.7 cells pretreated with LPS using Cm–Np-B@BSA (5/50 μM) as the probe. (A) Fluorescence imaging of living cells pretreated with 80 ng mL−1 of LPS; (B) control group of cells; (C) flow cytometric data of Raw 264.7 cells. λex = 405 nm, λem = 460–480 and 520–560 nm for blue and green channels, respectively. The images were visualized by confocal laser-scanning microscopy (scale bar = 10 μm). | ||
In summary, we developed an artificial protein-probe hybrid, Cm–Np-B@BSA, for ratiometric fluorescence detection and imaging of H2O2 in physiological buffers and live cells. Cm–Np-B@BSA was devised by assembling H2O2-responsive Cm–Np-B molecules with BSA via host–guest interactions. The strong binding of Cm–Np-B with BSA occurred at the preferred site I of BSA. In this complex, blue channel emission (λem = 460 nm) was activated (>145-fold enhancement) while the green channel (λem = 525 nm) remained weakly fluorescent. Cleavage of a borate group of Cm–Np-B@BSA by the H2O2-initialized reaction generated a new product Cm–Np@BSA, and enabled green channel emission observable as the result of FRET from Cm to Np. This change allowed Cm–Np-B@BSA to be used in ratiometric fluorescence detection of H2O2 in aqueous solution. This Cm–Np-B@BSA exhibited high sensitivity and selectivity for H2O2 detection with good biocompatibility, and demonstrated ratiometric fluorescence detection of H2O2 in live macrophage cells. Thus, the success in developing Cm–Np-B@BSA not only provides a useful tool to monitor H2O2 generation in live cells, but also offers a feasible way to design new artificial protein-probe hybrids for biomolecule detection and imaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the facilities and assistance of the Queensland Node of the Australian National Fabrication Facility (ANFF-Q), The University of Queensland. X. Zhang was financially supported by an award from China Scholarship Council (CSC) and an Australian Research Council Discovery Project grant (DP190103486).Notes and references
- A. Miyawaki and Y. Niino, Mol. Cell, 2015, 58, 632–643 Search PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- E. A. Rodriguez, R. E. Campbell, J. Y. Lin, M. Z. Lin, A. Miyawaki, A. E. Palmer, X. Shu, J. Zhang and R. Y. Tsien, Trends Biochem. Sci., 2017, 42, 111–129 CrossRef CAS PubMed.
- E. Eroglu, S. Charoensin, H. Bischof, J. Ramadani, B. Gottschalk, M. R. Depaoli, M. Waldeck-Weiermair, W. F. Graier and R. Malli, Free Radical Biol. Med., 2018, 128, 50–58 Search PubMed.
- Z.-j. Chen and H.-w. Ai, Biochemistry, 2014, 53, 5966–5974 CrossRef CAS PubMed.
- S. Sun, Y. Liu, J. Xia, M. Wang, R. Tang, C. Lei, Y. Huang, Z. Nie and S. Yao, Chem. Commun., 2019, 55, 2218–2221 Search PubMed.
- K. Fukunaga, T. Watanabe, D. Novitasari, H. Ohashi, R. Abe and T. Hohsaka, Chem. Commun., 2018, 54, 12734–12737 RSC.
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS PubMed.
- H. Shinoda, M. Shannon and T. Nagai, Int. J. Mol. Med., 2018, 19, 1548 Search PubMed.
- X. Liu, Z. Ye, W. Wei, Y. Du, J. Yuan and D. Ma, Chem. Commun., 2011, 47, 8139–8141 RSC.
- L. Tian, Z. Dai, X. Liu, B. Song, Z. Ye and J. Yuan, Anal. Chem., 2015, 87, 10878–10885 CrossRef CAS PubMed.
- J. Du, T. Zhu, Q. Gu, W. Cao, J. Fan and X. Peng, Sens. Actuators, B, 2018, 263, 661–667 Search PubMed.
- S. Yoo and M. S. Han, Chem. Commun., 2019, 55, 14574–14577 RSC.
- Y. Fu, H.-H. Han, J. Zhang, X.-P. He, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2018, 140, 8671–8674 CrossRef CAS PubMed.
- R. Zhang, B. Song and J. Yuan, TrAC, Trends Anal. Chem., 2018, 99, 1–33 CrossRef CAS.
- R. Sindhu, A. K. Tiwari, L. C. Mishra and M. M. Husain, Cancer Biother. Radiopharm., 2012, 27, 452–456 Search PubMed.
- T. Bayraktutan and Y. Onganer, Dyes Pigm., 2017, 142, 62–68 CrossRef CAS.
- S. C. Kampranis, N. A. Gormley, R. Tranter, G. Orphanides and A. Maxwell, Biochemistry, 1999, 38, 1967–1976 CrossRef CAS PubMed.
- S. I. Reja, I. A. Khan, V. Bhalla and M. Kumar, Chem. Commun., 2016, 52, 1182–1185 RSC.
- J. Park and Y. Kim, ChemBioChem, 2019, 20, 350–354 CrossRef CAS PubMed.
- H. H. Han, A. C. Sedgwick, Y. Shang, N. Li, T. Liu, B. H. Li, K. Q. Yu, Y. Zang, J. T. Brewster, M. L. Odyniec, M. Weber, S. D. Bull, J. Li, J. L. Sessler, T. D. James, X. P. He and H. Tian, Chem. Sci., 2020, 11, 1107–1113 RSC.
- H. Xiao, P. Li, S. Zhang, W. Zhang, W. Zhang and B. Tang, Chem. Commun., 2016, 52, 12741–12744 Search PubMed.
- H. Xiao, P. Li, X. Hu, X. Shi, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 6153–6159 Search PubMed.
- Y. Chen, X. Shi, Z. Lu, X. Wang and Z. Wang, Anal. Chem., 2017, 89, 5278–5284 CrossRef CAS PubMed.
- H. Wang, Z. He, Y. Yang, J. Zhang, W. Zhang, W. Zhang, P. Li and B. Tang, Chem. Sci., 2019, 10, 10876–10880 RSC.
- L. Chen, S. Xu, W. Li, T. Ren, L. Yuan, S. Zhang and X.-B. Zhang, Chem. Sci., 2019, 10, 9351–9357 RSC.
- W. Zhang, J. Zhang, P. Li, J. Liu, D. Su and B. Tang, Chem. Sci., 2019, 10, 879–883 RSC.
- W. Gao, Y. Zhao, X. Li, Y. Sun, M. Cai, W. Cao, Z. Liu, L. Tong, G. Cui and B. Tang, Chem. Sci., 2018, 9, 439–445 RSC.
- T. F. Brewer, F. J. Garcia, C. S. Onak, K. S. Carroll and C. J. Chang, Annu. Rev. Biochem., 2015, 84, 765–790 CrossRef CAS PubMed.
- Y. Hitomi, T. Takeyasu and M. Kodera, Chem. Commun., 2013, 49, 9929–9931 RSC.
- J. Zhang, L. Shi, Z. Li, D. Li, X. Tian and C. Zhang, Analyst, 2019, 144, 3643–3648 Search PubMed.
- M. Abo, Y. Urano, K. Hanaoka, T. Terai, T. Komatsu and T. Nagano, J. Am. Chem. Soc., 2011, 133, 10629–10637 CrossRef CAS PubMed.
- Y. Liu, C. Jiao, W. Lu, P. Zhang and Y. Wang, RSC Adv., 2019, 9, 18027–18041 Search PubMed.
- X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783–4804 RSC.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- V. Marx, Nat. Methods, 2017, 14, 949 CrossRef CAS PubMed.
- X. Liang, X. Xu, D. Qiao, Z. Yin and L. Shang, Chem. – Asian J., 2017, 12, 3187–3194 CrossRef CAS PubMed.
- C. Yik-Sham Chung, G. A. Timblin, K. Saijo and C. J. Chang, J. Am. Chem. Soc., 2018, 140, 6109–6121 Search PubMed.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, J. Chem. Educ., 2016, 93, 345–350 Search PubMed.
- S. Patra, K. Santhosh, A. Pabbathi and A. Samanta, RSC Adv., 2012, 2, 6079–6086 Search PubMed.
- M. S. Baptista and G. L. Indig, J. Phys. Chem. B, 1998, 102, 4678–4688 CrossRef CAS.
- X. Cao, Y. He, D. Liu, Y. He, X. Hou, Y. Cheng and J. Liu, RSC Adv., 2018, 8, 25519–25525 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00856g |
| This journal is © The Royal Society of Chemistry 2020 |