
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic strategies towards 1,3-polyol synthesis by enantioselective cascades creating multiple alcohol functions
Céline
Sperandio
,
Jean
Rodriguez
and
Adrien
Quintard
 *
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: adrien.quintard@univ-amu.fr; Web: http://ism2.univ-amu.fr/fr/annuaire/stereo/quintardadrien
First published on 14th January 2020
Abstract
This review highlights the different enantioselective catalyst-controlled cascades creating multiple alcohol functions through the formation of several carbon–carbon bonds. Through subsequent simple derivatization, these strategies ensure the rapid preparation of 1,3-polyols. Thanks to the use of efficient metal- or organo-catalysts, these cascades enable the selective assembly of multiple substrates considerably limiting operations and waste generation. For this purpose, several mono- or bi-directional approaches have been devised allowing successive C–C bond-forming events. The considerable synthetic economies these cascades enable have been demonstrated in the preparation of a wide variety of complex bioactive natural products, notably polyketides.
 Adrien Quintard | Adrien Quintard studied chemistry at a technical institute of University Toulouse III in Castres before obtaining a master's degree in 2007 at Lyon (CPE Lyon/University Lyon I). He then moved to Geneva for a PhD degree which he obtained in 2011 for his work on metal- and organo-catalysis with Prof. Alexandre Alexakis. After a first postdoc fellowship with Prof. Barry Trost at the University of Stanford, he joined Marseille in 2012 for a second postdoc fellowship before obtaining in 2013 an ANR starting grant to initiate a new research program on multicatalysis. He subsequently joined the CNRS as a senior researcher in 2014 with research interests encompassing a broad range of areas of organic chemistry (catalysis, synthesis and supramolecular chemistry). Recently, his contribution has been recognized by the award of the young researcher ‘Emergence’ prize from the Organic Division of the French Chemical Society. He was also awarded a Thieme Chemistry Journals award. |
Introduction
Catalytic cascades where starting materials can engage in multiple successive transformations directly forming elaborate scaffolds have changed chemists’ vision of complex molecule synthesis.1 These cascades can dramatically reduce operations, steps and waste associated with classical “stop and go” sequences notably as soon as the control of the stereochemistry of the final products is required. As a result, it is not surprising that a broad range of cascades have been developed to efficiently prepare a wide array of natural product scaffolds.Among these, extended 1,3-polyols are the most valuable and are present in a wide variety of natural products exemplified in the type 1 polyketides amphotericin and RK-397 of high bioactivity (Scheme 1a).2 The acyclic or pseudo-acyclic nature of these chemical entities makes the control of the multiple stereogenic centers a great challenge through a single cascade. While Nature takes advantage of decarboxylative cascade processes to efficiently prepare them (Scheme 1b), in the laboratory, chemists have for long relied on classical lengthy iterative sequences.3
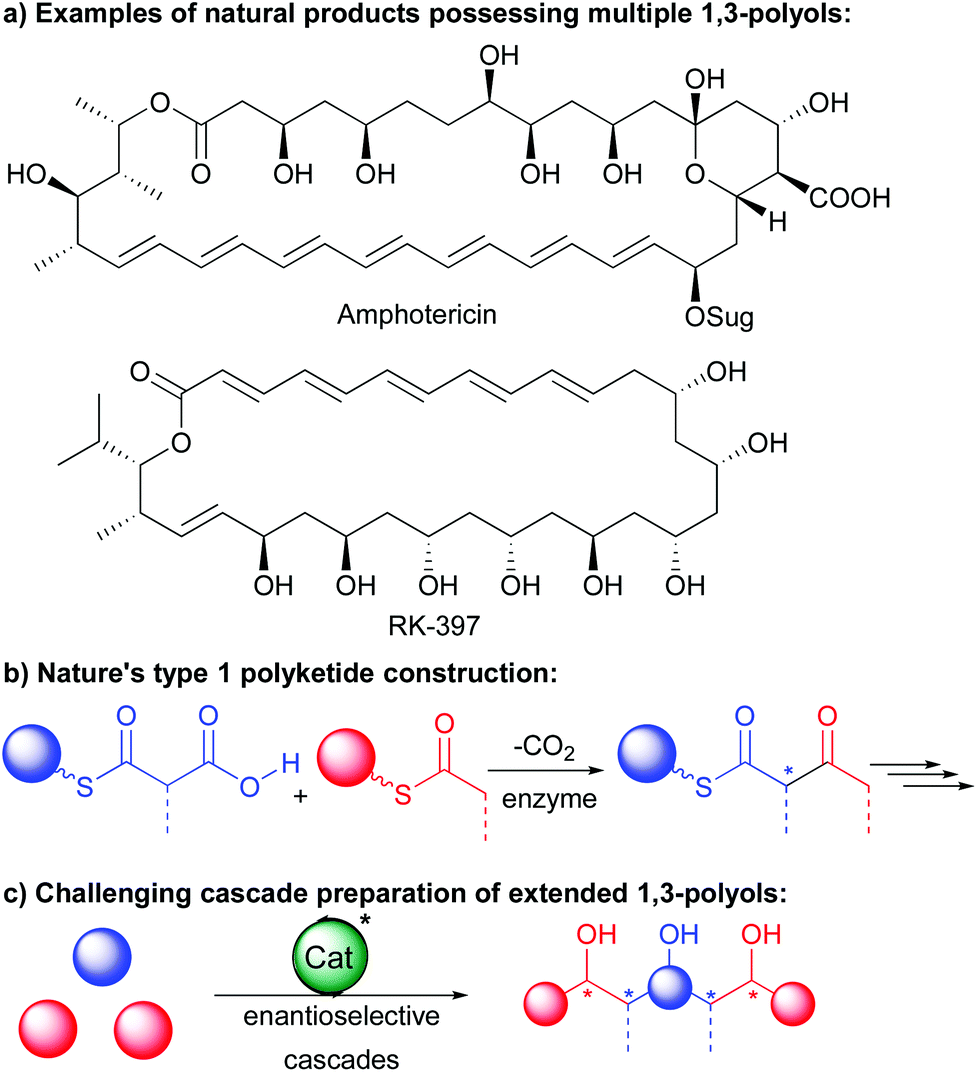 | ||
| Scheme 1 1,3-Polyols of interest and their challenging synthesis. | ||
As a result, the synthetic challenge associated with the preparation of these complex architectures has pushed organic chemists’ creativity notably towards the development of efficient enantioselective cascades controlling at the same time the formation of multiple alcohols (Scheme 1c).
The aim of this review is to highlight the different enantioselective catalyst-controlled cascades creating multiple alcohol functions through the formation of several carbon–carbon bonds. Simple derivatization of the obtained adducts enables the rapid preparation of 1,3-polyols. Through efficient catalytic activations, these cascades ensure the selective assembly of multiple substrates considerably limiting operation and waste generation.
For this purpose, several approaches have been devised allowing successive C–C bond forming events (Scheme 2). At first, extension of the functionalized carbon chain through a single direction can be achieved either by reacting twice an ambident substrate (a) or by gem-difunctionalization of a simple bis-pronucleophile (b). Alternatively, polyol synthesis can be based on bi-directional chain elongation.4 In these cases, either a central in situ generated bis-electrophile reacts with a twofold excess of a pronucleophile (c) or the central part acts as a bis-pronucleophile (d). For these bi-directional double additions, even though many symmetric products exist in Nature, a subsequent desymmetrization is often required to advance to the final bioactive molecules. For this purpose, several smart approaches have been devised and exemplified in this review.
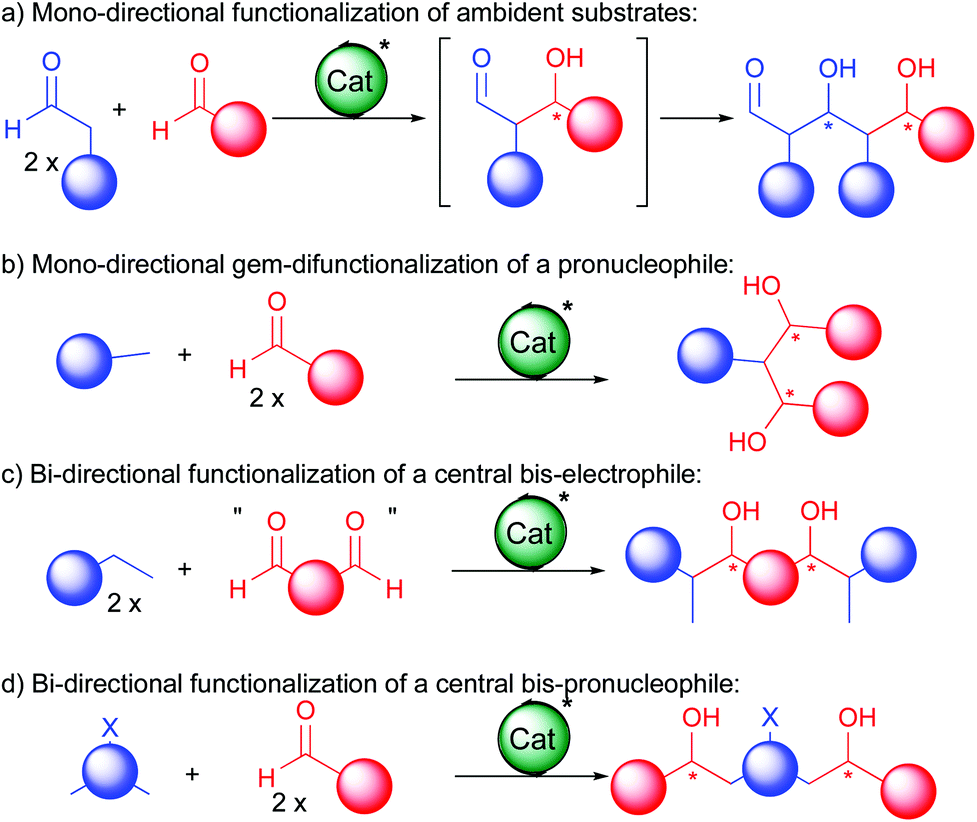 | ||
| Scheme 2 Enantioselective cascades towards 1,3-polyols. | ||
In the next sections, we will try to highlight the different strategies developed to try to minimize steps towards these crucial backbones. We will also discuss current limitations and future research directions in this challenging but exciting field.
Mono-directional functionalization of ambident substrates
One of the most classical enantioselective aldolization approaches involves enamine activation by chiral secondary amine catalysts. This reaction is extremely challenging since it requires the differentiation of the carbonyl precursors either as a donor or an acceptor.In 2002, the group of Barbas III reported that proline cat1 could catalyze the twofold condensation of propionaldehyde, acting consecutively as a pro-nucleophile and an electrophile, to other aliphatic aldehydes providing lactols 3 in a moderate yield after intramolecular hemiacetalization (Scheme 3).5 However, the reaction remains poorly selective forming mixtures of diastereomers with low enantiocontrol (33–43% ee) resulting from the reversibility of the first aldolization. In 2005, Cordova reported that changing the reaction conditions (rate of aldehyde addition and concentration) could in some cases slightly improve the reaction efficiency (up to 85% ee in one case).6 A solution to this challenge was later found by performing a sequential one-pot transformation, involving the addition of a second catalyst such as D-proline to promote the second aldolization event with success, leading to the products with high enantiocontrol.7
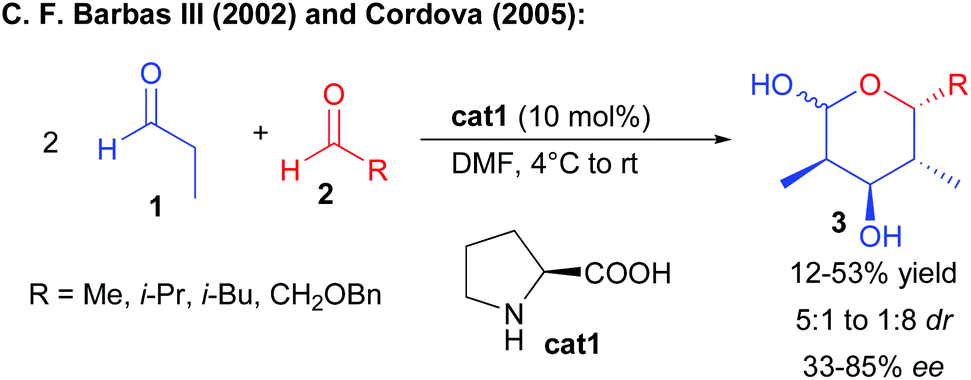 | ||
| Scheme 3 Proline-catalyzed double aldolization. | ||
Aside from enamine activation, different groups have attempted to discover another catalytic activation mode to promote chain elongation through multiple aldolization events from pro-nucleophilic aldehydes. However most of them rely on diastereoselective processes. Most notably, the group of Yamamoto was able to perform double or triple Mukaiyama aldolization using silyl enol ethers, albeit in a racemic form.8 The solution to induce enantiocontrol only came recently with the discovery by the group of Matsunaga and Kanai that a boron enolate acting successively as a pro-nucleophile and a pro-electrophile could engage in multiple enantioselective aldolizations catalyzed by a chiral copper complex (Scheme 4).9
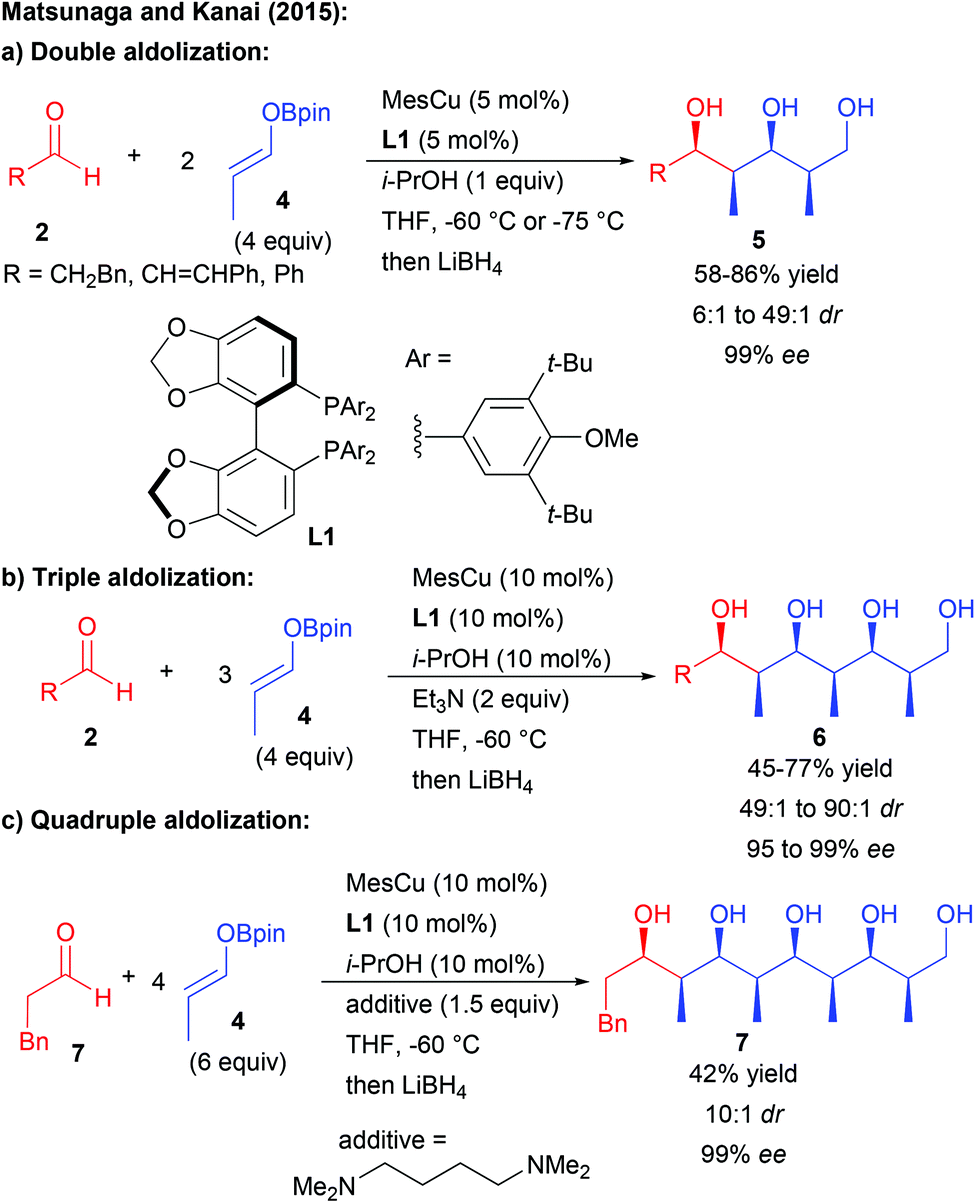 | ||
| Scheme 4 Copper-catalyzed multiple enantioselective aldolizations. | ||
Through this approach, the propionaldehyde derivative 4 underwent two to four consecutive aldolization steps under catalyst control. These multiple catalytic events explain the high levels of diastereo- and enantiocontrol obtained (typically 99% ee) through amplification of chirality phenomena. The number of aldolization events leading from 1,3,5- to 1,3,5,7,9-polyols depends on the equivalents of boron enolate used as well as on the amount of the catalyst and the nature of the additive.10
Overall, the cascade transformation enables the impressive formation of polyketides possessing up to 8 acyclic stereocenters efficiently controlled by the use of a single catalyst. The obtained structures are similar to some complex polyketide natural products, demonstrating the potential of this approach for the elaboration of biologically active molecules.
Mono-directional gem-difunctionalization of a pro-nucleophile
In complement, other strategies take advantage of a catalyst-controlled gem-difunctionalization of simple methylketones as pro-bisnucleophiles. In 2011, the group of Kotani and Nakajima reported an original enantioselective double aldolization of acetophenones with a twofold excess of non-enolizable aldehydes (Scheme 5).11 Such a process leads to the formation of a C2-symmetric 1,3-diol possessing two stereogenic centers and a central ketone. The reaction in the presence of an excess of SiCl4 and a tertiary amine is catalyzed by a chiral phosphine oxide Lewis base, activating a highly reactive in situ generated trichlorosilyl enol ether towards nucleophilic addition. Using BINAPO cat2, the reaction formed the diols with moderate to good enantiocontrol strongly depending on the pro-bisnucleophile/aldehyde combination used (56 to 97% ee).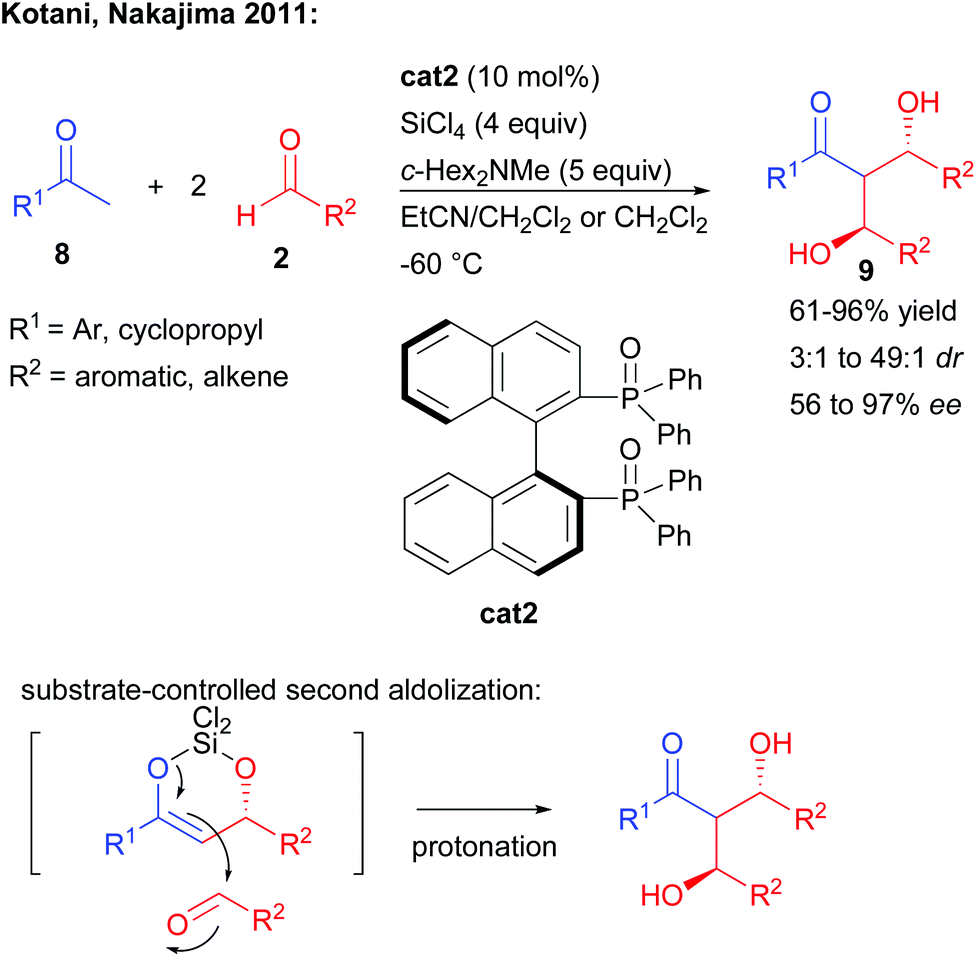 | ||
| Scheme 5 Initial development of a double mono-directional aldolization. | ||
Of importance, mechanistic investigations indicated that the second aldolization step was mostly under substrate-control through the formation of a cyclic enol ether with the generated alcohol.
The groups of Benaglia and Ding further investigated this transformation with acetophenones and aromatic aldehydes in order to improve its efficiency notably in terms of enantiocontrol (Scheme 6).12a,b While the bis-thiophene phosphine oxide cat3 resulted in no significant change, the spirocyclic analogue cat4 considerably raised the enantiocontrol regardless of the nature of the aromatic substituents (81 to >99% ee).
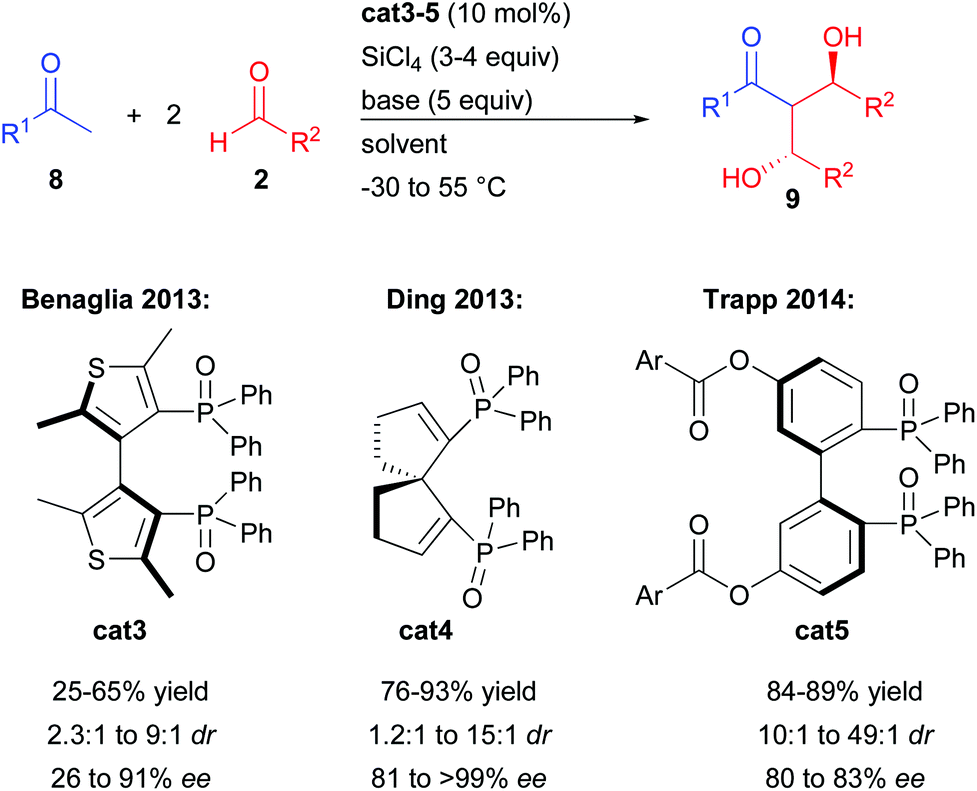 | ||
| Scheme 6 Development of other phosphine oxides for the double aldolization. | ||
Finally, the group of Trapp showed that stereo-labile interconverting cat5 could be isolated from the racemic mixture and directly engaged in the double aldolization process prior to catalyst racemization.12c Even if cat5 rapidly interconverts to the opposite enantiomer, performing the reaction at −55 °C allowed the isolation of the double aldol product with up to 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 83% ee.
:1 dr and 83% ee.
Even though the double aldolization does not obviously seem relevant in the context of natural product synthesis, it could be applied in the preparation of a useful C3 symmetric cage-like phosphite ligand 10 efficient in rhodium-catalyzed enantioselective hydrogenation (92% ee) (Scheme 7).
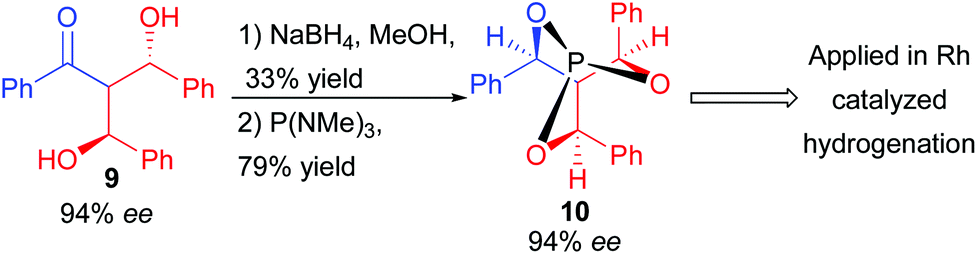 | ||
| Scheme 7 Application of the double aldolization to chiral phosphite synthesis. | ||
Bi-directional functionalization of a central bis-electrophile
One of the most synthetically relevant strategies to construct 1,3-polyols rapidly lies in the use of a central bis-electrophile. For this purpose the ideal substrate would be a 1,3-dialdehyde of poor stability. As a result, discovering 1,3-dialdehyde synthetic surrogates represents a great synthetic challenge. The group of Krische has developed in the last decade a broad range of metal-catalyzed enantioselective reductive couplings initiated by an initial alcohol dehydrogenation in the presence of an allylic pro-nucleophile.13 Expanding this reactivity to 1,3-diols and allylic acetates, the initial chiral iridium-catalyzed dehydrogenation triggers the formal formation of the key 1,3-dialdehyde (Scheme 8).14 The generated iridium hydride is able to insert on the allylic pro-nucleophile, generating a nucleophilic π-allyl complex performing the twofold bi-directional enantioselective addition on the 1,3-dialdehyde intermediate to create the expected chiral 1,3-diol. Once again, carrying out two cascade condensations under catalyst control ensures an amplification of chirality leading to excellent enantioselectivity of the final product (>99% ee).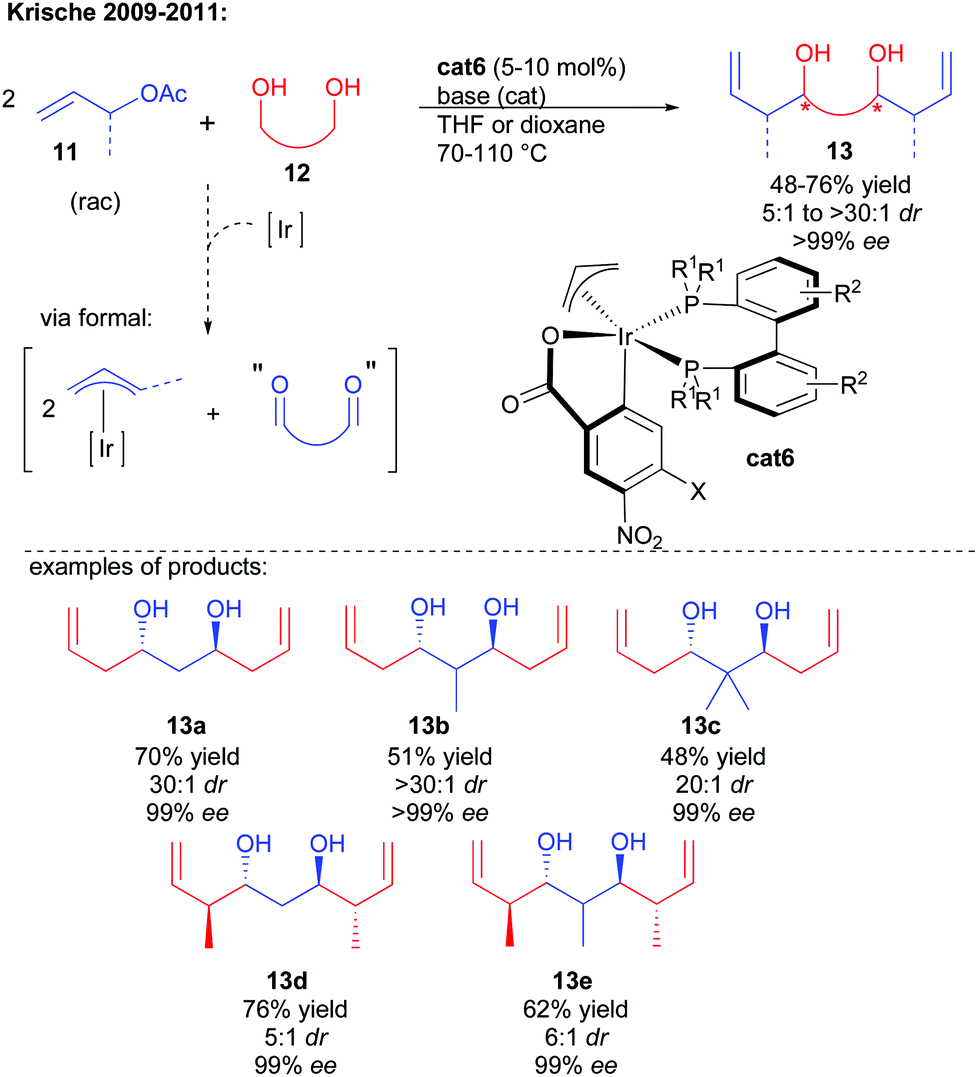 | ||
| Scheme 8 Krische's enantioselective approach towards 1,3-diols. | ||
Most notably, this cascade 1,3-diol direct synthesis has served as a strategic platform in the elaboration of a multitude of complex natural products.15 Given the efficiency of the cascade to rapidly prepare the core structure of these products, considerable synthetic economies (steps, protecting groups, and redox manipulation) are obtained through this bi-directional strategy. However, the C2-symmetric structure of the obtained compounds required the development of a broad range of desymmetrization techniques to advance towards more useful scaffolds as illustrated by the following examples.
For example, diol 13a could be derivatized by a chemoselective mono-acylation, providing 14, a precursor of cryptocaryol (Scheme 9).16
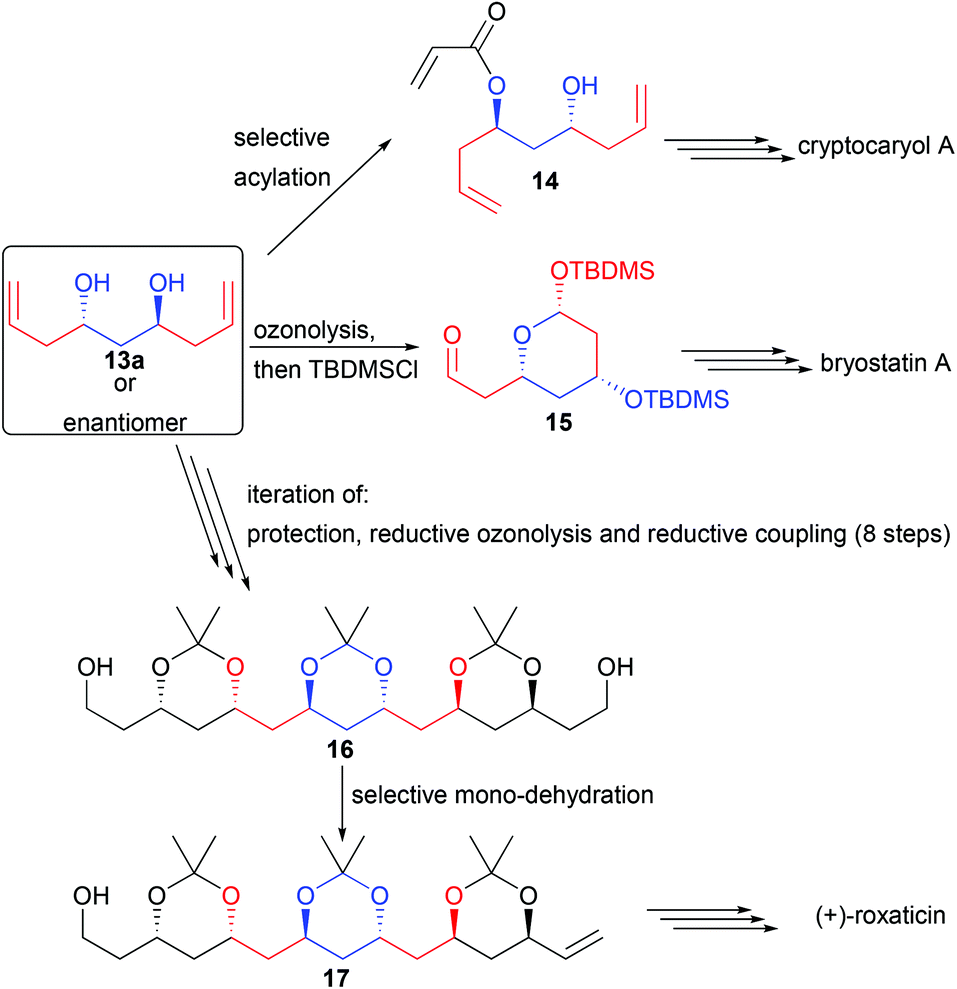 | ||
| Scheme 9 Application of 13a in natural product synthesis. | ||
Ozonolysis followed by protection of the in situ formed lactol also generates the precursor 15 of bryostatin, possessing 3 stereocenters in only 3 steps.17 Finally, iteration of alcohol protection, reductive ozonolysis and double reductive coupling could create in only 9 steps from commercial products the C2-symmetric 1,3,5,7,9,11,13,15-octanol 16. Mono-dehydration of one single terminal alcohol creates 17, a direct precursor of the macrolide roxaticin.18
Applying the C2-symmetric diol 13e leads to an additional challenge. Indeed, the central carbon center bearing the methyl substituent is prochiral and a desymmetrization step is required to reveal its chirality. For this purpose, two strategies involving the formation of pyran derivatives were reported. During the cyclization event, the methyl group prefers to reside in an equatorial position, forming either 18 by iodoetherification19 or 19 by palladium-catalyzed functionalization (Scheme 10).20 Most notably, pyran 18 created in only two steps from commercially available materials possesses 6 perfectly controlled stereogenic centers (>20:1 dr and >99.9% ee). This backbone was found to be crucial to create a broad range of natural products considerably short-cutting the known existing routes. Most notably, the pyran can easily be opened after iodine transmetallation allowing the rapid construction of acyclic complex structures.
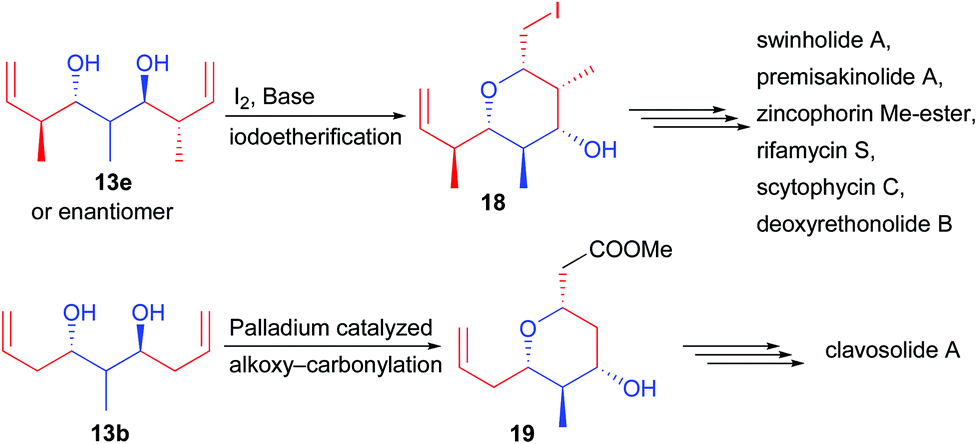 | ||
| Scheme 10 Desymmetrization of 13e and 13b towards natural product synthesis. | ||
Finally, 13c possessing a quaternary center could be desymmetrized to 20 through a cross-metathesis reaction creating two Michael acceptors (Scheme 11). One of them reacts through an oxa-Michael addition generating the pyran 20, a precursor of cyanolide A.21 Alternatively, alcohol mono-protection followed by ozonolysis and lactol protection provided 21, a precursor of psymberin.22
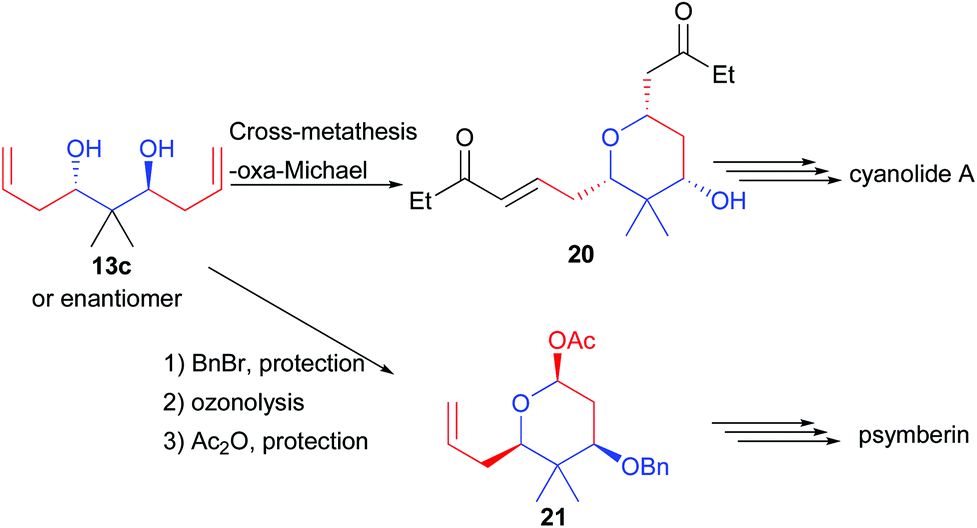 | ||
| Scheme 11 Application of 13c towards natural product synthesis. | ||
Overall, the bi-directional reductive coupling strategy has been applied in the preparation of more than 10 natural products. It showcases the potential of cascade reactions to prepare in the most straightforward manner and with high stereocontrol a broad range of key natural product architectures.
Bi-directional functionalization of a central bis-pronucleophile
This type of approach has for long been dominated by stoichiometric transformations notably based on boron reagents,23 but alternative catalytic methods have emerged, limiting the waste associated with such activating groups. Ideally, a central bis-pronucleophilic ketone is catalytically activated to react with two external aldehydes in a bi-directional fashion affording either 1,3-keto-diols or their surrogates, direct precursors of 1,3,5-triols, by simple reduction.Given the known efficiency of secondary amine organocatalysts to promote aldol reactions of aliphatic ketones, unsurprisingly, different groups have focused on the direct enamine-type double aldolization (Scheme 12). However, one of the main limitations lies in the limited Turn Over Frequency (TOF) of the catalytic cycles, disfavoring double addition pathways.24 Most notably, the slow second aldolization is in strong competition with a retro-aldol process from the first generated alcohol. As a result, these amino-catalyzed processes often provide moderate yields and are limited to the use of aromatic aldehydes (Scheme 12). Among the different aminocatalysts tested, the groups of Lipinski and Moyano showed that thio-prolinamide cat7 could provide decent yields and levels of enantiocontrol in this transformation, however, using electron-deficient aldehydes to facilitate the double aldolization process.25 In these aminocatalyzed processes, the H-bond donor (thioamide, amide or alcohol) is responsible for an activation of the aldehyde in the transition state (Scheme 12).
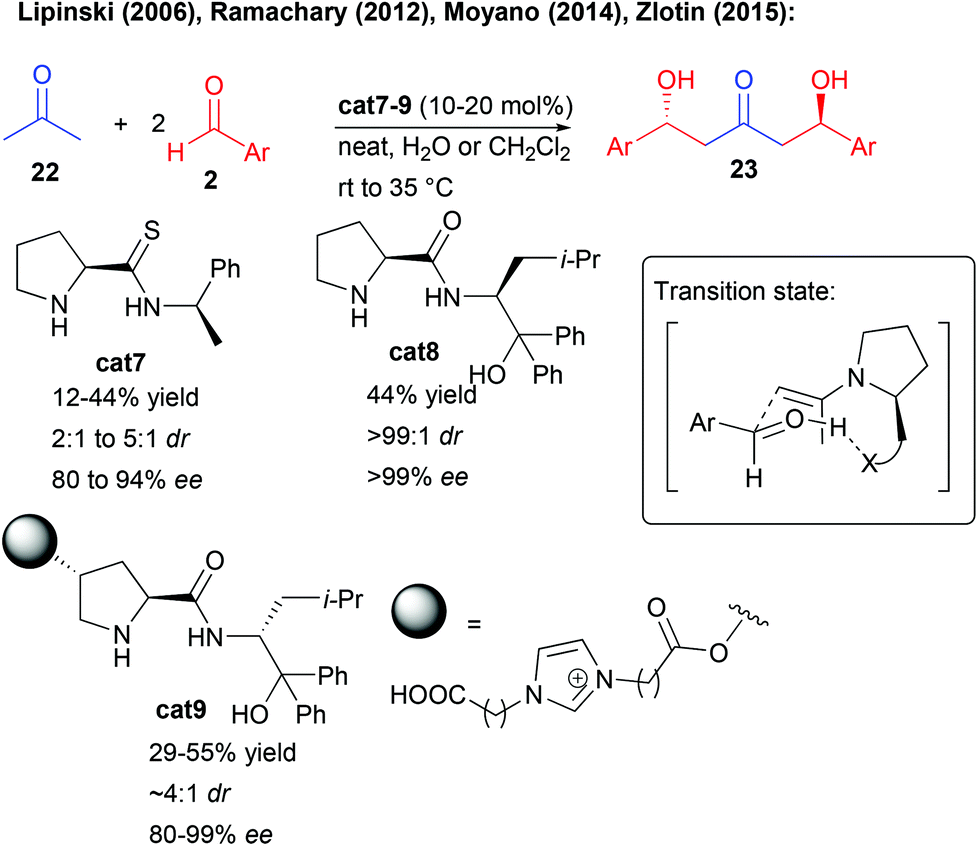 | ||
| Scheme 12 Double amino-catalyzed aldolization. | ||
Later, the group of Ramachary showed that the prolinamide derivative cat8 provided some of the best results in terms of activity and selectivity however on a single example using 2-ethynylbenzaldehyde.26 Finally, the group of Zlotin created a prolinamide-derived ionic-liquid-supported aminocatalyst cat9 performing an efficient reaction in water only with electron-deficient aldehydes.27 It must be pointed out that in these aminocatalyzed double aldolizations, the second catalyst-controlled reaction is responsible for an amplification of chirality associated with higher enantiocontrol of the double aldolization product than for the corresponding mono-aldolization one. Even though the reaction holds promises for the direct preparation of 1,3-keto-diols, a scaffold found in different natural products, the limited scope and reactivity hamper for now the applicability of this approach.
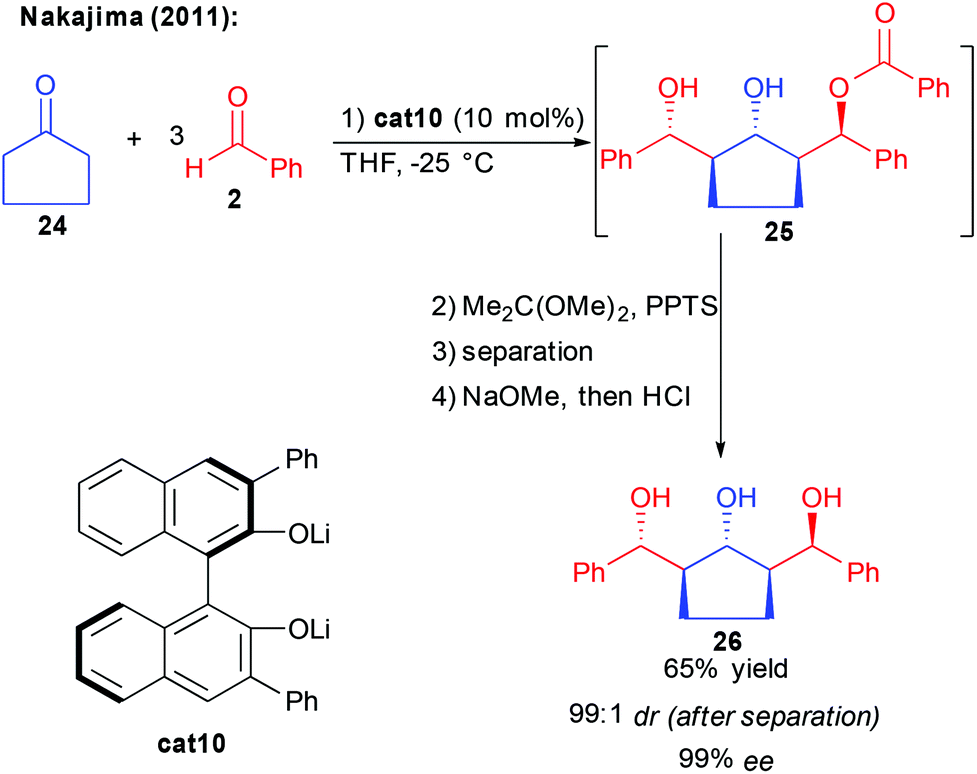
Following these reports, in 2011, the group of Nakajima observed the formation of a product of the bi-directional aldol/aldol-Tishchenko process 25, using cyclopentanone and an excess of benzaldehyde (Scheme 13).28
 | ||
| Scheme 13 Aldol/aldol-Tishchenko cascade from cyclopentanone. | ||
Thanks to a binuclear cat10 acting at the same time as a base activating the nucleophile and a Lewis acid activating the aldehyde, the twofold aldolization is followed by hydride transfer generating the third alcohol. From the in situ generated ester 25, acetalization, diastereomer separation and work-up provided the 1,3,5-triol possessing 5 stereogenic centers with 65% yield and 99% ee. Given the direct formation of complex 1,3,5-polyols in one-single cascade without an additional redox-step, this approach seems to be of great interest. Its limitation to the use of highly enolizable cyclopentanone, however, renders this reaction for now more a scientific curiosity than a general approach towards complex polyols.
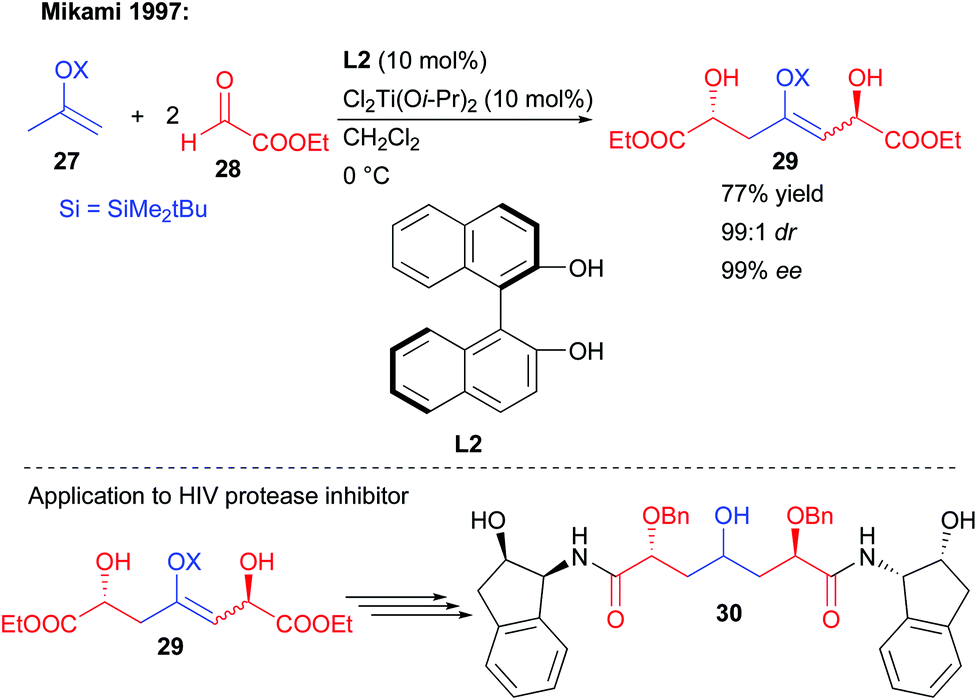
In an early report, Mikami and co-workers showed that a silyl enol ether could undergo a double nucleophilic addition on two equivalents of ethyl-glyoxylate 28 (Scheme 14).29 Through the use of a chiral BINOL/titanium complex, the creation of the two new alcohols could be controlled with virtually perfect diastereo- and enantioselectivity (99:1 dr, 99% ee). However, these conditions were limited to the single use of the highly electrophilic aldehyde 28, favoring the double addition pathway. Despite this limitation, the strategy could be applied to the preparation of a C2-symmetric bis-amide HIV protease inhibitor (Scheme 14).
 | ||
| Scheme 14 Double Mukaiyama type aldol cascade with ethyl glyoxylate. | ||
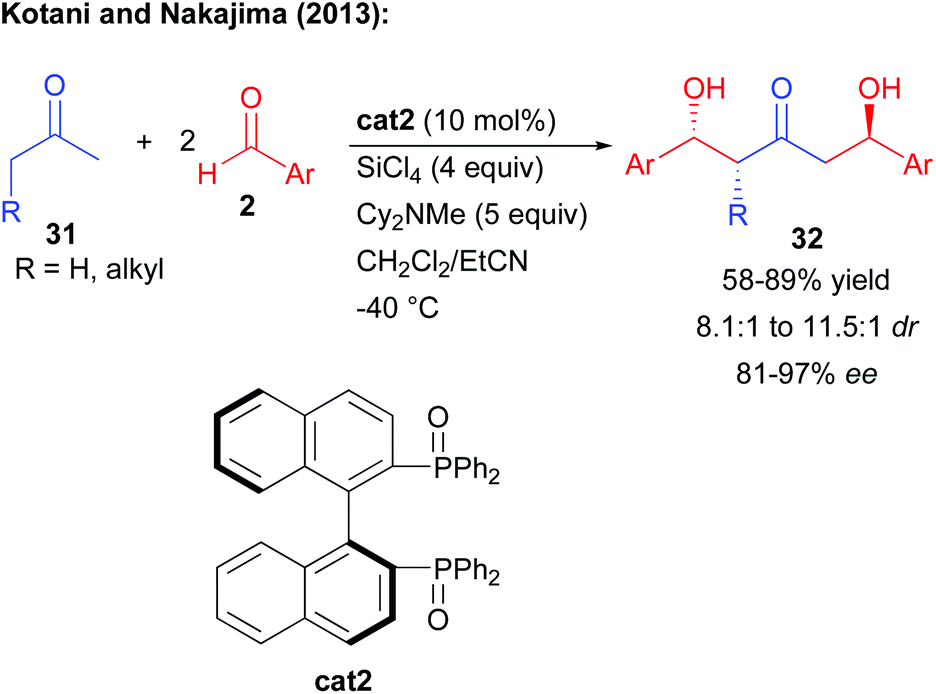
In order to find solutions to the challenge of performing an efficient double aldolization, the group of Kotani and Nakajima used their previously developed Mukaiyama type aldol process using BINAPO cat2 and SiCl4 to generate in situ the more reactive trichlorosilyl enol ether (Scheme 15).11,30 Different central ketones could react twice with two external benzaldehyde derivatives, constructing the expected 1,3-keto-diols with good yields and moderate to excellent stereocontrol. In this cascade, the first enantioselective aldolization occurs from the less substituted side of the ketone while the second aldolization occurs on the opposite side based on diastereoselective substrate control.
 | ||
| Scheme 15 Double Mukaiyama type aldol cascade on benzaldehyde derivatives. | ||
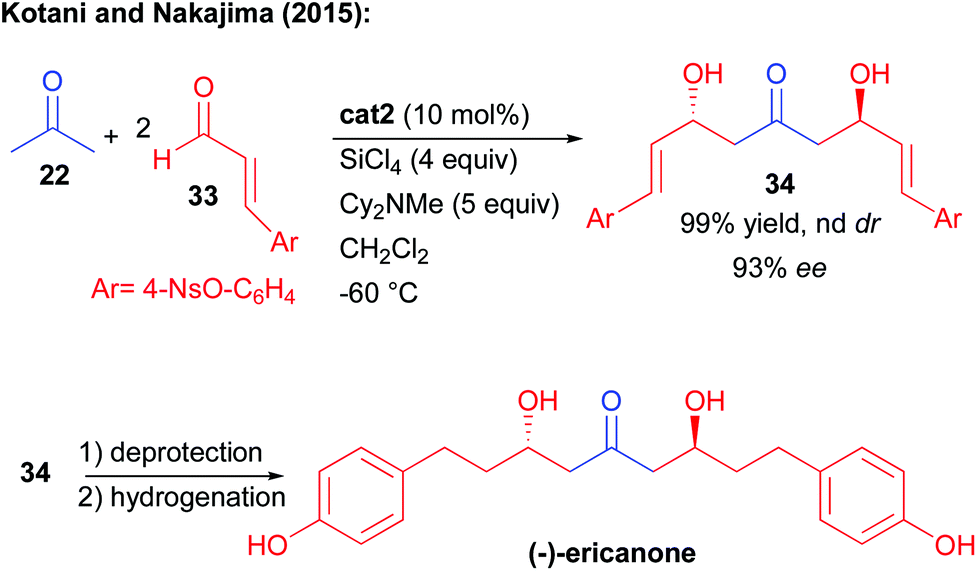
This reaction enables the direct construction of three stereogenic centers but is limited to the addition to sp2-branched aldehydes while requiring a large excess of activating reagents (4 equiv. of SiCl4 and 5 equiv. of Cy2NMe). However, its synthetic utility was demonstrated in the short preparation of ericanone, a natural C2-symmetric keto-diol (Scheme 16).31 Addition to α,β-unsaturated aldehydes 33 led to a selectivity challenge to avoid competing decomposition of these Michael acceptors in the presence of SiCl4. For this purpose, the use of para-nosyl protected phenol 33 was required leading under optimized conditions to the formation of keto-diol 34 with 99% yield and 93% ee. Further cleavage of the nosyl protecting group and hydrogenation of the two alkenes furnished the expected natural keto-diol.
 | ||
| Scheme 16 Application of the double Mukaiyama type aldol cascade to (−)-ericanone synthesis. | ||
More recently, the same team further optimized the process to create more complex keto-diols possessing four stereogenic centers (Scheme 17).32 The key to the success of this challenging cascade lies in the use of SiCl3OTf as silicon source. This additive is a more active silylating reagent, facilitating the double activation of the ketones 35 of increased substitution. Through this approach, the keto-diols possessing 4 controlled acyclic stereogenic centers were formed in a good yield. Moreover, the two catalyst-controlled Mukaiyama type aldolization events ensured excellent levels of enantioselectivity of the final keto-diols (93–99% ee). Reduction of the central ketones could provide the expected 1,3,5-triol.
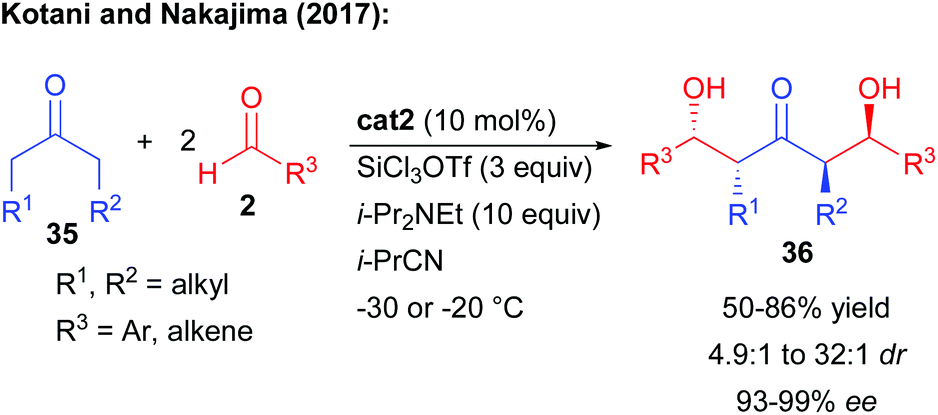 | ||
| Scheme 17 Application of the double Mukaiyama type aldol cascade to the formation of four stereogenic centers. | ||
More recently, the group of Quintard focused on an alternative method to construct 1,3-polyols notably possessing complementary aliphatic chains while modulating the polyol properties. Given the potential enhanced hydrogen bonding properties of 1,2-fluorohydrins and their limited reports in complex structures, the group devised a fluorination/double aldolization cascade, directly creating fluorinated keto-diols (Scheme 18).33 The sequence follows an aldehyde aminocatalyzed fluorination providing an unstable enantioenriched aldehyde with subsequent decarboxylative aldolization catalyzed by Cu(acac)2.34 The key to the success of this transformation creating four new bonds is the use of the bio-resourced 1,3-acetone-dicarboxylic acid derived from citric acid as a reactive acetone surrogate. The overall transformation can be performed in one pot adding the keto-acid after the initial fluorination or directly in a cascade. Excellent levels of enantiocontrol (98–99% ee) are observed through an amplification of chirality by the dimerization of the intermediate enantioenriched halogenated aldehydes. Reduction of the central ketone function leads to the construction of useful 1,3,5-triols in only two steps from commercial products.35
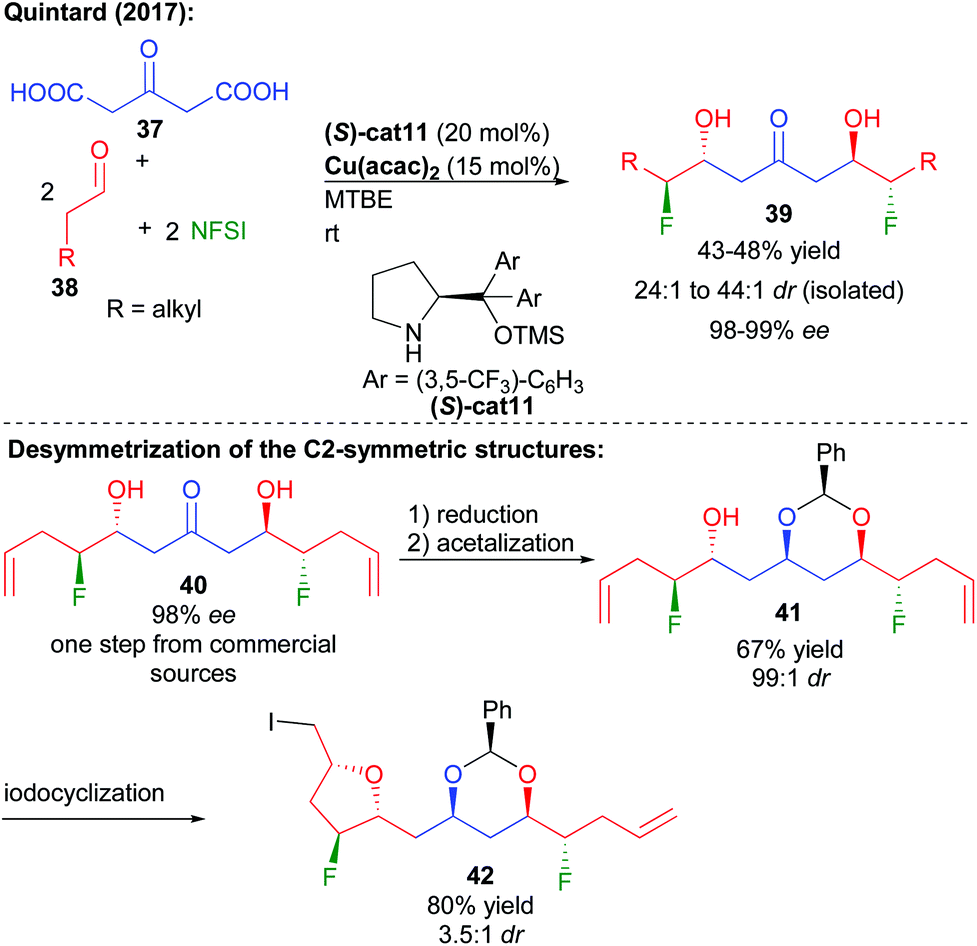 | ||
| Scheme 18 Multi-catalyzed bi-directional di-decarboxylative bis-fluoro-aldolization. | ||
Given the C2-symmetric structure of these 1,3,5-polyols, a desymmetrization is required to unveil the configuration of the central prochiral alcohol. Thermodynamically controlled formation of the more stable syn-acetal generated 41, possessing 6 controlled stereocenters and two differentiated terminal alkenes. From 41, iodoetherification furnished highly functionalized ether 42 with 3.5:1 dr.
This multi-catalyzed bi-directional di-decarboxylative bis-halo-aldolization could further be applied to the preparation of the fluorinated key analogue C15–C27 fragment of bastimolide A, a natural product showing promising antimalarial bioactivity (Scheme 19).36 In addition, the bi-directional process could be extended to the formation of the chlorinated fragment by the use of NCS instead of NFSI and L-prolinamide cat12. Reduction followed by TBDMS alcohol protecting group removal could provide rapidly and with excellent stereocontrol the expected halogenated polyol fragments. In addition to these halogenated analogues, chlorine removal could also provide access to the true non-halogenated natural product fragment. As a result, this approach is useful to fully study the effect of halogen insertion over these highly bioactive scaffold properties. Desymmetrization of the polyol through acetalization followed by oxidation could construct a more advanced intermediate of bastimolide A.
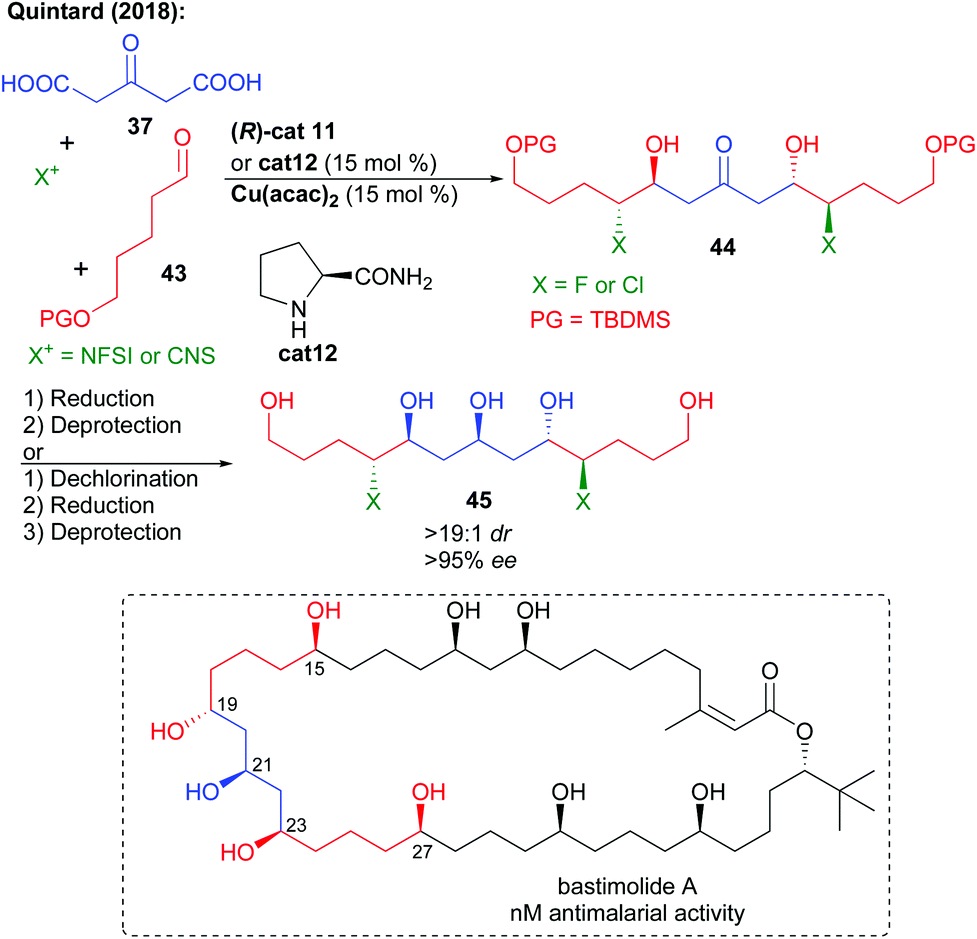 | ||
| Scheme 19 Multi-catalyzed approach towards bastimolide A. | ||
Recently, the same group reported that a direct enantioselective bi-directional decarboxylative aldolization could be applied to the preparation of perfluorinated keto-diols (Scheme 20).37 While different organocatalysts provided low levels of stereocontrol, the enantioselectivity of the process could be efficiently controlled by an appropriate combination between a copper salt and a bis-oxazoline ligand L3.38 Crucial fluorine–phenyl substituent interactions in the transition state were responsible for the high levels of enantiocontrol observed in the formation of the keto-diols (90–96% ee). Of particular importance, the triols obtained after reduction could represent excellent platforms for enantioselective anion binding, demonstrating the potential of polyols to effect crucial recognition mechanisms with high efficiency.
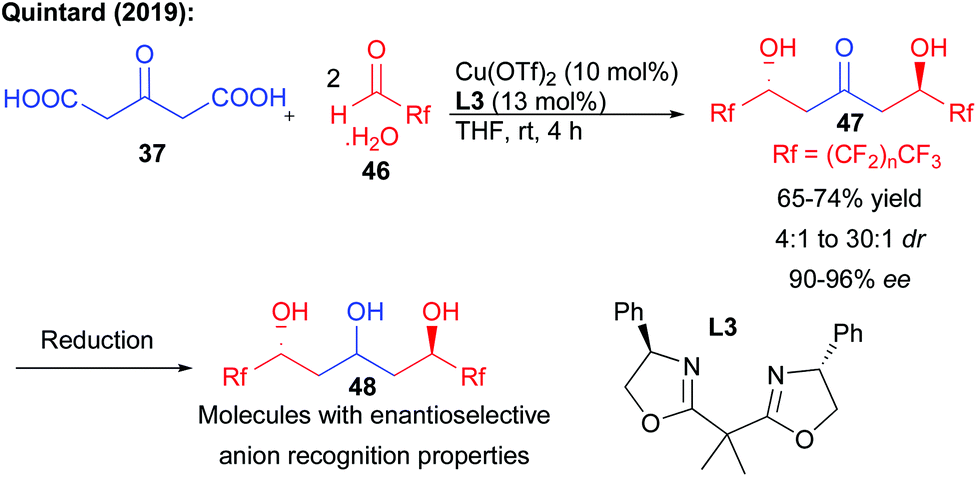 | ||
| Scheme 20 Bi-directional synthesis of perfluorinated 1,3,5-triols. | ||
In addition to perfluorinated chains, the same reaction could be performed on chloral hydrate 49 (Scheme 21). The hexachlorinated ketodiol 50 obtained with 90% ee and 68% yield is the scaffold of a natural product that could explain the observed toxicity of oxitropis common feed plants.39 In addition, it was already shown that this ketodiol could efficiently be desymmetrized, further highlighting the synthetic potential of this bi-directional process.39
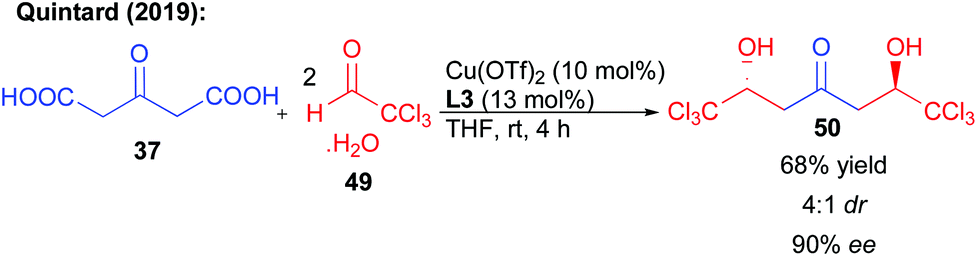 | ||
| Scheme 21 Bi-directional addition to chloral. | ||
Finally, following their interest in new bi-directional strategies, Quintard and collaborators devised an alternative more straightforward construction of aliphatic C2-symmetric 1,3,5-triols (Scheme 22).40 Taking advantage of the iridium reductive-coupling reactivity developed by the group of Krische, they found that commercially available 3-chloro-2-chloromethyl-1-propene 51 could serve as a perfect pro-bisnucleophile to condense in a single cascade with two aliphatic aldehydes. In this transformation, isopropanol acts as a source of hydrogen, ensuring the formation of a nucleophilic π-allyl complex able to undergo the double enantioselective addition. The process can also be performed sequentially in a diastereodivergent condensation to two different aldehydes, providing interesting non-symmetric structures. From the chiral homo-allylic 1,5-diols obtained with excellent stereocontrol through two successive events under catalyst control, reductive ozonolysis could provide in only two steps the desired 1,3,5-polyols of interest.
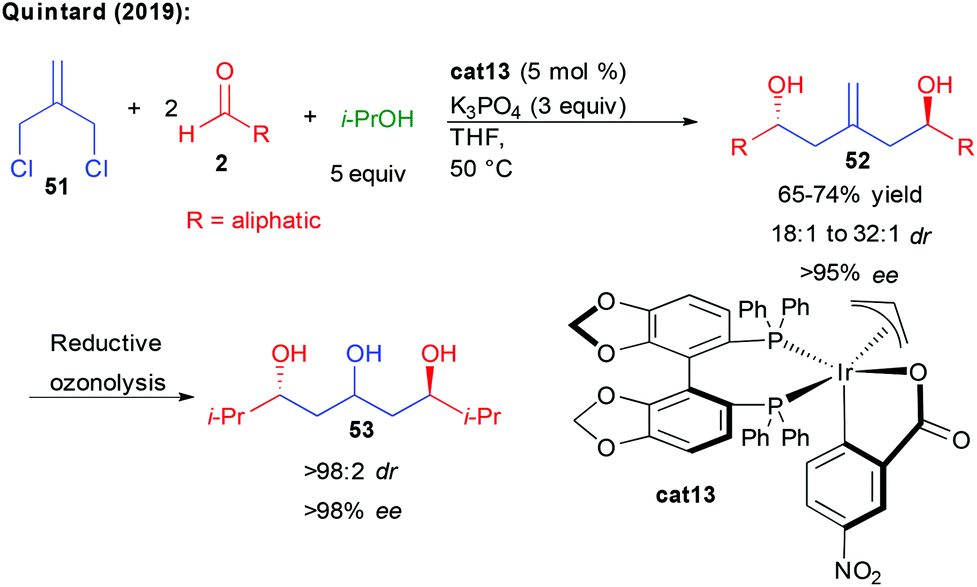 | ||
| Scheme 22 Bi-directional reductive coupling. | ||
Conclusions
Given their ubiquity in a wide array of drugs and bioactive natural products, the synthesis in a rapid and stereocontrolled manner of extended 1,3-polyols is crucial. This great importance has lead organic chemists to design and develop a broad range of catalytic cascades providing more reliable routes to these scaffolds. By efficiently engineering substrate combinations and catalyst structures, efficient enantioselective cascades have been devised generating through C–C bond formation multiple alcohol functions with high efficiency.Notably, through the appropriate use of the bi-directionality principle, diverse organo- or metal-catalysts have been shown to directly construct the core scaffolds of various families of natural products such as keto-diols or 1,3,5-polyols. Most notably, these catalysts are most of the time able to control multiple stereogenic centers, facilitating the preparation of these otherwise difficult to access acyclic structures. The efficiency of these protocols has been demonstrated in the preparation of numerous natural products and drugs, as well as chiral ligands or supramolecular hosts.
However, the field is far from mature and still faces crucial challenges. Many of the developed methods still require stoichiometric activating reagents, generating unnecessary waste. Thus discovering direct cascades from biosourced materials is highly desirable. The most synthetically relevant aliphatic aldehydes are in some cases reluctant to undergo efficient cascades due to their ease of enolization. Since most processes involve multiple substrates condensation, the ability of the catalytic methods to accommodate selective addition of structurally different pro-nucleophilic and pro-electrophilic partners would considerably facilitate and shorten further derivatization to the final molecules. Finally, the ultimate goal would be to develop stereodivergent cascade processes allowing the direct preparation of all possible stereoisomers of these acyclic structures through selective catalyst stereocontrol.41 Such a strategy might be conceivable in the future by the development of new multi-catalytic transformations.
As a conclusion, all future progress will be possible through the implementation of new strategies and innovative catalytic activation modes. The development of these new approaches should find direct useful implications by the synthetic community in the preparation of a broad array of key bioactive molecules.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Centre National de la Recherche Scientifique (CNRS) and Aix-Marseille Université are warmly acknowledged for financial support.Notes and references
- (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS; (b) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993 RSC; (c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (d) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- (a) D. O'Hagan, The Polyketide Metabolites, Ellis Horwood, Chichester, 1991 Search PubMed; (b) J. Rohr, Angew. Chem., Int. Ed., 2000, 39, 2847 CrossRef CAS; (c) A. M. P. Koskinen and K. Karisalmi, Chem. Soc. Rev., 2005, 34, 677 RSC.
- For general reviews on polyol synthesis: (a) T. Oishi and T. Nakata, Synthesis, 1990, 635 CrossRef CAS; (b) J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380 RSC; (c) S. E. Bode, M. Wolberg and M. Müller, Synthesis, 2006, 557 CAS; (d) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS PubMed; (e) T. Harschneck, S. Kirsch and F. Stefan, in Asymmetric Synthesis II, ed. M. Christmann and S. Brase, 2012, p. 179 Search PubMed; (f) D. Herkommer, B. Schmalzbauer and D. Menche, Nat. Prod. Rep., 2014, 31, 456 RSC; (g) A.-M. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504 RSC; (h) F. Schaefers, T. A. M. Gulder, C. Bressy, M. Smietana, E. Benedetti, S. Arseniyadis, M. Kalesse and M. Cordes, Polyketides, in From Biosynthesis to Total Synthesis, ed. A. L. Zografos, 2016, p. 21 Search PubMed; (i) J. Feng, Z. A. Kasun and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5467 CrossRef CAS; (j) P. Kumar, D. Tripathi, B. M. Sharma and N. Dwivedi, Org. Biomol. Chem., 2017, 15, 733 RSC.
- (a) S. R. Magnuson, Tetrahedron, 1995, 51, 2167 CrossRef CAS; (b) C. S. Poss and S. L. Schreiber, Acc. Chem. Res., 1994, 27(1), 9 CrossRef CAS.
- (a) N. S. Chowdari, D. B. Ramachary, A. Cordova and C. F. Barbas, III, Tetrahedron Lett., 2002, 43, 9591 CrossRef CAS. For general reviews on catalytic aldolization, see: (b) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (c) Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388 RSC.
- (a) A. Cordova, I. Ibrahem, J. Casas, H. Sunden, M. Engqvist and E. Reyes, Chem. – Eur. J., 2005, 11, 4772 CrossRef CAS PubMed; (b) A. Cordova, M. Engqvist, I. Ibrahem, J. Casas and H. Sunden, Chem. Commun., 2005, 2047 RSC.
- (a) J. Casas, M. Engqvist, I. Ibrahem, B. Kaynak and A. Cordova, Angew. Chem., Int. Ed., 2005, 44, 1343 CrossRef CAS PubMed. For another related approach involving a one-pot enamine Mukaiyama aldolization, see: (b) A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752 CrossRef CAS PubMed. Some aldolases have also been used for such a process: (c) H. J. M. Gijsen and C.-H. Wong, J. Am. Chem. Soc., 1995, 117, 7585 CrossRef CAS; (d) A. Szekrenyi, X. Garrabou, T. Parella, J. Joglar, J. Bujons and P. Clapés, Nat. Chem., 2015, 7, 724 CrossRef CAS PubMed.
- (a) B. J. Albert, Y. Yamaoka and H. Yamamoto, Angew. Chem., Int. Ed., 2010, 49, 2747 CrossRef CAS PubMed; (b) B. J. Albert, Y. Yamaoka and H. Yamamoto, Angew. Chem., Int. Ed., 2011, 50, 2610 CrossRef CAS PubMed; (c) J. Saadi, M. Akakura and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 14248 CrossRef CAS PubMed; (d) P. B. Brady and H. Yamamoto, Angew. Chem., Int. Ed., 2012, 51, 1942 CrossRef CAS PubMed.
- (a) L. Lin, K. Yamamoto, H. Mitsunuma, Y. Kanzaki, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2015, 137, 15418 CrossRef CAS PubMed. Recently, trapping of the final cascade products has been disclosed: (b) Y. Kanzaki, Y. Hirao, H. Mitsunuma and M. Kanai, Org. Biomol. Chem., 2019, 17, 6562 RSC.
- Interestingly, sequential aldolization changing the configuration of the ligand can lead to a diastereodivergent process.
- (a) Y. Shimoda, S. Kotani, M. Sugiura and M. Nakajima, Chem. – Eur. J., 2011, 17, 7992 CrossRef CAS PubMed. For an earlier example of such double aldolization in the racemic series using stoichiometric boron, see: (b) A. Abiko, J.-F. Liu, D. C. Buske, S. Moriyama and S. Masamune, J. Am. Chem. Soc., 1999, 121, 7168 CrossRef CAS. For recent reviews, see: (c) S. Kotani, M. Sugiura and M. Nakajima, Synlett, 2014, 25, 631 CrossRef CAS; (d) S. Kotani, M. Sugiura and M. Nakajima, Chem. Rec., 2013, 13, 362 CrossRef CAS PubMed; (e) S. Kotani, Chem. Pharm. Bull., 2019, 67, 519 CrossRef CAS PubMed.
- (a) A. Genoni, M. Benaglia, S. Rossi and G. Celentano, Chirality, 2013, 25, 643 CrossRef CAS PubMed; (b) P. Zhang, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 11054 CrossRef CAS PubMed; (c) F. Maier and O. Trapp, Angew. Chem., Int. Ed., 2014, 53, 8756 CrossRef CAS PubMed.
- For recent general reviews on reductive couplings, see: (a) J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142 CrossRef CAS; (b) A. Quintard and J. Rodriguez, Chem. Commun., 2016, 52, 10456 RSC; (c) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371 CrossRef CAS.
- (a) Y. Lu, I. S. Kim, A. Hassan, D. J. Del Valle and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 5018 CrossRef CAS; (b) X. Gao, H. Han and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 12795 CrossRef CAS; (c) A. Hassan, I. A. Townsend and M. J. Krische, Chem. Commun., 2011, 47, 10028 RSC.
- (a) A.-M. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504 RSC; (b) J. Feng, Z. A. Kasun and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5467 CrossRef CAS.
- F. Perez, A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2016, 55, 5049 CrossRef CAS.
- (a) Y. Lua and M. J. Krische, Org. Lett., 2009, 11, 3108 CrossRef; (b) Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876 CrossRef CAS.
- (a) Z. T. T. Kumpulainen, B. Kang and M. J. Krische, Org. Lett., 2011, 13, 2484 CrossRef PubMed; (b) B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559 CrossRef PubMed.
- (a) X. Gao, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 4223 CrossRef CAS PubMed; (b) Z. A. Kasun, X. Gao, R. M. Lipinski and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 8900 CrossRef CAS PubMed; (c) I. Shin and M. J. Krische, Org. Lett., 2015, 17, 4686 CrossRef CAS; (d) I. Shin, S. Hong and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14246 CrossRef CAS PubMed; (e) J. Willwacher and A. Fürstner, Angew. Chem., Int. Ed., 2014, 53, 4217 CrossRef CAS PubMed.
- See for example: (a) Z. Yang, B. Zhang, G. Zhao, J. Yang, X. Xie and X. She, Org. Lett., 2011, 13, 5916 CrossRef CAS PubMed; (b) J. M. Cabrera and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 10718 CrossRef CAS PubMed.
- A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2013, 52, 4470 CrossRef CAS.
- Y. Feng, X. Jiang and J. K. De Brabander, J. Am. Chem. Soc., 2012, 134, 17083 CrossRef CAS.
- (a) W. R. Roush, T. D. Bannister, M. D. Wendt, J. A. Jablonowski and K. A. Scheidt, J. Org. Chem., 2002, 67, 4275 CrossRef CAS PubMed; (b) S. E. Denmark and S. Fujimori, Org. Lett., 2002, 4, 3477 CrossRef CAS PubMed; (c) M. J. Mitton-Fry, A. J. Cullen and T. Sammakia, Angew. Chem., Int. Ed., 2007, 46, 1066 CrossRef CAS PubMed; (d) L. C. Dias, A. A. de Marchi, M. A. B. Feirrera and A. M. Aguilar, Org. Lett., 2007, 9, 4869 CrossRef CAS PubMed; (e) Y. Yamaoka and H. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5354 CrossRef CAS PubMed; (f) L. C. Dias, E. C. de Lucca Jr., M. A. B. Feirrera, M. D. C. Garcia and C. F. Tormena, Org. Lett., 2010, 12, 5056 CrossRef CAS PubMed.
- For the racemic version, see: (a) C. Ji, Y. Peng, C. Huang, N. Wang and Y. Jiang, Synlett, 2005, 985 Search PubMed; (b) S. Hu, T. Jiang, Z. Zhang, A. Zhu, B. Han, J. Song, Y. Xie and W. Li, Tetrahedron Lett., 2007, 48, 5613 CrossRef CAS.
- (a) D. Gryko and R. Lipinski, Eur. J. Org. Chem., 2006, 3864 CrossRef CAS; (b) G. Valero, J. M. Ribo and A. Moyano, Chem. – Eur. J., 2014, 20, 17395 CrossRef CAS PubMed.
- D. B. Ramachary, R. Mondal and R. Madhavachary, Org. Biomol. Chem., 2012, 10, 5094 RSC.
- A. S. Kucherenko, V. V. Gerasimchuk, V. G. Lisnyak, Y. V. Nelyubina and S. G. Zlotin, Eur. J. Org. Chem., 2015, 5649 CrossRef CAS.
- (a) T. Ichibakase and M. Nakajima, Org. Lett., 2011, 13, 1579 CrossRef CAS PubMed; (b) T. Ichibakase and M. Nakajima, Synthesis, 2012, 44, 3145 CrossRef CAS. For an earlier example of the aldol/Tishchenko process using stoichiometric Ti/cinchonine, see: (c) K. Rohr, R. Herre and R. Mahrwald, Org. Lett., 2005, 7, 4499 CrossRef CAS.
- K. Mikami, S. Matsukawa, M. Nagashima, H. Funabashi and H. Morishima, Tetrahedron Lett., 1997, 38, 579 CrossRef CAS.
- Y. Shimoda, T. Kubo, M. Sugiura, S. Kotani and M. Nakajima, Angew. Chem., Int. Ed., 2013, 52, 3461 CrossRef CAS PubMed.
- S. Kotani, K. Kai, Y. Shimoda, H. Hu, S. Gao, M. Sugiura, M. Ogasawara and M. Nakajima, Chem. – Asian J., 2016, 11, 37 CrossRef PubMed.
- S. Kotani, K. Kai, M. Sugiura and M. Nakajima, Org. Lett., 2017, 19, 3672 CrossRef CAS PubMed.
- (a) A. Quintard and J. Rodriguez, ACS Catal., 2017, 7, 551 CrossRef; (b) J. Rodriguez and A. Quintard, Chimia, 2018, 72, 580 CrossRef CAS PubMed.
- (a) A. Quintard and J. Rodriguez, Chem. – Eur. J., 2015, 21, 14717 CrossRef CAS PubMed; (b) A. Ricucci, J. Rodriguez and A. Quintard, Eur. J. Org. Chem., 2018, 3697 CrossRef CAS.
- These triols could serve as promising molecules given their improved supramolecular properties; see: (a) C. Sperandio, G. Quintard, J. V. Naubron, M. Giorgi, M. Yemloul, J.-L. Parrain, J. Rodriguez and A. Quintard, Chem. – Eur. J., 2019, 25, 15098 CrossRef CAS PubMed.
- A. Quintard, C. Sperandio and J. Rodriguez, Org. Lett., 2018, 20, 2574 CrossRef.
- (a) C. Sperandio, J. Rodriguez and A. Quintard, Chem. Sci., 2020 10.1039/c9sc05196a.
- (a) G. Lalic, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2003, 125, 2852 CrossRef CAS PubMed; (b) S. Orlandi, M. Benaglia and F. Cozzi, Tetrahedron Lett., 2004, 45, 1747 CrossRef CAS; (c) D. Magdziak, G. Lalic, H. M. Lee, K. C. Fortner, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2005, 127, 7284 CrossRef CAS PubMed; (d) K. C. Fortner and M. D. Shair, J. Am. Chem. Soc., 2007, 129, 1032 CrossRef CAS PubMed.
- (a) R. Yu, X. Li, T. Zhu, G. Yang, Z. Li and B. Yang, Shenyang Yaoxueyuan Xuebao, 1991, 8, 113 CAS; (b) R. E. Bowman and W. R. N. Williamson, J. Chem. Soc. C, 1970, 1, 101 RSC.
- A. Quintard and J. Rodriguez, Org. Lett., 2019, 21, 453 CrossRef CAS PubMed.
- S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |