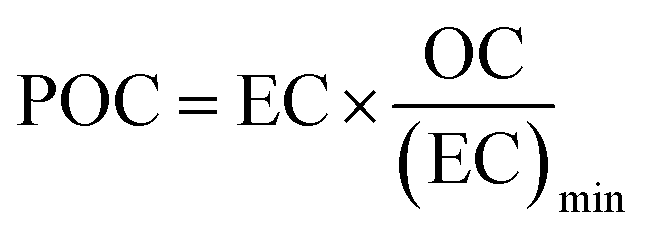Secondary organic aerosol tracers and related polar organic compounds between urban and rural areas in the Eastern Mediterranean region: source apportionment and the influence of atmospheric oxidants†
Received
27th May 2020
, Accepted 7th September 2020
First published on 8th September 2020
Abstract
Fine particle samples were collected during summer at an urban (LIM) and a rural/background (AGM) site of Cyprus. They were analyzed for pinene and isoprene secondary organic aerosol (PSOA–ISOA) tracers, linear dicarboxylic acids (DCAs), hydroxyacids (HAs), aromatic acids (AAs), monocarboxylic acids (MCAs) and levoglucosan by GC/MS with prior 3-step derivatization. DCAs, AAs, MCAs and levoglucosan exhibited significantly higher concentrations (p < 0.05) in LIM, PSOAs and ISOAs in AGM (p < 0.05), whereas mixed trends were found for HAs. Among DCAs, succinic was the most abundant in both sites, accounting for 42.5% and 36.5% of the total DCAs in LIM and AGM respectively, followed by adipic in LIM (20.1%) and azelaic in AGM (22.4%). Malic, phthalic and palmitic acids were the dominant HA, AA and MCA, respectively. Regarding PSOAs, significant differences were observed between the two sites, with the first-generation products accounting for 59.8% of the total measured PSOAs in AGM, but were remarkably lowered (10.3%) in LIM indicating that they were highly oxidized. 2-Methylerythritol was the dominant ISOA tracer in both sites; nevertheless the elevated relative abundance of 2-methylglyceric acid in LIM implies the influences of higher NOx levels. The increased O3 levels observed in AGM appear to have a significant impact on SOA formation. Source apportionment tools employed revealed factors related to secondary formation processes including oxidation of unsaturated fatty acids, pinene, isoprene and anthropogenic VOCs and factors associated with primary sources such as biomass burning, plant emissions and/or cooking and motor exhaust, with noteworthy differences observed between the two areas.
Environmental significance
Secondary organic aerosols (SOAs) are a significant component of airborne particulate matter. SOA yields consist of a great number of compounds formed by the reactions of biogenic and anthropogenic precursors with OH radicals, NO3, and O3 or via photolysis. Their formation is influenced by the type of each precursor and the ambient conditions. During the last decade, several studies have reported the impact of SOAs on global climate and human health. We present the spatial distribution of various SOA markers as well as other related polar compounds, in two Eastern Mediterranean sites, where photochemical processes are favored. The results provide useful insights into the sources and the formation pathways of SOA compounds under different atmospheric conditions.
|
1. Introduction
The organic fraction of airborne particulate matter (organic aerosol-OA) accounts for a significant portion of the total aerosol mass.1,2 OAs can be released into the atmosphere from multiple sources in two main ways; primary OAs (POAs) include the directly emitted OAs from natural (e.g. plant emissions, fungi spores, and sea spray) and anthropogenic sources (e.g. traffic and industrial emissions and biomass and waste burning).3–9 In contrast, secondary OAs (SOAs) are generated in situ in the atmosphere through multiple homogeneous or heterogeneous reactions, occurring in the gas or particle phase, of biogenic or anthropogenic volatile or semi-volatile organic compounds with O3, NOx, OH radicals, and chlorine atoms or via photolysis.10–14 SOA yields contain a great number of compounds as a result of the multivariate and diverse factors that affect SOA formation such as the type of each precursor along with the different ambient conditions, including temperature, relative humidity and the type and abundance of atmospheric oxidants (O3, NOx, etc.).11,15,16 SOAs are a remarkable component of atmospheric fine particles constituting a large to dominant (20–80%) percentage of their total organic mass;17 hence a comprehensive understanding of SOA formation mechanisms under different atmospheric conditions is essential.18,19
SOA marker compounds generally contain one or more oxygenated functional groups including hydroxyl (OH), carboxyl (HOC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and carbonyl (CO) groups, making them highly polar.11 SOAs have direct effects on climate as they can absorb and scatter solar radiation and indirect effects as they affect the cloud condensation nuclei (CCN) particles either by forming new particles or by growing the preexisting ones.16,20 SOA compounds can have adverse effects on human health. For example, short- and long-term exposure to dicarboxylic acids may lead to eye, nose and skin irritation, asthma and lung damage.21 Moreover, recent studies have associated the expression of oxidative stress response genes in human lung and bronchial epithelial cells with isoprene-derived compounds.22,23
O) and carbonyl (CO) groups, making them highly polar.11 SOAs have direct effects on climate as they can absorb and scatter solar radiation and indirect effects as they affect the cloud condensation nuclei (CCN) particles either by forming new particles or by growing the preexisting ones.16,20 SOA compounds can have adverse effects on human health. For example, short- and long-term exposure to dicarboxylic acids may lead to eye, nose and skin irritation, asthma and lung damage.21 Moreover, recent studies have associated the expression of oxidative stress response genes in human lung and bronchial epithelial cells with isoprene-derived compounds.22,23
Several organic compounds are considered as tracers for specific atmospheric reactions involving biogenic volatile organic compounds and atmospheric oxidants. For example, 2-methylerythritol, 2-methylthreitol and 2-methylglyceric acid are specific tracers of isoprene oxidation derived from both laboratory and field studies.24,25 In addition, pinic, pinonic, 3-hydroxyglutaric and 3-methylbutanetricarboxylic acids are known products from α/β pinene oxidation.26,27 In contrast, other polar organic compounds have more than one possible formation source. Particularly, dicarboxylic acids can be formed from the oxidation of various unsaturated hydrocarbons, such as cyclopentene, cyclohexene, cycloheptene, 1-methylcyclohexe, α/β-farnesene, limonene28–30 and fatty acids,31,32 although contributions from primary sources, including biomass and plastic waste burning, have been reported.33,34 Furthermore, the oxidation of aromatic volatile organic compounds and phthalates is a possible source of particle-bound aromatic carboxylic acids, such as phthalic and trimellitic acids.33,35,36 However, primary emissions, such as motor exhaust and biomass burning, are also reported to increase to aromatic acids' ambient concentrations.34,37,38
Eastern Mediterranean (EM) is a basin sensitive to climate changes,39,40 with increased levels of PM and ozone especially during summer.41,42 High ozone levels combined with intense solar radiation all-year round43 favor photochemical processes; hence causing SOA formation. For example, in Athens and Patra oxygenated organic aerosols were the dominant components of organic aerosols (65 and 78% respectively).44 In addition, more than 80% of the organic fraction of fine particles in Athens, during summer, has been attributed to secondary formation.45 In Cyprus, located on the east side of EM, significant amounts of SOA gas-phase precursors were presented in a recent study,47,48 which, combined with the high-ozone levels especially during summer,49 could lead to enhanced SOA yields. However, there are limited data about the individual SOA compounds and their levels in EM.18,46
This study focuses on the investigation of the abundance of SOA markers and other related polar organic compounds bound to fine particles, including seven biogenic SOA tracers, seven linear dicarboxylic acids, four monocarboxylic acids, four hydroxyacids, six aromatic acids and tracers of biomass burning (levoglucosan) in two Cypriot locations, representing an urban (Limassol) and a rural/background (Agia Marina Xyliatou) site during summer, while organic carbon and elemental carbon were also measured at the background area. For this purpose, a total of 88 samples were collected (n = 44 for each site) along with real time measurements of inorganic gases (O3, NOx and SO2). The results are presented and discussed regarding the spatial variation of the analytes, the potentially different sources between the two sites and the influence of atmospheric oxidants on SOA formation. As SOA research is limited in EM, this work can provide useful information about the SOA yields and sources in this region.
2. Methods
2.1 Site description & sample collection
The urban site is located at the city of Limassol (LIM) (34°40′ N, 33°2′ E; 10 m ASL) (Fig. 1), the second largest Cypriot city (235![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inhabitants, 2011 census). Samples were collected at a kerbside station with an approximate height of 4 m and at least 25 m from a major route. The rural/background site is located at the Cyprus Atmospheric Observatory (CAO) at Agia Marina Xyliatou (AGM), a remote location in the middle of the island (35.03°N, 33.05°E; 532 m ASL) (Fig. 1). CAO is 1 km south of the village of Agia Marina (population about 630) and more than 35 km away from the major Cypriot cities. The station is cooperated within the network of the European Monitoring and Evaluation Program (EMEP) by the Department of Labor Inspection (DLI). The AGM site is surrounded by extensive plant life, such as “maquis” and is also near oak and pine forests covering the Troodos mountain.50 Fine aerosol samples (n = 44 for LIM; n = 44 for AGM) were collected daily (00:00–23:59), on pre-weighted filters, with a diameter of 47 mm (Pall Tissuquartz 2500 QAT-UP), from 16 July 2018 to 28 August 2018 in AGM and to 30 August 2018 in LIM (samples on the 26th and 27th of July were lost due to power loss). Two low volume samplers (Leckel SEQ 47/50) were used to collect the PM2.5 (flow rate 2.3 m3 h−1) samples. Particle mass determination was carried out gravimetrically following the protocol of EN12341.51 After collection, each filter was stored in a Petri dish in a freezer (−21 °C). Weather stations (Campbell Scientific Europe, Antony, France) were located on the rooftop of the sampling stations and were used for the monitoring of real time meteorological parameters, such as atmospheric pressure (AP), atmospheric temperature (AT), relative humidity (RH), wind speed (WS), wind direction (WD) and solar radiation (SR) (5 min time resolution). Moreover, inorganic gases, i.e. NO, NO2, CO, SO2 and O3 (1 h time resolution) were measured in both sites for the whole sampling period. More details for the latter measurements are provided in Kleanthous et al. (2014).49 Data reported in this work regarding the meteorological parameters and inorganic gases originate from the air quality network of Cyprus operated by the Department of Labor Inspection (https://www.airquality.dli.mlsi.gov.cy/).
000 inhabitants, 2011 census). Samples were collected at a kerbside station with an approximate height of 4 m and at least 25 m from a major route. The rural/background site is located at the Cyprus Atmospheric Observatory (CAO) at Agia Marina Xyliatou (AGM), a remote location in the middle of the island (35.03°N, 33.05°E; 532 m ASL) (Fig. 1). CAO is 1 km south of the village of Agia Marina (population about 630) and more than 35 km away from the major Cypriot cities. The station is cooperated within the network of the European Monitoring and Evaluation Program (EMEP) by the Department of Labor Inspection (DLI). The AGM site is surrounded by extensive plant life, such as “maquis” and is also near oak and pine forests covering the Troodos mountain.50 Fine aerosol samples (n = 44 for LIM; n = 44 for AGM) were collected daily (00:00–23:59), on pre-weighted filters, with a diameter of 47 mm (Pall Tissuquartz 2500 QAT-UP), from 16 July 2018 to 28 August 2018 in AGM and to 30 August 2018 in LIM (samples on the 26th and 27th of July were lost due to power loss). Two low volume samplers (Leckel SEQ 47/50) were used to collect the PM2.5 (flow rate 2.3 m3 h−1) samples. Particle mass determination was carried out gravimetrically following the protocol of EN12341.51 After collection, each filter was stored in a Petri dish in a freezer (−21 °C). Weather stations (Campbell Scientific Europe, Antony, France) were located on the rooftop of the sampling stations and were used for the monitoring of real time meteorological parameters, such as atmospheric pressure (AP), atmospheric temperature (AT), relative humidity (RH), wind speed (WS), wind direction (WD) and solar radiation (SR) (5 min time resolution). Moreover, inorganic gases, i.e. NO, NO2, CO, SO2 and O3 (1 h time resolution) were measured in both sites for the whole sampling period. More details for the latter measurements are provided in Kleanthous et al. (2014).49 Data reported in this work regarding the meteorological parameters and inorganic gases originate from the air quality network of Cyprus operated by the Department of Labor Inspection (https://www.airquality.dli.mlsi.gov.cy/).
 |
| | Fig. 1 Eastern Mediterranean and Cyprus maps. Sampling sites are displayed. | |
2.2 Measurement of organic carbon and elemental carbon
Organic (OC) and elemental carbon (EC) were measured only for the samples collected at AGM. A 1 cm × 1.5 cm standard punch (1.5 cm2) of the aerosol filter samples and the field blanks was cut and analyzed for organic carbon (OC) and elemental carbon using an OC/EC Lab Instrument (Model 5, Sunset Laboratory Inc., USA), following the EUSAAR 2 temperature program. More details of the analysis can be found elsewhere.52 Secondary organic carbon (SOC) can't be measured directly, so it was measured using the method described by Castro et al. (1999).53 In particular, the minimum OC/EC ratio, from the measured OC and EC concentrations, was used to calculate primary organic carbon (POC) and SOC by applying the following equations eqn (1) and (2).| | 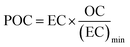 | (1) |
This method for SOC estimation has already been used in many rural/background and urban locations.19,54,55
2.3 Extraction, derivatization and GC-EI-MS determination
Details about filter pretreatment and the 3-step derivatization procedures are discussed in depth elsewhere.56 The rest of the filters collected at AGM, and the whole filters, collected at LIM, were subjected to a 30 mL extraction of (1:1) dichloromethane/methanol (Carlo Erba) in an ultrasonic bath for 20 min. Prior to the extraction, 5 μL of ketopinic acid (KPA) (Sigma Aldrich, 5 μg mL−1, in MeOH) was added as a surrogate standard. The extraction procedure was performed in triplicate, and then the combined extracts were filtered through a Pasteur pipette and concentrated at nearly 4 mL with a rotary evaporator (Buchi). Further condensation was performed with a gentle steam of nitrogen until dryness. The first step of the derivatization procedure starts with the addition of 60 μL of heated O-methylhydroxylamine hydrochloride (MHA) solution (Sigma Aldrich, 1 mg mL−1, in ACN). The reaction of carbonyl compounds with MHA took place for 60 minutes at 70 °C in a water bath. The second step includes the methylation of carboxylic acids to their methyl ester derivatives. This was achieved by adding 12 μL of (trimethylsilyl)diazomethane (TMSD) (Acros Organics) and 8 μL of methanol. Then, the vials were left in an ultrasonic bath for 20 minutes. Following this, 245 μL of N,O-bis(trimethylsilyl)-trifluoroacetamide (BSTFA) with 1% trimethylchlorosilane (TMCS) (TCI Chemicals) and 50 μL of pyridine were added in order to convert the hydroxyl groups of the species to trimethylsilyl ethers. The reaction was carried out in an oven for 60 minutes at 70 °C. After cooling to room temperature, 50 μL of the internal standard, tetracosane d-50 (Sigma Aldrich, 30 μg mL−1, in DCM), was added to each sample. A GC/MS system (6890N/5975B, Agilent Technologies, USA) was employed for both analyses. The GC instrument was equipped with a split/splitless injector and a HP-5ms [5%-(phenyl)-methylpolysiloxane] capillary column. High purity helium was the carrier gas with a velocity of 1.5 mL min−1. Pulsed splitless mode was used for the injection and the injector's temperature was set at 280 °C. The GC oven temperature program was: 84 °C (hold for 1 min) to 200 °C at 4 °C min−1, hold for 2 min, and then to 300 °C at 10 °C min−1 with final isothermal hold for 15 min. Inlet and MS source temperatures were 280 °C and 230 °C respectively. Selected ion monitoring (SIM) mode was used for the quantification of the analytes.
Most of the studied compounds were identified and quantified using available authentic standards. In particular, dicarboxylic acids including succinic (diC4), glutaric (diC5), adipic (diC6), pimelic (diC7), suberic (diC8), azelaic (diC9) and sebacic acid (diC10), monocarboxylic acids including palmitic (C16), margaric (C17), stearic (C18), and oleic (C18:1), and aromatic acids including benzoic (BEN), p-toluic (TOL), phthalic (PH), isophthalic (iPH), terephthalic (tPH) and trimellitic (TRIM) have been purchased from Sigma Aldrich. Pinene SOA tracers include pinonic acid (PNA), 3-hydroxyglutaric acid (3HGA) (Sigma Aldrich) and pinic acid (PA) (Chiron). The isoprene SOA tracer of 2-methylerythritol (MTL1) was purchased from Sigma Aldrich. Hydroxycarboxylic acids including malic (MAL), citric (CIT) and tartaric (TART) acids were obtained from TCI Chemicals and Sigma Aldrich. Finally, levoglucosan (LEV) was purchased from Sigma Aldrich. Apart from the compounds mentioned above, another five compounds, for which authentic standards were unavailable, were identified using MS data from the literature and with the help of the NIST library.57 Quantification was performed using the GC/MS response factors of the available standards. For example, 2-methylthreitol (MTL2), 2-methylglyceric acid (2MGA), 3-methyl-1,2,3-butanetricarboxylic acid (3MBTCA), and 2-hydroxyglutaric acid (2HGA) were quantified using the response factors of 2-methylerythritol, malic acid, suberic acid and 3-hydroxyglutaric acid, respectively. Detection limits ranged from 0.16 ng m−3 (suberic acid) to 0.62 ng m−3 (palmitic acid) and recoveries varied from 68% (pinonic acid) to 127% (stearic acid).
In Tables S1 and S2† the retention time and the ionic fragments used for the studied compounds are summarized. A total of six blank filters (three for each sampling site) were analyzed as real samples using the described procedure. If necessary, data were corrected appropriately for the field blanks but not for recoveries.
2.4 Statistical analysis
The Mann–Whitney test and principal component analysis (PCA) were applied to study the possible differences between sampling sites and for source apportionment purposes, respectively, using SPSS Software (IBM SPSS statistics, version 24). A value of p < 0.05 (95% confidence level) was considered to indicate a significant difference.
3. Results and discussion
In Table 1, the mean concentrations and concentration ranges of each individual analyte and inorganic gases are presented, as well as meteorological data values for both sampling sites. The measured OC and EC concentrations and the calculated SOC, regarding only the samples collected at AGM, are also shown in Table 1.
Table 1 Mean concentrations and concentration ranges of PM, DCAs, AAs, HAs, PSOAs, ISOAs, MCAs, LEV, inorganic gases, mean values and value ranges of meteorological data in both sampling locations and mean concentrations and concentration ranges of OC, EC, TC and SOC in AGM
| Species |
LIM (urban) (n = 44) |
AGM (background) (n = 44) |
p Value |
| Range |
Mean ± SD |
Range |
Mean ± SD |
|
Not detected.
Sum of PSOAs and ISOAs.
|
| CPM2.5 (μg m−3) |
10.2–21.1 |
15.4 ± 2.6 |
4.5–21.9 |
13.8 ± 4.2 |
0.111 |
|
|
DCAs (ng m
−3
)
|
| diC4 |
6.1–88.2 |
31.9 ± 21.0 |
4.6–51.5 |
18.1 ± 11.5 |
0.001 |
| diC5 |
3.0–30.5 |
6.8 ± 5.5 |
0.49–15.4 |
4.9 ± 3.7 |
0.102 |
| diC6 |
3.1–56.1 |
15.1 ± 11.9 |
0.38–18.6 |
6.8 ± 4.6 |
0.000 |
| diC7 |
0.69–16.1 |
3.1 ± 2.9 |
0.30–7.1 |
2.2 ± 1.3 |
0.253 |
| diC8 |
3.3–10.1 |
5.0 ± 1.7 |
0.60–9.9 |
4.1 ± 2.5 |
0.048 |
| diC9 |
6.2–41.6 |
12.9 ± 6.9 |
3.0–28.1 |
11.1 ± 5.5 |
0.841 |
| diC10 |
0.64–9.7 |
2.2 ± 1.6 |
Nda-8.1 |
2.3 ± 2.0 |
0.783 |
| ΣDCA |
31.7–252 |
76.9 ± 45.7 |
14.0–111 |
49.6 ± 23.6 |
0.001 |
|
|
AAs (ng m
−3
)
|
| BEN |
Nd-6.2 |
1.7 ± 1.6 |
Nd-2.9 |
0.96 ± 0.65 |
0.000 |
| TOL |
Nd-1.7 |
0.64 ± 0.35 |
Nd |
Nd |
— |
| PH |
3.7–52.0 |
17.1 ± 11.7 |
1.8–19.0 |
5.9 ± 3.6 |
0.000 |
| tPH |
1.6–6.9 |
2.8 ± 1.2 |
Nd-3.7 |
1.5 ± 0.95 |
0.000 |
| iPH |
0.78–3.2 |
1.5 ± 0.65 |
Nd-1.5 |
0.63 ± 0.40 |
0.000 |
| TRIM |
1.7–24.7 |
4.2 ± 3.8 |
Nd-4.2 |
1.2 ± 0.82 |
0.000 |
| ΣAA |
9.3–87.5 |
28.0 ± 18.4 |
2.8–24.3 |
10.3 ± 5.1 |
0.000 |
|
|
HAs (ng m
−3
)
|
| MAL |
2.6–58.7 |
22.3 ± 16.8 |
1.1–27.4 |
11.0 ± 6.4 |
0.009 |
| 2-HGA |
1.4–13.8 |
4.8 ± 3.3 |
Nd-14.9 |
4.6 ± 2.8 |
0.894 |
| TAR |
Nd-6.8 |
2.1 ± 2.1 |
1.2–12.3 |
3.9 ± 2.1 |
0.000 |
| CIT |
1.9–9.6 |
2.9 ± 1.4 |
0.39–4.8 |
2.3 ± 1.3 |
0.010 |
| ΣHCA |
7.1–79.0 |
32.1 ± 21.2 |
5.6–46.3 |
21.8 ± 9.4 |
0.102 |
|
|
PSOA tracers (ng m
−3
)
|
| PA |
Nd-4.7 |
0.72 ± 0.98 |
1.8–27.2 |
12.3 ± 5.8 |
0.000 |
| PNA |
0.32–2.5 |
1.1 ± 0.45 |
2.4–12.9 |
5.6 ± 2.3 |
0.000 |
| 3HGA |
1.3–16.8 |
6.0 ± 4.1 |
1.6–20.0 |
8.6 ± 4.3 |
0.002 |
| 3MBTCA |
1.9–31.2 |
10.2 ± 8.1 |
0.38–8.9 |
3.4 ± 2.1 |
0.000 |
| ΣPSOA |
3.9–49.2 |
18.0 ± 12.7 |
8.8–67.4 |
29.9 ± 13.3 |
0.000 |
|
|
ISOA tracers (ng m
−3
)
|
| MTL2 |
1.8–10.8 |
4.0 ± 2.0 |
4.3–20.8 |
7.3 ± 3.1 |
0.000 |
| MTL1 |
5.8–31.1 |
11.4 ± 5.4 |
7.8–57.5 |
17.9 ± 9.8 |
0.000 |
| 2MGA |
0.75–7.9 |
3.2 ± 2.4 |
0.53–3.9 |
0.93 ± 0.54 |
0.000 |
| ΣISOA |
9.1–49.9 |
18.6 ± 8.6 |
12.8–82.2 |
26.2 ± 13.2 |
0.000 |
| ΣBSOAb |
13.7–85.5 |
36.6 ± 17.3 |
27.2–95.9 |
56.1 ± 18.9 |
0.000 |
|
|
MCAs (ng m
−3
)
|
| C16 |
10.7–161 |
56.1 ± 34.6 |
6.1–77.5 |
32.7 ± 16.0 |
0.001 |
| C17 |
1.1–21.3 |
4.6 ± 4.2 |
Nd-8.8 |
2.7 ± 1.7 |
0.039 |
| C18 |
3.9–130 |
40.8 ± 28.9 |
2.7–41.2 |
17.1 ± 8.0 |
0.000 |
| C18:1 |
1.1–10.9 |
5.5 ± 2.5 |
0.82–3.2 |
1.6 ± 0.61 |
0.000 |
| ΣMCAs |
18.7–315 |
107 ± 67.0 |
9.8–126 |
54.1 ± 25.1 |
0.000 |
|
|
Biomass burning tracer (ng m
−3
)
|
| LEV |
0.65–21.1 |
3.3 ± 3.5 |
Nd-8.6 |
1.3 ± 1.6 |
0.000 |
|
|
Meteorological parameters, inorganic gases OC and EC
|
| T(oC) |
26.7–30.6 |
27.9 ± 1.0 |
23.3–30.5 |
26.3 ± 1.9 |
0.000 |
| RH (%) |
39.5–64.5 |
53.1 ± 7.0 |
29.8–76.0 |
55.2 ± 14.1 |
0.226 |
| O3 (μg m−3) |
40.9–70.6 |
53.8 ± 7.9 |
76.0–118 |
98.6 ± 10.3 |
0.000 |
| NO (μg m−3) |
1.6–13.0 |
4.4 ± 2.1 |
0.04–0.21 |
0.13 ± 0.04 |
0.000 |
| NO2 (μg m−3) |
9.9–48.4 |
23.9 ± 9.7 |
0.53–1.7 |
1.0 ± 0.23 |
0.000 |
| NOx (μg m−3) |
12.3–68.3 |
31.0 ± 11.7 |
0.63–1.9 |
1.2 ± 0.39 |
0.000 |
| SO2 (μg m−3) |
0.43–2.66 |
1.5 ± 0.61 |
0.35–2.2 |
0.91 ± 0.39 |
0.000 |
| OC (μg m−3) |
|
|
0.6–3.1 |
1.9 ± 0.45 |
|
| EC (μg m−3) |
|
|
0.08–0.46 |
0.19 ± 0.08 |
|
| TC (μg m−3) |
|
|
0.67–3.4 |
2.1 ± 0.50 |
|
| OC/EC |
|
|
5.5–18.8 |
11.0 ± 3.3 |
|
| SOC |
|
|
0.06–2.0 |
0.91 ± 0.41 |
|
| SOC/OC (%) |
|
|
2.9–70.9 |
46.8 ± 15.3 |
|
3.1 PM, OC, EC and SOC
Comparable levels of PM2.5 were observed at both sites, with those of LIM being slightly higher (15.4 ± 2.6 versus 13.8 ± 4.2 μg m−3) (Table 1). In general, in AGM, PM2.5 levels reach their maximum average concentrations during summer (July–August) as a result of increased SOA formation, limited precipitation and the Etesian winds which transport rich in fine particle air masses from Turkey.58 PM2.5 levels in LIM are also comparable with those measured at the same site during 2012–2013 (13.1 ± 4.7 μg m−3).59 Similar and higher concentrations (>20 μg m−3) have been reported in Akrotiri and Finokalia respectively, two sites of Crete, in Greece, during the summer months.60,61 In contrast, quite higher concentrations have been observed in three urban sites in Lebanon (from 27.6 to 40.9 μg m−3).62
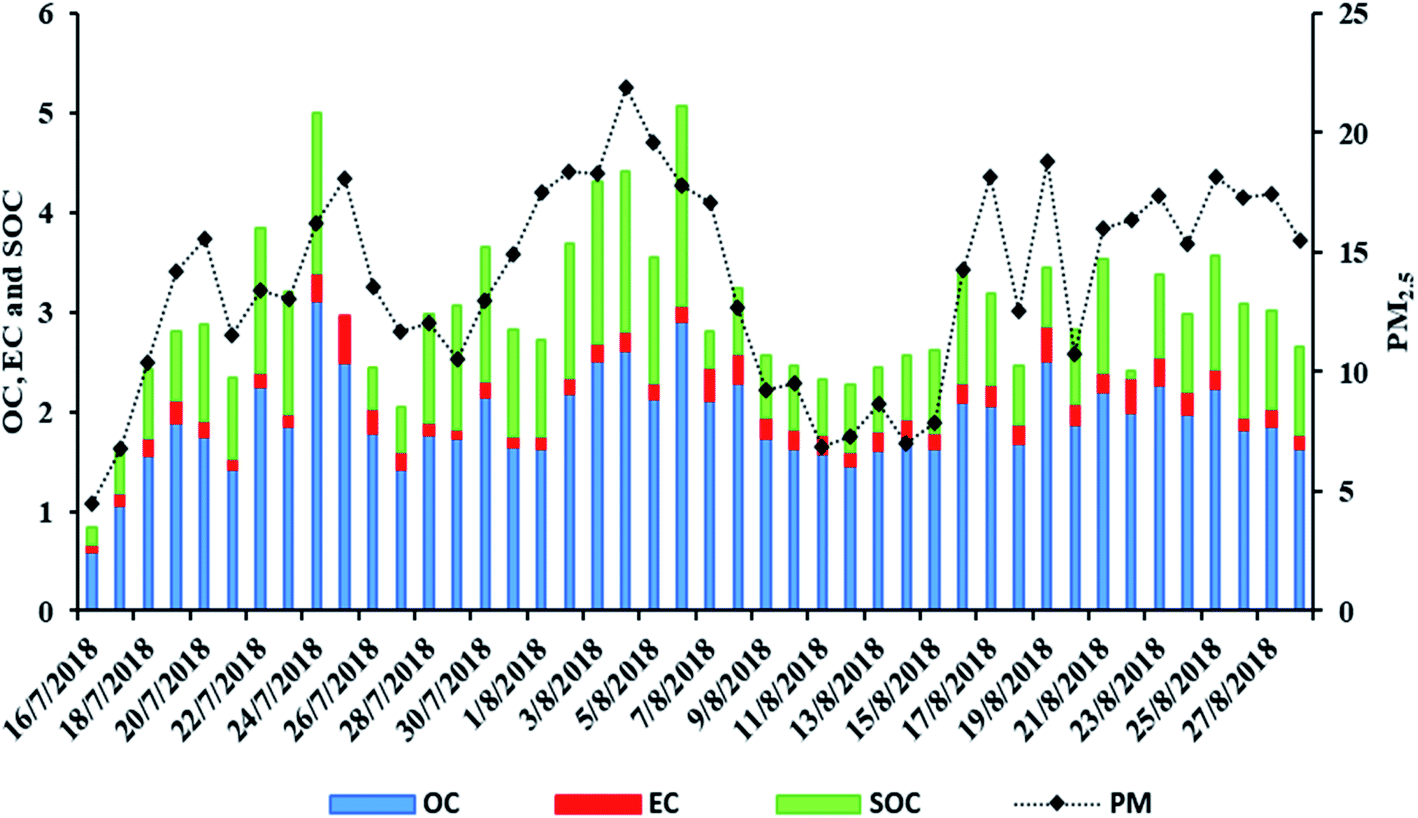
In AGM OC presented a mean concentration of 1.9 ± 0.45 μg m−3 which is quite higher than that measured in Azores, Portugal (0.33 ± 0.26) and Sonnblick, Austria (0.90 ± 1.0),63 but lower than 2.85 ± 1.63 μg m−3 measured in an urban-background site in Patra, Greece,64 and also lower than 3.5 ± 2.8 μg m−3 in a rural site in Brindisi, Italy55 and than 3.8 ± 3.7 μg m−3 found in Gongga Mountain, China.19 The average OC/EC ratio, in AGM, was 11.0 ± 3.3 which is nearly ten times higher than the ratio calculated in Thessaloniki (1.9 ± 1.4), an urban area of Eastern Mediterranean,65 and also higher than that at two urban-background Greek sites in Thessaloniki and Patra (8.1 and 7.5, respectively)64,65 but lower than those reported in Azores (13.5 ± 17.4).63 In general, the OC/EC ratio can be used to characterize the relative contribution of secondary versus primary sources, with values > 2 being indicative of significant SOA contributions.66,67 As a result, it appears that aerosols in AGM are significantly influenced by secondary sources. This can be further supported by the calculated SOC which contributes to a significant portion of the total OC (46.8 ± 15.3%). Particularly, SOC mean concentration was 0.91 ± 0.41 μg m−3 which, compared with SOC from other studies, calculated with the same method, is lower than that in Toulouse (3.04 μg m−3 and 65.85% of OC, during summer),68 and than 2.9 ± 3.4 μg m−3 in Gongga Mountain, China,19 while being at the same levels as in Brindisi, Italy (1.9 ± 2.2 μg m−3).55 The daily variations of PM, OC, EC and SOC are presented in Fig. 2.
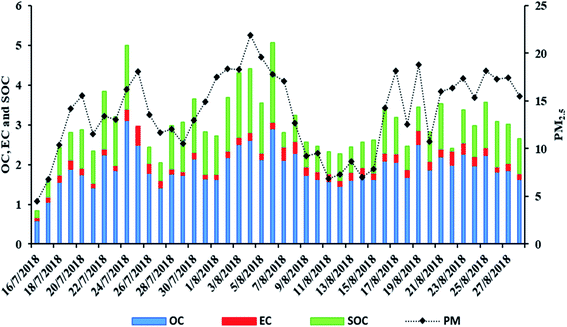 |
| | Fig. 2 Daily variations of PM2.5, OC, EC and SOC (μg m−3) of samples collected at AGM. | |
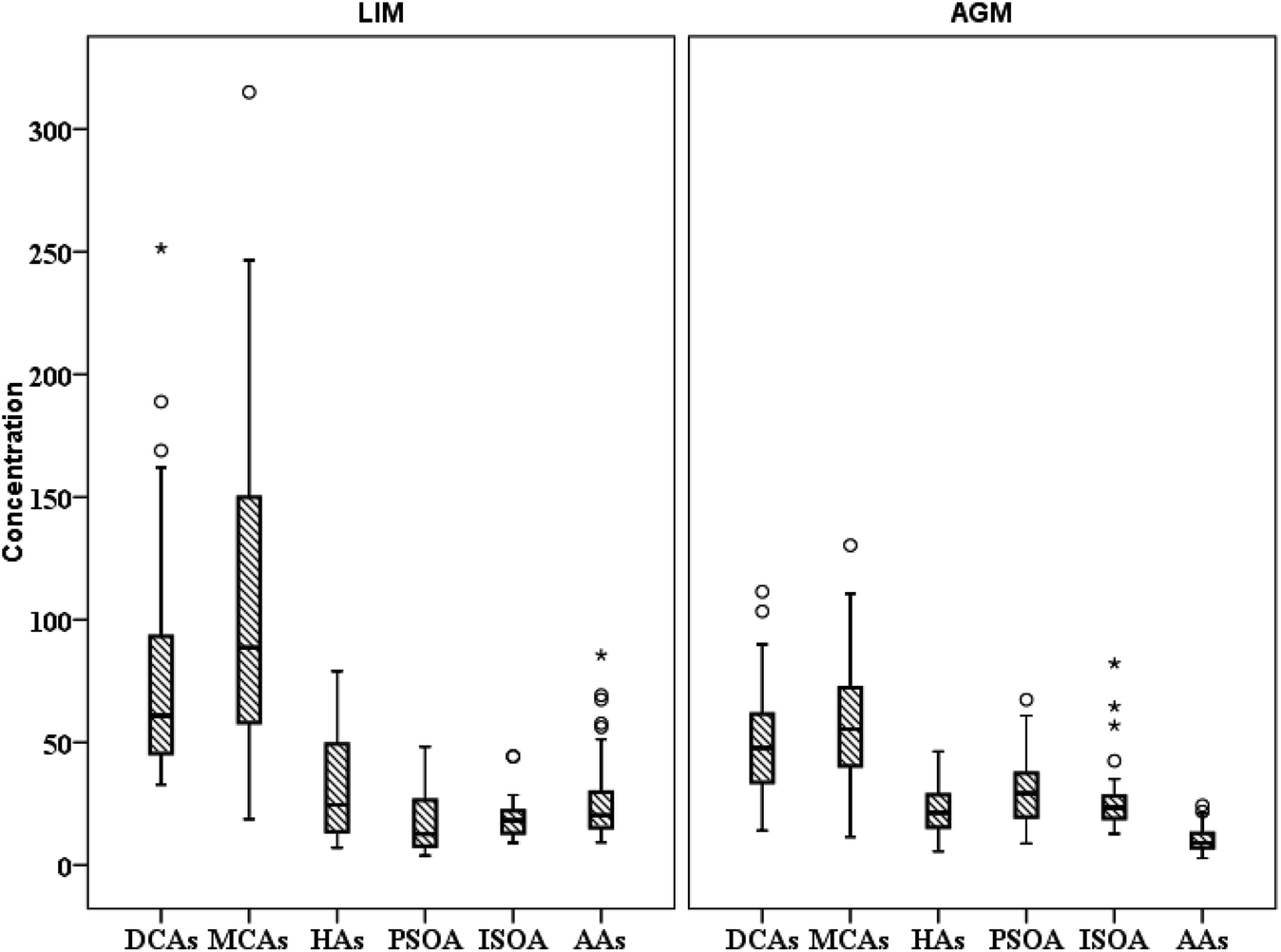
3.2 Dicarboxylic acids (DCAs)
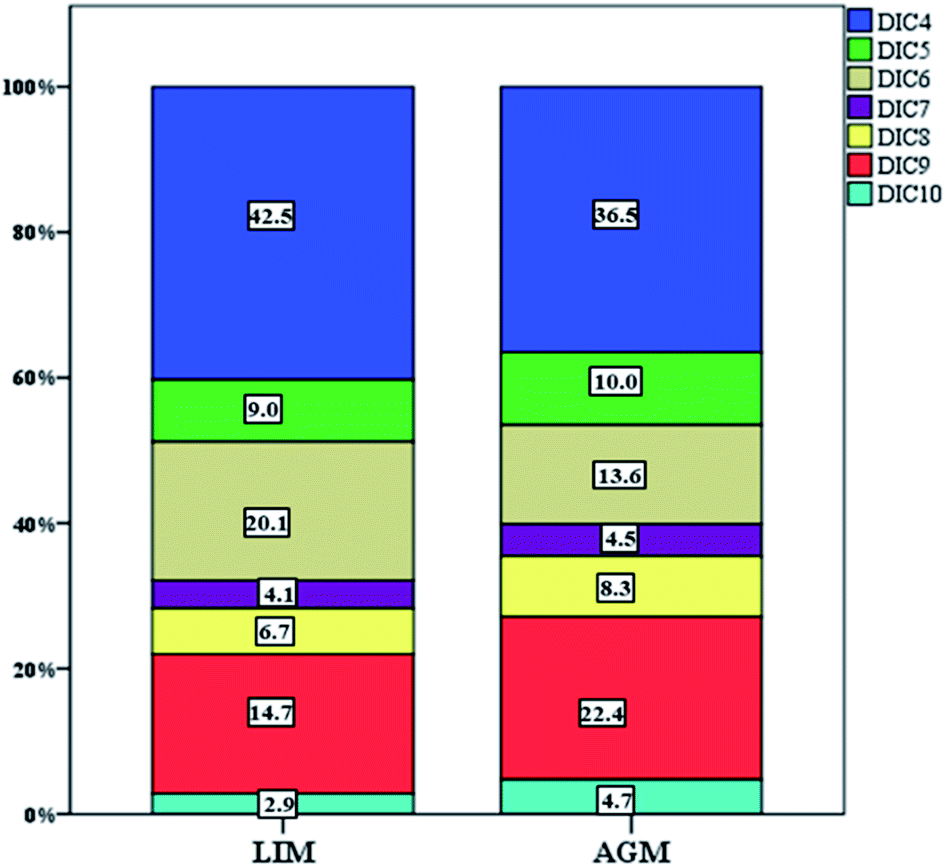
DCAs were abundant in both locations with LIM presenting significantly higher mean concentrations, 76.9 ± 45.7 ng m−3versus 49.6 ± 23.6 ng m−3 (Table 1, Fig. 3) measured in AGM. Considering that DCAs are formed by the oxidation of various precursors,69 it is possible that in urban atmospheres, where VOCs are more abundant, the concentrations of DCAs are elevated. This is in agreement with Zhao et al. (2014)70 and Guo et al. (2015),71 where DCA values were higher in urban sites than those in rural. In general, it has been reported that DCAs in urban aerosols are mostly secondarily produced.72 Although succinic acid was the dominant diacid in both sites, adipic acid presented the most intense difference as it was found over two times higher in LIM (15.1 ± 11.9 over 6.8 ± 4.6 ng m−3). Significantly higher concentrations were also observed for succinic, adipic and suberic acids. Glutaric and azelaic acids were found at higher, but not significantly, (p > 0.05) concentrations, whereas only sebacic acid was found at higher levels in AGM although the difference was also not statistically significant. The DCA profile of the two sites is displayed in Fig. 4. As is obvious, low molecular weight diacids (diC4–diC6) contribute almost 71.6% of the total measured diacids in LIM, whereas in AGM the percentage is reduced to 60%. The increased relative abundance of higher molecular weight DCAs in AGM can be explained by the elevated influence of biogenic sources.73 It is known that higher molecular weight diacids are oxidation products of biogenic unsaturated fatty acids,74,75 which are emitted from vegetation and the sea surface micro-layer as a result of phytoplankton activity,76 although anthropogenic primary sources have been reported, including automobile engine exhaust and cooking.77,78 A similar relative abundance was observed in six sites in the Pearl River Delta region of China, where in each site succinic acid was the most abundant DCA followed by azelaic acid,70 and in 2 sites in Oporto, Portugal.79 However, in other studies conducted at urban and background locations of China and Sweden, respectively, azelaic acid was the most abundant.73,80 In order to differentiate the relative contribution of anthropogenic versus biogenic sources, the ratios of diC6/diC9 and PH/diC9 can be used.81 Lower values indicate influence from biogenic sources, whereas higher values show the impact of anthropogenic sources. The mean ratio of diC6/diC9 for LIM (1.2 ± 0.79) was almost two times higher than that calculated for AGM (0.67 ± 0.42), while the mean ratios of PH/diC9 were respectively equal to 1.4 ± 0.94 and 0.57 ± 0.28 for LIM and AGM, revealing the stronger impact of anthropogenic sources in the urban site. However, in a study conducted at two sites in the Po Valley, Italy, during the deep-winter of 2013, slightly higher values of the mentioned ratios have been found for the rural/background site than those for the urban, indicating the impact of anthropogenic sources.82 Moreover, in the Gosan site of Jeju Island, South Korea, the increase of the diC6/diC9 and Ph/diC9 ratios during wintertime was attributed to the greater amounts of transported air pollutants from East Asia.83 A comparison with ratios from other studies around the world is presented in Table S3.†
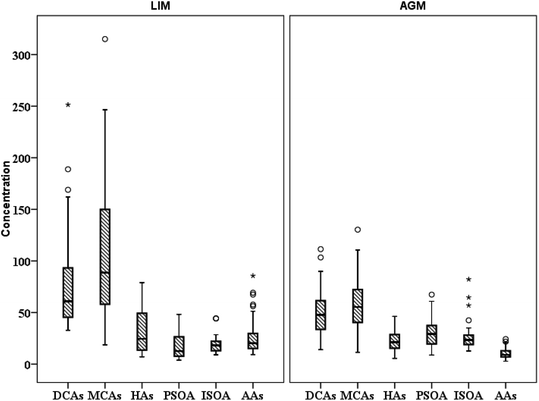 |
| | Fig. 3 Spatial distribution of the sum of studied polar organic compound groups (ng m−3) (○ refers to values above the 3rd quartile and * to outliers). | |
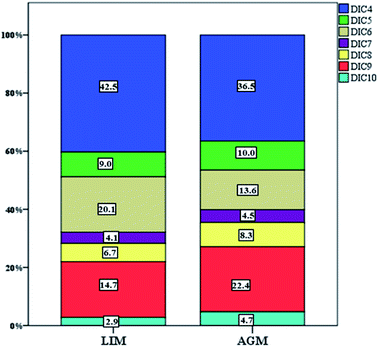 |
| | Fig. 4 Relative abundance of each individual DCA at both sampling sites. | |
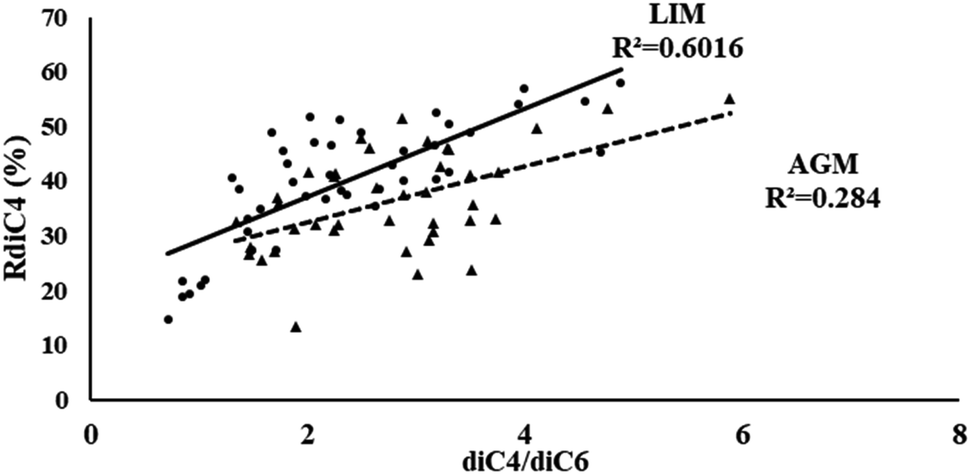
Another possible source of low molecular weight DCAs is the oxidation of their higher homologues. According to Yang et al. (2008),32 the photo-oxidation of azelaic acid can lead to the formation of lower homologues, including succinic and glutaric acids, as a result of the favorable cleavage of its C4–C5 bonds. Thus, such a relationship can be implied by the correlation of the diC4/diC6 ratio with the relative abundance of succinic acid (RdiC4%).84 As is obvious from Fig. 5, in LIM the stronger correlations (R2 = 0.60) between the aforementioned parameters could indicate another possible source of succinic acid, but in AGM such a relationship is much weaker (R2 = 0.28).
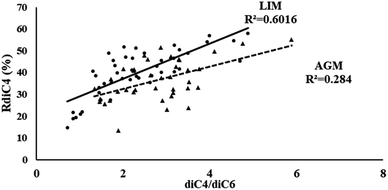 |
| | Fig. 5 Scatter plot of the diC4/diC6 ratio and the diC4 relative abundance (RdiC4%) in LIM (circle) and AGM (triangle). | |
3.3 Aromatic acids (AAs) and levoglucosan (LEV)
Six aromatic acids were studied in both sites including compounds with one (BEN and TOL), two (PH, iPH, and tPH) and three (TRIM) carboxyl groups in their molecules. The sum of aromatic acids presented almost three-fold higher mean concentrations in LIM (28.0 ± 18.4 ng m−3) than in AGM (10.3 ± 5.1 ng m−3) (p < 0.05) (Table 1, Fig. 3). PH was the dominant AA in both sites, although it was found almost three times higher in LIM (mean of 17.1 ± 11.7 versus 5.9 ± 3.6 ng m−3, p < 0.05). In other urban sites, PH was found at a considerably higher concentration such as in Thessaloniki (74 ± 55 ng m−3), with an average summer value of over 30 ng m−3.18 In Guangzhou and Hok Tsui, two sites of the Pearl River Delta region with severe air pollution, PH was measured to be respectively equal to 215 ± 86.1 and 130 ± 75.0 ng m−3 during summer.78 However, lower values have been measured on Jeju Island (4.9 ± 2.9 ng m−3, during summer)83 and in Vavihill, Sweden (2.4 ± 1.4 ng m−3).73 TRIM presented the biggest difference in concentration values between the two studied areas, as it was nearly 4 times higher in LIM than in AGM (4.2 ± 3.8 versus 1.2 ± 0.82 ng m−3, respectively). Higher TRIM levels have been reported in six Chinese sites with average concentrations ranging from 5.6 ± 2.5 to 9.9 ± 4.9 ng m−3.70 Quite lower concentrations have been observed in both areas for monocarboxylic aromatic acids (BEN and TOL) and especially in AGM, TOL was detected in lower than 10% of the samples. This can be explained by their higher volatilities favoring their presence in the gas phase,85,86 which can be further enhanced by the increased ambient temperature during the sampling period. However, significantly elevated concentrations of benzoic acid (>100 ng m−3) have been measured in PM2.5 samples at four different sites in the Pearl River Delta region.78 AAs can be emitted or formed in the atmosphere from a variety of primary and secondary sources.36,70,75,86,87 In our correlation analysis we have also included the biomass burning tracer of LEV. LEV is a definite tracer of biomass burning,88 which is often correlated with aromatic acids. For example, in Thessaloniki PH was strongly correlated with biomass burning,18 whereas, in Iowa, the secondary formation of PH was indicated as it was correlated with the toluene-SOA tracer, and tPH was associated mostly with biomass burning and iPH appeared to originate from mixed sources.89 Despite the fact that LEV was found at low levels, its presence confirms the contribution of biomass burning to atmospheric aerosols in Eastern Mediterranean either from regional wildfires or long range transport.90 In this study, the correlation analysis for LIM exported two groups of compounds that were strongly correlated (Table S4†). The first group includes PH, BEN, and TRIM (r = 0.72–0.95, p < 0.05) and is associated with secondary sources, while the second group includes LEV, tPH and TOL (r = 0.69–0.72, p < 0.05), which can be attributed to biomass burning. iPH presented moderate but significant correlations with PH (r = 0.58, p < 0.05) and BEN (r = 0.54, p < 0.05) suggesting contributions from secondary sources. Similar correlations were also observed in AGM (Table S5†) regarding BEN, PH and TRIM (r = 0.56–0.84, p < 0.05). However, iPH was strongly correlated with tPHA (r = 0.84, p < 0.05) indicating emissions from primary sources, which can be further supported by their significant correlations with LEV (r = 0.51 and 0.70, respectively, p < 0.05). Analogous correlations have been observed in a study conducted at Chinese urban and suburban sites, where tricarboxylic aromatic acids and PH were associated with secondary sources, whereas iPH and tPH with primary.36
3.4 Biogenic secondary organic aerosol (BSOA) tracers and hydroxyacids (HAs)
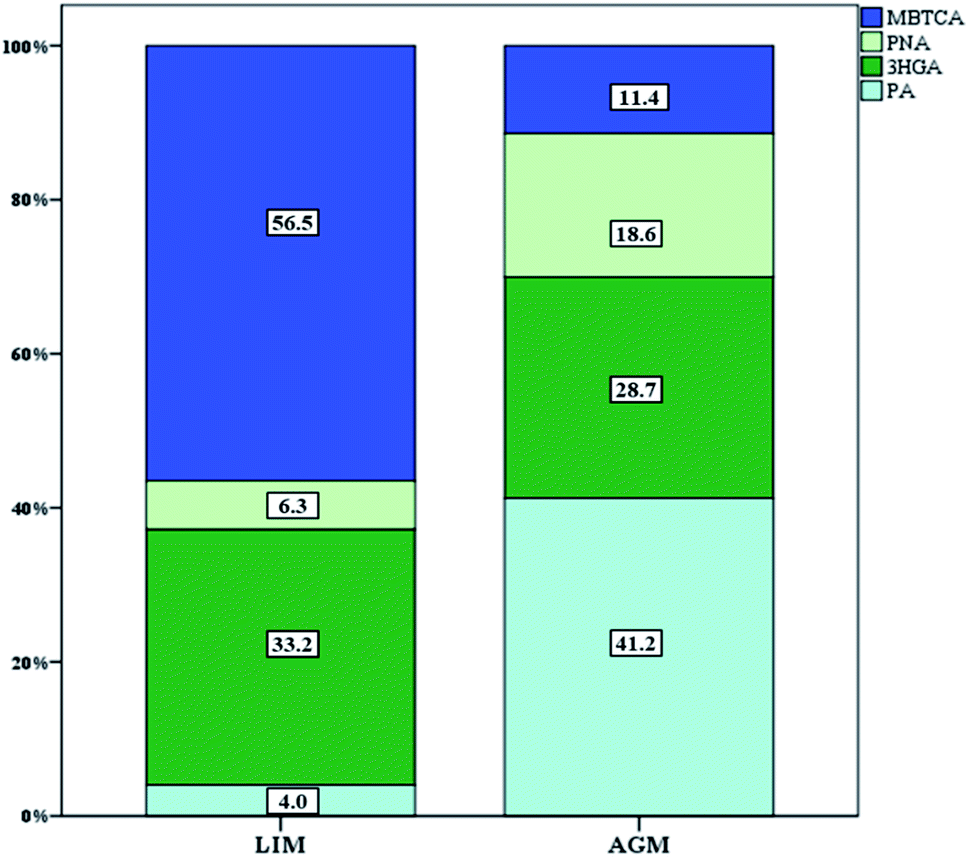
Seven BSOA tracer species were observed at both sites including four pinene oxidation products (PA, PNA, 3HGA and MBTCA) and three isoprene oxidation products (MTL1, MTL2 and 2MGA). Significantly higher BSOA concentrations were found in AGM, with a mean of 56.1 ± 18.9 ng m−3 compared with LIM, where BSOA average concentrations ranged from 13.7–85.5 ng m−3, with a mean of 36.6 ± 17.3 ng m−3 (Table 1, Fig. 3). This difference is not surprising as there is widespread vegetation enclosing the background site of AGM (maquis, oak and pine forests). In AGM PSOA tracers exhibited slightly higher concentrations than ISOAs (29.9 ± 13.3 versus 26.2 ± 13.2 ng m−3) which is in agreement with Debevec et al. (2018),48 who found, in the same site during June–July 2015, α/β-pinene levels higher than those of isoprene. In LIM, similar concentrations between ISOAs, ranging from 9.1–49.9 ng m−3 with a mean of 18.6 ± 8.6, and PSOAs, from 3.9 to 49.2 ng m−3 with an average of 18.0 ± 12.7, have been observed. A comparison with BSOA data from other studies is presented in Table 2. Among PSOA tracers in AGM, PA was the most abundant (12.3 ± 5.8 ng m−3), followed by 3HGA and PNA while MBTCA presented the lowest concentrations (3.4 ± 2.1 ng m−3). PA and PNA are first generation oxidation products of pinenes, via O3 or OH radicals, which can be further oxidized to 3HGA and MBTCA in the presence of NOx.91–93 Thus, the elevated relative abundance of PA and PNA (41.2 and 18.6%, respectively) (Fig. 6) can be explained by the increased O3 along with the low NOx levels favoring the formation of these products. However, PNA ambient concentrations are expected to be higher, as due to its volatility, it can be found in higher diameter particles94 or in the gas phase.95 In LIM, the PSOA profile was different as the most abundant tracer was MBTCA (10.2 ± 8.1 ng m−3 and 56.5% of the total PSOAs) followed by 3HGA (33.2%). PA and PNA contributed only 10.3% with PA being the least abundant (4.0%). It appears that the increased NOx levels have a significant impact on PSOA formation, as higher NO2 mixing ratios lead to a remarkable reduction in PA levels and to a moderate decrease of PNA;96 hence the formation of second generation PSOA products may be prevailed.19 This can be further supported by the ratios of MBTCA to PA plus PNA which are indicative of the aging degree of PSOAs.97 In AGM the latter ratio presented mean values of 0.18 ± 0.07 which are quite lower than those observed in LIM 5.6 ± 3.6 implying that SOAs in AGM are freshly produced, whereas in LIM they are highly oxidized. Furthermore, the 3HGA/MBTCA ratio can be applied to discriminate the contribution of the individual monoterpenes as α-pinene SOA yields have been reported to be significantly more enriched in MBCTA relative to 3HGA compared with those of β-pinene or δ-limonene.97–99 Our results suggest that β-pinene is a more important contributor in AGM (ratio of 0.67 ± 0.28) and α-pinene in LIM (3.2 ± 1.9), which is constant with the higher concentrations of β-pinene than α-pinene that were observed in AGM in a previous study.48
Table 2 Concentration ranges and mean concentrations of BSOA tracers in comparison with other studies. Each study refers to PM2.5 samples
| Species |
Location, time |
Concentration ng m−3 |
Reference |
| Mean ± SD |
| ΣPSOA |
AGM/LIM Cyprus, July–August 2017 |
29.9 ± 13.3/18.0 ± 12.7 |
This study |
| Mt Tai Mo Shan, Hong Kong, September–November 2010 |
26.3 ± 4.5 |
105
|
| Gongga Mountain, China, August 2011 |
3.6 ± 5.7 |
19
|
| Mt Wuyi, China, November 2015–July 2016 |
30.66 ± 24.44 |
97
|
| Marseille, France, July 2008 |
28.1 |
104
|
| Fairbanks, Alaska, June–September 2009 |
9.2 ± 8.7 |
106
|
| Beirut, Lebanon, winter 2012 |
19.3 |
46
|
| Yuen Long, Hong Kong, summer 2006 |
199 ± 291 |
107
|
| ΣISOA |
AGM/LIM Cyprus, July–August 2017 |
26.2 ± 13.2/18.6 ± 8.6 |
This study |
| Mt Tai Mo Shan, Hong Kong, September–November 2010 |
54.7 ± 22.7 |
105
|
| Gongga Mountain, China, August 2011 |
88.6 ± 106.1 |
19
|
| Mt Wuyi, China, November 2015–July 2016 |
45.28 ± 65.52 |
97
|
| Marseille, France, July 2008 |
3.7 |
104
|
| Fairbanks, Alaska, June–September 2009 |
41 ± 43 |
106
|
| Beirut, Lebanon, winter 2012 |
0.79 |
46
|
| Yuen Long, Hong Kong, summer 2006 |
29.5 ± 33.0 |
107
|
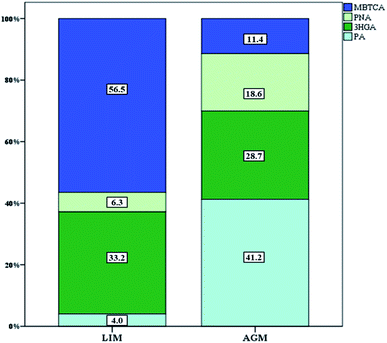 |
| | Fig. 6 Relative abundance of the individual PSOA tracer species. | |
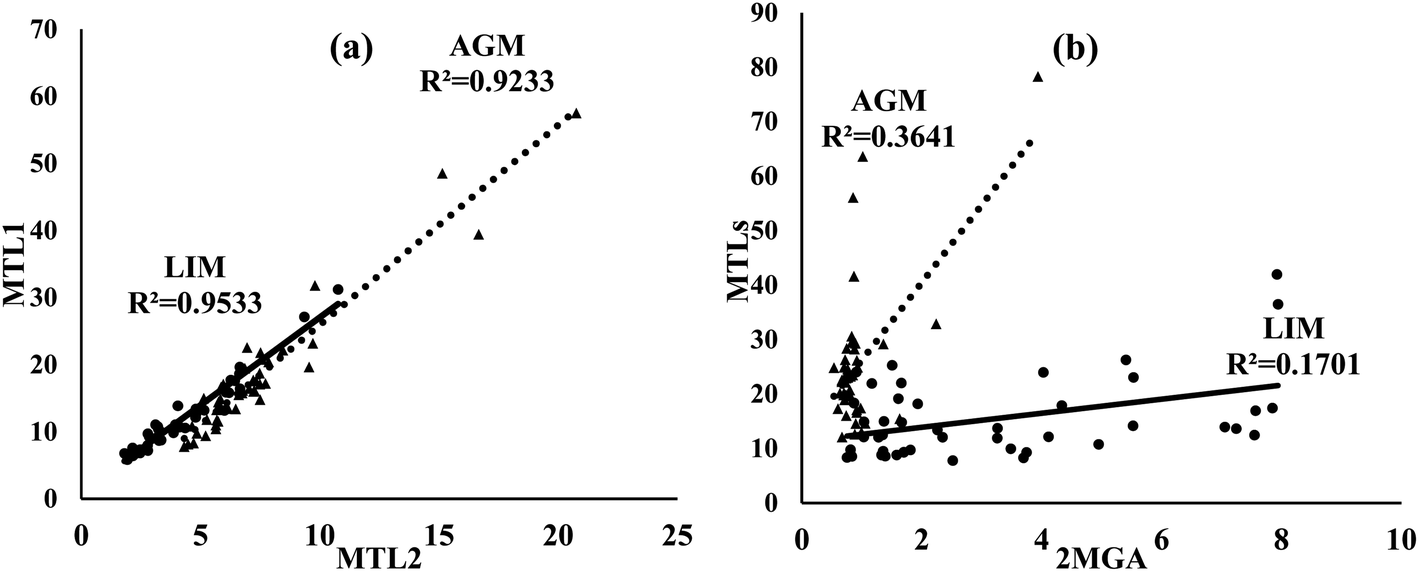
Of ISOA tracers, MTL1 was the most abundant in both sites followed by MTL2 and 2MGA (Table 1). Both MTLs were found at significantly (p < 0.05) higher concentrations in AGM. However, the concentrations of 2MGA were significantly increased in LIM (3.2 ± 2.4 versus 0.93 ± 0.54 ng m−3). MTLs can be generated from the oxidation of isoprene through the HO2 channel in the absence or at low levels of NOx, whereas 2MGA is formed under rich-NOx conditions by the oxidation of methacrolein, a gas phase first-generation product of isoprene's oxidation;24,100–103 therefore the use of the MTLs/2MGA ratio could evaluate the effect of NOx on ISOA formation, although factors including aerosol acidity, SO2, RH and the ratio of NOx/isoprene may also play an important role.103 The aforementioned ratio was almost four times higher in AGM (29.0 ± 11.5) than that in LIM (7.5 ± 5.6) revealing the influence of NOx on ISOA formation. Correspondingly, in a mountainous forest area of southeastern China, the winter-to-summer increase of MTLs/2MGA values was attributed to the higher NOx levels during fall–winter.97 Different formation mechanisms of ISOAs can also be implied as the excellent correlations observed between MTL1 and MTL2 (Fig. 7) at both sites indicate identical formation pathways, as also found in another Mediterranean site,104 whereas MTLs were moderately to weakly correlated with 2MGA (R2 = 0.36 in AGM; R2 = 0.17 in LIM).
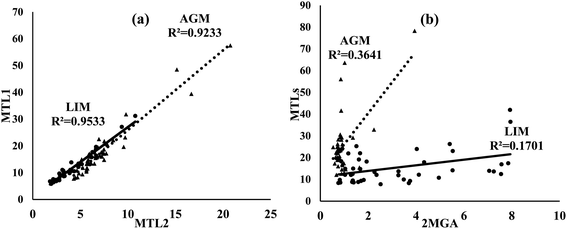 |
| | Fig. 7 Scatter plots of (a) MTL1 versus MTL2 and (b) MTLs (MTL1 + MTL2) versus 2MGA in AGM (triangle) and LIM (circle). Concentrations refer to ng m−3. | |
HAs including MAL, 2HGA, TAR and CIT have been considered as secondary products of various precursors. Based on field studies, MAL, TAR and 2HGA have been associated with isoprene's oxidation in Amazonian aerosols.108 In Alaskan aerosols TAR and CIT were considered, as well, to arise from isoprene's oxidation.106 Apart from an isoprene product, MAL has been suggested as a transformation product of several other VOCs such as monoterpenes, β-caryophyllene, toluene, etc.109 Recently, HAs including MAL, TAR, 2HGA and others were linked with photochemical oxidative chain reactions, as intermediates to the formation of oxalic acid from its higher homologues.110 MAL was the dominant HA in both sites although its mean concentration was over two times higher in LIM (22.3 ± 16.8 ng m−3) than in AGM (11.0 ± 6.4 ng m−3) (Table 1), which can be explained by the increased levels of its possible precursors in the urban area. TAR was the only HA found in significantly (p < 0.05) higher concentrations in AGM, which may indicate a biogenic origin. This can be reinforced as in AGM TAR was correlated strong and significantly (r = 0.77, p < 0.05) with MTLs (Table S6†) supporting the findings of Claeys et al. (2004)108 and Deshmukh et al. (2019).106 Moreover, CIT was well correlated with TAR (r = 0.72, p < 0.05) and MTLs (r = 65, p < 0.05) suggesting isoprene's oxidation as a potential source. On the other hand, MAL can be associated with pinene SOA formation due to its high correlation with PSOA tracer species (r = 0.72, p < 0.05), which is in accordance with Hu and Yu (2013),109 where PSOAs and MAL, during summer, were satisfactorily correlated (R2 = 0.89). Different sources can be indicated for 2HGA as it was mostly correlated with the sum of diC8-diC10, which is in agreement with the study of Gowda and Kawamura (2018),110 although the moderate correlation with PSOAs (r = 0.53, p < 0.05) could show a minor boost from pinenes' oxidation. On the other hand, in LIM, a different trend was noticed (Table S7†). HA individual compounds were strongly correlated with each other (r = 0.61–0.83, p < 0.05) attributed to the same origin. Moderate to strong correlations were observed with ΣdiC4–diC6 and ΣdiC8–diC10 (from 0.55 to 0.78) indicating their formation from the oxidation of unsaturated fatty acids or higher homologous diacids, as intermediates, through photochemical oxidative chain reactions.110 In this perspective, no significant associations have been found between HAs and BSOA except a moderate correlation of 2HGA with PSOA tracers (r = 0.53, p < 0.05).
3.5 Monocarboxylic acids (MCAs)
The group of MCAs is a remarkable component of atmospheric aerosols as it accounts for 46–80% of the total extractable organic compounds of urban PM2.5 aerosols.111 Several natural and anthropogenic sources have been reported for MCA emissions, including vehicular exhaust, cooking, biomass burning, fossil fuel combustion, and vegetative emissions and microbial activity.71,77,79,112,113 MCAs in LIM presented significantly higher concentrations than those measured in AGM with mean concentrations respectively equal to 107 ± 67.0 and 54.1 ± 25.1 ng m−3 (Table 1, Fig. 3), suggesting that anthropogenic contributions are more important. Anthropogenic emissions are enhanced in LIM, as during summer, population is significantly elevated due to international and domestic tourism.58 In both sites, C16 was the dominant MCA followed by C18, while C17 presented the lowest concentrations, which is consistent with the dominance of C16 over C18 measured in the Po Valley, Italy82 and in 6 sites of the Pearl River Delta region.70 In Beirut, Lebanon, MCAs were measured at quite higher concentrations, compared to our study, with C18 being the most abundant of the measured MCAs followed by C16.46 Aerosol aging can be indicated through the value of the C18:1/C18 ratio, with higher values implying fresher aerosols.34 C18:1 is more reactive than C18 due to the formers' double bond which makes it vulnerable to atmospheric oxidants including O3 and OH radicals.106 Fresher aerosols are indicated in LIM as mean C18:1/C18 values were significantly (p < 0.05) higher (0.19 ± 0.15) than those calculated in AGM (0.11 ± 0.05), probably because of (a) the increased anthropogenic emissions in the urban site and (b) the rapid degradation of C18:1 in AGM due to the almost two-fold higher concentrations of O3. The latter hypothesis can be reinforced by the significantly higher (p < 0.05) ratio of diC9/C18:1 (mean of 7.3 ± 3.4) observed in AGM, where the respective mean value in LIM was 3.1 ± 2.7 suggesting that the photochemical process is enhanced in AGM.114
3.6 Influence of atmospheric oxidants on SOA formation
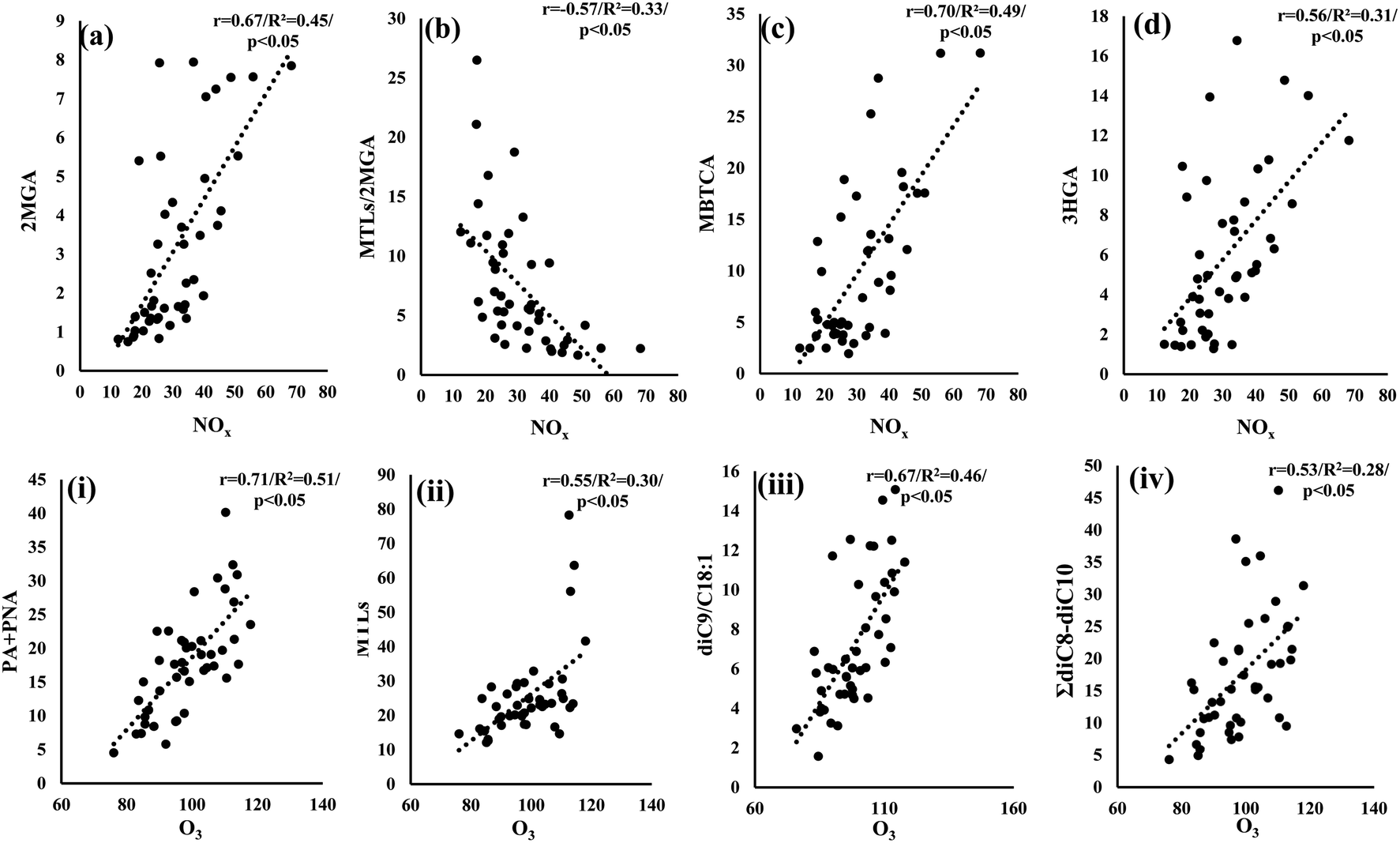
SOA formation and aging involve multiple and complex oxidation reactions, affected by the atmospheric abundance of OH radicals, O3, NOx, SO2, aerosol acidity, etc.11 Many laboratory studies have highlighted the influence of the aforementioned parameters on SOA formation.10,11,115,116 Considering that mostly NOx and SO2 are of anthropogenic origin, several field studies in the US have revealed their impact on the increase of biogenic secondary organic aerosols.117,118 As we mentioned above, the higher NOx levels observed in LIM than in AGM possibly affect PSOAs with the dominance of MBTCA in LIM and ISOAs with the significantly higher relative abundance of 2MGA. This hypothesis can be further supported by the respectively positive and negative significant correlations observed between NOx concentration values with 2MGA (r = 0.67, p < 0.05) and MTLs/2MGA (r = −0.57, p < 0.05) (Fig. 8a and b), indicating that isoprene's oxidation is influenced by NOx in LIM. In another urban Mediterranean site, NOx levels had a similar influence as a noticeable decrease of MTLs/2MGA was observed with the increase of NOx,104 which is also in accordance with the proposed pathway of isoprene's oxidation under high NOx conditions.24,102 The involvement of NOx in PSOA formation could be suggested due to the significant correlations of NOx with MBTCA (r = 0.70, p < 0.05) and HGA (r = 0.56, p < 0.05) (Fig. 8c and d). MBTCA was extensively detected in laboratory experiments from the ozonolysis and photo-oxidation of α-pinene under high NOx levels, although several N-containing products have been also detected.119 In addition, the increase of atmospheric oxidants including NO2 and O3 leads to elevated concentrations of 3HGA, other PSOA tracers.103 In other studies, positive correlations were observed between NO and NO2 with several other polar organic compounds including malic, maleic, succinic, phthalic acids, etc. indicating their secondary formation through various reactions.18
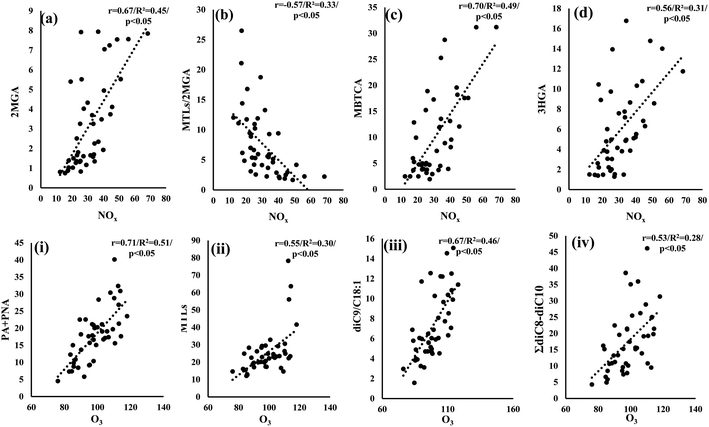 |
| | Fig. 8 Scatter plots of NOx (μg m−3) versus (a) 2MGA (ng m−3), (b) MTLs/2MGA, (c) MBTCA (ng m−3) and (d) 3HGA (ng m−3) in LIM and scatter plots of O3 (μg m−3) versus (i) PA + PNA (ng m−3), (ii) MTLs (ng m−3), (iii) diC9/C18:1 and (iv) ΣdiC8–C10 (ng m−3) in AGM. | |
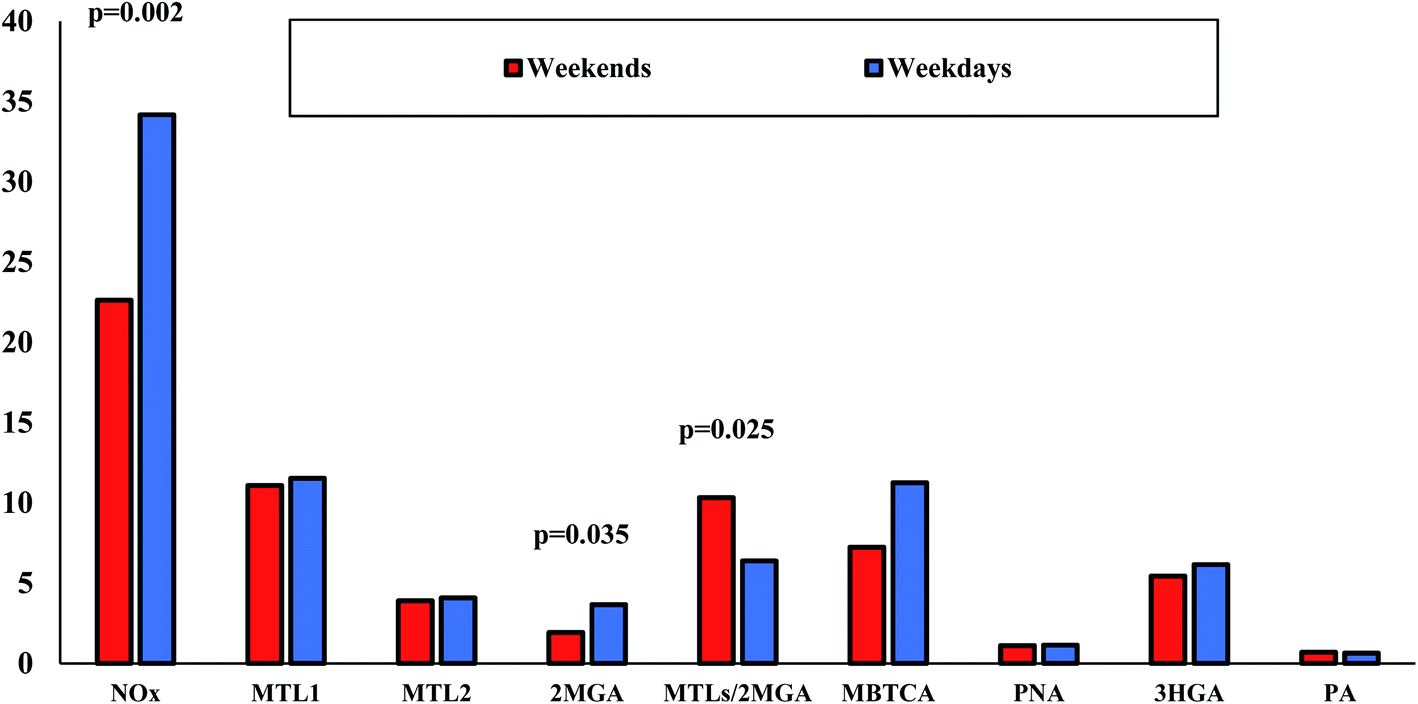
Further examination of NOx influence on BSOA was carried out through the results of the weekday–weekend variation on SOA tracer species for the reason that NOx sources are mostly anthropogenic.120 Weekday versus weekend variations in organic aerosol compositions and SOA tracers are limited.121 As is clear from Fig. 9, NOx concentrations were, unsurprisingly, significantly (p < 0.05) lower during weekends probably due to lower traffic emissions. Noteworthy differences were observed mostly for 2MGA as it was found almost two times lower during weekends than on weekdays (1.9 versus 3.7 ng m−3). At the same approach, the MTLs/2MGA ratio was significantly higher during weekends (1.6 times higher than on weekdays) indicating the lower impact of NOx to isoprene chemistry. Lower, but not significantly, concentrations were also found for MBTCA during weekends. However, due to the small number of weekend samples, this outcome should be further investigated with more samples.
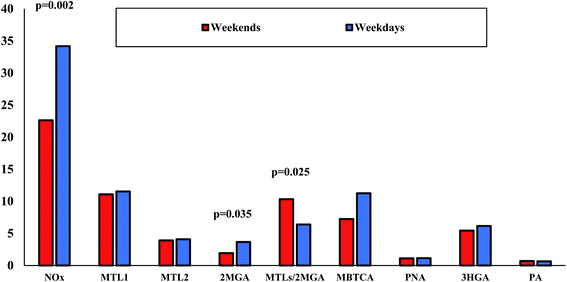 |
| | Fig. 9 Weekday (n = 32) versus weekend (n = 12) variations of NOx (μg m−3), BSOA tracers (μg m−3) and the ratio of MTLs/2MGA in LIM. | |
On the other hand, in AGM no significant correlations between NOx and the studied compounds have been observed. Nevertheless, the quite higher O3 levels appear to have an impact on secondary formation (Fig. 8). In particular, a significant (p < 0.05) increase of PSOA first generation products (PA + PNA) was observed with the increase of O3 (Fig. 8(i)), which is constant with the proposed α/β-pinene ozonolysis mechanisms.92,122 Another noteworthy correlation was observed between O3 and MTLs (Fig. 8(ii)) suggesting that the enhanced O3 levels could play an important role in isoprene's oxidation. Although isoprene's oxidation is initiated mostly by OH radicals, an ozonolysis pathway has been considered as a minor contributor to ISOAs in laboratory experiments.123 Recently, Riva et al. (2016)124 underlined the importance of ozonolysis on isoprene's transformation, as they found yields of MTLs and related isoprene organosulfates after the ozonolysis of isoprene in the presence of acidified sulfate aerosols. In addition to this, field studies have implied the impact of isoprene's ozonolysis through the moderate but significant correlations of O3 with MTLs.125,126 Our findings are in agreement with previous studies, as similar statistically significant correlations were observed, suggesting that isoprene's ozonolysis contributes to ISOA formation in AGM. Finally, high O3 levels could possibly affect the oxidation of unsaturated fatty acids as the sum of diC8–diC10 diacids, i.e. known products of the formers' photo-oxidation,31 was significantly (r = 0.53, p < 0.05) correlated with O3 (Fig. 8(iv)). Besides this, the ratio of diC9/C18:1 significantly (r = 0.67, p < 0.05) increases as the O3 concentration values increase (Fig. 8(iii)), indicating that higher molecular weight DCAs are formed by the oxidation of unsaturated fatty acids with O3, such as C18:1, emitted from the local ecosystem. A similar correlation, between the ratio of diC9/(C18:1 + C18:2) and O3, has been reported in a forest area, implying the secondary formation of diC9 from the forest's primary emissions.127
3.7 Principal component analysis (PCA)
Principal component analysis was applied to the dataset in order to assess any possible trends and to examine potential different sources between the two studied areas. PCA for AGM and LIM is illustrated in (Tables 3 and 4). TOL was not included in the PCA referring to AGM as it was detected in lower than 10% of the samples. Variable loadings >0.6 were considered as tightly clustered and are presented in bold.128 Six factors explained 80.1% and 82.7% of the variance in AGM and LIM data, respectively. Although the number of exported factors is the same for both sites, there are some noteworthy differences regarding the compounds clustered in each factor and the relative variance coverage. Particularly, in AGM-PCA (Table 3) the first factor (19.5% of the variance) was loaded with diC4, diC5, diC6, diC7, PH, BEN and TRIM indicating secondary formation from anthropogenic precursors. The same factor has been found in a background area of Sweden, which correlated low molecular weight dicarboxylic acids with phthalic acid.73 Factors 2 (14.1%) and 3 (13.3%) are associated with secondarily formed compounds from the oxidation of pinene and isoprene, respectively. Along with the PSOA tracer compounds (Factor 2), strong loadings of MAL were observed (0.76) indicating a possible biogenic source of MAL such as pinene oxidation, as also proposed by other studies.107,109 Similarly, in the ISOA factor (Factor 3), TAR and CIT are tightly clustered suggesting their potential origin from isoprene's transformation.106 Factor 4 (13.0%) is associated with the oxidation of unsaturated fatty acids due to the high positive values of diC8-diC10 and 2HGA, whereas Factors 5 and 6 (11.1 and 9.2%, respectively) are both linked with primary emissions local or regional, such as plant emissions, cooking and other anthropogenic sources70 (Factor 5) and biomass burning, as Factor 6 is loaded with the biomass burning tracer of LEV, iPH and tPh, which can also be emitted from biomass burning.89 In contrast, Factor 1 (26.4% of the variance) in LIM-PCA (Table 4) is heavily loaded with total DCAs and HAs attributed to the oxidation of unsaturated fatty acids or higher molecular weight DCAs with HAs as possible intermediates of these oxidative chain reactions leading to lower molecular weight DCAs.110 Factor 2 (14.5%) is linked with primary emissions probably from anthropogenic activities (vehicle exhausts, cooking, etc.) due to the tight clusters of MCAs, while Factor 4 (11.4%) reveals secondarily formed compounds from anthropogenic precursors including BEN, PH, and TRIM, as well as iPH which is different with that observed in AGM. These three factors have many similarities with those derived from Zhao et al. (2014),70 where MCAs, DCAs and polycarboxylic AAs were loaded in separate factors, connected with primary (MCAs) and secondary formation processes from different precursors (DCAs and AAs). Factor 5 (9.0%) is attributable to biomass burning through the strong loads of LEV. The presence of tPH in this factor confirms its primary origin although its other isomers (PH and iPH) are considered to be generated from secondary sources in LIM. Factors 3 and 6, with respective variance coverage of 13.9 and 7.5%, are associated with BVOC oxidation including pinene and isoprene respectively. Interestingly, although 2MGA is an isoprene's oxidation product, it is loaded in the pinene oxidation factor which may also reveal the influence of NOx levels on BSOA formation from the fact that the other heavily clustered compounds (MBTCA and 3HGA) are significantly affected by the NOX concentrations as already discussed in the previous section. Overall, the most interesting outcome exported from PCA is the significant contribution of the oxidation of unsaturated fatty acids in the urban site and the biogenic origin of hydroxyacids in the background site. In addition, the restriction on the application of iPH as an anthropogenic SOA tracer89 is supported as it was considered as a secondary product in LIM but mostly correlated with primarily emitted compounds in AGM.
Table 3 Varimax rotated component matrix of the studied compounds in AGM
| Variance (%) |
Component |
| 19.5 |
14.1 |
13.3 |
13.0 |
11.1 |
9.2 |
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
| DiC4 |
0.866
|
0.132 |
0.272 |
−0.018 |
−0.046 |
−0.080 |
| DiC5 |
0.891
|
0.111 |
0.282 |
−0.087 |
0.082 |
0.067 |
| DiC6 |
0.886
|
0.186 |
0.054 |
−0.007 |
0.092 |
−0.001 |
| DiC7 |
0.672
|
0.012 |
0.174 |
0.130 |
−0.156 |
0.209 |
| DiC8 |
0.083 |
0.184 |
0.018 |
0.855
|
0.158 |
0.244 |
| DiC9 |
0.156 |
0.231 |
0.066 |
0.833
|
0.252 |
0.260 |
| DiC10 |
0.269 |
0.344 |
0.008 |
0.797
|
0.107 |
0.093 |
| MAL |
0.096 |
0.763
|
0.138 |
0.241 |
0.109 |
0.112 |
| 2HGA |
0.132 |
0.107 |
0.047 |
0.783
|
0.353 |
0.135 |
| TAR |
0.236 |
0.154 |
0.825
|
0.291 |
−0.136 |
0.022 |
| CIT |
0.459 |
0.154 |
0.609
|
0.270 |
0.037 |
0.205 |
| LEV |
0.020 |
0.182 |
0.017 |
0.330 |
0.026 |
0.722
|
| MBTCA |
0.057 |
0.806
|
0.342 |
0.240 |
−0.020 |
0.241 |
| PNA |
0.181 |
0.849
|
0.224 |
−0.037 |
−0.087 |
0.055 |
| 3HGA |
0.161 |
0.785
|
0.324 |
0.331 |
−0.015 |
0.100 |
| PA |
0.290 |
0.819
|
0.082 |
0.204 |
−0.040 |
0.246 |
| MTL2 |
0.293 |
0.220 |
0.882
|
0.011 |
0.015 |
0.018 |
| MTL1 |
0.196 |
0.220 |
0.892
|
0.007 |
0.084 |
0.068 |
| 2MGA |
0.088 |
0.232 |
0.721
|
−0.230 |
−0.058 |
−0.076 |
| PH |
0.878
|
0.051 |
0.105 |
0.330 |
0.000 |
0.122 |
| TRIM |
0.659
|
0.059 |
−0.055 |
0.221 |
−0.052 |
0.194 |
| tPH |
0.164 |
0.209 |
0.030 |
0.239 |
0.113 |
0.881
|
| iPH |
0.230 |
0.163 |
0.056 |
0.059 |
0.265 |
0.837
|
| BEN |
0.842
|
0.237 |
0.074 |
0.165 |
0.013 |
0.036 |
| C16 |
−0.018 |
−0.101 |
0.091 |
0.167 |
0.913
|
0.015 |
| C17 |
−0.027 |
0.053 |
−0.020 |
0.215 |
0.814
|
0.332 |
| C18 |
0.012 |
−0.131 |
0.066 |
0.158 |
0.889
|
−0.035 |
| C18:1 |
−0.038 |
0.135 |
−0.194 |
0.087 |
0.452 |
0.116 |
Table 4 Varimax rotated component matrix of the studied compounds in LIM
| Variance (%) |
Component |
| 26.4 |
14.5 |
13.9 |
11.4 |
9.0 |
7.5 |
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
| DiC4 |
0.850
|
−0.183 |
0.187 |
0.126 |
0.107 |
0.175 |
| DiC5 |
0.905
|
−0.001 |
0.013 |
0.113 |
0.104 |
0.162 |
| DiC6 |
0.807
|
−0.096 |
0.173 |
0.227 |
0.107 |
0.129 |
| DiC7 |
0.769
|
0.485 |
−0.084 |
0.011 |
0.125 |
0.029 |
| DiC8 |
0.843
|
0.287 |
0.146 |
0.256 |
0.145 |
−0.007 |
| DiC9 |
0.732
|
0.591 |
−0.128 |
0.105 |
0.087 |
0.023 |
| DiC10 |
0.824
|
0.455 |
−0.072 |
0.130 |
0.060 |
0.099 |
| MAL |
0.683
|
−0.009 |
0.242 |
0.160 |
0.141 |
−0.103 |
| 2HGA |
0.702
|
0.208 |
0.513 |
0.123 |
0.043 |
0.042 |
| TAR |
0.713
|
0.185 |
0.398 |
0.199 |
0.098 |
−0.211 |
| CIT |
0.890
|
0.195 |
0.149 |
−0.084 |
0.035 |
−0.034 |
| LEV |
0.141 |
0.073 |
−0.019 |
0.222 |
0.905
|
0.022 |
| MBTCA |
0.132 |
−0.253 |
0.835
|
0.142 |
0.146 |
0.142 |
| PNA |
0.019 |
−0.260 |
0.611
|
0.157 |
0.345 |
−0.007 |
| 3HGA |
0.330 |
−0.210 |
0.848
|
0.086 |
0.134 |
−0.058 |
| PA |
0.175 |
−0.143 |
0.682
|
0.084 |
0.219 |
0.037 |
| MTL2 |
0.089 |
0.037 |
0.031 |
0.048 |
0.055 |
0.975
|
| MTL1 |
0.057 |
−0.013 |
0.071 |
−0.009 |
0.081 |
0.976
|
| 2MGA |
0.043 |
0.030 |
0.721
|
0.057 |
−0.116 |
0.035 |
| PH |
0.261 |
−0.097 |
0.087 |
0.912
|
0.164 |
−0.038 |
| TRIM |
0.233 |
−0.277 |
0.034 |
0.691
|
0.224 |
0.236 |
| tPH |
0.173 |
0.062 |
0.240 |
−0.033 |
0.831
|
0.210 |
| iPH |
0.015 |
0.228 |
0.145 |
0.841
|
−0.194 |
−0.010 |
| BEN |
0.254 |
−0.158 |
0.238 |
0.869
|
0.157 |
−0.060 |
| TOL |
0.263 |
−0.187 |
0.455 |
0.030 |
0.764
|
−0.071 |
| C16 |
0.253 |
0.860
|
−0.170 |
−0.041 |
0.029 |
0.019 |
| C17 |
0.322 |
0.734
|
−0.135 |
0.176 |
−0.154 |
0.066 |
| C18 |
0.199 |
0.882
|
−0.273 |
−0.079 |
0.029 |
−0.042 |
| C18:1 |
−0.097 |
0.847
|
−0.110 |
−0.322 |
0.001 |
−0.038 |
4. Conclusions
Twenty-eight polar organic species from six different compound classes and levoglucosan were studied in PM2.5 aerosol samples from an urban and a rural/background site of Cyprus during summer. Total DCAs, MCAs and AAs presented significantly higher concentrations in the urban site, and PSOAs and ISOAs were significantly lower, whereas a mixed trend was found for HAs. The ratios of selected compounds showed that aerosols in LIM are fresher and quite more affected by anthropogenic sources while PSOAs were found highly oxidized. The higher NOx levels in LIM appear to affect BSOA formation as they were significantly correlated with MBTCA, 3HGA and 2MGA. In contrast, the higher O3 observed in AGM enhances the formation of first-generation PSOAs and high molecular weight DCAs and contribute to ISOA formation as well. Correlation analysis revealed different potential sources or formation pathways for some of the studied compounds between the two sites, including MAL, TAR and CIT, which appear to be generated from BVOC oxidation in AGM, while in LIM they appear to be related to unsaturated fatty acids' or higher DCAs' oxidation, or iPH which was considered as a secondary product in LIM but associated with biomass burning in AGM. PCA analysis resulted in six factors for both studied areas with different, nevertheless, individual variance coverages and compounds loaded to each factor, including oxidation of unsaturated fatty acids, anthropogenic VOCs, isoprene and pinene along with primary emissions such as biomass burning, natural and anthropogenic related emissions (plant emissions, cooking, motor exhaust emissions, etc.)
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This research is co-financed by Greece and the European Union (European Social Fund-ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning» in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (IKY).
References
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang and J. H. Kroll,
et al., Evolution of organic aerosols in the atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS.
- J. L. Hand, B. A. Schichtel, M. Pitchford, W. C. Malm and N. H. Frank, Seasonal composition of remote and urban fine particulate matter in the United States, J. Geophys. Res. Atmos., 2012, 117, D05209 Search PubMed.
- L. P. Chrysikou and C. A. Samara, Seasonal variation of the size distribution of urban particulate matter and associated organic pollutants in the ambient air, Atmos. Environ., 2009, 43, 4557–4569 CrossRef CAS.
- P. Q. Fu, K. Kawamura, J. Chen, B. Charrière and R. Sempéré, Organic molecular composition of marine aerosols over the Arctic Ocean in summer: Contributions of primary emission and secondary aerosol formation, Biogeosciences, 2013, 10, 653–667 CrossRef.
- A. Piazzalunga, M. Anzano, E. Collina, M. Lasagni, F. Lollobrigida and A. Pannocchia,
et al., Contribution of wood combustion to PAH and PCDD/F concentrations in two urban sites in Northern Italy, J. Aerosol Sci., 2013, 56, 30–40 CrossRef CAS.
- C. Wiedinmyer, R. J. Yokelson and B. K. Gullett, Global Emissions of Trace Gases, Particulate Matter, and Hazardous Air Pollutants from Open Burning of Domestic Waste, Environ. Sci. Technol., 2014, 48, 9523–9530 CrossRef CAS.
- L. Ren, P. Fu, Y. He, J. Hou, J. Chen and C. M. Pavuluri,
et al., Molecular distributions and compound-specific stable carbon isotopic compositions of lipids in wintertime aerosols from Beijing, Sci. Rep., 2016, 6, 1–12 CrossRef.
- R. Zangrando, E. Barbaro, T. Kirchgeorg, M. Vecchiato, E. Scalabrin and M. Radaelli,
et al., Five primary sources of organic aerosols in the urban atmosphere of Belgrade (Serbia), Sci. Total Environ., 2016, 571, 1441–1453 CrossRef CAS.
- M. Kang, L. Ren, H. Ren, Y. Zhao, K. Kawamura and H. Zhang,
et al., Primary biogenic and anthropogenic sources of organic aerosols in Beijing, China: Insights from saccharides and n-alkanes, Environ. Pollut., 2018, 243, 1579–1587 CrossRef CAS.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson and M. Claeys,
et al., The formation, properties and impact of secondary organic aerosol: Current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- M. Huang, W. Zhang, X. Gu, C. Hu, W. Zhao and Z. Wang,
et al., Size distribution and chemical composition of secondary organic aerosol formed from Cl-initiated oxidation of toluene, J. Environ. Sci., 2012, 24, 860–864 CrossRef CAS.
- E. U. Emanuelsson, M. Hallquist, K. Kristensen, M. Glasius, B. Bohn and H. Fuchs,
et al., Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties, Atmos. Chem. Phys., 2013, 13, 2837–2855 CrossRef.
- J. E. Williams, P. F. J. van Velthoven and C. A. M. Brenninkmeijer, Quantifying the uncertainty in simulating global tropospheric composition due the variability in global emission estimates of Biogenic Volatile Organic Compounds, Atmos. Chem. Phys., 2013, 13, 2857–2891 CrossRef.
- R. Atkinson and J. Arey, Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther and J. L. Jimenez,
et al., Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 2017, 55, 509–559 CrossRef.
- A. G. Carlton, C. Wiedinmyer and J. H. Kroll, A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 2009, 9, 4987–5005 CrossRef CAS.
- D. Balla, D. Voutsa and C. Samara, Study of polar organic compounds in airborne particulate matter of a coastal urban city, Environ. Sci. Pollut. Res., 2018, 25, 12191–12205 CrossRef CAS.
- L. Li, W. Lai, J. Pu, H. Mo, D. Dai and G. Wu,
et al., Polar organic tracers in PM2.5 aerosols from an inland background area in Southwest China: Correlations between secondary organic aerosol tracers and source apportionment, J. Environ. Sci., 2018, 69, 281–293 CrossRef.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener and M. C. Facchini,
et al., Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
-
M. S. Parmar, in Encyclopedia of toxicology, 3rd Edition, ed. P. Wexler, Elsevier Inc, Academic Press, Amsterdam, 3rd edn, 2014, vol. 2, ch. Dicarboxylic acid, pp. 76–79 Search PubMed.
- Y. H. Lin, M. Arashiro, E. Martin, Y. Chen, Z. Zhang and K. G. Sexton,
et al., Isoprene-derived secondary organic aerosol induces the expression of oxidative stress response genes in human lung cells, Environ. Sci. Technol. Lett., 2016, 3, 250–254 CrossRef CAS.
- M. Arashiro, Y. H. Lin, Z. Zhang, K. G. Sexton, A. Gold and I. Jaspers,
et al., Effect of secondary organic aerosol from isoprene-derived hydroxyhydroperoxides on the expression of oxidative stress response genes in human bronchial epithelial cells, Environ. Sci.: Processes Impacts, 2018, 20, 332–339 RSC.
- J. D. Surratt, S. M. Murphy, J. H. Kroll, N. L. Ng, L. Hildebrandt and A. Sorooshian,
et al., Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A, 2006, 110, 9665–9690 CrossRef CAS.
- X. Ding, Q. F. He, R. Q. Shen, Q. Q. Yu, Y. Q. Zhang and J. Y. Xin,
et al., Spatial and seasonal variations of isoprene secondary organic aerosol in China: Significant impact of biomass burning during winter, Sci. Rep., 2016, 6, 1–10 CrossRef.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, M. Chan, K. A. Schilling and P. S. Chhabra,
et al., α-pinene photooxidation under controlled chemical conditions-Part 2: SOA yield and composition in low-and high-NOx environments, Atmos. Chem. Phys., 2012, 12, 7413–7427 CrossRef CAS.
- M. M. Haque, K. Kawamura and Y. Kim, Seasonal variations of biogenic secondary organic aerosol tracers in ambient aerosols from Alaska, Atmos. Environ., 2016, 130, 95–104 CrossRef CAS.
- S. Gao, M. Keywood, N. L. Ng, J. Surratt, V. Varutbangkul and R. Bahreini,
et al., Low-Molecular-Weight and Oligomeric Components in Secondary Organic Aerosol from the Ozonolysis of Cycloalkenes and α-Pinene, J. Phys. Chem. A, 2004, 108, 10147–10164 CrossRef CAS.
- S. Rossignol, L. Chiappini, E. Perraudin, C. Rio, S. Fable and R. Valorso,
et al., Development of a parallel sampling and analysis method for the elucidation of gas/particle partitioning of oxygenated semi-volatile organics: A limonene ozonolysis study, Atmos. Meas. Tech., 2012, 5, 1459–1489 CrossRef CAS.
- M. Jaoui, M. Lewandowski, J. H. Offenberg, K. S. Docherty and T. E. Kleindienst, Ozonolysis of α/β-farnesene mixture: Analysis of gas-phase and particulate reaction products, Atmos. Environ., 2017, 169, 175–192 CrossRef CAS.
- E. G. Stephanou and N. Stratigakis, Oxocarboxylic and α,ω-Dicarboxylic Acids: Photooxidation Products of Biogenic Unsaturated Fatty Acids Present in Urban Aerosols, Environ. Sci. Technol., 1993, 27, 1403–1407 CrossRef CAS.
- L. Yang, M. B. Ray and L. E. Yu, Photooxidation of dicarboxylic acids-Part II: Kinetics, intermediates and field observations, Atmos. Environ., 2008, 42, 868–880 CrossRef CAS.
- S. Kumar, S. G. Aggarwal, P. K. Gupta and K. Kawamura, Investigation of the tracers for plastic-enriched waste burning aerosols, Atmos. Environ., 2015, 108, 49–58 CrossRef CAS.
- F. Cao, S. C. Zhang, K. Kawamura, X. Liu, C. Yang and Z. Xu,
et al., Chemical characteristics of dicarboxylic acids and related organic compounds in PM2.5 during biomass-burning and non-biomass-burning seasons at a rural site of Northeast China, Environ. Pollut., 2017, 231, 654–662 CrossRef CAS.
- T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg and K. S. Docherty, The formation of SOA and chemical tracer compounds from the photooxidation of naphthalene and its methyl analogs in the presence and absence of nitrogen oxides, Atmos. Chem. Phys., 2012, 12, 8711–8726 CrossRef CAS.
- X. He, X. H. H. Huang, K. S. Chow, Q. Wang, T. Zhang and D. Wu,
et al., Abundance and Sources of Phthalic Acids, Benzene-Tricarboxylic Acids, and Phenolic Acids in PM2.5 at Urban and Suburban Sites in Southern China, ACS Earth Space Chem, 2018, 2, 147–158 CrossRef CAS.
- K. Kawamura and I. R. Kaplan, Motor Exhaust Emissions as a Primary Source for Dicarboxylic Acids in Los Angeles Ambient Air, Environ. Sci. Technol., 1987, 21, 105–110 CrossRef CAS.
- S. L. Mkoma and K. Kawamura, Molecular composition of dicarboxylic acids, ketocarboxylic acids, α-dicarbonyls and fatty acids in atmospheric aerosols from Tanzania, East Africa during wet and dry seasons, Atmos. Chem. Phys., 2013, 13, 2235–2251 CrossRef.
-
IPCC, Climate change 2007: Synthesis report, Intergovermental Panel on Climate Change, 2007 Search PubMed.
- U. Im, K. Markakis, A. Poupkou, D. Melas, A. Unal and E. Gerasopoulos,
et al., The impact of temperature changes on summer time ozone and its precursors in the Eastern Mediterranean, Atmos. Chem. Phys., 2011, 11, 3847–3864 CrossRef CAS.
- E. Gerasopoulos, G. Kouvarakis, P. Babasakalis, M. Vrekoussis, J. P. Putaud and N. Mihalopoulos, Origin and variability of particulate matter (PM10) mass concentrations over the Eastern Mediterranean, Atmos. Environ., 2006, 40, 4679–4690 CrossRef CAS.
- M. Kanakidou, N. Mihalopoulos, T. Kindap, U. Im, M. Vrekoussis and E. Gerasopoulos,
et al., Megacities as hot spots of air pollution in the East Mediterranean, Atmos, Environ, 2011, 45, 1223–1235 CrossRef CAS.
- U. Dayan, P. Ricaud, R. Zbinden and F. Dulac, Atmospheric pollution over the eastern Mediterranean during summer - A review, Atmos. Chem. Phys., 2017, 17, 13233–13263 CrossRef CAS.
- E. Kostenidou, K. Florou, C. Kaltsonoudis, M. Tsiflikiotou, S. Vratolis and K. Eleftheriadis,
et al., Sources and chemical characterization of organic aerosol during the summer in the eastern Mediterranean, Atmos. Chem. Phys. Discuss., 2015, 15, 3455–3491 CrossRef.
- I. Stavroulas, A. Bougiatioti, G. Grivas, D. Paraskevopoulou, M. Tsagkaraki and P. Zarmpas,
et al., Sources and processes that control the submicron organic aerosol composition in an urban Mediterranean environment (Athens): a high temporal-resolution chemical composition measurement study, Atmos. Chem. Phys., 2019, 19, 901–919 CrossRef CAS.
- A. Waked, C. Afif, J. Brioude, P. Formenti, S. Chevaillier and I. El Haddad,
et al., Composition and source apportionment of organic aerosol in Beirut, Lebanon, during winter 2012, Aerosol Sci. Technol., 2013, 47, 1258–1266 CrossRef CAS.
- C. Debevec, S. Sauvage, V. Gros, J. Sciare, M. Pikridas and I. Stavroulas,
et al., Origin and variability in volatile organic compounds observed at an Eastern Mediterranean background site (Cyprus), Atmos. Chem. Phys., 2017, 17, 11355–11388 CrossRef CAS.
- C. Debevec, S. Sauvage, V. Gros, K. Sellegri, J. Sciare and M. Pikridas,
et al., Driving parameters of biogenic volatile organic compounds and consequences on new particle formation observed at an eastern Mediterranean background site, Atmos. Chem. Phys., 2018, 18, 14297–14325 CrossRef CAS.
- S. Kleanthous, M. Vrekoussis, N. Mihalopoulos, P. Kalabokas and J. Lelieveld, On the temporal and spatial variation of ozone in Cyprus, Sci. Total Environ., 2014, 476–477, 677–687 CrossRef CAS.
- P. L. Fall, Modern vegetation, pollen and climate relationships on the Mediterranean island of Cyprus, Rev. Palaeobot. Palynol., 2012, 185, 79–92 CrossRef.
- EN 12341:2014, Ambient Air – Standard Gravimetric Measurement Method for the Determination of the PM10 or PM2.5 Mass Concentration of Suspended Particulate Matter.
- F. Cavalli, M. Viana, K. E. Yttri, J. Genberg and J. P. Putaud, Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: the EUSAAR protocol, Atmos. Meas. Tech., 2010, 3, 79–89 CrossRef CAS.
- L. M. Castro, C. A. Pio, R. M. Harrison and D. J. T. Smith, Carbonaceous aerosol in urban and rural European atmospheres: Estimation of secondary organic carbon concentrations, Atmos. Environ., 1999, 33, 2771–2781 CrossRef CAS.
- G. Wang, M. Xie, S. Hu, S. Gao, E. Tachibana and K. Kawamura, Dicarboxylic acids, metals and isotopic compositions of C and N in atmospheric aerosols from inland China: Implications for dust and coal burning emission and secondary aerosol formation, Atmos. Chem. Phys., 2010, 10, 6087–6096 CrossRef CAS.
- T. Siciliano, M. Siciliano, C. Malitesta, A. Proto, R. Cucciniello and A. Giove,
et al., Carbonaceous PM10 and PM2.5 and secondary organic aerosol in a coastal rural site near Brindisi (Southern Italy), Environ. Sci. Pollut. Res., 2018, 25, 23929–23945 CrossRef CAS.
- R. M. Flores and P. V. Doskey, Evaluation of multistep derivatization methods for identification and quantification of oxygenated species in organic aerosol, J. Chromatogr. A, 2015, 1418, 1–11 CrossRef CAS.
- A. I. Mologousi and E. B. Bakeas, Multivariate optimization of a simple and sensitive method for the determination of secondary biogenic organic compounds in airborne particles, Anal. Methods, 2016, 8, 4047–4055 RSC.
- M. Pikridas, M. Vrekoussis, J. Sciare, S. Kleanthous, E. Vasiliadou and C. Kizas,
et al., Spatial and temporal (short and long-term) variability of submicron, fine and sub-10 μm particulate matter (PM1, PM2.5, PM10) in Cyprus, Atmos. Environ., 2018, 191, 79–93 CrossRef CAS.
- S. Achilleos, J. M. Wolfson, S. T. Ferguson, C. M. Kang, D. G. Hadjimitsis and M. Hadjicharalambous,
et al., Spatial variability of fine and coarse particle composition and sources in Cyprus, Atmos. Res., 2016, 169, 255–270 CrossRef CAS.
- E. Gerasopoulos, E. Koulouri, N. Kalivitis, G. Kouvarakis, S. Saarikoski and T. Makela,
et al., Size-segregated mass distributions of aerosols over Eastern Mediterranean: seasonal variability and comparison with AERONET columnar size-distributions, Atmos. Chem. Phys., 2007, 7, 2551–2561 CrossRef CAS.
- M. Lazaridis, L. Dzumbova, I. Kopanakis, J. Ondracek, T. Glytsos and V. Aleksandropoulou,
et al., PM10 and PM2.5 Levels in the Eastern Mediterranean (Akrotiri Research Station, Crete, Greece), Water, Air, Soil Pollut., 2008, 189, 85–101 CrossRef CAS.
- N. A. Saliba, F. E. Jam, G. E. Tayar, W. Obeid and M. Roumie, Origin and variability of particulate matter (PM10 and PM2.5) mass concentrations over an Eastern Mediterranean city, Atmos. Res., 2010, 97, 106–114 CrossRef CAS.
- C. Pio, M. Cerqueira, R. M. Harrison, T. Nunes, F. Mirante and C. Alves,
et al., OC/EC ratio observations in Europe: Re-thinking the approach for apportionment between primary and secondary organic carbon, Atmos. Environ., 2011, 45, 6121–6132 CrossRef CAS.
- M. I. Manousakas, K. Florou and S. N. Pandis, Source Apportionment of Fine Organic and Inorganic Atmospheric Aerosol in an Urban Background Area in Greece, Atmosphere, 2020, 11, 330 CrossRef CAS.
- C. Samara, D. Voutsa, A. Kouras, K. Eleftheriadis, T. Maggos and D. Saraga,
et al., Organic and elemental carbon associated to PM10 and PM2.5 at urban sites of northern Greece, Environ. Sci. Pollut. Res., 2014, 21, 1769–1785 CrossRef CAS.
- B. Kunwar and K. Kawamura, One-year observations of carbonaceous and nitrogenous components and major ions in the aerosols from subtropical Okinawa Island, an outflow region of Asian dusts, Atmos. Chem. Phys., 2014, 14, 1819–1836 CrossRef.
- M. M. Haque, K. Kawamura, D. K. Deshmukh, C. Fang, W. Song and B. Mengying, et al., Characterization of organic aerosols from a Chinese megacity during winter: predominance of fossil fuel combustion, Atmos. Chem. Phys., 2019, 19, 5147–5164 CrossRef CAS.
- A. I. Calvo, V. Pont, C. Liousse, B. Dupre, A. Mariscal and C. Zouiten,
et al., Chemical composition of urban aerosols in Toulouse, France during CAPITOUL experiment, Meteorol. Atmos. Phys., 2008, 102, 307–323 CrossRef.
- K. Kawamura and S. Bikkina, A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 2016, 170, 140–160 CrossRef CAS.
- X. Zhao, X. Wang, X. Ding, Q. He, Z. Zhang and T. Liu,
et al., Compositions and sources of organic acids in fine particles (PM2.5) over the Pearl River Delta region, south China, J. Environ. Sci., 2014, 26, 110–121 CrossRef CAS.
- H. Guo, J. Zhou, L. Wang, Y. Zhou, J. Yuan and R. Zhao, Seasonal Variations and Sources of Carboxylic Acids in PM2.5 in Wuhan, China, Aerosol Air Qual. Res., 2015, 15, 517–528 CrossRef CAS.
- C. Cheng, G. Wang, B. Zhou, J. Meng, J. Li and J. Cao,
et al., Comparison of dicarboxylic acids and related compounds in aerosol samples collected in Xi’an, China during haze and clean periods, Atmos. Environ., 2013, 81, 443–449 CrossRef CAS.
- M. Hyder, J. Genberg, M. Sandahl, E. Swietlicki and J. Jönsson, Yearly trend of dicarboxylic acids in organic aerosols from south of Sweden and source attribution, Atmos. Environ., 2012, 57, 197–204 CrossRef CAS.
- K. Kawamura and R. B. Gagosian, Implications of ω-oxocarboxylic acids in the remote marine atmosphere for photo-oxidation of unsaturated fatty acids, Nature, 1987, 325, 330–332 CrossRef CAS.
- D. K. Deshmukh, K. Kawamura, M. Lazaar, B. Kunwar and S. K. R. Boreddy, Dicarboxylic acids, oxoacids, benzoic acid, α-dicarbonyls, WSOC, OC, and ions in spring aerosols from Okinawa Island in the western North Pacific Rim: Size distributions and formation processes, Atmos. Chem. Phys., 2016, 16, 5263–5282 CrossRef CAS.
- B. Kunwar and K. Kawamura, Seasonal distributions and sources of low molecular weight dicarboxylic acids, ω-oxocarboxylic acids, pyruvic acid, α-dicarbonyls and fatty acids in ambient aerosols from subtropical Okinawa in the western Pacific Rim, Environ. Chem., 2014, 11, 673–689 CrossRef CAS.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Measurement of emissions from air pollution sources. 1. C1 through C29 organic compounds from meat charbroiling, Environ. Sci. Technol., 1999, 33, 1566–1577 CrossRef CAS.
- K. F. Ho, S. S. H. Ho, S. C. Lee, K. Kawamura, S. C. Zou and J. J. Cao,
et al., Summer and winter variations of dicarboxylic acids, fatty acids and benzoic acid in PM2.5 in Pearl Delta River Region, China, Atmos. Chem. Phys., 2011, 11, 2197–2208 CrossRef CAS.
- C. Oliveira, C. Pio, C. Alves, M. Evtyugina, P. Santos and V. Goncalves,
et al., Seasonal distribution of polar organic compounds in the urban atmosphere of two large cities from the North and South of Europe, Atmos. Environ., 2007, 41, 5555–5570 CrossRef CAS.
- H. L. Yin, C. Y. Qiu, Z. X. Ye, S. P. Li and J. F. Liang, Seasonal variation and source apportionment of organic tracers in PM10 in Chengdu, China, Environ. Geochem. Health, 2015, 37, 195–205 CrossRef CAS.
- K. F. Ho, S. C. Lee, J. J. Cao, K. Kawamura, T. Watanabe and Y. Cheng,
et al., Dicarboxylic acids, ketocarboxylic acids and dicarbonyls in the urban roadside area of Hong Kong, Atmos. Environ., 2006, 40, 3030–3040 CrossRef CAS.
- M. C. Pietrogrande, D. Bacco, M. Visentin, S. Ferrari and V. Poluzzi, Polar organic marker compounds in atmospheric aerosol in the Po Valley during the Supersito campaigns - Part 1: Low molecular weight carboxylic acids in cold seasons, Atmos. Environ., 2014, 86, 164–175 CrossRef CAS.
- S. Kundu, K. Kawamura and M. Lee, Seasonal variations of diacids, ketoacids, and α-dicarbonyls in aerosols at Gosan, Jeju Island, South Korea: Implications for sources, formation, and degradation during long-range transport, J. Geophys. Res. Atmos., 2010, 115, D19307 CrossRef.
- S. Bikkina, K. Kawamura and Y. Miyazaki, Latitudinal distributions of atmospheric dicarboxylic acids, oxocarboxylic acids, and α-dicarbonyls over the western north pacific: Sources and formation pathways, J. Geophys. Res. Atmos., 2015, 120, 5010–5035 CAS.
- M. P. Fraser, G. R. Cass and B. R. T. Simoneit, Air quality model evaluation data for organics. 6. C3-C24 organic acids, Environ. Sci. Technol., 2003, 37, 446–453 CrossRef CAS.
- P. Q. Fu, K. Kawamura, C. M. Pavuluri, T. Swaminathan and J. Chen, Molecular characterization of urban organic aerosol in tropical India: Contributions of primary emissions and secondary photooxidation, Atmos. Chem. Phys., 2010, 10, 2663–2689 CrossRef CAS.
- P. Q. Fu, K. Kawamura, J. Chen, J. Li, Y. L. Sun and Y. Liu,
et al., Diurnal variations of organic molecular tracers and stable carbon isotopic composition in atmospheric aerosols over Mt. Tai in the North China Plain: An influence of biomass burning, Atmos. Chem. Phys., 2012, 12, 8359–8375 CrossRef CAS.
- B. R. T. Simoneit, Biomass burning - A review of organic tracers for smoke from incomplete combustion, Appl. Geochem., 2002, 17, 129–162 CrossRef CAS.
- I. M. Al-Naiema and E. A. Stone, Evaluation of anthropogenic secondary organic aerosol tracersfrom aromatic hydrocarbons, Atmos. Chem. Phys., 2017, 17, 2053–2065 CrossRef CAS.
- A. Bougiatioti, I. Stavroulas, E. Kostenidou, P. Zarmpas, C. Theodosi and G. Kouvarakis,
et al., Processing of biomass-burning aerosol in the eastern Mediterranean during summertime, Atmos. Chem. Phys., 2014, 14, 4793–4807 CrossRef.
- M. Claeys, R. Szmigielski, I. Kourtchev, P. Van Der Veken, R. Vermeylen and W. Maenhaut,
et al., Hydroxydicarboxylic acids: Markers for secondary organic aerosol from the photooxidation of α-pinene, Environ. Sci. Technol., 2007, 41, 1628–1634 CrossRef CAS.
- Y. Ma, T. R. Willcox, A. T. Russell and G. Marston, Pinic and pinonic acid formation in the reaction of ozone with α-pinene, Chem. Commun., 2007, 13, 1328–1330 RSC.
- R. Szmigielski, J. D. Surratt, Y. Gómez-González, P. van der Veken, I. Kourtchev and R. Vermeylen,
et al., 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 2007, 34, 2–7 CrossRef.
- J. J. Li, G. H. Wang, J. J. Cao, X. M. Wang and R. J. Zhang, Observation of biogenic secondary organic aerosols in the atmosphere of a mountain site in central China: Temperature and relative humidity effects, Atmos. Chem. Phys., 2013, 13, 11535–11549 CrossRef.
- Y. Y. Zhang, L. Müller, R. Winterhalter, G. K. Moortgat, T. Hoffmann and U. Pöschl, Seasonal cycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter, Atmos. Chem. Phys., 2010, 10, 7859–7873 CrossRef CAS.
- P. Fu, S. G. Aggarwal, J. Chen, J. Li, Y. Sun and Z. Wang,
et al., Molecular Markers of Secondary Organic Aerosol in Mumbai, India, Environ. Sci. Technol., 2016, 50, 4659–4667 CrossRef CAS.
- Z. Hong, H. Zhang, Y. Zhang, L. Xu, T. Liu and H. Xiao,
et al., Secondary organic aerosol of PM2.5 in a mountainous forest area in southeastern China: Molecular compositions and tracers implication, Sci. Total Environ., 2019, 653, 496–503 CrossRef CAS.
- M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg and E. O. Edney, Identification and Quantification of Aerosol Polar Oxygenated Compounds Bearing Carboxylic or Hydroxyl Groups. 2. Organic Tracer Compounds from Monoterpenes, Environ. Sci. Technol., 2005, 39, 5661–5673 CrossRef CAS.
- X. Ding, Q. He, R. Shen, Q. Yu and X. Wang, Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer, J. Geophys. Res. Atmos., 2014, 119, 11877–11891 CAS.
- M. Claeys, W. Wang, A. C. Ion, I. Kourtchev, A. Gelencser and W. Maenhaut, Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide, Atmos. Environ., 2004, 38, 4093–4098 CrossRef CAS.
- E. O. Edney, T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg and W. Wang,
et al., Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOX/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States, Atmos, Environ, 2005, 39, 5281–5289 CrossRef CAS.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. N. Chan, C. L. Loza and A. J. Kwan,
et al., Reactive intermediates revealed in secondary organic aerosol formation from isoprene, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 6640–6645 CrossRef CAS.
- Q. Yuan, S. Lai, J. Song, X. Ding, L. Zheng and X. Wang,
et al., Seasonal cycles of secondary organic aerosol tracers in rural Guangzhou, Southern China: The importance of atmospheric oxidants, Environ. Pollut., 2018, 240, 884–893 CrossRef CAS.
- I. El Haddad, N. Marchand, B. Temime-Roussel, H. Wortham, C. Piot and J.-L. Besombes,
et al., Insights into the secondary fraction of the organic aerosol in a Mediterranean urban area: Marseille, Atmos. Chem. Phys., 2011, 11, 2059–2079 CrossRef CAS.
- X. P. Lyu, H. Guo, H. R. Cheng, X. M. Wang, X. Ding and H. X. Lu,
et al., Observation of SOA tracers at a mountainous site in Hong Kong: Chemical characteristics, origins and implication on particle growth, Sci. Total Environ., 2017, 605–606, 180–189 CrossRef CAS.
- D. K. Deshmukh, M. Haque, Y. Kim and K. Kawamura, Organic tracers of fine aerosol particles in central Alaska: summertime composition and sources, Atmos. Chem. Phys., 2019, 19, 14009–14029 CrossRef CAS.
- D. Hu, Q. Bian, T. W. Y. Li, A. K. H. Lau and J. Z. Yu, Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006, J. Geophys. Res. Atmos., 2006, 113, D22206 CrossRef.
- M. Claeys, B. Graham, G. Vas, W. Wang, R. Vermeylen and V. Pashynska,
et al., Formation of Secondary Organic Aerosols through Photooxidation of Isoprene, Science, 2004, 303, 1173–1176 CrossRef CAS.
- D. Hu and J. Z. Yu, Secondary organic aerosol tracers and malic acid in Hong Kong: Seasonal trends and origins, Environ. Chem., 2013, 10, 381–394, DOI:10.1071/EN13104.
- D. Gowda and K. Kawamura, Seasonal variations of low molecular weight hydroxy-dicarboxylic acids and oxaloacetic acid in remote marine aerosols from Chichijima Island in the western North Pacific (December 2010–November 2011), Atmos. Res., 2018, 204, 128–135 CrossRef CAS.
- M. Zheng, M. Fang, F. Wang and K. L. To, Characterization of the solvent extractable organic compounds in PM2.5 aerosols in Hong Kong, Atmos. Environ., 2000, 34, 2691–2702 CrossRef CAS.
- W. F. Rogge, P. M. Medeiros and B. R. T. Simoneit, Organic marker compounds for surface soil and fugitive dust from open lot dairies and cattle feedlots, Atmos. Environ., 2006, 40, 27–49 CrossRef CAS.
- A. S. Shannigrahi, J. B. C. Pettersson, S. Langer, K. Arrhenius, M. Hagström and S. Janhäll,
et al., N-Alkanoic monocarboxylic
acid concentrations in urban and rural aerosols: Seasonal dependence and major sources, Atmos. Res., 2014, 143, 228–237 CrossRef CAS.
- K. F. Ho, S. C. Lee, S. S. H. Ho, K. Kawamura, E. Tachibana and Y. Cheng,
et al., Dicarboxylic acids, ketocarboxylic acids, α-dicarbonyls, fatty acids, and benzoic acid in urban aerosols collected during the 2006 Campaign of Air Quality Research in Beijing (CAREBeijing-2006), J. Geophys. Res. Atmos., 2010, 115, D19312 CrossRef.
- M. Sarrafzadeh, J. Wildt, I. Pullinen, M. Springer, E. Kleist and R. Tillmann,
et al., Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 2016, 16, 11237–11248 CrossRef CAS.
- D. Zhao, S. H. Schmitt, M. Wang, I. H. Acir, R. Tillmann and Z. Tan,
et al., Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 2018, 18, 1611–1628 CrossRef CAS.
- L. Xu, H. Guo, C. M. Boyd, M. Klein, A. Bougiatioti and K. M. Cerully,
et al., Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 37–42 CrossRef CAS.
- J. Liu, L. M. Russell, G. Ruggeri, S. Takahama, M. S. Claflin and P. J. Ziemann,
et al., Regional Similarities and NOx-Related Increases in Biogenic Secondary Organic Aerosol in Summertime Southeastern United States, J. Geophys. Res. Atmos, 2018, 123, 10620–10636 Search PubMed.
- J. H. Park, Z. B. Babar, S. J. Baek, H. S. Kim and H. J. Lim, Effects of NOx on the molecular composition of secondary organic aerosol formed by the ozonolysis and photooxidation of α-pinene, Atmos. Environ., 2017, 166, 263–275 CrossRef CAS.
- B. C. Mcdonald, T. R. Dallmann, E. W. Martin and R. A. Harley, Long-term trends in nitrogen oxide emissions from motor vehicles at National, State, and air basin scales, J. Geophys. Res. Atmos., 2012, 117, D00V18, DOI:10.1029/2012JD018304.
- D. R. Gentner, S. H. Jathar, T. D. Gordon, R. Bahreini, D. A. Day and I. El. Haddad,
et al., A review of urban secondary organic aerosol formation from gasoline and diesel motor vehicle emissions, Environ. Sci. Technol., 2017, 51, 1074–1093 CrossRef CAS.
- Y. Ma and G. Marston, Multifunctional acid formation from the gas-phase ozonolysis of β-pinene, Phys. Chem. Chem. Phys., 2008, 10, 6115–6126 RSC.
- T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, M. Jaoui and E. O. Edney, Ozone-isoprene reaction: Re-examination of the photochemically generate acidic aerosol, led to a substantial formation of secondary organic aerosol, Geophys. Res. Lett., 2007, 34, L01805 CrossRef.
- M. Riva, S. H. Budisulistiorini, Z. Zhang, A. Gold and J. D. Surratt, Chemical characterization of secondary organic aerosol constituents from isoprene ozonolysis in the presence of acidic aerosol, Atmos. Environ., 2016, 130, 5–13 CrossRef CAS.
- W. Rattanavaraha, K. Chu, S. H. Budisulistiorini, M. Riva, Y. H. Lin and E. S. Edgerton,
et al., Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 2016, 16, 4897–4914 CrossRef CAS.
- J. Li, G. Wang, C. Wu, C. Cao, Y. Ren and J. Wang,
et al., Characterization of isoprene-derived secondary organic aerosols at a rural site in North China Plain with implications for anthropogenic pollution effects, Sci. Rep., 2018, 8, 1–10 CrossRef.
- T. Mochizuki, K. Kawamura, Y. Miyazaki and R. Wada, Secondary formation of oxalic acid and related organic species from biogenic sources in a larch forest at the northern slope of Mt. Fuji,
Atmos. Environ.
, 2017, 166, 255–262 CrossRef CAS.
- K. G. Koukoulakis, E. Chrysohou, P. G. Kanellopoulos, S. Karavoltsos, G. Katsouras and M. Dassenakis,
et al., Trace elements bound to airborne PM 10 in a heavily industrialized site nearby Athens: Seasonal patterns, emission sources, health implications, Atmos. Pollut. Res., 2019, 10, 1347–1356 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0em00238k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a