
Glycopolymer decorated multiwalled carbon nanotubes for dual targeted breast cancer therapy†
Pinar Sinem
Omurtag Ozgen
 *a,
Sezen
Atasoy
b,
Belma
Zengin Kurt
c,
Zehra
Durmus
d,
Gulsah
Yigit
e and
Aydan
Dag
*cf
*a,
Sezen
Atasoy
b,
Belma
Zengin Kurt
c,
Zehra
Durmus
d,
Gulsah
Yigit
e and
Aydan
Dag
*cf
aDepartment of Analytical Chemistry, Faculty of Pharmacy, Istanbul Medipol University, 34815, Istanbul, Turkey. E-mail: psozgen@medipol.edu.tr
bDepartment of Biochemistry, Faculty of Pharmacy, Bezmialem Vakif University, 34093, Istanbul, Turkey
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Bezmialem Vakif University, 34093, Istanbul, Turkey. E-mail: adag@bezmialem.edu.tr
dBaglar Mah., Gunesli Konutlar, No: 38, D-24, 34212, Istanbul, Turkey
eDepartment of Biotechnology, Institute of Health Sciences, Bezmialem Vakif University, 34093, Istanbul, Turkey
fDrug Application and Research Center, Bezmialem Vakif University, 34093, Istanbul, Turkey
First published on 6th March 2020
Abstract
Carbon-based nanomaterials (CNMs) have attracted great attention in biomedical applications such as cancer imaging and therapy. CNMs, which are currently used in a wide range of applications, suffer from drawbacks of toxicity and low biocompatibility. Either noncovalent or covalent functionalization of CNMs with hydrophilic and biocompatible polymers which help to block hydrophobic interactivity between CNMs and cells can greatly increase their biocompatibility by eliminating their probable toxicity towards living organisms. In this report, we present a comparison of both noncovalent and covalent functionalization approaches in order to introduce a biocompatible glycoblock copolymer onto multi-walled carbon nanotubes (CNTs) in order to enhance their potential in therapies. An anticancer drug (doxorubicin, Dox) was conjugated with two different end functionalized poly(1-O-methacryloyl-β-D-fructopyranose-b-(2-methacryloxyethoxy))benzaldehyde glycoblock copolymers, which were synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization, by either noncovalent or covalent tethering. CNTs were coated separately with the synthesized drug-conjugated glycoblock copolymers and folic acid (FA) to obtain an efficient drug delivery platform for dual-targeting of glucose transporter protein (GLUT5) and folic acid receptors (FR) in breast cancer. A library of synthesized monomers, polymers and prepared glycoblock copolymer coated CNTs (hybrid-CNTs) using both approaches were comprehensively characterized by various techniques. Transmission electron microscopy measurements showed the homogeneous, smooth morphology of the prepared Dox-conjugated glycoblock copolymer coating of CNTs and confocal laser scanning microscopy images displayed successful cellular internalization of hybrid-CNTs in the MCF-7 and MDA-MB-231 human breast cancer cell lines. This research demonstrates the potential of hybrid-CNTs as a biocompatible drug delivery system as well as in vitro use of Dox-conjugated vehicles for dual receptor mediated breast cancer therapy.
Introduction
Mortality as a result of cancer is growing enormously worldwide. According to the latest report of the International Agency for Research on Cancer, breast cancer is the most frequently diagnosed cancer type among women; about 3.06 million diagnosed female breast cancer cases across all age groups will be expected in 2040.1–3Chemotherapy is one of the most common treatment strategies for breast cancer. Among several Food and Drug Administration (FDA) approved drugs, clinical studies show that doxorubicin (Dox) is a useful antineoplastic drug for breast cancer therapy. The working principle is that Dox kills cancer cells by DNA chelation, inhibition of topoisomerase II and hydroxyl radicals.4–6 Despite evidence of its therapeutic response, the chemotherapeutic drug can only be effective once it has accumulated in the right place with an appropriate therapeutic concentration. However, it is certainly difficult for the free chemotherapeutic drug to reach and locate specifically the tumor site by itself, and this causes serious side effects and ineffective chemotherapy.
Targeted drug delivery systems based on polymeric nano-carriers can overcome these challenges due to their capability to perform efficient delivery and sustained release of therapeutics to specific target areas. Several innovative nano-carrier systems or drug conjugates, including liposomes, micelles, capsules, dendrimers, and polymeric nanoparticles, have been identified as useful for their applications as targeted drug delivery systems.7,8 Such systems can be used together with carbon-based nanomaterials (CNMs), thanks to their unique high surface area to volume ratio for combination with molecules during drug delivery applications.9–12
Beyond the application of drug delivery systems, the usage of CNMs has been enlarged to a versatile platform of multiple therapeutic modalities with enhanced anticancer activity and reduced side effects. However, one of the significant drawbacks of CNMs is the highly hydrophobic nature of their carbon structure, which limits their biocompatibility, aqueous dispersibility and stability when delivered into the biological environment. Therefore, the application of novel biocompatible, biodegradable and multifunctional polymers together with CNMs enables the facile and controlled incorporation of a wide range of therapeutics.13–15
Among CNMs, carbon nanotubes (CNTs) can provide specific interaction with cancer cells by simply tuning the backbone of polymeric chains which are physically or chemically tethered on their surfaces. There are several reported synthetic procedures for the noncovalent and covalent functionalization of CNTs with small/macro molecules, in order to enhance the dispersibility and make them feasible for both therapy and early detection of cancer cells/tumors.16–22
Once their surface is hybridized, CNTs are found to be optimal nanocarriers and intermediaries for multimodal cancer therapies. Moreover, even simple surface functionalization can increase the therapeutic efficacy of the current treatment modalities.23,24 Also, the literature reported that the needle-like shape of CNTs provides an extra advantage in their uptake into cells.25 However, it is still a challenge to prepare a biocompatible hybrid system with a prolonged blood circulation time, enhanced tumor accumulation of anticancer drugs and effective anticancer efficacy.
Therefore, increased attention has been paid to creating drug-conjugated hybrid-CNTs by increasing the solubility/dispersibility of both drug molecules and CNTs in order to facilitate their smart response stimulation by specified actions, including the redox process, hypothermia, light and the pH level.14,26 Among biologically significant functionalities for the aqueous dispersion of CNTs, sugars in different configurations have proven to be particularly valuable, with the resulting sugar-functionalized carbon nanotubes exhibiting unusual features in their interactions with biological species.27–29
In essence, sugar-functionalized CNTs can be selectively internalized into cancer cells via glucose transporter (GLUT) receptor-mediated pathways, without them being internalized into normal cells. Among all of the isoforms of GLUTs, GLUT5 is a membrane protein that transports fructose into the cytoplasm and is primarily responsible for fructose absorption/metabolism.30,31 Moreover, many fructose-based glycopolymers are employed in bio-recognition systems. Zhao et al. prepared fructosylated diblock glycopolymers and fructosylated micelles, which could be selectively recognized by overexpressed GLUT5 receptors on the surfaces of breast cancer cells.32 Recently, a fructosylated polymer was grafted onto a nanodiamond via an amide bond followed by physical adsorption of Dox. According to 3D spheroid model studies, this hybrid system showed increased anticancer activity compared to free Dox after 8 days of incubation.33 As a consequence of the GLUT5 targeting ability of fructose moieties on the surface of CNTs, carbon nanomaterials can act as drug delivery vehicles with targeting functions that may enhance recognition, internalization and ligand transfer through a series of intracellular compartments towards breast cancer tissue.
Besides sugar molecules, folic acid (FA) is also a promising molecule to actively target the FA receptors overexpressing on the surface of breast cancer cells. A large number of studies have demonstrated that folic acid receptors (FR) are overexpressed in a variety of solid epithelial tumors, including breast cancer.34,35 Additionally, the heterogeneous expression of the FR in cancer tissues and its absence in non-neoplastic tissues makes FR a good target.36,37 Previously, it was shown that FA grafted CNTs have an increased cellular uptake through FR-mediated endocytosis, a highly regulated process of binding and internalization.38 In another study, Mehra et al. have show the in vitro and in vivo cancer targeting efficiency of a dual targeted delivery system for Dox that employs FA and estrone functionalized PEGylated multiwalled carbon nanotubes (MWCNTs).39 These studies outline the feasibility of folate decorated CNTs as an advancing area in breast cancer therapy.40,41
We hypothesized that the synergistic advantages of either noncovalent or covalent functionalized CNTs with biocompatible glycoblock copolymers that promote effective stimuli drug release could address the challenge of breast cancer cell recognition while simultaneously allowing dual-targeting breast cancer therapy.
Herein, we have introduced a novel combination of Dox-conjugated glycoblock copolymers with CNTs via one-step noncovalent or covalent functionalization strategies in order to design an efficient route for the nano-carrier system through a targeted drug delivery approach. Thus, the carboxylic acid-modified CNT surface was functionalized with poly(1-O-methacryloyl-β-D-fructopyranose-b-(2-methacryloxyethoxy)benzaldehyde)-Py/Dox via π–π stacking interactions or the alkynyl group decorated CNT surface with poly(1-O-methacryloyl-β-D-fructopyranose-b-(2-methacryloxyethoxy)benzaldehyde)-N3/Dox via a copper catalyzed azide alkyne click reaction (CuAAC) (Scheme 1). The goal was to improve the water solubility and biocompatibility as well as interactions between glycoblock copolymer decorated CNTs (hybrid-CNTs) and cancer cells. The hybrid-CNTs prepared by utilizing two different strategies were further coated individually with FA to investigate the possibility of using these systems in dual receptor mediated breast cancer therapy (Scheme 2). The hybrid-CNTs with tethered fructose and FA moieties presumably deliver anticancer drugs more efficiently into the cancer cell lines (MCF-7 and MDA-MB-231) after recognizing and binding selectively to their corresponding receptors. To the best of our knowledge, the functionalization of the CNT surface with an anticancer drug-carrying fructosylated glycoblock copolymer with the comparison of two different approaches has never been reported.
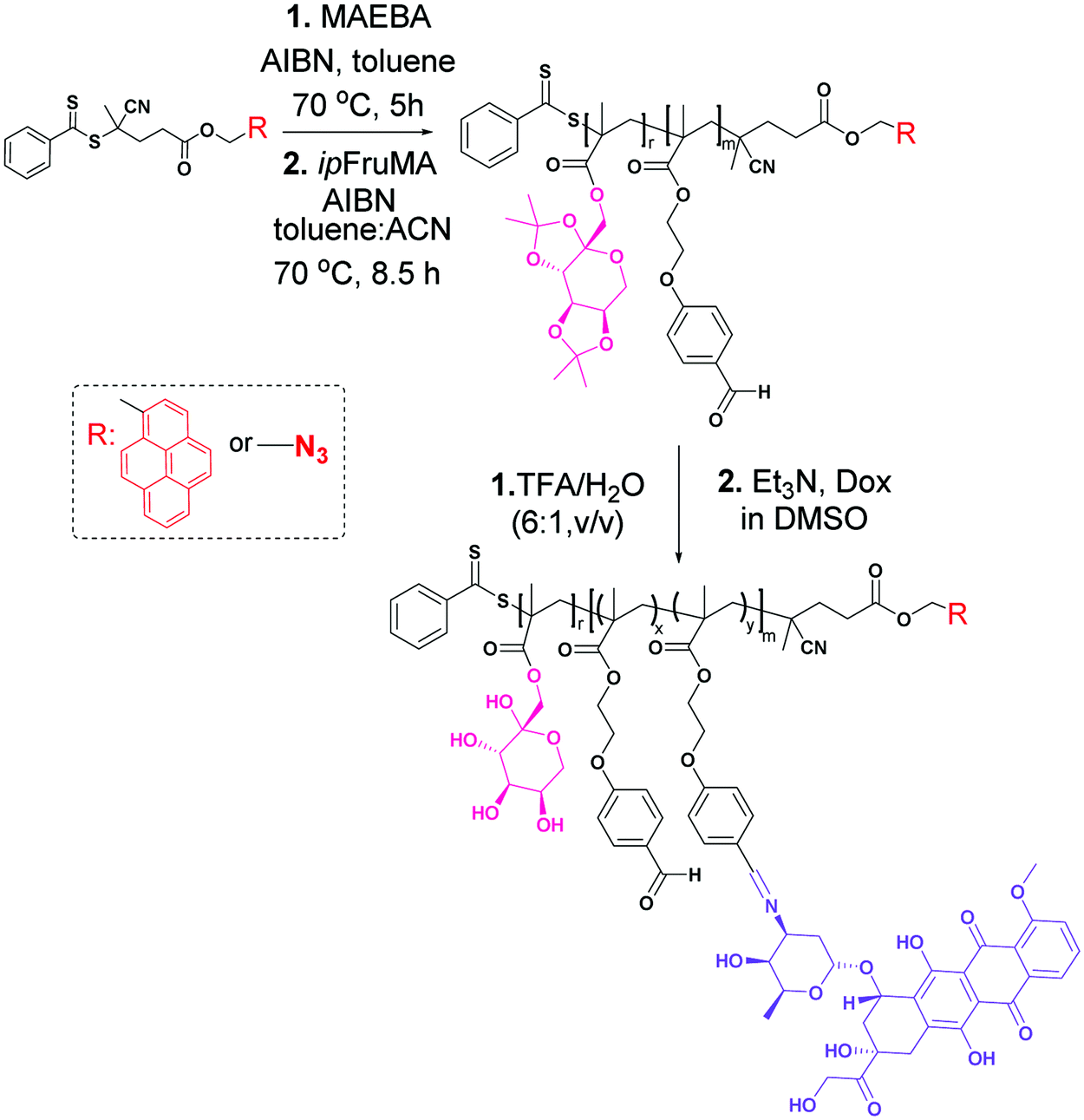 | ||
| Scheme 1 Schematic pathway for the synthesis of the Dox-conjugated glycoblock copolymers. | ||
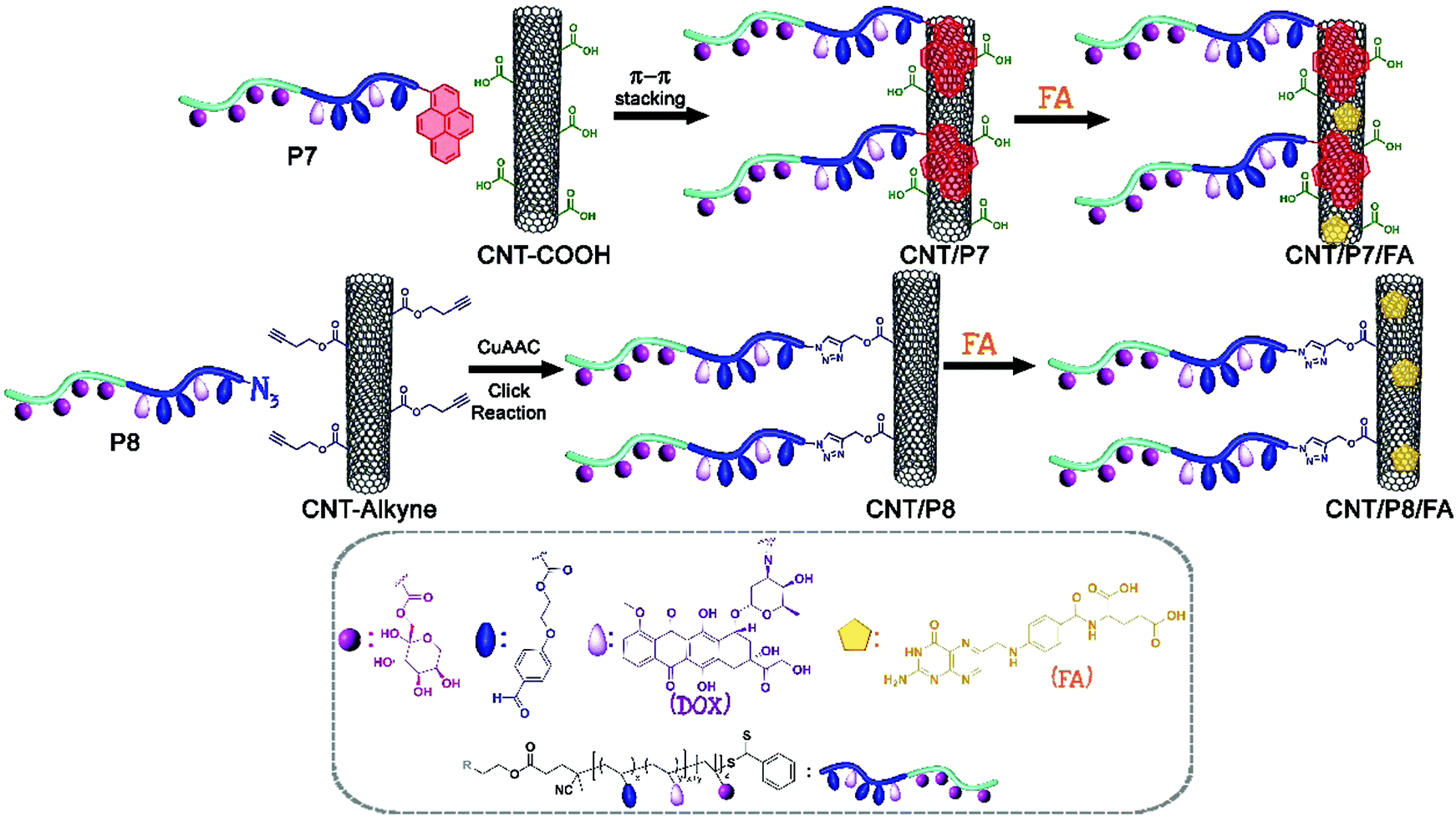 | ||
| Scheme 2 Schematic pathway for the modification of CNTs via π–π stacking and CuAAC click reaction strategies. | ||
Experimental section
Pristine MWCNTs (p-MWCNTs) were purchased from Sigma-Aldrich (Product Code: 724769). The surface of the pristine CNTs was oxidized with a mixture of H2SO4 and HNO3 to create carboxyl groups, which serve as chemical interaction points to attach different structures like propargyl alcohol to obtain alkyne functionalized carbon nanotubes, which are a precursor for the CuAAC click reaction.42–45 Unless otherwise specified, all other reagents were purchased from commercial suppliers and used as received. 4-Cyanopentanoic acid dithiobenzoate (CPADB),46p-(2-hydroxyethoxy) benzaldehyde (PEHBA),47p-(2-methacryloxyethoxy) benzaldehyde (MAEBA),47 2-azidoethanol48 and 1-O-methacryloyl-2,3:4,5-di-O-isopropylidene-β-D-fructopyranose (ipFruMA)32 were synthesized according to the reported literature and their characterization details have been given in the ESI† (Fig. S1, S2, S6 and S10–S17).Synthesis of the pyren-1-ylmethyl 4-cyano-4-((phenylcarbonothioyl)thio) pentanoate RAFT agent (CPADB-Py)
1-Pyrenemethanol (0.99 g, 4.29 mmol), CPADB (1.00 g, 3.57 mmol), and DMAP (0.43 g, 3.57 mmol) were dissolved in dry THF (20 mL). Then DCC (0.96 g, 4.65 mmol) was added and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was filtered to remove the white precipitate. The filtrate was concentrated enough to afford a crude red oil and the crude was further purified by silica column chromatography using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 EtOAc:hexane (v/v) to wash out the first two side products and 1:1 EtOAc:hexane (v/v) to wash out the target compound (yield: 1.28 g, 70%, Fig. S3–S5, ESI†).
:9 EtOAc:hexane (v/v) to wash out the first two side products and 1:1 EtOAc:hexane (v/v) to wash out the target compound (yield: 1.28 g, 70%, Fig. S3–S5, ESI†).
Synthesis of the 2-azidoethyl 4-cyano-4-((phenylcarbonothioyl)thio) pentanoate RAFT agent (CPADB-N3)
2-Azidoethanol (0.37 g, 4.29 mmol), CPADB (1.00 g, 3.57 mmol), and DMAP (0.43 g, 3.57 mmol) were dissolved in dry THF (20 mL). Then DCC (0.96 g, 4.65 mmol) was added and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was filtered to remove the white precipitate. The filtrate was concentrated enough to afford a crude red oil and the crude was further purified by silica column chromatography using 1:9 EtOAc:hexane (v/v) to wash out the first two side products and 1:1 EtOAc:hexane (v/v) to wash out the target compound (yield: 0.19 g, 41%, Fig. S7–S9, ESI†).
Synthesis of the pyrene-functionalized P((2-methacryloxyethoxy) benzaldehyde) homopolymer as a macro-RAFT agent (P(MAEBA)-Py, P1)
To a septa sealed round-bottom flask MAEBA (1.20 g, 5.12 mmol), azobisisobutyronitrile (AIBN) (2.62 mg, 16.0 × 10−3 mmol) and CPADB-Py (63.16 mg, 0.12 mmol) were added and dissolved in 5.12 mL toluene/acetonitrile mixture (5:1 v/v). Polymerization subsequently took place in a thermostatic silicone oil bath at 70 °C. The ratio between the monomer, reversible addition–fragmentation chain-transfer (RAFT) agent and AIBN ([M]0:[CTA]0:[I]0) in the polymerization medium was 40:1.0:0.125. The polymerization reaction was quenched after 5 h via rapid cooling and exposure to oxygen. Then, the mixture was precipitated in a 10-fold excess of hexane two times to remove the unreacted monomer and RAFT agent. Finally, the purified polymer was dried under a vacuum for 24 h (yield: 0.432 g; Mn,theo = 3.30 kDa, Mn,NMR = 4.20 kDa, Mn,GPC = 2.95 kDa, Đ = 1.13, relative to PS standards).
Synthesis of the azido-functionalized P((2-methacryloxyethoxy) benzaldehyde) homopolymer as a macro-RAFT agent (P(MAEBA)-N3, P2)
To a septa sealed round-bottom flask, MAEBA (2.0 g, 8.53 mmol), AIBN (4.30 mg, 26.28 × 10−3 mmol) and CPADB-N3 (74.36 mg, 0.21 mmol) were added and dissolved in 8.53 mL toluene/acetonitrile mixture (5:1 v/v). Polymerization subsequently took place in a thermostatic silicone oil bath at 70 °C. The ratio between the monomer, RAFT agent and AIBN ([M]0:[CTA]0:[I]0) in the polymerization medium was 40:1.0:0.125. The polymerization reaction was quenched after 5 h via rapid cooling and exposure to oxygen. Then, the mixture was precipitated in a 10-fold excess of methanol two times to remove the unreacted monomer and RAFT agent. Finally, the purified polymer was dried under a vacuum for 24 h (yield: 0.76 g; Mn,theo = 4.05 kDa, Mn,NMR = 4.80 kDa, Mn,GPC = 3.50 kDa, Đ = 1.06, relative to PS standards).
General procedure for preparation of the glycoblock copolymers via RAFT polymerization
In a general procedure, P1 was employed as a macro-RAFT agent for obtaining the glycoblock copolymer. Firstly, P1 (87.5 mg, 0.0206 mmol, Mn,NMR = 4.20 kDa), ipFruMA (1.689 g, 5.147 mmol), and AIBN (0.591 mg, 3.641 × 10−3 mmol) were added to a septa sealed round-bottom flask and dissolved in 4.20 mL toluene/acetonitrile mixture (2.5:1 v/v). The ratio between the monomer, macro-RAFT agent and AIBN ([M]0:[macro-RAFT]0:[I]0) in the polymerization medium was 250:1.0:0.175. The mixture was flushed with a nitrogen flow for an hour and allowed to stir in a silicone oil bath at 70 °C for 8.5 h. The glycoblock copolymer was purified by precipitation twice in methanol to remove the unreacted monomers and RAFT agent. Then it was dried under a vacuum overnight at 25 °C to give 0.928 g of a pale pink solid. The conversion of P(ipFruMA-b-MAEBA)-Py (P3) was 60.8% and the yield of the polymer was calculated as 55%. The same procedure was employed to synthesize P(ipFruMA-b-MAEBA)-N3 (P2). P2 (306 mg, 0.064 mmol, Mn,NMR = 4.80 kDa) was used as a macro-RAFT agent. Following the addition of the macro-RAFT agent to a septa sealed round-bottom flask, ipFruMA (5.23 g, 15.937 mmol) and AIBN (1.832 mg, 0.0112 mmol) were also added and dissolved in 12.18 mL toluene/acetonitrile mixture (2.5:1 v/v). The obtained polymer was 0.334 g of a pale pink solid. On the basis of the P(ipFruMA-b-MAEBA)-N3 (P4) conversion of 93.6%, the yield of the polymer was calculated as 85%. Both of the purified glycoblock copolymers were characterized by nuclear magnetic resonance (1H-NMR) and gel permeation chromatography (GPC) measurements (details are given in the ESI,† Fig. S19 and S23).
Deprotection of the glycoblock copolymers
The deprotection of the glycoblock copolymers was carried out as described by Dag et al.49 After the reaction, the polymer solution was dialyzed against Milli-Q water for 2 days (MWCO 3500) and then the deprotected polymers were freeze-dried. The resulting polymers (P5 and P6) were characterized by following the disappearance of characteristic isopropylidene signals by 1H-NMR (δ 1.2–1.6 ppm, Fig. S20 and S24, ESI†). The characteristic signals corresponding to the MAEBA block still exist, indicating that the aldehyde groups remain intact under this hydrolysis condition.Preparation of the glycoblock copolymer–Dox conjugates
The primary amine of doxorubicin (Dox, 8 equiv.) was covalently linked to the aldehyde group of the glycoblock copolymers (1 equiv.) in the presence of triethylamine (Et3N) in dimethyl sulfoxide (DMSO).50 The obtained Dox-conjugated glycoblock copolymers P(FruMA-b-MAEBA)-Py/Dox and P(FruMA-b-MAEBA)-N3/Dox are named P7 and P8, respectively.Protocol for π–π stacking between CNT-COOH and P(FruMA-b-MAEBA)-Py/Dox: (CNT/P7)
Noncovalent attachment between CNT-COOH and P(FruMA-b-MAEBA)-Py/Dox (P7) was carried out according to previous literature reports with slight modifications.30 Briefly, 70 mg of CNT-COOH was dispersed in 210 mL of dimethylformamide (DMF):ultrapure (UP) water (1:2, v/v) in an ultrasonic bath. After 10 minutes of dispersion, 350 mg Dox-conjugated pyrene end-functionalized glycoblock copolymer (P7) was added, and the mixture was stirred at ambient temperature for 96 h. The dispersion was filtered through a filter (PTFE membrane filter; pore size: 0.2 μm), to remove unloaded copolymer. The residual solid on the filter was sonicated using an excess amount of fresh ethanol and the solid filtered again until the unloaded copolymer could not be detected in the solvent (monitored by UV-vis). The recovered black powder was dried under a vacuum.
Protocol for the CuAAC click reaction between CNT-alkyne and P(FruMA-b-MAEBA)-N3/Dox: (CNT/P8)
70 mg of CNT-alkyne was dispersed in 210 mL of DMF:UP water (1:2, v/v) in an ultrasonic bath. After 10 minutes of dispersion, the Dox-conjugated azido end-functionalized glycoblock copolymer (P8, 350 mg), sodium ascorbate (10 mg), and CuSO4·5H2O (50 mg) were added, and the mixture was stirred at ambient temperature for 48 h after degassing with 3 freeze–pump–thaw cycles. The dispersion was filtered through a filter (PTFE membrane filter; pore size: 0.2 μm) to remove unreacted copolymer. The residual solid on the filter was sonicated using an excess amount of fresh ethanol and the solid filtered again until the unreacted copolymer could not be detected in the solvent (monitored by UV-vis). The recovered black powder was dried under a vacuum.
General protocol for FA loading of CNT/P7 and CNT/P8
20 mg of previously modified CNTs (CNT/P7, CNT/P8) were dispersed in 60 mL of DMF in an ultrasonic bath. After 10 minutes of dispersion, FA (100 mg) was added, and the mixture was stirred at ambient temperature for 48 h. The dispersion was filtered through a filter (PTFE membrane filter; pore size: 0.2 μm), to remove unloaded FA.51 The residual solid on the filter was sonicated using an excess amount of fresh ethanol and the solid filtered again until the unloaded FA could not be detected in the solvent (monitored by UV-vis). The recovered black powder was dried under a vacuum. The obtained FA-loaded carbon-based nanomaterials are named CNT/P7/FA and CNT/P8/FA.In vitro cell culture
Human breast cancer cell lines MCF-7 and MDA-MB-231 were obtained from the American Tissue Culture Collection. The cancer cell lines were grown in Dulbecco's modified Eagle's medium: nutrient mixture F-12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and 100 U mL−1 of penicillin–streptomycin at 37 °C in a humidified incubator with 5% CO2. After reaching 80% confluency, the cells were detached using 0.25% trypsin–ethylene diamine tetra acetic acid (EDTA). For further experiments, the cells were resuspended in the growth medium after collection and centrifugation.Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess cell viability as previously described.52 Briefly, 5 × 103 cells were seeded in flat-bottomed 96-well plates with growth medium. After 24 h incubation, treatment was performed with ten different decreasing doses of Dox, FA, P7, P8, CNT-COOH, CNT-alkyne, CNT/P7, CNT/P7/FA, CNT/P8, and CNT/P8/FA for 24 h and MTT assay was conducted. The absorbance value was read at 540 nm using a microplate reader. The relative %cell viability was calculated from the absorbance values according to following equation: relative percent cell viability = (test/control) × 100%. All experiments were carried out in triplicate; results are presented as means ± standard deviation (SD).Cellular internalization
The cellular uptake behavior of the hybrid-CNTs was measured using confocal laser scanning microscopy (Leica DMi8/SP8 TCS, CLSM) and flow cytometry. Cells were seeded at a density of 2 × 105 cells per dish for microscopy, and in 6-well plates at a density of 2 × 105 cells per well for flow cytometry. Following 24 h incubation with a working concentration of Dox, P7, CNT/P7, CNT/P7/FA, CNT/P8, and CNT/P8/FA, the cells were washed with phosphate buffered saline (PBS) and the cellular uptake was observed using CLSM. 4′,6-Diamidino-2-phenylindole (DAPI) staining was used for nuclei visualization. The determination of the total fluorescence intensity was described in a previous study.52 Briefly, fifteen images of internalized hybrid-CNTs were analysed using ImageJ software. The corrected total cell fluorescence (CTCF) was obtained by subtracting the background from the integrated density (IntDen). For flow cytometry analysis, MCF-7 and MDA-MB-231 cells were treated with Dox, P7, CNT/P7 and CNT/P7/FA. After 24 h incubation time, the medium was removed and the cells were washed with 0.1 M PBS solution to remove residual sample and resuspended into PBS. Then, the cells were analyzed with flow cytometry (BD Accuri C6, BD Biosciences, San Jose, USA). 50000 events comprising at least 3 separate experiments were acquired.
Competition assay with free fructose and FA
Further cellular mechanism of GLUT5 and the FR-based uptake of CNTs were estimated by flow cytometric measurements after stimulating the expression of GLUT5 and FR as described by Wang et al.53 Briefly, MCF-7 and MDA-MB-231 cells were incubated with medium, seeded into six-well culture plates at a density of 5 × 105 cells per well and incubated for a day before fructose and FA competition assay. After 24 h, the cells were washed with PBS and treated with medium containing 2.0 mM free fructose and/or 2.0 mM free FA. Also, Dox, CNT/P7, and CNT/P7/FA treatments were done for related wells. For the control, MCF-7 and MDA-MB-231 cells were cultured with medium. After 24 h incubation, the cells were collected using Trypsin/EDTA, washed with PBS and stained using Hoechst 33342 for 3 min. Competition assay was performed using the protocol as described for the flow cytometry analysis in the above session.54,55Apoptosis and cell cycle assay
The quantification of live, early, late apoptotic, and dead cells and also G0/G1, S, and G2/M phases of the cell cycle was performed by using a Muse™ Cell Analyzer (Merck-Millipore, Darmstadt, Germany). In general, both MCF-7 and MDA-MB-231 cells were treated with free-Dox, CNT/P7 and CNT/P7/FA containing equal amounts of Dox (fixed at 2.76 μM Dox). Untreated and free-Dox treated cells were used as the control. The effects of the samples were quantified and analyzed by the flow cytometry method using the Muse™ Annexin V and Dead Cell and Muse™ Cell Cycle Assay Kit as per the manufacturer's instructions.52,56 10000 events were analyzed for each condition. All cellular data were obtained in triplicate and the presented statistical data were analysed using Graphpad Prism v8.00 (GraphPad Software, La Jolla California USA).
Results and discussion
Preparation of glycoblock copolymer–dox conjugates
Two glycoblock copolymers were prepared via a two-step procedure. In the first step, two different pyrene and azido functionalized RAFT agents (pyren-1-ylmethyl 4-cyano-4-((phenylcarbonothioyl)thio)pentanoate (CPADB-Py) and 2-azidoethyl 4-cyano-4-((phenylcarbonothioyl)thio)pentanoate (CPADB-N3)) were employed separately to homopolymerize the p-(2-methacryloxyethoxy) benzaldehyde (MAEBA) monomer. These RAFT polymerization reactions were performed in toluene/acetonitrile (5:1 v/v) as a solvent at 70 °C for 5 h in the presence of the monomer, with a RAFT agent and AIBN ratio of 40:1.0:0.125. Then, the pyrene functionalized P(MAEBA)-Py homopolymer (P1) and azido functionalized P(MAEBA)-N3 homopolymer (P2) were purified by precipitation in methanol twice. The DPs were determined by 1H-NMR, comparing the integrated peak areas from the spectrum of the methylene proton signal next to the pyrene moiety (–CH2–Py) for P1 and the methylene proton signal (–CH2CH2COO–, 2.50 ppm) for P2 to that of the aldehyde signal (9.76 and 9.81 ppm) of the MAEBA repeating unit, as 15 and 19, respectively (Fig. S18 and S22, ESI†).
In the second step, in order to grow the second block, the previously synthesized homopolymers (P1 and P2) were used as macro-RAFT agents to polymerize the 1-O-methacryloyl-2,3:4,5-di-O-isopropylidene-β-D-fructopyranose (ipFruMA) monomer (Scheme 1). The copolymerization reactions were performed in a toluene/acetonitrile mixture (2.5:1 v/v) at 70 °C for 8.5 h. The ratio between the monomer, macro-RAFT agent and AIBN ([M]0:[macro-RAFT]0:[I]0) in the polymerization medium was 250:1.0:0.175. The obtained P(ipFruMA-b-MAEBA)-Py and P(ipFruMA-b-MAEBA)-N3 polymers are coded as P3 and P4, respectively. Well-defined block structures were also qualitatively determined by 1H-NMR analysis. In addition, the DPs of the fructose pendant blocks were calculated as 152 and 234 for the polymers P3 and P4 respectively (Fig. S19 and S23, ESI†).
Then, the protecting isopropylidene groups in P3 and P4 were hydrolyzed in trifluoroacetic acid (TFA) conditions to give deprotected polymers P(FruMA-b-MAEBA)-Py (P5) and P(FruMA-b-MAEBA)-N3 (P6) respectively.30 Additionally, before the deprotection reaction of the glycoblock copolymers (Fig. S19 and S23, ESI†), the peaks around δ = 3.5–5.2 ppm clearly showed the fructopyranose ring of the sugars, and those around δ = 6.5–7.2 ppm and δ = 9.81 ppm showed the phenyl groups and the aldehyde group in the P(MAEBA) block. Meanwhile, the peaks around δ = 1.41 ppm confirmed the presence of isopropylidene groups on the glycoblock copolymer backbone. After the deprotection reaction, the signals of the isopropylidene groups disappeared, showing the successful removal of the groups and the formation of unprotected P5 and P6 (Fig. S20 and S24, ESI†). FT-IR measurements of P5 and P6 were performed to confirm the deprotection of the isopropylidenyl groups (Fig. S21B and S25, ESI†). The spectrum also demonstrates the loss of the C–O absorption peak of the isopropylidene groups at 1216 cm−1 with the appearance of the characteristic broad absorption peak of hydroxyl groups at ν = 3340 cm−1. Moreover, the existence of end group fidelity after TFA hydrolysis of the polymers was confirmed by UV-vis and using an FT-IR spectrophotometer. The UV-vis spectra showed the characteristic peaks of the pyrene end group of P5 and its precursor polymers (Fig. S21, ESI†). The FT-IR spectra showed the characteristic peaks of the azide end groups of P6 and its precursor polymers (Fig. S25, ESI†). As can be seen in Fig. S27 (ESI†) and Table 1, the molecular weights of P5 and P6 are calculated from the 1H-NMR spectra as 42.05 and 63.05 kDa, respectively. Additionally, the unimodal peaks and molecular weight distributions of the P5 and P6 glycoblock copolymers indicate that no degradation or any side reactions occurred after TFA hydrolysis (Fig. S27, ESI†).
| Entry | Polymer | Conv.c (%) | M n,theo (kDa) | M n,NMR (kDa) | M n,GPC (kDa) | Đ |
|---|---|---|---|---|---|---|
| a Molecular weight (Mn) and Đ determined by THF-GPC. b M n and Đ values determined by DMF-GPC (MALS/RI/UV/dQLS detection). c Polymerization conversion determined gravimetrically. d Deprotection conversion determined based on integral ratios of distinct protons by 1H-NMR in DMSO-d6. e M n,theo = ([M]0/[I]0)·conversion %·MW of monomer + MW of initiator. f Determined on the basis of the integral ratios of distinct protons by 1H-NMR in CDCl3. g Determined on the basis of the integral ratios of distinct protons by 1H-NMR in DMSO-d6. | ||||||
| P1a | P(MAEBA)15-Py | 36 | 3.30 | 4.20f | 2.95 | 1.13 |
| P2a | P(MAEBA)19-N3 | 38 | 4.05 | 4.80f | 3.50 | 1.06 |
| P3a | P(ipFruMA152-b-MAEBA15)-Py | 55 | 49.35 | 54.10f | 22.30 | 1.15 |
| P4a | P(ipFruMA234-b-MAEBA19)-N3 | 85 | 74.55 | 81.60f | 40.00 | 1.21 |
| P5b | P(FruMA152-b-MAEBA15)-Py | 99d | 37.30 | 42.05g | 81.20 | 1.33 |
| P6b | P(FruMA234-b-MAEBA19)-N3 | 99d | 56.00 | 63.05g | 64.10 | 1.25 |
| P7b | P(FruMA152-b-MAEBA10)-Py/Dox5 | 62d | 40.00 | 44.75g | 90.80 | 1.48 |
| P8b | P(FruMA234-b-MAEBA13)-N3/Dox6 | 75d | 59.25 | 66.30g | 91.00 | 1.26 |
The condensation reaction of a primary amine with an aldehyde under mild conditions is well-known in organic chemistry and was used to conjugate the anticancer drug Dox onto P5 and P6 via an acid-labile imine linkage one by one.501H-NMR analysis was performed on P(FruMA-b-MAEBA)-Py/Dox (P7) and P(FruMA-b-MAEBA)-N3/Dox (P8) to elucidate the chemical composition of the aldehyde and Dox units in the copolymers. The number of conjugated Dox units was found to be 5 and 6, respectively. Besides the particular proton signals of the MAEBA and fructose units, the aromatic proton signals of Dox at 7.94–8.04 ppm and the methine proton signal (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) at 8.16–8.34 ppm are shown in Fig. 1 and 2. The FT-IR spectra of P7 and P8 revealed the stretching vibration bands of O–H at 3500 and 3300 cm−1 (Fig. S21B and S25, ESI†), respectively.
N) at 8.16–8.34 ppm are shown in Fig. 1 and 2. The FT-IR spectra of P7 and P8 revealed the stretching vibration bands of O–H at 3500 and 3300 cm−1 (Fig. S21B and S25, ESI†), respectively.
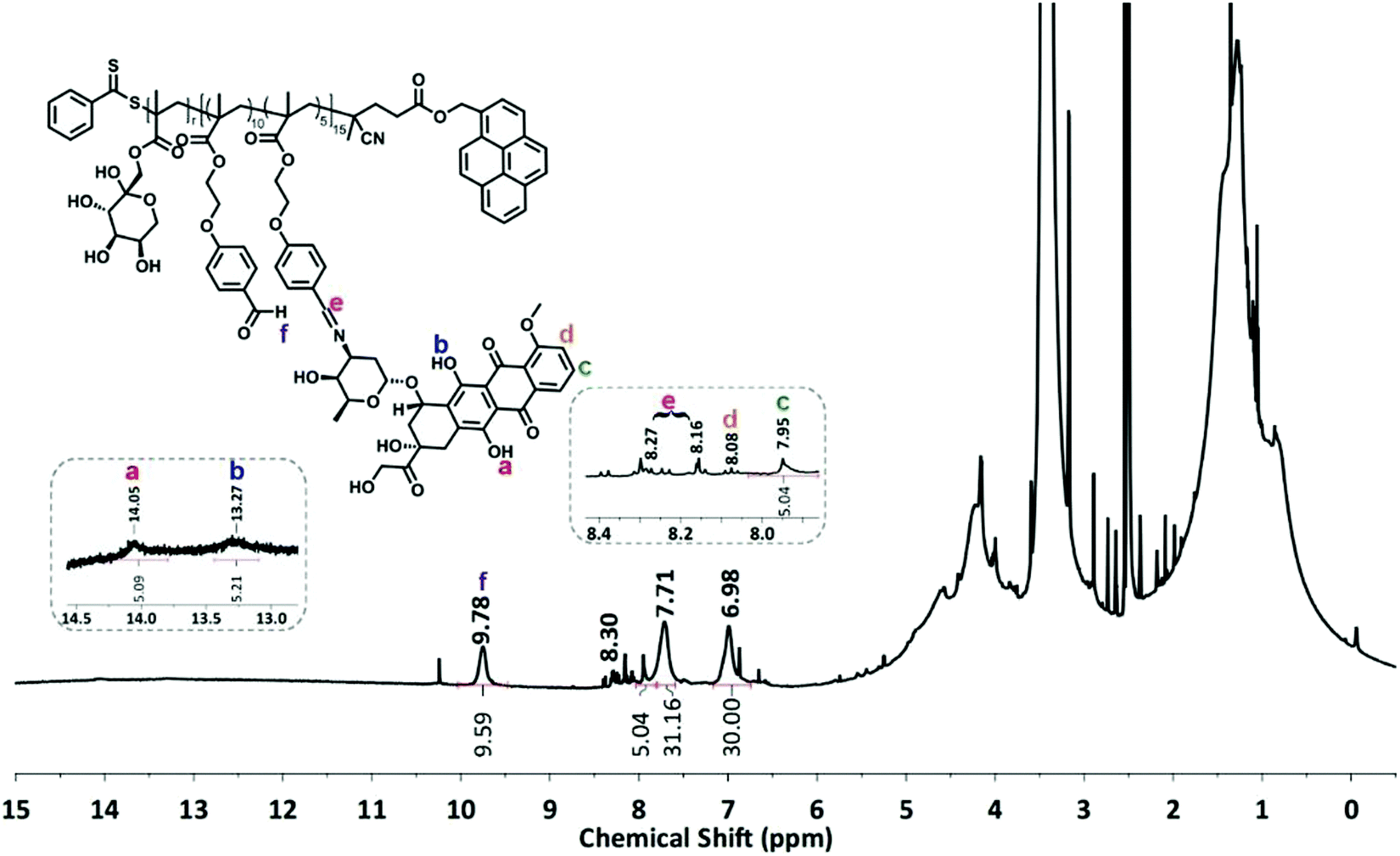 | ||
| Fig. 1 1H-NMR spectrum of P(FruMA-b-MAEBA)-Py/Dox (P7) in CDCl3 (500 MHz). | ||
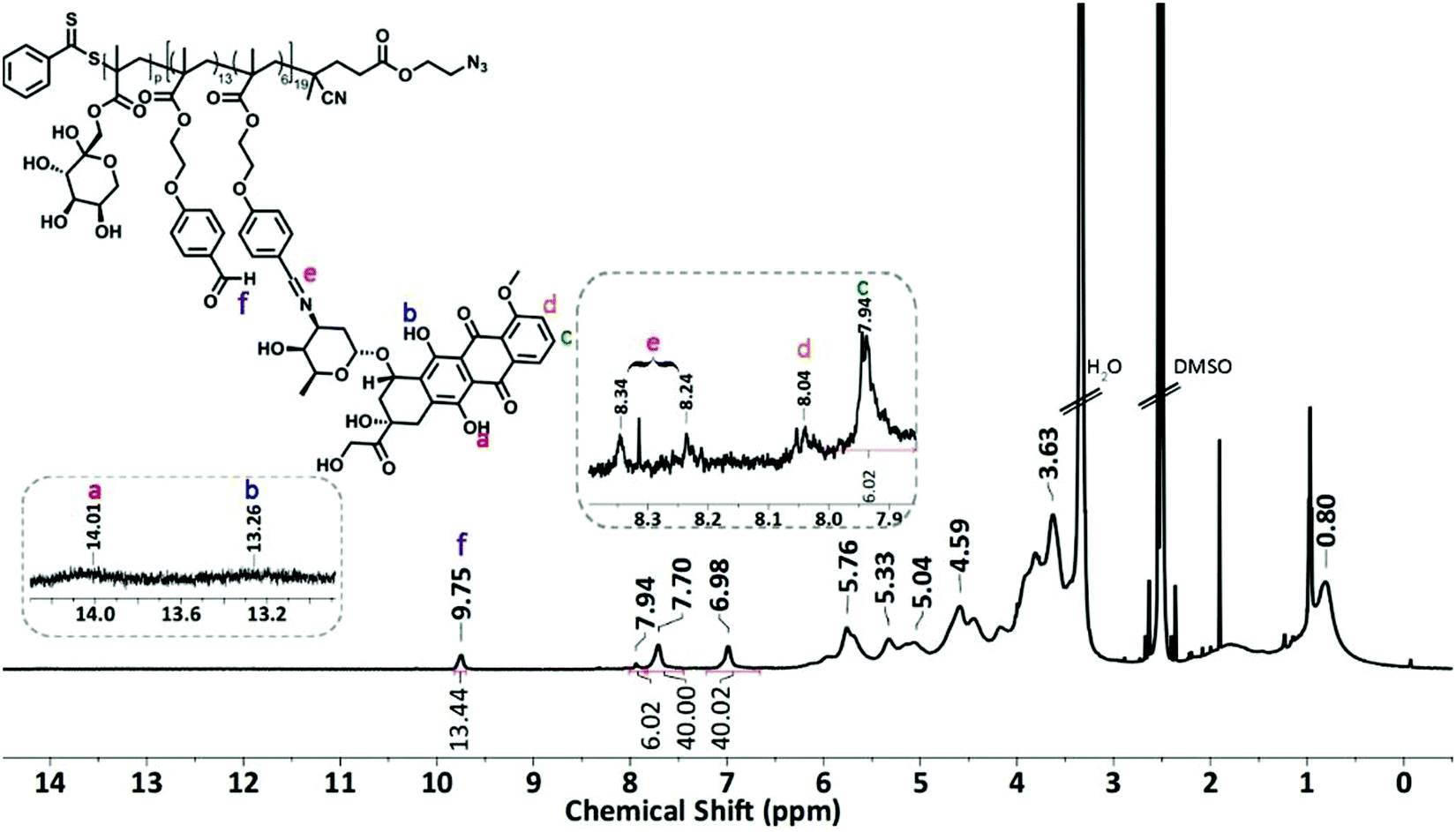 | ||
| Fig. 2 1H-NMR spectrum of P(FruMA-b-MAEBA)-N3/Dox (P8) in CDCl3 (500 MHz). | ||
The molecular weight characteristics of the glycoblock copolymers and their precursors are outlined in Table 1. An increase in molecular weight and narrow monomodal distributions are evident from the GPC traces (Fig. S26 and S27 and Table 1), which highlight the successful synthesis of the glycoblock copolymer–Dox conjugates.
Characterization of glycoblock copolymer Dox-conjugate decorated CNTs
P7 and P8 were utilized to cover CNT-COOH and CNT-alkyne separately via π–π stacking and a CuAAC click reaction, respectively, as described in the Experimental section.FT-IR spectra of p-MWCNTs and CNT-COOH are shown in Fig. S28 (ESI†). The FT-IR spectrum of p-MWCNTs shows characteristic peaks of CC and C–C bonds at 1582 cm−1 and 1160 cm −1, respectively.24 In the spectrum of CNT-COOH, peaks at 1705 cm−1 and 1638 cm−1 are present and can be assigned to carbonyl groups in the ester and ring structures. In the FT-IR spectrum of CNT-alkyne, the new signals at 1775 cm−1 (CO of ester), 2170 cm−1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch of alkyne), 2845 cm−1 (aliphatic hydrogens), and 2920 cm−1 (C–H stretch of alkyne) prove the attachment of alkyne groups via carboxylic groups onto the edges of the CNTs.57
C stretch of alkyne), 2845 cm−1 (aliphatic hydrogens), and 2920 cm−1 (C–H stretch of alkyne) prove the attachment of alkyne groups via carboxylic groups onto the edges of the CNTs.57
After the glycopolymer conjugation of CNTs with P7 and P8, the main absorption band characteristic of sugars was observed at 3300 cm−1, corresponding to the stretching vibration of the O–H bond, implying the successful functionalization of the CNTs.
The aromatic stretching of four-fused benzene rings, which consist of a pyrene unit, appears at 880 cm−1 as a fingerprint band in the spectra of P7 and CNT/P7. The absorptions at 1183 and 754 cm−1 are assigned to the out-of-plane vibrations of C–H bonds and the peaks at 1375 and 1602 cm−1 are associated with the CC bonds of the pyrene unit, which appear in both the P7 and CNT/P7 spectra (Fig. S29, ESI†).58 The FT-IR spectra of P2, P4, P6, and P8 showed the appearance of a band at around 2100 cm−1, which is associated with the azide group (C–N3) (Fig. S25, ESI†). The CuAAC click reaction of the azido-terminated Dox-conjugated glycoblock copolymer (P8) to CNT-alkyne was confirmed by the disappearance of the azide and alkyne moieties at 2160–2150 cm−1 and the appearance of the triazole ring at 1636 and 1457 cm−1 in the overlayed FT-IR spectra of CNT/P8 in Fig. S30 (ESI†).59,60
To ascertain that glycoblock copolymers were successfully synthesized and covered onto CNTs in a noncovalent and covalent manner, the resulting hybrid-CNTs were examined to confirm the quantitative functionalization via two strategies through thermogravimetric analysis (TGA). Fig. 3 shows the thermal degradation profiles of the untreated CNTs, acid-functionalized CNTs (CNT-COOH), and alkyne-functionalized CNTs (CNT-alkyne) together with those of the glycoblock copolymer and FA-modified hybrid-CNTs from room temperature to 700 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere. For all analysis of the CNTs, the initial weight loss of up to 150 °C is based on the removal of adsorbed water. The weight loss of p-MWNTs was 2% at 700 °C, which might arise from the decomposition of impurities. According to the TGA thermograms, significant weight loss occurs for both CNT/P7 and CNT/P8 higher than 250 °C.
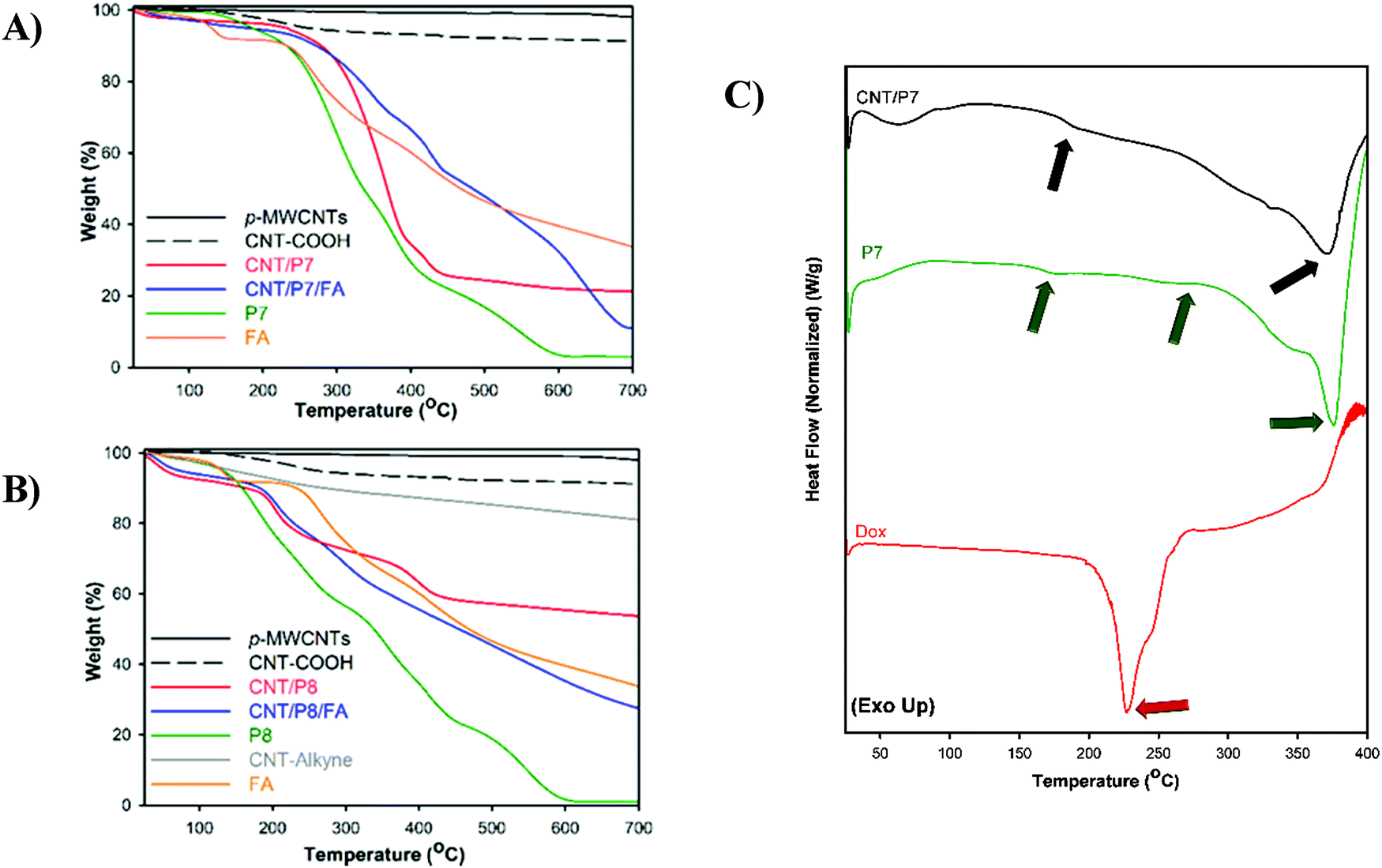 | ||
| Fig. 3 Thermogravimetric analysis of P7, P8, FA and CNTs under nitrogen flow at 10 °C min−1. (A) Modified hybrid-CNTs and their precursor via π–π stacking. (B) Modified hybrid-CNTs and their precursor via the CuAAC click reaction. Weight losses are listed in Table 2. (C) DSC thermogram of free-Dox, P7 and CNT/P7. | ||
The TGA profile for CNT/P8 shows a weight loss of 28% at 700 °C, which is less than that of CNT/P7 (69 wt%). Thus, the TGA data verifies that noncovalent attachment of glycoblock copolymers to CNTs is more effective than the click reaction between azido end-functionalized glycoblock copolymers and alkyne functionalized CNTs. Afterwards, the thermal behaviour of the FA loaded hybrid-CNTs (CNT/P7/FA, CNT/P8/FA) showed more thermal stability than their precursors. This behaviour associated with the FA structure improves the thermal stability of the hybrid carbon nanotubes.61Table 2 summarizes the thermogravimetric profiles of all hybrid-CNTs and their precursors.
| Material | Total weight lossa (%) | ζ mean valueb (mV) | Thicknessc (nm) | |
|---|---|---|---|---|
| a Weight loss fraction between 150 and 700 °C. b All samples are prepared in 1 mM PBS (pH 7.4) with a 0.5 mg mL−1 concentration. Data represented as mean ± SD (n = 3). c Obtained by TEM analysis. | ||||
| π–π stacking | CNT-COOH | 5 | −36.10 ± 0.92 | — |
| CNT/P7 | 69 | −20.47 ± 2.25 | 7.5 | |
| CNT/P7/FA | 79 | −19.77 ± 0.51 | 10 | |
| CuAAC click reaction | CNT-alkyne | 10 | −15.62 ± 0.23 | — |
| CNT/P8 | 28 | −22.83 ± 0.31 | 3 | |
| CNT/P8/FA | 53 | −19.83 ± 0.05 | 9 | |
The compatibility of the hybrid-CNTs and the Dox containing precursor polymer is also evaluated by differential scanning calorimetry analysis (DSC). The thermal properties provide insight into the interaction between components in the hybrid-CNTs. DSC curves of CNT/P7 and its precursors are shown in Fig. 3C and Fig. S31 (ESI†). Dox shows an endothermic peak at 156.9 °C (Fig. S31, ESI†) which was previously reported as reorganization of the Dox anhydrous phase62 and the melting temperature (Tm) was found to be 226.7 °C (Fig. 3C). The DSC thermogram of P7 shows several endothermic processes, which are attributed to the glass transition (Tg) of P7 at 176.7 °C, melting process of Dox at 250.9 °C and Tm of P7 at 372.3 °C. Similarly, two peaks can be found at 182.4 and 369.3 °C for CNT/P7 (Fig. 3C and Fig. S31, ESI†). It can be deduced that there is a good compatibility between Dox-conjugated glycoblock copolymer and CNT/P7.
The surface properties of hybrid-CNTs are also important parameters for good dispersion as well as cellular interaction. Zeta potential measurements (ζ) of the hybrid-CNTs were performed by dynamic light scattering (DLS) in order to ensure that the surface properties change significantly through noncovalent/covalent functionalization. The average ζ-potential value of CNT-COOH was found to be −36.10 ± 0.92 mV in 1 mM PBS (pH 7.4) due to the existence of –COOH groups, while the value of CNT/P7 varied to −20.47 ± 2.25 mV.63 This can be attributed to the P7 adsorbed onto the CNT walls. After FA conjugation, the surface ζ potential increased to −19.77 ± 0.51 mV for CNT/P7/FA, which is associated with the carboxyl groups of FA and the successful adsorption of FA onto CNT/P7 with electrostatic and π–π stacking interactions. By following the CuAAC click reaction, it was expectedly observed that the average ζ value of CNT/P8 decreased to −22.83 ± 0.31 mV with concomitant approval of the surface polarity of sugars. The further loading of FA onto CNT/P8 resulted in an increase in the ζ value to −19.83 ± 0.05 mV due to the presence of free amine groups on its surface (Table 2).
The transmission electron microscopy (TEM) images showed that the surface of the CNTs has been covered by the Dox-conjugated glycoblock copolymers (P7 and P8). CNT/P7 and CNT/P8 were thicker than their precursor CNTs (Fig. 4A and B and Fig. S32, ESI†). The TEM images of the samples revealed a relatively uniform coating ranging from 3 to 10 nm in thickness through the whole length of the nanotubes (Table 2). The lighter and thicker areas around the CNTs are intensified with glycoblock copolymer and FA attachment.
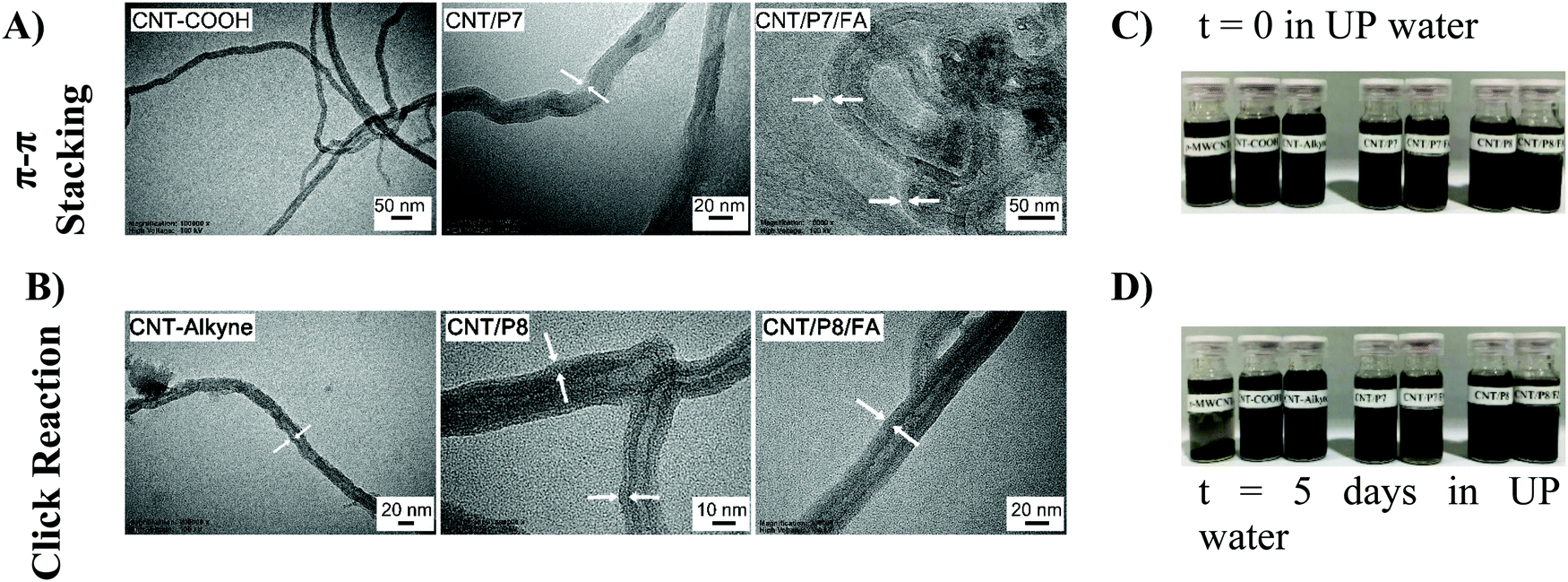 | ||
| Fig. 4 TEM images of (A) CNT-COOH, CNT/P7, and CNT/P7/FA and (B) CNT-alkyne, CNT/P8, and CNT/P8/FA. All dispersions of CNTs (C) and (D) were prepared through the sonication of 1 mg in 1.5 mL UP water for 15 minutes. | ||
1 mg of p-MWCNT, CNT-COOH, CNT-alkyne and the hybrid-CNTs (CNT/P7, CNT/P7/FA, CNT/P8, and CNT/P8/FA) in 1.5 mL of UP water were added into vials and then sonicated for 15 minutes to prepare the related dispersions (Fig. 4C). The obtained hybrid-CNTs were expected to be soluble in UP water. The samples were photographed after standing for 5 days at room temperature. Even after 5 days in UP water, the resulting dispersion was observed to have had negligible aggregation and to possess admissible stability (Fig. 4D). In addition, CNT/P8 showed relatively poor solubility in UP water. This could also be explained by the decrease in the modification (amount of weight loss) shown in the TGA results (Fig. 3).
In vitro drug release profile of CNT/P7
When delivering a drug, the desired release kinetics of the cargos must be considered. The pH of the extracellular matrix of cancer cells is lower than the intracellular matrix, so the therapeutic effect of anticancer drugs can be increased by using various pH-triggered drug delivery systems.64 In addition, acidic hydrolysis of Schiff bases such as benzylic imine linkages is well-known chemistry.65 The pH-sensitive imine linkages of the polymer/Dox scaffolds allowed triggered release of Dox.66,67 pH-sensitive cargo release was demonstrated to mimic the reducing conditions present in the cytoplasm environment of cancer cells. Experimental details for the determination of the in vitro drug release of CNT/P7 are described in the ESI.† UV-vis spectroscopy was used to observe the amount of Dox released from CNT/P7 in detail (Fig. S33, ESI†).68 The accomplishment of the Dox-conjugated glycoblock copolymer within CNT-COOH was observed with the absorption intensity at 480 nm (the characteristic absorption peak of Dox). The cumulative Dox release percentages were calculated in the first 8 h as 38.1% and 29.5% at pH 5.5 and 7.4, respectively. As expected, the results showed that the Dox amount released from CNT/P7 at pH 5.5 was considerably higher than at pH 7.4. These results suggested that CNT/P7 exhibits relative in vitro stability under a physiological environment (pH 7.4) but could intensely release Dox at pH 5.5.Cell viability assay and cellular internalization studies
The surface modification of CNTs plays a key role in cellular association and internalization. We examined the optimal surface modification in which the maximum difference in cellular internalization is achieved when the MCF-7 and MDA-MB-231 cell lines are compared. Noncovalently/covalently functionalized hybrid-CNTs of two types with/without FA coating, P7, P8 and free Dox were separately incubated with two different cancer cell lines to test their effects on cell viability and their targeting specificity.First of all, the cell viability of Dox, FA, P7, P8, CNT-COOH, CNT-alkyne, CNT/P7, CNT/P7/FA, CNT/P8, and CNT/P8/FA was assessed by MTT assay. Our results demonstrated that P5, P6 and FA alone were not associated with extraordinary cell toxicities, while the chemotherapeutic drug containing substances had higher cell death in the MCF-7 and MDA-MB-231 cell lines (Fig. 5 and Fig. S36, S37, ESI†). Moreover, interestingly CNT-COOH did not show high toxicity.69 Hence, carboxylation of p-MWCNT may enhance their dispersibility and biocompatibility. The MTT assay results showed that Dox-loaded glycoblock copolymer conjugation to CNTs with or without FA coating is more effective in terms of cytotoxicity towards both cell lines than free Dox. The cytotoxicity of the FA coated hybrid-CNTs was much higher than for the other substances, suggesting that FA has a critical effect on increasing the intracellular uptake of Dox. CNT/P7/FA was found to have a positive effect on the cell toxicity of MCF-7 and MDA-MB-231. Hybrid-CNT/FA may trigger the intracellular entrance of Dox, providing enhanced cellular damage leading to apoptosis of the cancer cells. The antitumor cytotoxic effect of CNT/P7/FA was higher for the MDA-MB-231 cell line. This result suggests that fructose and FA moieties improve the cytotoxicity of the conjugated chemotherapeutic drug. The existence of altered expression levels of GLUT5 in MDA-MB-231 compared to MCF-7 indicates that the accumulation of CNT/P7/FA was associated with fructose transporters. Hamann et al. examined the expression and function of GLUT5 in breast cancer cell lines and reported that GLUT5 mRNA and protein levels were higher in MDA-MB-231.70 Also, another study by Yang et al. showing the significant rise of FR in MDA-MB-231 further supports our findings.71
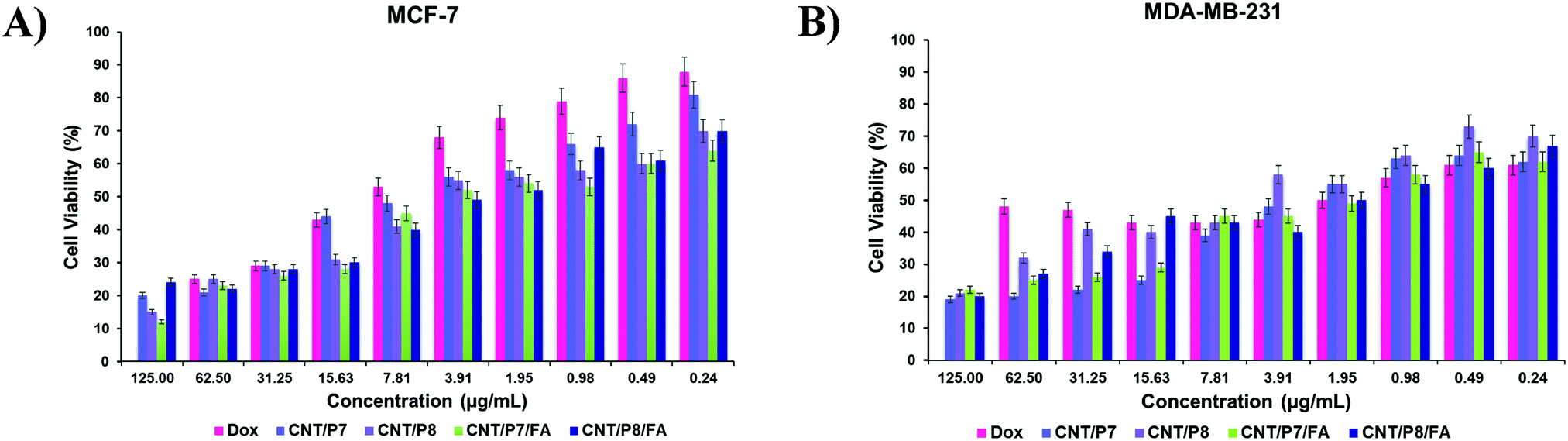 | ||
| Fig. 5 Cell viability (in %) after exposing cells to a solution containing Dox, CNT/P7, CNT/P8, CNT/P7/FA, and CNT/P8/FA for 24 h: (A) MCF-7 and (B) MDA-MB-231 cells. Data are presented as mean ± SD. | ||
To investigate the cellular internalization performance of the co-targeted therapy system, free Dox, P7, CNT/P7, CNT/P7/FA, CNT/P8 and CNT/P8/FA were employed to evaluate the influence of the glycoblock copolymer and FA. Both MCF-7 and MDA-MB-231 cells were treated with these samples for 24 h, stained with fluorescent dye DAPI, and imaged by CLSM to investigate whether Dox-conjugated hybrid-CNTs were able to enter cells (Fig. 6). The distribution ratios of Dox in the cells were determined by evaluating the CTCF values in the CLSM images. In that case, 15 images captured at the same exposure time of 500 ms were analysed using the Image J program as previously reported.52 Notably, as shown in Fig. S34 (ESI†), the treatment with P7 showed enhanced cancer cell internalization efficacy compared with the treatment with free Dox.
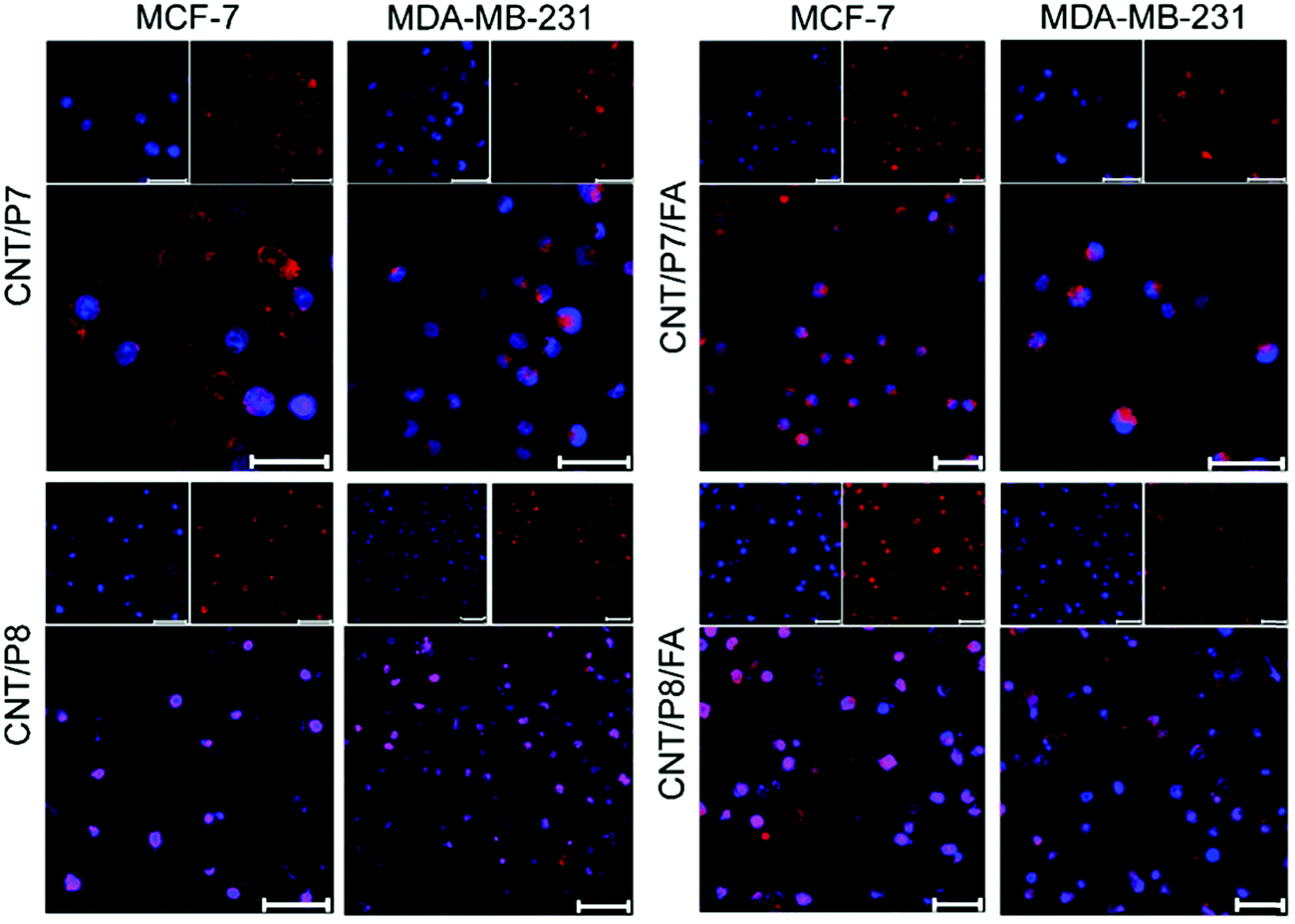 | ||
| Fig. 6 Cellular uptake of CNT/P7, CNT/P7/FA, CNT/P8, and CNT/P8/FA by MCF-7 and MDA-MB-231 cells after 24 h incubation (intracellular Dox shown in red; nuclei shown in blue; and merged images). Scale bar in the all images: 50 μm. | ||
This can be attributed to the ability of repeated fructose units in the glycoblock copolymer scaffold to play a critical role in enhancing the cellular uptake via GLUT5 targeting. Moreover, the FA modified hybrid-CNTs (CNT/P7/FA and CNT/P8/FA) showed much higher internalization in both MCF-7 and MDA-MB-231 cells (Fig. 6 and Fig. S35, ESI†).
Additionally, compared with the FA-unmodified hybrid-CNTs (CNT/P7 and CNT/P8), the cellular internalization of CNT/P7/FA and CNT/P8/FA had a roughly 1.5-fold and 1.2-fold increase in MCF-7 cells (Fig. S34A, ESI†), while achieving 1.8-fold and 1.3-fold improvements in MDA-MB-231 cells (Fig. S34B, ESI†). This reveals that FA has a stronger factor of enhancing cellular internalization.
Our study showed that CNT/P7/FA and CNT/P8/FA improved the delivery of Dox to the breast cancer cells. These results suggest that the FA enhanced drug uptake and initiated the death of cancer cells. This may be due to the enhancement of the sensitivities of breast cancer cells to the ligands caused by the combined actions on the co-targets of tumor cells. The further surface modification of the glycoblock copolymer coated CNTs with FA promotes the delivery of Dox to the co-targeted breast cancer cells that overexpress FR. In recent years, there has been an increasing amount of literature on how the raised expression of FR could be clinically useful for developing folate-targeted therapeutics or imaging.72 These unique advantages make the prepared multifunctional hybrid-CNTs promising for breast cancer diagnosis and therapy. In light of CLSM assessments, it is essential to quantify the cellular internalization of the hybrid-CNTs. Therefore, flow cytometry analyses were conducted for Dox, P7, CNT/P7 and CNT/P7/FA in two cell lines with an incubation time of 24 h.
Fig. 7A demonstrates a summary of data from the flow cytometry analysis. The detailed mean fluorescence intensity (MFI) of each sample is displayed in the bar graph. It is apparent that the MFI of MDA-MB-231 cells treated with CNT/P7/FA shows much higher cellular uptake (∼1.3 fold) than that of MCF-7 cells with the order of CNT/P7, P7 and free Dox. The MFI value of free Dox at 24 h was 5–6 times lower than for CNT/P7, and 4–5 times lower than that of CNT/P7/FA. Additionally, the MFI values for CNT/P7 and CNT/P7/FA were 1.2 times higher for MDA-MB-231, and 1.1 times higher for MCF-7. These results are consistent with the CLSM analyses. Our findings indicate that hybrid-CNTs with fructose and FA units possess good breast cancer cell internalization ability, could effectively release Dox and improve cytotoxicity, and thus would be an efficient drug delivery platform for breast cancer therapy.
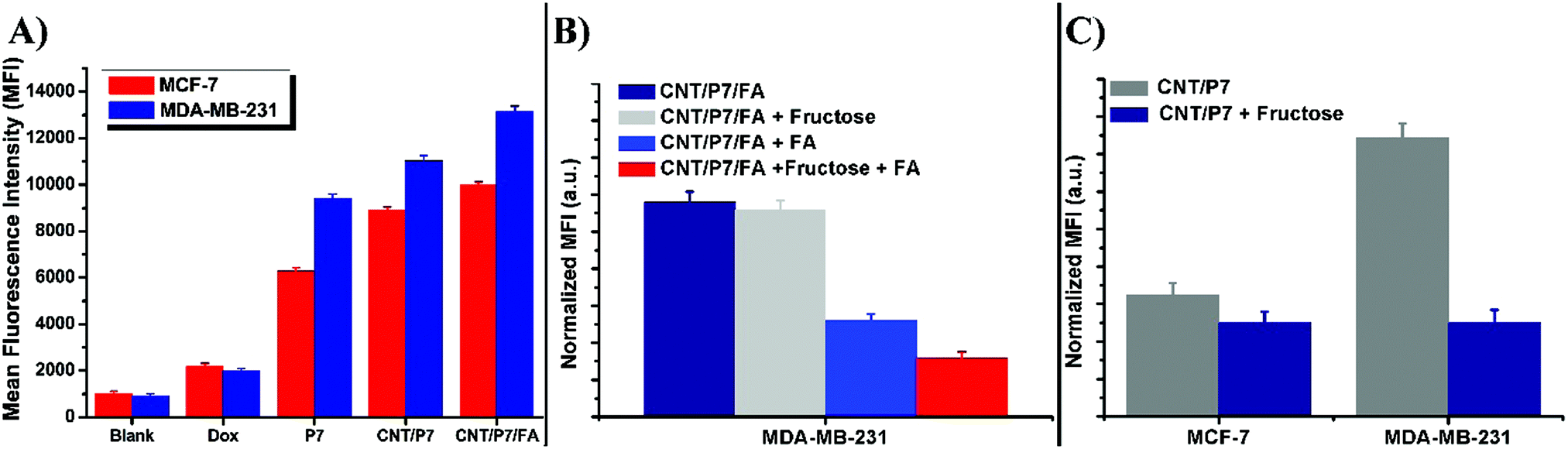 | ||
| Fig. 7 (A) Mean fluorescence intensity (MFI) determined by flow cytometry and normalized to non-treated cells of MCF-7 and MDA-MB-231. Competition studies of (B) CNT/P7/FA in the presence of free FA and/or fructose in MDA-MB-231 cells and (C) CNT/P7 in the presence of free fructose in MCF-7 and MDA-MB-231 cells. | ||
Transportation of fructose and FA through the cell membrane into the cytoplasm is regulated by membrane located receptors, which are upregulated in the MCF-7 and MDA-MB-231 cell lines.32,34,35 To understand the mechanism of receptors involved in fructose and FA moiety intake, we also performed fructose and FA competition assay. For this assay, MCF-7 and MDA-MB-231 cells were treated with either CNT/P7 or CNT/P7/FA in the presence of free fructose and/or FA for 24 h. As shown in Fig. 7C, recognition of MCF-7 and MDA-MB-231 cells which had been exposed to 2.0 mM free fructose caused a considerable decrease in cellular internalization of CNT/P7, exhibiting GLUT5-dependent recognition. In the folate competition studies, a slightly lower intracellular uptake of CNT/P7/FA was observed in MDA-MB-231 cells after incubation with free fructose in the same conditions. In addition, the cellular uptake of CNT/P7/FA in MDA-MB-231 cells was significantly lower than that seen in MDA-MB-231 after incubation with only free FA and/or fructose for the same period. This could be explained by the presence of sugar and FA layers on the outside of the CNTs, which increases the impassibility of the cell surface. Thus, the differences observed in the levels of cellular uptake could be induced directly by the binding of fructose/FA to GLUT5/FR. Consistently, it is notable that the free fructose and/or FA were shown to reduce the levels of cellular internalization of CNT/P7 and CNT/P7/FA.
Cell apoptosis and cell cycle analysis
For further evaluation of the efficacy of CNT/P7 and CNT/P7/FA, apoptosis and cell cycle analysis were conducted. Laser-based fluorescence detection using a cell analyzer was used for investigation of apoptotic cells and phases of the cell cycle. After 4 h treatment with free Dox, CNT/P7 and CNT/P7/FA, a major cell population displayed elevated percentages of apoptosis. 70.07% of MCF-7 and 63.36% of MDA-MB-231 cells were apoptotic after CNT/P7/FA treatment (Table 3). Besides, 24 h incubation further proved that the tested compounds progressed apoptosis in both cell lines (Table 3 and Fig. S38, ESI†). After treatment with CNT/P7 and CNT/P7/FA for 24 h in the MDA-MB-231 cell line, the total apoptotic cells were significantly higher than cells treated with free Dox (p < 0.0107 and p < 0.0038, respectively). It was clearly shown that treatment with CNT/P7/FA decreased viability by leading cells to apoptosis. CNT/P7/FA exhibited enhanced toxicity, particularly in MDA-MB-231 for 24 h treatment, demonstrating that fructose and FA units improve transportation of Dox into the cells, causing boosted effects of the chemotherapeutic drug.| MCF-7 | MDA-MB-231 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Live (%) | Early apop. (%) | Total apop. (%) | Live (%) | Early apop. (%) | Total apop. (%) | |||||||
| Sample | 4 h | 24 h | 4 h | 24 h | 4 h | 24 h | 4 h | 24 h | 4 h | 24 h | 4 h | 24 h |
| Blank | 78.43 | 66.62 | 3.10 | 19.47 | 20.38 | 31.81 | 84.97 | 79.75 | 8.46 | 7.31 | 12.29 | 13.73 |
| Dox | 44.97 | 55.57 | 47.63 | 28.37 | 54.66 | 42.68 | 59.62 | 26.68 | 18.31 | 33.16 | 39.44 | 72.80 |
| CNT/P7 | 39.30 | 15.00 | 54.59 | 47.44 | 60.70 | 85.00 | 38.24 | 18.47 | 34.25 | 36.65 | 60.91 | 81.24 |
| CNT/P7/FA | 29.71 | 12.20 | 59.21 | 25.97 | 70.07 | 87.46 | 36.25 | 3.37 | 32.22 | 34.34 | 63.36 | 96.58 |
We also performed cell cycle analysis based on the DNA content of the cells treated with free Dox, CNT/P7 and CNT/P7/FA. The cell cycle distributions of the untreated cell lines showed that 49.6% of MCF-7 cells were in G0/G1, 28.1% in S, and 16.6% in G2/M phases while 39.2% of MDA-MB-231 cells were in G0/G1, 30.4% in S, and 20.5% in G2/M phases. As shown in Fig. 8, in both cell lines treatment with CNT/P7 and CNT/P7/FA caused accumulation of cells in the S phase by allowing Dox to intercalate DNA, leading to an increase in apoptosis.73 The cells progressing into the S phase increase their DNA synthesis and energy metabolism, which are necessary for cell proliferation. For tissue growth and homeostasis, doubling the amount of DNA via cell cycle progression is essential.74,75 The differences between the untreated and treated cells indicated that the hybrid-CNTs with fructose and FA units were lethal for the demonstrated cell lines and increased the number of cells arrested in the S phase (39.5% and 39.6% for MCF-7 and 32.1% and 39.2% for MDA-MB-231, respectively) and also decreased the proportion of cells in the G2/M phase. Delaying or inhibiting the progression of the G2/M phase of the cell cycle with CNT/P7 and CNT/P7/FA was significantly more effective than the free Dox for both cell lines (p < 0.0217 and p < 0.0237, respectively, for MCF-7 and p < 0.0352 and p < 0.0412, respectively, for MDA-MB-231). From this data, it can be seen that the drug conjugated hybrid-CNTs were related to either significant blockage of the G2/M transition or a delay in the S phase during the cell cycle. As shown in Fig. 8 and Fig. S39 (ESI†), the Dox-loaded CNTs consistently had superior harmful effects on the cancer cell lines than the free Dox. Our findings indicate that hybrid-CNTs with fructose and FA units possess good breast cancer cell internalization ability, could effectively release Dox and improve cytotoxicity, and thus would be an efficient drug delivery platform for breast cancer therapy.
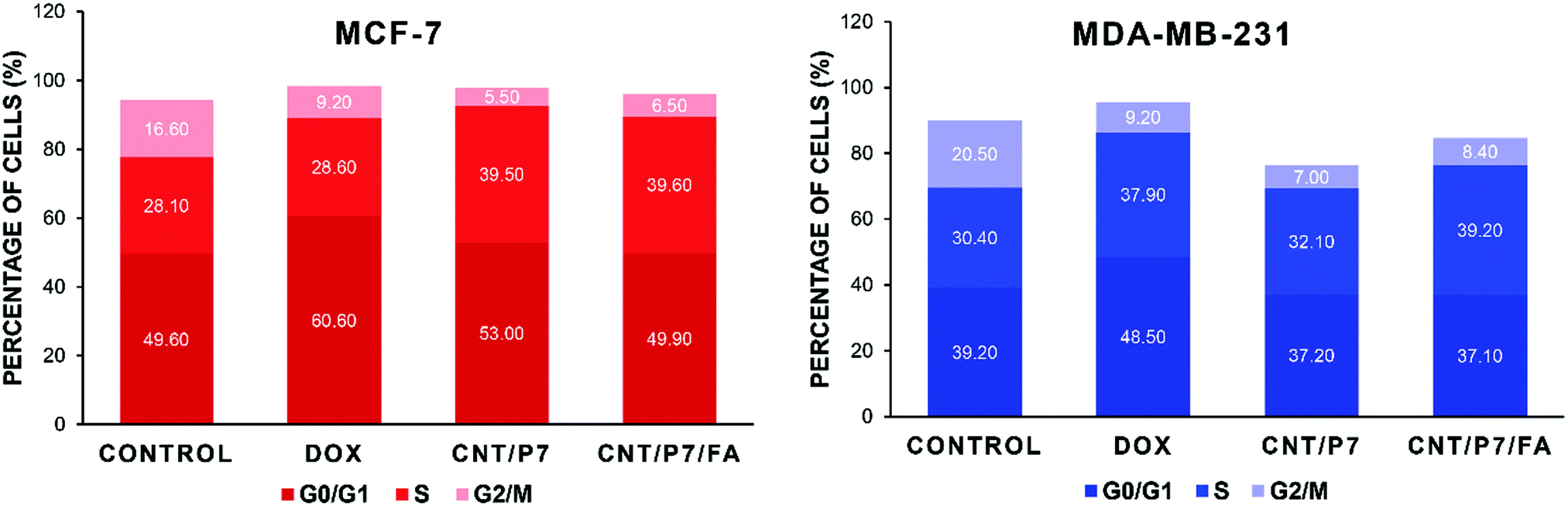 | ||
| Fig. 8 Cell cycle distributions displayed as the percentage of each cell cycle phase. | ||
Conclusion
We have used noncovalent and covalent approaches to produce new hybrid-CNTs by combining them with two different Dox-conjugated glycoblock copolymers. The strategy involves two major steps: first, Dox-conjugated pyrene-end functionalized glycoblock copolymers are attached onto CNTs via π–π stacking. Second, alkyne functionalized CNTs are clicked with Dox conjugated azido end-functionalized glycoblock copolymers by a CuAAC click reaction. The prepared hybrid-CNTs were further coated with FA to investigate both GLUT5 and FR mediated targeting. The obtained hybrid-CNTs (CNT/P7/FA and CNT/P8/FA) show excellent stability under various physiological conditions. By targeting dual receptors, the prepared hybrid-CNTs enhanced the stability of both the CNTs and anticancer drugs along with increasing the accumulation of Dox in the tumor tissue, thereby increasing the anticancer efficacy. The effective cellular uptake demonstrates the promise of hybrid-CNTs for co-targeted drug delivery to breast cancer cells.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to DEVA for the generous donation of Doxorubicin·HCl. The authors wish to thank Prof. Dr Gülaçtı Topçu for access to the Biotek Synergy H1 Hybrid Multi-Mode reader and Dr Beyza Goncu for providing the apoptosis and cell-cycle analysis. We would like to thank TETRA Teknolojik Sistemler A.Ş. for their support with TEM images and Istanbul Medipol University Innovative Polymer Nano-Therapeutics Research Lab (IPONT) for TGA measurements.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- B. E. Wilson, S. Jacob, M. L. Yap, J. Ferlay, F. Bray and M. B. Barton, Lancet Oncol., 2019, 20, 769–780 CrossRef PubMed.
- R. E. Hendrick, J. A. Baker and M. A. Helvie, Cancer, 2019, 125, 1482–1488 CrossRef PubMed.
- M. I. Nounou, F. ElAmrawy, N. Ahmed, K. Abdelraouf, S. Goda and H. Syed-Sha-Qhattal, Breast Cancer: Basic Clin. Res., 2015, 9(S2), 17–34 CAS.
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS PubMed.
- G. Wang, Y. Ma, Z. Wei and M. Qi, Chem. Eng. J., 2016, 289, 150–160 CrossRef CAS.
- Q. Sun, M. Radosz and Y. Shen, J. Controlled Release, 2012, 164, 156–169 CrossRef CAS PubMed.
- S. Aula, S. Lakkireddy, K. Jamil, A. Kapley, A. V. N. Swamy and H. R. Lakkireddy, RSC Adv., 2015, 5, 47830–47859 RSC.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Chen, Q. Yang, D. W. Felsher and H. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668–7672 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- C. Ménard-Moyon, E. Venturelli, C. Fabbro, C. Samorì, T. Da Ros, K. Kostarelos, M. Prato and A. Bianco, Expert Opin. Drug Discovery, 2010, 5, 691–707 CrossRef PubMed.
- M. Lucafò, S. Pacor, C. Fabbro, T. Da Ros, S. Zorzet, M. Prato and G. Sava, J. Nanopart. Res., 2012, 14, 830 CrossRef.
- W. Zhang, Z. Guo, D. Huang, Z. Liu, X. Guo and H. Zhong, Biomaterials, 2011, 32, 8555–8561 CrossRef CAS PubMed.
- V. D. Punetha, S. Rana, H. J. Yoo, A. Chaurasia, J. T. McLeskey, M. S. Ramasamy, N. G. Sahoo and J. W. Cho, Prog. Polym. Sci., 2017, 67, 1–47 CrossRef CAS.
- S. Shukla and N. F. Steinmetz, Exp. Biol. Med., 2016, 241, 1116–1126 CrossRef CAS PubMed.
- D. Ag, M. Seleci, R. Bongartz, M. Can, S. Yurteri, I. Cianga, F. Stahl, S. Timur, T. Scheper and Y. Yagci, Biomacromolecules, 2013, 14, 3532–3541 CrossRef CAS PubMed.
- M. Yuksel, D. G. Colak, M. Akin, I. Cianga, M. Kukut, E. I. Medine, M. Can, S. Sakarya, P. Unak, S. Timur and Y. Yagci, Biomacromolecules, 2012, 13, 2680–2691 CrossRef CAS PubMed.
- H. Ali-Boucetta, K. T. Al-Jamal, D. McCarthy, M. Prato, A. Bianco and K. Kostarelos, Chem. Commun., 2008, 459–461 RSC.
- X. Lou, C. Detrembleur, V. Sciannamea, C. Pagnoulle and R. Jérôme, Polymer, 2004, 45, 6097–6102 CrossRef CAS.
- Y. Wan, W. Deng, Y. Su, X. Zhu, C. Peng, H. Hu, H. Peng, S. Song and C. Fan, Biosens. Bioelectron., 2011, 30, 93–99 CrossRef CAS PubMed.
- J. J. Khandare, A. Jalota-Badhwar, S. D. Satavalekar, S. G. Bhansali, N. D. Aher, F. Kharas and S. S. Banerjee, Nanoscale, 2012, 4, 837–844 RSC.
- S. Sharma, N. K. Mehra, K. Jain and N. K. Jain, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1851–1860 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- K. H. Son, J. H. Hong and J. W. Lee, Int. J. Nanomed., 2016, 11, 5163–5185 CrossRef CAS PubMed.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J.-P. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- P. Anilkumar, K. N. Tackett, F. Lu, P. G. Luo, L. Gu, A. Anderson and Y.-P. Sun, in Petite and Sweet: Glyco-Nanotechnology as a Bridge to New Medicines, ed. J. J. B. X. Huang, American Chemical Society, 2011, vol. 1901, ch. 8, pp. 123–141 Search PubMed.
- L. Gu, P. G. Luo, H. Wang, M. J. Meziani, Y. Lin, L. M. Veca, L. Cao, F. Lu, X. Wang, R. A. Quinn, W. Wang, P. Zhang, S. Lacher and Y.-P. Sun, Biomacromolecules, 2008, 9, 2408–2418 CrossRef CAS PubMed.
- B. K. Gorityala, J. Ma, X. Wang, P. Chen and X.-W. Liu, Chem. Soc. Rev., 2010, 39, 2925–2934 RSC.
- S. Barone, S. L. Fussell, A. K. Singh, F. Lucas, J. Xu, C. Kim, X. Wu, Y. Yu, H. Amlal, U. Seidler, J. Zuo and M. Soleimani, J. Biol. Chem., 2009, 284, 5056–5066 CrossRef CAS PubMed.
- A. R. Manolescu, K. Witkowska, A. Kinnaird, T. Cessford and C. Cheeseman, Physiology, 2007, 22, 234–240 CrossRef CAS PubMed.
- J. Zhao, K. Babiuch, H. Lu, A. Dag, M. Gottschaldt and M. H. Stenzel, Chem. Commun., 2014, 50, 15928–15931 RSC.
- J. C. Zhao, H. W. Lai, H. X. Lu, M. H. Stenzel and P. Xiao, J. Controlled Release, 2017, 259, E107–E107 CrossRef.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- F. Sonvico, S. Mornet, S. Vasseur, C. Dubernet, D. Jaillard, J. Degrouard, J. Hoebeke, E. Duguet, P. Colombo and P. Couvreur, Bioconjugate Chem., 2005, 16, 1181–1188 CrossRef CAS PubMed.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS PubMed.
- A. Gangrade and B. B. Mandal, ACS Biomater. Sci. Eng., 2019, 5, 2365–2381 CrossRef CAS.
- H. Huang, Q. Yuan, J. S. Shah and R. D. K. Misra, Adv. Drug Delivery Rev., 2011, 63, 1332–1339 CrossRef CAS PubMed.
- N. K. Mehra and N. K. Jain, Mol. Pharmaceutics, 2015, 12, 630–643 CrossRef CAS PubMed.
- M. Habibizadeh, K. Rostamizadeh, N. Dalali and A. Ramazani, Mater. Sci. Eng., C, 2017, 75, 1527 CrossRef PubMed.
- L. Yang, M. Liu, K. Huang, X. Ai, C. Li, J. Ma, T. Jin and X. Liu, Appl. Surf. Sci., 2017, 392, 189–195 CrossRef CAS.
- T. A. Saleh, Appl. Surf. Sci., 2011, 257, 7746–7751 CrossRef CAS.
- C. Lynam, N. Gilmartin, A. I. Minett, R. O’Kennedy and G. Wallace, Carbon, 2009, 47, 2337–2343 CrossRef CAS.
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS.
- Y. Jing, H. Tang, G. Yu and P. Wu, Polym. Chem., 2013, 4, 2598–2607 RSC.
- Y. Jiang, H. Lu, Y. Y. Khine, A. Dag and M. H. Stenzel, Biomacromolecules, 2014, 15, 4195–4205 CrossRef CAS PubMed.
- A. W. Jackson, C. Stakes and D. A. Fulton, Polym. Chem., 2011, 2, 2500–2511 RSC.
- P. S. Omurtag, B. Alkan, H. Durmaz, G. Hizal and U. Tunca, Polymer, 2018, 141, 213–220 CrossRef CAS.
- A. Dag, M. Callari, H. X. Lu and M. H. Stenzel, Polym. Chem., 2016, 7, 1031–1036 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- J. J. Castillo, T. Rindzevicius, L. V. Novoa, W. E. Svendsen, N. Rozlosnik, A. Boisen, P. Escobar, F. Martínez and J. Castillo-León, J. Mater. Chem. B, 2013, 1, 1475–1481 RSC.
- A. Dag, P. S. Omurtag Ozgen and S. Atasoy, Biomacromolecules, 2019, 20, 2962–2972 CrossRef PubMed.
- Y. Wang, B. B. Newell and J. Irudayaraj, J. Biomed. Nanotechnol., 2012, 8, 751–759 CrossRef CAS.
- M. Lu, Y. Y. Khine, F. Chen, C. Cao, C. J. Garvey, H. Lu and M. H. Stenzel, Biomacromolecules, 2019, 20, 273–284 CrossRef CAS PubMed.
- Y.-Z. Du, L.-L. Cai, J. Li, M.-D. Zhao, F.-Y. Chen, H. Yuan and F.-Q. Hu, Int. J. Nanomed., 2011, 6, 1559 CrossRef CAS PubMed.
- S. Dhivya, A. Keshav Narayan, R. Logith Kumar, S. Viji Chandran, M. Vairamani and N. Selvamurugan, Cell Proliferation, 2018, 51, e12408 CrossRef PubMed.
- M. Namvari and H. J. J. o. M. S. Namazi, J. Mater. Sci., 2015, 50, 5348–5361 CrossRef CAS.
- L. Zhang, C. Li, A. Liu and G. Shi, J. Mater. Chem., 2012, 22, 8438–8443 RSC.
- K.-C. Mei, N. Rubio, P. M. Costa, H. Kafa, V. Abbate, F. Festy, S. S. Bansal, R. C. Hider and K. T. Al-Jamal, Chem. Commun., 2015, 51, 14981–14984 RSC.
- H. Li, Q. Zheng and C. Han, Analyst, 2010, 135, 1360–1364 RSC.
- A. Vora, A. Riga, D. Dollimore and K. S. Alexander, Thermochim. Acta, 2002, 392-393, 209–220 CrossRef CAS.
- A. Neacşu, Thermochim. Acta, 2018, 661, 51–58 CrossRef.
- D. Wang, Y. Ren, Y. Shao, D. Yu and L. Meng, Bioconjugate Chem., 2017, 28, 2815–2822 CrossRef CAS PubMed.
- E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS PubMed.
- H. Saito, A. S. Hoffman and H. I. Ogawa, J. Bioact. Compat. Polym., 2007, 22, 589–601 CrossRef CAS.
- P. S. Pramod, R. Shah and M. Jayakannan, Nanoscale, 2015, 7, 6636–6652 RSC.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- C. Sanson, C. Schatz, J. F. Le Meins, A. Soum, J. Thevenot, E. Garanger and S. Lecommandoux, J. Controlled Release, 2010, 147, 428–435 CrossRef CAS PubMed.
- R. Wang, C. Mikoryak, S. Li, D. Bushdiecker, I. H. Musselman, P. Pantano and R. K. Draper, Mol. Pharmaceutics, 2011, 8, 1351–1361 CrossRef CAS.
- I. Hamann, D. Krys, D. Glubrecht, V. Bouvet, A. Marshall, L. Vos, J. R. Mackey, M. Wuest and F. Wuest, FASEB J., 2018, 32, 5104–5118 CrossRef CAS PubMed.
- T. Yang, F. Xu, D. Fang and Y. Chen, Sci. Rep., 2015, 5, 16733 CrossRef CAS.
- M. D. A. Salazar and M. Ratnam, Cancer Metastasis Rev., 2007, 26, 141–152 CrossRef CAS.
- J. M. Dobson, A. E. Hohenhaus and A. E. Peaston, in Small Animal Clinical Pharmacology, ed. J. E. Maddison, S. W. Page and D. B. Church, W.B. Saunders, 2008, ch. 15, pp. 330–366 Search PubMed.
- C. Bertoli, J. M. Skotheim and R. A. de Bruin, Nat. Rev. Mol. Cell Biol., 2013, 14, 518–528 CrossRef CAS PubMed.
- W. K. Yong, Y. F. Ho and S. N. Malek, Pharmacogn. Mag., 2015, 11, S275–S283 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The materials section, the characterization procedures, NMR, FT-IR, UV-vis, GPC, TEM, DSC, Dox-release, fluorescence intensity, MTT, cell apoptosis and cell cycle distribution data. See DOI: 10.1039/c9tb02711d |
| This journal is © The Royal Society of Chemistry 2020 |