
Exploiting the mechanical bond for molecular recognition and sensing of charged species
Krzysztof M.
Bąk
 ab,
Kyriakos
Porfyrakis†
b,
Jason J.
Davis
c and
Paul D.
Beer
*a
ab,
Kyriakos
Porfyrakis†
b,
Jason J.
Davis
c and
Paul D.
Beer
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
bDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
cPhysical & Theoretical Chemistry Laboratory, South Parks Road, Oxford, OX1 3TA, UK
First published on 24th December 2019
Abstract
The unique properties of the mechanical bond have been increasingly used for the purpose of molecular recognition. The recent progress in the development of cation and anion template strategies for the construction of mechanically interlocked molecules (MIMs) have resulted in a variety of ion binding catenane and rotaxane host structures. The appropriate integration of reporting redox- and photo-active centres into their structural frameworks can result in prototype molecular sensors for targeting charged species and molecular switches for potential nanotechnological applications. This review presents progress in the field of MIM hosts for ion recognition and sensing since 2014, focusing on the synthetic approaches employed and mechanisms of host–guest binding and detection.
 Krzysztof M. Bąk | Krzysztof M. Bąk graduated from the University of Warsaw with a BSc (cum laude) and MSc (cum laude) in chemistry and BSc in physics. In 2019 he obtained PhD degree under the supervision of Dr Michał Chmielewski. He is currently working on the incorporation of fullerenes into interlocked structures and developing anion sensors as Postdoctoral Research Assistant in the Porfyrakis group and the Beer group at the University of Oxford. |
 Kyriakos Porfyrakis | Professor Kyriakos Porfyrakis FRSC holds the Chair of Materials and Chemical Engineering at the Faculty of Engineering and Science, University of Greenwich, UK. He previously held the post of Associate Professor of Materials at the Department of Materials, University of Oxford. He is also a visiting Professor at the Aristotle University of Thessaloniki, Greece. Prof. Porfyrakis has established a world-leading laboratory for the production and purification of both nitrogen-containing and metal-containing endohedral fullerene molecules. His research interests include the synthesis and functionalization of endohedral fullerenes for energy, biomedical and nanoelectronics applications. |
 Jason J. Davis | Professor Jason Davis is a Professor of Chemistry and Fellow of Christ Church, Oxford. He studied Chemistry at King's College London, then undertook a DPhil in Chemistry at Oxford (1994–1998). He was elected to an Extraordinary Junior Research Fellowship at The Queen's College in 1998, a Royal Society University Research Fellowship in 1999, a University Lectureship in 2003, a University Readership in 2008 and Full Professorship in 2014. His research is primarily focussed on the design/utilisation of advanced functional interfaces, particularly those associated with Diagnostics, Sensing, Molecular Switches and Imaging, where his group have published more than 160 papers. |
 Paul D. Beer | Professor Paul Beer obtained a PhD from King's College London in 1982 with Dr C. Dennis Hall. After a Royal Society European Post-doctoral Fellowship with Professor J.-M. Lehn and a Demonstratorship at the University of Exeter, he was awarded a Lectureship at the University of Birmingham in 1984. In 1990, he moved to the University of Oxford, where he was made a University Lecturer and Tutorial Fellow at Wadham College, and became a Professor of Chemistry in 1998. His research interests include coordination and supramolecular chemistry. |
Introduction
Mechanically interlocked molecules (MIMs) such as rotaxanes and catenanes have been the subject of intense research due to their non-trivial architectures and unique dynamic properties which can be exploited in an ever increasing range of applications.1–10 In recent years their potential in molecular guest recognition has also been realised. Various template strategies facilitate the synthesis of MIMs containing unique three dimensional cavities decorated with complementary donor/acceptor motifs required for the selective binding of a target guest species.11–15 Many interlocked structures have proved to be excellent molecular hosts displaying a notably enhanced selectivity in comparison to analogous non-interlocked systems.16 The incorporation of redox-active and photo-active reporting groups within such MIM hosts results in optical and/or electrochemical guest sensing capabilities.17 Moreover, successful recognition provides a means of control for the relative positions of interlocked components (co-conformation) in molecular switches.In this article we focus on the progress in the development of ion binding and sensing MIMs since the last reviews on this subject in 2014.18–20 Firstly, we discuss cation binding MIM host structures which remain in the minority, despite the popularity of metal-directed strategies employed in the synthesis of interlocked structures. Secondly, attention is turned to anion binding MIMs, which has been a research topic of special interest to us.
Mechanically interlocked molecules for cation recognition and sensing
The metal templated synthesis of MIMs, as pioneered by Sauvage, has become one of the most powerful tools for the formation of mechanical bonds.21 Removal of the template, typically a transition metal, provides a well-defined cavity in which the cation can be bound again. This passive template methodology usually leads to interlocked host structures which coordinatively saturate a bound metal. In 2006, Leigh and co-workers demonstrated another approach for the synthesis of MIMs, relying on a kinetic metal templating effect.22 In this active metal template (AMT) strategy, a transition metal cation plays a dual role of a template, typically bound within the cavity of a macrocycle, and a catalyst of the reaction leading to the formation of the mechanical bond. These two approaches are the most common methods employed in the synthesis of a range of MIM host structures subsequently capable of transition metal cation recognition.Goldup, Roessler and co-workers used an active template variant of Cu(I)-mediated alkyne–azide cycloaddition (CuAAC) to obtain a series of [2]rotaxanes 1–3 with an increasing number of N donors within the interlocked cavity (Fig. 1).23 Binding studies with transition metal ions such as Co2+, Ni2+, Cu2+ and Zn2+ revealed that the mechanical bond not only enhanced strength of cation binding but also enforced unusual coordination geometries of the metals, inaccessible with analogous non-interlocked ligands. Rotaxane 3, for example, formed unprecedented five-coordinate Co2+ and Ni2+ complexes in the solid state and solution as a result of the exclusion of additional ligands due to steric hindrance imbued by the rotaxane's binding pocket. In addition, as demonstrated by Sauvage in his original phenanthroline containing [2]catenanes,24 the metal's redox properties were shown to be significantly altered. For instance, the electrochemical stability of the 3·Cu2+ complex was dramatically increased in comparison to its non-interlocked analogue, by preventing a drastic reorganisation of ligands around the metal (ligand disproportionation).
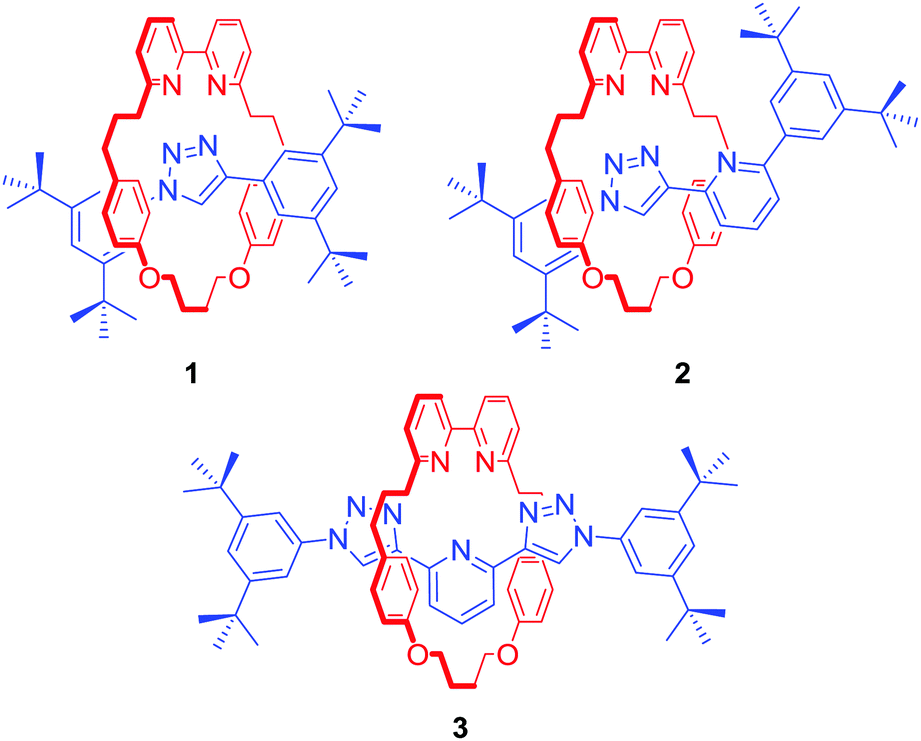 | ||
| Fig. 1 Goldup and Roessler's rotaxane hosts 1–3 for recognition of transition metals. | ||
In many cases the binding cavities of MIMs are not static but able to reorganise, allowing the encapsulation of various guests. In an elegant example, Loeb prepared a [2]rotaxane 4 containing a bis-benzimidazole axle component (Fig. 2) by a hydrogen bond template ring-closing metathesis (RCM) of a crown ether macrocyclic precursor.25 The addition of metal cations with different coordination preferences such as Cu+ and Li+ resulted in a rotation of the macrocyclic component around the axle to access the most suitable set of donors in solution and in the solid state. In the case of Cu+ the olefin moiety of the macrocycle was directly involved in cation binding. Whilst, in the case of Li+, the macrocycle was bound to the cation only via oxygen atoms. Further studies of this system showed that Ag+ can lead to remarkably different co-conformations of 4 in the solid state.26
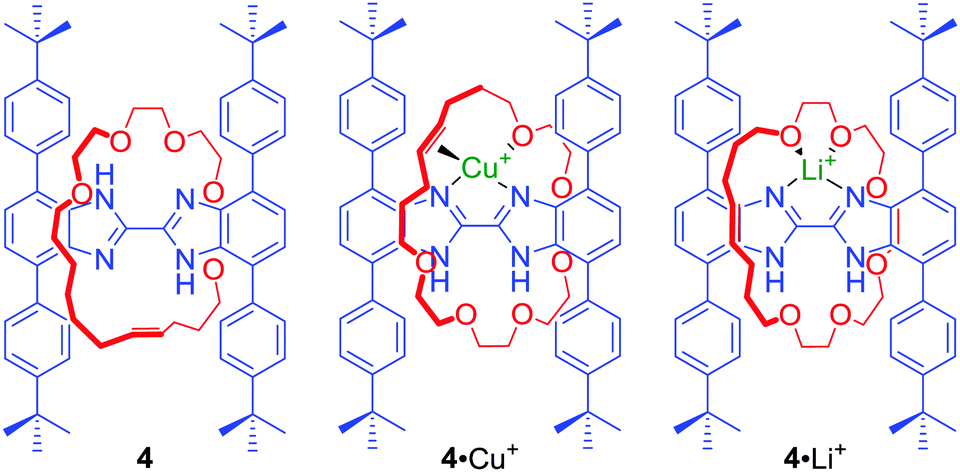 | ||
| Fig. 2 Loeb's rotaxane 4 and its complexes with Cu+ and Li+. | ||
Attaching a reporting group in close proximity to a binding pocket is a common approach to obtain ion sensing MIMs. For example Goldup, Watkinson and co-workers used the AMT-CuAAC strategy to obtain a series of [2]rotaxanes containing a fluorescent 1,8-naphthalimide derivative in the axle component (Fig. 3).27 It was found that the nature of the metal cation guest led to profound changes of the optical output. For instance, rotaxane 5 displayed a selective switch-on response upon Hg2+ binding, while 6 was selective for Zn2+ over various other transition metal cations including Cd2+ in MeCN/H2O 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Solid state studies suggested that the optical response was the result of altering macrocycle position relative to fluorophore upon binding inside the cavity. Importantly, the cavity had substantial influence over binding selectivity.
:2. Solid state studies suggested that the optical response was the result of altering macrocycle position relative to fluorophore upon binding inside the cavity. Importantly, the cavity had substantial influence over binding selectivity.
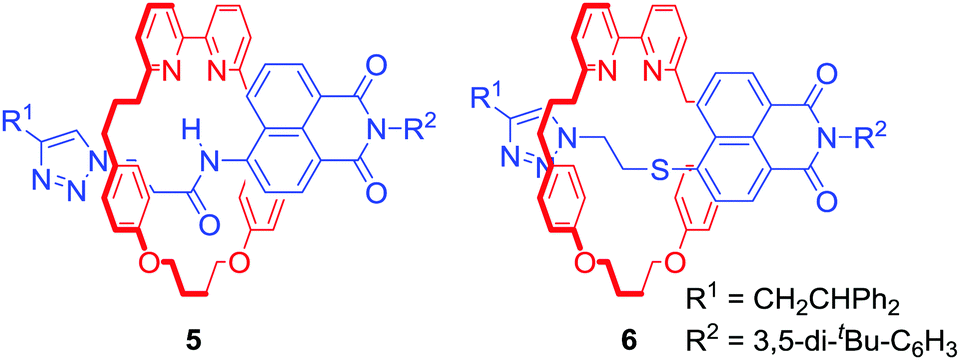 | ||
| Fig. 3 [2]Rotaxanes containing fluorescent 1,8-naphthalimide derivative in the axle component. | ||
Another approach to transition metal sensing was explored by Lin and co-workers who obtained rotaxane 7 (Fig. 4) by stoppering of a Pd2+ templated pseudorotaxane.28 One of its stopper components consisted of a tetraphenylethylene moiety which is known for exhibiting strong aggregation induced emission (AIE) behaviour. For that reason, rotaxane 7 generated a bright blue emission in the presence of more than 20% of water. Interestingly, it was selectively quenched by the addition of Fe3+ in contrast to various alkali and other transition metal ions. Additionally, emission quenching of the axle component by Fe3+ in the same solvent was significantly weaker. The authors claimed that this difference in behaviour was caused by the formation of the 1:1 stoichiometric complex between rotaxane 7 and Fe3+ due to the binding inside the cavity, which prevented aggregation of the rotaxane.
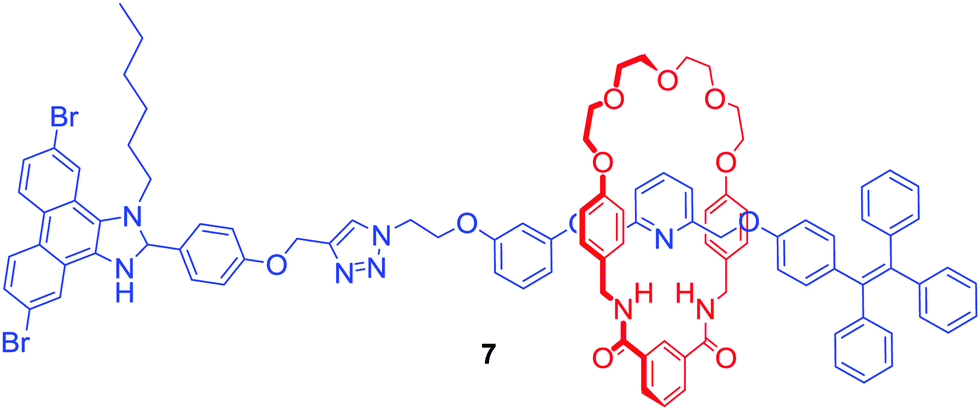 | ||
| Fig. 4 [2]Rotaxane exhibiting aggregation induced emission behaviour. | ||
Leung and co-workers used fluorescent [2]rotaxanes 8 and 9 for sensing of trivalent transition metals, particularly Au3+ (Fig. 5).29 The axle components of this system were stoppered with fluorophores, such as anthracene or BODIPY, whose fluorescence was quenched via photoinduced electron transfer (PET) by proximal macrocyclic components. Importantly, the dynamic behaviour of the macrocycle imine bonds allowed for metal induced hydrolysis of the rings, releasing the axle component and producing a turn-on fluorescent response. Interestingly, reduction of imine bonds to amine led to [2]rotaxanes which exhibited a strong turn-on fluorescent response upon addition of Au3+. The authors proposed that this behaviour was caused by the metal being encapsulated within the cavity of the rotaxanes.
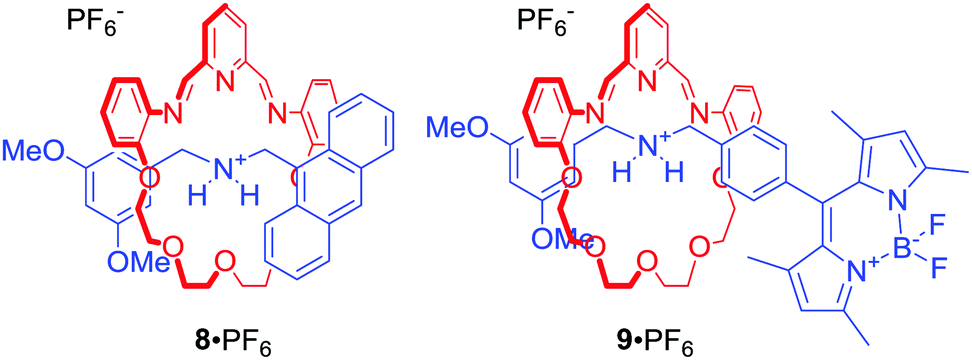 | ||
| Fig. 5 [2]Rotaxanes sensing Au3+ due to metal induced release of the axle component. | ||
Although lanthanides have been exploited as metal templates in the synthesis of catenanes,30 rotaxanes31 and molecular knots,32,33 their recognition by MIMs has only been recently investigated. Ghosh and co-workers used Cu(I) to template the formation of [2]catenane 10 which was shown to bind lanthanide metal ions such as Eu3+ and Gd3+via coordination to phenanthroline and ester groups inside an interlocked cavity formed by two macrocycles, leading to the increase of photoluminescence intensity of lanthanides in CH3CN/CHCl3 9:1 (Fig. 6).34
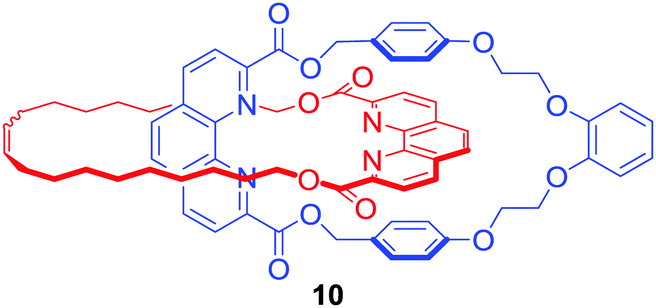 | ||
| Fig. 6 [2]Catenane host for lanthanide recognition. | ||
The interlocked nature of catenanes and rotaxanes provide an attractive environment for potential enantioselective binding. Niemeyer and co-workers obtained [2]catenane (S,S)-11 containing a BINOL-phosphate motif (Fig. 7) via ring closing metathesis of a Ca2+ templated orthogonal complex.35 The tetrabutylammonium (TBA) salt of 11 bound various doubly protonated chiral diamines such as C-protected amino acids Lys-OMe and Arg-OMe, or 1,2-diaminocyclohexane (DACH) in d6-DMSO. Most importantly, [2]catenane 11 exhibited a significantly higher degree of stereodiscrimination between enantiomers than its macrocyclic component demonstrating that incorporation of chiral bulky groups adjacent to the binding cavity of MIM is an effective strategy for the design of enantioselective receptors.
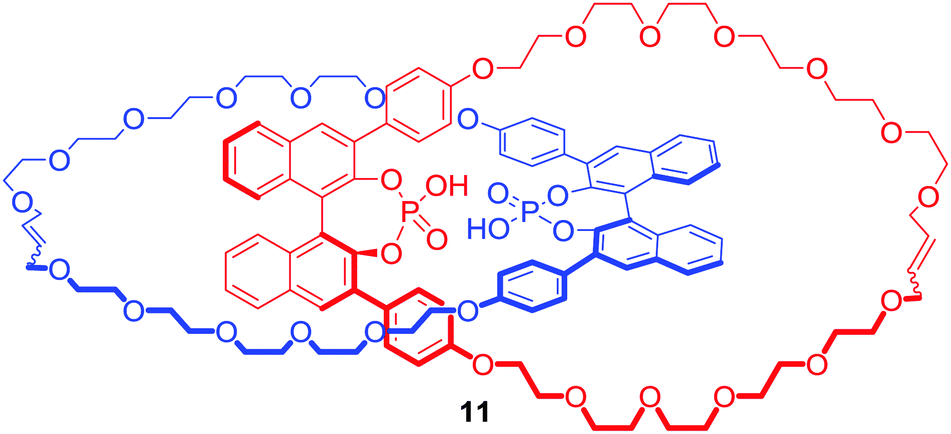 | ||
| Fig. 7 [2]Catenane host capable of enantioselective recognition. | ||
It should be noted that cation binding MIMs have also emerged as promising ligands in various transition metal catalysed reactions or even as catalysts themselves. For example, Leigh's group reported [2]rotaxane 12 containing a chiral trans-N,N′-dialkyl-1,2-cyclohexanediamine macrocycle (Fig. 8), which endotopically coordinated Cu+ during active metal Goldberg formation of C–N bond.36 The interlocked ligand was used in Ni2+ catalysed enantioselective Michael addition of diethyl malonate to trans-β-styrene with good yield (>98% conversion) and enantioselectivity (93:7 enantiomeric ratio) reported.
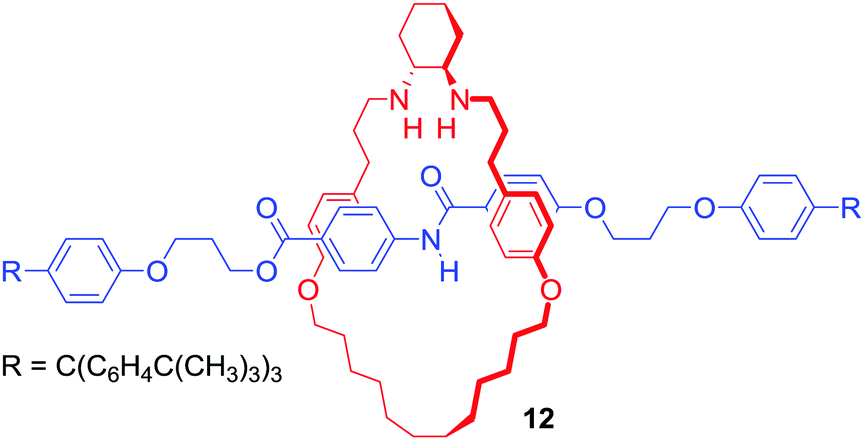 | ||
| Fig. 8 [2]Rotaxane ligand for Ni2+ catalysed Michael addition. | ||
Goldup and co-workers presented a catalytic [2]rotaxane 13 (Fig. 9) which required a Cu(I) cofactor to induce activity in a gold-catalysed cyclopropanation reaction.37 The authors suggested that the activity limiting interaction of the macrocycle with the axle incorporated Au+ catalytic centre was disrupted by copper coordination to a bipyridyl moiety. Further work on this system allowed the Goldup group to obtain a mechanically planar chiral ligand 14 (Fig. 9) for the enantioselective variant of cyclopropanation. Stereoselectivities observed in the presence of rotaxane 14 were comparable with a conventional covalent catalyst demonstrating the unexplored potential of mechanical chirality in enantioselective catalysis.38
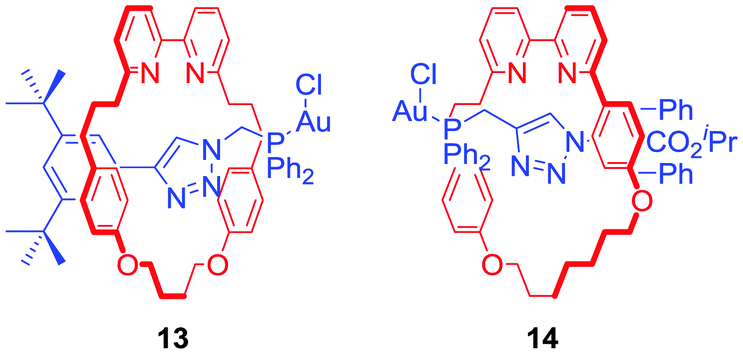 | ||
| Fig. 9 [2]Rotaxanes exhibiting catalytic activity upon Cu+ complexation. | ||
Chen, Chiu and co-workers prepared [2]rotaxane 15 (Scheme 1) with cation-induced catalytic properties via alkali metal ion templated clipping of an imino macrocycle and subsequent reduction.39 In its free state, 15 was not able to catalyse the Michael addition of diethyl malonate to nitrostyrene. However, upon complexation with Na+ its activity was immediately turned on. Interestingly, the authors reported that they were able to stop and restart the reaction by alternate additions of NaBArF4 and [2.2.2]cryptand sequestering Na+. In the noncomplexed state, 15 adopted conformations in which the oxygen atoms of the macrocyclic component formed hydrogen bonds with the amide proton located on the axle. The binding of a sodium cation resulted in a rotation of the ring to the co-conformation in which Na+ was simultaneously bound by the crown ether oxygens of the macrocycle and amide carbonyl of the axle (Scheme 1). This change enabled the activation of styrene in a newly formed cavity on the other side of the axle.
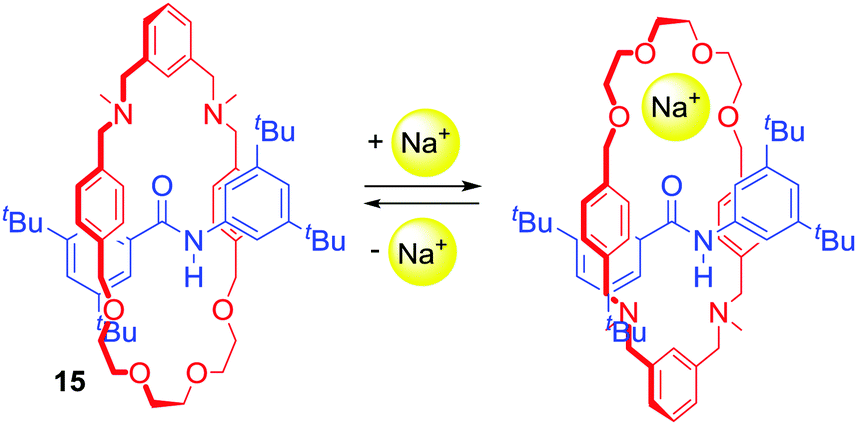 | ||
| Scheme 1 [2]Rotaxane catalyst activated by Na+ recognition. | ||
The examples presented above show the breadth of potential applications for cation binding interlocked structures. The unique cavities provide the environment required for selective binding of a range of metal cations or larger chiral guests and present a platform for the construction of selective optical sensors and novel catalysts.
Mechanically interlocked molecules for anion recognition and sensing
The selective binding of anions represents a significant scientific challenge due to their intrinsic properties such as complex geometries, low charge densities, pH-dependency and high hydration energies. At the same time anionic species play important roles in environmental, industrial and biological processes, which require methods for their detection and sequestering by artificial systems.40,41 During the past few decades, a plethora of acyclic and macrocyclic receptors capable of anion binding have been reported. In an effort to raise the bar in selectivity, we sought to exploit the potential of the unique three-dimensional binding cavities of MIMs for anion recognition and sensing applications.In 2001 our group demonstrated the formation of an orthogonal complex assembly between a hydrogen bond donating 3,5-bis-amide pyridinium group and a neutral isophthalamide motif via chloride anion coordination.42 Since then this anion template strategy has been employed in the synthesis of numerous interlocked structures, which importantly were demonstrated to bind anions with increased thermodynamic stability and selectivity in comparison to non-interlocked macrocyclic or acyclic receptor homologues. The vast majority of these MIMs bound anions via hydrogen bonds (HBs). In more recent years, halogen bonding (XB), the attractive interaction between an electron-deficient halogen atom and a Lewis base, has been used in anion host design.43–45 The comparable strength to HB and stringent linear directionality has made the XB interaction a particularly valuable tool in solid state crystal engineering and materials chemistry. Notably however, exploiting halogen bonding in the solution phase has proved to be an effective strategy for anion recognition particularly in highly competitive aqueous media. Indeed, anion recognition in water remains a key challenge in modern anion supramolecular chemistry46–49 and our group has constructed various MIMs incorporating halogen bond donors which demonstrate this capability. For the purpose of this review, the following examples of anion binding MIMs are divided into three main categories dictated by the non-covalent interactions used to achieve anion recognition: hydrogen bonding systems, mixed hydrogen–halogen bonding systems, and systems dominated by halogen bonding.
HB MIM host systems
The majority of MIMs prepared via the anion templation protocol have used chloride as the template. However, synthesis of [2]catenane 16·NO3 (Fig. 10) via RCM was an unprecedented example of using a nitrate anion template.50 Nitrate recognition by host systems is difficult due to its complex trigonal planar geometry, low affinity for hydrogen bond donors and relatively strong solvation. It is, thus, rarely used as a template. Importantly, the complementary tridentate binding cavity of 16·PF6 was selective for nitrate and chloride over a range of mono-charged oxoanions in CDCl3/CD3OD/D2O 45:45:10. Nitrate templation was also successfully used in the synthesis of [2]rotaxane 17·2PF6 (Fig. 10).51 This doubly charged bis-triazole MIM bound nitrate even more strongly than 16·PF6, however the degree of selectivity over halides was lower.
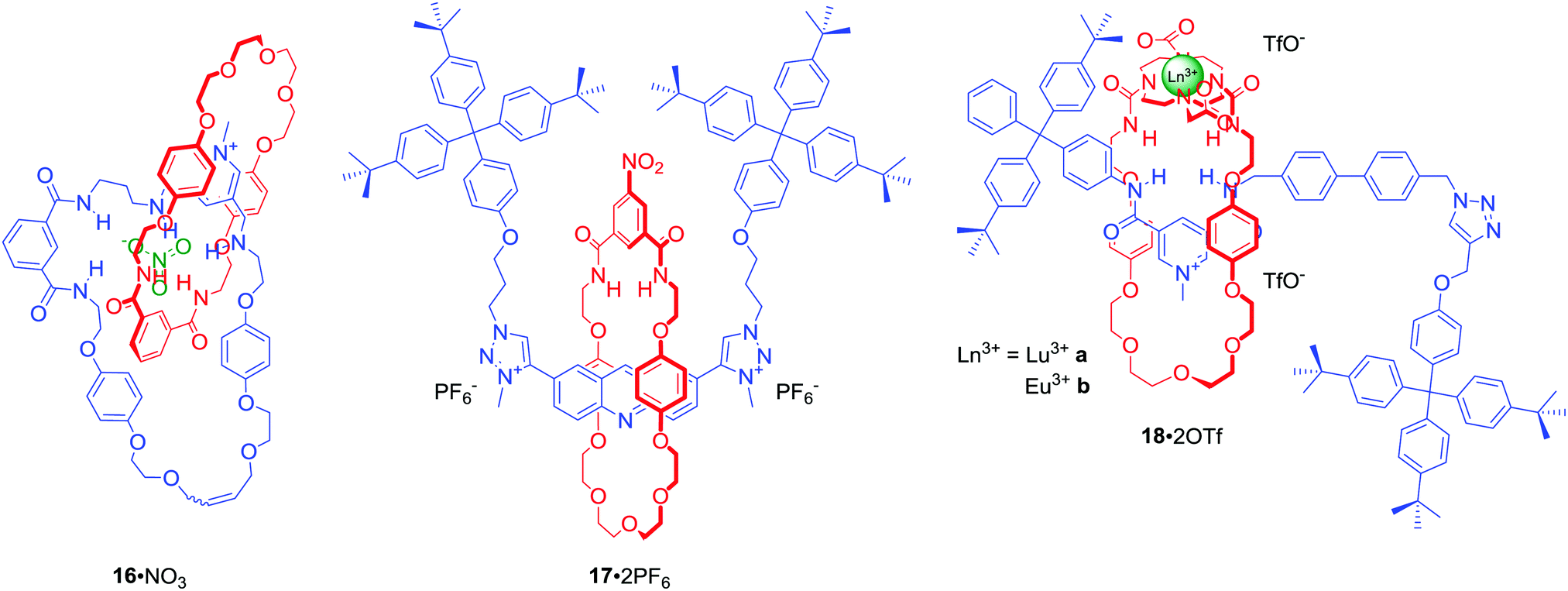 | ||
| Fig. 10 Interlocked structures for nitrate (16, 17) and nitrite recognition (18). | ||
Binding of the nitrite anion (NO2−) by molecular hosts is even more uncommon than that of nitrate. Nevertheless NO2− was used as a unique template, exploiting direct anion-lanthanide coordination in concert with HB, in the synthesis of luminescent [2]rotaxanes 18a·2OTf and 18b·2OTf in ca. 50% yields (Fig. 10).52 Monitoring the photophysical behaviour of the DOTA encapsulated europium cation enabled selective luminescence sensing of fluoride in (CH3)2CO/H2O 99:1. Binding of F− led to a drastic emission quenching due to direct coordination of the halide anion to the metal centre and a displacement of water from the inner coordination sphere of the metal. In contrast, acetate and nitrite caused only moderate quenching while the addition of chloride led to no changes of the lanthanide emission.
Among many MIMs obtained via chloride templation are various structures containing polarized C–H hydrogen bond donors such as triazole or triazolium moieties. [2]Rotaxane 19·PF6 (Fig. 11) was designed to incorporate amide and triazolium functionalities in the axle component.53 The resulting interlocked host demonstrated halide selectivity (Cl− > Br−) over oxoanions such as H2PO4− or AcO− in CDCl3/CD3OD 1:1. Importantly, this selectivity was more pronounced than in the cases of analogous rotaxanes 20·PF6 or 21·PF6 incorporating pyridinium bis-amide or pyridinium bis-triazole axle components.
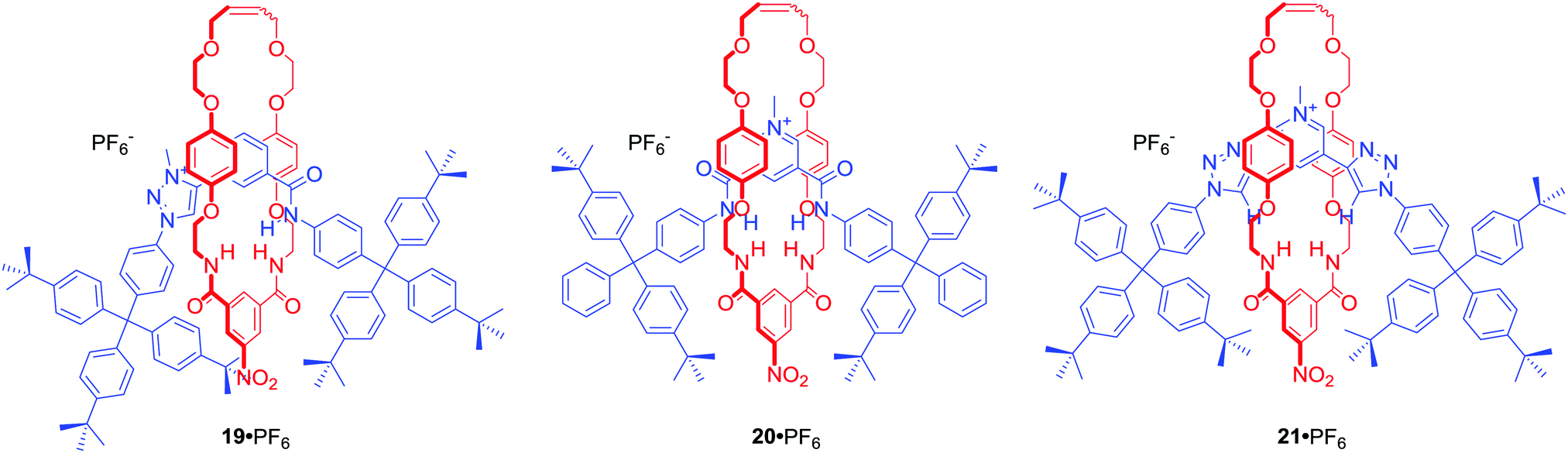 | ||
| Fig. 11 Anion binding MIMs incorporating iodotriazoloum and/or pyridinium motif. | ||
Most of the anion binding interlocked hosts obtained via anion templation have been positively charged. 22a–d were the first neutral redox-active [2]rotaxanes containing a ferrocene-appended macrocycle unit enabling sensing by means of electrochemical methodologies (Fig. 12).54 These rotaxanes were capable of binding halides (Cl− > Br− > I−) over H2PO4− or AcO− in a competitive CDCl3/CD3OD/D2O 45:45:10 solvent mixture and detecting chloride via a significant cathodic shift of the ferrocene/ferrocenium redox couple.
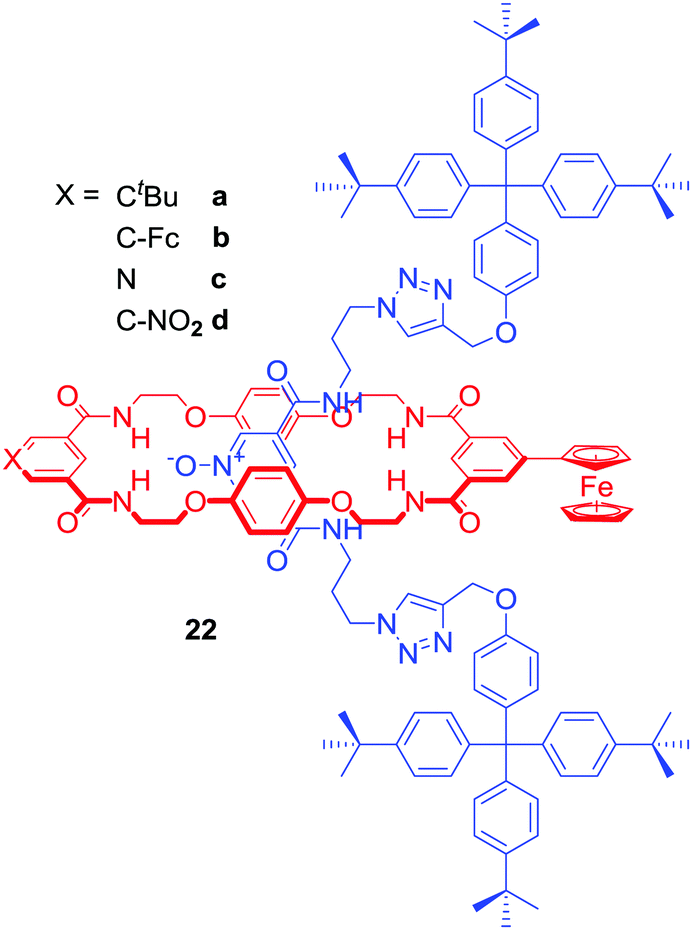 | ||
| Fig. 12 Neutral redox-active [2]rotaxanes. | ||
Metalloporphyrins have been utilised as optically responsive reporter groups in interlocked hosts such as anion binding [2]catenane 23·PF6 (Fig. 13).55 The solid state structure of 23·PF6 clearly demonstrated that the pyridine–zinc coordinate bond within the interlocked structure preorganizes the anion binding cavity for chloride recognition. Interestingly, despite the considerable chloride affinity only modest detectable changes in the metalloporphyrin component's UV/Vis and fluorescence spectra were observed.
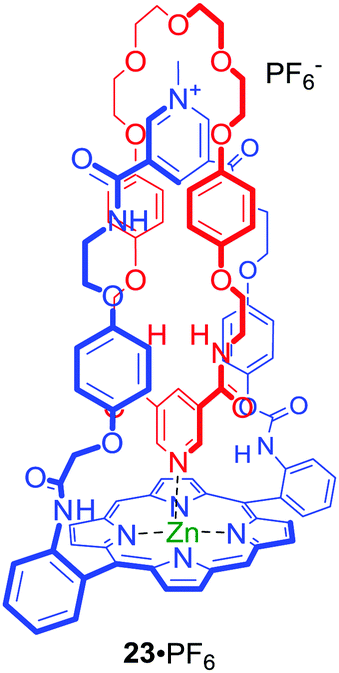 | ||
| Fig. 13 Metalloporphyrin containing [2]catenane. | ||
Anion template directed synthesis is not the only method of producing anion binding MIMs. Jolliffe and Goldup used the active metal template strategy to obtain fluorescent [2]rotaxane 24 containing a urea motif directly linked to a naphthalimide group in the axle component (Fig. 14).56 Strong intramolecular hydrogen bonds between the urea and bipyridyl moiety of the macrocycle prevented anion binding. However, protonation of the bipyridyl unit translocated the macrocycle facilitating binding of anions to the urea. In CDCl3/CD3CN 1:1 the determined anion association constant values of 24·H+ followed the trend: Cl− > Br− > MsO− > HSO4− > TsO− > I− which reflects a combination of the H-bond accepting properties of anions57 and size preference of the rotaxane's cavity. The binding event was accompanied by a switch-on fluorescence response of the axle naphthalimide group.
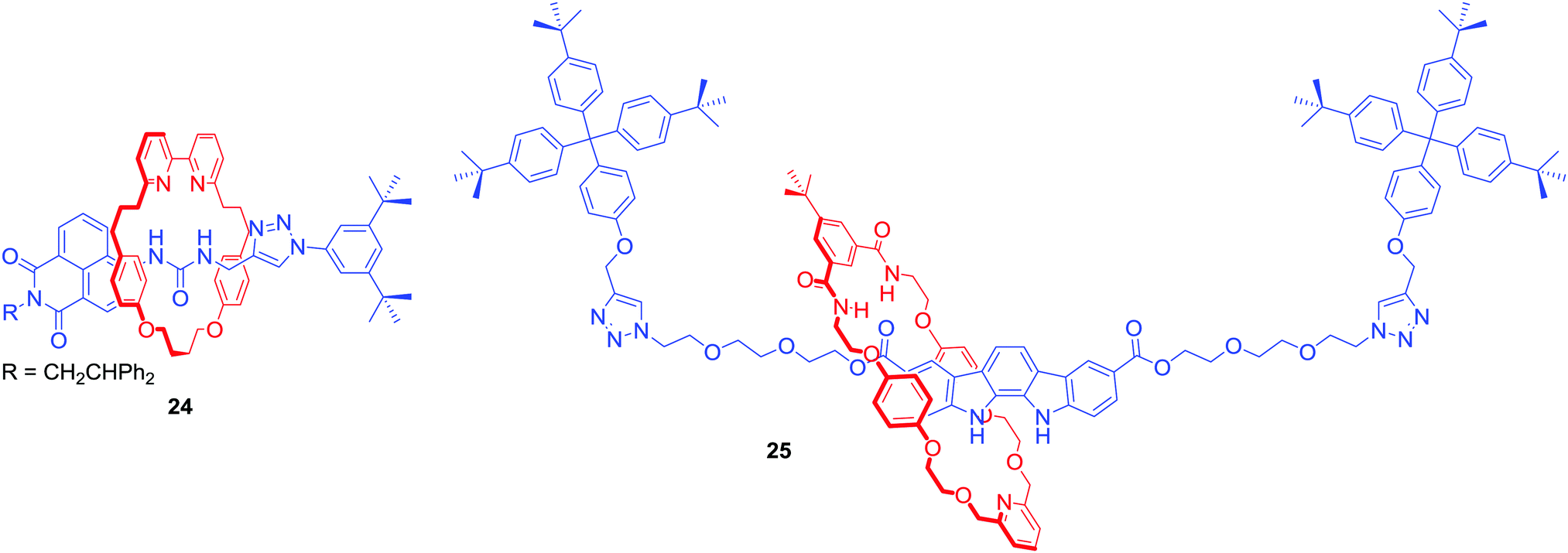 | ||
| Fig. 14 Fluorescent [2]rotaxanes obtained via AMT-CuAAC strategy. | ||
AMT was also used to synthesise neutral [2]rotaxane 25 containing a fluorescent indolocarbazole-based axle component and an isophthalamide functionalised macrocycle (Fig. 14).58 Binding studies in (CD3)2CO/D2O 95:5 revealed this MIM host system to exhibit a preference for AcO− and H2PO4− binding over halides. The rotaxane's axle indolocarbazole moiety facilitated the selective fluorescent sensing of chloride by significant enhancement of the heterocycle fluorophore's emission, whilst addition of other anions such as H2PO4−, AcO− or F− led to substantial quenching. Selective quenching of fluorescence upon addition of H2PO4− was also observed by Lin and co-workers in BODIPY-stoppered [2]rotaxanes containing two triazolium moieties.59,60
Another approach allowed Byrne, Gunnlaugsson and co-workers to obtain an anion binding [2]catenane via self-complementary hydrogen bond formation.61 In this case RCM was exploited to perform a double cyclisation to produce the homo catenane 26 (Fig. 15) which was shown to selectively bind H2PO4− over Cl−, SO42− or NO3− in competitive solvents like d6-DMSO.
 | ||
| Fig. 15 Anion binding MIM obtained via self-complementary hydrogen bond formation. | ||
HB/XB MIM host systems
Since the first example of a halogen bonding rotaxane 27·PF6 (Fig. 16),62 the iodotriazolium motif has been widely exploited in our group to construct MIMs with enhanced binding and sensing properties. For example, shortening of the axle component in rotaxane 28·PF6 resulted in a higher degree of preorganisation leading to a significant increase of halide binding strength and enhanced selectivity for iodide in the competitive solvent mixture CDCl3/CD3OD/D2O 45:45:10.63 The iodotriazolium motif was also used to improve halide recognition, in particular of bromide and iodide, in the binding cavity of rotaxane 29·2PF6 in concert with amide HB donors.64
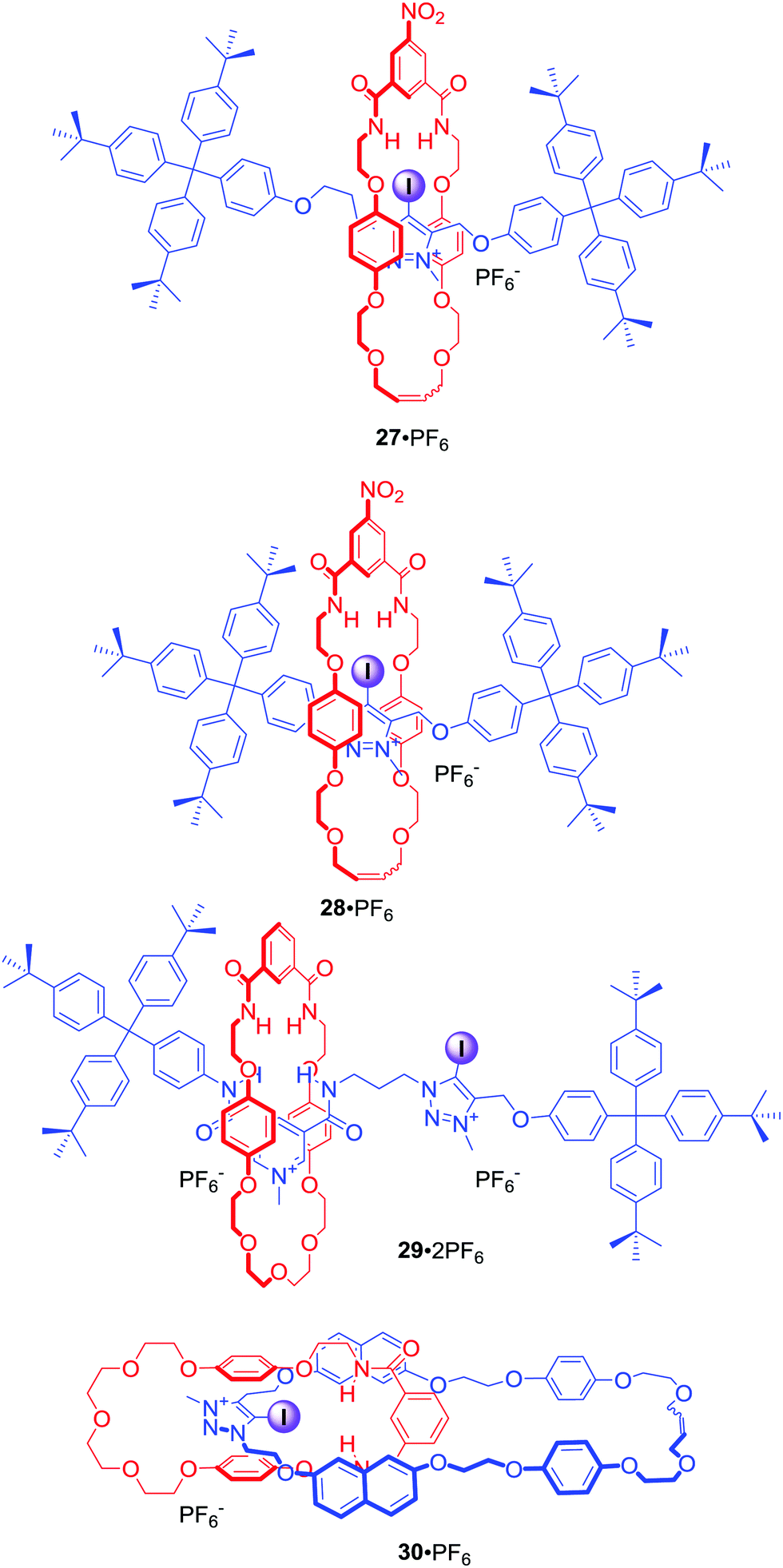 | ||
| Fig. 16 Anion binding MIMs incorporating iodotriazoloum motif. | ||
[2]Catenane 30·PF6 consisted of the iodotriazolium based macrocycle containing naphthalene groups in close proximity to the binding pocket (Fig. 16).65 Addition of various anions, particularly acetate and dihydrogen phosphate, to a solution of 30·PF6 in acetonitrile caused an increase of the naphthalene monomer emission bands and the concomitant decrease in intensity of an excimer emission band. The observed sensory response was attributed to the increased rigidity reducing the number of available vibrational and rotational non-radiative decay pathways upon anion binding.
In 2014 we demonstrated for the first time XB anion recognition in water and showed that incorporating XB donors into a rotaxane structure results in a significant enhancement of anion binding strength and selectivity in water compared to hydrogen bond analogues.66 [2]Rotaxanes 31·2NO3 and 32·2NO3 consisted of a hydrogen bond donating bis-amide pyridinium macrocycle and the axle component stoppered on both sides with permethylated β-cyclodextrins, which imparted solubility in water (Fig. 17). The binding constant of I− in water by bis-iodotriazole rotaxane 31b·2NO3 was two orders of magnitude larger than that of the rotaxanes 31a·2NO3 and 32·2NO3 containing bis-triazole or bis-amide motifs in their axles. Thermodynamic binding data showed that the large association constant for I− with iodotriazole rotaxane 31b·2NO3 was the sole result of favourable enthalpy manifesting extraordinary properties of XB in water. In contrast iodide binding of both rotaxanes 31a·2NO3 and 32·2NO3 was entropically driven and enthalpically disfavoured. Further studies of the hydrogen bonding analogue 33·2NO3 containing a wider, secondary rim functionalised cyclodextrin stoppers showed no significant difference in anion binding comparing to 32·2NO3.67 This observation suggested that the influence of the bulky cyclodextrin derivatives on a MIM binding cavity was negligible.
 | ||
| Fig. 17 Anion binding MIMs containing permethylated β-cyclodextrin stoppers. | ||
The excellent binding properties of 31b·2NO3 in aqueous media became the basis for the development of the first MIM host system capable of the selective sensing of iodide in water. The integration of a bipyridyl motif into the macrocyclic component led to rotaxane 34·3NO3 containing the photoactive [Ru(bipy)2]2+ group (Fig. 17).68 It exhibited a further increase in anion binding strength and facilitated the luminescent sensing of iodide in water via an enhancement of metal-to-ligand charge transfer (MLCT) emission intensity upon binding. Further structural elaboration of rotaxane 31·2NO3 included the insertion of electron withdrawing perfluoroaryl spacers between the iodotriazole motif and cyclodextrin stoppers, which sought to further increase the strength of XB–anion interactions.69 This modification allowed for the synthesis of rotaxane 35b·PF6 (Fig. 17) via chloride templated amide condensation in an unprecedented near quantitative 91% yield. Monocationic 35b·PF6 was able to bind iodide with an association constant of >105 M−1 in D2O/(CD3)2CO 1:1, which was at least three orders of magnitude greater in comparison to a hydrogen bonding rotaxane host analogue 35a·PF6.
The above examples demonstrated the potency of the bis-iodotriazole pyridinium moiety working in concert with hydrogen bond donors in rotaxane structures. Similar binding motifs were also incorporated into the structure of [2]catenane 36·PF6 (Fig. 18), which was able to selectively bind I− and Br− over Cl−.70 Moreover, a range of oxoanions showed no affinity towards 36·PF6 in solution with a subsequent crystal analysis confirming that sulfate was too large to be accommodated by the interlocked binding cavity. Importantly, analysis of halide complexes of catenane 36 by X-ray Absorption Spectroscopy (XAS) provided for the first time a direct measure of the degree of covalency in the XB–anion interaction, which was found to be comparable to the degree of covalency in transition metal complexes of chloride.
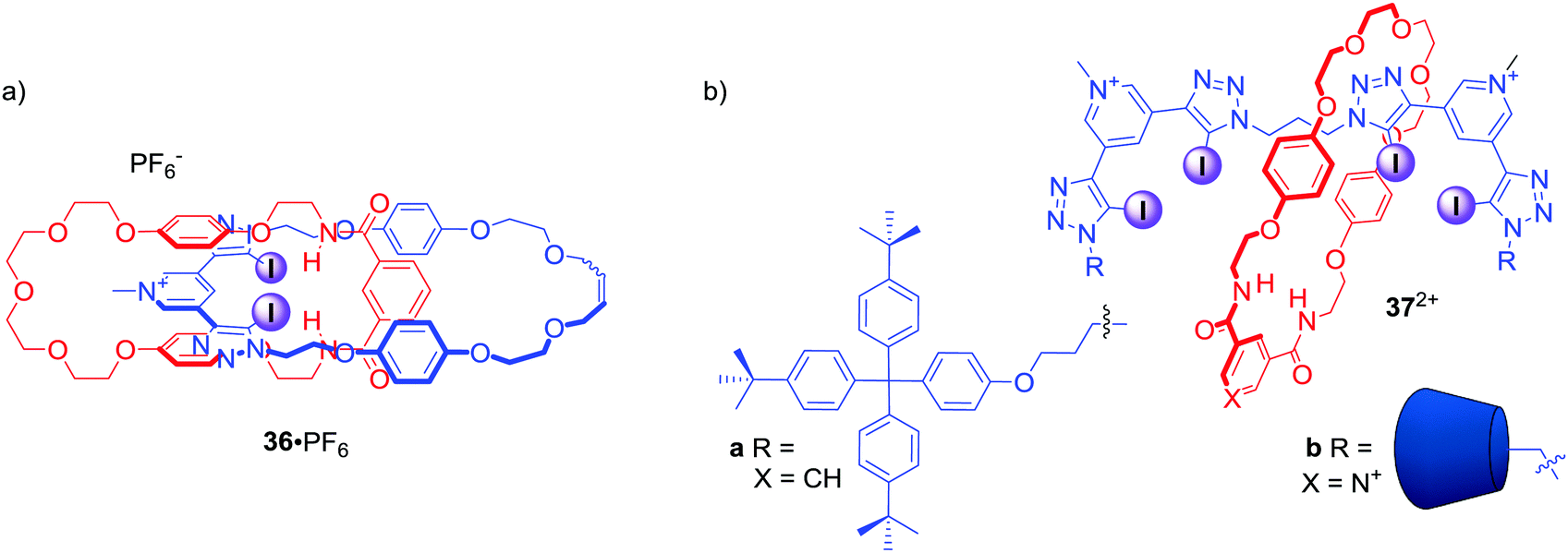 | ||
| Fig. 18 (a) Anion binding [2]catenane containing bis-iodotriazole pyridinium motif. (b) Anion binding [2]rotaxanes containing two bis-iodotriazole pyridinium motifs in the axle. | ||
Further work on the interlocked anion receptors containing bis-iodotriazole pyridinium motif involved covalently linking two of these moieties as in the axle of rotaxanes 37a·2PF6 and 37b·3OTf (Fig. 18).71 Receptor 37a·2PF6 was able to bind chloride with significantly higher affinity than nitrate in CDCl3/CD3OD/D2O 45:45:10, whilst the β-cyclodextrin stoppered rotaxane 37b·3OTf displayed almost identical association constants with Cl− and NO3− in D2O/(CD3)2CO 9:1.
In a related work two bis-iodotriazole pyridinium moieties were linked by a fluorescent (S)-BINOL group in the axle component of a chiral XB [3]rotaxane (Fig. 19). 38·2PF6, consisting of two mechanically bonded HB macrocycles, enabled discrimination between dicarboxylic enantiomers (R) vs. (S)-N-Boc-Glu2− and between geometric isomers malonate vs. fumarate. These dicarboxylate anion guest species were simultaneously bound by the extended cavity of the [3]rotaxane in a sandwich type complex.72 Computational modelling investigations suggested that the impressive geometric isomer selectivity of 38·2PF6 for fumarate (Kfumarate/Kmalonate = 4.4) in CHCl3/CH3OH/H2O 60:39:1 was mainly attributed to anion solvation effects inside the binding cavity. In contrast, the notably high enantioselectivity of 38·2PF6 for (S)-N-Boc-Glu2−arose primarily from host–guest structural complementarity (KS/KR = 5.7). Moreover, the binding of dicarboxylates could be readily sensed by fluorescence quenching of the rotaxane's axle BINOL fluorophore emission.
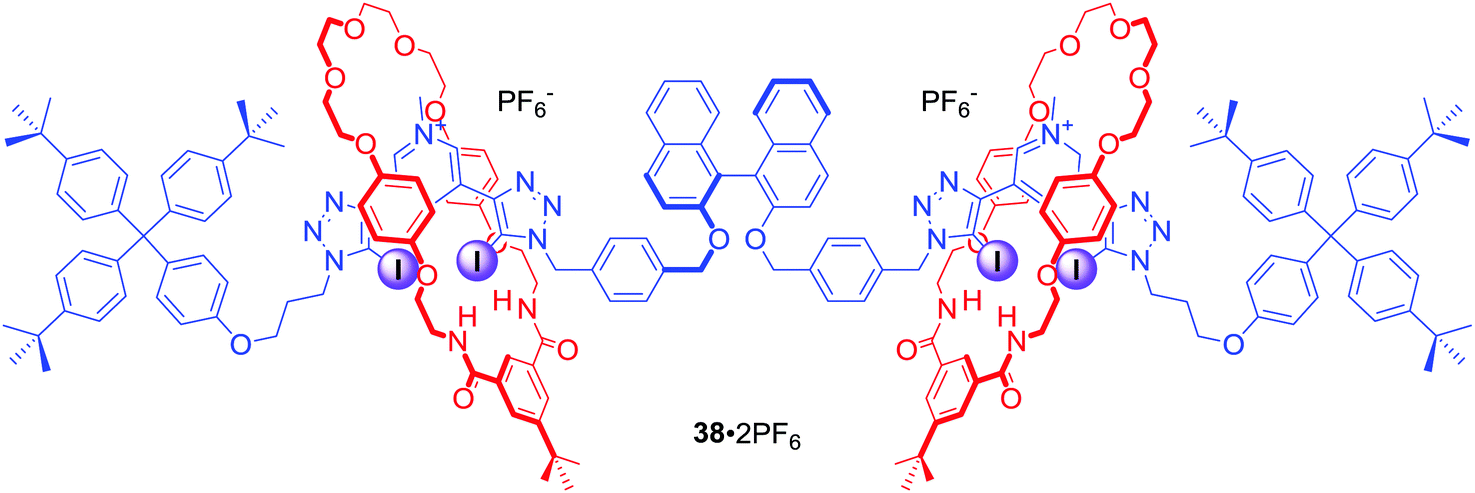 | ||
| Fig. 19 Chiral [3]catenane capable of enantioselective anion sensing. | ||
Recently, a novel XB tetra(iodotriazole)-pyridinium group was incorporated into MIM structures of catenane 39·PF6 and rotaxane 40·PF6 (Fig. 20).73 Interestingly, significant differences in anion binding strengths were observed for these MIMs. Although both 39 and 40 are monocationic, rotaxane 40·PF6 bound iodide almost three times stronger than catenane 39·PF6 in CDCl3/CD3OD/D2O 45:45:10. Moreover, catenane 39·PF6 bound iodide even more weakly than its analogue 36·PF6 with only two halogen bond donors. This difference might be a result of steric constraints restricting access to the binding cavity.
 | ||
| Fig. 20 Anion binding MIMs containing tetra(iodotriazole)-pyridinium group. | ||
Other motifs used in the construction of mixed halogen and hydrogen bond donating interlocked anion receptors included 4-bromopyridinium or 4-iodopyridinium in [2]catenanes 41·PF6,74 bis-iodotriazolium carbazole in [2]rotaxane 42·2PF6,75 and bis-iodotriazole amine in electroneutral [2]rotaxanes 43 and 44 (Fig. 21).76 The last of these are particularly interesting since conformational flexibility of combined Lewis basic and Lewis acidic groups facilitates rotaxanes 43 and 44 to strongly bind transition metal cations via bis-tridentate N-donor coordination and to bind anions weakly through XB and HB interactions. Protonation of the central amine moiety, however, reverses binding properties allowing for much stronger anion binding whilst negating metal cation recognition.
 | ||
| Fig. 21 Anion binding MIMs containing XB and HB donors. | ||
In a similar manner to using electron-deficient heavy halogen atoms for XB anion recognition, electron-deficient heavy chalcogen atoms can be exploited to bind anions via favourable chalcogen bonding (ChB) interactions. The first examples of chalcogen bonding MIMs were [2]rotaxanes 45a–b and 46a·3PF6 (Fig. 22) synthesised by AMT CuACC reaction, in which Cu(I) was coordinated via the chalcogen atoms themselves.77 Anion binding studies in d6-acetone revealed the electroneutral rotaxane 45b containing Te atoms to be a more potent MIM host in comparison to the hydrogen bonding and Se analogues 45c and 45a. A comparison of anion binding properties of triply charged rotaxanes 46a·3PF6 and 46b·3PF6 in (CD3)2CO/D2O 4:1 also showed significant differences. For instance, the chalcogen rotaxane bound halides with affinities decreasing in the order I− > Br− > Cl−, while 46b·3PF6 showed significant selectivity for Br−. The biggest difference was observed for acetate binding with an enhancement in association constant over 2 orders of magnitude larger for the HB host 46b·3PF6.
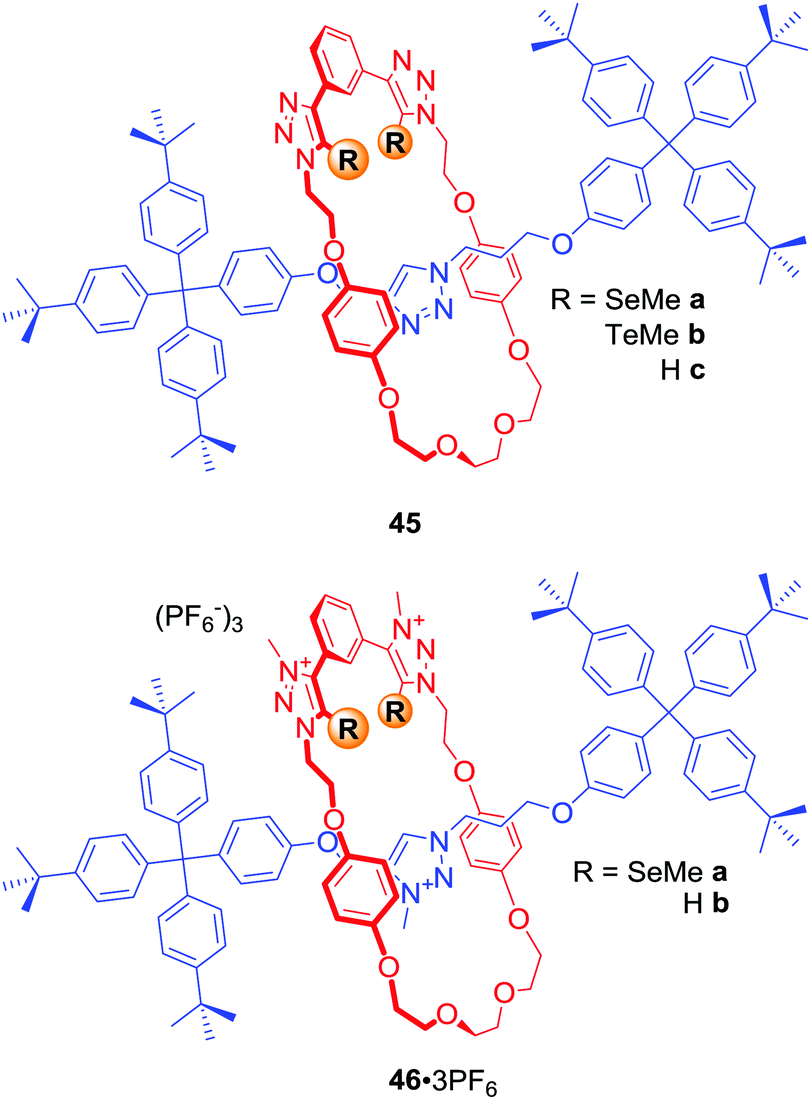 | ||
| Fig. 22 Chalcogen bonding [2]rotaxanes. | ||
All XB MIM host systems
The first halogen bonding MIM which did not contain strong hydrogen bond donors (like amide NH) was [2]catenane 47·2PF6 (Fig. 23) prepared by bromide anion templation.78 The first all-halogen bonding rotaxane was [2]rotaxane 48·2PF6 (Fig. 23) obtained by chloride templation.79 Its axle component was based on a bis-(iodo)triazolium carbazole moiety while the macrocycle contained a photoactive rhenium(I) bipyridyl entity. Quenching of its MLCT emission band allowed for selective fluorescent sensing of halides, in particular I−, over a range of oxoanions in aqueous solvent mixtures containing up to 50% water. | ||
| Fig. 23 First all XB examples of catenane and rotaxane. | ||
The CuACC-AMT strategy has proved to be particularly useful in the preparation of anion binding interlocked hosts containing iodotriazole motifs as halogen bond donors. The first example of such a structure was [2]rotaxane 49 (Fig. 24), whose macrocyclic component contained a bis-iodotriazole group capable of forming an endotopic complex with Cu(I).80 Removal of the copper template and subsequent metalation with a more sterically demanding rhenium(I) complex inverted the geometry of the macrocycle, forcing the transition metal to adopt an exotopic orientation and directing XB-donors into the rotaxane's cavity. Although this rotaxane was not luminescent, a decrease in the MLCT absorption band could be observed upon addition of anions in CHCl3. Association constants of this electrically neutral rotaxane with anions followed the trend Cl− > Br− > I− > AcO−. The positively charged analogue 50·PF6 was shown to exclusively bind bromide over chloride, iodide and oxoanions in (CD3)2CO/D2O 9:1, whereas the corresponding metal-free rotaxane 51·PF6 displayed Hofmeister bias halide selectivity: I− > Br− > Cl−.81
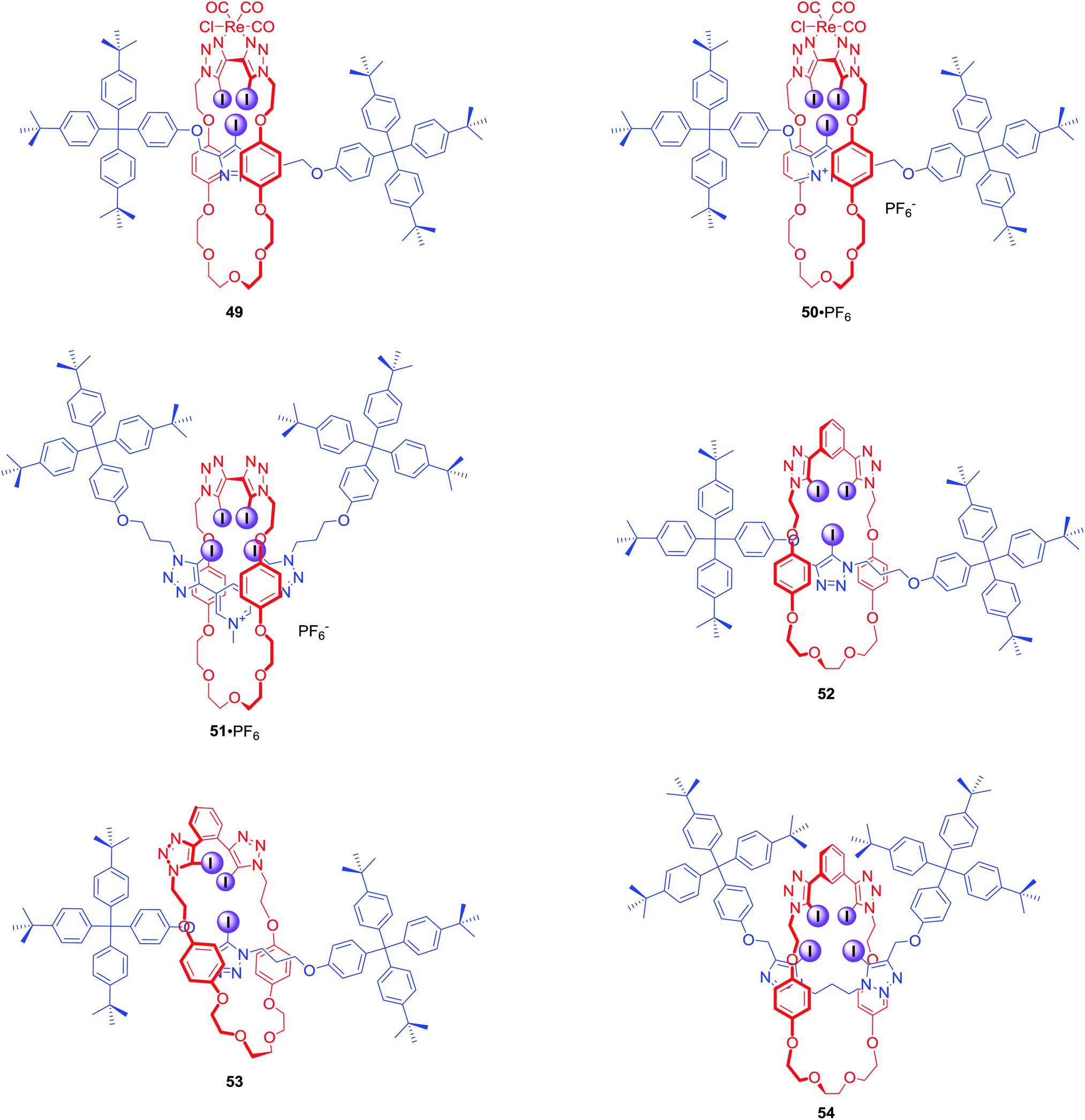 | ||
| Fig. 24 Anion binding MIMs obtained via CuAAC-AMT. | ||
The AMT strategy was also employed in the synthesis of a variety of neutral XB rotaxanes 52–54 (Fig. 24) with increasing number of XB donors inside the binding cavity.82 Rotaxanes 52 and 53 bound anions in d6-acetone following the selectivity trend of SO42− > Cl− > Br− > I−, which corresponds to a decreasing charge density and basicity of the guest anion. Rotaxane 52 bound anions much more strongly than 53 by up to two orders of magnitude, demonstrating the importance of the spatial separation of iodotriazole groups in the receptors. Interestingly, in a (CD3)2CO/D2O 98:2 solvent mixture the reverse anion binding selectivity was observed: I− > Br− > Cl− > SO42−. In the case of 54, binding of bromide was stronger than that of iodide. However, this preference disappeared upon increasing water content to 5%, underscoring the importance of anion hydration in the binding process.
A redox-active ferrocene appended [2]rotaxane 55·PF6 containing four halogen bond donors in its cavity was also obtained via the AMT-CuAAC strategy (Fig. 25).83 Anion binding studies in CD3CN/(CD3)2CO/D2O 45:45:10 revealed that rotaxane 55 displayed Hofmeister anion binding preference: I− > Br− > Cl− > SO42−. This trend was also reflected in the electrochemical sensing behaviour of 55·PF6 where a larger magnitude of cathodic perturbation of the ferrocene/ferrocenium redox couple for Br− over Cl− in the presence of water was demonstrated.
 | ||
| Fig. 25 Electrochemically active XB [2]rotaxane. | ||
Four halogen bond donors were also present in the structure of [2]rotaxanes 56–58·PF6 (Fig. 26).84 The macrocyclic component of 56–58·PF6 contained two iodotriazole motifs separated by either (S)-BINOL (56·PF6 and 57·PF6) or an achiral biphenyl group (58·PF6). The axle component incorporated a bis-iodotriazole pyridinium moiety stoppered by a chiral serine derivative (56·PF6 and 58·PF6) or an achiral group (57·PF6). Two chiral components in rotaxane 56·PF6 formed a unique binding pocket which allowed for significant enantioselectivity in binding of N-Boc amino acid derivatives: Leu, Pro, Trp and BINOLPO4 in (CD3)2CO/D2O 98:2. Rotaxane 56·PF6 showed selectivity for the S-enantiomer of amino acids and the R-enantiomer of BINOL-phosphate. Surprisingly, 57·PF6 (chiral macrocycle and achiral axle) exhibited the reverse selectivity for R amino acids, whilst 58·PF6 (achiral macrocycle and chiral axle) showed practically no enantioselectivity. This suggested that the chiral XB macrocycle component of the rotaxane exerted a dominating influence on the effectiveness of chiral discrimination, which may be partly due to the greater rigidity and larger steric bulk of the macrocycle's chiral (S)-BINOL group as compared to the point-chiral (S)-serine-derived asymmetric units of the axle.
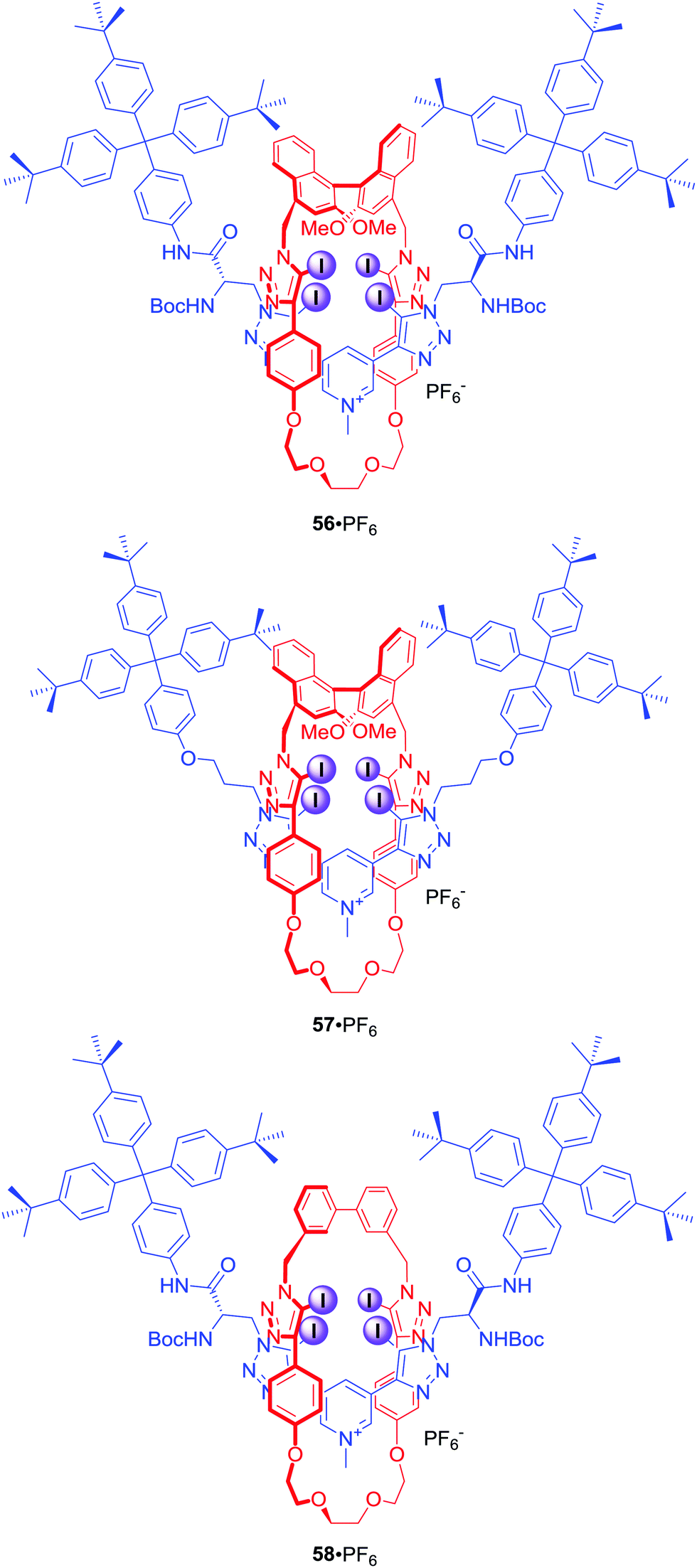 | ||
| Fig. 26 Chiral XB [3]rotaxanes. | ||
Dynamic MIM systems for anion recognition and sensing
In the above examples anion binding by MIMs in general was achieved via anion guest encapsulation within their unique three dimensional preorganized binding cavities. Any conformational changes experienced by these molecules upon a binding event were relatively minor and limited mostly to rigidifying the interlocked structure. Dynamic MIMs designed to undergo large amplitude changes of their co-conformation triggered by anion recognition are discussed as a separate category in this section.Halogen bonding MIMs in most cases exhibit a strong preference for iodide binding over chloride in competitive protic solvent mixtures. Our group prepared rotaxane 59·2PF6 (Scheme 2), whose axle component contained XB-iodotriazolium and HB-triazolium stations separated by a naphthalene moiety.85 The contrasting halide anion binding affinities of these stations allowed shuttling of the macrocyclic component along the axle (Scheme 2). In the presence of Cl−, the macrocycle displayed 100% occupancy of the HB-triazolium station in CDCl3/CD3OD 1:1. The high affinity of iodide to XB–iodotriazolium caused shuttling of the macrocycle in response to iodide addition. Rotaxane 59·2PF6 was the first example of using XB in the solution phase to control MIM molecular motion.
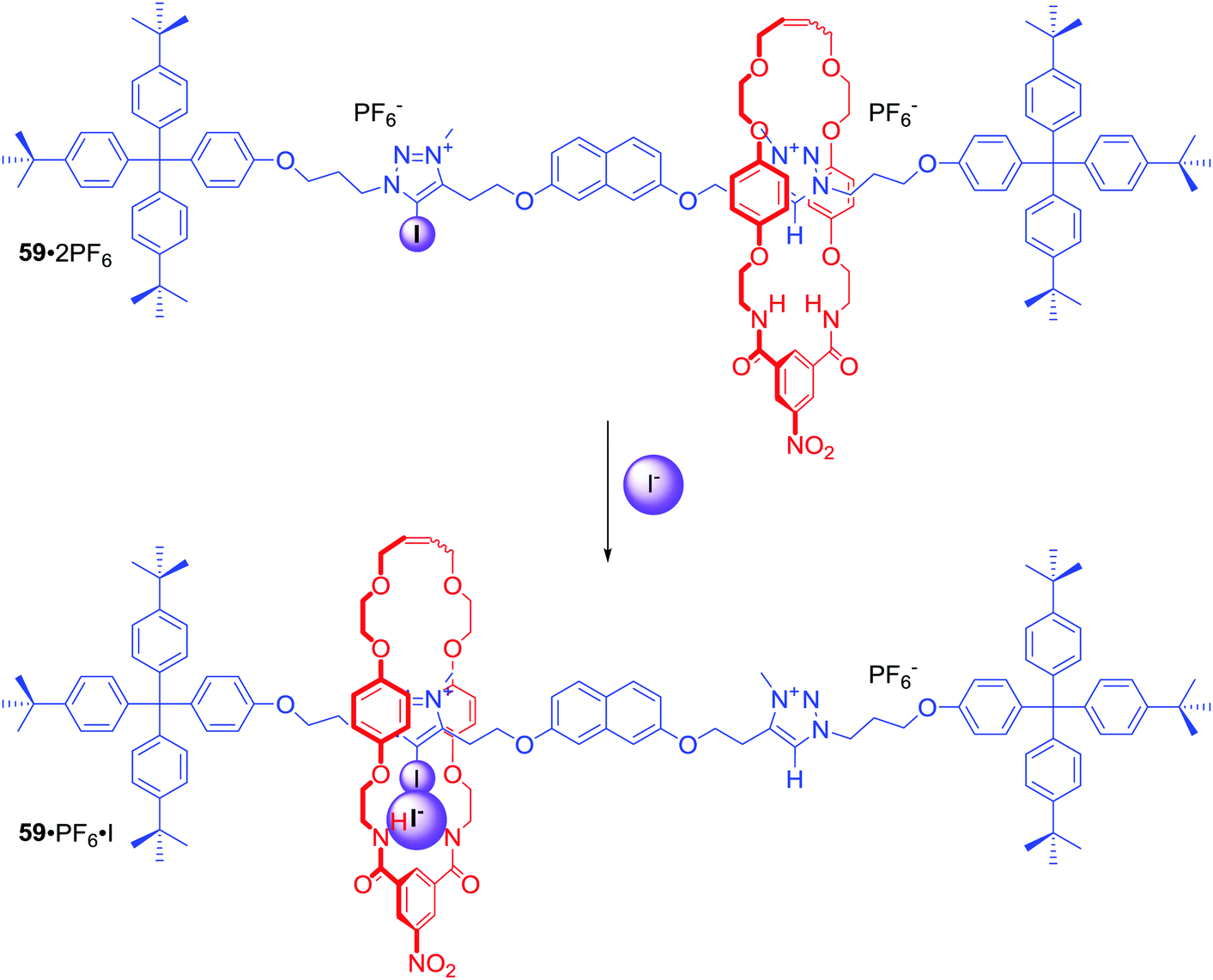 | ||
| Scheme 2 Iodide-driven shuttling of [2]rotaxane. | ||
[2]Rotaxanes 60a·PF6 and 60b·PF6 were examples of dynamic anion binding MIMs capable of colorimetric sensing (Scheme 3).86 The axle component of these systems contained an electron deficient naphthalene diimide (NDI) motif exhibiting aromatic donor–acceptor charge transfer interactions with the electron rich hydroquinone groups of the macrocycle component. The second station on the axle was a positively charged XB iodo- or HB proto-triazolium group which could participate in anion binding. In the presence of chloride, the macrocyclic component resided over the axle's triazolium station as a result of anion coordination to both rotaxane's components. Exchange of chloride to hexafluorophosphate resulted in the macrocycle shuttling to the axle NDI station which was manifested by a naked-eye detectable colour change in CDCl3 solution (Scheme 3). Detailed NMR studies of rotaxanes 60a, 60b and their non-interlocked analogues allowed for the estimation of station occupancy by the macrocycle component in the presence of Cl−, I− and PF6− in various solvent mixtures. In CDCl3 triazolium station was occupied almost 100% in complexes of 60a and 60b with Cl− and I−. Exchange for PF6− led to 62% occupancy of the NDI station for the XB rotaxane, but only 24% in the case of the HB rotaxane. Increasing polarity of solvent hampered anion binding, therefore in CDCl3/CD3OD 1:1 solutions of 60a·Cl, 60b·I and 60a·Cl, NDI was the most occupied station and anion exchange to PF6− only led to a modest increase of NDI occupancy. Only in 60b·I complex the macrocycle resided mostly over the triazolium station. These results clearly demonstrated that the XB rotaxane switch was a superior example of synthetic molecular shuttle over its HB analogue in both CDCl3 and CDCl3/CD3OD solvent mixtures.
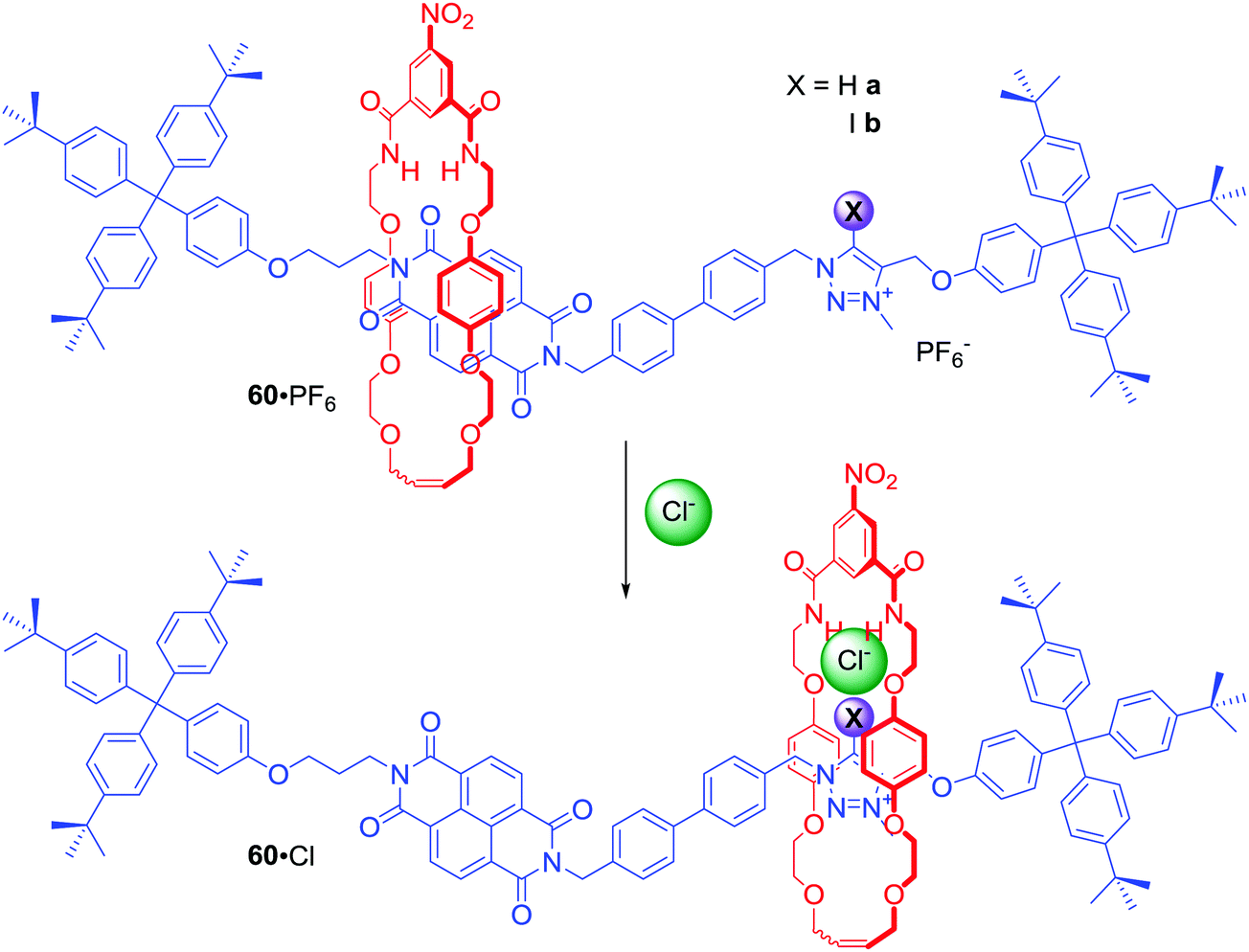 | ||
| Scheme 3 Chloride-driven shuttling of [2]rotaxane. | ||
Two NDI and two triazolium stations were incorporated in the axle component of [3]rotaxanes 61a·2PF6 and 61b·2PF6 (Scheme 4).87 Similar to 60a–b, exchange of PF6− for chloride led to the two macrocycles shuttling from the NDI to the triazolium moieties causing a colour change of solution. Interestingly, anion association constant values for XB rotaxane 61a·2PF6 were significantly higher than for HB rotaxane 61b·2PF6 in 1:1 CDCl3/CD3OD and the XB host system exhibited an enhanced association strength of oxoanions over the HB system. Moreover, both rotaxanes exhibited the rare selectivity for NO3− over acetate, bicarbonate, dihydrogen phosphate and notably chloride, resulting from a distinctive binding mode in which nitrate is bound in a sandwich-like complex between the two macrocycles (Scheme 4).
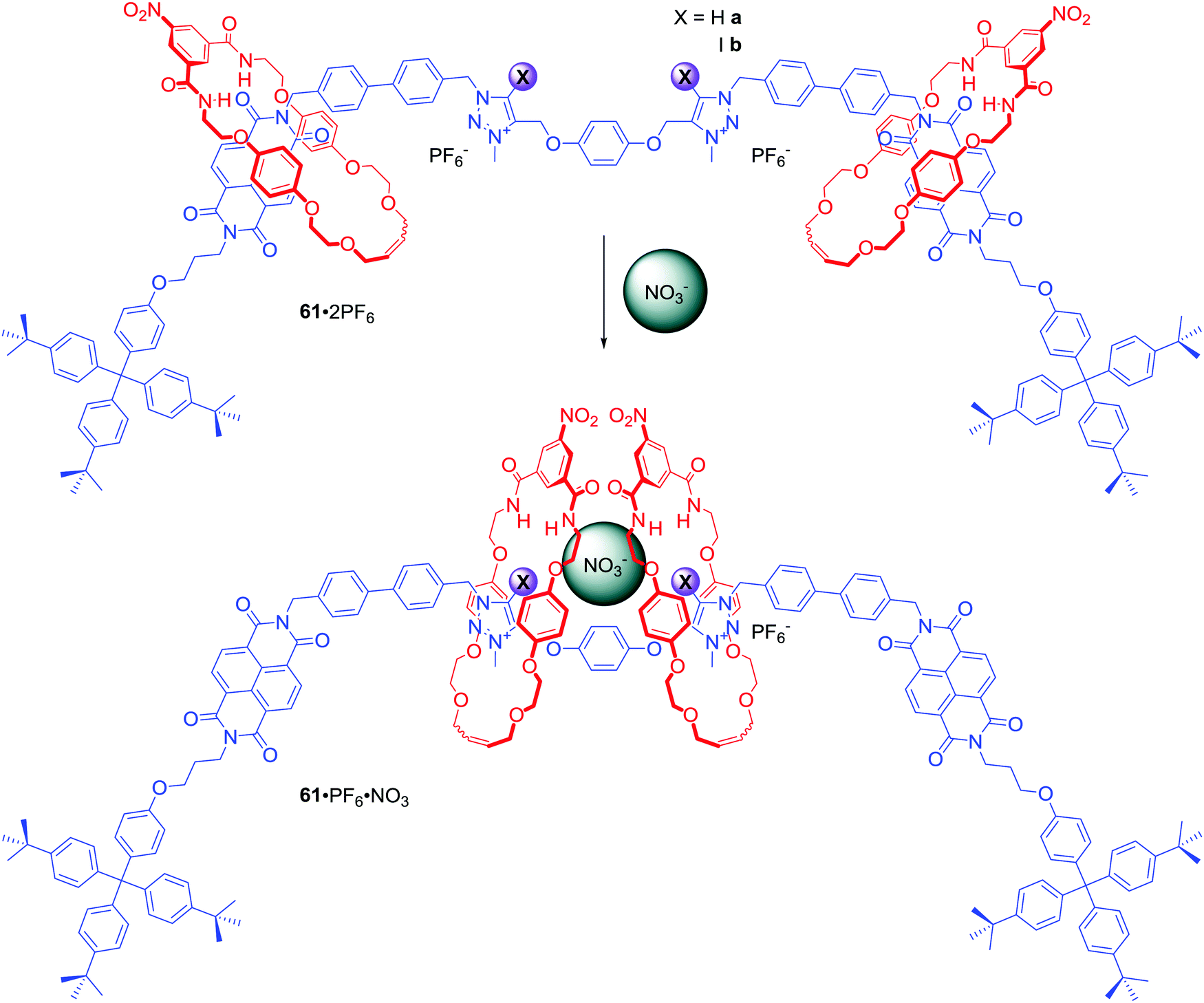 | ||
| Scheme 4 Nitrate-driven shuttling of [3]rotaxane. | ||
The above design was further extended in [3]rotaxane 62·2PF6 by incorporation of C60 fullerene, a strong electron acceptor with redox properties, in the centre of the axle (Fig. 27).88 Additionally, isophthalamide macrocycles were appended by a ferrocene group, which is a strong electron donor. This elaborate interlocked system was demonstrated to adopt two distinct anion-dependent co-conformations that interconvert via translational motion of both ferrocenyl macrocycles between the peripheral NDI and central triazolium-bridged C60 axle stations. Anion exchange of PF6− for Cl− triggered a naked-eye switch-on response due to the macrocycles shuttling to the centre of the axle. This change precluded electron transfer to NDI, causing an increase of NDI fluorescence emission and concomitant formation of a C60 fullerene-based charge-separated state. Ultimately, selectivity of the initially populated charge-separated state in [3]rotaxane 62·2PF6 was enabled by anion-induced co-conformational changes causing unprecedented alteration of communication pathways operating between the electron donor and acceptor motifs.
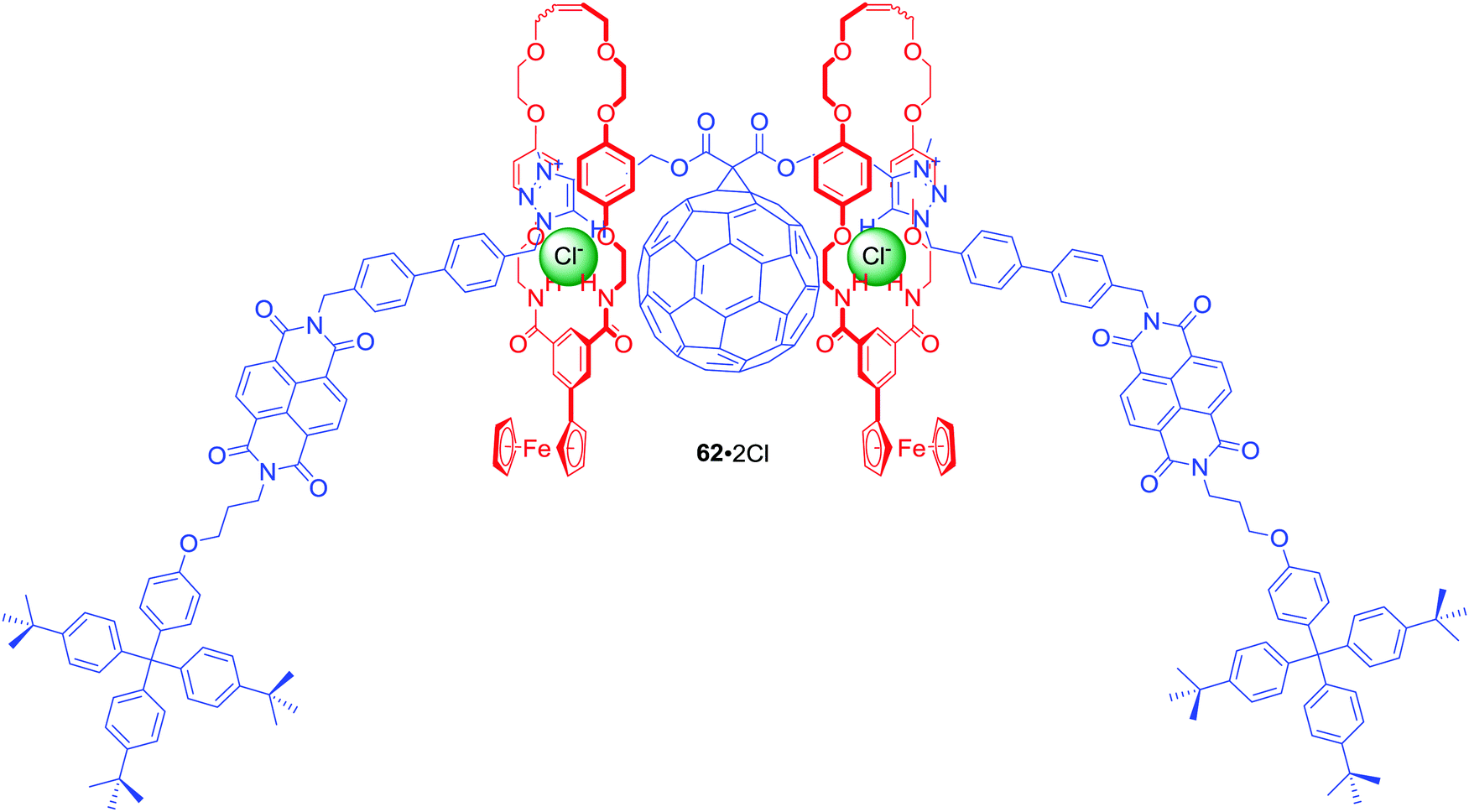 | ||
| Fig. 27 Chloride complex of [3]rotaxane incorporating fullerene in the axle component. | ||
[3]Catenane 63·2PF6 (Scheme 5) consisted of a large macrocycle incorporating two triazolium stations and tetrachloro-functionalized perylene diimide (PDI).89 Similar to NDI, PDI is an electron deficient scaffold with excellent chromophoric, emissive, redox properties and a strong tendency to aggregate. However, tetrachlorination of PDI bay positions introduces steric bulk that forces the aromatic framework to twist from planarity, limiting aggregation. The smaller isophthalamide macrocycles of catenane 63·2PF6 could not pass over either the twisted PDI or bulky tetraphenyl spacer unit and so their position was restricted to one half of the larger central ring. Exchange of PF6− for chloride in CDCl3/CD3OD 3:1 caused circumrotary motion of the smaller macrocyclic rings from the PDI motif to the two triazolium groups (Scheme 5). Dynamic behaviour of this unique system enabled colorometric and fluorescence anion sensing in CHCl3/CH3OH/H2O 45:45:10.
 | ||
| Scheme 5 Chloride-driven shuttling of [3]catenane. | ||
[2]Rotaxane 64·PF6 was a relatively simple interlocked system containing a halogen bonding benzimidazole-iodotriazole station directly conjugated to a naphthalamide station in the axle component (Fig. 28).90 In its neutral state, the macrocyclic component resided over the axle naphthalamide group. Protonation of the benzimidazole moiety and subsequent addition of chloride led to translocation of the macrocycle towards the benzimidazolium-iodotriazole station due to anion coordination by halogen and hydrogen bond donors. This change of co-conformation could be observed via naked-eye colour change of solution, as well as an increase in fluorescence emission intensity.
 | ||
| Fig. 28 Anion sensitive fluorescent rotaxanes. | ||
Anion sensing MIMs do not always rely on recognition using weak interactions. Jasti et al. obtained, via AMT, [2]rotaxane 65 with fluorescent pyridyl-embedded cycloparaphenylene motif and the axle stoppered with a fluorescent quenching 3,5-dinitro phenyl group at the one end and fluoride-cleveable triisoproylsilyl group on the other.91 Addition of 1 equivalent of TBA+F− to a solution of rotaxane in chloroform led to a dramatic 123-fold increase in emission intensity due to the dethreading of the macrocycle from the destoppered axle. Although such a chemodosimeter is not reversible, it provided a sensitive method for F− sensing.
In a recent example Baroncini, Credi and co-workers presented [2]rotaxane 66·H·I3 consisted of an axle with C∞v symmetry surrounded by a macrocycle with a Cs symmetry (Scheme 6).92 In the protonated form of 66, dibenzeno[24]crown-8 encircled ammonium centre in an achiral co-confromation. Addition of base, however, caused the shift of the macrocycle towards triazolium station leading to desymmetrized mechanically planar chiral states, which have been observed via1H NMR. Interestingly, addition of chiral anion, (1S)-(+)-10-camphorsulfonate, induced a significant difference in the population of the stations due to its coordination to triazolium moiety of the axle. Apparently, the ring-axle arrangement in the rotaxane created a nonsymmetric environment enabling enantioselective anion recognition. This example vividly shows the unexplored potential of mechanical chirality of MIMs in the development of new receptors, sensors and molecular devices.
 | ||
| Scheme 6 Acid/base switching of a mechanically planar chirality in a [2]rotaxane molecular shuttle. | ||
Mechanically interlocked molecules for ion-pair recognition and sensing
Ion-pair receptors are designed to combine the functionalities of cation and anion receptors for cooperative binding.93–95 This increases the synthetic complexity of the host structure which may be the reason behind the scarcity of ion-pair binding MIMs.
67 was the first example of a neutral heteroditopic rotaxane host system capable of binding alkali metal halides as axle-separated ion-pairs (Scheme 7).96 Its macrocyclic component consisted of a calix[4]diquinone cation recognition site covalently linked to an isophthalamide anion binding motif, while the axle component consisted of a bis-amide pyridine N-oxide moiety. In the presence of sodium cation, rotaxane 67 adapted a co-conformation in which Na+ was bound by calix[4]diquinone and the oxygen atom of N-oxide (Scheme 7). This resulted in the formation of a binding cavity containing four amide NH groups. Removal of the sodium cation induced a pirouetting motion of the macrocycle due to formation of hydrogen bonds between the bis-amide group of the macrocycle and the N-oxide group of the axle. Such behaviour was responsible for remarkable cooperativity factors for chloride and bromide binding in the presence of sodium in CDCl3/CD3OD 4:1 solution. A similar approach to the design of MIM ion pair receptors led to rotaxanes 68 and 69 which were able to complex Zn2+ and various anions in competitive solvent mixture: CDCl3/CD3OD/D2O 45:45:10 (Fig. 29).97
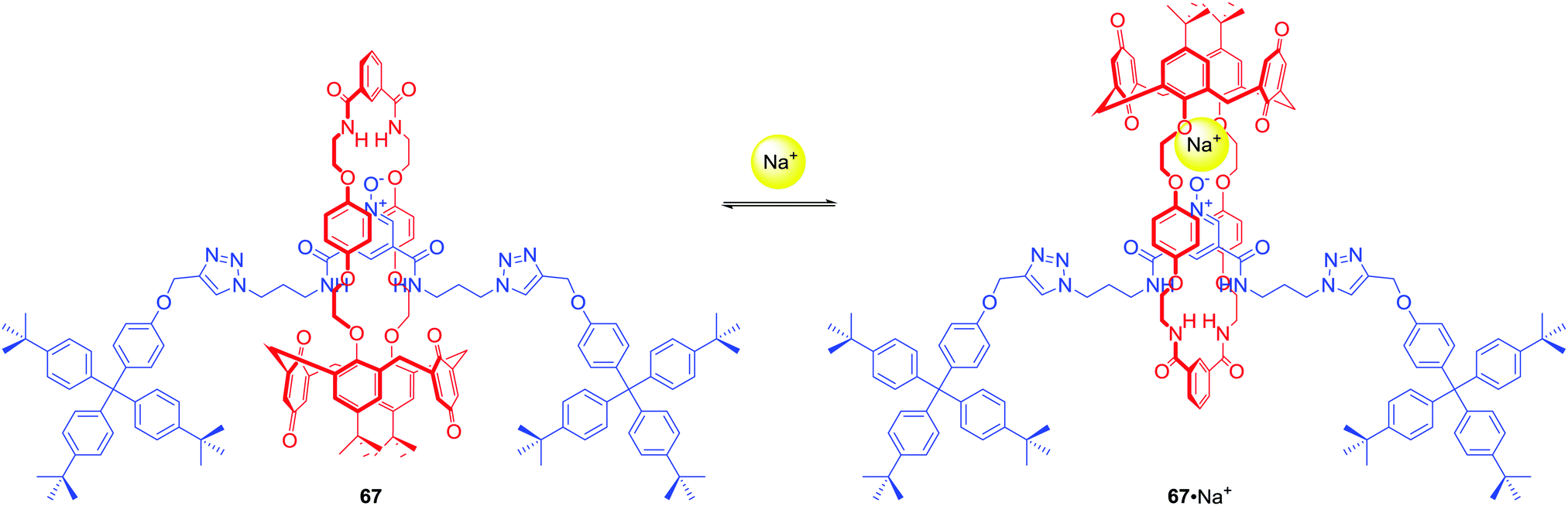 | ||
| Scheme 7 Sodium cation-driven pirouetting motion of ion pair binding [2]rotaxane. | ||
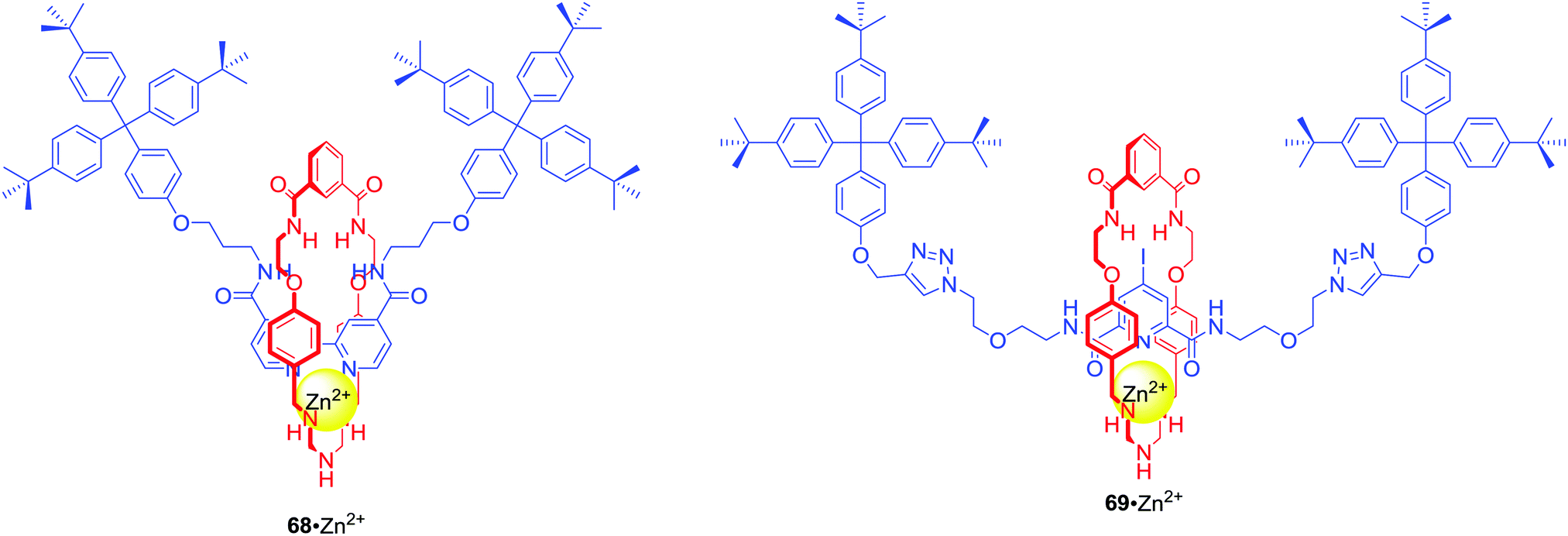 | ||
| Fig. 29 Ion pair binding [2]rotaxanes. | ||
Ballester and co-workers obtained [2]rotaxane 70 which also incorporated a bis-amide pyridine N-oxide moiety in the axle and bis(calix[4]pyrrole) as the macrocyclic component (Fig. 30).98 In chloroform solution, rotaxane 70 functioned as a heteroditopic receptor for tetraalkylammonium salts of chloride, nitrate and cyanate forming 1:1 complexes in which cation and anion were separated by the macrocyclic component.
 | ||
| Fig. 30 Ballester's ion pair binding [2]rotaxane. | ||
Conclusions
Mechanically interlocked molecules have proved to be excellent platforms for the construction of ion binding receptors and sensors due to the unique properties of their mechanically bonded cavities. Furthermore, the increasing number of synthetic approaches makes MIMs more and more available.In particular, active metal templation has emerged recently as a useful methodology leading to both transition metal cation and anion binding MIMs. On the other hand, the anion template strategy has evolved over the last 20 years from a mere curiosity to a versatile tool allowing for the synthesis of elaborate structures capable of anion recognition, sensing and induced shuttling. In more recent years the incorporation of halogen bond donors into interlocked structures have resulted in MIM host systems capable of anion binding in water with enhanced thermodynamic stability and selectivity in comparison to HB MIM and non-interlocked host analogues.
However, the full potential of ion binding MIMs has yet to be realised. For example, only recently have the unique spatial properties of the mechanical bond begun to be explored in enantioselective recognition. The integration of chiral groups into MIM binding cavities or exploitation of chirality arising from the mechanical bond is still rare but such host systems have the potential to demonstrate enhanced chiral recognition and to play an important role in asymmetric catalysis. In addition, the application of MIM hosts for extraction and membrane transport applications are areas to be investigated. Moreover, the increasing number of ion recognition driven shuttling MIMs illustrate the exciting future development of sophisticated responsive nanotechnological devices and materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KMB thanks the EPSRC for postdoctoral funding (EPSRC grant number EP/P033490/1). The authors thank Dr Jessica Pancholi and Andrew Docker, University of Oxford, for helpful discussions.Notes and references
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, New Jersey, 2016 Search PubMed.
- J. F. Stoddart, The chemistry of the mechanical bond, Chem. Soc. Rev., 2009, 38, 1802 RSC.
- E. A. Neal and S. M. Goldup, Chemical consequences of mechanical bonding in catenanes and rotaxanes: isomerism, modification, catalysis and molecular machines for synthesis, Chem. Commun., 2014, 50, 5128 RSC.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Development of pseudorotaxanes and rotaxanes: from synthesis to stimuli-responsive motions to applications, Chem. Rev., 2015, 115, 7398 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial molecular machines, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Catenanes: Fifty years of molecular links, Angew. Chem., Int. Ed., 2015, 54, 6110 CrossRef PubMed.
- J. E. M. Lewis, M. Galli and S. M. Goldup, Properties and emerging applications of mechanically interlocked ligands, Chem. Commun., 2017, 53, 298 RSC.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Artificial molecular motors, Chem. Soc. Rev., 2017, 46, 2592 RSC.
- M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, An autonomous chemically fuelled small-molecule motor, Nature, 2016, 534, 235 CrossRef CAS PubMed.
- S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Rotary and linear molecular motors driven by pulses of a chemical fuel, Science, 2017, 358, 340 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Active metal template synthesis of rotaxanes, catenanes and molecular shuttles, Chem. Soc. Rev., 2009, 38, 1530 RSC.
- K. D. Hänni and D. A. Leigh, The application of CuAAC ‘click’ chemistry to catenane and rotaxane synthesis, Chem. Soc. Rev., 2010, 39, 1240 RSC.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Strategies and tactics for the metal-directed synthesis of rotaxanes, knots, catenanes, and higher order links, Angew. Chem., Int. Ed., 2011, 50, 9260 CrossRef CAS PubMed.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Metal ions in the synthesis of interlocked molecules and materials, Chem. Soc. Rev., 2017, 46, 2577 RSC.
- M. Denis and S. M. Goldup, The active template approach to interlocked molecules, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- G. T. Spence and P. D. Beer, Expanding the scope of the anion templated synthesis of interlocked structures, Acc. Chem. Res., 2013, 46, 571 CrossRef CAS PubMed.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Interlocked host rotaxane and catenane structures for sensing charged guest species via optical and electrochemical methodologies, Org. Biomol. Chem., 2009, 7, 415 RSC.
- M. J. Langton and P. D. Beer, Rotaxane and catenane host structures for sensing charged guest species, Acc. Chem. Res., 2014, 47, 1935 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Progress in the synthesis and exploitation of catenanes since the Millennium, Chem. Soc. Rev., 2014, 43, 4658 RSC.
- A. Caballero, F. Zapata and P. D. Beer, Interlocked host molecules for anion recognition and sensing, Coord. Chem. Rev., 2013, 257, 2434 CrossRef CAS.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Une nouvelle famille de molecules: les metallo-catenanes, Tetrahedron Lett., 1983, 24, 5095 CrossRef CAS.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, Catalytic ‘click’ rotaxanes: a substoichiometric metal-template pathway to mechanically interlocked architectures, J. Am. Chem. Soc., 2006, 128, 2186 CrossRef CAS PubMed.
- M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, Rotaxane-based transition metal complexes: effect of the mechanical bond on structure and electronic properties, J. Am. Chem. Soc., 2019, 141, 879 CrossRef CAS PubMed.
- C. Dietrich-Buchecker, J. P. Sauvage and J. M. Kern, Synthesis and electrochemical studies of catenates: stabilization of low oxidation states by interlocked macrocyclic ligands, J. Am. Chem. Soc., 1989, 111, 7791 CrossRef CAS.
- G. Baggi and S. J. Loeb, Rotationally active ligands: dialing-up the co-conformations of a [2]rotaxane for metal ion binding, Angew. Chem., Int. Ed., 2016, 55, 12533 CrossRef CAS PubMed.
- G. Baggi and S. J. Loeb, Rotationally active ligands: dialing-up multiple interlocked co-conformations for silver(I) coordination, Chem. – Eur. J., 2017, 23, 14163 CrossRef CAS PubMed.
- M. Denis, J. Pancholi, K. Jobe, M. Watkinson and S. M. Goldup, Chelating rotaxane ligands as fluorescent sensors for metal ions, Angew. Chem., Int. Ed., 2018, 57, 5310 CrossRef CAS PubMed.
- T. Shukla, A. K. Dwivedi, R. Arumugaperumal, C.-M. Lin, S.-Y. Chen and H.-C. Lin, Host-guest interaction of rotaxane assembly through selective detection of ferric ion: insight into hemin sensing and switching with sodium ascorbate, Dyes Pigm., 2016, 131, 49 CrossRef CAS.
- S.-M. Chan, F.-K. Tang, C.-S. Kwan, C.-Y. Lam, S. C. K. Hau and K. C. F. Leung, Water-compatible Fluorescent [2]rotaxanes for Au3+ detection and bioimaging, Mater. Chem. Front., 2019, 3, 2388 RSC.
- C. Lincheneau, B. Jean-Denis and T. Gunnlaugsson, Self-assembly formation of mechanically interlocked [2]- and [3]catenanes using lanthanide ion [Eu(III)] templation and ring closing metathesis reactions, Chem. Commun., 2014, 50, 2857 RSC.
- F. Zapata, O. A. Blackburn, M. J. Langton, S. Faulkner and P. D. Beer, Lanthanide cation-templated synthesis of rotaxanes, Chem. Commun., 2013, 49, 8157 RSC.
- J. F. Ayme, G. Gil-Ramírez, D. A. Leigh, J. F. Lemonnier, A. Markevicius, C. A. Muryn and G. Zhang, Lanthanide template synthesis of a molecular trefoil knot, J. Am. Chem. Soc., 2014, 136, 13142 CrossRef CAS PubMed.
- G. Zhang, G. Gil-Ramírez, A. Markevicius, C. Browne, I. J. Vitorica-Yrezabal and D. A. Leigh, Lanthanide template synthesis of trefoil knots of single handedness, J. Am. Chem. Soc., 2015, 137, 10437 CrossRef CAS PubMed.
- M. Nandi, S. Bej, T. K. Ghosh and P. Ghosh, A multifunctional catenated host for the efficient binding of Eu3+ and Gd3, Chem. Commun., 2019, 55, 3085 RSC.
- R. Mitra, M. Thiele, F. Octa-Smolin, M. C. Letzel and J. Niemeyer, A bifunctional chiral [2]catenane based on 1,1′-binaphthyl-phosphates, Chem. Commun., 2016, 52, 5977 RSC.
- S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, Goldberg active template synthesis of a [2]rotaxane ligand for asymmetric transition-metal catalysis, J. Am. Chem. Soc., 2015, 137, 7656 CrossRef CAS PubMed.
- M. Galli, J. E. M. Lewis and S. M. Goldup, A stimuli-responsive rotaxane-gold catalyst: regulation of activity and diastereoselectivity, Angew. Chem., Int. Ed., 2015, 54, 13545 CrossRef CAS.
- A. Heard and S. Goldup, A mechanically planar chiral rotaxane ligand for enantioselective catalysis, ChemRxiv, 2019 DOI:10.26434/chemrxiv.9701789.v1.
- Y. J. Lee, K. S. Liu, C. C. Lai, Y. H. Liu, S. M. Peng, R. P. Cheng and S. H. Chiu, Na+ ions induce the pirouetting motion and catalytic activity of [2]rotaxanes, Chem. – Eur. J., 2017, 23, 9756 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Applications of supramolecular anion recognition, Chem. Rev., 2015, 115, 8038 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Advances in anion supramolecular chemistry: from recognition to chemical applications, Angew. Chem., Int. Ed., 2014, 53, 11716 CrossRef CAS PubMed.
- J. A. Wisner, P. D. Beer and M. G. B. Drew, A demonstration of anion templation and selectivity in pseudorotaxane formation, Angew. Chem., Int. Ed., 2001, 40, 3606 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Halogen bonding in supramolecular chemistry, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- A. Brown and P. D. Beer, Halogen bonding anion recognition, Chem. Commun., 2016, 52, 8645 RSC.
- J. Y. C. Lim and P. D. Beer, Sigma-hole interactions in anion recognition, Chem, 2018, 4, 731 CAS.
- S. Kubik, Anion recognition in water, Chem. Soc. Rev., 2010, 39, 3648 RSC.
- M. J. Langton, C. J. Serpell and P. D. Beer, Anion recognition in water: recent advances from a supramolecular and macromolecular perspective, Angew. Chem., Int. Ed., 2016, 55, 1974 CrossRef CAS.
- S. Kubik, Anion recognition in aqueous media by cyclopeptides and other synthetic receptors, Acc. Chem. Res., 2017, 50, 2870 CrossRef CAS.
- M. A. Yawer, V. Havel and V. Sindelar, A bambusuril macrocycle that binds anions in water with high affinity and selectivity, Angew. Chem., Int. Ed., 2015, 54, 276 CrossRef CAS.
- M. J. Langton and P. D. Beer, Nitrate anion templated synthesis of a [2]catenane for nitrate recognition in organic-aqueous solvent media, Chem. Commun., 2014, 50, 8124 RSC.
- V. Martí-Centelles and P. D. Beer, Nitrate anion recognition in organic-aqueous solvent mixtures by a bis(triazolium)acridine-containing [2]rotaxane, Chem. – Eur. J., 2015, 21, 9397 CrossRef PubMed.
- M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Nitrite-templated synthesis of lanthanide-containing [2]rotaxanes for anion sensing, Angew. Chem., Int. Ed., 2014, 53, 11463 CrossRef CAS PubMed.
- N. G. White, A. R. Colaço, I. Marques, V. Félix and P. D. Beer, Halide selective anion recognition by an amide-triazolium axle containing [2]rotaxane, Org. Biomol. Chem., 2014, 12, 4924 RSC.
- J. Y. C. Lim, M. J. Cunningham, J. J. Davis and P. D. Beer, Neutral redox-active hydrogen- and halogen-bonding [2]rotaxanes for the electrochemical sensing of chloride, Dalton Trans., 2014, 43, 17274 RSC.
- A. Brown, M. J. Langton, N. L. Kilah, A. L. Thompson and P. D. Beer, Chloride-anion-templated synthesis of a strapped-porphyrin-containing catenane host system, Chem. – Eur. J., 2015, 21, 17664 CrossRef CAS.
- M. Denis, L. Qin, P. Turner, K. Jolliffe and S. M. Goldup, A fluorescent ditopic rotaxane ion pair host, Angew. Chem., Int. Ed., 2018, 57, 5315 CrossRef CAS.
- S. J. Pike, J. J. Hutchinson and C. A. Hunter, H-Bond acceptor parameters for anions, J. Am. Chem. Soc., 2017, 139, 6700 CrossRef CAS PubMed.
- A. Brown, T. Lang, K. M. Mullen and P. D. Beer, Active metal template synthesis of a neutral indolocarbazole-containing [2]rotaxane host system for selective oxoanion recognition, Org. Biomol. Chem., 2017, 15, 4587 RSC.
- R. Arumugaperumal, V. Srinivasadesikan, M. V. Ramakrishnam Raju, M. C. Lin, T. Shukla, R. Singh and H. C. Lin, Acid/base and H2PO4− controllable high-contrast optical molecular switches with a novel BODIPY functionalized [2]rotaxane, ACS Appl. Mater. Interfaces, 2015, 7, 26491 CrossRef CAS PubMed.
- R. Arumugaperumal, P. Venkatesan, T. Shukla, P. Raghunath, R. Singh, S. P. Wu, M. C. Lin and H. C. Lin, Multi-stimuli-responsive high contrast fluorescence molecular controls with a far-red emitting BODIPY-based [2]rotaxane, Sens. Actuators, B, 2018, 270, 382 CrossRef CAS.
- J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Formation of self-templated 2,6-bis(1,2,3-triazol-4-yl)pyridine [2]catenanes by triazolyl hydrogen bonding: selective anion hosts for phosphate, Angew. Chem., Int. Ed., 2016, 55, 8938 CrossRef CAS PubMed.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, Enhancement of anion recognition exhibited by a halogen-bonding rotaxane host system, J. Am. Chem. Soc., 2010, 132, 11893 CrossRef CAS.
- J. M. Mercurio, R. C. Knighton, J. Cookson and P. D. Beer, Halotriazolium axle functionalised [2]rotaxanes for anion recognition: investigating the effects of halogen-bond donor and preorganisation, Chem. – Eur. J., 2014, 20, 11740 CrossRef CAS.
- A. E. Hess and P. D. Beer, Chelated charge assisted halogen bonding enhanced halide recognition by a pyridinium-iodotriazolium axle containing [2]rotaxane, Org. Biomol. Chem., 2016, 14, 10193 RSC.
- J. M. Mercurio, A. Caballero, J. Cookson and P. D. Beer, A halogen- and hydrogen-bonding [2]catenane for anion recognition and sensing, RSC Adv., 2015, 5, 9298 RSC.
- M. J. Langton, S. W. Robinson, I. Marques, V. Felix and P. D. Beer, Halogen bonding in water results in enhanced anion recognition in acyclic and rotaxane hosts, Nat. Chem., 2014, 6, 1039 CrossRef CAS PubMed.
- M. Řezanka, M. J. Langton and P. D. Beer, Anion recognition in water by a rotaxane containing a secondary rim functionalised cyclodextrin stoppered axle, Chem. Commun., 2015, 51, 4499 RSC.
- M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Iodide recognition and sensing in water by a halogen-bonding ruthenium(II)-based rotaxane, Chem. – Eur. J., 2016, 22, 185 CrossRef CAS PubMed.
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, A potent halogen-bonding donor motif for anion recognition and anion template mechanical bond synthesis, Angew. Chem., Int. Ed., 2019, 58, 13823 CrossRef CAS PubMed.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, Evidence for halogen bond covalency in acyclic and interlocked halogen-bonding receptor anion recognition, J. Am. Chem. Soc., 2015, 137, 499 CrossRef CAS PubMed.
- S. W. Robinson and P. D. Beer, Halogen bonding rotaxanes for nitrate recognition in aqueous media, Org. Biomol. Chem., 2017, 15, 153 RSC.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, A chiral halogen bonding [3]rotaxane for recognition and sensing of biologically-relevant dicarboxylate anions, Angew. Chem., Int. Ed., 2018, 57, 584 CrossRef CAS PubMed.
- H. A. Klein and P. D. Beer, Iodide discrimination by tetra-iodotriazole halogen bonding interlocked hosts, Chem. – Eur. J., 2019, 25, 3125 CAS.
- L. C. Gilday and P. D. Beer, Halogen- and hydrogen-bonding catenanes for halide-anion recognition, Chem. – Eur. J., 2014, 20, 8379 CrossRef CAS PubMed.
- B. R. Mullaney, B. E. Partridge and P. D. Beer, A halogen-bonding bis-triazolium rotaxane for halide-selective anion recognition, Chem. – Eur. J., 2015, 21, 1660 CrossRef CAS PubMed.
- X. Li, J. Y. C. Lim and P. D. Beer, Acid-regulated switching of metal cation and anion guest binding in halogen-bonding rotaxanes, Chem. – Eur. J., 2018, 24, 17788 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Félix and P. D. Beer, Chalcogen bonding macrocycles and [2]rotaxanes for anion recognition, J. Am. Chem. Soc., 2017, 139, 3122 CrossRef CAS PubMed.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, A halogen-bonding catenane for anion recognition and sensing, Angew. Chem., Int. Ed., 2012, 51, 1876 CrossRef CAS PubMed.
- B. R. Mullaney, A. L. Thompson and P. D. Beer, An all-halogen bonding rotaxane for selective sensing of halides in aqueous media, Angew. Chem., Int. Ed., 2014, 53, 11458 CrossRef CAS PubMed.
- M. J. Langton, Y. Xiong and P. D. Beer, Active-metal template synthesis of a halogen-bonding rotaxane for anion recognition, Chem. – Eur. J., 2015, 21, 18910 CrossRef CAS PubMed.
- X. Li, J. Y. C. Lim and P. D. Beer, Cationic all-halogen bonding rotaxanes for halide anion recognition, Faraday Discuss., 2017, 203, 245 RSC.
- J. Y. C. Lim, T. Bunchuay and P. D. Beer, Strong and selective halide anion binding by neutral halogen-bonding [2]rotaxanes in wet organic solvents, Chem. – Eur. J., 2017, 23, 4700 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Electrochemical bromide sensing with a halogen bonding [2]rotaxane, Eur. J. Org. Chem., 2019, 3433 CrossRef CAS.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Enantioselective anion recognition by chiral halogen-bonding [2]rotaxanes, J. Am. Chem. Soc., 2017, 139, 12228 CrossRef CAS.
- A. Caballero, L. Swan, F. Zapata and P. D. Beer, Iodide-induced shuttling of a halogen- and hydrogen-bonding two-station rotaxane, Angew. Chem., Int. Ed., 2014, 53, 11854 CrossRef CAS.
- T. A. Barendt, S. W. Robinson and P. D. Beer, Superior anion induced shuttling behaviour exhibited by a halogen bonding two station rotaxane, Chem. Sci., 2016, 7, 5171 RSC.
- T. A. Barendt, A. Docker, I. Marques, V. Félix and P. D. Beer, Selective nitrate recognition by a halogen-bonding four-station [3]rotaxane molecular shuttle, Angew. Chem., Int. Ed., 2016, 55, 11069 CrossRef CAS.
- T. A. Barendt, I. Rašović, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, Anion mediated photophysical behavior in a C60 fullerene [3]rotaxane shuttle, J. Am. Chem. Soc., 2018, 140, 1924 CrossRef CAS PubMed.
- T. A. Barendt, L. Ferreira, I. Marques, V. Felix and P. D. Beer, Anion- and solvent-induced rotary dynamics and sensing in a perylene diimide [3]catenane, J. Am. Chem. Soc., 2017, 139, 9026 CrossRef CAS PubMed.
- H. A. Klein, H. Kuhn and P. D. Beer, Anion and pH dependent molecular motion by a halogen bonding [2]rotaxane, Chem. Commun., 2019, 55, 9975 RSC.
- J. M. Van Raden, B. M. White, L. N. Zakharov and R. Jasti, Nanohoop rotaxanes from active metal template syntheses and their potential in sensing applications, Angew. Chem., Int. Ed., 2019, 58, 7341 CrossRef CAS PubMed.
- S. Corra, C. De Vet, J. Groppi, M. La Rosa, S. Silvi, M. Baroncini and A. Credi, Chemical on/off switching of mechanically planar chirality and chiral anion recognition in a [2]rotaxane molecular shuttle, J. Am. Chem. Soc., 2019, 141, 9129 CrossRef CAS PubMed.
- S. K. Kim and J. L. Sessler, Ion pair receptors, Chem. Soc. Rev., 2010, 39, 3784 RSC.
- A. J. McConnell and P. D. Beer, Heteroditopic receptors for ion-pair recognition, Angew. Chem., Int. Ed., 2012, 51, 5052 CrossRef CAS PubMed.
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Macrocycles as ion pair receptors, Chem. Rev., 2019, 119, 9753 CrossRef CAS PubMed.
- R. C. Knighton and P. D. Beer, Axle component separated ion-pair recognition by a neutral heteroditopic [2]rotaxane, Chem. Commun., 2014, 50, 1540 RSC.
- A. Brown, K. M. Mennie, O. Mason, N. G. White and P. D. Beer, Copper(II)-directed synthesis of neutral heteroditopic [2]rotaxane ion-pair host systems incorporating hydrogen and halogen bonding anion binding cavities, Dalton Trans., 2017, 46, 13376 RSC.
- J. R. Romero, G. Aragay and P. Ballester, Ion-pair recognition by a neutral [2]rotaxane based on a bis-calix[4]pyrrole cyclic component, Chem. Sci., 2017, 8, 491 RSC.
Footnote |
| † Current address: Faculty of Engineering and Science, University of Greenwich, Central Avenue, Chatham Maritime, Kent, ME4 4TB, UK. |
| This journal is © the Partner Organisations 2020 |