
pH-Responsive protein nanoparticles via conjugation of degradable PEG to the surface of cytochrome c†
Elena
Steiert‡
a,
Johannes
Ewald‡
b,
Annika
Wagner
a,
Ute A.
Hellmich
 a,
Holger
Frey
b and
Peter R.
Wich
*acd
a,
Holger
Frey
b and
Peter R.
Wich
*acd
aInstitute of Pharmacy und Biochemistry, Johannes Gutenberg-University Mainz, 55128 Mainz, Germany. E-mail: p.wich@unsw.edu.au
bInstitute of Organic Chemistry, Johannes Gutenberg-University Mainz, 55128 Mainz, Germany
cAustralian Centre for NanoMedicine, University of New South Wales, Sydney, NSW 2052, Australia
dSchool of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia
First published on 17th October 2019
Abstract
Proteins represent a versatile biopolymer material for the preparation of nanoparticles. For drug delivery applications an acid-triggered disassembly and payload release is preferred. Herein, we present a protein nanoparticle system based on cytochrome c, which is surface-modified with acid-degradable polyethylene glycol (PEGylation). pH-Sensitivity was obtained through vinyl ether moieties distributed in the polyether backbone. When PEGylated, cytochrome c shows a different solubility behaviour in organic solvents, which allows for particle preparation using an emulsion-based solvent evaporation method. The resulting particles are stable under physiological conditions but degrade at acidic pH values. Fluorescence-labelled dextran was successfully encapsulated as a hydrophilic model payload in these degradable nanoparticles and a release under acidic conditions was observed.
Introduction
Polyethylene glycol (PEG) is the current gold standard for the protective surface modification of proteins. More than forty years ago it was discovered that the conjugation of proteins with PEG (“PEGylation”) leads to better immunogenic properties and increased circulation half-life compared to the unmodified protein.1,2 Several protein–polymer conjugates are FDA approved, which highlights the importance of these systems in therapeutic applications.3 In addition to the reduced immunogenicity and improved pharmacokinetic properties, protein–PEG conjugates show enhanced stability and solubility in biologically relevant environments.4–6 However, concerns were raised due to the lack of degradability which can cause accumulation in human tissue when higher molecular weight PEGs are used.7 PEGylation has also been shown to impair the catalytic activity of enzyme conjugates.8 For these reasons, increased efforts are made to develop PEG alternatives and degradable analogs.7,9,10So far, most approaches focus on the use of degradable linkers between protein and polymer, e.g. via hydrazine,11 azidomethyl-methylmaleic anhydride12 or disulfide13 linkages, where the protein–polymer bond can be cleaved under acidic or reductive conditions. There are only a few protein–polymer conjugates which make use of a degradable polymer backbone. Acetals14 or poly(phosphates)15 in the polymer backbone can result in polymer degradation under acidic or hydrolytic conditions. A recent work by Pelegri-O'Day et al. demonstrated the use of a degradable PEG analogue for reversible PEGylation of lysozyme. However, this approach requires a non-physiological Grubbs III catalyst for the degradation by depolymerisation of the polymer.16
A very promising cleavable moiety for degradable PEG are vinyl ethers, as they combine a fast hydrolysis rate at physiologically relevant acidic ranges of pH 4–5 with excellent stability at pH 7.4.17 This structure has first been implemented in PEG by Hawker et al. via copolymerization of ethylene oxide (EO) and epichlorohydrin (ECH), followed by subsequent elimination of chloride.18 However, these materials still show rather high dispersity and no defined end groups as a result of their triethylaluminium-catalysed polymerization. Furthermore, as recently published by Danner et al., the respective copolymerization shows a strong reactivity difference between EO and ECH, which results in a pronounced monomer gradient and consequently an uneven distribution of the predetermined cleaving sites along the backbone and therefore considerable dispersity of the hydrolysed product.19 The Frey group recently reported another approach to vinyl ether containing PEG.17 Classic anionic ring opening copolymerization (AROP) of EO and 3,4-epoxy-1-butene (EPB) to obtain P(EG-co-EPB), followed by isomerization of allylic ether moieties to vinyl ethers (P(EG-co-isoEPB)) proved to be the method of choice to combine low polydispersity, well-defined end groups, tailorable molecular weight and adjustable content of cleavable moieties in the polyether backbone. This material may serve as a promising substitute for PEG for various therapeutic and biomedical applications in the future, as it expands the favourable properties of well-known and well-established PEG with degradability at physiologically relevant pH values on reasonable timescales.
In this work we apply this material to prepare a new type of degradable protein–PEG conjugate that can be formulated into acid-responsive nanoparticles. The general concept of preparing nanoparticles based on protein–polymer conjugates has attracted considerable interest in recent years.20,21 For example, we reported a protein-based nanoparticle system using highly PEGylated lysozyme (LYZ) for the delivery of anticancer drugs. The LYZ-PEG conjugate is soluble in dichloromethane (DCM) and at the same time, the protein structure is preserved and protected. Nanoparticles are prepared using a mild emulsion-based method that allows the encapsulation of hydrophilic and hydrophobic payloads.22–24 While the particles are capable of releasing their payload via passive diffusion, a triggered release would be preferred. For this purpose, the introduction of stimuli-responsiveness is desired, in order for the drug delivery system to release their payload in a controlled manner.25 For example, De Geest et al. developed a pH-sensitive polymer–protein (BSA) conjugate that self-assembles into nanoparticles. The polymer chains include dioxolane groups that can switch under acidic conditions from a hydrophobic form to hydrophilic diol groups. This results in the loss of the self-assembly behaviour of this system and the disassembly of the particles.26 A drawback of the system is the permanent linkage of the polymers to the protein and the highly acidic conditions (pH 1) required for particle degradation that are, with the exception of the gastrointestinal tract, not accessible in a biological system.
Wang et al. developed a multi-stimuli responsive nanoparticle system consisting of hydrazine-modified BSA proteins that were conjugated with aldehyde-functionalized thermo-responsive copolymers. The resulting hydrazone bond between the protein and polymer is acid-sensitive. After the temperature-induced self-assembly of the protein–polymer conjugate above its lower critical solution temperature (LCST) the particles needed further stabilisation by crosslinking the BSA using cysteamine. The resulting disulfide bonds between the proteins introduced sensitivity under reductive conditions. Apart from an elaborate multistep synthesis to obtain the final particles, another disadvantage of this system is that the particles do not disassemble in acidic conditions alone. For complete particle degradation, both acidic and reductive microenvironments have to be present at the same time.27
In this work, we present a novel class of pH-sensitive protein nanoparticles using an intrinsic degradable PEG copolymer. In straightforward synthesis steps, we prepare a particle system that is stable without crosslinking and can degrade and disassemble at physiologically relevant acidic pH values. For this purpose, an active ester of fully degradable vinyl ether PEGs was synthesized and conjugated to the surface of cytochrome c (Cyt). This new type of protein–polymer conjugate (CytdegPEG) is soluble in organic solvents and particles can be prepared by forming an emulsion under mild conditions.
Results and discussion
Synthesis of mP(EG81-co-isoEPB6)-NHS
For the synthesis of the acid degradable PEG copolymer, triethylene glycol monomethyl ether was chosen as a monofunctional initiator due to its convenient structure, which readily enables determination of molecular weight and comonomer content by NMR integration. The precursor molecule, the allyl ether containing PEG copolymer mP(EG81-co-EPB6) was synthesised by AROP of EO and EPB based on a procedure recently reported by Worm et al. (Fig. 1).17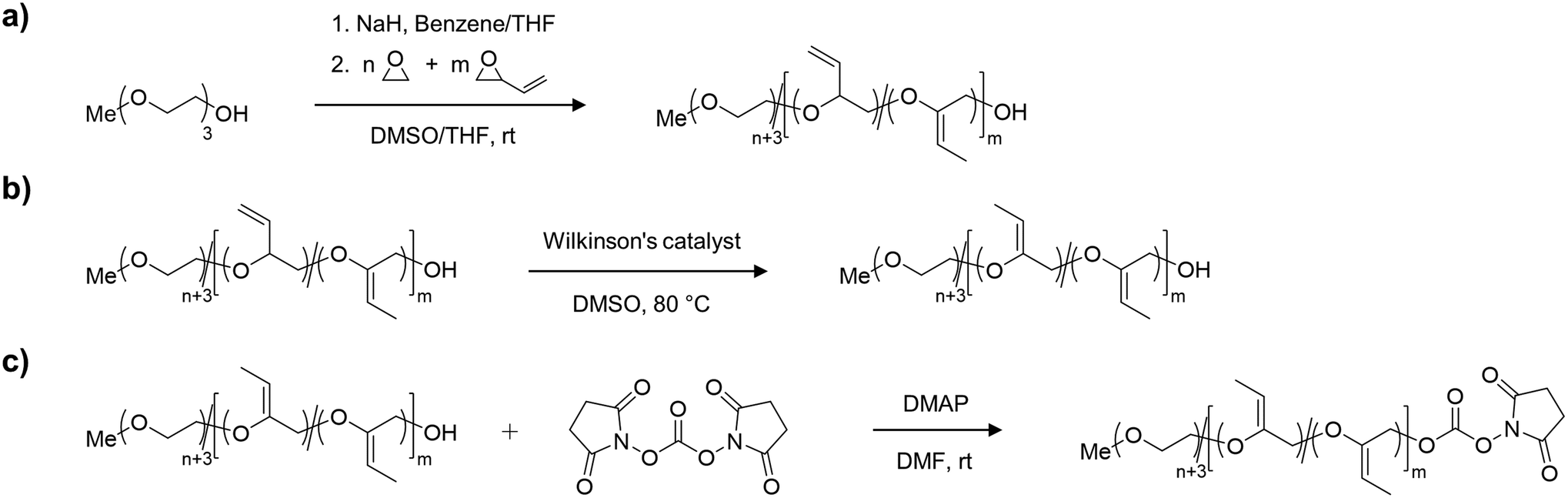 | ||
| Fig. 1 Synthesis route to obtain mP(EG81-co-isoEPB6)-NHS; (a) copolymerization of EO and EPB to obtain mP(EG81-co-EPB6); (b) isomerization of mP(EG81-co-EPB6) to mP(EG81-co-isoEPB6); (c) activation of hydroxyl group to NHS active ester m(P(EG81-co-isoEPB6)-NHS) using NHS-DSC. | ||
The resulting allylic ether moieties distributed at the polymer backbone were isomerized quantitively to acid-labile vinyl ethers to obtain mP(EG81-co-isoEPB6), while the structure and integrity of the copolymer chain was retained (Fig. 1b). To target nucleophilic lysine residues at the surface of cytochrome c, the hydroxyl end group of mP(EG81-co-isoEPB6) was reacted with N,N′-disuccinimidyl carbonate (NHS-DSC) to obtain the activated carbonate ester.
A molecular weight of Mn = 4000 g mol−1 with 12 mol% EPB was targeted. 1H-NMR integration of the allylic moieties (between 6.0 and 4.5 ppm), the ether backbone (between 4.0 and 3.3 ppm) and the methyl ether end group (3.24 ppm), confirmed this molecular weight (see ESI Fig. S1†).
An incorporation 7 mol% of EPB was found, which is lower than targeted but still more than sufficient to guarantee the degradation of the polymer chain (Table 1). Interestingly, already during the copolymerization a small amount of vinyl ether moieties (around 10% of total allylic units) was formed, as can be seen by NMR. Quantitative isomerization was confirmed by the absence of distinct allylic signals in 1H-NMR spectrum and appearance of vinyl ether signals (4.8–4.6 ppm) as reported previously after this step as well.17
| Sample | M n, NMR (g mol−1) | M n, SEC (g mol−1) | M n, MALDI (g mol−1) | Đ SEC | mol% EPB |
|---|---|---|---|---|---|
| SEC: DMF, RI Detector, PEG standards. | |||||
| mP(EG81-co-isoEPB6) | 3815 | 3380 | 3620 | 1.04 | 7 |
Fig. 2b shows the molecular weight distribution of mP(EG81-co-EPB6) at all stages referred to in this work. Analysis by SEC after polymerization shows a well-defined copolymer with very narrow weight distribution (Đ = 1.03–1.04) and a molecular weight of Mn = 3380 g mol−1. It should be noted, that SEC underestimates the molecular weight of these copolymers, as the hydrodynamic radius of PEG copolymers differs from that of PEG standards. During the post-polymerization steps, the molecular weight distribution remained low (Đ = 1.03–1.04), indicating that no unwanted cleavage or crosslinking of the polymer occurred.
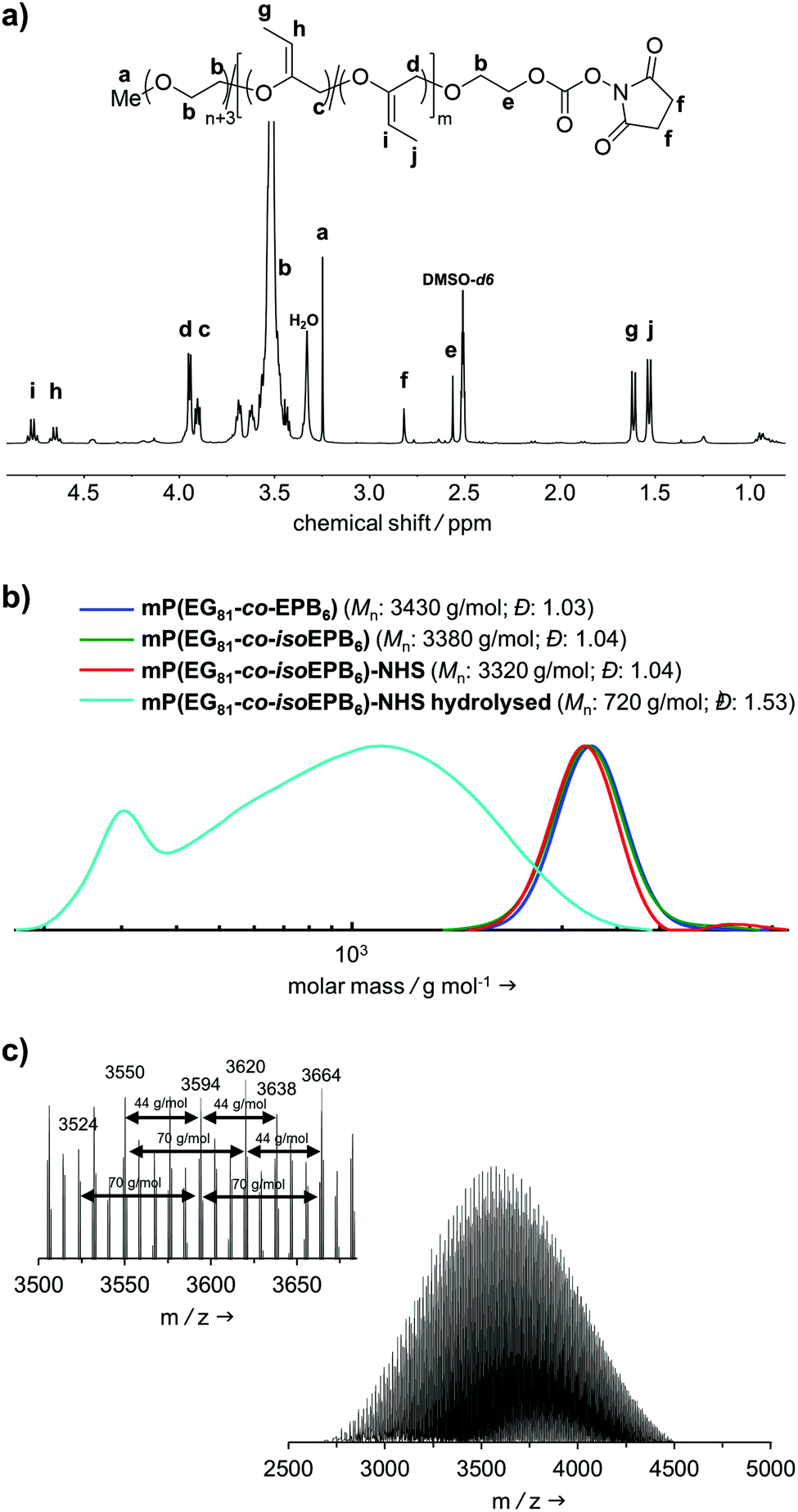 | ||
| Fig. 2 (a) 1H-NMR spectrum (400 MHz, DMSO-d6) of mP(EG81-co-isoEPB6)-NHS; (b) SEC analysis of mP(EG81-co-EPB6) copolymers at different stages referred to in this work (eluent: DMF, RI detector, PEG standard); (c) MALDI-ToF analysis of mP(EG81-co-isoEPB6). | ||
The successful copolymerization was also confirmed by MALDI-ToF, resulting in distinct intervals of 44 g mol−1 for EG and 70 g mol−1 for EPB units between the signals as well as a monomodal distribution with Mn = 3620 g mol−1 (Fig. 2c).
The degree of functionalization to NHS ester reached 37%. Most post-functionalization approaches for PEGylating agents targeting lysine residues usually involve the use of carboxylic acids, carboxylic anhydrides or acyl chlorides to form an ester or amide bond between the polymer and a linker to the reactive group used for PEGylation. As this would include the occurrence of acidic protons (which is not favourable in the case of an acid-labile polymer), we decided to use NHS-DSC as the most convenient way to form an activated carbonate ester for targeting of lysine residues. While this reaction did not result in quantitative functionalization of the polymer, this could easily be overcome by increasing the amount of polymer per protein during PEGylation. Importantly, the non-functionalized PEG chains do not interfere with the protein PEGylation and the procedure additionally includes a washing step for the removal of excess PEG.
In an acidic environment, the vinyl ether moieties in the polymer backbone are cleaved by a two-step mechanism: initially, a hydronium ion is transferred to the substrate, followed by the addition of water to form a hemiacetal, which then decomposes.28 The fast rate of hydrolysis of this type of copolymer has already been reported by 1H-NMR in situ kinetics in our previous work.17 The result of the hydrolysis of the copolymer is demonstrated by SEC (Fig. 2b). After incubation of mP(EG81-co-isoEPB6) under acidic conditions, the molecular weight shifted from Mn = 3320 g mol−1 to Mn = 720 g mol−1, while the molecular weight distribution increased to Đ = 1.53.
Protein modification
The activated, degradable PEG was coupled to cytochrome c (Cyt) as a model protein, due to the relatively high amount of addressable nucleophilic amine groups at the surface (19×) compared to its small size (12.4 kDa). Furthermore, since Cyt is part of the apoptotic system,29 catalytically active nanoparticles based on this enzyme could be used as future therapeutic nanomaterials. This may be of particular interest e.g. for cancer treatment. Upon attachment of a sufficient number of polymer chains to the protein surface, the special characteristic of PEG, to have a broad solubility in a variety of solvents, is transferred to the underlying protein. This renders the protein–polymer conjugate soluble in dichloromethane and further allowing for nanoparticle formation using a solvent evaporation method.23,24The cytochrome c modification takes place in buffered aqueous solution with a 17-fold excess of mP(EG81-co-isoEPB6)-NHS per protein (which corresponds to 6.3 equivalents of NHS-functionalized polymer per protein) (Fig. 3a).
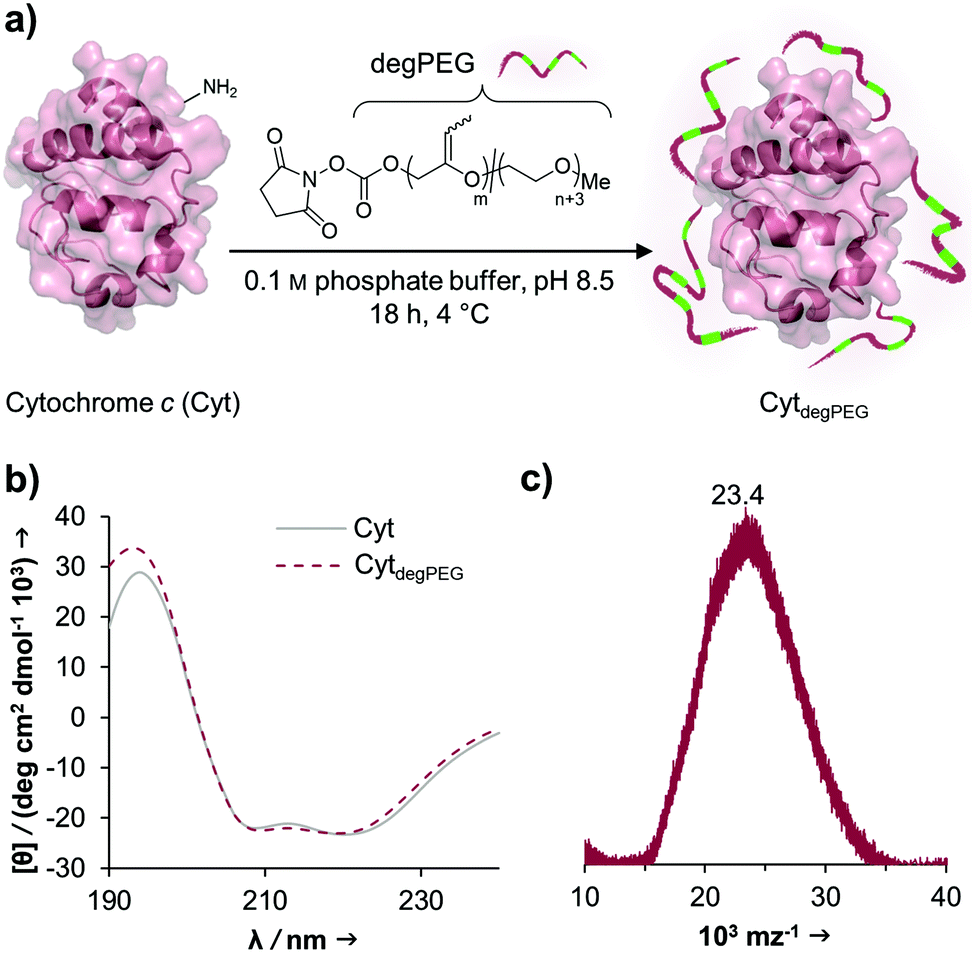 | ||
| Fig. 3 (a) Cyt modification with mP(EG81-co-isoEPB6)-NHS. The wavy line in the polymer sidechain represents both possible isomerization products. The attachment point of the polymer to the protein is the activated carbonate ester. The resulting in CytdegPEG is a schematic illustration and does not reflect the final amount and the actual attachment point of polymer chains; (b) CD spectra of native Cyt (solid line) and CytdegPEG (dotted line); (c) MALDI-ToF of CytdegPEG. | ||
After overnight reaction, the resulting Cyt-polymer conjugate (CytdegPEG) was purified by SEC. The extent of conjugation was qualitatively analysed by SDS-PAGE, which shows a broad band extending from around 23 to 170 kDa (Fig. 4b and ESI section 2.1†). This is a known behaviour of PEGylated protein conjugates, which is commonly explained by interactions between PEG and SDS.30 Most importantly, the SDS-PAGE confirmed that all proteins were modified. Additional MALDI-ToF MS characterization shows a molecular weight increase from 12.4 to 23 kDa upon PEGylation (Fig. 3c). This indicates that on average 3 polymer chains per protein molecule were successfully attached to the protein surface. We performed circular dichroism (CD) spectroscopy to assess whether the modification of Cyt leads to protein structural changes. Only minor alterations (<2%) in the secondary structure elements between modified and unmodified Cyt can be seen (Fig. 3b and ESI Table S1†). We further analysed the catalytic activity of the Cyt–PEG conjugate compared to the native enzyme using an ABTS assay. Importantly, PEGylation does not affect CytdegPEG activity, as the modified protein retained 91 ± 5% activity compared to native Cyt (see ESI section 2.3†). Together, CD spectroscopy and the enzymatic activity assay confirm that neither protein structure nor activity is majorly affected by PEGylation.
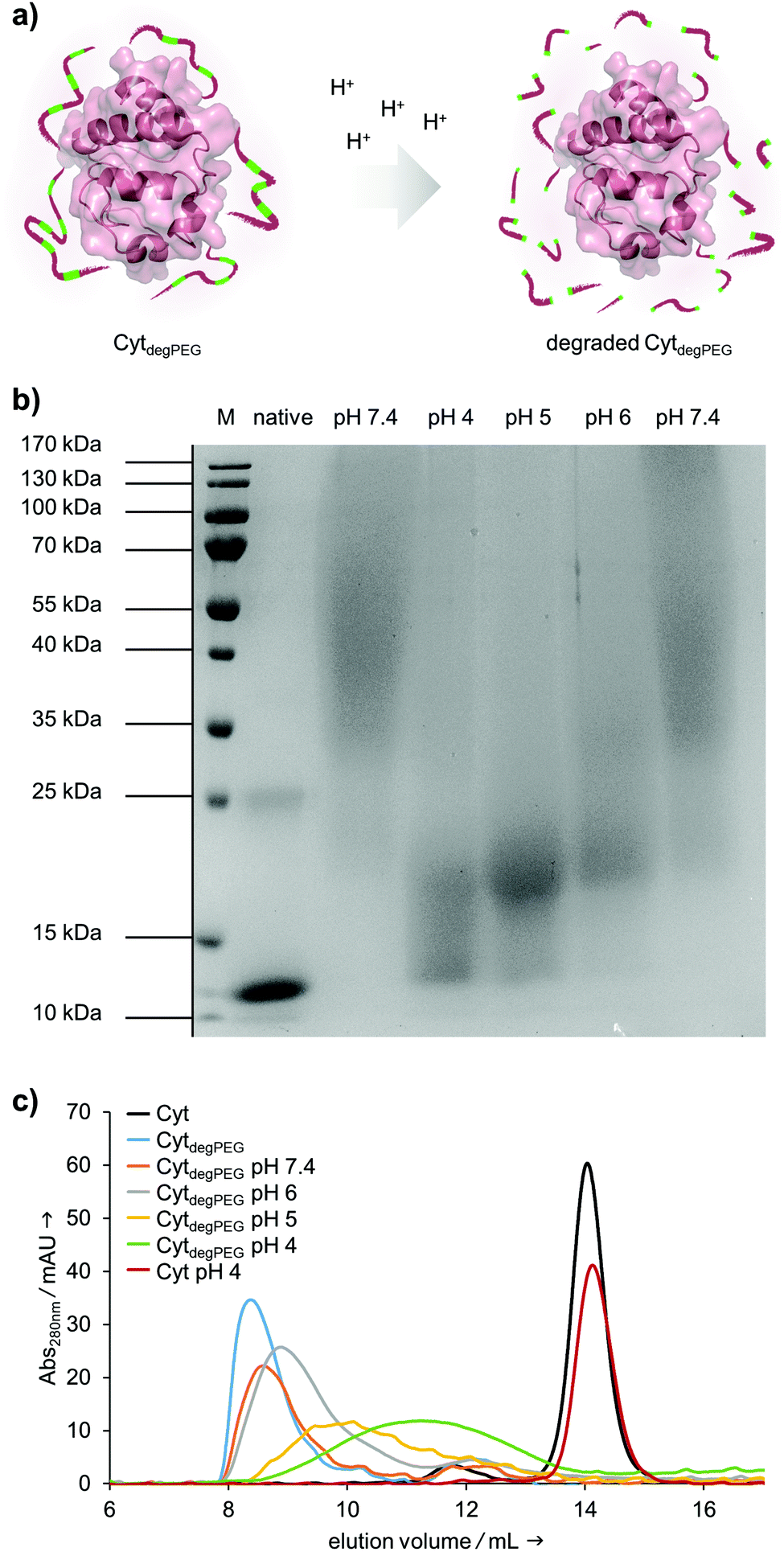 | ||
| Fig. 4 (a) Degradation of the vinyl ether moieties in the polyether backbone of CytdegPEG under acidic conditions; (b) SDS-PAGE (15%) of Cyt (native) and CytdegPEG (left pH 7.4 lane) which were freshly dissolved in dd-H2O. CytdegPEG was incubated at various pH values at 37 °C for 24 h and subsequently analysed through SDS-PAGE (pH 4, pH 5, pH 6 and pH 7.4); (c) SEC analysis of freshly dissolved Cyt and CytdegPEG (black and blue curves) compared to CytdegPEG incubated at various pH values at 37 °C for 24 h. Native Cyt shows no change in elution volume when freshly dissolved or incubated at pH 4 for 24 h (black and red curves). | ||
After the successful modification of Cyt, we investigated degradability of the attached PEG polymer (Fig. 4a). For this, the CytdegPEG conjugate was dissolved in different acidic buffers, incubated at 37 °C for 24 h and characterised via SDS-PAGE. The incubation at pH 4 leads to nearly complete degradation of the polymer linked to the protein surface, as demonstrated by the down-shifted band on the gel, almost back to the same running height as the non-modified enzyme (Fig. 4b). As expected, the increase in pH led to slightly reduced extents of PEG degradation. Nonetheless, even mildly acidic conditions at pH 6 were sufficient to induce a noticeable gel shift and thus PEG degradation. The Cyt-polymer conjugate is stable at neutral conditions (pH 7.4) during the incubation time of 24 h, there is no shift recognizable between freshly dissolved and incubated CytdegPEG in the SDS-PAGE (Fig. 4b, left and right pH 7.4 lane). Efficient, pH-dependent PEG cleavage was further confirmed by SEC, where consecutively later elution volumes with samples incubated at increasing acidity are observed (Fig. 4c).
Nanoparticle preparation, degradation and payload release
For the nanoparticle preparation, we adapted a solvent evaporation method described previously.22 Briefly, the surface modification of Cyt results in a solubility switch and allows the dissolution of CytdegPEG in dichloromethane. For the emulsion preparation, this solution was covered with an aqueous phase containing Oregon-Green™488-dextran (OGD) as a fluorescent model compound to test the loading efficiency and later the release under acidic conditions. After a first sonication step, additional PBS (pH 7.4) was added, and a second sonication step was carried out, resulting in an w/o/w emulsion. This entraps the hydrophilic payload OGD in the inner water droplet, which is surrounded by the DCM layer that contains the CytdegPEG particle material. Subsequent evaporation of the organic solvent leads to the formation of a particle suspension (CytdegPEG-NP) where the dehydrated PEGylated material forms a tight and stable network (ESI, Fig. S10†).Free OGD was removed by dialysis. After purification, we determined an encapsulation efficiency of 50% compared to the initial OGD feed (see ESI†). The encapsulated OGD concentration was 2.41 μM (0.04 mol OGD per 1 mol particle material). This corresponds to a loading content of 1.72 wt% of OGD compared to the total weight of the nanoparticle material (see ESI section 3.1†).
Nanoparticle tracking analysis (NTA) showed an average diameter of 140 nm for the particles, which was confirmed by transmission electron microscopy (TEM) measurements (Fig. 5b and c). The surface charge of the nanoparticles was analysed by ζ-potential measurements, showing nearly neutral CytdegPEG-NPs (−2 mV, see ESI section 3.3†). This is most likely a result of the PEGylation, which shields remaining amino acids on the surface of the individual enzymes.
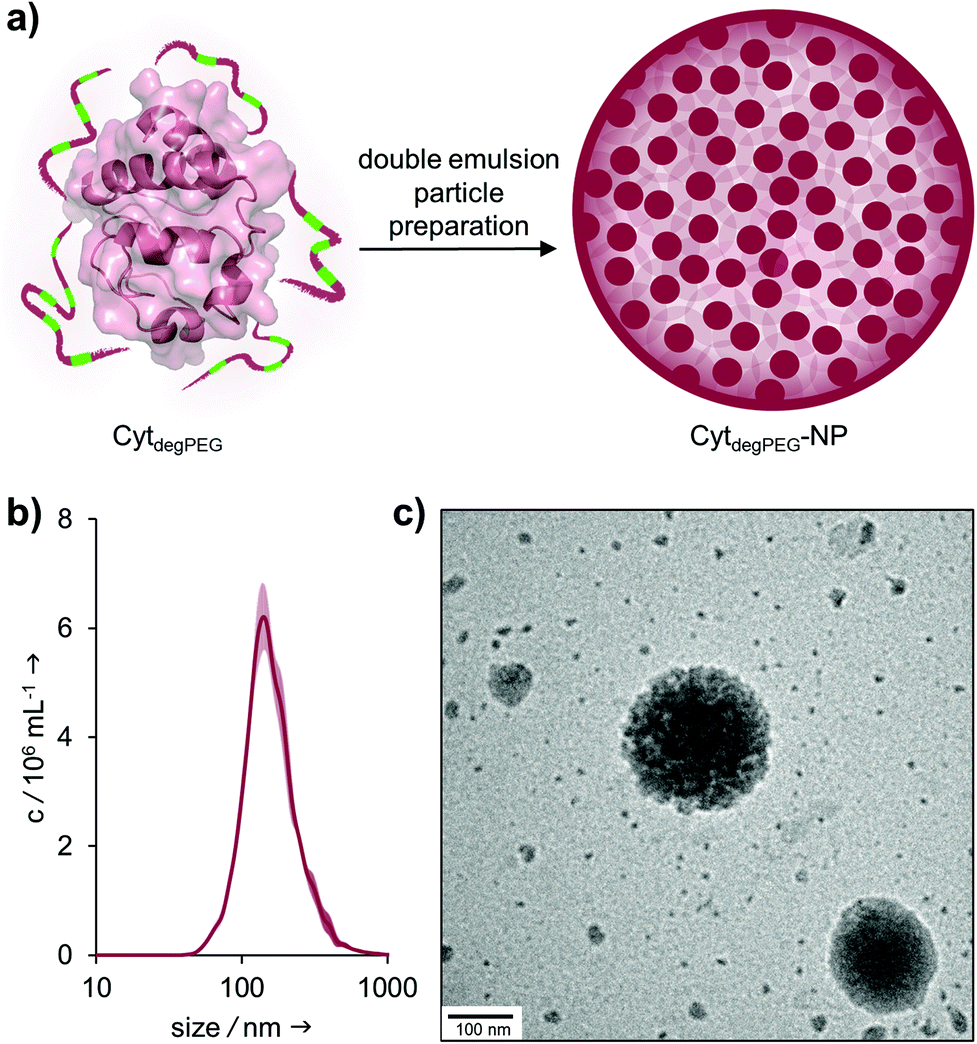 | ||
| Fig. 5 (a) Schematic representation of CytdegPEG-NP preparation; (b) NTA measurements of CytdegPEG-NP show a size around 140 nm; (c) TEM image of CytdegPEG-NP. | ||
Our particles are designed to be stable at neutral pH values but should degrade in an acidic environment due to the intrinsic cleavability of the vinyl ether moieties of the PEG conjugates. This results in the cleavage of the majority of the PEG material with only very small non-degradable PEG segments remaining on the surface of the protein. With this, the protein regains its native hydrophilic solubility, and the individual hydrophobic protein assemblies are broken up, i.e. the overall particle complex falls apart (Fig. 6a).
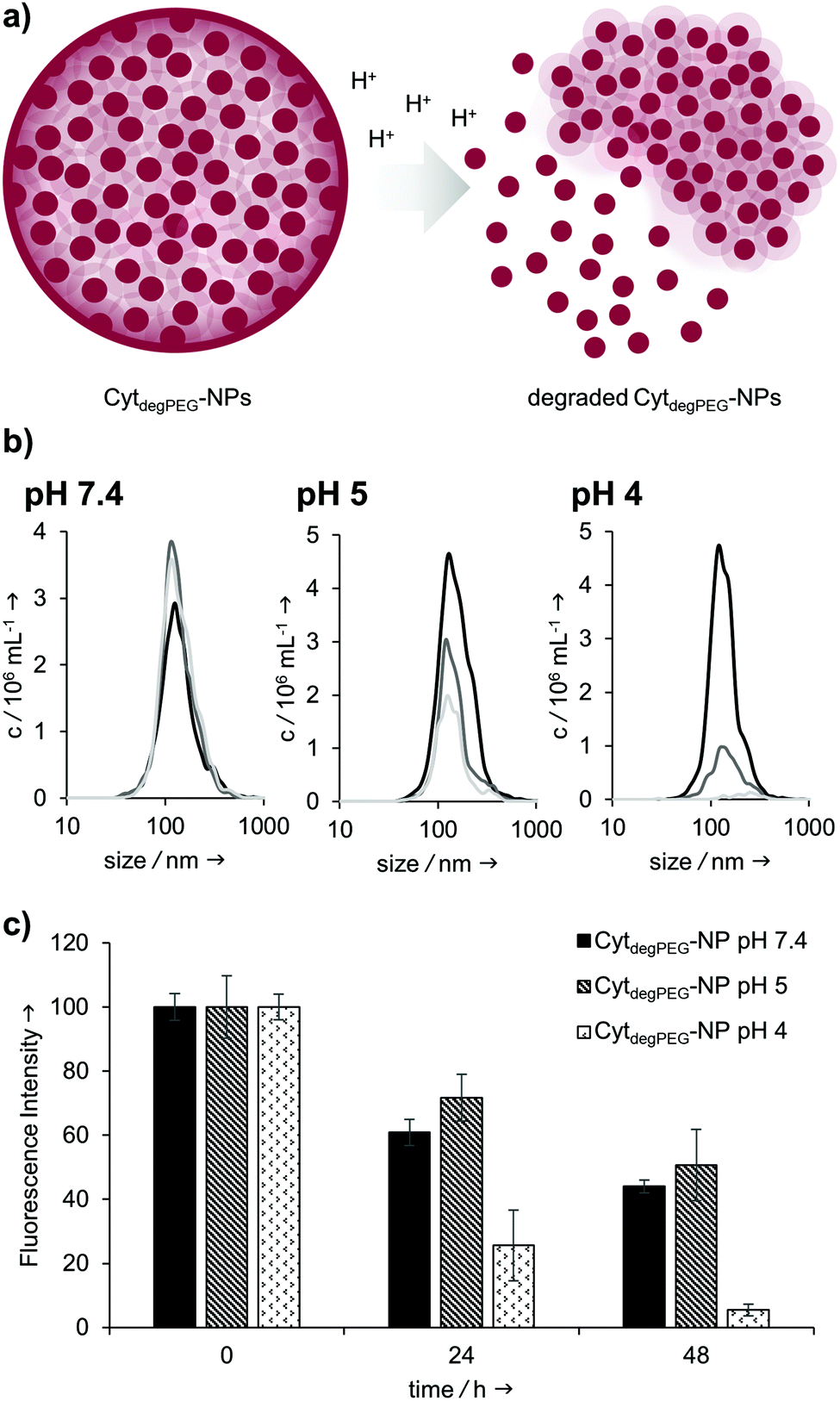 | ||
| Fig. 6 (a) CytdegPEG-NP disassembly under acidic conditions; (b) NTA and (c) fluorescence measurements of CytdegPEG-NP degradation and OGD release over time. Purified particle suspensions were mixed with different buffers and dialysed against corresponding buffers. (b) Particle size measurements (NTA curves) at pH 7.4, pH 5 and pH 4 at different time points of the dialysis experiment. Black curves: measurements directly after mixing the nanoparticles suspensions (0 h); grey curves: after 24 h; light grey curves: after 48 h; (c) fluorescence measurement of the nanoparticle suspension at different time points of the dialysis experiment and under various pH conditions. The lower the bars, the more fluorescent payload was released due to the disassembly of the particle system. | ||
We monitored the particle degradation in a dialysis experiment. For this purpose, nanoparticle suspensions were mixed with different buffers (pH 4, 5 and pH 7.4) and dialysed against corresponding buffers at 37 °C. Particle degradation and OGD release was analysed over time with NTA (Fig. 6b) and fluorescence measurements (Fig. 6c).
Nanoparticles incubated in neutral buffer are stable over a period of 48 h, as can be seen in the concentration of particles over time in NTA measurements (Fig. 6b). At pH 5 the concentration of particles decreased by 50% after 48 h, and at pH 4 almost no particles are visible at the end of the experiment.
Looking at the OGD release over time, it can be seen in the fluorescence measurements (Fig. 6c) that both at pH 7.4 and pH 5 some of the payload is released. This is most likely due to passive diffusion, which is common for hydrophilic payloads, as reported previously.22 However, at pH 4, a clear difference in fluorescence intensity can be seen at 24 and 48 h, due to the full disassembly of the particles and the active OGD release.
For biological applications, the effect of our nanoparticles on human cells needs to be determined. We thus analysed the toxicity of the CytdegPEG material and the nanoparticles on HeLa cells (see Fig. 7b and ESI section 3.5†). The cells were incubated with native Cyt, CytdegPEG and CytdegPEG-NPs. Native Cyt and the CytdegPEG material show no toxicity (Fig. 7a). It has been previously reported, that native Cyt cannot cross the cell membrane.31 The result of the MTT-assay indicates that this is also the case for CytdegPEG. In contrast, CytdegPEG-NPs are most likely taken up by cells by an endosomal pathway which results in a lower cell survival with increasing material concentrations. This can be explained by the high amount of Cyt in the cytosol of the cells where it can perform its native function. Physiologically, Cyt is part of the intrinsic apoptotic pathway in the cytosol since it binds the protease-activating factor-1 (APAF1) which is part of the apoptosome.29 Therefore, our results indicate that the PEG-conjugates are also cleaved in vitro, and the unobstructed enzymes might initiate to some extent apoptosis. Future experiments will focus on the detailed analysis of the particle uptake, protein pathways and the remaining enzymatic activity.
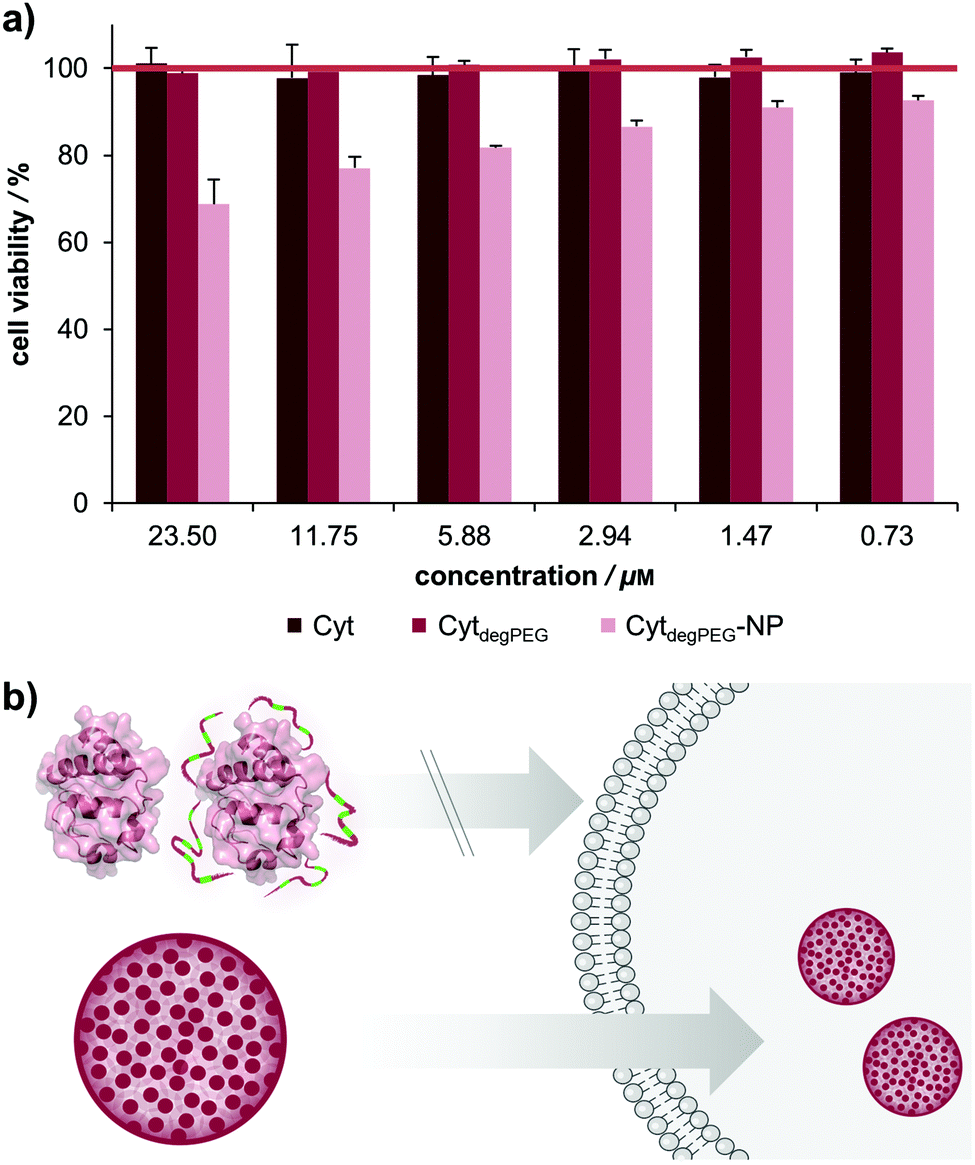 | ||
| Fig. 7 (a) Results of cell viability tests using HeLa cells after incubation with native Cyt (dark red), CytdegPEG (red) and CytdegPEG-NP (light red) in concentrations ranging from 0.73–23.50 μM incubated for 48 h. (b) MTT results indicate that native Cyt and CytdegPEG show no membrane permeability, unlike CytdegPEG-NPs which can be taken up by cells, most likely by endocytic pathways. | ||
Conclusions
We introduce a degradable PEG copolymer that shows intrinsic acid-degradability and can be used for the PEGylation of proteins. The degradability is achieved by the incorporation of vinyl ether moieties into the backbone of PEG. These groups are acid-cleavable at physiologically relevant pH values. Relying on this degradable polyether structure, we developed a new class of pH-responsive protein nanoparticles using surface-modified cytochrome c. The PEGylation renders our model protein soluble in organic solvents while preserving its activity and secondary structure. This approach should in principle be feasible for any protein of choice. Using a mild emulsion-based method we successfully prepared nanoparticles that are stable at pH 7.4 but can degrade and disassemble at mildly acidic conditions, as can be found in endolysosomal compartments, tumour environments or inflamed tissue.Introducing responsiveness into these protein nanoparticles not only allows for a triggered release of therapeutic payloads, but also almost completely recovers the initial protein starting material. The first in vitro results indicate that the underlying enzymatic activity of the particle system might be as important as the payload itself. Hence, for future nanomedicine applications we will further explore the activity of the protein nanoparticles in combination with their capability to deliver drugs.
Experimental
Copolymerization of mP(EG81-co-EPB6)
In a flame dried Schlenk flask, sodium hydride (15 mg, 0.62 mmol) and triethylene glycol monomethyl ether (253 mg, 1.54 mmol) were dissolved in benzene (5 mL) and dry THF (5 mL), stirred under slightly reduced pressure at 60 °C for 30 min and subsequently dried in high vacuum for 16 h. The resulting initiator was dissolved in dry DMSO (7 mL) and dry THF (3 mL, stored over sodium). To this solution, ethylene oxide (5 mL, 110 mmol) was cryo-transferred using a graduated ampule. EPB (1.21 mL, 18.5 mmol, dried over CaH and freshly distilled) was added via syringe and the polymerization was carried out for 5 days at room temperature. After quenching (3 mL methanol), dialysis was performed against methanol for 24 h (MWCO 1000 Da) and the polymer dried in high vacuum. (Yield 70%) 1H NMR (400 MHz, DMSOd6) δ [ppm] 5.72 (ddd, 5.51H, 3JAB = 17.2 Hz, 3JAB = −10.5 Hz, 3JAB = 6.7 Hz, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.26 (m, 11.41H, 3JAB = 17.2 Hz, 3JAB = 2.1 Hz, 3JAB = 1.1 Hz, –CHHH), 4.76 (q, 0.44H, J = 6.7 Hz, CCH–CH3 (E isomer)), 4.57 (bs, 0.77H, OH), 3.96–3.87 (m, 7.38H, CHO–CHCH2, OCH2–CCH–CH3 and CH2O–CCH–CH3), 3.71–3.35 (m, 336H, CH2O), 3.24 (s, 3H, O–CH3), 1.52 (d, 1.35H, J = 6.7 Hz, CCH–CH3 (E isomer)).
CH2), 5.26 (m, 11.41H, 3JAB = 17.2 Hz, 3JAB = 2.1 Hz, 3JAB = 1.1 Hz, –CHHH), 4.76 (q, 0.44H, J = 6.7 Hz, CCH–CH3 (E isomer)), 4.57 (bs, 0.77H, OH), 3.96–3.87 (m, 7.38H, CHO–CHCH2, OCH2–CCH–CH3 and CH2O–CCH–CH3), 3.71–3.35 (m, 336H, CH2O), 3.24 (s, 3H, O–CH3), 1.52 (d, 1.35H, J = 6.7 Hz, CCH–CH3 (E isomer)).
Isomerization of mP(EG81-co-isoEPB6)
mP(EG81-co-EPB6) (500 mg) was dissolved in DMSO (0.5 mL) and degassed by 3 freeze–pump–thaw cycles. RhCl(PPh3)3 (20 mg, 0.022 mmol) and BHT (3 mg) were added under argon atmosphere and the resulting solution was stirred at 80 °C for 24 h. Subsequently, the polymer was twice precipitated in acetone/diethylether (50/50 vol%) and dried in high vacuum. (Yield 90%) 1H NMR (400 MHz, DMSOd6): δ [ppm] 4.77 (q, 2.47H, J = 6.7 Hz, CCH–CH3 (E isomer)), 4.65 (q, 1.84H, J = 6.7 Hz, CCH–CH3 (E isomer)), 4.57 (bs, 0.45H, OH), 3.99–3.88 (m, 14H, CHO–CHCH2, OCH2–CCH–CH3 and CH2O–CCH–CH3), 3.71–3.35 (m, 314H, CH2O), 3.24 (s, 3H, O–CH3), 1.61 (d, 5.56H, J = 6.7 Hz, CCH–CH3 (Z isomer)), 1.52 (d, 7.46H, J = 6.7 Hz, CCH–CH3 (E isomer)).
Functionalization to mP(EG81-co-isoEPB6)-NHS
To a solution of mP(EG81-co-isoEPB6) (300 mg, 0.075 mmol) in dioxane (2 mL) N,N′-hydroxysuccinimide carbonate (192 mg, 0.75 mmol, dissolved in 2 mL DMF) and DMAP (92 mg, 0.75 mmol, dissolved in 2 mL DMF) were slowly added via syringe. The resulting solution was stirred for 24 h at room temperature and subsequently precipitated three times in acetone/diethylether (50/50 vol%). The obtained polymer was dried in high vacuum and stored routinely in a refrigerator at −20 °C. (Yield 85%) 1H NMR (400 MHz, DMSOd6): δ [ppm] 4.77 (q, 2.40H, J = 6.7 Hz, CCH–CH3 (E isomer)), 4.65 (q, 1.78H, J = 6.7 Hz, CCH–CH3 (Z isomer)), 3.99–3.88 (m, 14H, CHO–CHCH2, OCH2–CCHCH3 and CH2O–CCH–CH3), 3.71–3.35 (m, 317H, CH2O), 3.24 (s, 3H, O–CH3), 2.82 (s, 1.47 h, –CO–CH2–CH2–CO–)1.61 (d, 5.53H, J = 6.7 Hz, CCH–CH3 (Z isomer)), 1.52 (d, 7.45H, J = 6.7 Hz, CCH–CH3 (E isomer)).
PEGylation of cytochrome c
5 mg horse heart cytochrome c (0.4 μmol) and 27.42 mg mP(EG81-co-isoEPB6)-NHS (6.9 μmol, 37% functionalized) were dissolved in 1 mL 0.1 M phosphate buffer pH 8.5 and stirred overnight at 4 °C. The mixture was purified by column chromatography (Sephadex® G-75 medium, Sigma Aldrich, buffer: dd-H2O). After collecting the first fraction yielding the product, the modified protein was concentrated with Amicon® Ultra 4 centrifugal filter membranes (regenerated cellulose, MWCO 10 kDa, 7500g, 10 min, 4 °C, Merck Millipore). The product was lyophilized (ALPHA 1-2 LD plus).Activity of PEGylated enzyme
The enzymatic activity of native Cyt, CytdegPEG and CytdegPEG-NPs were analysed using an 2,2′-azinobis-(2-ethylbenzthiazoline-6-sulfonate (ABTS)-assay. 10 mM phosphate buffer pH 7.4 was used as working buffer. 100 μL of ABTS solution (2 mM) were mixed with 50 μL of samples (native Cyt or CytdegPEG, 22.6 μM) in a 96-well-microplate (flat bottom). The reaction was initiated by adding 50 μL of an H2O2 solution (2 mM in dd-H2O). Subsequently, the change in absorption at 405 nm was measured for 3 minutes (Infinite® 200 PRO (Tecan) plate reader). The absorption of the background (100 μL ABTS with 50 μL sample, but instead of H2O2 solution only 50 μL dd-H2O) was subtracted from each measurement (see ESI section 2.3†).Analysis of CytdegPEG degradation by SDS-PAGE
CytdegPEG was dissolved in 0.1 M sodium acetate buffer pH 4 or pH 5 or 0.1 M phosphate buffer pH 6 or pH 7.4 in concentrations of 8 mg mL−1. These samples were incubated for 24 h at 37 °C and shaken with 1000 rpm. Native Cyt (1 mg mL−1) and CytdegPEG (8 mg mL−1) were freshly dissolved in dd-H2O. 15 μL of the protein-solutions were denaturized by adding 5 μL of Roti®-Load 1 (Carl Roth) and heated in a boiling water bath for 15 min. The different samples were analysed with a 15% polyacrylamide gel (see ESI section 2.1†).Analysis of CytdegPEG degradation by SEC
CytdegPEG was dissolved in a concentration of 150 μM in 0.1 M sodium acetate buffer (pH 4, pH 5) or in 0.1 M phosphate buffer (pH 6, pH 7.4). Similarly, native Cyt (150 μM) was dissolved in 0.1 M sodium acetate buffer pH 4 as control. These samples were incubated for 24 h at 37 °C and shaken at 1000 rpm. For comparison, native Cyt (150 μM) and CytdegPEG (150 μM) were freshly dissolved in dd-H2O and measured directly. 150 μL of the protein-solutions were analysed with size exclusion chromatography. The SEC runs were performed on a 24 mL high-resolution size exclusion column (Superdex™ 75 10/300 GE Healthcare Bio-Sciences AB) at 4 °C with a flow rate of 0.8 mL in PBS pH 7.4.Nanoparticle preparation by double emulsion
CytdegPEG (2.75 mg) was dissolved in 0.4 mL DCM. After adding 50 μL Oregon-Green™488-dextran (OGD) solution (10 kDa dextran, 2 mg mL−1 in PBS pH 7.4), the two phases were sonicated on ice for 15 s. To the resulting emulsion 2 mL of ice-cold PBS buffer (pH 7.4) was added and treated again with ultrasonic pulses for 30 s. The DCM of the w/o/w emulsion was evaporated by stirring at RT overnight, resulting in an opaque nanoparticle suspension.Nanoparticle degradation and OGD release
The degradation of CytdegPEG-NP and the release of OGD under acidic conditions was analysed using a dialysis experiment. OGD-loaded CytdegPEG-NP were freshly prepared and purified by dialysis (Float-a-Lyzer®G2 Dialysis Device, MWCO 100 kDa, Spectrum Labs) for 4 h to remove not encapsulated OGD. The particles were then mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with either 0.1 M sodium acetate buffer pH 4, 0.1 M sodium acetate buffer pH 5 or PBS pH 7.4 as control. 100 μL of these mixtures were dialysed (Xpress Micro Dialyzer MD100, MWCO 140 kDa, Serva) against the corresponding buffer mix (1:1 mix of 0.1 M sodium acetate buffer pH 4 and PBS pH 7.4, 0.1 M sodium acetate buffer pH 5 and PBS pH 7.4 or PBS pH 7.4 alone) at 37 °C. The particle degradation and OGD release were analysed by NTA size measurements (see ESI section 3.2†) and a fluorescence readout (Ex: 490 nm, Em: 527 nm; Infinite® 200 PRO (Tecan) plate reader) over a period of 48 h. The data for the 0 hour value was measured directly after mixing of the nanoparticle suspension with the appropriate buffer.
:1 ratio with either 0.1 M sodium acetate buffer pH 4, 0.1 M sodium acetate buffer pH 5 or PBS pH 7.4 as control. 100 μL of these mixtures were dialysed (Xpress Micro Dialyzer MD100, MWCO 140 kDa, Serva) against the corresponding buffer mix (1:1 mix of 0.1 M sodium acetate buffer pH 4 and PBS pH 7.4, 0.1 M sodium acetate buffer pH 5 and PBS pH 7.4 or PBS pH 7.4 alone) at 37 °C. The particle degradation and OGD release were analysed by NTA size measurements (see ESI section 3.2†) and a fluorescence readout (Ex: 490 nm, Em: 527 nm; Infinite® 200 PRO (Tecan) plate reader) over a period of 48 h. The data for the 0 hour value was measured directly after mixing of the nanoparticle suspension with the appropriate buffer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr P. Besenius, Prof. Dr T. Efferth and Prof. Dr P. Langguth (all JGU Mainz, Germany), for the kind permission to use some of their analytical equipment. We also thank Dr E. Berger-Nicoletti for the help with the MALDI-ToF measurements.Notes and references
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- A. Abuchowski, T. Van Es, N. Palczuk and F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- S. N. S. Alconcel, A. S. Baas and H. D. Maynard, Polym. Chem., 2011, 2, 1442–1448 RSC.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- T. A. Wright, R. C. Page and D. Konkolewicz, Polym. Chem., 2019, 10, 434–454 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS.
- J. A. Rodríguez-Martínez, I. Rivera-Rivera, R. J. Solá and K. Griebenow, Biotechnol. Lett., 2009, 31, 883–887 CrossRef.
- Y. Qi and A. Chilkoti, Curr. Opin. Chem. Biol., 2015, 28, 181–193 CrossRef CAS.
- M. Barz, R. Luxenhofer, R. Zentel and M. J. Vicent, Polym. Chem., 2011, 2, 1900–1918 RSC.
- A. Grotzky, Y. Manaka, T. Kojima and P. Walde, Biomacromolecules, 2011, 12, 134–144 CrossRef CAS.
- K. Maier and E. Wagner, J. Am. Chem. Soc., 2012, 134, 10169–10173 CrossRef CAS.
- C. G. Decker and H. D. Maynard, Eur. Polym. J., 2015, 65, 305–312 CrossRef CAS.
- A. Yurkovetskiy, S. Choi, A. Hiller, M. Yin, C. McCusker, S. Syed, A. J. Fischman and M. I. Papisov, Biomacromolecules, 2005, 6, 2648–2658 CrossRef CAS.
- T. Steinbach, G. Becker, A. Spiegel, T. Figueiredo, D. Russo and F. R. Wurm, Macromol. Biosci., 2017, 17, 1600377 CrossRef.
- E. M. Pelegri-O'Day, N. M. Matsumoto, K. Tamshen, E. D. Raftery, U. Y. Lau and H. D. Maynard, Bioconjugate Chem., 2018, 29, 3739–3745 CrossRef.
- M. Worm, D. Leibig, C. Dingels and H. Frey, ACS Macro Lett., 2016, 5, 1357–1363 CrossRef CAS.
- P. Lundberg, B. F. Lee, S. A. van den Berg, E. D. Pressly, A. Lee, C. J. Hawker and N. A. Lynd, ACS Macro Lett., 2012, 1, 1240–1243 CrossRef CAS.
- A.-K. Danner, D. Leibig, L.-M. Vogt and H. Frey, Chin. J. Polym. Sci., 2019, 37, 912–918 CrossRef CAS.
- C. Boyer, X. Huang, M. R. Whittaker, V. Bulmus and T. P. Davis, Soft Matter, 2011, 7, 1599–1614 RSC.
- K. DeFrates, T. Markiewicz, P. Gallo, A. Rack, A. Weyhmiller, B. Jarmusik and X. Hu, Int. J. Mol. Sci., 2018, 19, 1717 CrossRef.
- E. Steiert, L. Radi, M. Fach and P. R. Wich, Macromol. Rapid Commun., 2018, 39, 1800186 CrossRef.
- M. Fach, L. Radi and P. R. Wich, J. Am. Chem. Soc., 2016, 138, 14820–14823 CrossRef CAS.
- L. Radi, M. Fach, M. Montigny, E. Berger-Nicoletti, W. Tremel and P. R. Wich, MedChemComm, 2016, 7, 1738–1744 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- N. Vanparijs, R. De Coen, D. Laplace, B. Louage, S. Maji, L. Lybaert, R. Hoogenboom and B. G. De Geest, Chem. Commun., 2015, 51, 13972–13975 RSC.
- L. Wang, L. Liu, B. Dong, H. Zhao, M. Zhang, W. Chen and Y. Hong, Acta Biomater., 2017, 54, 259–270 CrossRef CAS.
- A. J. Kresge, D. S. Sagatys and H. L. Chen, J. Am. Chem. Soc., 1977, 99, 7228–7233 CrossRef CAS.
- Y.-L. P. Ow, D. R. Green, Z. Hao and T. W. Mak, Nat. Rev. Mol. Cell Biol., 2008, 9, 532 CrossRef CAS.
- C. Y. Zheng, G. Ma and Z. Su, Electrophoresis, 2007, 28, 2801–2807 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01162e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |