
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed three-component olefin reductive dicarbofunctionalization to access alkylborates†
Xiao-Xu
Wang
,
Xi
Lu
 *,
Shi-Jiang
He
and
Yao
Fu
*
*,
Shi-Jiang
He
and
Yao
Fu
*
Hefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Urban Pollutant Conversion, Anhui Province Key Laboratory of Biomass Clean Energy, iChEM, University of Science and Technology of China, Hefei 230026, China. E-mail: luxi@mail.ustc.edu.cn; fuyao@ustc.edu.cn
First published on 8th July 2020
Abstract
We report a three-component olefin reductive dicarbofunctionalization for constructing alkylborates, specifically, nickel-catalyzed reductive dialkylation and alkylarylation of vinyl boronates with a variety of alkyl bromides and aryl iodides. This reaction exhibits good coupling efficiency and excellent functional group compatibility, providing convenient access to the late-stage modification of complex natural products and drug molecules. Combined with alkylborate transformations, this reaction could also find applications in the modular and convergent synthesis of complex compounds.
Introduction
Olefins are fundamental chemicals in organic synthesis. They frequently occur in natural products, are produced in enormous quantities in the petroleum industry, and are prepared through a variety of synthetic methods in the laboratory. The reactive double bonds make olefins attractive substrates for high-complexity synthesis. Among the well-developed olefin functionalization strategies,1,2 the rising reductive dicarbofunctionalization has already been proven to be a powerful and straightforward method (Scheme 1a).3 For example, Nevado and co-workers realized nickel-catalyzed intermolecular olefin reductive alkylarylation, in which one C(sp3)–C(sp3) bond and one C(sp3)–C(sp2) bond were formed.4 Chu and co-workers reported an example of intermolecular olefin reductive carboacylation with fluoroalkyl iodides and acyl chlorides.5 However, the development of important dialkylation processes6 is relatively limited and still relies on organometallics that are sensitive to many functional groups.7,8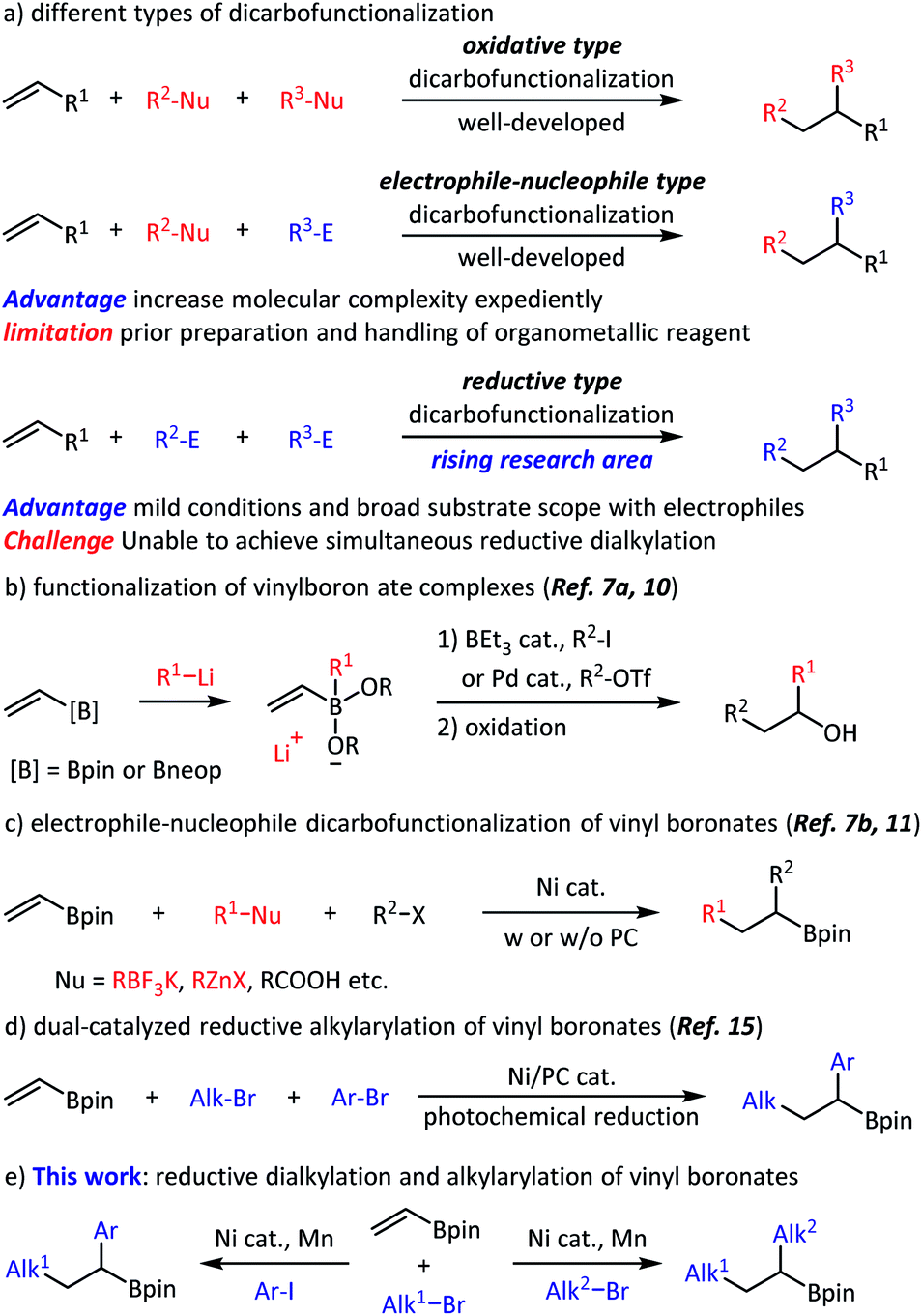 | ||
| Scheme 1 Dicarbofunctionalization of vinyl boronates to access alkylborates. B2pin2 = bis(pinacolato)diboron. B2neop2 = bis(neopentyl glycolato)diboron. Tf = triflyl. Nu = nucleophile. E = electrophile. w = with. w/o = without. PC = photoredox catalysis. Ar = aryl. Alk = alkyl. | ||
Recently, dicarbofunctionalization of commercially available vinyl boronates has been applied to the diversified synthesis of alkylborates.9 For example, Morken, Studer and Aggarwal independently achieved functionalization of a vinylboron ate complex using an organolithium reagent and another electrophile (Scheme 1b).7a,10 Most recently, the electrophile-nucleophile dicarbofunctionalization of a vinyl boronate has been achieved with appropriate radical precursor (Scheme 1c).7b,11 In addition, hydroalkylation and hydroarylation of alkenyl boronic esters has also been realized.12 Despite these great successes, general and modular methods to access alkylborates without using any organometallic reagents are still desirable. With our focus on olefin reductive coupling13 and alkylborate synthesis,14 we set out to realize the regioselective dicarbofunctionalization of vinyl boronates, taking advantage of nickel-catalyzed reductive coupling and classical Giese-type addition. Very recently, Martin and co-workers reported an efficient alkylarylation of vinyl boronates through the nickel/photoredox dual-catalyzed reductive cross-coupling (Scheme 1d).15 Our work extended the approach towards an alkyl,alkyl-difunctionalization. Intermolecular three-component reductive olefin dialkylation, especially alkyl,alkyl-difunctionalization of vinyl boronates, could find many applications in organic synthesis and medicinal chemistry; however, to the best of our knowledge, is yet to be reported, and is addressed in this work. Herein, we report a three-component olefin reductive dicarbofunctionalization for constructing alkylborates, specifically, nickel-catalyzed reductive dialkylation and alkylarylation of vinyl boronates with a variety of alkyl bromides and aryl iodides (Scheme 1e). This reaction shows good coupling efficiency, excellent functional group compatibility, and a high degree of regioselectivity. From the point of view of alkylborate transformations,16 this reaction might find a number of applications in modular and convergent synthesis of complex, densely functionalized compounds.
Results and discussion
We began this study with the synthesis of alkylborate 4 through the proposed dialkylation (Table 1). We systematically screened all the reaction parameters (see the ESI† for more details), and desired product 4 was obtained in 88% gas chromatography (GC) yield and 83% isolated yield in the presence of NiBr2(diglyme), a dipyrazolpyridine ligand (L), a Mn(0) reductant, and a NaI additive in DMAc (entry 1). A number of other nitrogen-containing ligands were compared: all bidentate ligands, bipyridine (L1 and L2), pyridine-oxazoline (L3), and bioxazoline (L4) were inefficient; tridentate tripyridine (L5) yielded only a small amount of the desired products; and pyridine-oxazoline ligands (L6 and L7) produced moderate yields. In the absence of nickel catalysts or ligands, dialkylation could not proceed (entry 2). Other nickel sources, including NiCl2(PPh3)2, NiI2, and Ni(COD)2 could also be used instead of NiBr2(diglyme); however, they led to different decreases in coupling efficiency (entry 3). Metal Zn13c,17 and diboron13b,18 were potential reductants for this transformation (entries 4–6),19 but our previously used nickel-silane reductive system13a,d was incompetent (entry 7). Amide solvents were critical: DMF and NMP resulted in comparable yields (entry 8) to that of DMAc (optimal conditions), but the reaction was completely inhibited in THF, 1,4-dioxane, CH3CN, and DMSO (entry 9). The performance of primary alkyl iodide 5 was barely satisfactory (entry 10), and a significant amount of homocoupling product 6 was observed. Tertiary alkyl bromides were irreplaceable: the corresponding iodides and chlorides provided no dialkylation product (entry 11), even in the presence of activator reagents (entry 12). Finally, the iodide ion additive and ratio of starting material were also carefully selected (entries 13–16).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
| a Standard conditions: 1 (0.1 mmol, 1.0 equiv.), 2 (0.2 mmol, 2.0 equiv.), 3 (0.2 mmol, 2.0 equiv.), NiBr2(diglyme) (0.01 mmol, 10 mol%), L (0.012 mmol, 12 mol%), Mn (0.3 mmol, 3.0 equiv.), NaI (0.05 mmol, 0.5 equiv.), DMAc (0.5 mL, 0.2 M), argon, room temperature (r.t.), 12 h. GC yield. 4,4′-Dimethoxybenzophenone was used as an internal standard. b Isolated yield. Bz = benzoyl. Diglyme = 2-methoxyethyl ether. DMAc = N,N-dimethylacetamide. COD = cis,cis-1,5-cyclooctadiene. DEMS = diethoxymethylsilane. DMF = N,N-dimethylformamide. NMP = 1-methyl-2-pyrrolidinone. THF = tetrahydrofuran. DMSO = dimethyl sulfoxide. Cp2TiCl2 = titanocene dichloride. TBAI = tetrabutylammonium iodide. | ||
| 1 | None | 88 (83b) |
| 2 | W/o NiBr2(diglyme) or w/o L | N.R. |
| 3 | NiCl2(PPh3)2, NiI2, or Ni(COD)2 instead of NiBr2(diglyme) | 49–83 |
| 4 | 3.0 eq. Zn instead of Mn | 76 |
| 5 | 3.0 eq. B2pin2 and 3.0 eq. LiOMe instead of Mn | 44 |
| 6 | 3.0 eq. B2pin2 and 3.0 eq. K3PO4 instead of Mn | 50 |
| 7 | 3.0 eq. DEMS and 3.0 eq. Na2CO3 instead of Mn | 11 |
| 8 | DMF, or NMP instead of DMAc | 72–87 |
| 9 | THF, 1,4-dioxane, CH3CN, or DMSO instead of DMAc | <2 |
| 10 | 5 instead of 3 | 54 |
| 11 | tBuI or tBuCl instead of tBuBr | <2 |
| 12 | tBuCl with 20% Cp2TiCl2 instead of tBuBr | <2 |
| 13 | 30% TBAI instead of 50% NaI | 82 |
| 14 | 20% NaI instead of 50% NaI | 45 |
| 15 | Ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 = 1:1:1 instead of 1:2:2 :2:3 = 1:1:1 instead of 1:2:2 |
52 |
| 16 | Ratio of 1:2:3 = 1:1.5:2 instead of 1:2:2 |
76 |

|
||

With suitable conditions in hand, we set out to evaluate the scope of this olefin dialkylation reaction. As shown in Scheme 2, a variety of primary alkyl bromides delivered desired products 7–25 in moderate to good yields (60–84%). Because of mild reductive cross-coupling conditions, this reaction exhibited good compatibility with a wide range of synthetically useful functional groups, such as ester (4), ether (7–8), aryl fluoride (10), trifluoromethyl (11), and trifluoromethoxy (12) groups. Satisfactory chemoselectivity was observed in compounds 13–15; in these cases, aryl chlorides and bromides were proven to be less reactive than alkyl bromides. This chemoselectivity provided a profitable platform for further manipulations at the surviving aryl electrophilic sites. Both base-sensitive ketone (16) and cyano (17) groups and acid-sensitive acetal (18) groups posed no problem during this transformation. Several heterocycles such as phthalimide (19), thiophene (20), furan (21), morpholine (22), and indole (23) moieties, were well tolerated. Finally, this reaction also performed well in the presence of amide-possessing N–H bonds (24) and unprotected alcohol (25) groups.
 | ||
| Scheme 2 Substrate scope of primary alkyl bromides. Standard conditions: as shown in Table 1, entry 1, 0.2 mmol scale. Isolated yield. a The product was isolated after the oxidization of the corresponding alkylborate. Isolated yield. b Nuclear magnetic resonance (NMR) yield for the corresponding alkylborate. Dibromomethane was used as an internal standard. | ||
The versatility of this reaction was further demonstrated in terms of the tertiary alkyl partners (Scheme 3). Both acyclic (26–30) and cyclic (31–33) tertiary alkyl bromides were successfully converted to the desired products. With respect to the acyclic substrates, dramatically different steric hindrances only resulted in a slight influence on the coupling efficiencies. Finally, tertiary alkyl bromides containing ester (32), ether (33), acetal (34), and C(sp3)–Cl (35) groups were indeed good substrates during the transformation and afforded the corresponding products with moderate to good isolated yields.
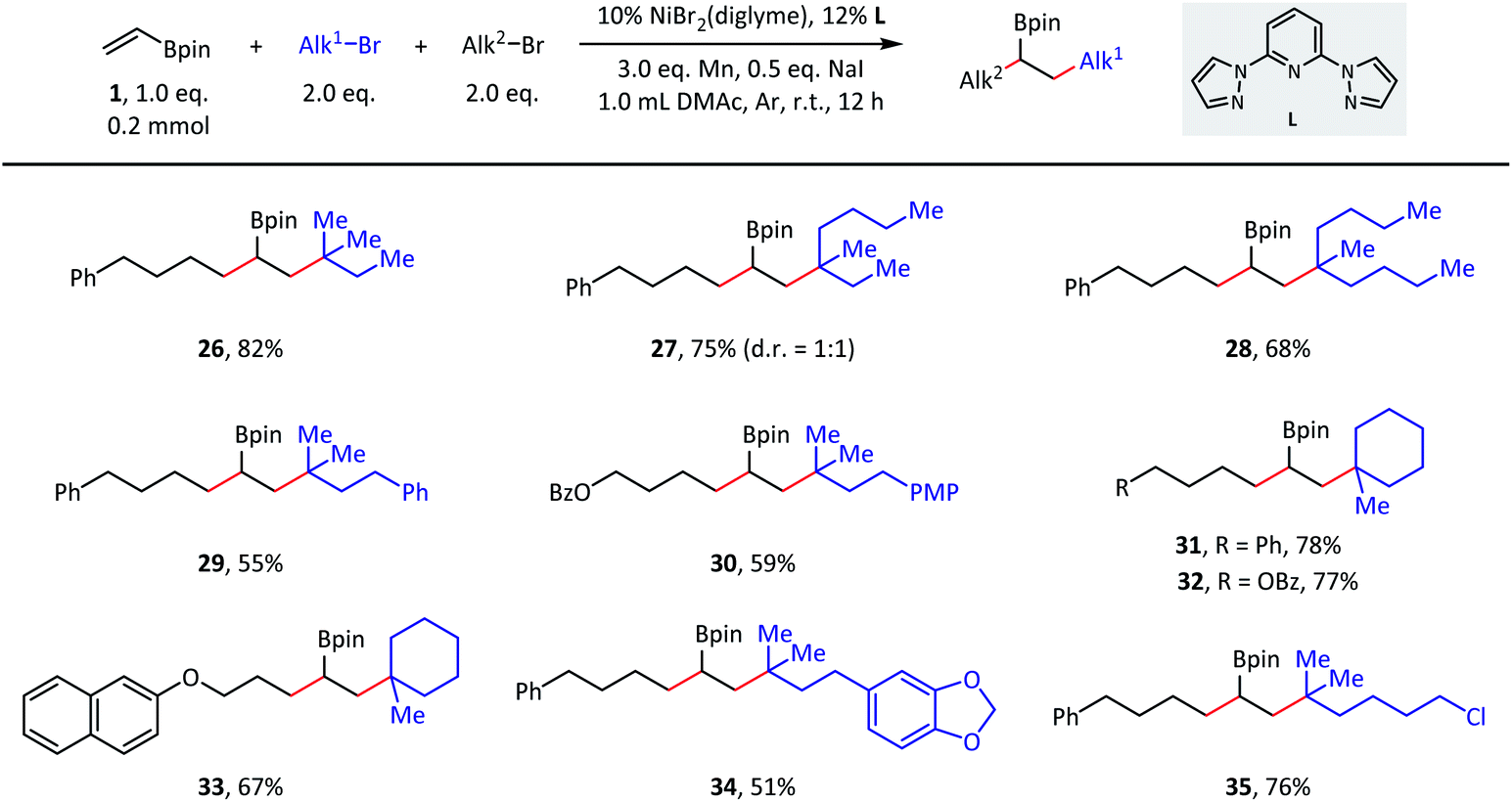 | ||
| Scheme 3 Substrate scope of tertiary alkyl bromides. Standard conditions: as shown in Table 1, entry 1, 0.2 mmol scale. Isolated yield. PMP = p-methoxyphenyl. | ||
Although the primary focus of this study was olefin reductive dialkylation, the optimized conditions could also be extended to alkylarylation (Scheme 4). Benzylic boronates were obtained conveniently with the simultaneous formation of one aryl–alkyl bond and one alkyl–alkyl bond. With respect to aryl coupling partners, both electron-donating (36) and electron-withdrawing (37–39) substituents were well tolerated in the meta- and para-positions and afforded the corresponding products in moderate (39–50%) isolated yields. In addition, this transformation is orthogonal to classical Suzuki cross-coupling procedures, as the C(sp2)–B bond remained intact in substrate 40. Finally, different tertiary alkyl bromides were also explored (41–43), in which the desired alkylarylation products were delivered smoothly.
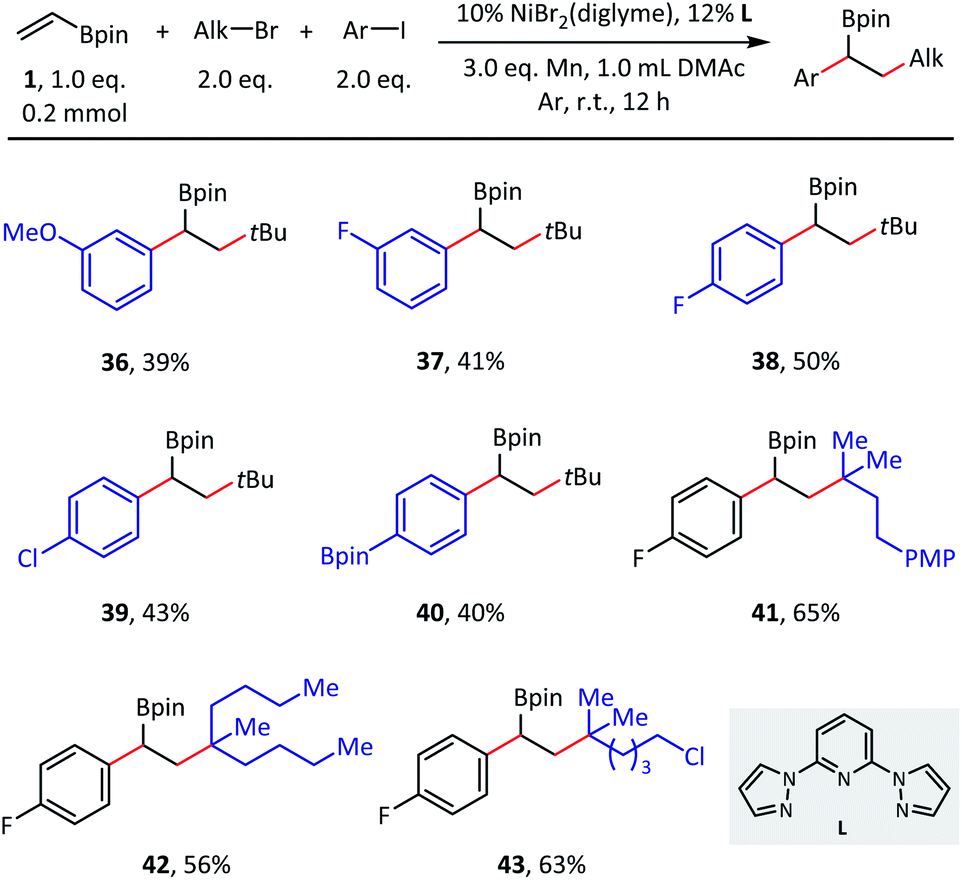 | ||
| Scheme 4 Substrate scope of olefin reductive alkylarylation. Conditions: as shown in Table 1, entry 1, without NaI, 0.2 mmol scale. Isolated yield. | ||
In a scale-up reaction, we successfully obtained reductive dialkylation product 9 with a satisfactory 84% isolated yield (Scheme 5a), which highlights the practicality of this new alkylborate synthetic method. Combined with alkylborate transformations, our method provided a modular strategy for the synthesis of complex compounds (Scheme 5b). For example, structurally complicated alcohols (45–46),11a diaryl alkanes (47–48),20 and alkyl iodide (49)21 were created via such an assembly-line synthetic route. Finally, we used this method for the late-stage functionalization of complex natural products and drug molecules (Scheme 5c). The efficient conversion of glucose (50), indomethacin (51), and oleanic acid (52) derivatives to the desired products demonstrated a high degree of tolerance to diverse functional groups.
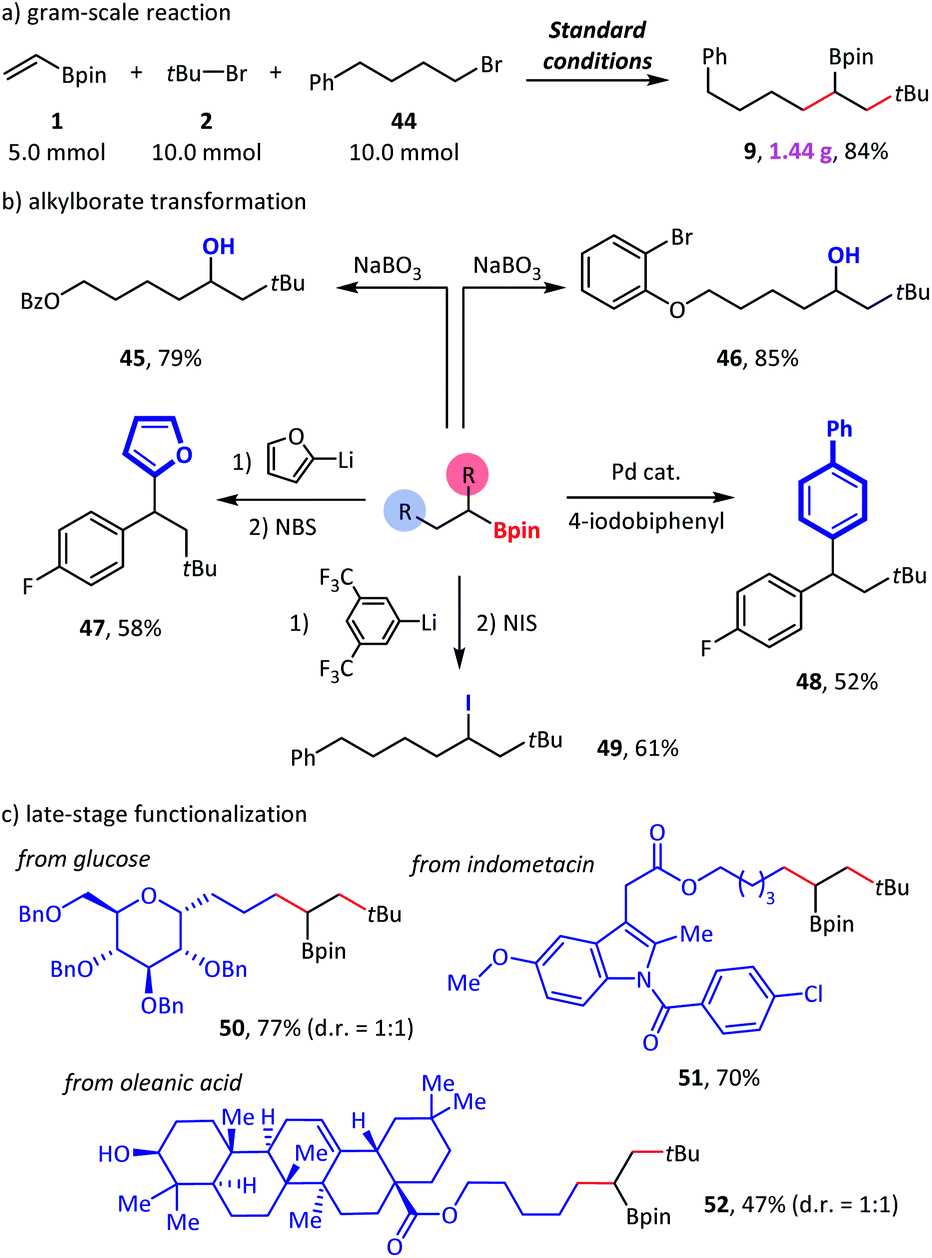 | ||
| Scheme 5 Synthetic applications. Standard conditions: as shown in Table 1, entry 1. Isolated yield. See the ESI† for more details. NBS = N-bromosuccinimide. NIS = N-iodosuccinimide. | ||
To examine the reaction mechanism, we carried out competition experiments (Scheme 6a). 4-Bromobut-1-ene (53) was subjected to standard conditions, and desired product 54 was obtained in 63% isolated yield, with the terminal alkene group retained. In the competition reaction between vinyl boronate (1) and dec-1-ene (55), the vinyl boronate dialkylation product (4) was formed in 70% GC yield. However, the dialkylation product (56) of dec-1-ene was not observed, with 77% recovery of the starting material dec-1-ne (55). The competition reaction between vinyl boronate (1) and acrylamide (57) was also conducted, and both the vinyl boronate dialkylation product (9) and the acrylamide alkylation product (58) were observed. Thus, electron-deficient olefins were more reactive in this reaction, and no reaction occurred for the electron-rich olefins. In addition, tertiary alkyl bromides exhibited higher radical addition reactivity than the primary alkyl bromides (see ESI† for more details). The radical clock experiment was tested using (bromomethyl)cyclopropane (60), and we obtained only ring opening product 54 in 42% yield, which revealed the radical activation of primary alkyl bromides (Scheme 6b). Finally, the nonmetallic reductant TDAE was used instead of Mn(0) and resulted in a decent 25% GC yield. We deduced that the activation of alkyl bromides was a single-electron-transfer (SET) process, but not the in situ formation of alkylmanganese reagents (Scheme 6c).
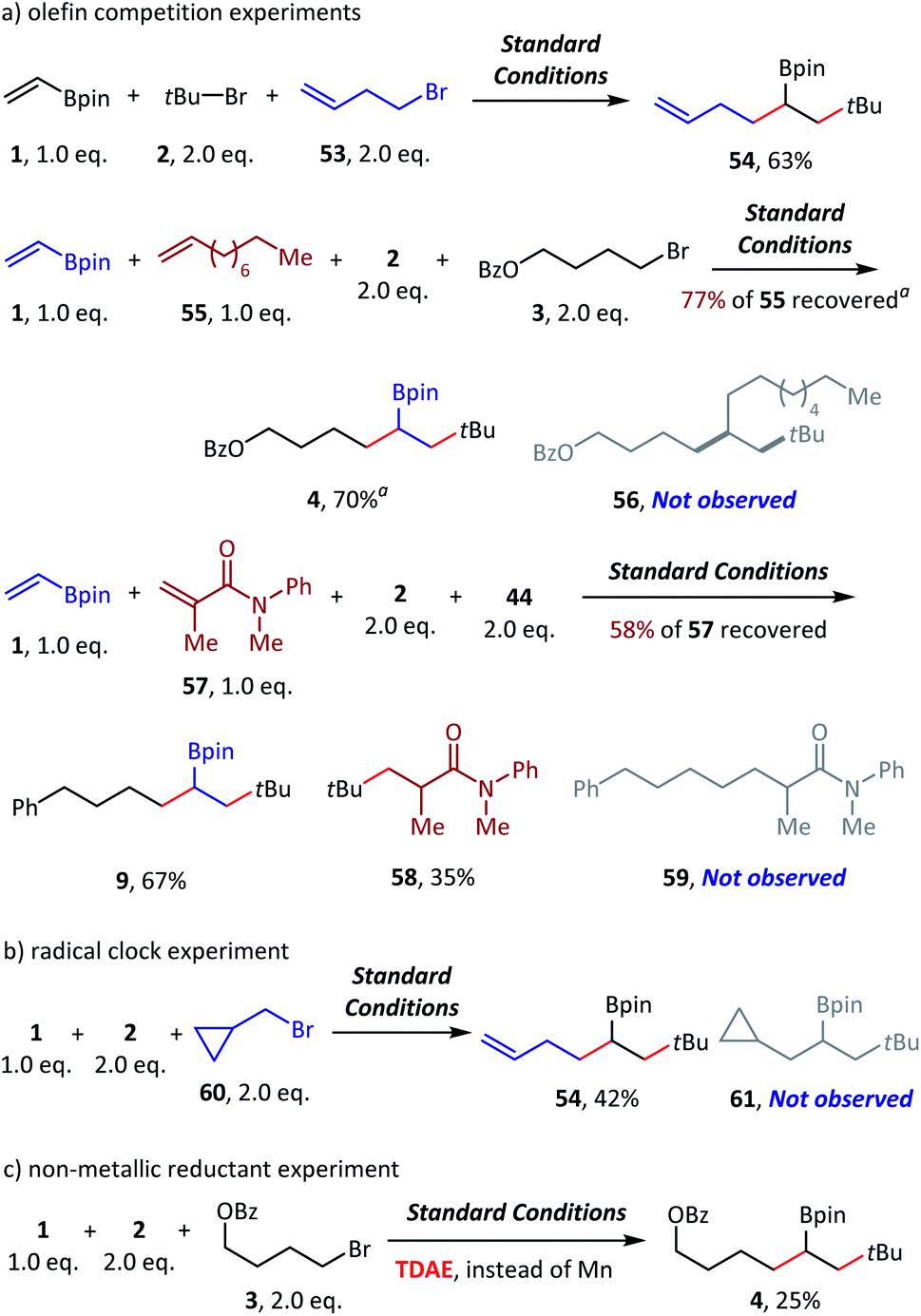 | ||
| Scheme 6 Mechanistic probes. Standard conditions: as shown in Table 1, entry 1, 0.2 mmol scale. Isolated yield. a GC yield. 4,4′-Dimethoxybenzophenone was used as an internal standard. See the ESI† for more details. TDAE = tetrakis(dimethylamino)ethylene. | ||
Based on the aforementioned experimental observations and previous literatures,4a,11a,22 an envisioned mechanism for this olefin reductive dicarbofunctionalization was proposed (Scheme 7). We had sufficient evidences to prove that this reaction was initiated with the formation of a nucleophilic tert-alkyl radical (I), and added to the vinyl boronate (1). Then the resulting sec-alkyl radical (II), which was stabilized by contiguous boron atom, might be trapped by a nickel catalyst, and then to finish the cross-coupling. The mechanistic basis of our reaction design, namely the formation of a boron atom stabilized radical, was in consistent with Martin's work.15 It should also be pointed out that the actual mechanism might be more complicated due to a number of changeable valence states of nickel catalysts under reductive conditions.
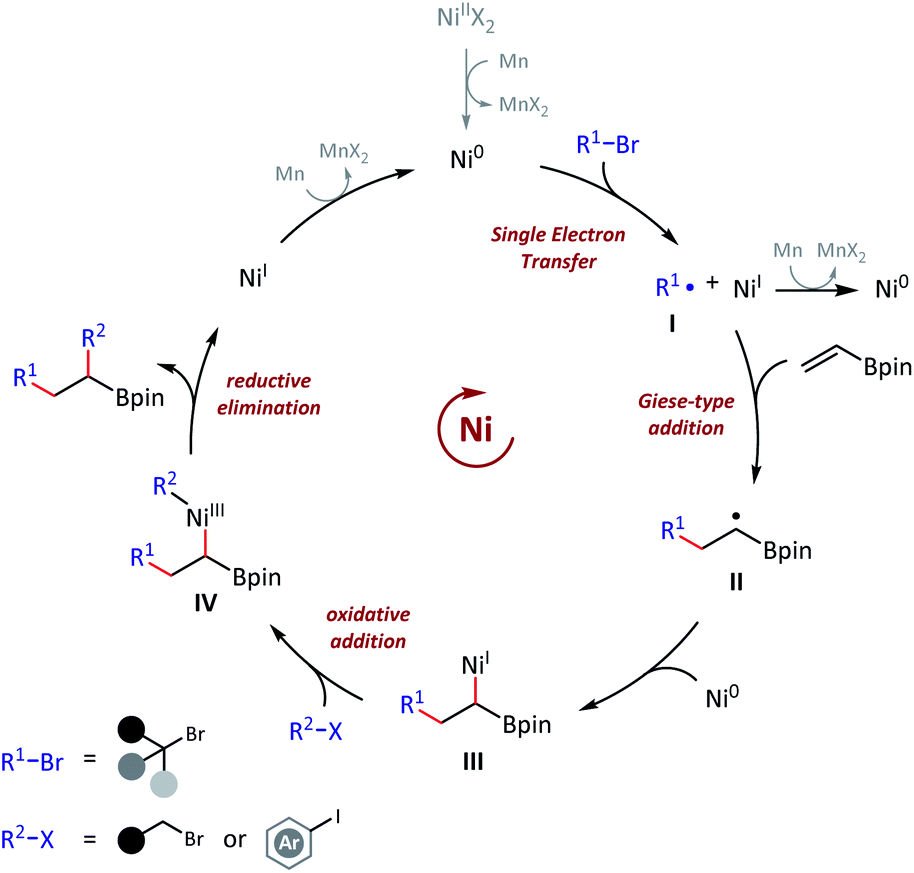 | ||
| Scheme 7 Envisioned mechanism. R1 = tert-alkyl. R2 = prim-alkyl or aryl. X = halogen. | ||
Conclusions
We reported a convenient method to access alkylborates through nickel-catalyzed olefin reductive dialkylation and alkylarylation of vinyl boronates. A variety of alkyl bromides and aryl iodides were converted to the corresponding products with both good coupling efficiency and excellent functional group compatibility. This reaction is practical and useful in the late-stage modification of natural products and the modular synthesis of complex compounds. Our next challenge is the improvement of stereochemical control.23Conflicts of interest
There is no conflict of interest to report.Acknowledgements
The authors acknowledge the generous grants from the National Natural Science Foundation of China (Grants 21732006, 21702200, 51821006, 21927814, and 51961135104), the National Key R&D Program of China (Grant 2017YFA0303502), the Strategic Priority Research Program of CAS (Grant XDB20000000), and the Fundamental Research Funds for the Central Universities.Notes and references
- For selected reviews on olefin dicarbofunctionalization, see: (a) R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314 CrossRef CAS PubMed; (b) J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem.–Asian J., 2018, 13, 2277 CrossRef CAS PubMed; (c) Y. Ping, Y. Li, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562 CrossRef CAS PubMed; (d) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013 CrossRef CAS PubMed.
- For selected examples on olefin dicarbofunctionalization, see: (a) K. B. Urkalan and M. S. Sigman, Angew. Chem., Int. Ed., 2009, 48, 3146 CrossRef CAS PubMed; (b) A. Trejos, A. Fardost, S. Yahiaoui and M. Larhed, Chem. Commun., 2009, 7587 RSC; (c) M. S. McCammant, L. Liao and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 4167 CrossRef CAS PubMed; (d) Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122 CrossRef CAS PubMed; (e) B. Shrestha, P. Basnet, R. K. Dhungana, S. Kc, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653 CrossRef CAS PubMed; (f) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC; (g) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657 CrossRef CAS PubMed; (h) P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. Kc, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782 CrossRef CAS PubMed; (i) H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed; (j) W. You and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 14578 CrossRef CAS PubMed; (k) J. A. Walker, K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228 CrossRef CAS PubMed; (l) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (m) J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270 CrossRef CAS PubMed; (n) L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904 CrossRef CAS PubMed; (o) M. P. Watson and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 12594 CrossRef CAS PubMed; (p) W. You and M. K. Brown, J. Am. Chem. Soc., 2014, 136, 14730 CrossRef CAS PubMed; (q) L. Zhou, S. Li, B. Xu, D. Ji, L. Wu, Y. Liu, Z.-M. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 2769 CrossRef CAS PubMed; (r) D. Huang, D. Olivieri, Y. Sun, P. Zhang and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 16249 CrossRef CAS PubMed; (s) Z.-M. Zhang, B. Xu, L. Wu, Y. Wu, Y. Qian, L. Zhou, Y. Liu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14653 CrossRef CAS; (t) M. Koy, P. Bellotti, F. Katzenburg, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 2375 CrossRef CAS PubMed.
- For selected examples on olefin reductive dicarbofunctionalization, see: (a) D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198 CrossRef CAS PubMed; (b) J. Li, Y. Luo, H. W. Cheo, Y. Lan and J. Wu, Chem, 2019, 5, 192 CrossRef CAS; (c) K. Wang, Z. Ding, Z. Zhou and W. Kong, J. Am. Chem. Soc., 2018, 140, 12364 CrossRef CAS PubMed; (d) C.-S. Yan, Y. Peng, X.-B. Xu and Y.-W. Wang, Chem.–Eur. J., 2012, 18, 6039 CrossRef CAS PubMed; (e) Z.-X. Tian, J.-B. Qiao, G.-L. Xu, X. Pang, L. Qi, W.-Y. Ma, Z.-Z. Zhao, J. Duan, Y.-F. Du, P. Su, X.-Y. Liu and X.-Z. Shu, J. Am. Chem. Soc., 2019, 141, 7637 CrossRef CAS PubMed.
- (a) W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, J. Am. Chem. Soc., 2019, 141, 13812 CrossRef CAS PubMed; (b) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed.
- X. Zhao, H.-Y. Tu, L. Guo, S. Zhu, F.-L. Qing and L. Chu, Nat. Commun., 2018, 9, 3488 CrossRef PubMed.
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- For selected examples of olefin dialkylation using organometallics, see: (a) M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Science, 2017, 355, 936 CrossRef CAS PubMed; (b) M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 14245 CrossRef CAS PubMed; (c) J. Terao, K. Saito, S. Nii, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1998, 120, 11822 CrossRef CAS; (d) K. Wakabayashi, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2001, 123, 5374 CrossRef CAS PubMed; (e) J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liu and K. M. Engle, Chem. Sci., 2018, 9, 5278 RSC; (f) C. Xu, Z.-F. Yang, L. An and X. Zhang, ACS Catal., 2019, 9, 8224 CrossRef CAS.
- For a recent example of two-component olefin reductive dialkylation, see: Q. Lin and T. Diao, J. Am. Chem. Soc., 2019, 141, 17937 CrossRef CAS PubMed.
- (a) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155 CrossRef CAS PubMed; (b) B. Zhao, Z. Li, Y. Wu, Y. Wang, J. Qian, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 9448 CrossRef CAS PubMed.
- (a) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70 CrossRef CAS PubMed; (b) M. Silvi, C. Sandford and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 5736 CrossRef CAS PubMed.
- (a) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS PubMed; (b) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375 CrossRef CAS PubMed.
- (a) Y. Zhang, B. Han and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 13860 CrossRef CAS PubMed; (b) S. Bera and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 13854 CrossRef CAS PubMed.
- (a) X. Lu, B. Xiao, Z.-Q. Zhang, T.-J. Gong, W. Su, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed; (b) X. Lu, Y. Wang, B. Zhang, J.-J. Pi, X.-X. Wang, T.-J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632 CrossRef CAS PubMed; (c) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809 RSC; (d) S.-J. He, J.-W. Wang, Y. Li, Z.-Y. Xu, X.-X. Wang, X. Lu and Y. Fu, J. Am. Chem. Soc., 2020, 142, 214 CrossRef CAS PubMed.
- (a) W. Su, T.-J. Gong, X. Lu, M.-Y. Xu, C.-G. Yu, Z.-Y. Xu, H.-Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957 CrossRef CAS PubMed; (b) L. Li, T.-J. Gong, X. Lu, B. Xiao and Y. Fu, Nat. Commun., 2017, 8, 345 CrossRef PubMed; (c) X. Lu, Z.-Q. Zhang, L. Yu, B. Zhang, B. Wang, T.-J. Gong, C.-L. Tian, B. Xiao and Y. Fu, Chin. J. Chem., 2019, 37, 11 CrossRef CAS.
- S.-Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370 CrossRef CAS PubMed.
- (a) B. S. L. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 11700 CrossRef CAS PubMed; (b) J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk and G. C. Fu, Science, 2016, 354, 1265 CrossRef CAS PubMed; (c) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481 RSC.
- (a) L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454 CrossRef CAS PubMed; (b) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146 CrossRef CAS PubMed.
- H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022 RSC.
- (a) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC; (b) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828 CrossRef CAS PubMed.
- (a) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584 CrossRef CAS PubMed; (b) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024 CrossRef CAS PubMed.
- Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 6703 CrossRef CAS PubMed.
- Y. Jin and C. Wang, Chem. Sci., 2019, 10, 1780 RSC.
- A moderate 40% enantiomeric excess was obtained using a pyridine-oxazoline ligand (see the ESI† for more details).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02054k |
| This journal is © The Royal Society of Chemistry 2020 |