
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the mechanism of the surface functionalization dependent neurotoxicity of graphene family nanomaterials†
Zhiling
Guo
 abc,
Peng
Zhang
*c,
Andrew J.
Chetwynd
c,
Heidi Qunhui
Xie
abd,
Eugenia
Valsami-Jones
c,
Bin
Zhao
*abd and
Iseult
Lynch
c
abc,
Peng
Zhang
*c,
Andrew J.
Chetwynd
c,
Heidi Qunhui
Xie
abd,
Eugenia
Valsami-Jones
c,
Bin
Zhao
*abd and
Iseult
Lynch
c
aState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: binzhao@rcees.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Geography, Earth and Environmental Sciences, University of Birmingham, Birmingham B15 2TT, UK. E-mail: p.zhang.1@bham.ac.uk
dInstitute of Environment and Health, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 18th August 2020
Abstract
Graphene family nanomaterials (GFNs) have shown great potential for biological and environmental applications; however, their future use has been debated due to their reported potential neurotoxicity. Moreover, the effects of surface functionalization on their biological end points are largely unknown. Here, we compared the effects of reduced graphene oxide (RGO), and carboxylated (G-COOH), hydroxylated (G-OH) and aminated (G-NH2) graphene nanosheets on human neuroblastoma cells (SK-N-SH). All GFNs inhibited cellular growth at concentrations of 0.1–10 mg L−1 after 24 h exposure. The toxicity was attenuated over longer exposure times, with the exception of G-NH2. Although the overall acute toxicity followed the order: G-OH ≈ G-COOH > RGO > G-NH2, G-NH2 induced more persistent toxicity and more metabolic disturbance compared to the other GFNs, with lipid and carbohydrate metabolism being the most affected. The potential for physical disruption of the lipid membrane and oxidative damage induced by GFNs varied with different functionalization, which accounts for the observed differences in neurotoxicity. This study provides significant insights into the neurological effects of GFNs, and suggests that G-NH2 is not as safe as reported in many previous studies. The neurological effect of GFNs over longer term exposure should be considered in future studies.
1. Introduction
Graphene based nanomaterials (GFNs) have attracted great research interest in recent years due to their unique optical, electronic, mechanical and thermal properties.1 GFNs have great potential in the biomedical sector for drug delivery, bio-imaging, phototherapy etc.2 As a 2-D material, graphene has been reported as an ideal substrate to support cell attachment, proliferation and differentiation.3 Recent studies have shown that GFNs might also be used as a platform to enhance cellular growth such as that of neuronal cells.4 However, the potential neurotoxicity of GFNs has also been reported in different biological models such as human neuroblastoma cells (SH-SY5Y),5Caenorhabditis elegans,6 or zebrafish.7 For example, graphene oxide (100–800 nm) at concentrations higher than 80 mg L−1 can cause neurotoxicity to SH-SY5Y cells.5 Graphene oxide at environmentally relevant concentrations (0.01–1 μg L−1) can induce Parkinson's disease-like symptoms in zebrafish larvae.7 Clearly, more studies are required to fully understand the neurological effects of GFNs before they can be used safely for clinical and tissue engineering applications.GFN-induced cytotoxicity mainly involves physical damage and oxidative stress. The sharp edge of GFNs may penetrate the cell membrane and extract membrane lipids, thereby causing membrane damage.8 GFNs may also generate free radicals9 or oxidize antioxidants10 in cells, causing oxidative stress. The biological effects of GFNs and the mechanisms of action are highly dependent upon their physicochemical properties such as lateral size, shape, thickness, surface charge, and surface functionalization.11 Graphene oxide with larger lateral sizes can wrap around the cell surface in nutrient rich medium, thereby preventing cells from acquiring essential nutrients from the culture medium.12 Positively charged GFNs might bind more easily to the negatively charged phosphate headgroups in the phospholipid membrane, causing cell membrane damage.13
The aim of this study is to understand how surface functionalization affects the biological outcomes of GFNs on neuronal cells. Specifically, we compared the effects of reduced graphene oxide (RGO), and carboxylated (G-COOH), hydroxylated (G-OH) and aminated (G-NH2) graphene nanosheets on the growth of human neuroblastoma cells (SK-N-SH). The potential of GFNs to induce physical disruption of the cell membranes and cellular oxidative damage was evaluated to understand the difference in the mechanisms of action between different functionalisations. Moreover, the metabolic disturbance to the cells induced by the four GFNs was evaluated using a metabolomics approach. All four GFNs showed neurotoxicity to the SK-N-SH cells although to different extents. Both physical disruption and oxidative stress were involved in the toxicity. G-NH2 showed less acute but more persistent toxicity and induced more metabolic disturbance of the cells than the other GFNs.
2. Results and discussion
RGO, G-OH, G-COOH and G-NH2 all have similar lateral sizes which were 0.14 ± 0.04 μm, 0.16 ± 0.06 μm, 0.14 ± 0.04 μm and 0.15 ± 0.03 μm, respectively (Fig. 1A; Table S1†). The thicknesses of RGO (0.91 ± 0.15 nm), G-OH (0.90 ± 0.16 nm), G-COOH (0.89 ± 0.23 nm), and G-NH2 (0.95 ± 0.14) obtained from the AFM images were also similar (Fig. 1A), suggesting that all the four GFNs were triple layered. The defects and disorder in the materials, indicated by Id/Ig ratios (high ratio indicates high disorder and defect14) in the Raman spectra followed the order: G-NH2 > G-COOH > G-OH > RGO (Fig. S1†). The hydrodynamic sizes of the four GFNs in deionized water showed no significant difference (Table S1†), suggesting that their agglomeration states were similar. The RGO, G-OH and G-COOH were negatively charged, while G-NH2 was positively charged in deionized water (Table S1†), due to the presence of different functional groups. XPS analysis showed that the RGO, G-OH and G-COOH were composed of C and O, and G-NH2 was composed of C, O and N (Fig. 1B). The atomic percent of C, N and O in the materials is shown in Table S1.† Since the primary lateral size, thickness and hydrodynamic size of the four types of graphene were all similar, any difference in the cellular effects between the GFNs can be attributed to the difference in the functional groups.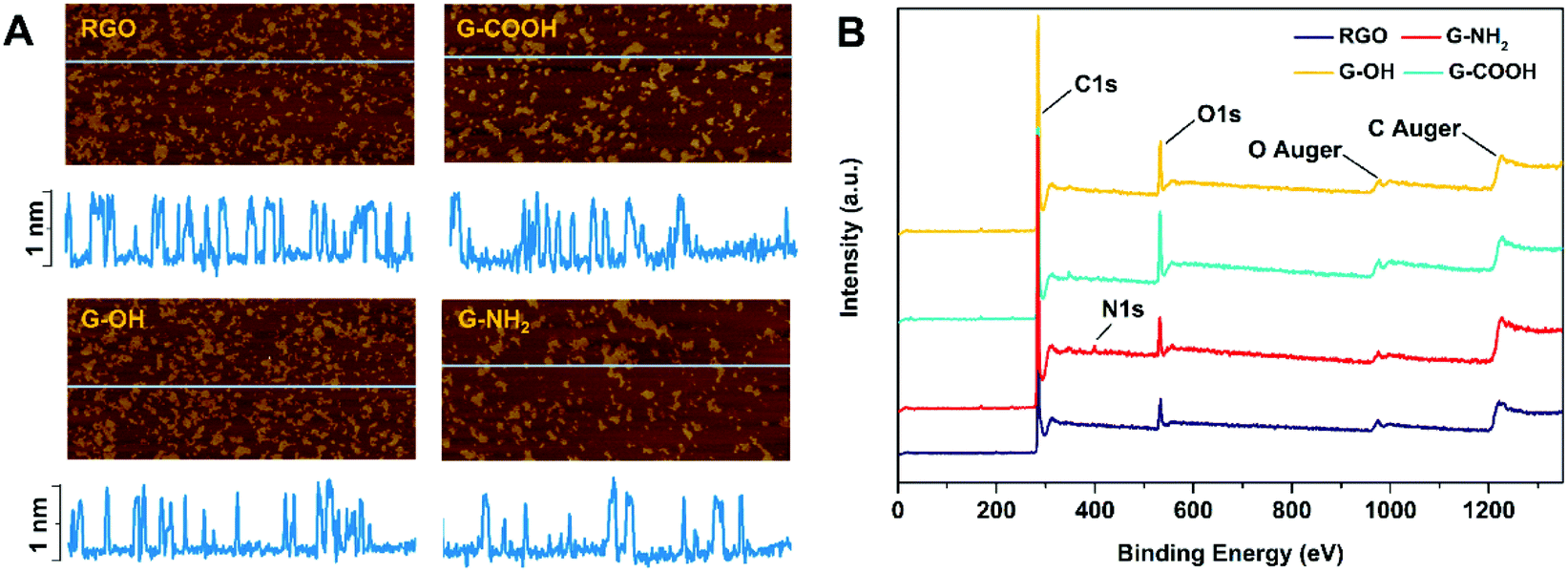 | ||
| Fig. 1 AFM image and height profile (A) and the XPS spectra (B) of the four GFNs. The yellow plaques in A indicate that the GFNs were dispersed on silicon wafers. The blue horizontal lines (with a length of 2 μm) in the AFM images indicate the regions analyzed for the height profiles. | ||
We initially compared the neurotoxic potential of the GFNs over 72 h, with cell viability tested at 24, 48 and 72 h (Fig. 2A). All materials showed inhibitory effects on the cellular growth of SK-N-SH cells at a concentration of 0.1 mg L−1. G-NH2 showed the lowest toxic response with 76% cell survival at 24 h, while only 54–59% of cells survived in the RGO, G-OH, and G-COOH groups. Interestingly, with increased exposure time, the cell viability in the RGO, G-OH, and G-COOH groups increased, while for the G-NH2 group it remained stable (76–80%). After 72 h exposure, the cell viability in the RGO and G-COOH groups increased to ∼100%, while the cell viability in the G-OH group increased to 77.6%. The cell viability did not show a significant change when the exposure concentration was increased to 1 mg L−1 in the RGO, G-OH, and G-NH2 groups, while the cell viabilities in the G-COOH group decreased by 13% and 8% at 48 and 72 h, respectively, compared with that at 0.1 mg L−1. A further increase of the exposure concentration to 10 mg L−1 caused a significant decrease of the cell viability, with 42%, 28% and 23% survival rate at 24 h for RGO, G-OH, and G-COOH, respectively, and increased to 66%, 43% and 49%, respectively, after 72 h exposure. However, increasing the G-NH2 exposure concentration did not affect the cell viability compared to the initial 0.1 mg L−1 exposure.
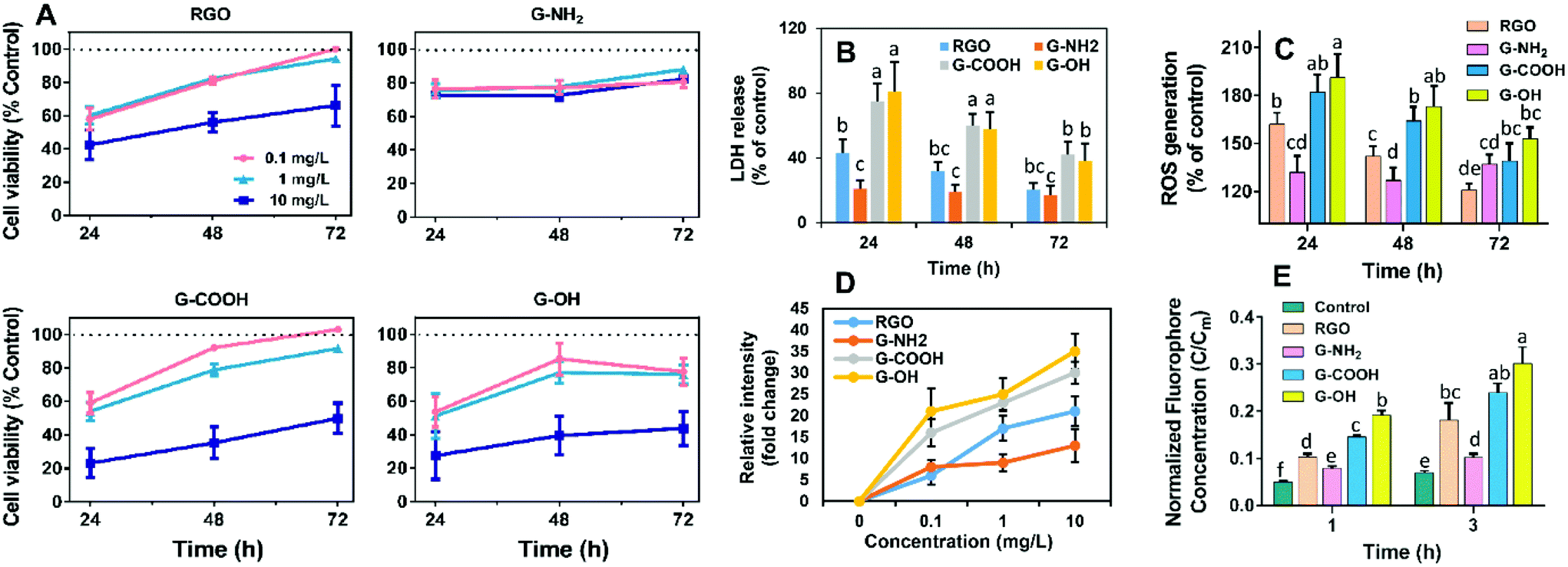 | ||
| Fig. 2 (A) Cell viability after treatment with four GFNs for 24, 48 and 72 h. (B) LDH release from SK-N-SH cells. (C) ROS accumulation (% of control) in SK-N-SH cells. (D) Oxidative potential of the GFNs by measuring the time-dependent oxidation of DCFH. (E) Potential of graphene-induced physical disruption of lipid membranes by measuring the fluorophore leakage from the liposome vesicles. All data are expressed as mean ± SD (n = 6). Different lowercase letters indicate significant difference at p < 0.05. | ||
In summary, the neurotoxic potential of the four GFNs followed the order: G-OH ≈ G-COOH > RGO > G-NH2. The lower toxicity observed for G-NH2 is consistent with previous studies which showed that positively charged aminated graphene was more favorable for neuronal growth.15 It was proposed that amine modification can reduce the charge transfer to cells, thus reducing the toxicity of graphene.16 However, in our study, G-NH2 showed sustained neurotoxicity over the exposure period, which was distinct from the adaptive response of the SK-N-SH cells observed for other three GFN groups, suggesting that G-NH2 may cause persistent neurotoxicity.
To further elucidate the different patterns of the neurotoxicity induced by the GFNs, we examined whether exposure to the GFNs induces membrane damage and/or oxidative stress in the cells. All of the GFNs induced membrane damage as demonstrated by the leakage of LDH (Fig. 2B) and lipid peroxidation (Fig. S2†). The extent of the damage followed the same trend as the cell viability assay: G-OH ≈ G-COOH > RGO > G-NH2, suggesting that the graphenes caused different degrees of membrane damage. Adaptive responses were also observed, with the membrane damage being alleviated after 72 h of exposure to G-OH, G-COOH and RGO; however, G-NH2 caused sustained LDH release throughout the 72 h exposure period.
The membrane damage may be due to a reaction between membrane lipids and excess free radicals or be the result of physical disruption by the sharp edge of the graphene sheets. Indeed, overaccumulation of ROS in the cells was observed in all the treatments in the same patterns as the cell viability (Fig. 2C). GFNs may also cause oxidative damage due to their intrinsic oxidative potential.17,18Fig. 2D shows that the four GFNs exhibited intrinsic oxidative potential, evidenced by their ability to oxidize DCFH. G-NH2 demonstrated a higher oxidative potential than the other materials, which is consistent with its higher disorder and the defects observed by Raman spectroscopy (Fig. S1†). However, this is inconsistent with the results of the cellular toxicity of G-NH2 being lower than the other GFNs, suggesting that the mechanism of neurotoxicity is not solely explained by the direct oxidation of cells by GFNs. Previous studies reported that graphene may cause membrane damage by direct physical insertion.19 We therefore examined this possibility and found that the GFNs have different capacities for causing membrane damage to liposome vesicles. The extent of leakage of the fluorophore from vesicles followed the same trend as that of cell viability and membrane damage (Fig. 2E). These results suggest that physical disruption of the membrane lipids, rather than the direct oxidation by the GFNs, is the main mechanism of neurotoxicity, and that surface functionalization affected their capacity for disrupting lipid membranes and the intrinsic oxidative potentials of GFNs.
To obtain additional molecular mechanisms for the differential patterns of neurotoxicity triggered by the GFNs, non-targeted metabolomics was carried out. The score plot of the partial least-squares discriminant analysis (PLS-DA) showed a clear separation between the control and treatment groups, indicating that all treatments caused perturbation in the metabolic profiles of SK-N-SH cells. Moreover, all treatment groups could be discriminated from each other except for G-COOH versus G-OH (Fig. 3A), which correlates with the cytotoxicity data indicating that G-COOH and G-OH induced similar inhibitory effects on neuronal growth. The discriminating metabolites that lead to the separation of the control and treatment groups (VIP > 1 and p < 0.05) were isolated and identified (Fig. S3†). More metabolites were altered by RGO and G-NH2 than G-COOH and G-OH (Fig. S3†). In total, 35, 31, 8 and 14 metabolites were significantly altered by RGO, G-NH2, G-COOH, and G-OH, respectively. Following exposure to RGO, G-COOH, and G-OH, all the altered metabolites were downregulated, while for the G-NH2 exposed cells, 19 metabolites were upregulated and 12 were downregulated (Fig. S3†). Only a few metabolites (1–6) overlapped among the treatments (Fig. 3B). Treatment-specific metabolites accounted for 65.7% and 67.7% in RGO and G-NH2 groups, respectively (Fig. 3B). The perturbed metabolites were predominantly lipids, carbohydrates, nucleotides, and amino acids (Fig. 3C). Functional enrichment analysis showed that the disturbed metabolites in each treatment participate in carbohydrate metabolism, lipid biosynthesis, and antioxidant metabolism (Fig. S4†). The four GFNs induced different patterns of alterations in metabolites that are related to those pathways (Fig. 3D). In particular, metabolites related to energy production were differentially altered by the GFNs. Glycolytic intermediates such as glucose, glycerol 3-phosphate, and UDP-N-acetylglucosamine were significantly decreased in the RGO group, while they were not affected by the other GFNs. Metabolites that participate in oxidative phosphorylation were downregulated in the RGO (e.g. ADP, nicotinamide, 1-methylnicotinamide) and G-COOH (e.g. 1-methylnicotinamide) groups but were upregulated in the G-NH2 group (e.g. ATP, FMN, FAD) (Fig. 3D). Additionally, all treatments induced decreased lipid biosynthesis although the discriminating lipids were different. Moreover, the antioxidant activity was differently affected. For example, L-gulonic gamma lactone, an essential substrate for the biosynthesis of the antioxidant ascorbic acid,20 was decreased by RGO treatment. Taurine, a critical component involved in antioxidative defense,21 was decreased in the G-COOH group but increased in the G-NH2 group. These results indicate that different surface treatments of otherwise identical GFNs exert different effects on the metabolism of SK-N-SH cells.
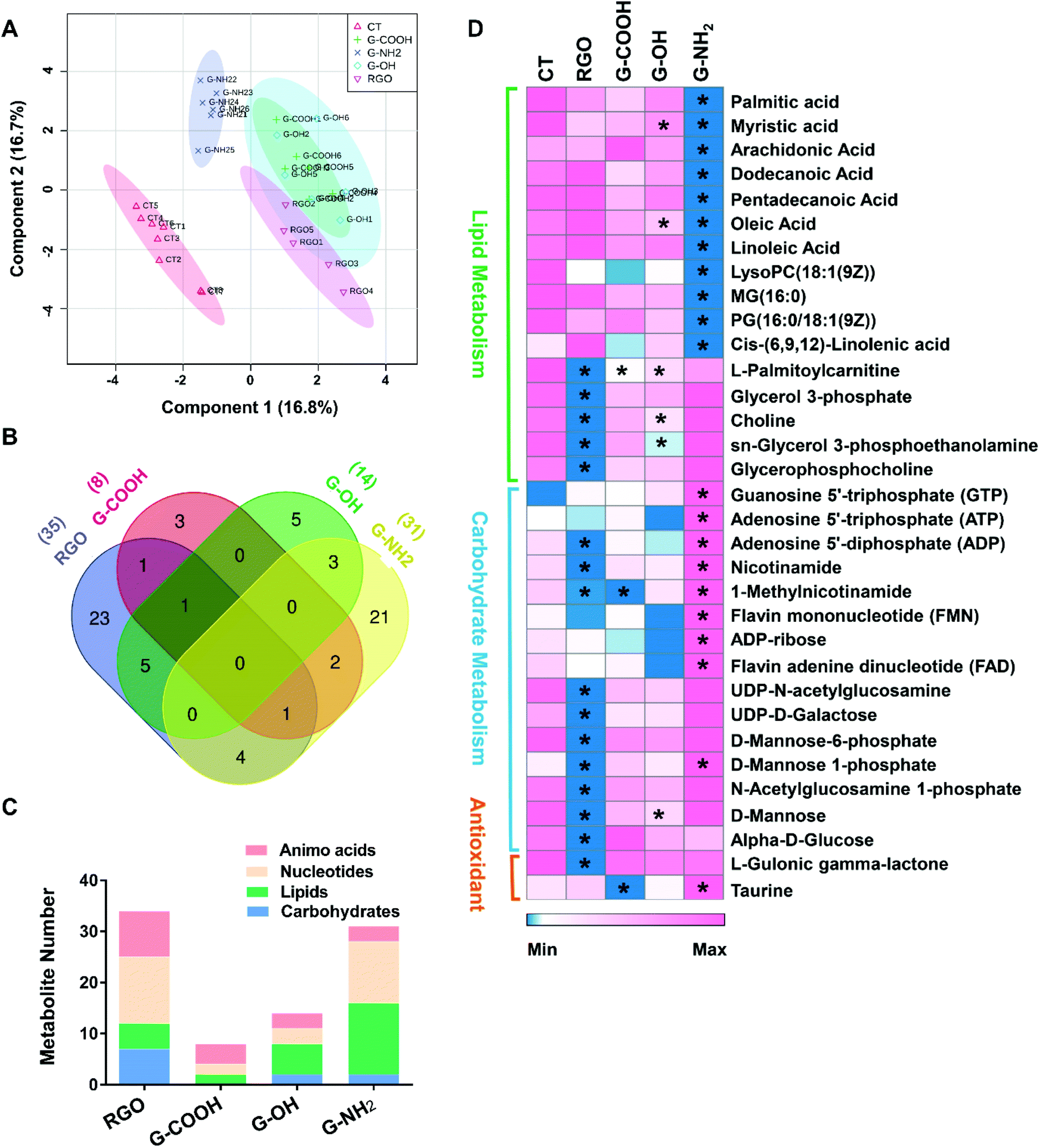 | ||
| Fig. 3 PLS-DA score plot of metabolic profiles in SK-N-SH cells after 72 h treatment with 10 mg L−1 of the four GFNs (A). Venn diagram analyzed using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) shows the overlap of perturbed metabolites in the different treatments (B). The number of perturbed amino acids, nucleotides, lipids and carbohydrates in each treatment (C). Relative levels of metabolites involved in lipid biosynthesis, carbohydrate metabolism, and antioxidant metabolism after GFN exposure. Rows indicate metabolites. The degree of change in the metabolite concentration following GFN exposure is color-coded. * indicates significant difference when compared with the control (CT). | ||
Previous studies suggest that GFNs may trigger the mitochondrial pathway and activate caspase-3 and PARP, which eventually lead to cell apoptosis.22 The mitochondria generated superoxide can result in the loss of mitochondrial membrane potential, which impairs the electron transfer chain and energy metabolism. It was reported that few-layer graphene can increase the cytoplasm Ca2+ concentration and thus cause depolarization of the mitochondrial membrane and increase the membrane permeability and eventually initiate apoptosis.23 Additional signaling pathways including MAPKs and TGF-β might be also triggered by GFNs.22 GFNs may trigger JNK which activates the Bc1–2 protein family proapoptotic members (Bim and Bax) followed by the activation of MOMP and caspase-3 and subsequent cell apoptosis.24 These signaling pathways may also be involved in the neurotoxicity of the GFNs observed in this study, which need further studies in the future.
3. Conclusion
In summary, this study found that surface functionalization affects the neurotoxic potential of graphene. All GFNs caused neurotoxicity even at the lowest concentration (0.1 mg L−1). The toxicity is greater than that previously reported by Lv et al. where the authors found the neurotoxicity of GO to SH-SY5Y cells (a subclone of the SK-N-SH cells) at a concentration higher than 80 mg L−1.5 This might be due to the different sizes and surface oxygen contents of the GFNs in the two studies, as well as different sensitivities of the cell lines that were used. It is interesting that G-COOH and G-OH caused more severe acute toxicity than RGO and G-NH2, while RGO and G-NH2 caused more disturbance in the cellular metabolism. Moreover, the toxicity of G-COOH, G-OH and RGO was all alleviated over longer exposure times; however, the toxicity of G-NH2 remained constant, suggesting that G-NH2 may cause longer chronic neurotoxicity than the other materials. Our results indicate that caution should be taken in developing and testing GFNs as a general enhancer for cellular growth. Physicochemical properties such as the particle size, surface functionalization and sensitivity of the specific cell lines should all be considered during the study for safe-by-design of GFNs for environmental and biological application. Moreover, longer term studies are required to evaluate the chronic effects of GFNs on cell growth.4. Methods
4.1. Materials and characterization
RGO, G-COOH, G-OH and G-NH2 nanosheets were procured from Chengdu Organic Chemicals Co. Ltd (Chengdu, China). The morphology, lateral size and thickness of the GFNs were measured by atomic force microscopy (AFM) (Veeco Dimension 3100, USA). The chemical structures were determined by XPS (ESCALAB 250Xi, Thermo Scientific, USA) equipped with a twin-crystal monochromatic X-ray source. The hydrodynamic sizes of the GFNs (10 mg L−1, suspension in deionized water) were measured using Dynamic Light Scattering (DLS) and the ζ potentials of the GFN suspensions were measured using a ZetaSizer Nano ZS system (Malvern Instruments, UK).4.2. Cell culture
SK-N-SH cells were purchased from the cell resource center of the Chinese Academy of Medical Sciences (Beijing, China). Cells (passage 3-passage 5 after defrosting) were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 100 IU mL−1 penicillin and 100 μg mL−1 streptomycin, and incubated at 37 °C in a water-saturated 5% CO2 incubator. All reagents for cell culture were obtained from Invitrogen (Carlsbad, CA, USA).4.3. Cell counting kit-8 (CCK-8) assay
For cell viability tests, cultured SK-N-SH cells in 96-well plates (1 × 104 cells per well) were treated with the four GFNs at concentrations of 0, 0.1, 1, and 10 mg L−1 for 24, 48 and 72 h. Cell viability was measured using the CCK-8 kit (Abcam, UK) according to the manufacturer's instructions.4.4. ROS content
Total reactive oxygen species (ROS) contents in the cells were measured by the DCFH-DA (2′,7′-dichlorodihydrofluorescein diacetate) staining method according to the manufacturer's instruction (Nanjing Jiancheng Ltd, China).4.5. Dye-leakage experiment
Physical damage of the cell membrane by the GFNs was evaluated via a dye-leakage experiment following the method described previously.25 Briefly, monodisperse DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) liposomes of 100 nm encapsulating carboxyfluorescein dye were prepared using extrusion methods (Avanti Polar Lipids, Inc). The liposomes were then exposed to 10 mg L−1 of the different GFNs. The physical disruption of the liposome membrane was evaluated by measuring the concentration of the carboxyfluorescein leaking out from the liposomes. Details of the experiments are presented in the ESI.†4.6. Intrinsic oxidative potential
The intrinsic oxidative potential of the GFNs was evaluated by a method described previously by Zhang et al.18 Briefly, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was hydrolyzed in 1 M NaOH for 30 min in the dark. The solution was then neutralized with phosphate buffer solution (PBS, pH 7.4). The obtained DCFH (30 μM) was then mixed with GFN suspensions (0.1, 1, 10 mg L−1) for 24 h. The fluorescence spectra and intensity were then measured at 500–600 nm on a microplate reader (TECAN Infinite 200 Pro, Switzerland) at an excitation wavelength of 490 nm.4.7. Metabolite profiling
The cells were seeded in a 6-well-plate at 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well 24 h before exposure to GFNs. Four different GFNs were employed individually at a concentration of 10 mg L−1. Seventy-two hours after treatment, the cells were washed three times with phosphate buffered saline (1 × PBS), followed by addition of 1 mL of methanol/acetonitrile/H2O (2:2:1, v/v/v). Samples were ultrasonicated for 30 min at 4 °C twice, and then kept at −20 °C for 1 h for protein precipitation. The precipitates were removed by centrifugation at 13000 rpm at 4 °C for 15 min. The supernatants were collected, lyophilized, and stored at −80 °C prior to metabolomics analysis. QC samples were prepared by mixing equal volumes of each sample. Untargeted metabolomic analysis was carried out using ultraperformance liquid chromatography quadrupole time-of-flight tandem mass spectrometry (UHPLC-QTOF-MS/MS). Details of the instrumentation and data analysis are described in the ESI.†
000 cells per well 24 h before exposure to GFNs. Four different GFNs were employed individually at a concentration of 10 mg L−1. Seventy-two hours after treatment, the cells were washed three times with phosphate buffered saline (1 × PBS), followed by addition of 1 mL of methanol/acetonitrile/H2O (2:2:1, v/v/v). Samples were ultrasonicated for 30 min at 4 °C twice, and then kept at −20 °C for 1 h for protein precipitation. The precipitates were removed by centrifugation at 13000 rpm at 4 °C for 15 min. The supernatants were collected, lyophilized, and stored at −80 °C prior to metabolomics analysis. QC samples were prepared by mixing equal volumes of each sample. Untargeted metabolomic analysis was carried out using ultraperformance liquid chromatography quadrupole time-of-flight tandem mass spectrometry (UHPLC-QTOF-MS/MS). Details of the instrumentation and data analysis are described in the ESI.†
Author contributions
Z. G., P. Z. and B. Z. designed the study. Z. G. and P. Z. performed the experiments and wrote the manuscript. I. L., A. C., H. Q. X. and E. V. J. revised the manuscript. B. Z. provided the funding support.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21836004, 91543204, and 21525730), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB14030400), and the National Key Research and Development Program of China (Grant No. 2018YFA0901103). The Marie Skłodowska-Curie Individual Fellowship (NanoBBB Grant Agreement No. 798505 to Z. Guo) under the European Union's Horizon 2020 research program was acknowledged. Technical support from the Shanghai Applied Protein Technology facility (Shanghai, China) is gratefully acknowledged.References
- C. Soldano, A. Mahmood and E. Dujardin, Carbon, 2010, 48, 2127–2150 CrossRef CAS.
- S. Song, H. Shen, Y. Wang, X. Chu, J. Xie, N. Zhou and J. Shen, Colloids Surf., B, 2020, 185, 110596 CrossRef CAS.
- O. N. Ruiz, K. S. Fernando, B. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y.-P. Sun and C. E. Bunker, ACS Nano, 2011, 5, 8100–8107 CrossRef CAS.
- E. C. Qin, M. E. Kandel, E. Liamas, T. B. Shah, C. Kim, C. D. Kaufman, Z. J. Zhang, G. Popescu, M. U. Gillette and D. E. Leckband, Acta Biomater., 2019, 90, 412–423 CrossRef CAS.
- M. Lv, Y. Zhang, L. Liang, M. Wei, W. Hu, X. Li and Q. Huang, Nanoscale, 2012, 4, 3861–3866 RSC.
- P. Li, T. Xu, S. Wu, L. Lei and D. He, J. Appl. Toxicol., 2017, 37, 1140–1150 CrossRef CAS.
- X. Hu, Z. Wei and L. Mu, Carbon, 2017, 117, 182–191 CrossRef CAS.
- Y. Li, H. Yuan, A. Von Dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef CAS.
- W. Zhang, C. Wang, Z. Li, Z. Lu, Y. Li, J. J. Yin, Y. T. Zhou, X. Gao, Y. Fang and G. Nie, Adv. Mater., 2012, 24, 5391–5397 CrossRef CAS.
- M. Zhou, X. Gong, Y. Wang, C. Liu, M. Hong, L. Wang and F. Hong, Biol. Trace Elem. Res., 2011, 142, 760–772 CrossRef CAS.
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef CAS.
- I. E. M. Carpio, C. M. Santos, X. Wei and D. F. Rodrigues, Nanoscale, 2012, 4, 4746–4756 RSC.
- H. Zhu, L. Gao, X. Jiang, R. Liu, Y. Wei, Y. Wang, Y. Zhao, Z. Chai and X. Gao, Chem. Commun., 2014, 50, 3695–3698 RSC.
- T. Palaniselvam, H. B. Aiyappa and S. Kurungot, J. Mater. Chem., 2012, 22, 23799–23805 RSC.
- Q. Tu, L. Pang, Y. Chen, Y. Zhang, R. Zhang, B. Lu and J. Wang, Analyst, 2014, 139, 105–115 RSC.
- A. Sasidharan, L. Panchakarla, P. Chandran, D. Menon, S. Nair, C. Rao and M. Koyakutty, Nanoscale, 2011, 3, 2461–2464 RSC.
- H. Pieper, S. Chercheja, S. Eigler, C. E. Halbig, M. R. Filipovic and A. Mokhir, Angew. Chem., Int. Ed., 2016, 55, 405–407 CrossRef CAS.
- Q. Zhang, X. Liu, H. Meng, S. Liu and C. Zhang, Environ. Sci.: Nano, 2018, 5, 1361–1371 RSC.
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan and H. Fang, Nat. Nanotechnol., 2013, 8, 594 CrossRef CAS.
- N. Smirnoff, Vitam. Horm., 2001, 61, 241–266 CrossRef CAS.
- M. Sinha, P. Manna and P. C. Sil, BMB Rep., 2008, 41, 657–663 CrossRef CAS.
- Y. Li, Y. Liu, Y. Fu, T. Wei, L. Le Guyader, G. Gao, R.-S. Liu, Y.-Z. Chang and C. Chen, Biomaterials, 2012, 33, 402–411 CrossRef CAS.
- A. Sasidharan, S. Swaroop, P. Chandran, S. Nair and M. Koyakutty, Nanomedicine, 2016, 12, 1347–1355 CrossRef CAS.
- S. Zhou, Y. Wang and J.-J. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 7674–7682 CrossRef CAS.
- X. Lu, X. Feng, J. R. Werber, C. Chu, I. Zucker, J.-H. Kim, C. O. Osuji and M. Elimelech, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9793–E9801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Raman spectra, size, height, hydrodynamic size and zeta potential of the GFNs, MDA content, differentially regulated metabolites, and metabolite set enrichment analysis. See DOI: 10.1039/d0nr04179c |
| This journal is © The Royal Society of Chemistry 2020 |