
Tuning of endosomal escape and gene expression by functional groups, molecular weight and transfection medium: a structure–activity relationship study†
Friederike
Richter
a,
Liam
Martin
a,
Katharina
Leer
a,
Elisabeth
Moek
a,
Franziska
Hausig
a,
Johannes C.
Brendel
 ab and
Anja
Traeger
*ab
ab and
Anja
Traeger
*ab
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstrasse 10, 07743 Jena, Germany. E-mail: anja.traeger@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
First published on 8th April 2020
Abstract
The use of genetic material by non-viral transfer systems is still in its initial stages, but there are high expectations for the development of targeted therapies. However, nucleic acids cannot enter cells without help, they must be well protected to prevent degradation and overcome a variety of biological barriers, the endosomal barrier being one of the greatest cellular challenges. Herein, the structure–property-relationship was investigated in detail, using well-defined polymers. Polyacrylamides were synthesized via RAFT polymerization resulting in a polymer library of (i) different cationic groups as aminoethyl acrylamide (AEAm), dimethylaminoethyl acrylamide (DMAEAm), dimethylaminopropyl acrylamide (DMAPAm) and guanidinopropyl acrylamide (GPAm); (ii) different degree of polymerization; and investigated (iii) in different cell culture settings. The influence of molar mass and cationic moiety on complex formation with pDNA, cytotoxicity and transfection efficiency of the polymers were investigated. The systematic approach identified a pH-independent guanidinium-containing homopolymer (PGPAm89) as the polymer with the highest transfection efficiency and superior endosomal release under optimal conditions. Since PGPAm89 is not further protonated inside endosomes, common escape theories appear unsuitable. Therefore, the interaction with bis(monoacryloylglycerol)phosphate, a lipid specific for endosomal vesicles, was investigated. Our research suggests that the interactions between amines and lipids may be more relevant than anticipated.
Introduction
Gene therapy is a powerful approach towards treating genetic disorders, cancer and other diseases. Due to an instability against nucleases and reduced uptake ability of naked genetic material, carriers are needed for effective protection and transportation.1 Carriers designed by nature are known as viruses, some of which have already been approved by different agencies.2 However, viruses pose a high risk due to unexpected side-effects, show limitations in modification as well as in the amount of delivered genetic material, and are not amenable to scale-up. Polymers are thus an extremely appealing alternative.3,4 To protect their genetic cargo, the polymers form stable complexes with the negatively charged phosphate groups of the genetic material and are, as such, typically positively charged.5 Moreover, the cationic charge aids in overcoming gene delivery barriers like the cellular membrane and lysosomal entrapment. Especially the endosomal escape is important to prevent recycling or enzymatic degradation and to transport the genetic material closer to its site of action.6–8 A well-known cationic polymer is linear poly(ethylene imine) (LPEI) which shows high transfection efficiency, but also low biocompatibility, promoting the development of less toxic alternatives.9–11Beyond PEI, various cationic polymers with a range of backbone chemistries have been studied as non-viral gene delivery vectors. Polymers based on vinyl backbones are of particular interest, since they can possess a broad range of functional pendant groups, and may be readily synthesized by various polymerization techniques, including radical polymerization. For example, poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) shows a propensity for gene delivery and is extensively studied. PDMAEMA possesses tertiary amine pendant groups and a pKa which renders it pH-responsive within a physiologically useful pH window (≈7.4).12,13 While there are many other examples of vinylic cationic polymers in polymer-based gene delivery, such as polybutylamino vinyl ethers (PBAVE),14,15 work has mostly been limited to polymethacrylates or polymethacrylamides. The polyacrylamides (PAms) represent an interesting alternative since they are hydrolytically stable (unlike many polymethacrylates) and are considered to possess more hydrophilic polymeric backbones than their acrylate, methacrylate and methacrylamide counterparts. Therefore, they are more stable to store and less likely to be modified in the body. Regarding their polymerization, acrylamide monomers possess a comparatively high rate constant of propagation (kp) and are typically less prone to side reactions of transfer during radical polymerization, which makes them well suited for reversible deactivation radical polymerization (RDRP) techniques such as reversible addition–fragmentation chain transfer (RAFT) polymerization.16–19 Concerning gene delivery, Nakayama and co-workers developed cationic star polymers based on poly(N,N-dimethylaminopropyl acrylamide) (PDMAPAm) and showed higher transfection efficiency than PEI in COS-1 cells.20–23 The group of Young studied linear PAms bearing primary amine pendant groups with varying spacer length and also reported promising transfection efficiency with primary amine/imidazole functional polymers, with or without the presence of the stealth polymer poly(ethylene glycol) (PEG).24–26 However, to the best of our knowledge, no further systematic studies have been performed with polyacrylamides as cationic homopolymers of linear architecture for gene delivery. In addition to the polymer backbone chemistry, there are a number of other properties which influence gene delivery potential. The molar mass of a given cationic polymer has been demonstrated in several cases to play an important role. Increasing molar masses typically correspond to increased transfection efficiency, but also increased cytotoxicity.27–31
Moreover, the nature of the cationic moiety is of crucial importance, since it is a vital feature for DNA binding, cellular uptake, endosomal escape and DNA release. There are only few systematic studies focusing on the investigation of the correlation between the nature of the cationic moiety and the transfection mechanism; and they were not conducted with polyacrylamides. Reineke and coworkers investigated methacrylamide-based RAFT copolymers bearing primary, secondary, tertiary or ternary amine and carbohydrate pendant groups in different copolymer compositions (block vs. statistical copolymer, monomer ratio).32,33 More recently, a library of methacrylate-based homo- and co-polymers (bearing primary, secondary and/or tertiary amines) identified primary amine-based polymers to possess high potential.34 However, the spacing between the cationic moiety and the polymer backbone also had an effect on the physicochemical properties.12,29,35
Most studies focused on pH-responsive polymers that were (partially) protonated at physiological pH values.12,34,36 pH-independent polymers with quaternized amines, for example, have rarely been investigated so far. They showed the efficiency-effectiveness dilemma: less toxicity but also less efficiency compared to their pH-dependent analogs. The assumed reasons are an inefficient DNA release and/or a lower endosomal release.13,33,37,38 Another interesting cationic moiety is the guanidinium group, that is not pH-dependent in a physiological context (pKa > 12). Guanidine occurs in many different biomolecules and contributes to protein denaturation, DNA-synthesis inhibition or in the amino acid arginine in active sites of enzymes.39 Many cell penetrating peptides, such as the TAT peptide, are rich in arginine residues.40 However, while their proficiency for intracellular trafficking is well known, their mechanism of cellular entry, i.e. via transduction or endocytosis, remains a topic for debate.41–46 Assuming endocytosis, the mechanism of endosomal escape for guanidinium containing cell penetrating peptides (CPP) has to be considered. In studies with membrane lipids of the endolysosomal pathway, arginine containing CPPs were shown to bind bis(monoacryloyl glycerol)phosphate (BMP), a lipid present at the inner side of the membrane of intra late endosomal vesicles (ILEV) and to disrupt BMP-containing liposomes, indicating a possible pathway for the endosomal escape.47,48 Since this feature is of great importance in applications such as drug or gene delivery, the guanidinium group is frequently exploited for the development of synthetic vectors.46,49–53 Regarding examples possessing vinylic polymeric backbones, Funhoff et al. reported promising transfection and uptake in COS-7 cells for poly(3-guanidinopropyl methacrylate) (PGPMA) homopolymers prepared via free radical polymerization.38 However, well-defined homopolymers of guanidinium-functional PAms have not been systematically studied as gene delivery vectors, nor compared with other cationic moieties. Still, they are of vital importance for the potential utilization of nature-inspired specific cation–lipid-interactions.
In this work, we synthesized a library of well-defined cationic PAm homopolymers of varying molar mass bearing either primary amine, tertiary amine or guanidinium pendant groups via RAFT polymerization in order to assess their potential for gene delivery. The influence of the different properties on the transfection efficiency of the polymers was investigated and they were further characterized regarding the media influence, their pDNA binding capability, uptake and different types of toxicity. Due to the outstanding performance of the guanidinium functionalized polymer, the underlying endosomal escape mechanism was investigated in more detail using calcein release and lipid–polymer binding assays.
Material and methods
Materials, instruments, further methods and calculations can be found in the ESI.†RAFT polymerization
Experimental details of all polymerizations are provided in the Tables S1 and S2 (ESI†).
Polyplex preparation
For the preparation of polyplexes, plasmid DNA (pDNA) and different amounts of polymer dissolved in water were mixed in HBG buffer (20 mM 4-(2-hydroxethyl)piperazine-1-ethanesulfonic acid (HEPES) and 5% (w/v) glucose, pH 7.2) to give a final pDNA concentration of 15 μg mL−1, with varying N*/P ratios (molar ratio of protonatable nitrogen atoms to phosphates of pDNA, see ESI†). Immediately after combination, the mixtures were vortexed for 10 s at maximum speed (3200 rpm) and incubated at room temperature for 15 min to ensure complex formation.Ethidium bromide quenching (EBA) and heparin dissociation assays (HRA)
The formation of polyplexes with pDNA was identified via quenching of ethidium bromide (EtBr) fluorescence by polymers interacting with pDNA. Briefly, 15 μg mL−1 pCMV-GFP pDNA in a total volume of 100 μL HBG buffer were incubated with EtBr (1 μg mL−1) at room temperature for 10 min. Subsequently, polyplexes with different quantities of polymer stock solutions (various N*/P ratios) were prepared in black 96-well plates (Nunc, Thermo Fisher, Germany) and incubated at 37 °C for 15 min before measuring the fluorescence intensity at λEx = 525 nm/λEm = 605 nm. A sample containing only pDNA and EtBr was defined as maximum fluorescence (100%).For the heparin dissociation assay, heparin was added to the formed polyplex–EtBr mixtures using the dispenser of the microplate reader to obtain the indicated concentrations (Table S4, ESI†). After each addition, the plate was shaken, incubated at 37 °C for 10 min and fluorescence intensity was measured.
The percentage of EtBr displaced upon polyplex formation or re-intercalating following pDNA release by heparin was calculated using eqn (1):
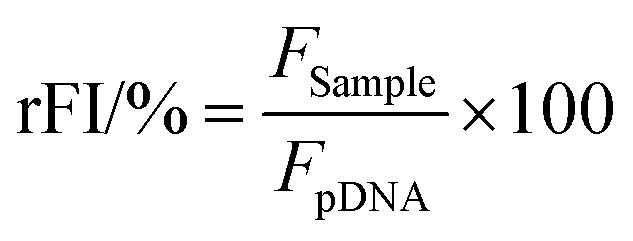 | (1) |
For a summarized depiction of the EBA results, the % of bound pDNA was calculated as 100% – rFI%. The heparin concentration needed to release the maximum of pDNA was calculated with OriginPro, Version 2018b (OriginLab Corporation, US) which can be found in the ESI.†
Determination of cytotoxicity
Cytotoxicity studies were performed with the mouse fibroblast cell line L929 (CLS, Germany), as recommended by ISO10993-5. For cytotoxicity of the polymers in HEK293T cells, refer to ESI.† The L929 cells were cultured in low glucose Dulbecco's modified eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS, Capricorn), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified 5% (v/v) CO2 atmosphere. In detail, cells were seeded at 104 cells per well in a 96-well plate without using the outer wells and incubated in medium containing 10 mM HEPES for 24 h. 1 h after medium change, cells were treated in sixtuplicates with polymers at different concentrations, ranging from 5 μg mL−1 to 500 μg mL−1, and incubated for additional 24 h. The medium was replaced by a 10% (v/v) alamarBlue solution in fresh culture medium, prepared according to the manufacturer's instructions. Following an incubation for 4 h at 37 °C, the fluorescence was measured at λEx = 570/λEm = 610 nm. Non-treated control cells on the same plate were referred to as 100% viability. Values below 70% were regarded as cytotoxicity. Data are expressed as mean ± SD of at least three independent determinations or as CC50 values ± 95% confidence interval (CI). Information regarding CC50 calculations can be found in the ESI.†Transfection of adherent cells
Transfection studies were performed with the human embryonic kidney cell line HEK293T (CLS, Germany). The cells were routinely cultured in DMEM medium (1 g L−1 glucose) supplemented with 10% FCS, 100 μg mL−1 streptomycin and 100 U mL−1 penicillin at 37 °C in a humidified 5% (v/v) CO2 atmosphere.For transfection studies, HEK293T cells were seeded at a density of 105 cells per well in 24-well plates and incubated at 37 °C (5% CO2) for 24 h. One hour prior to transfection, the medium was replaced by 450 μL serum reduced Opti-MEM™ or fresh growth medium (DMEM + 10% FCS + 10 mM HEPES) to reduce pH variance during the experiments. Polyplexes were prepared as described above with isolated pEGFP-N1 pDNA and added to the cells diluting the polyplexes 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in the cell culture medium. If incubated in Opti-MEM™, the supernatant was replaced by fresh growth medium after 4 h and incubated for further 20 h. When transfections were performed in growth medium, the cells were incubated with polyplexes for 24 h. For analysis via flow cytometry, cells were harvested by trypsinization and resuspension in Hanks’ Balanced Salt Solution, supplemented with 2% FCS and 20 mM HEPES (FC-buffer). For determination of transfection efficiency, cells were analyzed as described in the instrumentation section (see ESI†). Viable cells showing EGFP signal higher than the control cells incubated with pDNA only were gated as % of cells expressing EGFP and the relative mean fluorescence intensity (rMFI) of all viable cells was calculated in relation to the control. The experiments were performed at least three times and data are expressed as mean ± SD.
:10 in the cell culture medium. If incubated in Opti-MEM™, the supernatant was replaced by fresh growth medium after 4 h and incubated for further 20 h. When transfections were performed in growth medium, the cells were incubated with polyplexes for 24 h. For analysis via flow cytometry, cells were harvested by trypsinization and resuspension in Hanks’ Balanced Salt Solution, supplemented with 2% FCS and 20 mM HEPES (FC-buffer). For determination of transfection efficiency, cells were analyzed as described in the instrumentation section (see ESI†). Viable cells showing EGFP signal higher than the control cells incubated with pDNA only were gated as % of cells expressing EGFP and the relative mean fluorescence intensity (rMFI) of all viable cells was calculated in relation to the control. The experiments were performed at least three times and data are expressed as mean ± SD.
Polyplex uptake
To study the uptake of polymers over time, cells were seeded at a density of 105 cells per well in 24-well plates and cultured for 24 h in growth medium. One hour prior to the addition of the polyplexes, the medium was changed to serum reduced Opti-MEM™ or fresh growth medium. Polyplexes were prepared as described above after labelling 1 μg pCMV-GFP pDNA with 0.027 nmol YOYO-1 iodide. Subsequently, the polymer–pDNA-solutions were added to the cells, diluting the polyplexes 1:10 in cell culture medium. Following incubation for 4 h, cells were harvested by trypsinization and resuspension in FC-buffer. Trypan blue solution (0.4%) was added to a final concentration of 0.04% to quench fluorescence of polyplexes outside the cells. Cells were analyzed via flow cytometry as described in the instrumentation section (see ESI†). Viable cells showing YOYO-1 signal higher than the control cells, which were incubated with YOYO-1-pDNA only, were gated as % of cells that have taken up pDNA and the relative mean fluorescence intensity (rMFI) of all viable cells was calculated in relation to the control cells. The experiments were performed at least three times.
Hemolysis assay
The interaction of polymers with cellular membranes was examined by analyzing the release of hemoglobin from erythrocytes as published before.36,54 Blood from human donors, collected in tubes with citrate, was obtained from the Department of Transfusion Medicine of the University Hospital, Jena. The blood was centrifuged at 4500 × g for 5 min, and the pellet was washed three times with cold phosphate buffered saline (PBS, pH 7.4). Following a 10 times dilution with PBS (either pH 7.4 or pH 6.0), 500 μL aliquots of erythrocyte suspension were mixed 1:1 with the polymer solutions, which were prepared with PBS pH 7.4 or pH 6.0, and incubated at 37 °C for 60 min. After centrifugation at 2400 × g for 5 min, the supernatant was transferred to a clear flat bottomed 96-well plate (VWR, Germany) and the hemoglobin release was determined as the hemoglobin absorption at λ = 544 nm. Absorption at λ = 630 nm was used as reference. Complete hemolysis (100%) was achieved using 1% Triton X-100 as positive control. Pure PBS was used as negative control (0% hemolysis). The hemolytic activity of the polycations was calculated as follows (2):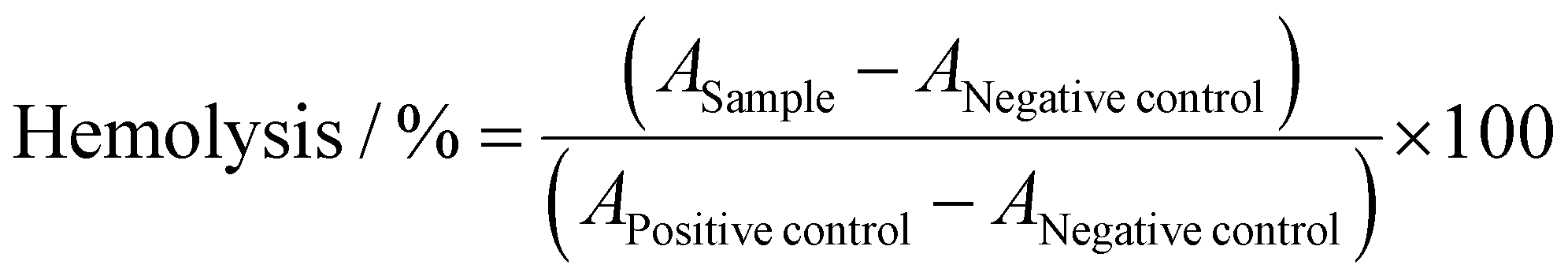 | (2) |
control and APositivecontrol are the absorption values of a given sample, the PBS treatment and the Triton X-100 treatment, respectively. A value less than 2% hemolysis rate was classified as non-hemolytic, 2 to 5% as slightly hemolytic and values >5% as hemolytic. Experiments were run in technical duplicates and were performed with three different blood donors.
Erythrocyte aggregation
Erythrocytes were isolated as described above. For determining the aggregation, 100 μL of the erythrocyte-polymer suspension were transferred to a clear flat bottomed 96-well plate (VWR, Germany). The cells were incubated at 37 °C for 2 h, and the absorbance was measured at λ = 645 nm. Cells treated with PBS served as negative control and cells treated with 50 μg mL−1 10 kDa BPEI were used as positive control. Aggregation potential of the polymers was calculated as follows (3):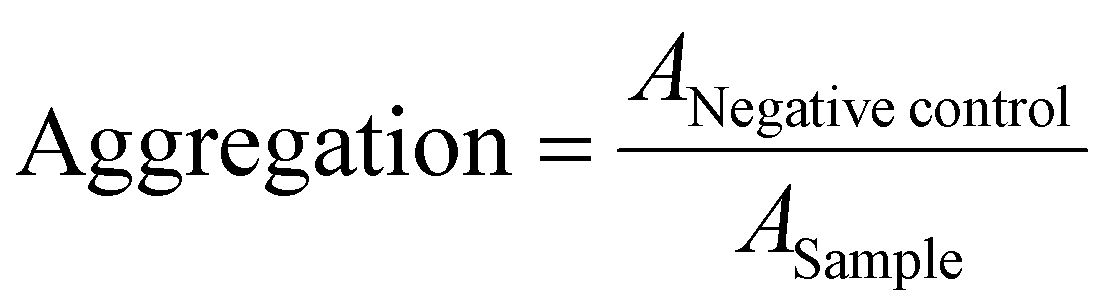 | (3) |
control are the absorption values of a given sample and the PBS treatment, respectively. Experiments were run in technical duplicates and were performed with blood from three different blood donors.
Calcein release assay
To determine the endosomal escape efficiency of the polymers, a calcein release assay was performed. HEK293T cells were seeded at a density of 105 cells per well in 24-well plates and cultured for 24 h in growth medium. They were treated with polyplexes as described for transfection studies. Just before the addition of polyplexes, the non-cell-permeable dye calcein was added to the cells to give a final concentration of 25 μg mL−1. Following incubation for 4 h, cells were carefully washed twice with cold PBS and harvested by trypsinization and resuspension in FC-buffer. Via flow cytometry, cells were analyzed as described in the instrumentation section (ESI†). Viable cells showing a calcein signal higher than the control cells incubated with calcein only were gated as % of cells that show strong calcein signal and the relative mean fluorescence intensity (rMFI) of all viable cells was calculated in relation to the control cells. The experiments were performed three times.Lipid–polymer binding assay
To investigate the polymer–membrane-interaction in more detail, the binding of the polymers to several lipids specific for different membrane stages along the endolysosomal pathway was determined using a lipid–polymer binding assay which was modified according to Erazo-Olivereas et al. 2016.47 The lipids were dissolved in hexane and the DY635-labeled polymers (see ESI†) were diluted in acetate buffer (0.1 M, pH 5.7) at equal amine concentrations (≙ N*/P 30, 1.2–1.4 mM). The lipid and polymer solutions were vigorously mixed at a ratio of 1:1 in 200 μL for one minute. Following incubation at −20 °C for 10 min, all samples were centrifuged in a microcentrifuge at 6000 rpm (Carl Roth, Germany) to allow phase separation. To determine the amount of polymer binding to the lipids, 50 μL of the aqueous phase were diluted 1:2 with acetate buffer, transferred to a black 96-well plate (Nunc, Thermo Fisher, Germany) and fluorescence intensity of DY635 was measured at λEx = 633/λEm = 680 nm in a plate reader. The mixture of pure hexane and the respective labeled polymer in the aqueous phase was used as the negative control (0% lipid binding). The polymer–lipid interaction was calculated as follows (4):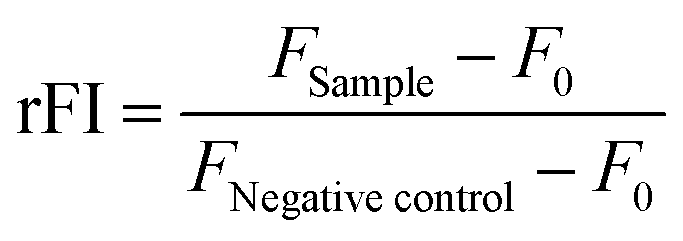 | (4) |
control and F0 represent the fluorescence intensity of the aqueous phase following lipid incubation, the fluorescence intensity of the aqueous phase following pure hexane incubation and the fluorescence intensity of pure buffer, respectively.
Statistics
To determine the statistical significance, analysis of variance (ANOVA) was performed. If the ANOVA revealed significant differences (p < 0.05), post hoc analyses with a Bonferroni correction were applied. All statistical analyses with data of n ≥ 3 and the determination of the Pearson's correlation coefficient were performed with Origin, Version 2018b (OriginLab Corporation, US). Further details can be found in the ESI.†Results and discussion
Monomer synthesis and characterization
Four acrylamide-based monomers were selected to prepare the cationic polymer library. Dimethylamino ethyl acrylamide (DMAEAm) is the acrylamide analogue of widely studied DMAEMA, while dimethylamino propyl acrylamide (DMAPAm) is a pertinent addition since PDMAPAm is a promising instance of cationic polyacrylamides used in gene delivery.20–23 While both monomers bear the same cationic moiety, the different length of the alkyl chain should influence the hydrophobicity and the pKa of the resulting polymers, which in turn influences their interactions with DNA and cells, and ultimately their propensity for transfection.12 Boc-protected forms of the primary amine- and guanidinium-functional monomers were used in this work since it permits the use of typical RAFT polymerization conditions, circumventing the need to conduct polymerizations in aqueous buffer. The Boc-protected primary amine-functional monomer, Boc-aminoethyl acrylamide (AEAmBoc), was synthesized according to literature (A),55 while a newly reported di-Boc-protected guanidinium-functional monomer, di-Boc-Guanidine propyl acrylamide (GPAmBoc), was synthesized in a two-step synthesis adapted from literature (Scheme 1B).56 Characterization of the monomers may be found in the ESI.†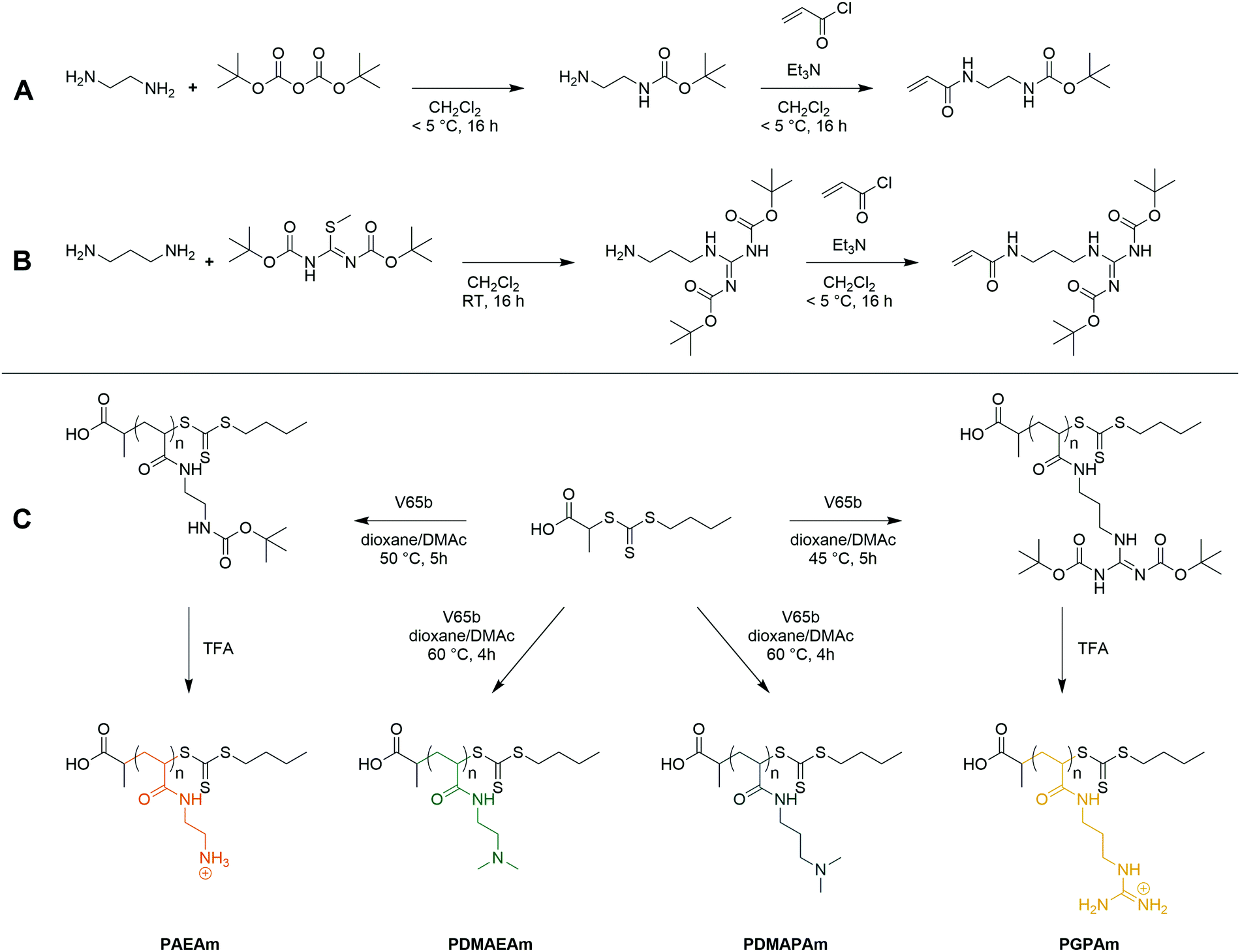 | ||
| Scheme 1 Synthesis of the AEAmBoc (A) and GPAmBoc (B) monomers used in this work and synthetic route towards cationic polyacrylamide library (C). | ||
Polymer synthesis and characterization
For each monomer, homopolymers with average degrees of polymerization (DP) ranging from 10 to 100 were targeted, in order to assess the influence of the polymer molar mass on biological activity. The library of cationic polymers was synthesized by RAFT polymerization using PABTC or its NHS activated ester derivative NHS-PABTC as CTA, with 1,4-dioxane/DMAc as a solvent system and V65B as azoinitiator (Scheme 1C). PABTC has been extensively used for the controlled polymerization of acrylamide and acrylate monomers.17,19 Moreover, the COOH R-group may be used to functionalize the polymers at the α-chain end. The solvent system was found to be suitable for the polymerization of all four monomers, while V65B (10 h half-life temperature of 51 °C in toluene) offers a reasonable rate of radical generation across the range of polymerization temperatures employed (45–60 °C) in this solvent system.Polymerizations of DMAPAm were stopped at monomer conversions of 70–80% in order to reduce the occurrence of side reactions, while for the other monomers, polymerizations were able to reach higher conversions (typically 85–95% as determined via1H NMR). SEC analysis of the polymers revealed monomodal populations with narrow molar mass distributions (Đ ≤ 1.3) in most cases (Table 1, Fig. 1E–H and Table S2, ESI†). The experimental molar masses (MnSEC) were slightly different to the theoretically determined values in most cases, which may be attributed to differences in the hydrodynamic radii of the polymers and the standards used to calibrate the respective SEC systems (PMMA for DMAc-SEC and P2VP for Aq.-SEC).
| Polymer | DPa | M n,th | M n,th | M n,SEC | Đ |
|---|---|---|---|---|---|
| (kg mol−1) | |||||
| a Determined via 1H NMR. b Determined using eqn (6), ESI. c Excluding mass of counter-ion. d Determined via Aq.-SEC with P2VP standards. | |||||
| PGPAm8 | 8 | 2.0 | 1.7 | 1.8 | 1.12 |
| PGPAm22 | 22 | 4.8 | 4.0 | 3.4 | 1.12 |
| PGPAm43 | 43 | 9.1 | 7.6 | 5.0 | 1.18 |
| PGPAm94 | 94 | 19.6 | 16.3 | 8.5 | 1.27 |
| PAEAm9 | 9 | 2.3 | 1.3 | 2.6 | 1.09 |
| PAEAm24 | 24 | 5.7 | 3.0 | 4.5 | 1.08 |
| PAEAm45 | 45 | 7.0 | 5.4 | 7.0 | 1.15 |
| PAEAm96 | 96 | 14.6 | 11.3 | 10.0 | 1.17 |
| PDMAEAm8 | 8 | 1.4 | — | 3.3 | 1.13 |
| PDMAEAm22 | 22 | 3.4 | — | 4.2 | 1.13 |
| PDMAEAm45 | 45 | 6.6 | — | 5.6 | 1.50 |
| PDMAEAm88 | 88 | 12.7 | — | 10.0 | 1.48 |
| PDMAPAm11 | 11 | 2.0 | — | 3.5 | 1.19 |
| PDMAPAm24 | 24 | 4.0 | — | 5.1 | 1.13 |
| PDMAPAm38 | 38 | 6.2 | — | 6.2 | 1.41 |
| PDMAPAm71 | 71 | 11.3 | — | 9.7 | 1.26 |
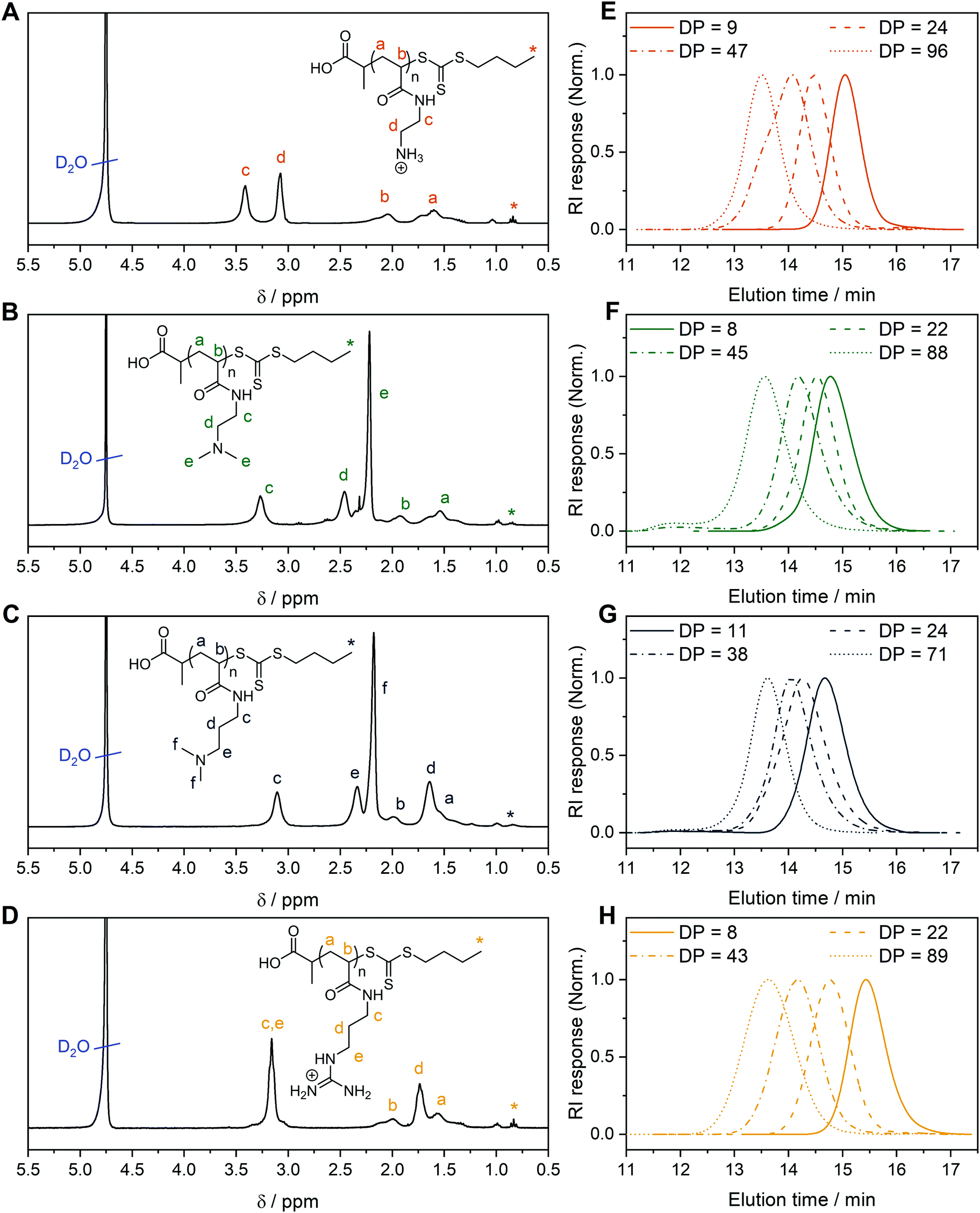 | ||
| Fig. 1 1H NMR of (A) PAEAm24, (B) PDMAEAm22, (C) PDMAPAm24, (D) PGPAm22 in D2O. Aqueous (0.1% TFA + 0.1 mol L−1 NaCl) SEC traces of (E) PAEAm, (F) PDMAEAm, (G) PDMAPAm, (H) PGPAm polymers synthesized via RAFT polymerization. | ||
Deprotection of the PGPAmdiBoc and PAEAmBoc polymers to give well-defined guanidinium and primary amine polymers, respectively, was performed using TFA. Quantitative removal of the Boc-protecting groups was confirmed using 1H NMR (Fig. 1A and D), while Aq.-SEC of the resulting polymers revealed monomodal populations with narrow molar mass distributions in all cases (Đ ≤ 1.3) (Fig. 1E and H).
To better understand the behavior of these cationic homopolymers at different pH values, the pKa values of the highest and lowest molar mass polymers from each set were determined. Acidified solutions (∼pH 2) of the polymers in 125 mM NaCl were titrated with 0.1 mol L−1 NaOH (0.5 mol L−1 in the case of PGPAm polymers) and pKa values were determined using the Henderson–Hasselbalch equation ((S5) and Fig. S4–S6, ESI†). Fig. S8 (ESI†) shows the calculated degree of protonation of the polymers at different pH values. All measured polymers possessed a pKa of 7.8 or above, while for the PGPAm polymers the pKa could not be determined with the available system (assumed to be 12 or above, Fig. S7, ESI†). All polymers are mainly positively charged under physiological conditions (pH 7.4) promoting the complexation with nucleic acids. A small molar mass dependence on pKa value was observed for each cationic polymer type, with the lowest molar mass polymer possessing the higher pKa in each case. This trend is consistent with simulations conducted by Nová and co-workers, where the difference in pKa showed little variation after a DP of ∼50.57 However, it was also observed that the titration curves become increasingly non-ideal with increasing polymer length (again up to a DP of ∼50), due to local effects of the neighboring monomers. There is a clear trend in the pKa of the different cationic moieties, with PDMAEAm (7.8–8.0) < PAEAm (8.3–8.5) < PDMAPAm (8.7–8.9) < PGPAm (assumed ≈ 12). The values for PDMAEAm and PDMAPAm are comparable to those obtained for methacrylamide-based systems.12,58 The significant difference between PDMAEAm and PDMAPAm can likely be attributed to the distance between protonatable groups, where increasing distance would reduce electrostatic repulsion between charged groups, leading to higher pKa values.59
Transfection efficiency and cytotoxicity
Since all polymers contain positively charged amines suitable for gene delivery, the polymer library was investigated regarding their potential for protein expression (Fig. 2A). HEK293T cells were incubated with polyplexes at N*/P 30 of pDNA encoding the EGFP protein using different transfection protocols: (i) adding polyplex to growth medium for 24 h or (ii) adding polyplex to serum-reduced Opti-MEM™ for 4 h and further incubation in growth medium for 20 h. For the different types of cationic polymer an increased EGFP-expression was observed with increasing molar mass, but to different extents for each type of cationic polymer. The transfection efficiency in Opti-MEM™ was increased compared to growth medium for LPEI (28% vs. 12% EGFP-positive cells) and PGPAm (30% vs. 3% EGFP-positive cells) with the highest increase of about ten-fold by PGPAm89 (p < 0.001). The highest transfection efficiency was achieved with the PGPAm89-based polyplex using Opti-MEM™ (30% EGFP-positive cells), which showed no significant difference to the positive control LPEI (p = 1.000). Taking the different DP of these polymers into account (89 for PGPAm vs. 600 for LPEI) the observed transfection efficiency showed the potential of the PGPAm polymers. All other polymers in the PAm library revealed less transfection efficiency compared to commercial LPEI (p < 0.05). Moderate transfection efficiency of PAEAm96 (7% EGFP-positive cells) was observed in serum-reduced medium. In growth medium, PAEAm96 showed the highest transfection efficiency among the PAm polymers (7% EGFP-positive cells, p = 1.000 compared to LPEI). Enhanced transfection efficiency in Opti-MEM™ is well-known and was also found for other polymer-based transfection agents.34,36,60 The lower transfection efficiency in growth medium could indicate interaction with serum proteins.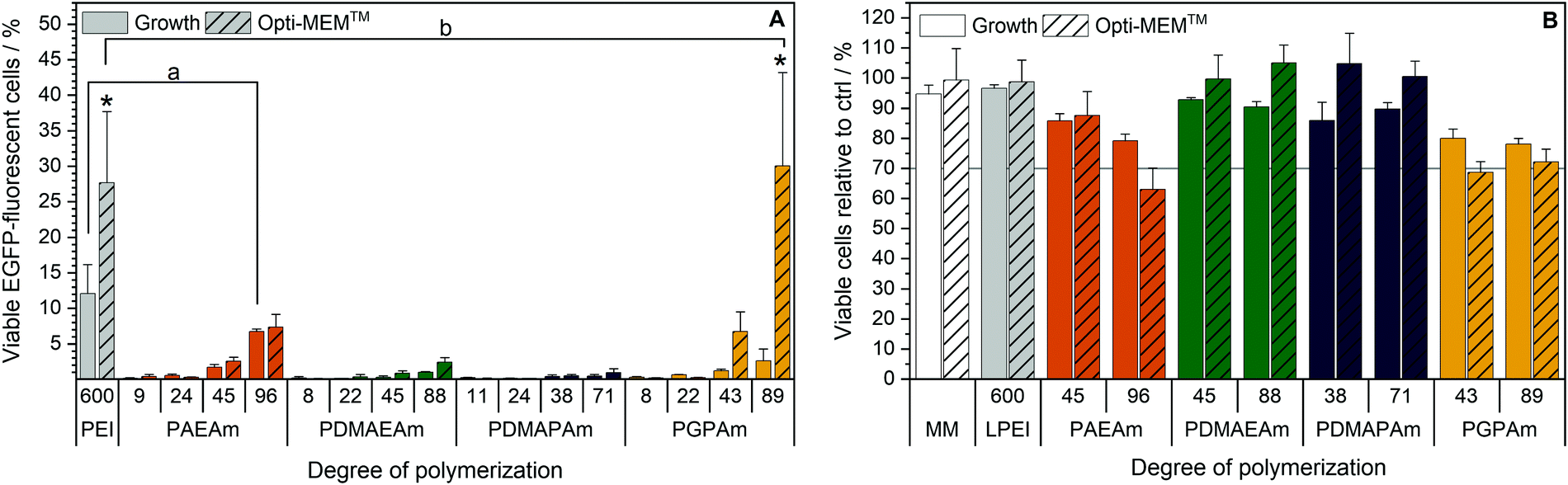 | ||
| Fig. 2 Transfection efficiency and toxicity of PAm homopolymers in HEK293T cells. (A) Transfection efficiency: cells were incubated with polyplexes of pEGFP-N1 pDNA and polymers at N*/P 30 (Table S6, ESI†). EGFP expression of viable cells was analyzed via flow cytometry. Two different transfection protocols were applied; either 24 h in growth medium (DMEM + 10% FCS + 10 mM HEPES) or 4 h in serum-reduced Opti-MEM™ followed by medium change to growth medium and further incubation for 20 h. Values represent mean ± SD (n ≥ 3). a: no significant difference (p > 0.05) to LPEI in growth medium, b: no significant difference (p > 0.05) to LPEI in Opti-MEM™, *: significant difference (p < 0.001) to same polymer in growth medium. (B) Cytotoxicity of PAm homopolymers in HEK293T cells. Metabolic activity was measured in HEK293T cells using the alamarBlue assay following incubation with indicated polymers at equal amine concentrations (≙N*/P 30) for 4 h. Values represent mean ± SD (n = 3). | ||
Polymers for gene delivery, in particular homopolymes, are known to reduce the viability of cells due to their cationic charges affecting the integrity of the cellular membrane.29 Therefore, the alamarBlue assay was performed in L929 cells according to ISO10993-5 (Fig. S9 and S10, ESI†) and of the interesting polymers with longer DP values in HEK293T cells (B). All tested acrylamide-based cationic polymers were found to be less toxic than the commercial gold standard LPEI (CC50 = 26 μg mL−1; see ESI†) and showed a reduced viability with increasing molar mass and concentration. The type of the cationic group influenced the viability and the following trend of cytotoxicity was observed: PDMAEAm < PDMAPAm < PAEAm = PGPAm, indicating the polymers with tertiary amines as least cytotoxic. The length of the side chain (propyl vs. ethyl) appeared to have a slight influence on the cytotoxic profile of the dimethylamino functional polymers. However, the concentrations used for further biological investigations (N*/P 30; 19–24 μg mL−1) showed high viability (≥90%) for all polymers (Table S6, ESI†). Regarding the cytotoxicity of N*/P 30 polyplexes in HEK293T cells, a similar trend was observed but with slightly less viability of PAEAm and PGPAm polymers (65–85%; Fig. 2B).
To the best of our knowledge, this is the first systematic study of PAm homopolymers of different molar mass, cationic moiety and hydrophobicity for gene delivery. However, some structure–property-relationships were already described in literature. The molar mass dependency of transfection efficiency and cytotoxicity has been shown for a variety of polymers such as PEI, PDMAEMA or lysine-functionalized methacrylamides.28,29,31,61–66 This dependency on molar mass could be attributed to the charge distribution in relation to the cell membrane by the polymers: in high molar mass polymers the positive charge is present in one large coiled molecule focusing the charge at one spot of the cellular membrane which could lead to its disruption. In low molar mass polymers, the same amount of charges is distributed within several small molecules and therefore spread over a larger membrane area. Regarding different cationic moieties, an increased toxicity for polymers with primary amines compared to the tertiary analogs was also shown for poly(2-oxazolines).67 In our study, the polymers with tertiary amines showed slight differences in toxicity and in protein expression with the ethyl spacer polymers performing slightly better than polymers with propyl spacer. This was also observed in studies of other vinyl polymers and could be due to increased interactions between propyl spacer polymers and DNA leading to a slow release of the genetic material inside the cytosol.12,24
Guanidinium-containing polymers are inspired by nature, more precisely by the amino acid arginine, which is abundant in well-known CPPs such as TAT or R8.68,69 However, the known polymer backbones differ to the polymers investigated herein and the guanidinium group is often used in combination with other functional moieties. Relatively low molar mass guanidinium-bearing poly(methacrylamides) (DP of 20) offered transfection efficiency of about 50% of that of jetPEI in HEK293T cells in serum free medium and 48 h post transfection.70 On the other hand, a guanidinium-bearing polymethacrylate with an approximately twofold higher number average molar mass (25 kg mol−1) compared to PGPAm89 and a 42.4 kDa poly-arginine exhibited lower transfection efficiency than PDMAEMA in COS-7 cells and serum-free medium.38 In another study a similar poly-arginine showed transfection efficiency comparable to lipofectamine in mixed cortical cells.71 The observed toxicity was also described with comparable guanidinium functional polymers of different backbone chemistry and spacer length.38,56,72
In the case of PGPAm polymers with low molecular weight, comparisons are only possible with oligo-arginines. Oligo-arginines ranging from 5 to 11 residues in length showed transfection of about 50% of that of BPEI (25 kDa) in A549 cells in serum free medium.73
All in all, the high molar mass guanidinium functional polyacrylamide PGPAm89 led to promising transfection results, comparable to commercial LPEI, which nicely demonstrates the potential of the controlled synthesis of this polymer class. However, the mechanism for the pDNA delivery of PGPAm89 remains to be investigated, as the guanidinium functionality was used to support gene delivery in random studies before, but successful protein expression was not shown with a homopolymer. The common design of polymers for gene delivery is based on the pH-sensitive character of the polymers, which changes the protonation and thus partly also the hydrophilicity in the endosome.74,75 However, the influence of the buffer capacity of the polymer was also discussed contrarily.76 Since the protonation of PGPAm89 does not change at endosomal pH (Fig. S8, ESI†), more detailed investigations of the transfection mechanism can help to design more efficient polymers.
Polyplex formation and characterization
To investigate the transfection mechanism, different bottle necks were investigated, starting at the formation of polyplexes. Therefore, the interaction between polymer and pDNA was investigated using the ethidium bromide quenching assay (EBA, Fig. 3A). The formation of polyplexes is indicated by displacement of intercalated ethidium bromide from the pDNA due to hydrophobic and electrostatic interactions of pDNA with the polymer, resulting in a decrease of ethidium bromide fluorescence. Various N*/P ratios were analyzed to determine the optimal conditions for sufficient pDNA binding (Fig. S11A, ESI†). In the EBA, all polymers reduced the fluorescence intensity of the pDNA-ethidium bromide solution with increasing N*/P ratios, plateauing at values of N*/P 5 or above. However, while the molar mass of the polymer within each cation set had no impact on the value of the plateaus, the nature of the cationic group showed an influence. At N*/P 30, PDMAEAm polymers led to a binding of about 75% of the pDNA, whereas all other polymer groups, including LPEI, bound about 85–90% of the pDNA.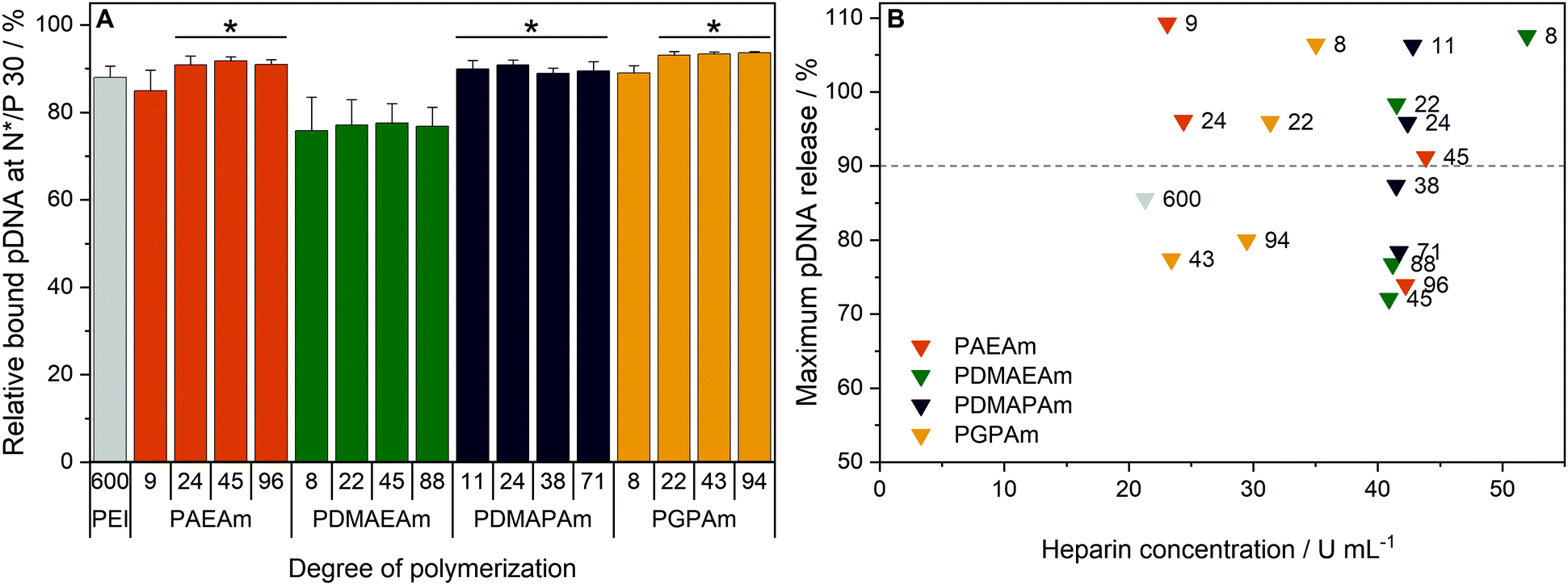 | ||
| Fig. 3 Polyplex formation and stability tests with pDNA and PAm homopolymers. (A) EBA of all polymers at N*/P 30 in HBG buffer. Values represent mean ± SD (n ≥ 3). *: significant difference to all PDMAEAm polymers (p < 0.05). (B) HRA of polymers at N*/P 30. Values were calculated as the heparin concentration needed to release the maximum amount of pDNA (defined as the beginning of the plateau, see Fig. S1 and S11B, ESI†) following fitting of a piecewise equation to the respective data (n = 3) for each polymer. Numbers in plot represent the degree of polymerization. | ||
Subsequently, the HRA and pH dependent EBA were used to further investigate the influence of cationic moiety, side chain length and DP on polyplex properties. In the case of the HRA, the formed polyplexes were incubated with heparin, a competing polyanion disrupting the electrostatic interaction between pDNA and polymer, which leads to re-intercalation of ethidium bromide and therefore increased rFI. The release of genetic material was observed with all investigated polymers, albeit with different release profiles and plateaus. At N*/P 30, only the polymers with lower molar mass (DP < 50) were able to release the pDNA completely, as indicated by an increase of the rFI above 90% (B). Interestingly at DP < 25, the PAEAm and PGPAm polymers required less heparin (20–35 U mL−1) to release the same amount of pDNA than PDMAEAm and PDMAPAm polymers with tertiary amines (40–50 U mL−1). Regarding the higher molar mass polymers with DP > 50, only PGPAm94 (30 U mL−1) released the pDNA at low heparin concentrations comparable to LPEI (21 U mL−1). This molar mass dependency was not observed for PDMAEAm and PDMAPAm polymers. In contrast to the EBA, these results showed a dependency on the polymer length for PAEAm and PGPAm and on the property of the side chain (Fig. S11B, ESI†). Furthermore, the results identify the polyplexes with PGPAm43 and PGPAm94 to be promising polymers, showing strong binding but no full release of pDNA by electrostatic competitors.
Additionally, a pH-dependent EBA (pH value 5 to 9) was performed and differences between PGPAm and the other PAms were observed (Fig. S12, ESI†). The PGPAm polyplexes showed a strong and pH-independent polyplex formation, whereas the other polymers showed less pDNA binding at higher pH values.
The investigation of polyplex formation and stability indicated that PAEAm and PGPAm polymers bind pDNA very well (Fig. 3A), complexing pDNA to a slightly greater extent than LPEI and releasing it at moderate heparin concentrations (B). The good binding of PGPAm polymers could be attributed to the nature of the bidentate binding of guanidinium to the phosphate of the pDNA displacing EtBr more efficiently than the other polymers.77 On the other hand, the low heparin concentration needed to partially release the PGPAm could indicate a high affinity of guanidinium for the sulphate groups of heparin compared to the phosphate groups of the pDNA.78,79 In the case of the longer PGPAm, the inefficient release of pDNA by heparin may also be due to further, non-electrostatic interactions of the polymers with the DNA. In contrast, pDNA binding with PDMAEAm appeared to be weaker despite higher concentration of heparin being required for release of the genetic material (Fig. 3). The other tertiary amine-based system, PDMAPAm, showed the same strong pDNA–polymer interaction once the polyplex was formed. This difference in pDNA binding affinity between primary and tertiary amine moieties has been observed previously with polymers comprising methacrylate backbones.34 The higher pDNA complexation by PDMAPAm polymers compared to PDMAEAm could be due to the increased hydrophobicity conferred by the propyl spacer of the side chain.80,81 Moreover, Van de Wetering et al. explained this reduced affinity for the phosphates of the pDNA with the reduced steric availability of the tertiary amines of ethyl spacer polymers.12 In the end, the complexation of less pDNA by PDMAEAm compared to the other PAm polymers could also be explained by the partial protonation of the PDMAEAm polymers, which possess the lowest pKa of this library, at pH 7.4 leading to a decrease in protonated amines available for pDNA binding (see Fig. S8 and S12, ESI†).
To further characterize the formed polyplexes, their size (hydrodynamic diameter) was investigated (Table S7 and Fig. S13, S14, ESI†). Indeed, several studies have reported that polyplex sizes below 100 nm offer increased transfection efficiency.74,82 The size of polyplexes formed at N*/P 30 was assessed via dynamic light scattering (DLS). The Z-average diameter of all polyplexes ranged from 32 to 69 nm with only PAEAm9 and PDMAEAm22 showing polyplex sizes of 127 and 115 nm, respectively. Therefore, the main size-population was in the favored size range for all polyplexes.
In summary, no significant influence of hydrophobicity or type of cationic moiety on the polyplex size was found. The molar mass of the polymers showed only a slight influence on the size of the polyplexes. These results correspond very well to conclusions of other research groups using various cationic polymeric materials.12,29,62,83 So far, the only difference that corresponds to high transfection efficiency is the low heparin concentration required to release a high amount of the pDNA. Hydrophobic interactions might be a reason for the incomplete release by heparin. They were also promoted for other gene carrier systems.84
Cellular internalization of PAm homopolymers
To further investigate the difference in transfection efficiency between the PAm polymers, pDNA uptake properties were studied using flow cytometry or confocal laser scanning microscopy (CLSM) with HEK293T cells following incubation of the cells with polyplexes of YOYO-1 iodide labeled pDNA and polymers at N*/P 30 for 4 h. This method was used to assess the influence of temperature, media and molar mass of the polymers.To visualize the uptake, HEK293T cells were incubated with YOYO-1-labeled polyplexes containing the highest molar mass polymers or LPEI at N*/P 30 in Opti-MEM™ for 4 h and imaged with CLSM (Fig. 4A). Hoechst 33342 was added 10 min before imaging to stain the nuclei and trypan blue was used to quench the fluorescence of YOYO-1-labeled pDNA outside the cells. All tested polymers led to a punctate pattern of green fluorescence within the cells, whereas the control with YOYO-1 labeled pDNA and no polymer did not show green fluorescence. These results indicate an efficient uptake of YOYO-1-labeled pDNA by the PAm polymers or LPEI.
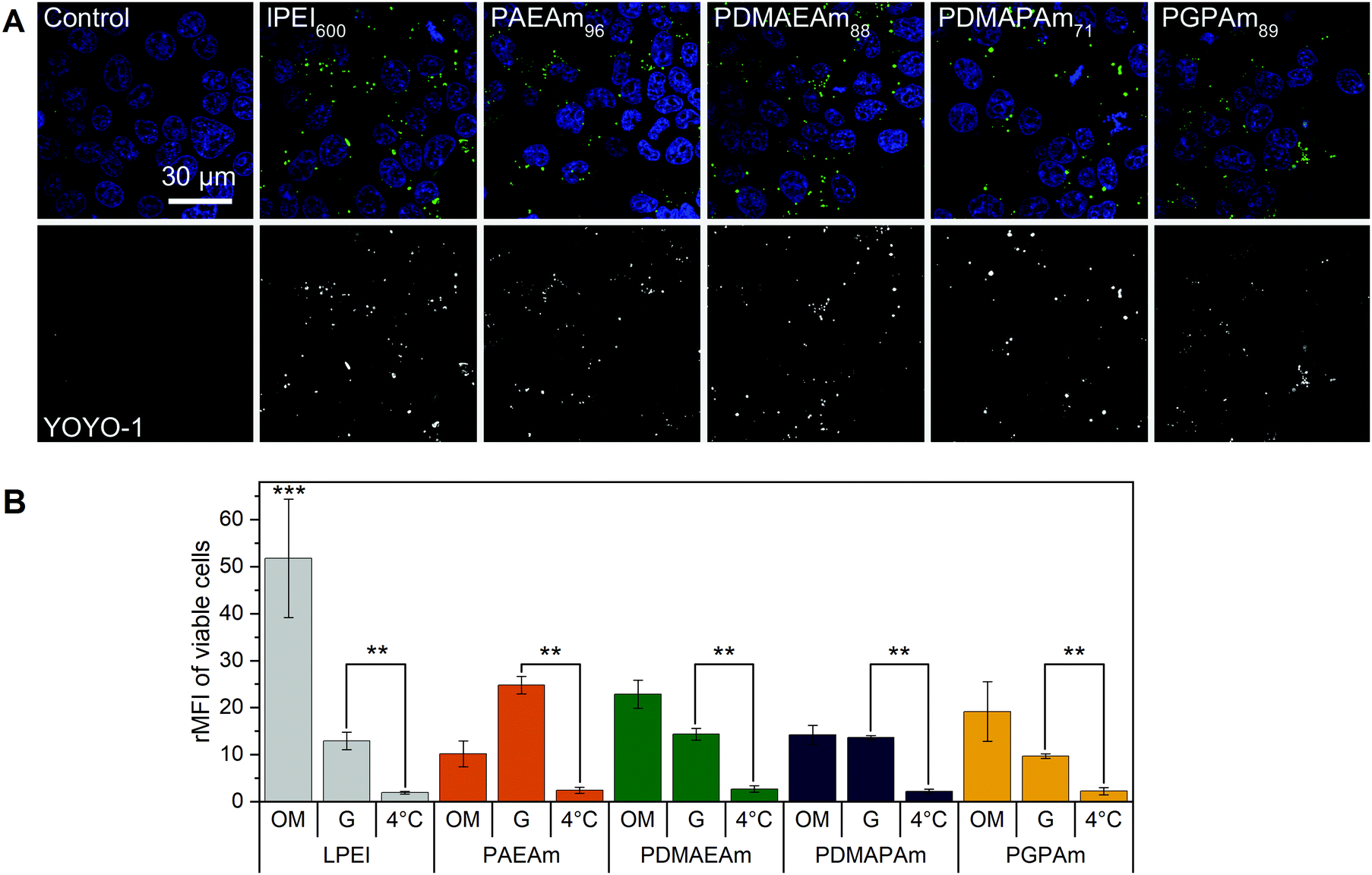 | ||
| Fig. 4 Polyplex uptake with PAm polymers (A) CLSM: HEK293T cells were incubated with polyplexes of YOYO-1-labeled pDNA and polymers at N*/P 30 (Table S6, ESI†) in Opti-MEM™ for 4 h. Nuclei were stained with Hoechst33342 and YOYO-1 fluorescence was quenched with trypan blue. (B) Flow cytometry: HEK293T cells were incubated with polyplexes of YOYO-1-labeled pDNA and LPEI, PAEAm96, PDMAEAm88, PDMAPAm71 or PGPAm94 at N*/P 30. Incubation was in growth medium at 37 °C for 4 h (G), in serum-reduced Opti-MEM™ at 37 °C for 4 h (OM) or in growth medium at 4 °C for 4 h (4 °C). Cells incubated with labeled pDNA served as control (rMFI = 1). Values represent mean ± SD (n ≥ 3). **: significant difference to indicated sample (p < 0.001). ***: significant difference to all other samples (p < 0.001). | ||
The uptake was investigated in more detail by flow cytometry (Fig. 4B). First, the common method of incubating cells at low temperature was used to find out whether the polyplexes were taken up via energy-dependent processes like clathrin-mediated endocytosis, often proposed for nanoparticles below 200 nm, or by translocation across the membrane.82 Therefore, HEK293T cells were incubated with the polyplexes in growth medium at 4 °C for 4 h, to inhibit all energy-dependent processes. All tested polymers showed a significant decrease of pDNA uptake compared to that observed in growth medium at 37 °C (p < 0.001). Furthermore, no difference in pDNA uptake was observed between the different polyplexes at 4 °C (p = 0.937).
Subsequently, the influence of the used transfection media on cellular uptake was studied. All polymers showed increased rMFI in both media. When incubated in Opti-MEM™, the quantity of internalized pDNA was slightly increased compared to growth medium, but only for LPEI significance (p < 0.001) was found. The highest increase in uptake of pDNA in Opti-MEM™ was observed for all PDMAEAm polymers and LPEI with rMFI of up to 25 and 51.8, respectively. However, LPEI-polyplexes showed a threefold higher pDNA uptake compared to all PAm homopolymers (p < 0.001). In growth medium, the highest increase in rMFI was observed for PAEAm96, indicating a possible explanation for the higher transfection efficiency of the polymer in the presence of serum. Interestingly, molar mass dependence was only observed for the PAEAm polymers in growth medium (Fig. S15, ESI†).
The temperature dependent uptake and a punctuate uptake pattern in CLSM studies, demonstrate that polyplexes (pDNA) were taken up via an energy-dependent mechanism. Although this was not previously investigated for PAm homopolymers, it is known for other cationic polymers used for gene delivery.34,36,85 Regarding the guanidinium functional polymers, previous studies of other research groups showed contradictory results of temperature-independent and temperature-dependent uptake, respectively.38,70 This inconsistency is also known for guanidinium-containing peptides,86 indicating that there are other factors additional to the type of functional group determining the way of internalization and should therefore be considered for novel polymers.
A reduced uptake of pDNA in the presence of growth medium was also observed by other groups.87,88 In the presence of serum, the cationic charged polyplexes tend to interact with negatively charged proteins, leading to aggregation and therefore reduced uptake.6,89,90 It could also be assumed that the interaction with extracellular matrix components such as heparan sulfate proteoglycans is less pronounced due to competition with serum proteins, so that less pDNA can be uptaken.91–94 However, in our study, the uptake of pDNA did not correlate well to the observed EGFP expression, where PGPAm96 showed the best performance in Opti-MEM™ whereas the other PAm polymers exhibited only slight EGFP expression. This was also observed in previous studies using methacrylate-based polymers.34 Therefore, further mechanistic assays were performed to find out, why PDMAEAm delivered as much pDNA into the cells as PGPAm, but showed nearly no transfection efficiency.
Interaction of polymers with cellular membranes
To further investigate the structure–property relationship of polymers for efficient gene delivery, the interaction with membranes, representing the main biological barriers, was analyzed. The influence of the polymers on the membrane integrity was tested via hemolysis and aggregation assays using human erythrocytes, which are well known for studies regarding membrane–polymer-interaction. The cells were washed with PBS and incubated with the polymers at equal amine concentrations (≙N*/P 30) before either the release of hemoglobin from the cells as indicator for cell lysis or the absorption of light by the cells as indicator for cell aggregation was measured.The low molar mass polymers can be considered as non-aggregating at the tested conditions (Fig. 5A). Meanwhile, the highest molar mass polymers (additionally PGPAm43) exhibited a potential for aggregation of erythrocytes (p < 0.001). The influence of the pH value was dependent on the type of cationic polymer, whereby only PAEAm96 (p < 0.001) and PDMAEAm88 (p = 0.02) showed significant dependence.
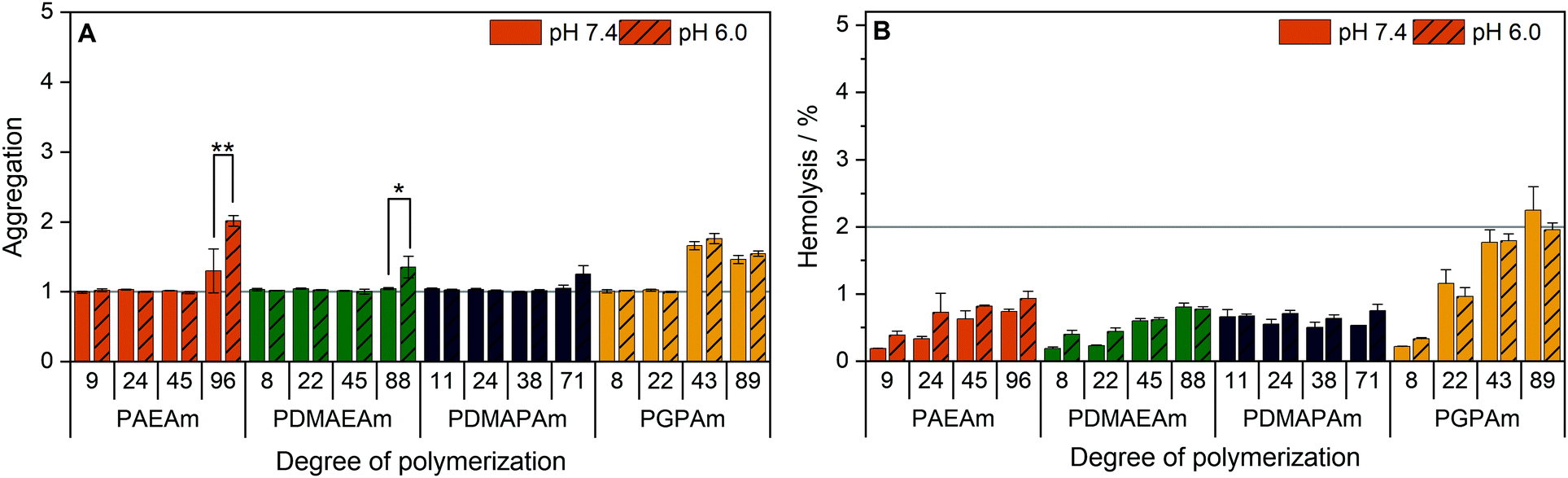 | ||
| Fig. 5 Interaction of PAms with erythrocyte membranes. Human erythrocytes were washed and incubated with polymers at equal amine concentrations (≙N*/P 30, Table S6, ESI†) in PBS of different pH values present in blood/cytoplasm (pH 7.4) or endosomal compartments (pH 6). (A) Aggregation of indicated polymers measured as light absorption by erythrocytes. Values are calculated as the negative control (PBS value) relative to the sample value and represent mean ± SD (n = 3). *: significant difference (p < 0.05), **: significant difference to indicated sample (p < 0.001). (B) Hemolysis as the amount of released hemoglobin calculated relative to 1% Triton X-100 as positive control (100% hemolysis). Values represent mean ± SD (n = 3) and are classified as slightly hemolytic between 2% and 5%, as non- or hemolytic if lower or higher than 2% or 5%, respectively. | ||
Beside the aggregation of erythrocytes, the potential of the polymers to induce membrane leakage was tested (Fig. 5B). It was found that all polymers were non-hemolytic (values below 2%). Moreover, the pH value had no significant influence on the hemolytic activity.
The increased aggregation of the red blood cells by the high DP polymers does not necessarily relate to membrane destruction but rather to membrane interaction via the high positive charge density of cationic polymers.95 The findings of this study indicate that, at concentrations equal to N*/P 30, no severe lysis of the erythrocyte membrane occurred in the presence of any of the polymers and also the pH values showed no influence on the membrane leakage potential of the polymers.
Since the membrane composition of erythrocytes differs from that of the cells used for transfection, the influence of polyplexes on membranes of HEK293T cells was studied (Fig. S16, ESI†).96,97 Therefore, a LDH assay was performed following incubation of the cells with polyplexes of the highest DP polymers at N*/P 30 in growth medium or in Opti-MEM™ for 4 h. If the polyplex or polymer decreases the integrity of the cellular membrane, the enzyme LDH will be released to the medium, which can be measured indirectly via the conversion of its substrates into fluorescent molecules. All polymers showed higher LDH release profiles in Opti-MEM™ compared to growth medium with only PAEAm96 and PGPAm89 showing significant differences (p < 0.001). The tertiary amine polymers and LPEI caused minor increases in both media (up to 5% relative to Triton X-100), whereas PAEAm96 and PGPAm89 in Opti-MEM™ showed an increase of about 26 and 31%, respectively (p < 0.001), indicating membrane-lytic activity. In growth medium, the LDH release by PGPAm89 was comparable to that of the tertiary amine polymers (2% relative to Triton X-100), whereas PAEAm96 exhibited the highest LDH-release of 14% (p = 1.000). This correlates well with the results for transfection efficiency and could again point out a medium dependency of PGPAm.
Endosomal release of polymers
To elucidate the mechanism of transfection for the polymer library, in particular for PGPAm, further investigations were required. The PGPAm polymers showed high transfection and only slight membrane destruction, while being pH unresponsive. The common hypotheses for endosomal release “proton sponge” as well as the “membrane permeability and pore formation” are based on the concept of pH-dependent increased protonation of the polymers during endosomal maturation.74 These hypotheses do not fit for polymers such as PGPAm exhibiting very high pKa values (≈12). To study the endosomal release of the polymer library, a calcein release assay was performed. Calcein is a non-cell-permeable, fluorescent dye taken up via endocytic pathways resulting in the formation of a punctuated pattern inside the cytoplasm. If polymers are able to destabilize the endosomal membrane, calcein is released into the cytoplasm giving a diffused fluorescence pattern.98,99 This effect can be detected by flow cytometry as an increase in fluorescence intensity as well as in an altered histogram (see Fig. S18, ESI†). Based on the previous results, only the polymers with the highest molar masses were screened using HEK293T cells following incubation with the polyplexes at N*/P 30 and calcein (25 μg mL−1) for 4 h (Fig. 6A).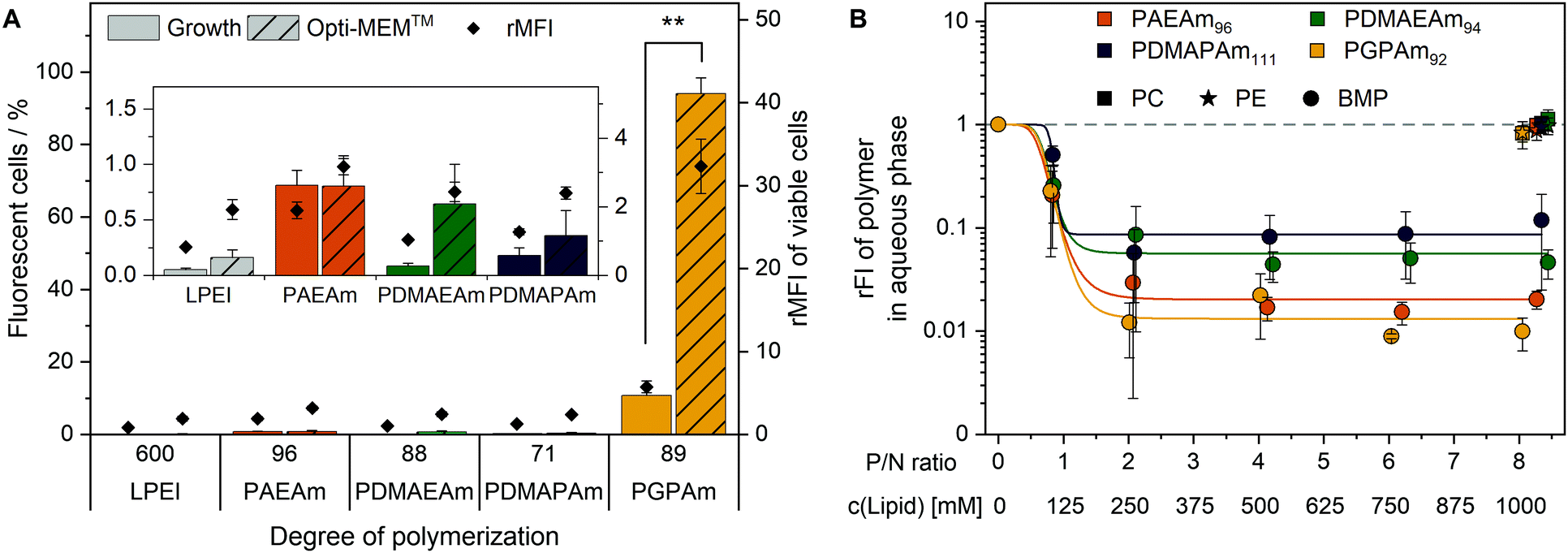 | ||
| Fig. 6 Endosomal escape of PAm homopolymers. (A) Calcein release assay: HEK293T cells were incubated with indicated polymers at N*/P 30 (Table S6, ESI†) for 4 h at 37 °C and the mean fluorescence intensity relative to the calcein control as well as the number of viable cells with higher fluorescence than the calcein control (%) were analyzed via flow cytometry. Values represent mean ± SD (n = 3). **: significant difference to indicated sample (p < 0.001). (B) Lipid binding assay: DY635-labeled PAm polymers in acetate buffer (pH 5.7) were incubated with different lipids in hexane at indicated concentrations and P/N ratios (lipid-phosphate to polymer-amine). Following phase separation, FI of the aqueous phase was measured and rFI calculated relative to the control with no lipids in the hexane phase. A decrease in rFI indicated partitioning of the DY635-labeled polymer into the hexane phase. Dots represent mean ± SD (n = 3). Lines represent a logistic equation fitted to the values of each replicate. | ||
A significant increase in calcein fluorescence was observed following incubation with PGPAm89-containing polyplexes (p < 0.001). All other tested polyplexes caused only a slight increase in rMFI. Interestingly, the endosomal release of PGPAm89-polyplexes was again influenced by the medium. The calcein fluorescence in growth medium was about 80% lower than in Opti-MEM™ and only 10.7% of cells showed higher fluorescence than the calcein control (p < 0.001).
The results of the calcein release assay demonstrate the potential of PGPAm89-polyplexes to escape the endosome. In serum-reduced and growth medium, the endosomal release of polyplexes with PGPAm89 outperformed all other tested polyplexes including those with LPEI. The difference between transfection and calcein release of LPEI-polyplexes could be explained with the higher pDNA uptake with LPEI compared to all other polymers, so that few calcein release from a higher number of endosomes was sufficient to yield a high transfection efficiency. The endosomal release potential for PDMAEMA, the methacrylate analog to PDMAEAm and PDMAPAm, was also found to be low.100 However, for guanidinium-containing CPPs an efficient endosomal release is known.41 To our knowledge, the high level of calcein release achieved with polyplexes of PGPAm89 homopolymers in this study was not shown for guanidinium functional polymers before. The results indicate an efficient and pH-independent endosomal release for PGPAm89.
Since the PGPAm polymers were not pH-sensitive but able to escape the endosome, the endosomal escape mechanism of those polymers was investigated in more detail. For guanidinium-containing CPPs, the endosomal release was proposed to occur via binding to BMP, a lipid present in the membranes of ILEV, but not in the limiting membrane of late endosomes or lysosomes.47,101,102 Therefore, the lipid–polymer binding assay was conducted to investigate the interaction of the PAm homopolymers with this lipid (Fig. 6B). DY635-labeled PAm polymers were diluted in acetate buffer (pH 5.7) to equal amine concentrations and mixed thoroughly with different concentrations of BMP in hexane. For comparison to other phosphate-containing but neutral lipids, PC and PE were used in the hexane phase. Following phase separation and isolation of the aqueous phase, the fluorescence of the aqueous phase was measured. A decrease in fluorescence intensity indicated the removal of the polymer from the aqueous phase and therefore lipid binding.
The incubation of the polymers with PC or PE in the hexane phase caused a negligible decrease in relative fluorescence intensity (rFI) by all tested polymers. When incubated with BMP, all tested polymers showed a decrease in fluorescence intensity with increasing P/N ratio (lipid-phosphate to polymer-amine). PGPAm exhibited the highest decrease in rFI indicating a slightly stronger binding than PAEAm and the tertiary amine analogues. The results are comparable to those for dfTAT of Erazo-Oliveras et al., who propose BMP-binding as the mechanism for endosomal escape of this peptide.47 In our study however, the non-calcein-releasing polymers also showed BMP-binding properties, albeit not as strong as PGPAm. This might be due to the higher density of cationic moieties in the PAm homopolymers compared to the density in dfTAT. The investigations indicate a multifactorial endosomal escape of PGPAm.
Conclusions
In this study, a library of cationic PAm homopolymers was synthesized and investigated for their transfection efficiency. RAFT polymerization was used to yield a series of well-defined polymers with narrow dispersity and narrow molar mass distributions. The polymers differ in their properties regarding (i) molar mass (DP10-100), (ii) cationic moiety (primary, tertiary, guanidinium) and (iii) length of alkyl spacer in the side chain (ethyl, propyl). The primary and tertiary amine functional polymers possessed pKa values slightly above physiological pH, whereas for PGPAm the pKa value could not be determined. Therefore, all PAm polymers in principle possess a high degree of protonation (≥70%) at physiological pH values and exhibited good pDNA binding (≥80%) as determined via EBA.Toxicity and efficiency are the main characteristics of transfection polymers. Therefore, the polymer library was investigated to identify interesting candidates. All PAm homopolymers were found to be less cytotoxic than LPEI in L929 cells, but only the highest molar mass guanidinium polymer, PGPAm94, was able to achieve a transfection efficiency as high as LPEI. The primary amine functional PAEAm96 polymers also resulted in notable transfection efficiency. For a better understanding of the transfection mechanism of the polymers, further investigations were performed, in detail: polyplex uptake, membrane interaction and endosomal release. The results showed beneficial effects of increasing molar mass and the presence of guanidinium- as well as primary amine-functional groups on transfection relevant aspects.
There was evidence for an endocytic uptake with a punctuate pattern of YOYO-1-labeled pDNA in CLSM studies and no uptake of all PAm polymers at 4 °C (inhibition of ATP-dependent uptake). Furthermore, there was a strong correlation with increased lysis of cytoplasmic membranes (erythrocyte, HEK293T) and efficient endosomal release (Fig. 7).
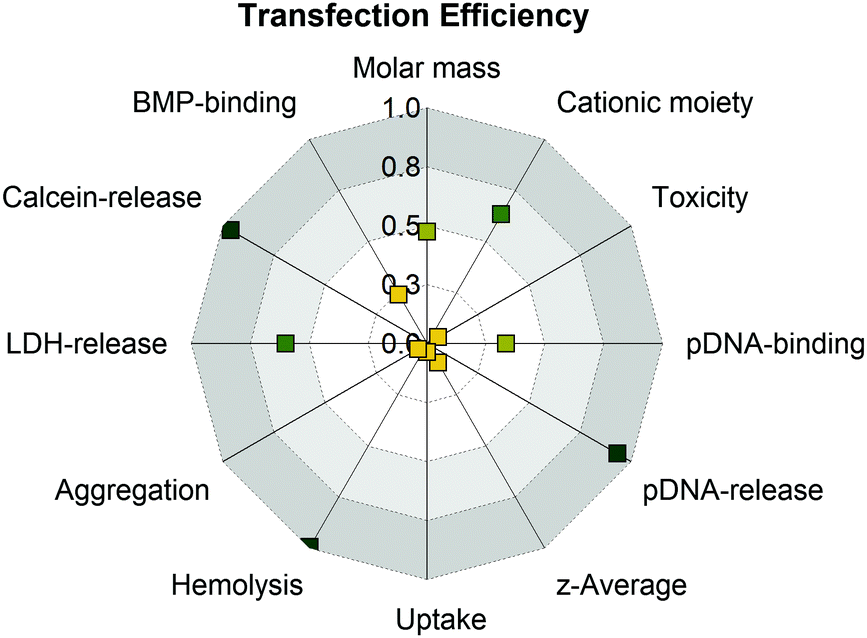 | ||
| Fig. 7 Analysis of structure–property-correlations for PAms using the squared Pearson's correlation coefficient (R2) to determine linearity of the correlation. Values around 1.0 indicate strong positive (green) and values around 0 indicate no correlation (yellow). For a detailed overview which data were used for correlation see Table S8 (ESI†). | ||
Interestingly, PGPAm89 exhibited superior endosomal release properties, although it is not pH responsive. Therefore, we postulate a strong interaction of the polymer with the endolysosomal membrane as a mechanism for endosomal escape. However, the lipid–polymer binding assay investigating the binding of the PAms to BMP revealed that all tested polymers were able to bind BMP, albeit PGPAm was the most efficient. A possible explanation could be the difference to the composition of natural ILEV consisting of more than just one lipid or a more effective mechanism of guanidinium polymers to leave the endolysosome once the polymers escaped the ILEV. Further aspects should also be considered. The pDNA release could be a further crucial step in the delivery process, since the results showed a strong correlation between the amount of heparin needed to achieve an incomplete pDNA release and transfection efficiency (Fig. 7).
Finally, with a transfection efficiency as high as that of LPEI and superior calcein release properties, the guanidinium functional PAm polymers present a promising class of polymers for gene delivery.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
The authors gratefully acknowledge the Bundesministerium für Bildung und Forschung (BMBF, Germany, #13XP5034A PolyBioMik), the German Research Foundation (DFG) for generous funding within the Emmy-Noether Programme (Projekt ID: 358263073), and the support by the DFG-funded Collaborative Research Centre PolyTarget (SFB 1278, project B06, project ID: 316213987). The authors thankfully acknowledge Karina Rost for performing toxicity assays and Prof. U. S. Schubert for providing excellent facilities. Furthermore, the authors acknowledge Bärbel Beringer-Siemers, Carolin Kellner, Elisabeth Preußger and Jana Solomun for their excellent technical assistance and discussions.References
- D. Lechardeur, K. Sohn, M. Haardt, P. Joshi, M. Monck, R. Graham, B. Beatty, J. Squire, H. O’brodovich and G. Lukacs, Gene Ther., 1999, 6, 482 CrossRef CAS PubMed.
- K. Lundstrom, Diseases, 2018, 6, 42 CrossRef PubMed.
- R. Gardlik, R. Palffy, J. Hodosy, J. Lukacs, J. Turna and P. Celec, Med. Sci. Monit., 2005, 11, Ra110–Ra121 CAS.
- B. Kealy, A. Liew, J. M. McMahon, T. Ritter, A. O'Doherty, M. Hoare, U. Greiser, E. E. Vaughan, M. Maenz and C. O'Shea, Tissue Eng., Part C, 2009, 15, 223–231 CrossRef CAS PubMed.
- D. He and E. Wagner, Macromol. Biosci., 2015, 15, 600–612 CrossRef CAS PubMed.
- R. J. Christie, N. Nishiyama and K. Kataoka, Endocrinology, 2010, 151, 466–473 CrossRef CAS PubMed.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef PubMed.
- Z. Zhou, X. Liu, D. Zhu, Y. Wang, Z. Zhang, X. Zhou, N. Qiu, X. Chen and Y. Shen, Adv. Drug Delivery Rev., 2017, 115, 115–154 CrossRef CAS PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS PubMed.
- H. K. de Wolf, J. Luten, C. J. Snel, G. Storm and W. E. Hennink, Mol. Pharmaceutics, 2008, 5, 349–357 CrossRef CAS PubMed.
- M. Neu, D. Fischer and T. Kissel, J. Gene Med., 2005, 7, 992–1009 CrossRef CAS PubMed.
- P. van de Wetering, E. E. Moret, N. M. E. Schuurmans-Nieuwenbroek, M. J. van Steenbergen and W. E. Hennink, Bioconjugate Chem., 1999, 10, 589–597 CrossRef CAS PubMed.
- S. Agarwal, Y. Zhang, S. Maji and A. Greiner, Mater. Today, 2012, 15, 388–393 CrossRef CAS.
- D. B. Rozema, D. L. Lewis, D. H. Wakefield, S. C. Wong, J. J. Klein, P. L. Roesch, S. L. Bertin, T. W. Reppen, Q. Chu, A. V. Blokhin, J. E. Hagstrom and J. A. Wolff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12982–12987 CrossRef CAS PubMed.
- D. H. Wakefield, J. J. Klein, J. A. Wolff and D. B. Rozema, Bioconjugate Chem., 2005, 16, 1204–1208 CrossRef CAS PubMed.
- J. Schrooten, I. Lacík, M. Stach, P. Hesse and M. Buback, Macromol. Chem. Phys., 2013, 214, 2283–2294 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- L. Martin, G. Gody and S. Perrier, Polym. Chem., 2015, 6, 4875–4886 RSC.
- I. Mizoguchi, Y. Ooe, S. Hoshino, M. Shimura, T. Kasahara, S. Kano, T. Ohta, F. Takaku, Y. Nakayama and Y. Ishizaka, Biochem. Biophys. Res. Commun., 2005, 338, 1499–1506 CrossRef CAS PubMed.
- Y. Nakayama, C. Kakei, A. Ishikawa, Y.-M. Zhou, Y. Nemoto and K. Uchida, Bioconjugate Chem., 2007, 18, 2037–2044 CrossRef CAS PubMed.
- Y. Nemoto, A. Borovkov, Y.-M. Zhou, Y. Takewa, E. Tatsumi and Y. Nakayama, Bioconjugate Chem., 2009, 20, 2293–2299 CrossRef CAS PubMed.
- N. Yasuhide, M. Takeshi, N. Makoto, H. Michiko, O. Moto and H.-S. Mariko, Curr. Drug Delivery, 2005, 2, 53–57 CrossRef PubMed.
- J.-H. Ke, M.-F. Wei, M.-J. Shieh and T.-H. Young, J. Biomater. Sci., Polym. Ed., 2011, 22, 1215–1236 CrossRef CAS PubMed.
- J.-H. Ke, M.-F. Wei, M.-J. Shieh and T.-H. Young, J. Biomater. Sci., Polym. Ed., 2011, 22, 1753–1775 CrossRef CAS PubMed.
- J.-H. Ke and T.-H. Young, Biomaterials, 2010, 31, 9366–9372 CrossRef CAS PubMed.
- A. Aied, U. Greiser, A. Pandit and W. Wang, Drug Discovery Today, 2013, 18, 1090–1098 CrossRef CAS PubMed.
- J. Cai, Y. Yue, D. Rui, Y. Zhang, S. Liu and C. Wu, Macromolecules, 2011, 44, 2050–2057 CrossRef CAS.
- A. C. Rinkenauer, S. Schubert, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 7477–7493 RSC.
- M. Wagner, A. C. Rinkenauer, A. Schallon and U. S. Schubert, RSC Adv., 2013, 3, 12774–12785 RSC.
- C. V. Synatschke, A. Schallon, V. Jérôme, R. Freitag and A. H. E. Müller, Biomacromolecules, 2011, 12, 4247–4255 CrossRef CAS PubMed.
- D. Sprouse and T. M. Reineke, Biomacromolecules, 2014, 15, 2616–2628 CrossRef CAS PubMed.
- H. Li, M. A. Cortez, H. R. Phillips, Y. Wu and T. M. Reineke, ACS Macro Lett., 2013, 2, 230–235 CrossRef CAS PubMed.
- A. K. Trutzschler, T. Bus, M. Reifarth, J. C. Brendel, S. Hoeppener, A. Traeger and U. S. Schubert, Bioconjugate Chem., 2018, 29, 2181–2194 CrossRef PubMed.
- C. Zhu, S. Jung, G. Si, R. Cheng, F. Meng, X. Zhu, T. G. Park and Z. Zhong, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2869–2877 CrossRef CAS.
- T. Bus, C. Englert, M. Reifarth, P. Borchers, M. Hartlieb, A. Vollrath, S. Hoeppener, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2017, 5, 1258–1274 RSC.
- C. Arigita, N. J. Zuidam, D. J. A. Crommelin and W. E. Hennink, Pharm. Res., 1999, 16, 1534–1541 CrossRef CAS PubMed.
- A. M. Funhoff, C. F. van Nostrum, M. C. Lok, M. M. Fretz, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 2004, 15, 1212–1220 CrossRef CAS PubMed.
- E. D. Raczyńska, M. K. Cyrański, M. Gutowski, J. Rak, J. F. Gal, P. C. Maria, M. Darowska and K. Duczmal, J. Phys. Org. Chem., 2003, 16, 91–106 CrossRef.
- C. Bechara and S. Sagan, FEBS Lett., 2013, 587, 1693–1702 CrossRef CAS PubMed.
- J. K. Allen, D. J. Brock, H. M. Kondow-McConaghy and J.-P. Pellois, Biomolecules, 2018, 8, 50 CrossRef PubMed.
- F. Madani, S. Lindberg, Ü. Langel, S. Futaki and A. Gräslund, J. Biophys., 2011, 414729 Search PubMed.
- A. Pantos, I. Tsogas and C. M. Paleos, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 811–823 CrossRef CAS PubMed.
- M. Silhol, M. Tyagi, M. Giacca, B. Lebleu and E. Vivès, Eur. J. Biochem., 2002, 269, 494–501 CrossRef CAS PubMed.
- S. Futaki and I. Nakase, Acc. Chem. Res., 2017, 50, 2449–2456 CrossRef CAS PubMed.
- J. D. Ramsey and N. H. Flynn, Pharmacol. Ther., 2015, 154, 78–86 CrossRef CAS PubMed.
- A. Erazo-Oliveras, K. Najjar, D. Truong, T. Y. Wang, D. J. Brock, A. R. Prater and J. P. Pellois, Cell Chem. Biol., 2016, 23, 598–607 CrossRef CAS PubMed.
- S.-T. Yang, E. Zaitseva, L. V. Chernomordik and K. Melikov, Biophys. J., 2010, 99, 2525–2533 CrossRef CAS PubMed.
- Y. Kim, S. Binauld and M. H. Stenzel, Biomacromolecules, 2012, 13, 3418–3426 CrossRef CAS PubMed.
- N. Schmidt, A. Mishra, G. H. Lai and G. C. Wong, FEBS Lett., 2010, 584, 1806–1813 CrossRef CAS PubMed.
- M. Ahmed, Biomater. Sci., 2017, 5, 2188–2211 RSC.
- K. J. Abd Karim, R. H. Utama, H. Lu and M. H. Stenzel, Polym. Chem., 2014, 5, 6600–6610 RSC.
- C. Cokca, L. Zartner, I. Tabujew, D. Fischer and K. Peneva, Macromol. Rapid Commun., 2020, 41, 1900668 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed.
- L. Martin, R. Peltier, A. Kuroki, J. S. Town and S. Perrier, Biomacromolecules, 2018, 19, 3190–3200 CrossRef PubMed.
- L. Nová, F. Uhlík and P. Košovan, Phys. Chem. Chem. Phys., 2017, 19, 14376–14387 RSC.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed.
- M. Ullner, B. Jönsson, B. Söderberg and C. Peterson, J. Chem. Phys., 1996, 104, 3048–3057 CrossRef CAS.
- D. M. Drake, R. K. Keswani and D. W. Pack, Pharm. Res., 2010, 27, 2457–2465 CrossRef CAS PubMed.
- J. M. Layman, S. M. Ramirez, M. D. Green and T. E. Long, Biomacromolecules, 2009, 10, 1244–1252 CrossRef CAS PubMed.
- A. C. Rinkenauer, A. Vollrath, A. Schallon, L. Tauhardt, K. Kempe, S. Schubert, D. Fischer and U. S. Schubert, ACS Comb. Sci., 2013, 15, 475–482 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 1999, 45, 268–275 CrossRef CAS PubMed.
- R. N. Johnson, D. S. H. Chu, J. Shi, J. G. Schellinger, P. M. Carlson and S. H. Pun, J. Controlled Release, 2011, 155, 303–311 CrossRef CAS PubMed.
- B. D. Monnery, M. Wright, R. Cavill, R. Hoogenboom, S. Shaunak, J. H. G. Steinke and M. Thanou, Int. J. Pharm., 2017, 521, 249–258 CrossRef CAS PubMed.
- C. J. Bishop, K. L. Kozielski and J. J. Green, J. Controlled Release, 2015, 219, 488–499 CrossRef CAS PubMed.
- A. C. Rinkenauer, L. Tauhardt, F. Wendler, K. Kempe, M. Gottschaldt, A. Traeger and U. S. Schubert, Macromol. Biosci., 2015, 15, 414–425 CrossRef CAS PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and U. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, Cell. Mol. Life Sci., 2005, 62, 1839–1849 CrossRef CAS PubMed.
- Z. Tan, Y. K. Dhande and T. M. Reineke, Bioconjugate Chem., 2017, 28, 2985–2997 CrossRef CAS PubMed.
- J.-B. Kim, J. S. Choi, K. Nam, M. Lee, J.-S. Park and J.-K. Lee, J. Controlled Release, 2006, 114, 110–117 CrossRef CAS PubMed.
- A. Kuroki, A. Kengmo Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock, M. Unnikrishnan and S. Perrier, Biomaterials, 2019, 217, 119249 CrossRef CAS PubMed.
- N. A. Alhakamy and C. J. Berkland, Mol. Pharmaceutics, 2013, 10, 1940–1948 CrossRef CAS PubMed.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- A. M. Funhoff, C. F. van Nostrum, G. A. Koning, N. M. E. Schuurmans-Nieuwenbroek, D. J. A. Crommelin and W. E. Hennink, Biomacromolecules, 2004, 5, 32–39 CrossRef CAS PubMed.
- D. M. Perreault, L. A. Cabell and E. V. Anslyn, Bioorg. Med. Chem., 1997, 5, 1209–1220 CrossRef CAS PubMed.
- P. Juhasz and K. Biemann, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4333–4337 CrossRef CAS PubMed.
- K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67–114 CrossRef CAS PubMed.
- Z. Liu, Z. Zhang, C. Zhou and Y. Jiao, Prog. Polym. Sci., 2010, 35, 1144–1162 CrossRef CAS.
- A. Alshamsan, A. Haddadi, V. Incani, J. Samuel, A. Lavasanifar and H. Uludag, Mol. Pharming, 2009, 6, 121–133 CrossRef CAS PubMed.
- S. Prabha, G. Arya, R. Chandra, B. Ahmed and S. Nimesh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 83–91 CrossRef CAS PubMed.
- I. Tabujew, C. Cokca, L. Zartner, U. S. Schubert, I. Nischang, D. Fischer and K. Peneva, J. Mater. Chem. B, 2019, 7, 5920–5929 RSC.
- E. Mastrobattista and W. E. Hennink, Nat. Mater., 2012, 11, 10–12 CrossRef CAS PubMed.
- T. Lühmann, M. Rimann, A. G. Bittermann and H. Hall, Bioconjugate Chem., 2008, 19, 1907–1916 CrossRef PubMed.
- E. Wexselblatt, J. D. Esko and Y. Tor, J. Org. Chem., 2014, 79, 6766–6774 CrossRef CAS PubMed.
- A. K. Trutzschler, T. Bus, M. Sahn, A. Traeger, C. Weber and U. S. Schubert, Biomacromolecules, 2018, 19, 2759–2771 CrossRef PubMed.
- S. Pan, D. Cao, R. Fang, W. Yi, H. Huang, S. Tian and M. Feng, J. Mater. Chem. B, 2013, 1, 5114–5127 RSC.
- R. Kircheis, S. Schüller, S. Brunner, M. Ogris, K. H. Heider, W. Zauner and E. Wagner, J. Gene Med., 1999, 1, 111–120 CrossRef CAS PubMed.
- M. Ogris, P. Steinlein, M. Kursa, K. Mechtler, R. Kircheis and E. Wagner, Gene Ther., 1998, 5, 1425–1433 CrossRef CAS PubMed.
- I. Kopatz, J.-S. Remy and J.-P. Behr, J. Gene Med., 2004, 6, 769–776 CrossRef CAS PubMed.
- K. A. Mislick and J. D. Baldeschwieler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12349–12354 CrossRef CAS PubMed.
- P. C. Billings and M. Pacifici, Connect. Tissue Res., 2015, 56, 272–280 CrossRef CAS PubMed.
- G. W. Doorley and C. K. Payne, Chem. Commun., 2011, 47, 466–468 RSC.
- E. Moreau, M. Domurado, P. Chapon, M. Vert and D. Domurado, J. Drug Targeting, 2002, 10, 161–173 CrossRef CAS PubMed.
- A. A. Spector and M. A. Yorek, J. Lipid Res., 1985, 26, 1015–1035 CAS.
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef CAS PubMed.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS PubMed.
- Y. Hu, T. Litwin, A. R. Nagaraja, B. Kwong, J. Katz, N. Watson and D. J. Irvine, Nano Lett., 2007, 7, 3056–3064 CrossRef CAS PubMed.
- R. A. Jones, M. H. Poniris and M. R. Wilson, J. Controlled Release, 2004, 96, 379–391 CrossRef CAS PubMed.
- T. Kobayashi, M.-H. Beuchat, J. Chevallier, A. Makino, N. Mayran, J.-M. Escola, C. Lebrand, P. Cosson, T. Kobayashi and J. Gruenberg, J. Biol. Chem., 2002, 277, 32157–32164 CrossRef CAS PubMed.
- W. Mobius, E. van Donselaar, Y. Ohno-Iwashita, Y. Shimada, H. F. Heijnen, J. W. Slot and H. J. Geuze, Traffic, 2003, 4, 222–231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Material, additional methods and results, Fig. S1–S18, supplementary Tables S1–S8. See DOI: 10.1039/d0tb00340a |
| This journal is © The Royal Society of Chemistry 2020 |