DOI:
10.1039/D0CP04139D
(Perspective)
Phys. Chem. Chem. Phys., 2020,
22, 22852-22863
Recent progress in use and observation of surface hydrogen migration over metal oxides
Received
4th August 2020
, Accepted 28th September 2020
First published on 28th September 2020
Abstract
Hydrogen migration over a metal oxide surface is an extremely important factor governing the activity and selectivity of various heterogeneous catalytic reactions. Passive migration of hydrogen governed by a concentration gradient is called hydrogen spillover, which has been investigated broadly for a long time. Recently, well-fabricated samples and state-of-the-art measurement techniques such as operando spectroscopy and electrochemical analysis have been developed, yielding findings that have elucidated the migration mechanism and novel utilisation of hydrogen spillover. Furthermore, great attention has been devoted to surface protonics, which is hydrogen migration activated by an electric field, as applicable for novel low-temperature catalysis. This article presents an overview of catalysis related to hydrogen hopping, sophisticated analysis techniques for hydrogen migration, and low-temperature catalysis using surface protonics.

Kota Murakami
| Kota Murakami received his degree from Waseda University. He is a research associate at Waseda University. His research interests are in the areas of catalysts, ammonia synthesis, CO2 capture processes, and DFT calculations for catalysis. |

Yasushi Sekine
| Yasushi Sekine received his PhD from The University of Tokyo in 1998. He was promoted to Professor in 2012. His research interests are in the areas of catalysts for hydrogen production, methane activation, carbon dioxide utilization, biomass conversion and catalysis in electric fields. |
1 Overview
Hydrogen migration plays an important role in various catalyses such as hydrogenation–dehydrogenation of hydrocarbon,1–5 CO2 reduction6–9 and NH3 synthesis.10–17 However, the utilisation of hydrogen migration is not limited to catalysis. It is also fundamentally important for catalyst preparation,18,19 hydrogen storage,20,21 fuel cells and sensors.22,23
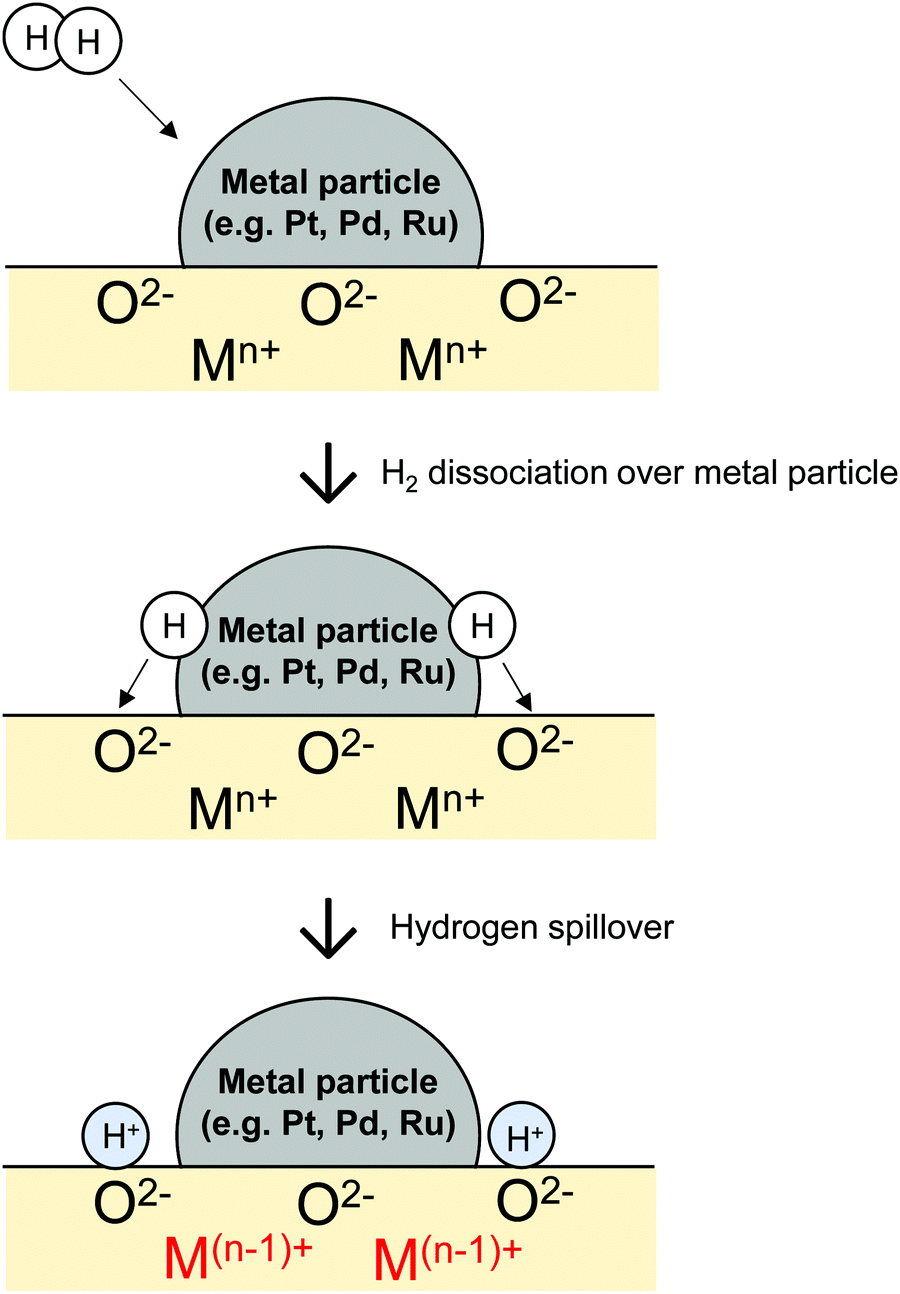
Commonly, hydrogen migration is driven by the concentration gradient. Supplied gaseous H2 is dissociated on active metals. H atoms migrate onto adjacent hydrogen-poor surfaces such as metal oxide, which is not able to activate H2 under the same conditions. This phenomenon is designated as hydrogen spillover.24,25 Spillover was first reported by Khoobiar et al. in 1964.26 They observed that WO3 was reduced by H2 into blue HxWO3 at 323 K only when it was in contact with Pt-loaded metal oxides (Pt/γ-Al2O3). This result suggests that H atoms over Pt particles spill over onto physically connected WO3. Since that seminal report, many researchers studying catalysts have shown keen interest in hydrogen spillover.
A well-balanced and comprehensive review of hydrogen spillover was written by Prins in 2012.24 The review also explained some points of uncertainty in the discussion about hydrogen spillover onto non-reducible metal oxides such as SiO2 and Al2O3. Over reducible metal oxides, fast hydrogen spillover proceeds via coupled proton electron transfer, as presented in Fig. 1.27–33 In contrast, this mechanism is not approved over non-reducible metal oxides. The main controversies related to this concept involve the lack of a detailed understanding of migration mechanism and direct proof of the catalysis using migrating H atoms. After publication of the review by Prins, in-depth investigation using well-fabricated samples, operando spectroscopy and density functional theory (DFT) have been reported.29,32–37 Choi et al. presented a review about one of them: encapsulated metal catalyst.38
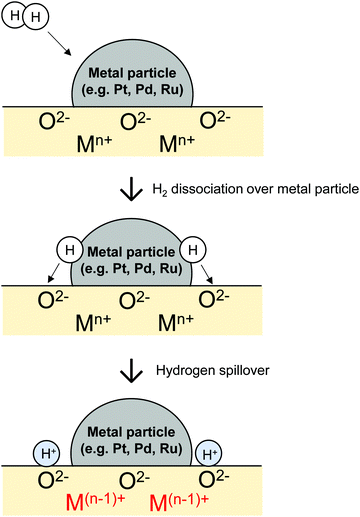 |
| | Fig. 1 Schematic images of spillover of H atoms from metal particles to reducible metal oxides. | |
Furthermore, most recently, we have actively facilitated hydrogen migration by application of an electric field.39–41 This active migration is called surface protonics. It has been revealed that surface protonics plays a key role in activating the cleavage of strong bonds, as in the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in N2,13–17,42,43 and the C–H bond in hydrocarbons, as in CH442–46 and methylcyclohexane (MCH).4,5 Those activations make it possible to catalyze hydrogen production and formation of the hydrogen carrier, even at low temperatures (<500 K). Therefore, we have eagerly investigated low-temperature catalysis using surface protonics in an electric field as promising novel catalytic systems.39–41
N bond in N2,13–17,42,43 and the C–H bond in hydrocarbons, as in CH442–46 and methylcyclohexane (MCH).4,5 Those activations make it possible to catalyze hydrogen production and formation of the hydrogen carrier, even at low temperatures (<500 K). Therefore, we have eagerly investigated low-temperature catalysis using surface protonics in an electric field as promising novel catalytic systems.39–41
In this review, we provide an overview of recent reports about catalytic reactions and operando analysis related to hydrogen spillover and active facilitation of the reaction using surface protonics. We first discuss catalysis involving hydrogen spillover and recent progress in that field in chapter 2. In chapter 3, state-of-the-art evaluation techniques of hydrogen migration using well-fabricated samples, operando spectroscopy, DFT calculations and electrochemical measurements are also introduced. Finally, novel low-temperature catalyses achieved by virtue of surface protonics are presented in chapter 4.
2. Recent reports of catalyses related to hydrogen spillover
Hydrogen spillover has been applied for various catalytic reactions. Its benefits are attributable to two main processes. One process is the supply of H atoms from one active site to other sites where there is no ability to dissociate H2. In Sections 2.1–2.3, such benefits are explained. Especially, the change of net migration by doping is shown in Section 2.3. The other process is removal of H atoms from sites that are hindered by excess amount of H atoms, as introduced in Section 2.4.
2.1 Enhancement of atom efficiency
Noble metal nanoparticles are well known to be active to several reactions, but high atomic efficiency is necessary because of their scarcity on Earth. Recently, to enhance atomic efficiency, great efforts have been undertaken for specific examination of size reduction of the supported metal particles to single atoms, which can achieve 100% atomic efficiency. Studies of single-atom catalysts (SAC) were described by the Liu group.47,48 Those catalysts have been used for various reactions such as CO oxidation,49 water gas shift reaction,50 and O2 or CO2 electroreduction.51,52 However, they do not function as a catalyst for ketone–aldehyde reductions, which are widely used and fundamental reactions in chemical engineering. Difficulties arise from a low reaction rate of H2 dissociation over single-atom sites. H2 dissociates heterogeneously at single-atom sites. The dissociation barrier is much higher than a homolytic reaction over metal nanoparticle surfaces. Yan et al. reported the H2 reaction order as about 1.2 for the hydrogenation of 1,3-butadiene over Pd single-atom-supported catalysts, indicating hydrogen dissociation as the rate-determining step.53
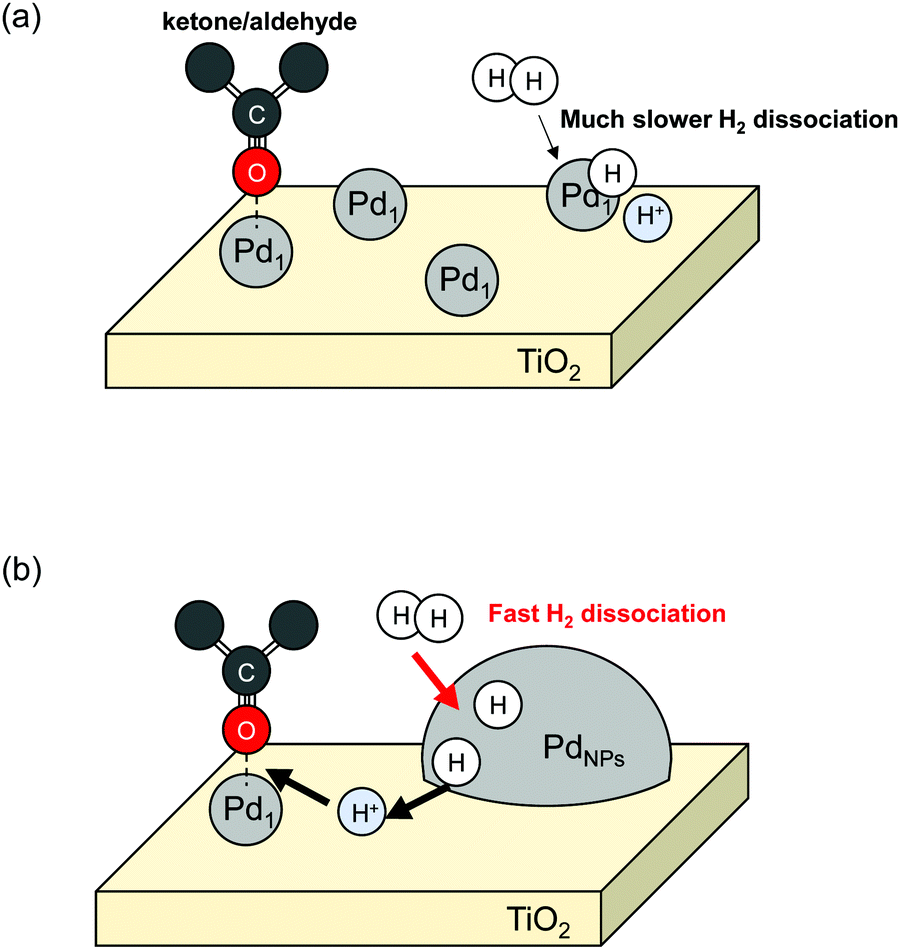
Kuai et al. solved this limitation by hydrogen spillover and achieved high atomic efficiency of Pd-based catalysts for ketone–aldehydes into alcohols.54 They supported both Pd single atoms (Pd1) and Pd nanoparticles (PdNPs) over mesoporous TiO2 (Pd1+NPs/TiO2) using spray-assisted method, developed by the same group.54,55 They compared 4-methylacetophenone (MAP) hydrogenation activities among Pd1+NPs/TiO2, Pd1/TiO2 and PdNPs/TiO2. A schematic image is presented in Fig. 2. Results revealed that Pd1+NPs/TiO2 synergetic catalyst exhibited much higher activity than either Pd1/TiO2 or PdNPs/TiO2. Regarding the activity per exposed Pd atom (turn over frequency, TOF), Pd1+NPs/TiO2 and PdNPs/TiO2 showed almost equal values (4361 h−1 for Pd1+NPs/TiO2 and 4565 h−1 for PdNPs/TiO2), although Pd1/TiO2 exhibited much lower TOF (645 h−1). Those results clarified a synergetic working mechanism of Pd1 and PdNPs. Gaseous H2 splits over PdNPs into H atoms. The H atoms migrate over TiO2 surface. They are supplied to ketone–aldehyde adsorbed on Pd1 sites. By virtue of that migration, Pd1 can function as a reaction site for the hydrogenation of ketone–aldehydes. Consequently, hydrogen spillover can expand the possibilities of SAC usage. Although the TOF over Pd1 and PdNPs changed slightly in this report, some reports have described single atoms as more positively charged rather than nanoparticles. The charge difference influences the reactivity.8,49 Therefore, this research by Kuai et al.54 can facilitate not only atom efficiency for various reactions but also utilisation of SAC reactivity.
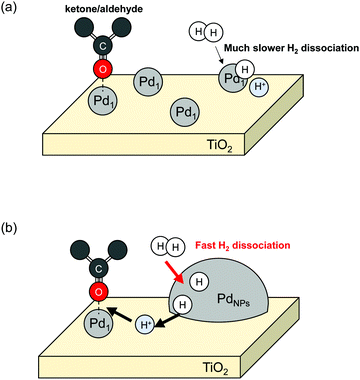 |
| | Fig. 2 Schematic image of the ketone/aldehydes hydrogenation over Pd1/TiO2 (a) and Pd1+NPs/TiO2 (b). | |
2.2 Selective hydrogenation
Synergy of the two reaction sites, which are called tandem catalysts or dual sites catalysts, using hydrogen spillover have enabled highly selective hydrogenation.
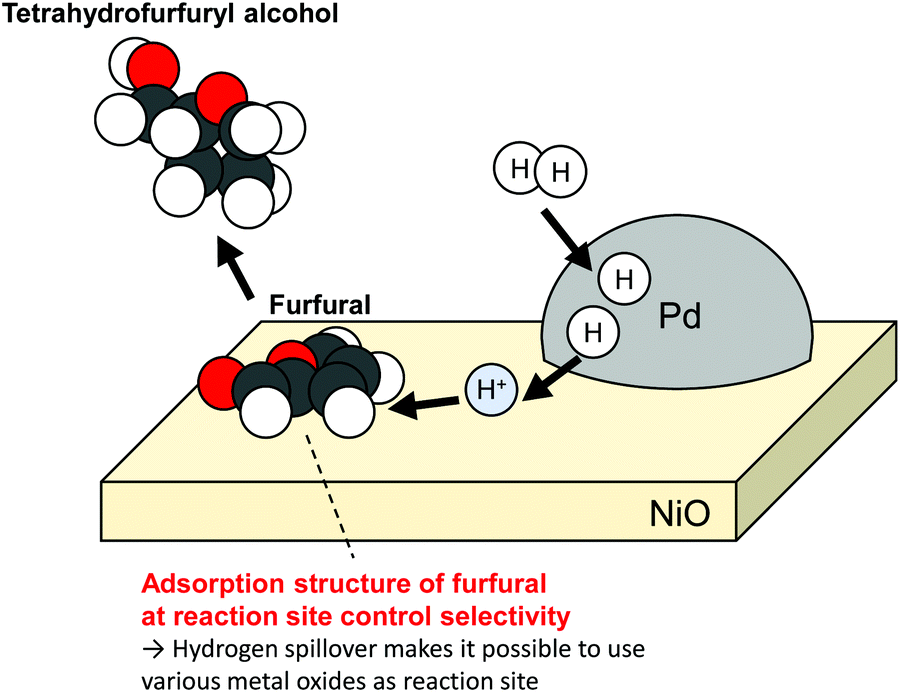
Campisi et al. reported that Pd/NiO can catalyze the selective conversion of furfural to tetrahydrofurfuryl alcohol (72%), although Pd/TiO2 favours furfuryl alcohol (68%).2 Furfural is regarded as a promising platform molecule to various products in a biorefinery. Its selective conversion using tailored catalysts is desired. Without Pd loading, TiO2 did not exhibit observable activity; also, NiO showed minor activity. Actually, Pd/NiO retained its high activity even with a tiny amount of Pd loading. The activity of 0.1 wt%Pd/NiO exhibited almost identical activity to that of 1 wt%Pd/NiO. In contrast, 0.1 wt%Pd/TiO2 showed much smaller activity than 1 wt%Pd/TiO2. Those results suggest that furfuryl over NiO was converted by H atoms supplied from Pd particles. Reportedly, the product distribution of furfural hydrogenation depends on the adsorption structure of the reactant over reaction sites.56,57 Campisi et al. also confirmed distinct differences of adsorption structures over each surface using DFT calculations. No clear role of the adsorption structures has been revealed yet. However, they reported that the parallel adsorption of a furan ring and alcohol group over NiO support led to selective formation of tetrahydrofurfuryl alcohol (Fig. 3). As described in this report, hydrogen spillover makes it possible to use various metal oxides surface as reaction sites.
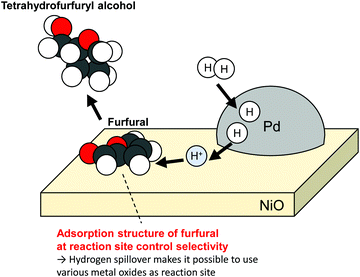 |
| | Fig. 3 Selective hydrogenation of furfural over NiO using hydrogen spillover from Pd. | |
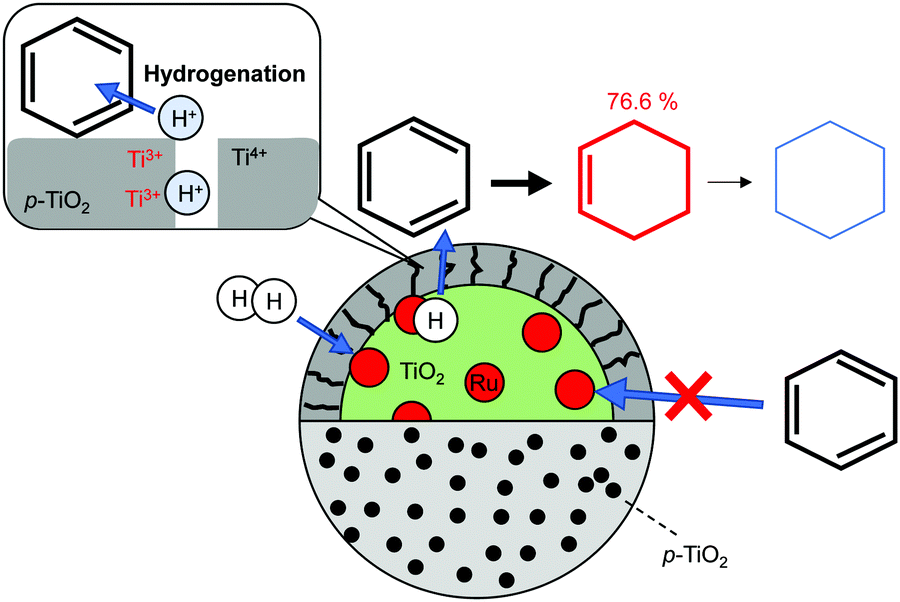
Preparation of outstanding catalysts that have two totally separated active sites has been investigated for selective hydrogenation. Xue et al. constructed a novel Ru/TiO2 catalyst encapsulated by porous TiO2 (denoted as (Ru/TiO2)@p-TiO2) using an earlier reported method58 with some modification, and achieved selective hydrogenation of benzene to cyclohexene (98.1% benzene conversion, 76.6% cyclohexene selectivity).3 The partial hydrogenation of benzene is of great importance in cyclohexene production: it is a key intermediate for various fine chemicals because of its reactive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Ru-based catalysts have been investigated widely for this hydrogenation. However, the selectivity remains limited because of the favourable deep hydrogenation into cyclohexane over a Ru surface.59,60 Xue et al. overcame this limitation by capping a Ru surface with porous TiO2. Fig. 4 presents a schematic image of this catalyst and selective hydrogenation of benzene. Results obtained from H2-TPD and CO-TPD revealed that only H2 can diffuse into interior Ru particles. Access of other reactants was inhibited by the TiO2 shell. During hydrogenation, H2 dissociates over Ru particles and H atoms spill over to TiO2 surface. Formation of the hydroxyl species was confirmed using FT-IR measurement. The hydroxyl species migrate toward the external surface via coupled proton electron transfer as shown in Fig. 1. Finally, over the external surface of the TiO2 shell, the hydroxyls react with hydrocarbons along with the charge recombination. Thanks to the weak adsorption of cyclohexene and high reaction barrier of the hydrogenation of cyclohexene on the TiO2 surface, (Ru/TiO2)@p-TiO2 exhibited high selectivity, as indicated by DFT calculations.
C bond. Ru-based catalysts have been investigated widely for this hydrogenation. However, the selectivity remains limited because of the favourable deep hydrogenation into cyclohexane over a Ru surface.59,60 Xue et al. overcame this limitation by capping a Ru surface with porous TiO2. Fig. 4 presents a schematic image of this catalyst and selective hydrogenation of benzene. Results obtained from H2-TPD and CO-TPD revealed that only H2 can diffuse into interior Ru particles. Access of other reactants was inhibited by the TiO2 shell. During hydrogenation, H2 dissociates over Ru particles and H atoms spill over to TiO2 surface. Formation of the hydroxyl species was confirmed using FT-IR measurement. The hydroxyl species migrate toward the external surface via coupled proton electron transfer as shown in Fig. 1. Finally, over the external surface of the TiO2 shell, the hydroxyls react with hydrocarbons along with the charge recombination. Thanks to the weak adsorption of cyclohexene and high reaction barrier of the hydrogenation of cyclohexene on the TiO2 surface, (Ru/TiO2)@p-TiO2 exhibited high selectivity, as indicated by DFT calculations.
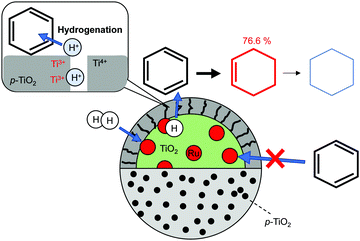 |
| | Fig. 4 Selective hydrogenation of benzene over (Ru/TiO2)@p-TiO2. | |
2.3 Change of reaction mechanism by doping because of hydrogen migration enhancement
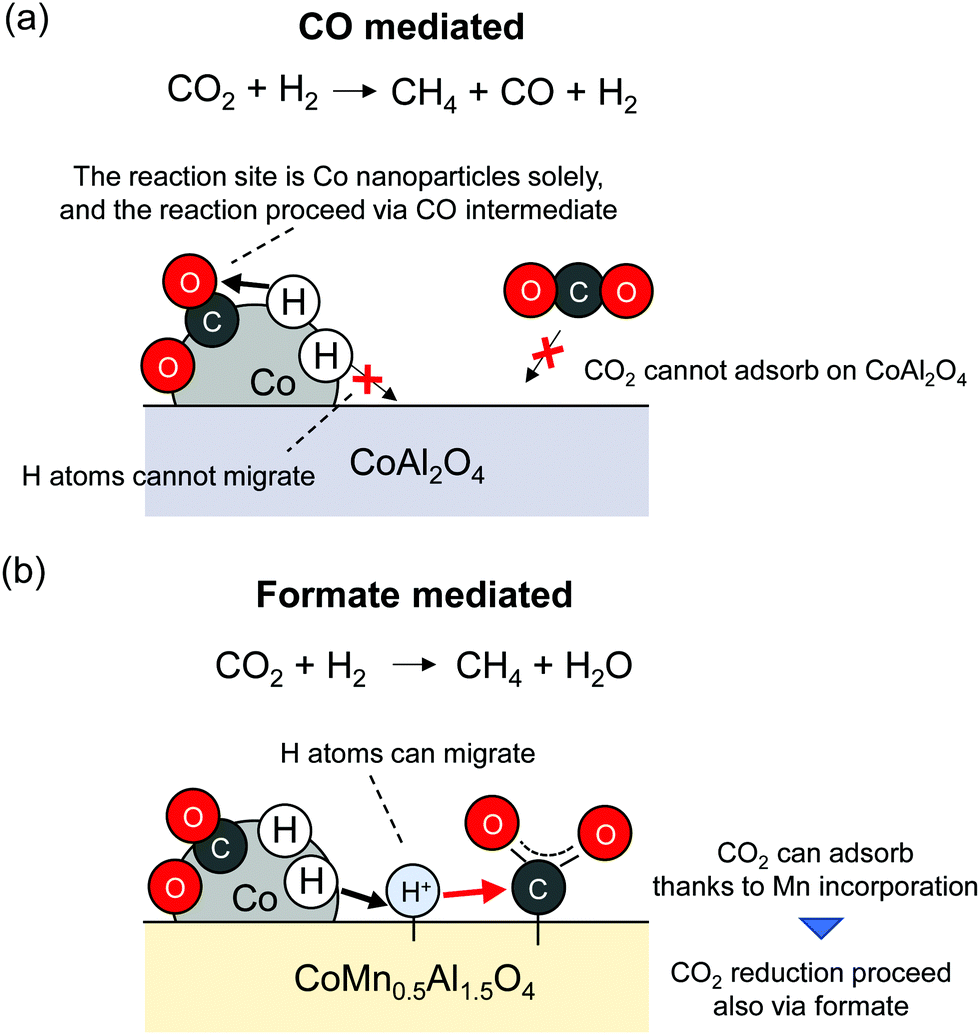
It can be readily inferred that doping into metal oxides can change the extent of hydrogen spillover. Franken et al. reported that Mn addition into CoAl2O4 facilitated hydrogen spillover, leading to the high CO2 methanation activity.6 Because CO2 is a main greenhouse gas component, the development of CO2 capture and storage (CCS) or capture and utilisation (CCU) methods is urgently necessary. The reaction mechanism of CO2 reduction, which is complex, has given rise to some conflicting views. Their details will not be explained herein because comprehensive reviews on the topic have been published.61,62 Franken et al. examined the utilisation of mixed metal oxides (CoMnxAl2−xO4) with pre-reduction for this reaction. The Co nanoparticles are formed by pre-reduction of CoMnxAl2−xO4; the particles then function as active sites. They found that Mn incorporation led to the strong CO2 adsorption over supports. Furthermore, hydrogen spillover was facilitated because of the redox ability of doped Mn cations. These two effects of Mn incorporation decreased the activation energy from 108 kJ mol−1 (CoAl2O4) to 69 kJ mol−1 (CoMn0.5Al1.5O4). In line with operando DRIFTS measurements, it was inferred that the reaction sites and reaction mechanism are changed by Mn incorporation effects as presented in Fig. 5. Without Mn addition, Co nanoparticles function as CO2 reduction sites solely, and CO is formed as a reaction intermediate. In contrast, with Mn addition, supports can function as main reaction sites. Hence, the reaction over CoMn0.5Al1.5O4 proceeds via formate species, where the adsorbed CO2 over metal oxides is reduced by H atoms supplied from Co nanoparticles. The shifts of reaction sites and mechanisms by Mn addition resulted in suppressing the formation of CO as a by-product. This report describes the possibility of hydrogen spillover on various metal oxides by the doping of cations with redox ability.
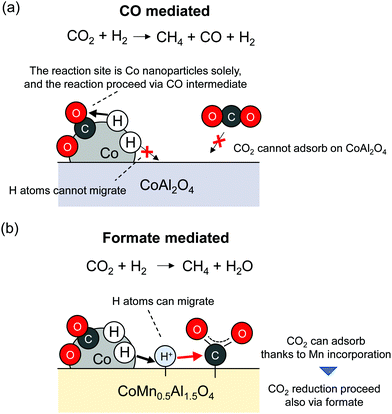 |
| | Fig. 5 Change of CO2 methanation mechanism by addition of Mn. | |
2.4 Suppression of hydrogen poisoning
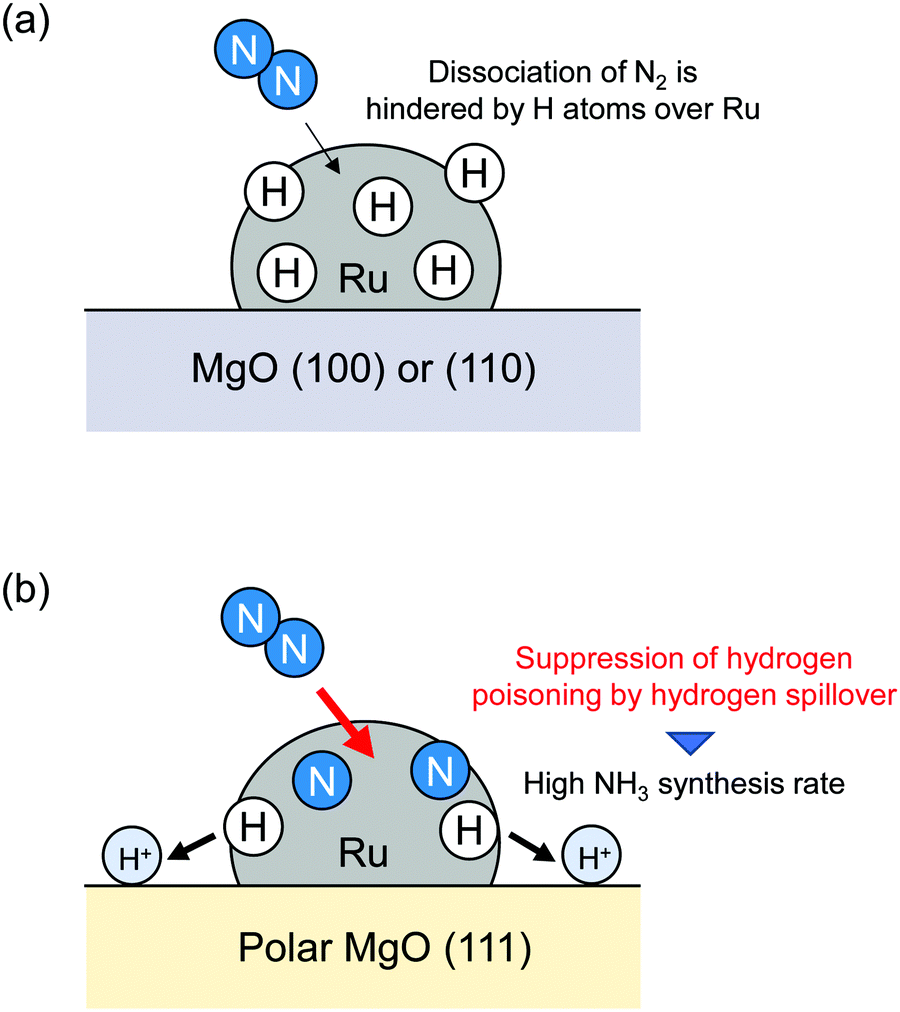
Wu et al. reported hydrogen spillover effects on NH3 synthesis.10 It is widely known that NH3 serves as an important nitrogen source for chemical industry purposes, but it is also a promising candidate as a hydrogen carrier by virtue of its high hydrogen content (17.8 wt%). Therefore, many studies of NH3 synthesis have been developed. Ozaki and Aika et al. reported that Ru-based catalysts had much higher intrinsic activity than widely used doubly promoted iron catalyst (Fe–Al2O3–K2O).63,64 Since those reports were published, various important Ru-based catalysts have been developed.65–68 It is said conventionally that N2 direct dissociation over a Ru surface is the rate-determining step.69 In addition, Ru-based catalysts are poisoned easily by H2 under high H2 pressure. Typical H2 reaction orders for NH3 synthesis over Ru-based catalysts are well known to be negative.70,71 Therefore, the H atom removal from Ru surfaces are also fundamentally important for the total NH3 synthesis rate. Wu et al. reported that hydrogen poisoning of the Ru surface was suppressed by simple replacement of the polycrystalline MgO(110) or (100) with polar MgO(111).10 Comparison of the NH3 synthesis rate revealed that Ru/MgO(111) has a much higher NH3 synthesis rate than that of Ru/MgO(110) or (100), although the N2 reaction order was almost identical (Ru/MgO(111): 0.9, Ru/MgO(110): 0.9, Ru/MgO(100): 0.8). In addition, the H2 reaction order became a positive value (0.6) over Ru/MgO(111) from negative values over other MgO surfaces (Ru/MgO(110): −0.2, Ru/MgO(100): −0.5). Therefore, suppression of hydrogen poisoning over Ru loaded over MgO(111) was confirmed to cause a high NH3 synthesis rate. 1H NMR, in situ FTIR measurements and DFT calculations clarified the high stability of H atoms over polar MgO(111). They concluded that H atom stability on MgO(111) led to removal of hydrogen poisoning over Ru, as depicted in Fig. 6. Their report did not address the net migration of H atoms and the reaction of H atoms after migration. However, to reduce hydrogen coverage over Ru by hydrogen spillover, their details must be considered. Therefore, in-depth consideration of relations among H atom stability, net migration rates, hydrogen coverage over loaded metals and NH3 synthesis rate using sophisticated methods are recommended, as introduced in the following chapter.
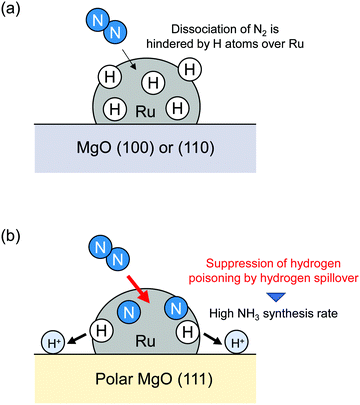 |
| | Fig. 6 Hydrogen poisoning suppression by hydrogen spillover. | |
3. Novel technique for observing hydrogen migration
3.1
In situ FT-IR measurement
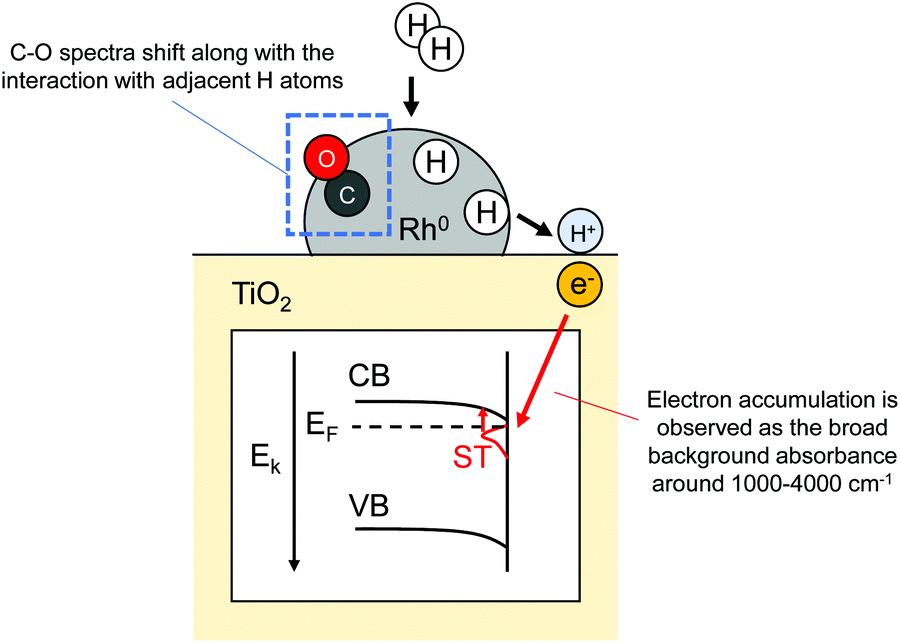
Reportedly, after H2 interaction with metal nanoparticles (Cu, Ru, Pt, Au, Rh) supported reducible metal oxides such as ZnO72,73 and TiO2,30,31,33 broad-band IR background absorbance around 1000–4000 cm−1 is observed. Such signals are assumed to be unrelated to the vibration of metal oxide lattices but are related to their respective electronic characters. Panayotov et al. explained the case of H2 reaction with Au- or Rh-supported TiO2 as follows.31,33 First, supplied gaseous H2 dissociates over metal nanoparticles. Then, H atoms spill over onto the TiO2 surface. At that time, H atoms protonate the TiO2. Upon formation of hydroxyl over the TiO2 surface, electrons are donated to shallow trap (ST) state resulting in n-doping. Doped electrons can be trapped in ST state or can be excited thermally and by IR radiation into the conduction band (CB). Such electron transfer is induced by Fermi level difference between TiO2 and H atoms. Consequently, n-doping by hydrogen spillover on semiconductor supports can be detected sensitively using IR spectroscopic method. Furthermore, they obtained in-depth understandings of hydrogen spillover procedure over Rh/TiO2 by simultaneous monitoring of adsorbed CO (carbonyl species) and a broad-band IR background absorbance in accordance with electron accumulation (Fig. 7).33 Actually, CO adsorption over metallic Rh (Rh0) works as a sensitive probe for the detection of co-adsorbed H atoms.32 The prepared Rh/TiO2 was partially covered with CO and supplied H2. Dissociative adsorption of H2 over the Rh0 surface can be detected as a shift of peaks assigned to carbonyl species. They also investigated the time dependence of the charge accumulation amount in TiO2. Obtained time-resolved spectra revealed the existence of two kinetic branches: fast accumulation occurring upon initial 10 min H2 exposure and subsequent slower accumulation for longer exposure times. Initially, H atoms are supplied quickly from Rh0 nanoparticles to TiO2, followed by an immediate response of electron accumulation. Progression of TiO2 n-doping, the conduction band edge of TiO2 (at Rh-TiO2 interface) bends down, meaning the decrease of Fermi level difference between TiO2 and H atoms. This approach lowers the probability of subsequent n-doping. As discussed up to this point, in situ FT-IR measurements for charge accumulation and CO probe have provided some important mechanistic insights into hydrogen spillover.
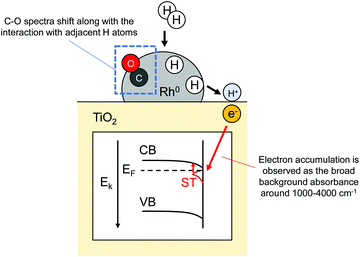 |
| | Fig. 7 Illustration of FT-IR measurement for detecting H atoms adsorption over Rh0 and n-doping into TiO2. | |
3.2 X-ray adsorption spectromicroscopy with clearly separated iron oxide and Pt pairs
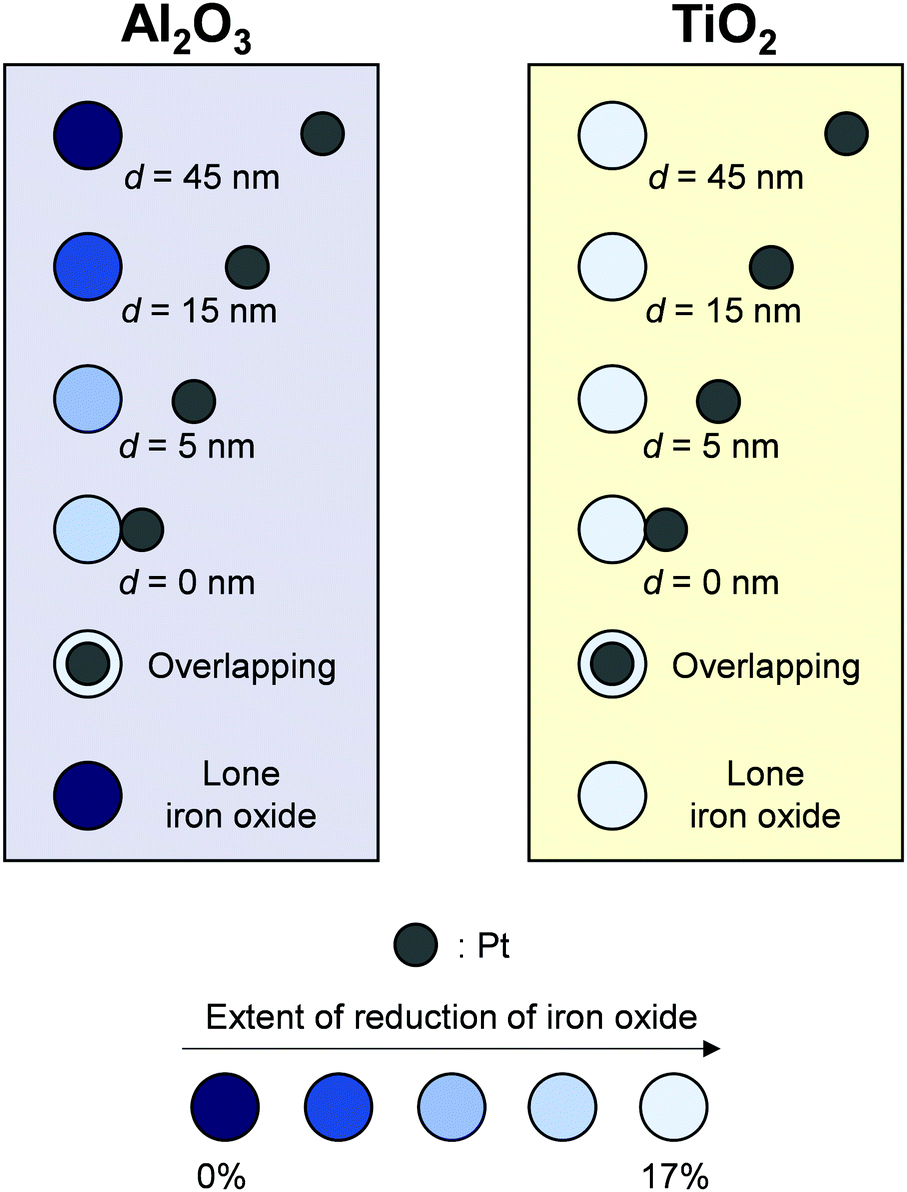
Karim et al. visually quantified hydrogen spillover onto both non-reducible and reducible metal oxides using well-defined samples prepared and confirmed based on results of electron beam lithography74 and STM measurement.29 They arranged clearly separated pairs of iron oxides and Pt nanoparticles over TiO2- and Al2O3-support, precisely controlling the distance of 0–45 nm. Subsequently, they measured iron oxides reduction by H atoms supplied from Pt nanoparticles using in situ X-ray absorption spectromicroscopy (XAS).74 This investigation elicited information about the hydrogen spillover length, as presented in Fig. 8. Regarding Al2O3, 15 nm was the limitation. In contrast, irrespective of the distance between iron oxides and Pt particles, all iron oxide particles were reduced over TiO2, clearly indicating long-range hydrogen spillover. Additionally, they confirmed three-coordinated surface Al adsorption sites,75 which can adsorb H atoms.76 Their works clearly illustrate differences of hydrogen spillover on reducible metal oxides and non-reducible metal oxides. They quantitatively revealed the limitation of spillover distance on Al2O3.
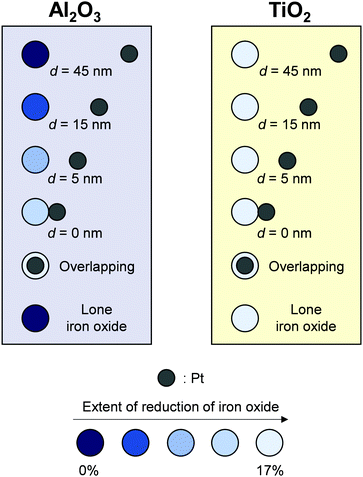 |
| | Fig. 8 Spillover on Al2O3 and TiO2 traced using Pt-iron oxide pairs. | |
3.3 Surface enhanced Raman spectroscopy using Au/TiO2/Pt sandwich sample
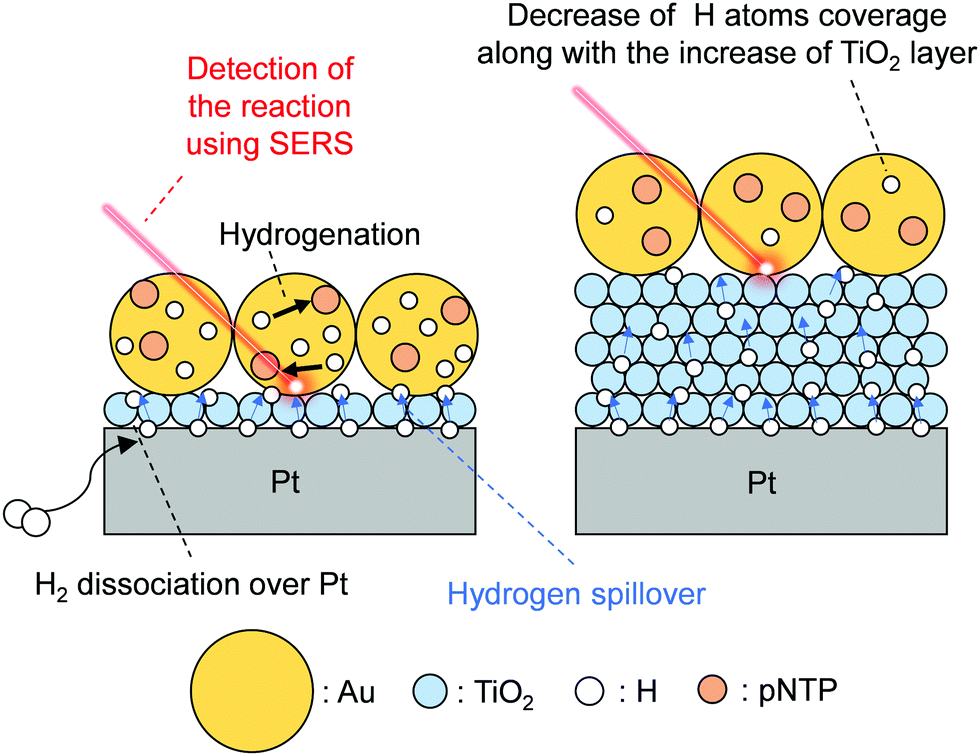
Wei et al. investigated hydrogen spillover and its role in catalytic hydrogenation using surface enhanced Raman spectroscopy (SERS) and precisely tuned Au/TiO2/Pt sandwich nanostructures.34 Reportedly, we can sensitively analyze the vibration of adsorbed molecules over rough metals using SERS by virtue of Raman signal enhancement by a strong electromagnetic field.77–79 Therefore, it is a promising method for tracking catalytic reactions in situ.80,81 The well-fabricated sample (Au/TiO2/Pt sandwich) designed by Wei et al. has spatially isolated active sites (Fig. 9): a hydrogen activation site (Pt), hydrogen migration site (TiO2), and hydrogenation site (Au). For Au, they prepared large particles that cannot dissociate H2.82 They prepared a monolayer of TiO2 nanoparticles (10 nm) using Langmuir–Blodgett (LB) method and loaded it on Pt film. The TiO2 layer height was controlled by increasing the deposition layers. Finally, a monolayer of Au nanoparticles (55 nm) with probe molecule (para-nitrothiophenol, pNTP) was deposited. Using those finely fabricated samples, they performed hydrogenation of pNTP. The reaction was traced by SERS. Consequently, efficient pNTP conversion was detected over Au/10 nm-TiO2/Pt, indicating that H atoms can migrate from Pt to Au through the TiO2 surface. The activation energy increases along with increasing TiO2 thickness. Results show that reaction proceeds only slightly for TiO2 thickness of 50 nm. Furthermore, the selectivity changed by TiO2 thickness because of the change of H atom coverage over Au, which is related to H atom reactivity over Au. Consequently, in situ SERS with well-fabricated sample can achieve direct confirmation of the hydrogen spillover length effects on the reactivity and selectivity of hydrogenation.
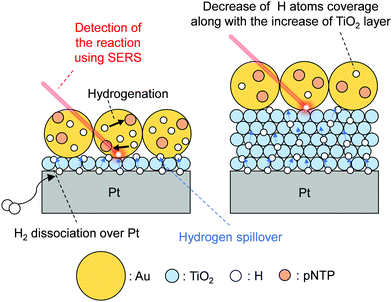 |
| | Fig. 9 Schematic image of hydrogen spillover on a Au/TiO2/Pt sandwich sample. | |
3.4 Encapsulated sample and DFT calculation
As Prins reported in his review, hydrogen spillover on non-reducible materials such as Al2O3, SiO2 and zeolites are controversial issues.24 Recently, direct experimental evidence of the hydrogen spillover on a zeolite surface was found using encapsulated samples.35,36 Details of experiments using zeolite capsules were explained by Choi et al.38 The concept itself is the same as that of the research explained in Section 2.2. Therefore, for this discussion, only the hydrogen migration mechanism over zeolite revealed by DFT calculations is introduced.35,36 Im et al. and Shin et al. reported that dynamic defect generation and annihilation cause and influence long-range hydrogen migration over aluminosilicate with sufficient Brønsted acid sites. Fig. 10 shows the schematic image of H atoms migration mechanism over aluminosilicate. Al placed at the left end in the figure represents the external support surface. It has three covalent bands. At the first stage without defect, one H atom at Brønsted acid sites can migrate to adjacent sites, and the activation energy of migration (through Si atom) is 0.98 eV (Fig. 10(a)).35 Additionally, it was revealed that Pd6 cluster deposition can facilitate this initial defect formation step with 0.52 eV activation energy.37 This first migration generates [AlO4H2]+ and [AlO4]− simultaneously. Once these defect pairs are formed, subsequent hydrogen migration proceeds effectively at both sites (Fig. 10(b)). The [AlO4]− site can accept H radical (H˙) yielding [AlO4]− H˙. Also, [AlO4]− H˙ has a three-centred bond (O–H–O) with local structural distortion compared with Brønsted acid sites [AlO4]− H+ because of spatially localised unpaired electron (i.e. polaron). This unpaired electron located at [AlO4]− readily diffuses into neighbouring Brønsted acid sites, forming another three-centred O–H–O bond (i.e. polaron conduction) (Fig. 10(c)). When H+ reached the cation defect at the external support surface, the H+ recombines with the electron (Fig. 10(c)). It was also confirmed that the cation defect can function as a reaction site of benzene hydrogenation with the reached H+.
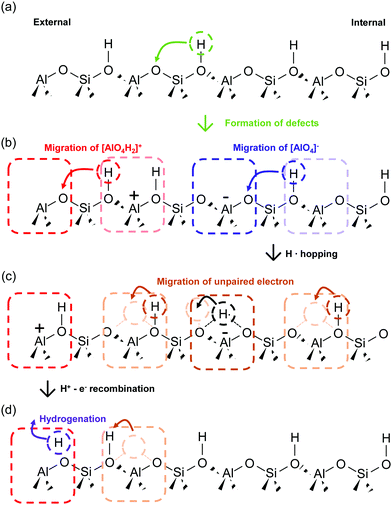 |
| | Fig. 10 Schematic images of H atoms migration mechanism over aluminosilicate. (a) Generation of [AlO4H2]+ and [AlO4]− by H+ migration. (b) Subsequent H atoms migration at both [AlO4H2]+ and [AlO4]−. As for the migration at [AlO4]−, [AlO4]− H˙ is formed. (c) Migration of unpaired electron. H+ reached to the external support surface recombines with the electron. (d) Hydrogenation of hydrocarbons using H+ at the external support. | |
3.5 Electrochemical impedance spectroscopy using porous pellets
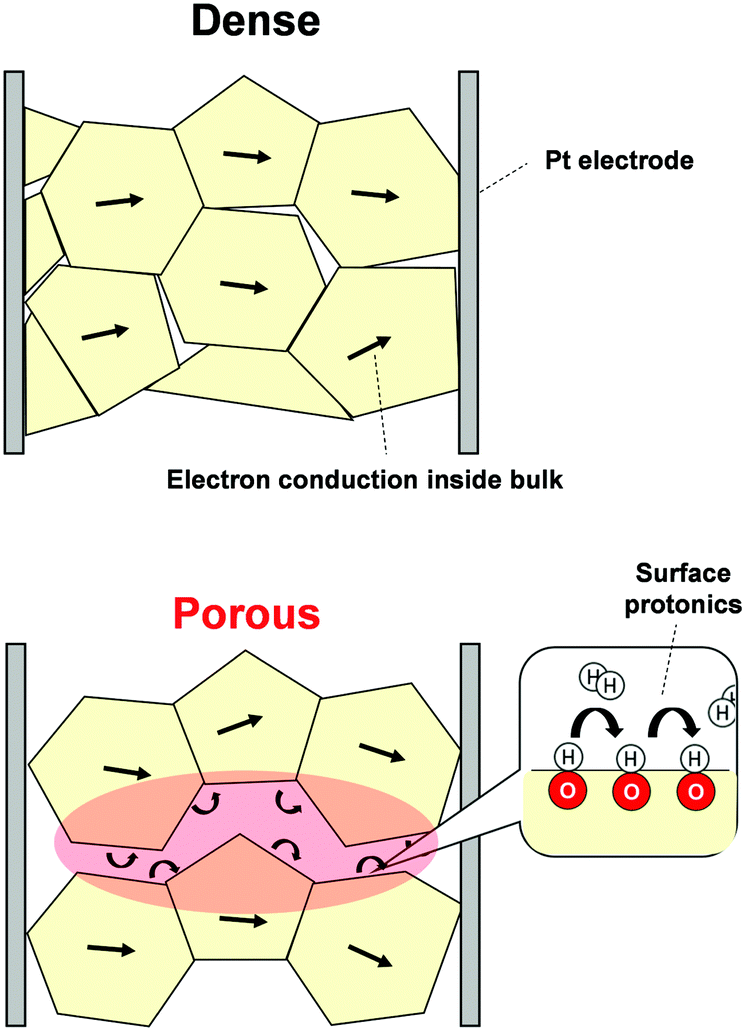
Recently, evaluation methods for surface protonics under H2O atmosphere have been developed using electrochemical impedance spectroscopy (EIS).22,23,41 Furthermore, we have observed surface protonics under H2 atmosphere using this technique.83 First, we prepared two density-controlled SrZrO3 pellets: porous pellets with 60% relative density and dense samples with 90% relative density. With dense samples, the detected conductivity was dominated by resistance of the inner bulk. Conversely, with porous samples, the conduction of adsorbent also becomes necessary. Schematic images of conduction over each sample are presented in Fig. 11. Thanks to open pores, EIS measurements using porous samples provide information about conduction relevant to the adsorbed species. Based on this idea, we conducted EIS measurements under N2 or N2 + H2 atmospheres using density-changed pellets. The apparent conductivity of dense pellets was unaffected by H2 interaction, although that of porous pellets was increased clearly by the H2 supply. Such a completely different response to H2 supply between two pellets can be attributed to the dissociative adsorption of H2 and the formation of conductive species. Additionally, we investigated H/D isotope effects on apparent conductivities to ascertain conductive species under a H2 (D2) atmosphere. Only the porous sample exhibited a H/D isotope effect, indicating that H+ (D+) was the dominant conductive species over porous pellets. In this manner, detection of surface protonics using EIS was demonstrated.
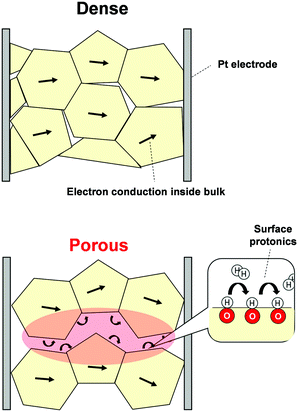 |
| | Fig. 11 Schematic images of EIS measurements using dense and porous samples. | |
4. Low-temperature catalysis using surface hydrogen migration in an electric field
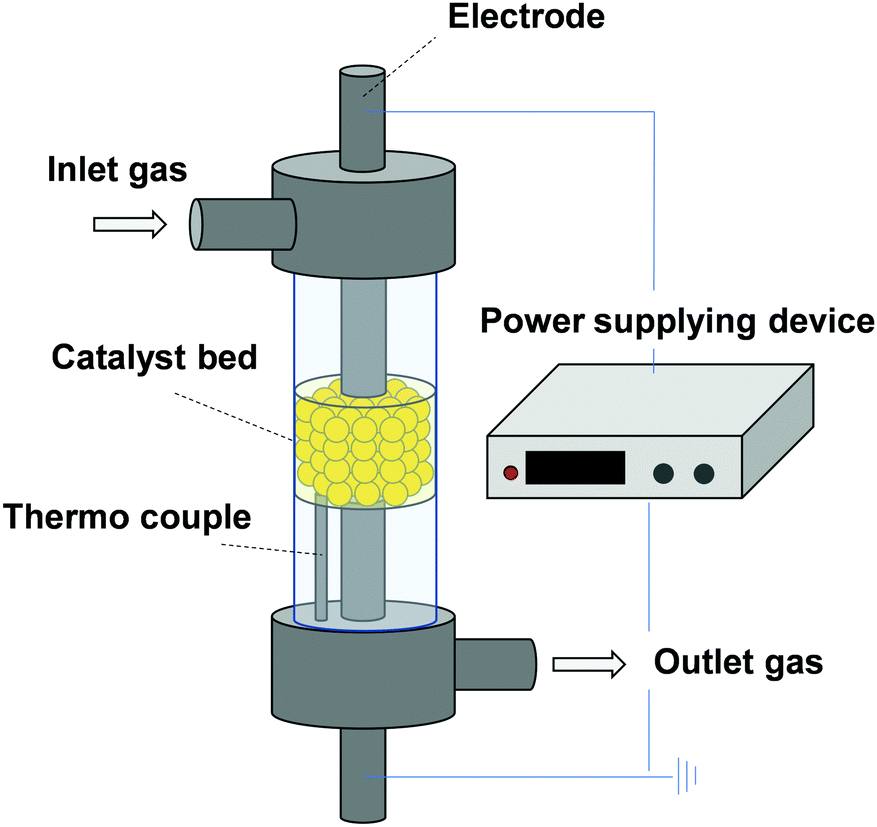
Recently, low-temperature catalysis is attracting great attention for on-site and on-demand processes, which are important for sustainable chemistry. Consequently, moderation of catalyst working conditions using external stimuli such as photonic, magnetic, and electric fields is becoming a key research topic. We have specifically wrestled with enhancement of catalytic activity at low-temperature regions by application of the electric field on catalyst beds. Using this approach, two electrodes are attached on catalyst granules. Weak direct current is applied as presented in Fig. 12. To date, this method has been applied to various catalytic reactions, revealing that surface ion conduction participates in low-temperature catalysis.39,40,84 Herein, we have described reaction mechanisms that are relevant to surface protonics under a H2 atmosphere: NH3 synthesis proceeds via N2H11–17 and dehydrogenation of methylcyclohexane (MCH) facilitated by H+ collision. Both compounds are promising hydrogen carrier candidates. Their efficient synthesis at moderate conditions is needed.85
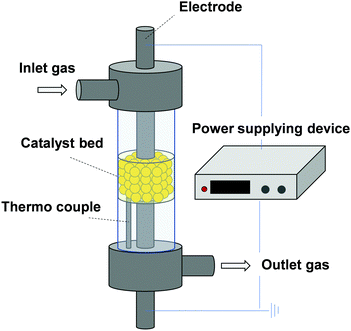 |
| | Fig. 12 Schematic image of the apparatus for catalytic reaction in the electric field. | |
4.1 NH3 synthesis in an electric field
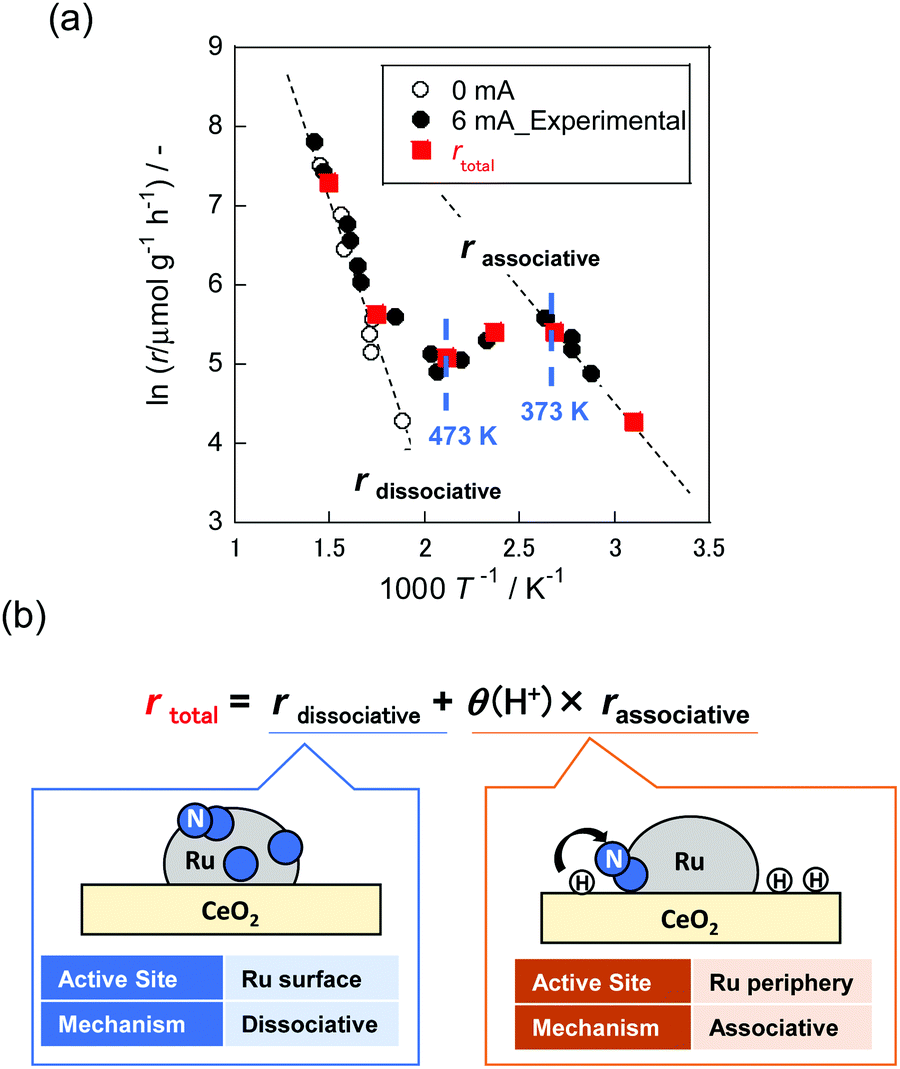
It is noteworthy that NH3 synthesis using Ru/CeO2 as a catalyst in the electric field exhibited anti-Arrhenius-like behaviour at 373–473 K, at which temperature the NH3 synthesis rate increased with lowering of the catalyst bed temperature (Fig. 13(a), black closed plot).16 Furthermore, partial pressures of H2 positively influenced the reaction rate at this temperature region, where anti-Arrhenius-like behaviour was observed. As described in Section 2.4, Ru-based catalysts conventionally show negative values on the H2 reaction order because of H2 poisoning.70,71 Therefore, we assumed that the NH3 synthesis mechanism in the electric field would be different from conventional dissociative mechanisms (eqn (1)–(3)).
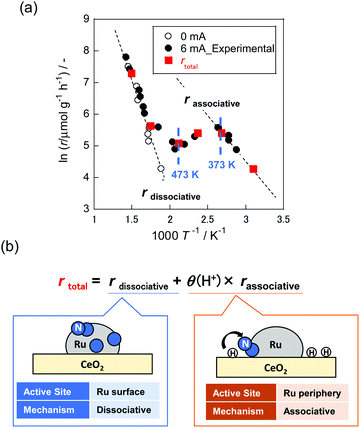 |
| | Fig. 13 Temperature dependence of NH3 synthesis rate (r) without the electric field ((a), open plot), in the electric field ((a), black closed plot) and overall reaction rate ((a), red closed plot), calculated based on the concept shown in (b). | |
Those reactions proceed via a novel mechanism by which H+ derived from the supplied H2 facilitate N2 dissociation (eqn (4)–(8)), which is designated as an associative mechanism.
| | | N2ad + Had+ + e− →N2Had | (6) |
| | | N2Had + Had+ + e− →N2H2ad | (7) |
| | | N2H2ad + Had+ + e− →Nad + NH3 | (8) |
Hereinafter, “ad” denotes adsorbed species. Had+ sits on lattice oxygen of supports (Olat), and N2Hx ad (x = 0–2) represents adsorbed species over supported metals. This hypothesis was elucidated quantitatively using in situ FT-IR measurements. The spectroscopic examination revealed that H+ coverage over supports decreased as the temperature increased. It suggested that H+ coverage over metal oxides played a key role in the anti-Arrhenius-like behaviour. Based on these results, the overall reaction rate (rtotal) was formulated as presented in Fig. 13(b). The first term represents the rate of NH3 synthesis process via dissociative mechanisms over the Ru surface (rdissociative). The second term shows that via associative mechanisms at the Ru periphery (rassociative). These two terms are divisible because of the separation of reaction sites. Furthermore, rassociative was multiplied by H atom coverage over supports (θ(H+)) because the rate-determining step in this reaction mechanism is N2H formation.12 As presented in Fig. 13(a), the reaction rates (red closed plot) calculated based on assumption described above show great agreement with the experimentally obtained values (black closed plot). Consequently, specific low-temperature catalysis using actively promoted hydrogen migration was confirmed.
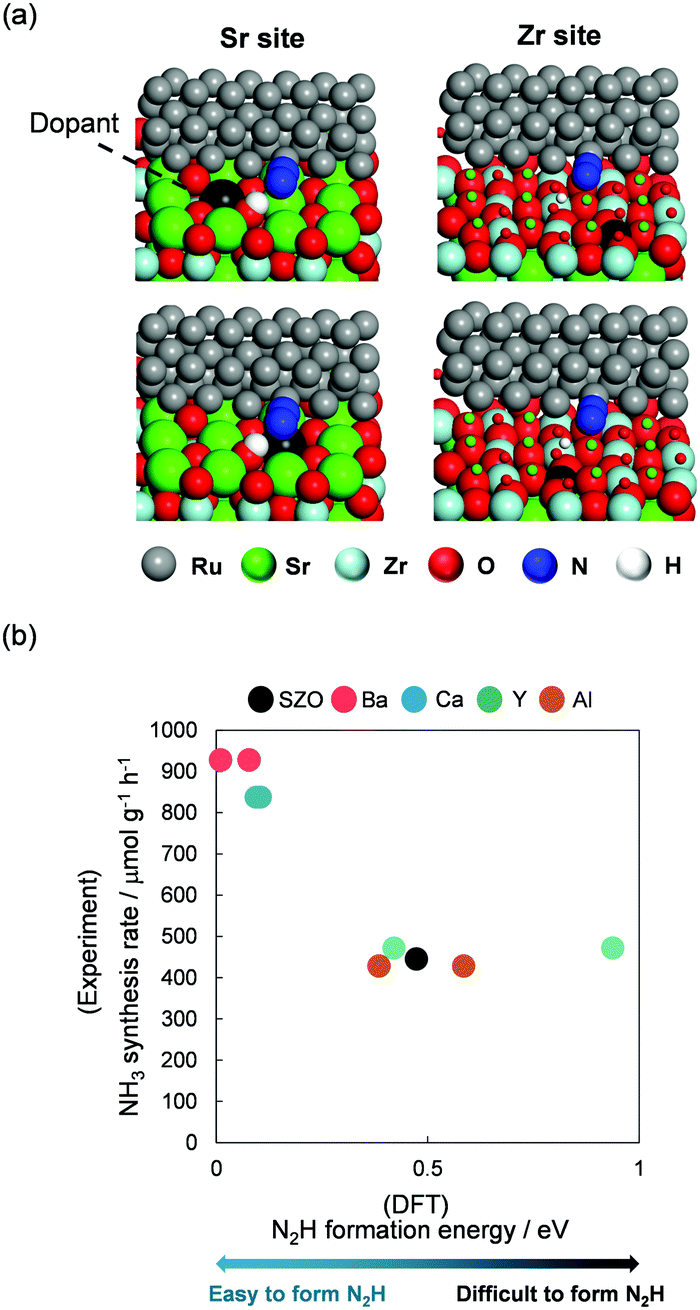
This novel reaction mechanism via N2H was also elucidated based on the relation between the NH3 synthesis rate and N2H formation energy from the viewpoints of supports and active metals. First, we examined the support role in NH3 synthesis in the electric field combining experiments and DFT calculations.12 Ru was chosen as a supported metal. Ru particle was expressed by a Ru rod having a periodic structure in DFT calculation, as presented in Fig. 14(a). This model can keep the experimentally confirmed facets of Ru: (1011) and (0001).11 SrZrO3 was selected as a host support, and the A site (Sr) or B site (Zr) cations were partially replaced by other cations (A site, Ba, Ca; B site, Al, Y). Doped surfaces were expressed by substitution of Sr (or Zr) around a Ru rod during DFT calculations (Fig. 14(a)). Using those as model supports and an active metal, correlation between DFT calculated N2H formation energy at Ru periphery and experimentally obtained NH3 synthesis rate was regarded as presented in Fig. 14(b). Results revealed that the NH3 synthesis rate increased as the N2H formation became feasible. In addition, analyses of the electronic density and adsorption energies of intermediates using DFT calculations revealed that both the electron and proton donation ability of supports were fundamentally important for high NH3 synthesis rates in the electric field.
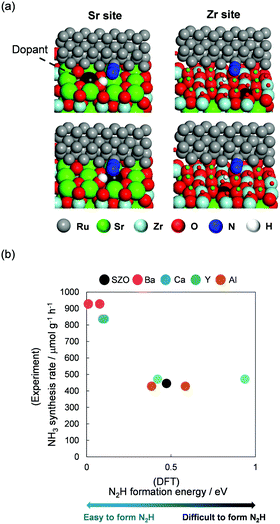 |
| | Fig. 14 (a) DFT calculation models for Ru-supported SrZrO3 with dopants, and (b) correlation between the experimentally obtained NH3 synthesis rate in the electric field and DFT calculated N2H formation energy. | |
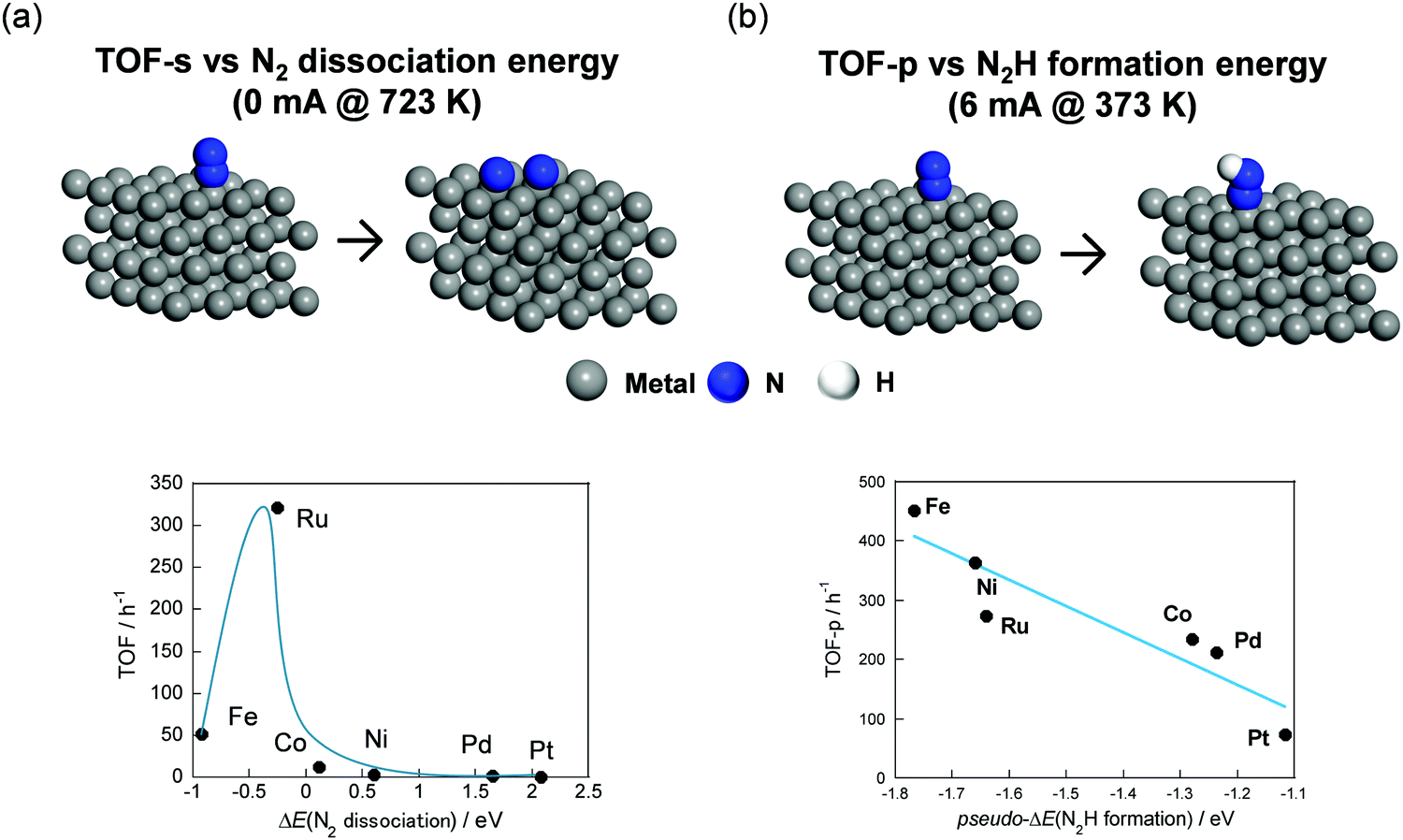
Secondly, the effects of active metals on NH3 synthesis rate in the electric field were investigated using CeO2 as a support. Fig. 15 portrays schematic images of DFT calculation models and the relation between experimentally obtained TOF and DFT calculated N2 dissociation energy or N2H formation energy. Here, TOF-s represents the activity per exposed metal atom. Also, TOF-p represents the activity per exposed periphery of loading metal. TOF depends on the N2 dissociation energy without the electric field (Fig. 15(a)). Then, Ru showed the highest performance. This trend corresponded closely to descriptions in earlier reports.86 A surprising finding is that Fe and Ni showed higher TOF than Ru did in the electric field. The result was found to depend on the N2H formation energy (Fig. 15(b)). As described above, a novel reaction mechanism via N2H was confirmed from various analyses.
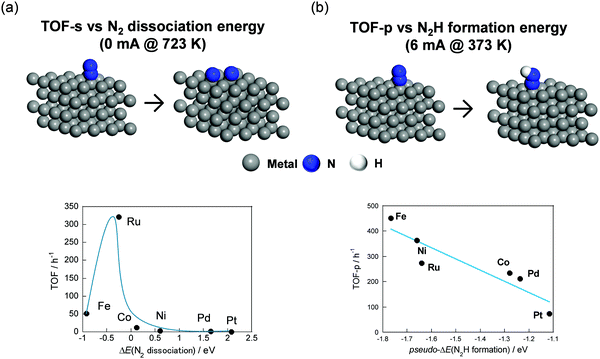 |
| | Fig. 15 Illustration of DFT calculation models and relation between TOF-s without the electric field and N2 dissociation energy (a), TOF-p with the electric field, and N2H formation energy (b). | |
4.2 MCH dehydrogenation in the electric field
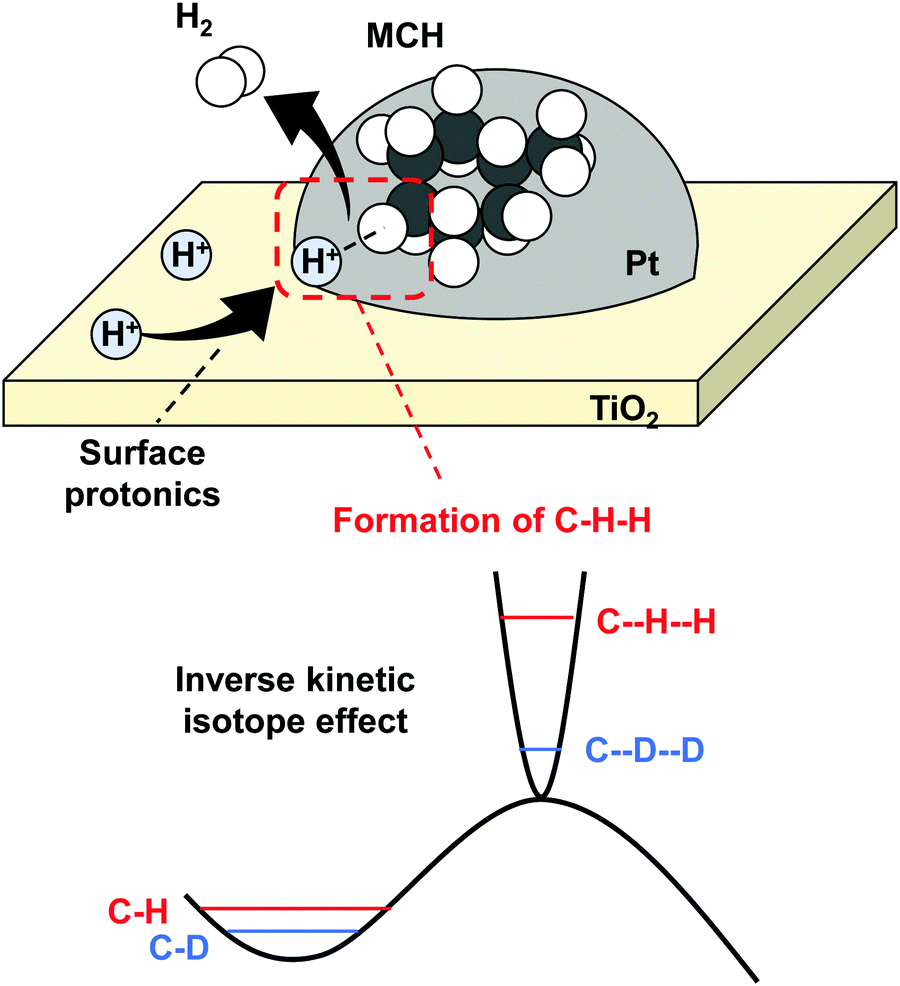
MCH dehydrogenation in the electric field also proceeds at low temperatures by virtue of surface protonics. Even in this reaction, the H2 reaction order was positive in the electric field. Commonly, an opposite trend is observed in dehydrogenation.87 Therefore, we considered that the MCH dehydrogenation rate was also enhanced by surface protonics. The facilitation mechanism is little different from that of NH3 synthesis in the electric field. In this reaction, the rate-determining step is C–H bond cleavage, which is promoted by the collision of H+ in the electric field, as presented in Fig. 16. We clarified a similar mechanism in CH4 steam reforming with the electric field.42,43 In this reaction mechanism, H+ and MCH form three atoms (C–H–H) structures at the transition state. This peculiar mechanism was elucidated by an inverse kinetic isotope effect, which was observed when MCH (C7H14) was replaced by MCHD (C7D14). Reportedly, C–H–H at the transition state has stronger bond than C–H has,88–90 meaning the potential curve at the transition state is steeper than that at the initial state. The zero-point energy difference between C–H–H and C–D–D is large than that between C–H and C–D. Therefore, the activation energy decreased when the hydrogen was exchanged with heavier deuterium.
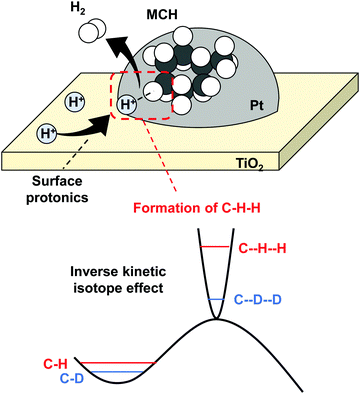 |
| | Fig. 16 Schematic image of MCH dehydrogenation facilitated by surface protonics, and inverse kinetic isotope effect. | |
5. Perspective
This review described recent investigations for the utilisation and observation of surface hydrogen migration over metal oxides.
Catalytic reactions related to the hydrogen spillover were highlighted. Migration provides various benefits. The hydrogen spillover effects are divisible into two types. One is the supply of H atoms to sites where we cannot activate H2. The other is the expulsion of H atoms from sites that have excess H atoms. The former is worthwhile from various perspectives. Net migration of H atoms from metal nanoparticles to single atom sites expanded the usage of single atom catalysts that have high atomic efficiency. Hydrogenation over metal oxides with H atoms supplied by hydrogen spillover can hydrogenate hydrocarbons selectively. The latter is simpler. The benefit is hydrogen poisoning suppression, as we introduced with examples of NH3 synthesis.
We have demonstrated various cutting-edge analyses that have supported our in-depth understanding of hydrogen spillover. In situ FT-IR measurements are the most powerful method to detect as-made complicated catalysts that are usually used in various situations. This method can detect the accumulation of electrons supplied from H atoms to the shallow state of semiconductors. XAS and SERS measurements with well-fabricated samples can detect the hydrogen spillover directly and can provide fundamental migration mechanisms. EIS measurements with porous pellets can evaluate the electrochemical conductivity of H atoms over metal oxide surfaces, which is important for the utilisation of surface protonics for fuel cells and sensors.
Finally, we introduced novel low-temperature catalytic reactions using surface protonics, which is hydrogen migration activated by the electric field. Regarding NH3 synthesis in the electric field, N2 is protonated by active H+. The reaction proceeds via N2H intermediate. Furthermore, C–H bonds in MCH are dissociated by collision of H+ and the formation of the C–H–H structure. This novel concept, utilisation of actively migrated H atoms, has great potential because it is readily applicable to various reactions.
Understanding of hydrogen spillover and novel concepts of catalysis using surface proton hopping have been expanding the usage of surface hydrogen migration. Therefore, in the field of catalysis, conceptual design of catalysts and in-depth knowledge of individual reaction systems involving hydrogen migration are promising. Furthermore, doping into metal oxides can control the surface hydrogen migration amounts, but the governing factors of the surface hydrogen conductivity remain unclear. Therefore, their elucidation is a crucially important theme for future analyses.
Conflicts of interest
There are no conflicts to declare.
References
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef.
- S. Campisi, C. E. Chan-Thaw, L. E. Chinhilla, A. Chutia, G. A. Botton, K. M. H. Mohammed, N. Dimitratos, P. P. Wells and A. Villa, ACS Catal., 2020, 10, 5483–5492 CrossRef CAS.
- X. Xue, J. Liu, D. Rao, S. Xu, W. Bing, B. Wang, S. He and M. Wei, Catal. Sci. Technol., 2017, 7, 650–657 RSC.
- K. Takise, A. Sato, K. Murakami, S. Ogo, J. G. Seo, K. Imagawa, S. Kado and Y. Sekine, RSC Adv., 2019, 9, 5918–5924 RSC.
- M. Kosaka, T. Higo, S. Ogo, J. G. Seo, K. Imagawa, S. Kado and Y. Sekine, Int. J. Hydrogen Energy, 2020, 45(1), 738–743 CrossRef CAS.
- T. Franken, J. Terreni, A. Borgschulte and A. Heel, J. Catal., 2020, 382, 385–394 CrossRef CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, N. Kruse and G. A. Somorjai, Nano Lett., 2014, 14(8), 4792–4796 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y. W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- T. Yabe, K. Yamada, T. Oguri, T. Higo, S. Ogo and Y. Sekine, ACS Catal., 2018, 8, 11470–11477 CrossRef CAS.
- S. Wu, Y.-K. Peng, T.-Y. Chen, J. Mo, A. Large, I. McPherson, H.-L. Chou, I. Willkinson, F. Venturini, D. Grinter, P. F. Escorihuela, G. Held and S. C. E. Tsang, ACS Catal., 2020, 10, 5614–5622 CrossRef CAS.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, A. Ishikawa, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- A. Gondo, R. Manabe, R. Sakai, K. Murakami, T. Yabe, S. Ogo, M. Ikeda, H. Tsuneki and Y. Sekine, Catal. Lett., 2018, 148(7), 1929–1938 CrossRef CAS.
- K. Murakami, R. Manabe, H. Nakatsubo, T. Yabe, S. Ogo and Y. Sekine, Catal. Today, 2018, 303, 271–275 CrossRef CAS.
- K. Murakami, Y. Tanaka, S. Hayashi, R. Sakai, Y. Hisai, Y. Mizutani, A. Ishikawa, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki, H. Nakai and Y. Sekine, J. Chem. Phys., 2019, 151, 064708 CrossRef.
- K. Murakami, Y. Tanaka, R. Sakai, K. Toko, K. Ito, A. Ishikawa, T. Higo, T. Yabe, S. Ogo, M. Ikeda, H. Tsuneki, H. Nakai and Y. Sekine, Catal. Today, 2020, 351, 119–124 CrossRef CAS.
- K. Murakami, Y. Tanaka, R. Sakai, Y. Hisai, S. Hayashi, Y. Mizutani, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki and Y. Sekine, Chem. Commun., 2020, 56, 3365–3368 RSC.
- R. Sakai, K. Murakami, Y. Mizutani, Y. Tanaka, S. Hayashi, A. Ishikawa, T. Higo, S. Ogo, H. Tsuneki, H. Nakai and Y. Sekine, ACS Omega, 2020, 5(12), 6846–6851 CrossRef CAS.
- A. Aitbekova, E. D. Goodman, L. Wu, A. Boubnov, A. S. Hoffman, A. Genc, H. Cheng, L. Casalena, S. R. Bare and M. Cargnello, Angew. Chem., Int. Ed., 2019, 58, 17451–17457 CrossRef CAS.
- S. Masuda, K. Shun, K. Mori, Y. Kuwahara and H. Yamashita, Chem. Sci., 2020, 11, 4194–4203 RSC.
- A. Lueking and R. T. Yang, J. Catal., 2002, 206, 165–168 CrossRef CAS.
- F. H. Yang and R. T. Yang, Carbon, 2002, 40(3), 437–444 CrossRef CAS.
- S. Miyoshi, Y. Akao, N. Kuwata, J. Kawamura, Y. Oyama, T. Yagi and S. Yamaguchi, Chem. Matter, 2014, 26(18), 5194–5200 CrossRef CAS.
- S. Ø. Stub, E. Vøllestad and T. Norby, J. Phys. Chem. C, 2017, 121, 12817–12825 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- S. Khoobiar, J. Phys. Chem., 1964, 68, 411–412 CrossRef CAS.
- L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2008, 112(6), 1755–1758 CrossRef CAS.
- X. Sha, L. Chem, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2009, 113(26), 11399–11407 CrossRef CAS.
- W. Karim, C. Spreafic, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS.
- D. A. Panayotov and J. T. Yates, J. Phys. Chem. C, 2007, 111(7), 2959–2964 CrossRef CAS.
- D. A. Panayotov, S. P. Burrows, J. T. Yates and J. R. Morris, J. Phys. Chem. C, 2011, 115(45), 22400–22408 CrossRef CAS.
- D. Panayotov, M. Mihaylov, D. Nihitianova, T. Spassov and K. Hadjiivanov, Phys. Chem. Chem. Phys., 2014, 16, 13136–13144 RSC.
- D. Panayotov, E. Ivanova, M. Mihaylov, K. Chakarova, T. Spassov and K. Hadjiivanov, Phys. Chem. Chem. Phys., 2015, 17, 20563–20573 RSC.
- J. Wei, S.-N. Qin, J.-L. Liu, X.-Y. Ruan, Z. Guan, H. Yan, D.-Y. Wei, H. Zhang, J. Cheng, H. Xu, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2020, 59, 1–6 CrossRef.
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Nat. Commun., 2014, 5, 3370 CrossRef.
- S. Lee, K. Lee, J. Im, H. Kim and M. Choi, J. Catal., 2015, 325, 26–34 CrossRef CAS.
- H. Shin, M. Choi and H. Kim, Phys. Chem. Chem. Phys., 2016, 18, 7035–7041 RSC.
- M. Choi, S. Yook and H. Kim, ChemCatChem, 2015, 7, 1048–1057 CrossRef CAS.
- M. Torimoto, K. Murakami and Y. Sekine, Bull. Chem. Soc. Jpn., 2019, 92(10), 1785–1792 CrossRef CAS.
-
Y. Sekine and R. Manabe, Faraday Discuss., 10.1039/C9FD00129H.
- R. Manabe, S. Stub, T. Norby and Y. Sekine, Solid State Commun., 2018, 270, 45–49 CrossRef CAS.
- R. Manabe, S. Okada, R. Inagaki, K. Oshima, S. Ogo and Y. Sekine, Sci. Rep., 2016, 6, 38007 CrossRef CAS.
- S. Okada, R. Manabe, R. Inagaki, S. Ogo and Y. Sekine, Catal. Today, 2018, 307, 272–276 CrossRef CAS.
- M. Torimoto, S. Ogo, D. Harjowinoto, T. Higo, J. G. Seo, S. Furukawa and Y. Sekine, Chem. Commun., 2019, 55, 6693–6695 RSC.
- A. Takahashi, R. Inagaki, M. Torimoto, Y. Hisai, T. Matsuda, Q. Ma, J. G. Seo, T. Higo, H. Tsuneki, S. Ogo, T. Norby and Y. Sekine, RSC Adv., 2020, 10, 14487–14492 RSC.
- M. Torimoto, S. Ogo, Y. Hisai, N. Nakano, A. Takahashi, Q. Ma, J. G. Seo, H. Tsuneki, T. Norby and Y. Sekine, RSC Adv., 2020, 10, 26418–26424 RSC.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46(8), 1740–1748 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7(1), 34–59 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Marvrikakis and M. F. Stephanopoulos, Science, 2014, 346(6216), 1498–1501 CrossRef CAS.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wui and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. F. Stephanopoulos, G. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902–2907 CrossRef CAS.
- H. Yan, H. Lv, H. Yi, W. Liu, Y. Xia, X. Huang, W. Huang, S. Wei, X. Wu and J. Lu, J. Catal., 2018, 366, 70–79 CrossRef CAS.
- L. Kuai, S. Liu, S. Cao, Y. Ren, E. Kan, Y. Zhao, N. Yu, F. Li, X. Li, Z. Wu, X. Wang and B. Geng, Chem. Matter, 2018, 30(16), 5534–5538 CrossRef CAS.
- R. Liu, Y. Yang, N. Sun and L. Kuai, Chem. – Eur. J., 2019, 25, 15586–15593 CrossRef.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3(12), 2655–2668 CrossRef CAS.
- X. Meng, Y. Yang, L. Chen, M. Xu, X. Zhang and M. Wei, ACS Catal., 2019, 9(5), 4226–4235 CrossRef CAS.
- W. Li, J. Yang, Z. Wu, J. Wang, B. Li, S. Feng, Y. Deng, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2012, 134(29), 11864–11867 CrossRef CAS.
- G. Zhou, Y. Pei, Z. Jiang, K. Fan, M. Qiao, B. Sun and B. Zong, J. Catal., 2014, 311, 393–403 CrossRef CAS.
- J. Liu, S. Xu, W. Bing, F. Wang, C. Li, M. Wei, G. Evans and X. Duan, ChemCatChem, 2015, 7(5), 846–855 CrossRef CAS.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS.
- A. Ozaki, K. Aika and H. Hori, Bull. Chem. Soc. Jpn., 1971, 44(11), 3216 CrossRef CAS.
- K. Aika, H. Hori and A. Ozaki, J. Catal., 1972, 27(3), 424–431 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS.
- M. Kitano, J. Kujirai, K. Ogasawara, S. Matsuishi, T. Tada, H. Abe, Y. Niwa and H. Hosono, J. Am. Chem. Soc., 2019, 141(51), 20344–20353 CrossRef CAS.
- Y. Ogura, K. Sato, S. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555–558 CrossRef CAS.
- K. Aika, Catal. Today, 2017, 286, 14–20 CrossRef CAS.
- H. Bielawa, O. Hinrichsen, A. Birkner and M. Muhler, Angew. Chem., Int. Ed., 2001, 40, 1061–1063 CrossRef CAS.
- F. Boccuzzi, A. Chiorino, G. Ghiotti and E. Guglielminotti, Langmuir, 1989, 5(1), 66–70 CrossRef CAS.
- E. Guglielminotti, F. Boccuzzi, G. Ghiotti and A. Chiorino, Surf. Sci., 1987, 189–190, 331–338 CrossRef CAS.
- W. Karim, A. Kleibert, U. Hartfelder, A. Balan, J. Gobrecht, J. A. van Bokhoven and Y. Ekinci, Sci. Rep., 2016, 6, 18818 CrossRef CAS.
- J. A. van Bokhoven, A. M. J. van der Eerden and D. C. Koningsberger, J. Am. Chem. Soc., 2003, 125, 7435–7442 CrossRef CAS.
- R. Wischert, P. Laurent, C. Copéret, F. Delbecq and P. Sautet, J. Am. Chem. Soc., 2012, 134, 14430–14449 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- S. Nie and R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fun, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS.
- V. Joseph, C. Engelbrekt, J. Zhang, U. Gernert, J. Ulstrup and J. Kneipp, Angew. Chem., Int. Ed., 2012, 51, 7592–7596 CrossRef CAS.
- W. Xie, R. Grzeschik and S. Schlüker, Angew. Chem., Int. Ed., 2016, 55, 13729–13733 CrossRef CAS.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109(30), 14581–14587 CrossRef CAS.
- Y. Hisai, K. Murakami, Y. Kamite, Q. Ma, E. Vøllestad, R. Manabe, T. Matsuda, S. Ogo, T. Norby and Y. Sekine, Chem. Commun., 2020, 56, 2699–2702 RSC.
- S. Ogo and Y. Sekine, Chem. Rec., 2017, 17(8), 726–738 CrossRef CAS.
- K. Takise and Y. Sekine, Acc. Mater. Surf. Res., 2019, 4(3), 115–134 Search PubMed.
- S. Dahl, A. Logadottir, C. J. H. Jacobsen and J. K. Nørskov, Appl. Catal., A, 2001, 222, 19–29 CrossRef CAS.
- F. Alhumaidan, D. Cresswell and A. Garforth, Energy Fuels, 2011, 25, 4217–4234 CrossRef CAS.
- K. B. Wiberg, Chem. Rev., 1955, 55(4), 713–743 CrossRef CAS.
- B. Kerkeni and D. C. Clary, Phys. Chem. Chem. Phys., 2006, 8, 917–925 RSC.
- M. J. Kurylo, G. A. Hollinden and R. B. Timmons, J. Chem. Phys., 1970, 52, 1773 CrossRef CAS.
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*