
Twenty-five years of bis-pentafluorophenyl borane: a versatile reagent for catalyst and materials synthesis
Evan A.
Patrick
 and
Warren E.
Piers
*
and
Warren E.
Piers
*
Department of Chemistry, University of Calgary, 2500 University Drive N.W., Calgary, Alberta T2N 1N4, USA. E-mail: wpiers@ucalgary.ca
First published on 17th December 2019
Abstract
In 1995, the synthesis, properties and remarkable hydroboration activity of bis-pentafluorophenyl borane was first reported. Its reactivity stems from the ready accessibility of the monomeric borane and its high Lewis acidity. In the intervening twenty five years, this reagent has been widely exploited as a means of incorporating Lewis acidic –B(C6F5)2 groups into complex structures for a range of applications. In this “25th Anniversary” Feature article, we highlight the synthetic methods to the borane, its fundamental properties and chemistry as well as the diverse array of uses of this borane. These include self-activating olefin polymerization catalysts, frustrated Lewis pair generation, small molecule activation, bond cleavage reactions, Lewis acid catalysis and modification of organic materials.
Introduction
Perfluoroarylboranes1 are an important class of organoboranes that have been employed extensively as Lewis acids for a variety of chemical applications. The parent compound, B(C6F5)3, was reported in the early 1960s,2,3 essentially as a curiosity, but its remarkable properties led eventually to its extensive exploitation in 1990s as an olefin polymerization co-catalyst.4 This resulted in its widespread commercial availability and gave chemists the opportunity to explore applications as a Lewis acid catalyst for many other useful transformations.4–9 Key to its versatility are the properties imparted by the three electron withdrawing C6F5 groups, including both high Lewis acidity (comparable to boron trihalides BF3 and BCl38) and B–C bonds that are remarkably resistant to protic cleavage. Combined with a ready solubility in organic solvents, B(C6F5)3 is a near ideal organometallic Lewis acid.5Though B(C6F5)3 has shown exceptional utility, new applications frequently require a tailored approach for construction of more complex and function-specific molecules. Accordingly, in the early 1990's we targeted the synthesis of bis-pentafluorophenyl borane, (C6F5)2BH, 1, envisioning it as a potentially useful synthon for incorporating –B(C6F5)2 groups into molecules via hydroboration or sigma bond metathesis protocols. Since the first publication describing its synthesis 25 years ago in 1995,10 bis-pentafluorophenylborane has seen widespread use and (thanks to the organic chemistry community's penchant for naming reactions and reagents!) has come to be known in the literature as “Piers’ borane”.11 It not only retains high Lewis acidity but provides a reactive function in the hydride for hydroboration and small molecule activation. Originally intended for generating soluble, self activating Ziegler–Natta-type olefin polymerization catalysts,12 this exceptional hydroboration reagent13,14 has found a broadened scope of application to fields ranging from frustrated Lewis pair synthesis, to Lewis acid functionalised materials, to small molecule activation, and metal-free catalysis. In this feature article, we highlight the chemistry and some of the many applications of 1 reported in the past 25 years.
Synthesis and properties of (C6F5)2BH
Wagner and co-workers have provided a cogent overview of the synthetic methods to bis-pentafluorophenylborane,15 which we summarize and augment here. In our original synthesis, [(C6F5)2BH]2 was generated through the transmetallation of (C6F5)2SnMe2 with BCl3 to form the monomeric chloroborane (C6F5)2BCl, which was converted to 1 using Me2Si(Cl)H as a hydride source (Scheme 1a, red route).10 This procedure is highly reliable but has been justifiably described as being “synthetically demanding”.16 Given the hazards associated with fluorinated organolithium reagents (which can be circumvented by using the less explosive C6F5MgBr) and organotin compounds, an alternative route involving reaction of Et3SiH with commercially available B(C6F5)3 at 60 °C (Scheme 1a, green route) was developed, providing 1 in 69% yield.13 Though the cost of B(C6F5)3 is a disadvantage, this is a more convenient synthesis of gram quantities of 1, and seems to be the route of choice for most researchers. Care must be taken not to over-reduce the borane and samples prepared in this way can be contaminated with small amounts (2–4%) of [(C6F5)BH(μ-H)2B(C6F5)2]. This was confirmed in a recently published careful spectroscopic and kinetic study on the reactions of B(C6F5)3 with various silanes and germanes.17 Interestingly, the haloboranes (C6F5)2BX (X = Cl, Br) are now available cleanly and in very high yield from 1 by treatment of the borane with trityl chloride or bromide.18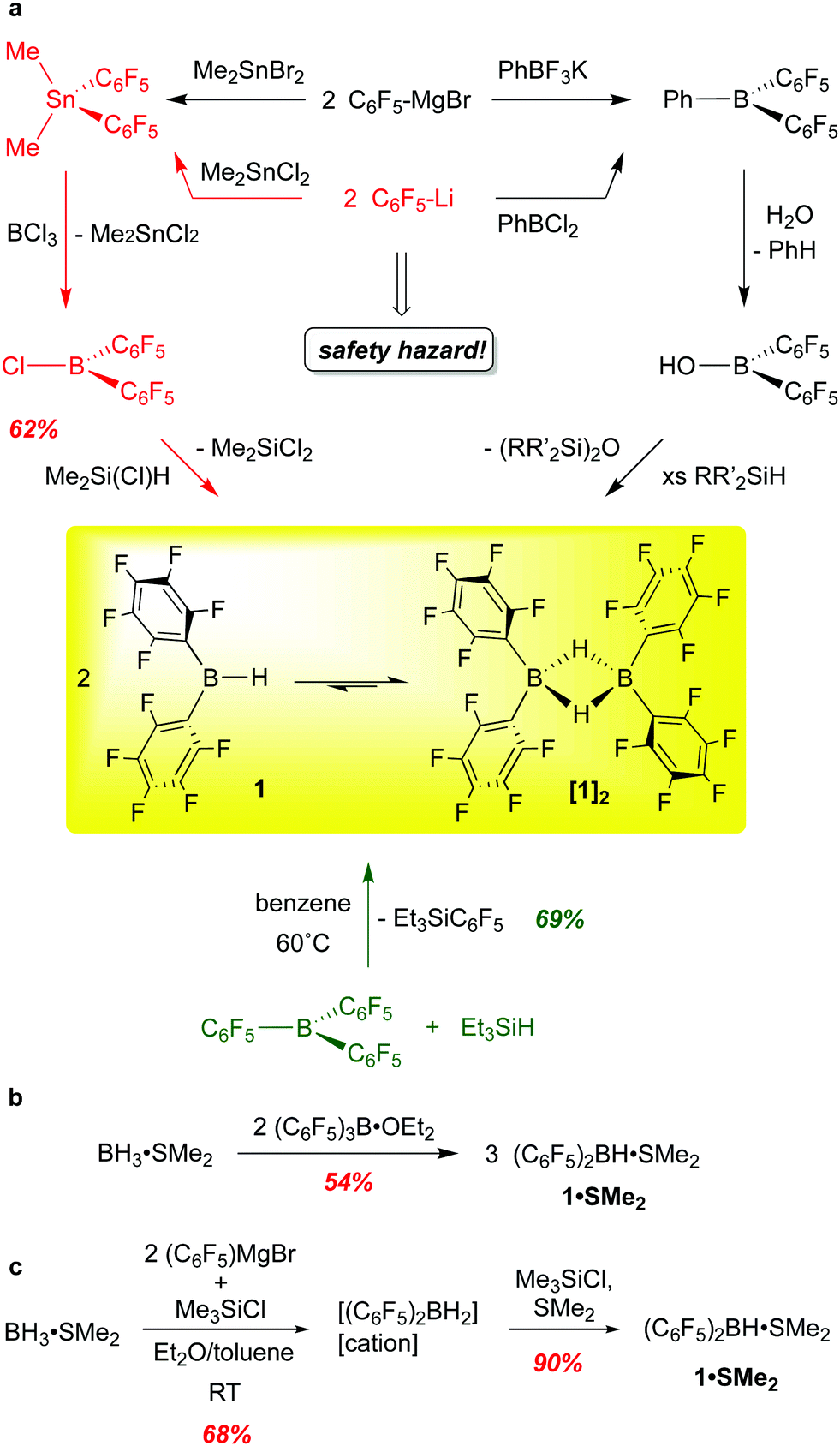 | ||
| Scheme 1 Synthetic routes to HB(C6F5)2, 1, and the SMe2 adduct 1·SMe2. | ||
Other researchers have developed methods to prepare 1in situ via the borinic acid (C6F5)2BOH (Scheme 1a, black route) or as the SMe2 adduct (Scheme 1b and c). Chang and co-workers take advantage of selective protonolysis of the non-fluorinated aryl B–C bond in (C6F5)2BPh to generate the borinic acid, which can be converted to 1 with various silanes.19 This reaction appears to be quantitative as determined by NMR spectroscopy but has not been utilized to make 1 preparatively; this would seem to bear some further investigation. The groups of Lancaster16 and Wagner15,20 have accessed (C6F5)2BH·SMe2 using a ligand redistribution strategy, taking advantage of the facile exchange of –C5F5 and hydride groups at boron. Scheme 1b shows Lancaster's route from B(C6F5)3 and Me2S·BH3, while Wagner's strategy (Scheme 1c) utilizes the more readily available pentafluorophenyl Grignard reagent to generate a mixture of hydridoborates dominated by [(C6F5)2BH2]−[cation]+. Subsequent hydride abstraction/salt elimination with Me3SiCl in the presence of SMe2 delivers 1·SMe2. Both preparations give decent yields of the SMe2 adduct of 1, and procedures provide straightforward and relatively safe access to a bis-pentafluorophenyl boron synthon. Unfortunately, 1·SMe2 does not have the same broad utility of base-free borane. Although 1·SMe2 has been shown to hydroborate alkenes and alkynes,21 the substrate scope is quite narrow, possibly due to the decreased access to the monomeric borane from the SMe2 adduct compared to the free borane [(C6F5)2BH]2. Furthermore, in some instances, the presence of the Lewis base led to unwanted side-products due to nucleophilic attack by free SMe2.22
When prepared from B(C6F5)3 and Et3SiH, 1 is isolated as a moisture sensitive, white, microcrystalline solid, [(C6F5)2BH]2 that can be stored at room temperature in an inert atmosphere for months without losing any of its activity.10 In the solid state, the borane is dimeric with two bridging hydrides, typical of [R2BH]2 structures. In arene solution, however, a monomer–dimer equilibrium is observed, with up to 10–15% of the borane speciation in the monomeric form; a 5 kcal mol−1 barrier to dissociation has been estimated.13,23 The favouring of monomer to this extent is unusual for diarylboranes and accounts partially for the compound's exceptional hydroboration activity and reactivity.
The substitution of a C6F5 substituent for a hydride dampens the Lewis acidity in 1vs. B(C6F5)3 somewhat, but bis-pentafluorophenyl borane retains significant electrophilicity. Gas phase calculations have determined the electron affinity of (C6F5)2BH is over 2.7 times greater than for (C6H5)2BH.24 Accordingly, in addition to high hydroboration activity, [(C6F5)2BH]2 also demonstrates catalytic H/D exchange with H2 and various deuterated silanes.25 Though the borane was calculated to lack sufficient Lewis acidity for direct binding of H2, experimental and computational evidence indicate that a facile sigma-bond metathesis type reaction is operative in activating both H–H and Si–H bonds at room temperature. Such reactivity is usually associated with highly Lewis acid centres.26
While other fluorinated diaryl boranes have been prepared,27–29 they tend to be less reactive than 1 for electronic30 or steric31 reasons and have not been utilized nearly to the same extent as 1 since we reported it 25 years ago.10 The high reactivity of monomeric 1 towards a variety of nucleophilic and unsaturated functions has made it the prime synthon for incorporating Lewis acidic –B(C6F5)2 units into organometallic, organic and materials structures, for applications ranging from polymerization catalysis to FLP generation to small molecule activation. Before discussing these applications, we review its reactivity with a variety of hydrocarbyl and other coordinated ligands.
Reactions of 1 with coordinated ligands
Because of the efficacy of perfluoroarylboranes as olefin polymerization co-catalysts we undertook a systematic study of the reactions of 1 towards archetypical metal hydrocarbyl functions, i.e. metal alkyls and alkylidenes, to determine the compatibility of the borane with these groups. These studies were later extended to include akylidyne and carbide functions and a summary of the reactivity observed is shown in Scheme 2. Although primarily exploratory in nature, discoveries of both fundamental and practical significance emanated from this line of research.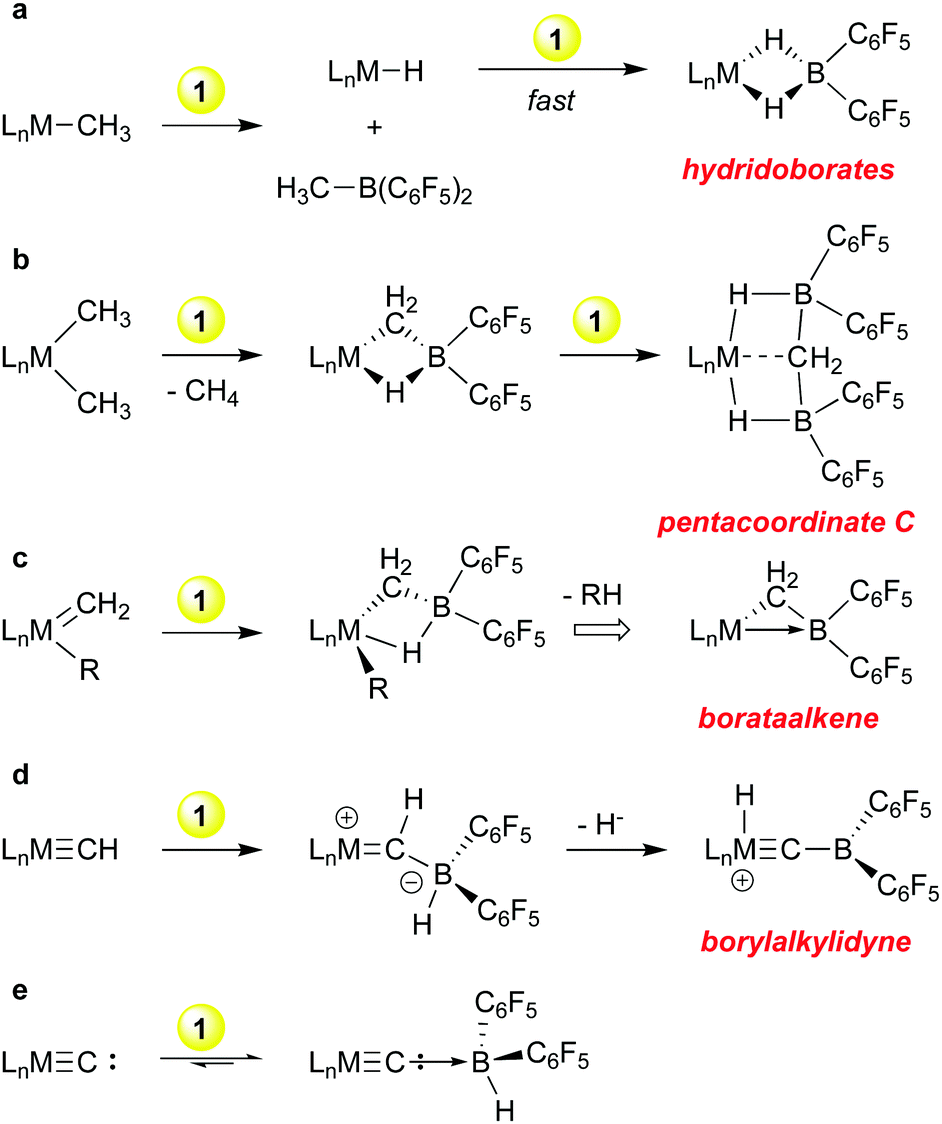 | ||
| Scheme 2 Reactions of HB(C6F5)2, 1, with archetypical hydrocarbyl ligands. | ||
With early transition metal alkyl compounds, borane 1 undergoes a rapid H/R exchange process that formally has the appearance of a sigma bond metathesis reaction (Scheme 2a). The process is generally rapid and mechanistic possibilities range from a fully concerted four-centred process26 to a stepwise alkide abstraction/hydride back transfer sequence. In any case, the products are alkyl boranes RB(C6F5)2 and “LnM–H”; the latter is often highly reactive towards further equivalents of 1 leading to the bis-pentafluorophenyl hydridoborates shown. This has been observed for M = Mg,32 Sc,33 Ti,34 Zr35–38 and Zn39 and in general, these hydridoborates are thermodynamic wells that do not provide access to either monomeric 1 nor “LnM–H”. In the case of dimethyl zirconocene, in hexanes a competing path in which methane is eliminated upon treatment with 1 was observed; the putative product of this reaction is the Tebbe reagent-like40 species shown in Scheme 2b, in which a Cp2Zr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 is stabilized by complexation with HB(C6F5)2.35 This compound rapidly picks up a second equivalent of 1 to give the intriguing pentacoordinate carbon compound41 in which the methylidene is stabilized by two equivalents of borane but retains significant interaction with the metal centre.42 These observations led us to explore the reactions of 1 with a bona fide methylidene compound, Schrock's classic tantalocene methylidene methyl.43 In these studies, it was found that 1 readily complexes MCH2 units44 and the products can be further converted into “borataalkene” complexes45 that exhibit olefin like reactivity at the tantalum centre46 but can also be formulated as Z-type ligands47 as shown in Scheme 2c.
CH2 is stabilized by complexation with HB(C6F5)2.35 This compound rapidly picks up a second equivalent of 1 to give the intriguing pentacoordinate carbon compound41 in which the methylidene is stabilized by two equivalents of borane but retains significant interaction with the metal centre.42 These observations led us to explore the reactions of 1 with a bona fide methylidene compound, Schrock's classic tantalocene methylidene methyl.43 In these studies, it was found that 1 readily complexes MCH2 units44 and the products can be further converted into “borataalkene” complexes45 that exhibit olefin like reactivity at the tantalum centre46 but can also be formulated as Z-type ligands47 as shown in Scheme 2c.
Scheme 2d and e outline further transformations that occur when a metal methylidyne and a metal carbide is reacted with borane 1. For the former, we turned again to a Schrock compound, the tungsten methylidyne (dmpe)2W(Cl)CH,48 while for the latter we utilized Heppert's remarkable ruthenium carbide,49 derived from Grubb's olefin metathesis catalysts. The Lewis basic nature of the methylidyne and carbide ligands in these compounds resulted in facile adduct formation with 1. In the tungsten system, the methylidyne hydrogen is transferred to the metal centre upon abstraction of the borohydride from the ligated borane, resulting in novel borylalkylidyne structures.50 In the ruthenium carbide system, which is notably less basic, reversible adduct formation is observed, but the adduct of a bis-phosphine derivative was structurally characterized.23 The adduct itself was prone to side reactions that involved the phosphine ancillary ligands and these observations eventually led us to explore the protonation of ruthenium carbides derived from first and second generation Grubb's olefin metathesis catalysts, a line of research that resulted in the discovery of highly active, rapidly initiating olefin metathesis catalysts.51
These fundamental studies aimed at mapping the reaction types possible when treating various hydrocarbyl groups with the highly reactive borane 1 thus resulted in insights of significant theoretical interest (hypervalent carbon, novel bonding in borataalkenes) and gave rise to new developments in catalyst development for an important reaction class (olefin metathesis). Indeed, researchers continue to explore the reactivity of 1 with coordinated ligands in complexes from across the d-block, with a view to activation of catalysts and/or small molecules. Select recent examples are given in Scheme 3.
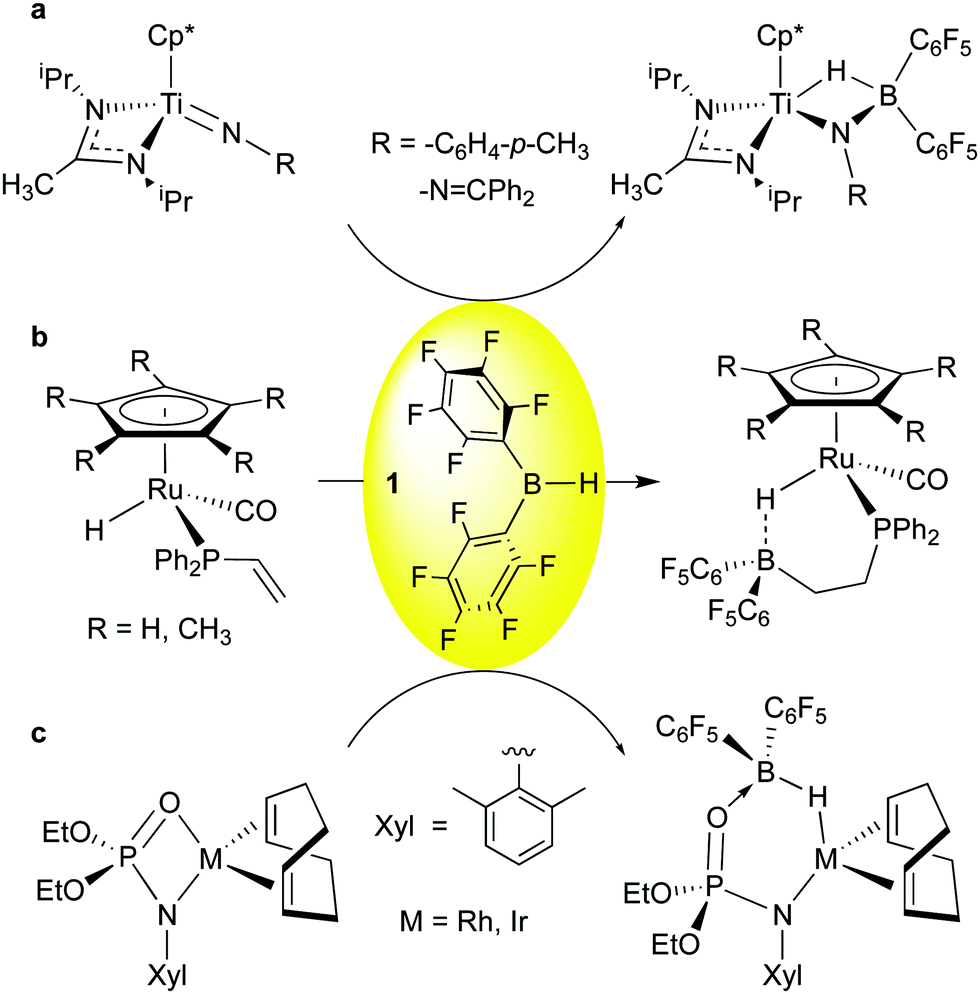 | ||
| Scheme 3 Reactions of HB(C6F5)2, 1, with other ligands. | ||
Addition of 1 to TiN imido bonds was observed by Mountford and co-workers (Scheme 3a);52 here, as in the other examples, the greater basicity of the heteroatom-based ligands means that other, less Lewis acidic boranes like 9-BBN also engage in this reactivity. However, because of the higher Lewis acidity of 1, incomplete transfer of hydride to Ti is observed, and the borane-complexed imido species, similar in nature to the “Tebbe-like” compounds of Scheme 2b and c, results. Interestingly, for the hydrazido-derived imide (R = –NCPh2) kinetic complexation of 1 to the β-nitrogen is immediate, and isomerization to the more thermodynamically stable final product occurs over the course of several hours. The Klankermayer group reported the facile hydroboration of a pendant vinyl phosphine group in piano stool ruthenium complexes using 1 (Scheme 3b).53 Here the bis-pentafluorophenyl group loosely coordinates the Ru-hydride moiety and the Ru–H⋯B bridge can be opened via protonation or by methanol to yield highly reactive compounds capable of delivering an H2 equivalent as a hydride/proton pair. Finally, Love and Schafer et al. have shown in the group 9 metal phosphoramidinate complexes depicted in Scheme 3c,54 borane 1 adds across the M–O bond, leading to M–H–B moieties that can be further elaborated by B–H activation. These examples reflect the rich chemistry possible by reaction of 1 with coordinated ligands in metal complexes.
Hydroboration with 1 for catalyst synthesis
As a means of incorporating –B(C6F5)2 units into organic and organometallic structures, bis-pentafluorophenylborane is an unparalleled reagent due to its hydroboration activity and high reactivity towards M–R functions. It has been used extensively in this regard, especially in the context of olefin polymerization catalysis and frustrated Lewis pair genesis.Indeed, 1 was originally conceptualized as a reagent for hydroborating pendant unsaturated groups on early transition metal organometallic compounds for the development of “self-activating” or one-component olefin polymerization catalysts.55 While moderate success was achieved in generating active zwitterionic catalysts12,56 with borates affixed to the active alkyl groups in the wedge of metallocene catalysts (“girdle” zwitterions) using some of the reactions described above (Scheme 2a),38,56,57 truly self-activating catalysts attained by incorporating the Lewis acid activator into the catalyst structure (“ring” or “bridge” zwitterions) via hydroboration of pendant unsaturated groups were more elusive. In addition to our efforts, the Erker group also made significant contributions and some of the systems studied are summarized in Scheme 4. The synthesis and characterization of zirconium dichloride catalyst precursors with dangling –B(C6F5)2 groups proved relatively straightforward since 1 is unreactive towards early transition metal M–Cl bonds. All examples in Scheme 4 (except a)58 were prepared by hydroboration of alkene (b–f)55,59–61 or alkyne (g)62 functions on the periphery of the metallocene or post-metallocene ligand framework. However, hydroboration of dimethyl or dialkyl versions of these precursors was less clean due to competition between olefin hydroboration and the aforementioned reactivity of 1 with these functions (Scheme 2). Furthermore, alkylation of the metallocene or post-metallocene halides of Scheme 4 with alkyllithium reagents was hampered by competing reactions with the Lewis acidic borane centres. Erker and co-workers were able to overcome this problem in system f by protecting the –B(C6F5)2 centre with N-methylimidazole Lewis bases.63 Here, alkylation proceeded smoothly with LiCH2SiMe3 but self-activation through alkide abstraction was not observed. In instances where zwitterionic – [linker(alkyl)B(C6F5)2]− borates were thought to have been generated, facile alkyl or linker exchange processes lowered catalyst stability as the necessary ion pair, and so side reactions lowered turnover numbers in these systems. Thus, generally in cases where polymerization activity was examined, alkylation was done in the standard way using alkylaluminum co-catalysts and the borane function was thought to simply serve as another substituent on the ligand rather than an active co-catalytic participant.58,60,62
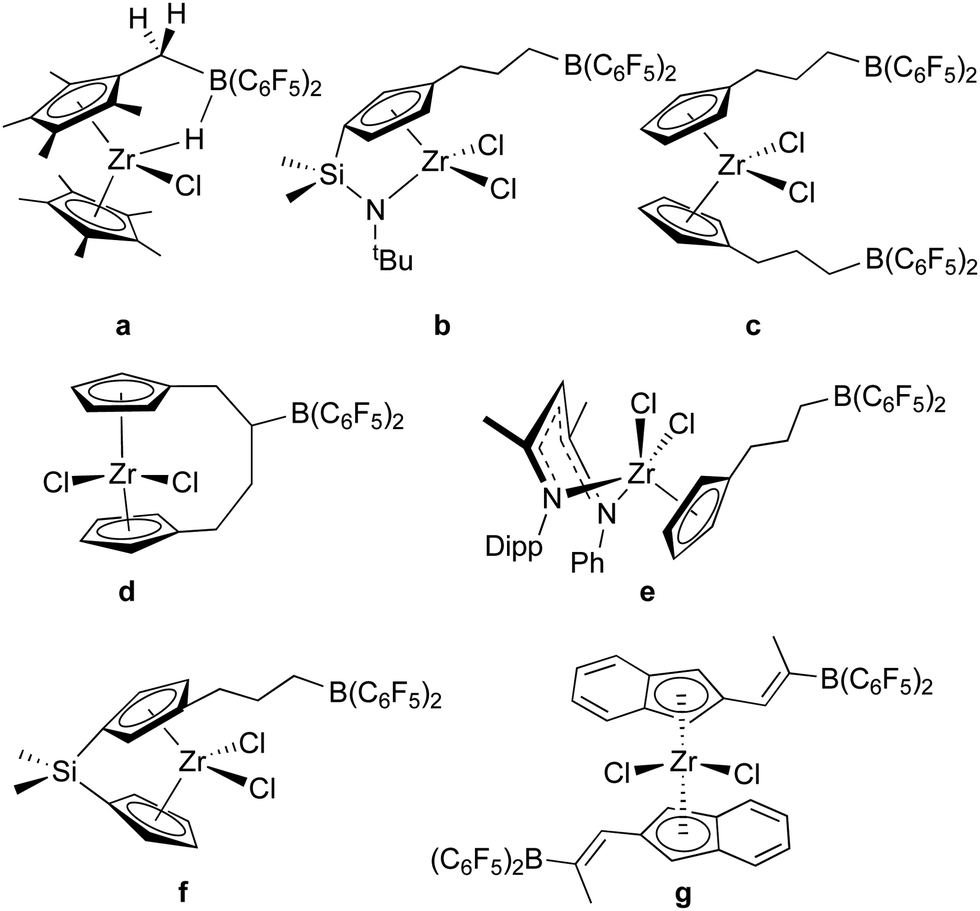 | ||
| Scheme 4 Hydroboration of pendant olefin and alkyne groups using 1. | ||
While limited success in the arena of olefin polymerization was attained, the use of 1 as a hydroboration agent for the generation of frustrated Lewis pairs has been one of bis-pentafluorophenylborane's most important applications in the past decade. The original vicinal, intramolecular FLP was synthesized by hydroboration of Mes2P-CHCH2 with 1,64 and Erker and co-workers have demonstrated this species to be a versatile FLP for the activation or bonding of a number of small molecules (Scheme 5). While most of these examples mirror reactions in intermolecular FLPs,65,66 the incorporation of the Lewis acid and base moiety in one molecule accelerates the rate by which the FLP reaction occurs. Thus, the heterolyic splitting of H2 (blue reaction) for this 1,2-vicinal P/B FLP is facile at room temperature, and the compound is highly active as a hydrogenation catalyst for imine functions.67 The α,β unsaturated ynone substrate shown in the black reaction adds in a 1,4 fashion,68 forming the novel enol allene product, while carbonyl functions, olefins,69 and the small molecules CO270 and SO271 add in a 1,2-sense to give the products shown in the green reactions. For CO2, bonding is reversible upon exposure of the product to reduced pressures of CO2. Finally, substrates like azides,72 CO73 and NO74 are bound in a 1,1 mode (red reactions). The bound CO can be further functionalized (vide infra) and the nitrosyl radical formed from bonding of NO to the FLP is a potent hydrogen atom acceptor whose chemistry the Erker and Warren groups have explored extensively.75 The 1,1 bonding chemistry is not as prevalent in intermolecular FLPs and is therefore unique to the vicinal systems formed from borane 1 and vinyl or alkynyl functionalized Lewis bases via hydroboration.
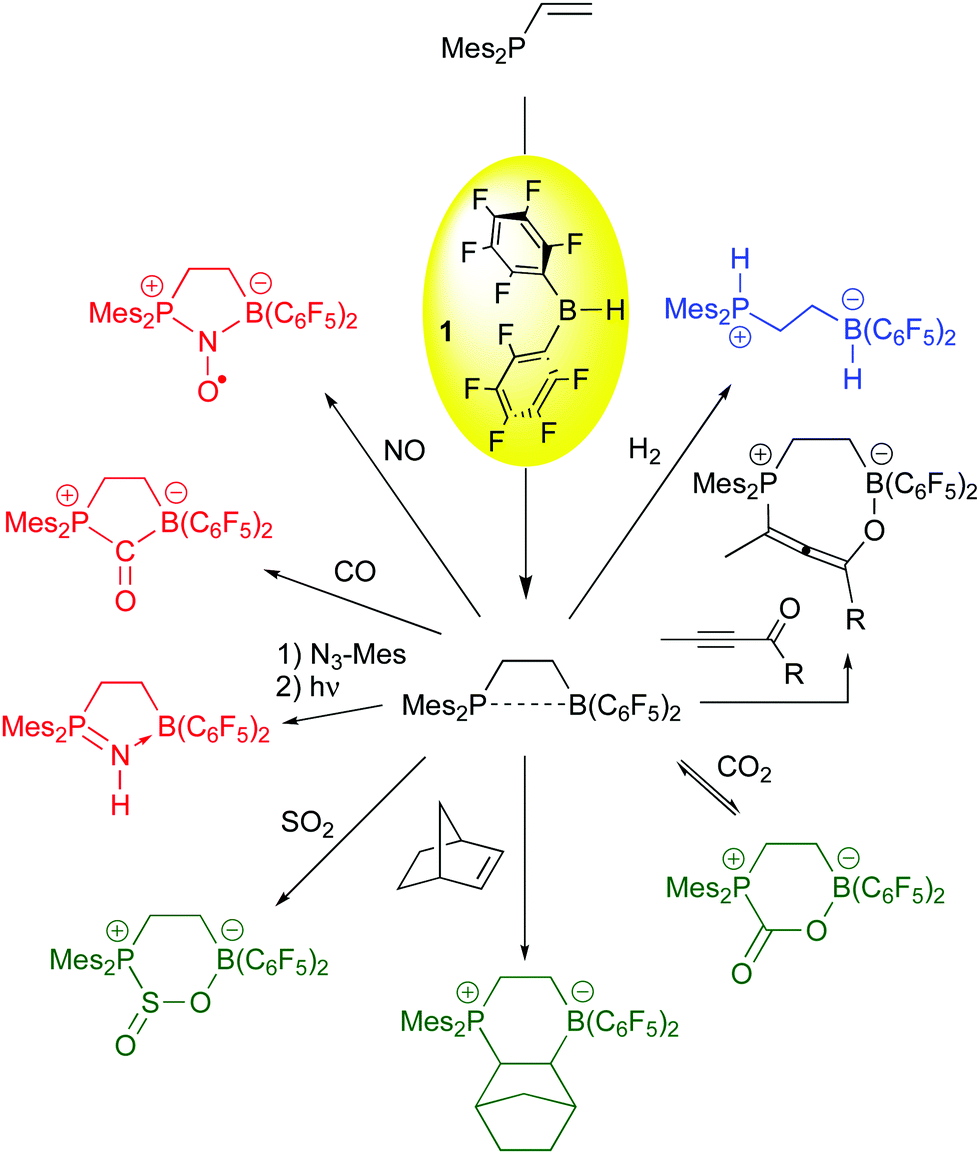 | ||
| Scheme 5 Hydroboration synthesis of Erker's 1,2-vicinal P/B FLP with 1 and its reactivity with small molecules. | ||
The use of FLPs for the catalytic hydrosilation76,77 or hydrogenation67,78 of unsaturated functions was a major breakthrough and inspired a number of groups to develop the field of transition metal free catalysis. A significant challenge for these metal free systems in comparison to traditional transition metal catalysts concerns the issue of stereoselectivity. The intermolecular FLPs lack the rigidity of the intramolecular systems of general formula R2PCH2CH2B(C6F5)2 and consequently the latter have been more efficacious in asymmetric hydrogenations. Again, 1 has proven to be a useful reagent in generating chiral boron Lewis acids for asymmetric hydrogenations via the FLP mechanism (Scheme 6). Our original paper showed that α-pinene could be hydroborated diastereoselectively,10 and isomerized to a thermodynamic isomer (Scheme 6, a); the Klankermeyer group used this borane to obtained 13% ee in the asymmetric hydrogenation of the N-phenyl ketimine substrate shown at the bottom of Scheme 6, using a bulky phosphine as the Lewis base partner in the FLP.79 While somewhat disappointing selectivity was observed, the experiment did demonstrate that asymmetric induction was possible using this strategy. Subsequent studies using diastereomeric catalysts b and c, formed via non-selective hydroboration of a phenyl substituted camphor-derived substrate, demonstrated ee values of up to 79%.80 Other derivatives of this catalyst family were subsequently demonstrated to give >80% ee in the FLP type hydrosilation76,77 of imines81 in excellent conversions.82 Erker et al. used a planar chiral ferrocenyl complex (d) to generate an intramolecular FLP capable of modest enantioselectivity in the hydrogenation of the ketimine substrate shown (53% ee) as well as others.83
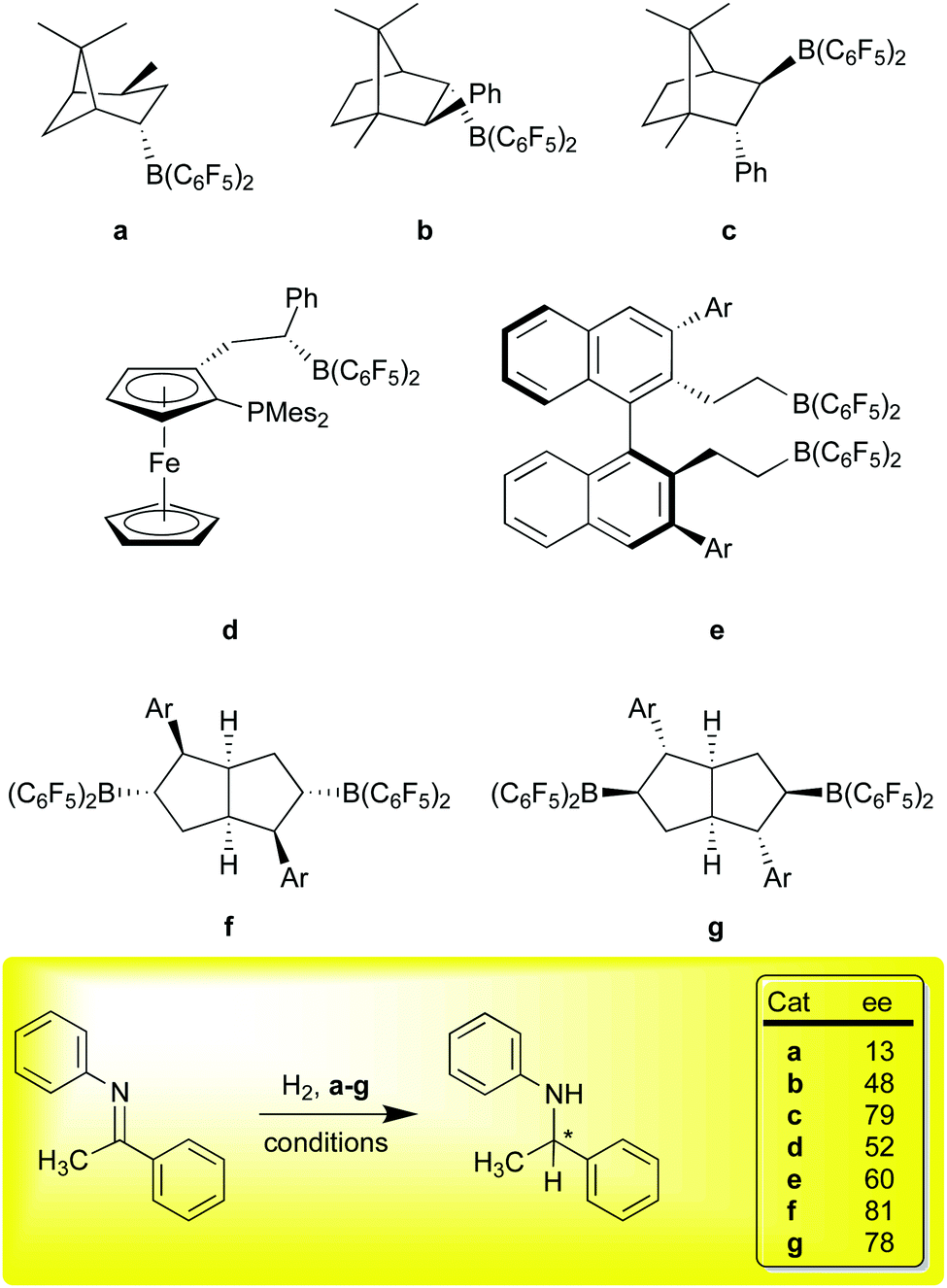 | ||
| Scheme 6 Hydroboration synthesis of chiral Lewis acids using 1 for asymmetric FLP hydrogenation and hydrosilation. | ||
All of the systems a–d, have the disadvantage of (potentially) producing diastereomeric mixtures of catalysts that either must be separated, or may even be in equilibrium due to facile retrohydroboration/hydroboration sequences characteristic of this borane. To overcome this problem, Du and co-workers developed an extensive family of catalysts based on the binaphthyl framework represented by catalyst e;84 in these systems, the substrate imines served as the Lewis base partner and no added phosphine was required. In the generation of these catalysts, the hydroboration cannot produce diastereomers, and indeed, these catalysts are simply formed in situ from 1 and the requisite divinyl binaphthyl catalyst precursor via rapid hydroboration. A number of examples, varying in the 3,3′-aryl groups, have been prepared and ee values of up to 60% were observed for the test ketimine substrate discussed here. Higher ee values were observed for other imine substrates, highlighting the effectiveness of this family of catalysts for asymmetric hydrogenation catalysis. Finally, in a very recent example, Wang and co-workers have utilized 1 to selectively hydroborate C2-symmetric bicyclic [3.3.0] dienes substituted with Ar groups of increasing steric bulk to give catalysts f (kinetic products at room temperature) or g (thermodynamic products at 80 °C).30 This chemistry takes advantage of the high regioselectivity of addition of 1 to trisubstituted aryl olefins shown in our original report13 and the facility of retrohydroboration paths to thermodynamic isomers. Remarkably, ee values of about 80% were achieved at ambient temperatures and further improvements to above 90% were obtained as temperatures were lowered to −40 °C. This catalyst family is thus the best performing metal free asymmetric hydrogenation catalyst for imine hydrogenation using an FLP type mechanism to date.
All of the above examples utilize chiral boranes to induce asymmetric reduction of the imine CN bond. Du has also explored the use of a chiral base in a novel transfer hydrogenation catalyst system utilizing 1 and an asymmetric tert-butyl sulfinimide as the chiral transfer agent (Scheme 7).84,85 In stoichiometric reactions, the ketimine substrate is reduced with high conversion (99%) and ee (90%), with the hydrogen being supplied by 1 and (S)-tert-butylsulfinimide. The boron-containing product is as shown in Scheme 7. For catalytic processes, dihydrogen proved an inefficient hydrogen source, but the use of ammonia borane was highly effective for reaction turnover and optimization of the reaction conditions eventually lead to a system in which a variety of imines were reduced with 85–95% ee.
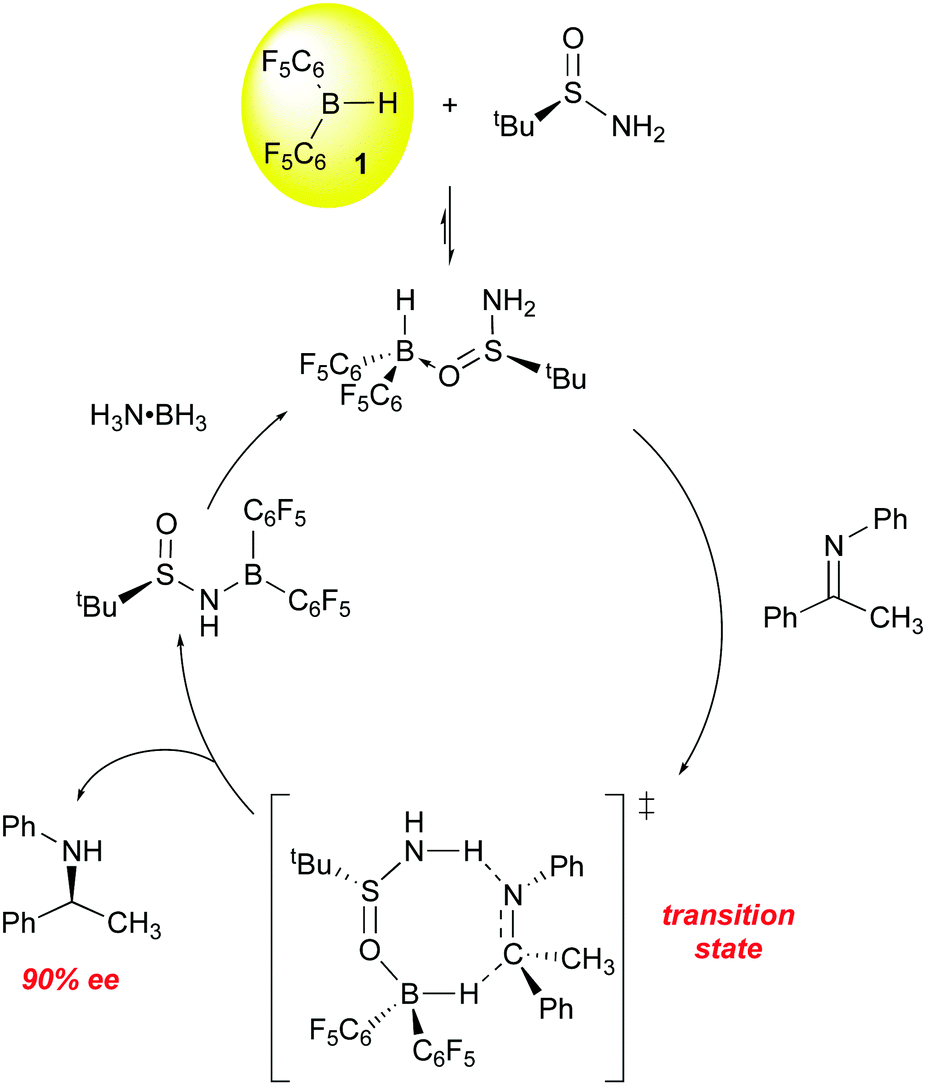 | ||
| Scheme 7 Transfer hydrogenation of imines using 1/(S)-tert-butylsulfinimide and ammonia borane. | ||
The hydroborative role of 1 in FLP genesis has been an important one in the last 15 years, but recent reports highlight its use in other types of catalyst activation. A particularly mechanistically intriguing one involves the observation that 1 catalyses the addition of HBpin (a typically unreactive borane with respect to hydroboration) to alkynes. This is a reaction for which there are many catalysts, mostly transition metal-based. However, Stephan et al. showed in 2016 that 1 is also an effective catalyst precursor for this transformation (Scheme 8a).86 Under mild conditions, several internal and terminal alkynes are smoothly converted to the syn-Bpin hydroboration products.
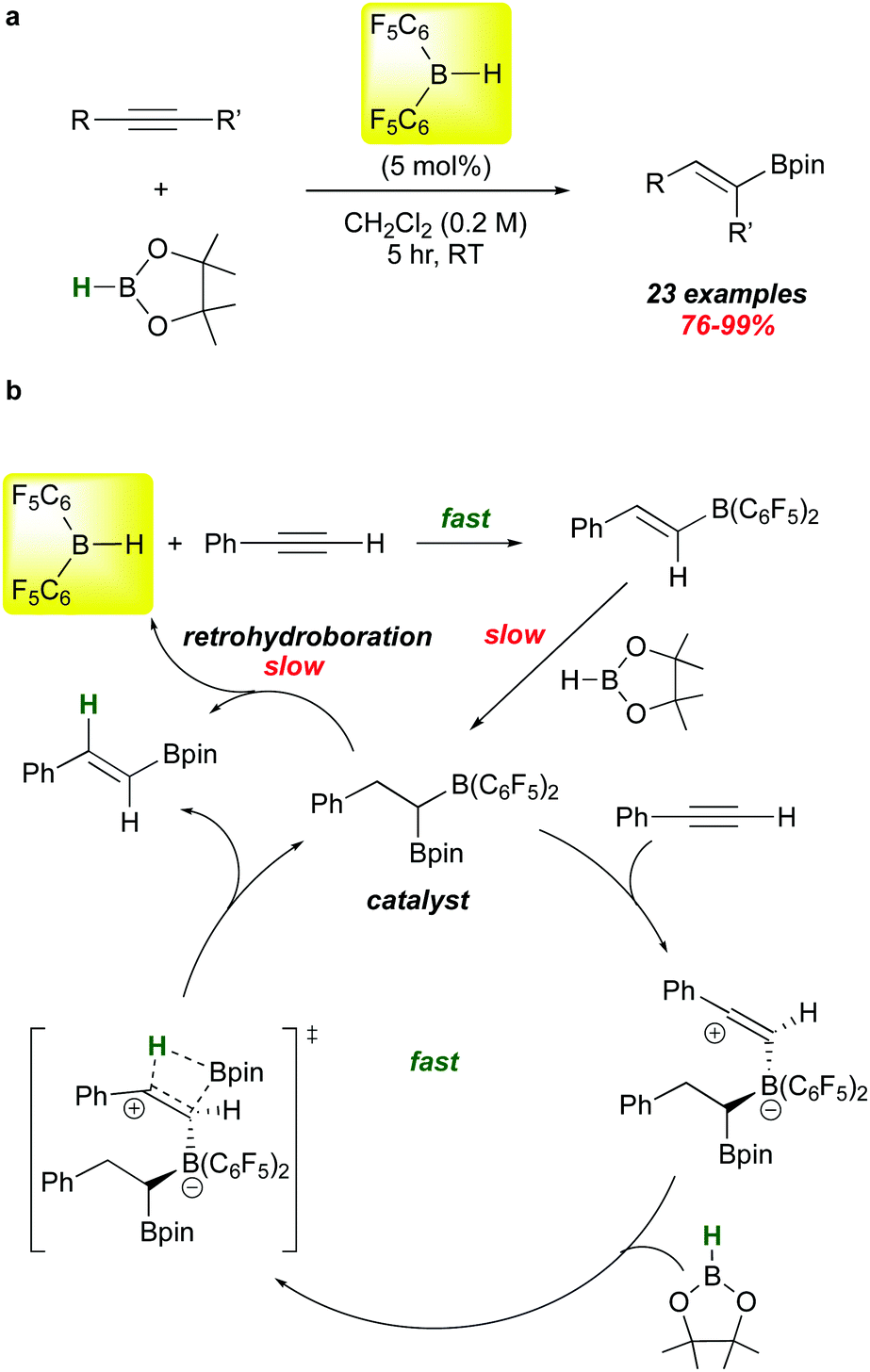 | ||
| Scheme 8 Catalytic HBpin hydroboration of alkynes using 1 as a catalyst precursor. | ||
The proposed mechanism of this catalytic reaction is unusual (Scheme 8b). Clearly, the 5% loading of 1 rapidly hydroborates the substrate to give the alkenyl borane as shown.10 Reaction of this species with HBpin, while slower, is relatively facile and forms the mixed 1,1-diboryl species, which is believed to be the active catalyst on the basis of several control experiments. These studies show that retrohydroboration to eliminate 1 and release product is not kinetically viable enough to account for the observed rates of these reactions. Rather, the authors propose that the –B(C6F5)2 unit in the diboryl catalyst electrophilically activates the alkyne substrate,87 triggering addition of HBpin across the alkyne via the proposed transition state depicted. Product release regenerates the active catalyst, which can be separately synthesized and proven to be a viable mediator of the process at kinetically similar rates.
Erker and co-workers recently reported that 1 can be employed to selectively trimerize allene and cyclohexylallene to the 1,3,5-trimethylenecyclohexane product88 (Scheme 9a), an isomer not usually accessible via transition metal catalysed processes. Mechanistic experiments suggest that the first step is hydroboration of the allene substrate, followed by successive allylborations of the other two allene equivalents. Product release is via retrohydroboration, which also regenerates catalyst 1; this step requires higher temperatures and is likely the factor that most limits the reaction to ≈10 turnovers. Nonetheless, this chemistry allows for access to workable quantities of these trialkene cyclohexanes, and the parent compound can be further converted to the interesting triborane shown in Scheme 9bvia exhaustive hydroboration with 1. This triboryl species can serve as an FLP hydrogenation catalyst for imines, ligates three equivalents of tert-butyl nitrile, and undergoes three 1,1-carboborations89 with PhCCSiMe3. In a follow up study, the Erker group showed that, in contrast to the two substrates in Scheme 9, aryl allenes are stochiometrically dimerized by 1.90
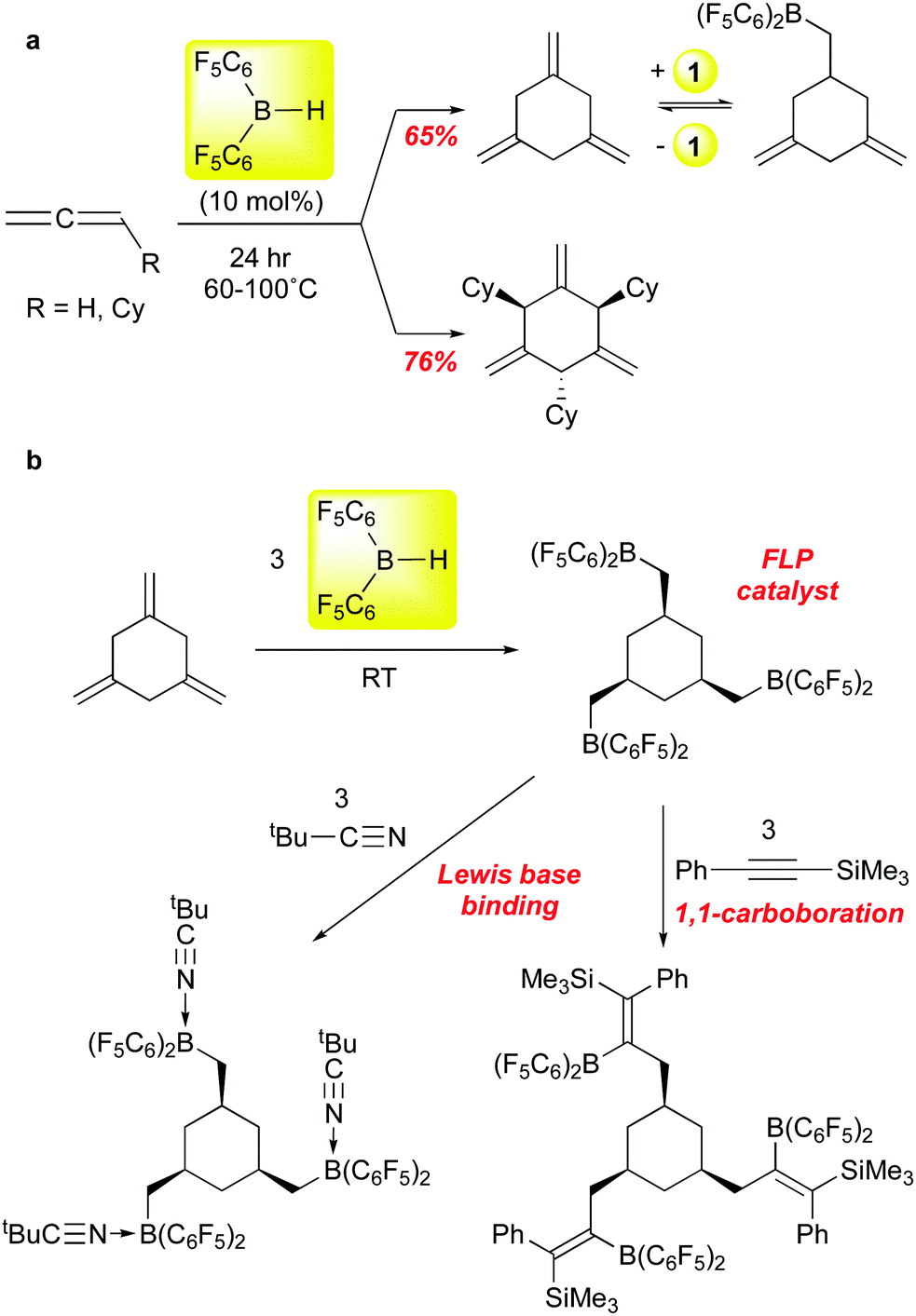 | ||
| Scheme 9 Catalytic trimerization of allenes using 1 and hydroboration of 1,3,5-trimethylenecyclohexane using 1 to form trifunctional pentafluorophenyl boranes. | ||
It is clear from all the examples discussed in this section that bis-pentafluorophenylborane is a powerful reagent for incorporating Lewis acidic –B(C6F5)2 units into molecular catalyst structures. These units can serve as intramolecular weakly coordinating ions in cationic olefin polymerization catalysts or, more interestingly, as the Lewis acidic partner in vicinal frustrated Lewis pairs. The many reactions of the parent intramolecular FLP depicted in Scheme 5 (and more) have been repeated for several related FLP systems,91–95 all formed using 1 as a synthon. While these reactions received justifiable attention, many did not go beyond the binding of the substrate, with subsequent transformations elusive. In the next section, we highlight examples wherein further reactivity is observed, exhibiting true small molecule and bond activations.
Small molecule and bond activations with 1
While 1 has been a key reagent for generating a number of FLPs capable of binding small molecules, in a few instances chemistry involving further functionalization of the small molecule was observed. A particularly intriguing example, reported in a series of papers from the Erker group, involves the reduction of CO to formyl moieties through FLP binding of CO activated by ligation to 1 (Scheme 10). A series of three internal FLP molecules were each shown to bind CO73 (as depicted in Scheme 5), but this binding is relatively weak and occurs at low temperature and moderate pressures of CO, so further functionalization of the bound CO is difficult. However, in the presence of another equivalent of 1, the FLP stabilized formyl borane products a–c shown in Scheme 10 form readily.96,97 Through detailed DFT computations and experimental studies, the authors show that, remarkably, the reactions proceed via FLP bonding of carbon monoxide coordinated to 1 in a rare example of a borane–CO adduct. Compound 1·CO forms reversibly at low temperature and could be characterized in situ via spectroscopic methods and single crystal X-ray diffraction. Conversion to the formyl borane isomer via insertion of CO into the B–H bond of 1 was not observed and this process was determined to be thermodynamically disfavoured by DFT. Thus, when 1·CO forms in the presence of an internal FLP, the coordinated CO ligand is cooperatively bound by the phosphine–borane moiety, which triggers a 1,2 hydrogen shift of the hydride from boron to carbon, reducing the CO to a formyl group. While thermodynamically uphill in the absence of the FLP, formation of the formyl borane moiety is driven by the stabilization provided by the P–C and B–O bonds in the FLP adduct of (C6F5)2B–C(H)O. The norbornene-based complex b was shown to undergo further reactivity with H2 or the Lewis base pyridine (Scheme 11).97 Both reactions can be explained by the weak B–O bond in the FLP-stabilised formato borane which can dissociate reversibly. The resulting species can be trapped by pyridine to yield the adducts of both the original FLP and the formyl borane that is not thermodynamically favoured to form from 1 and CO (see Scheme 10). This pyridine adduct of (C6F5)2B–C(H)O is isolable and was fully characterized by multinuclear NMR spectroscopy and X-ray crystallography and undergoes a number of reactions, including with further equivalents of 1. It is remarkably resistant to deinsertion chemistry which implies that pyridine dissociation is disfavored. Alternatively, when b is treated with a high pressure of H2, the dissociated form can act as a B–O FLP that heterolytically cleaves dihydrogen as shown in Scheme 11. This intermediate irreversibly converts to the product in which the C–O bond of the original carbon monoxide has been cleaved.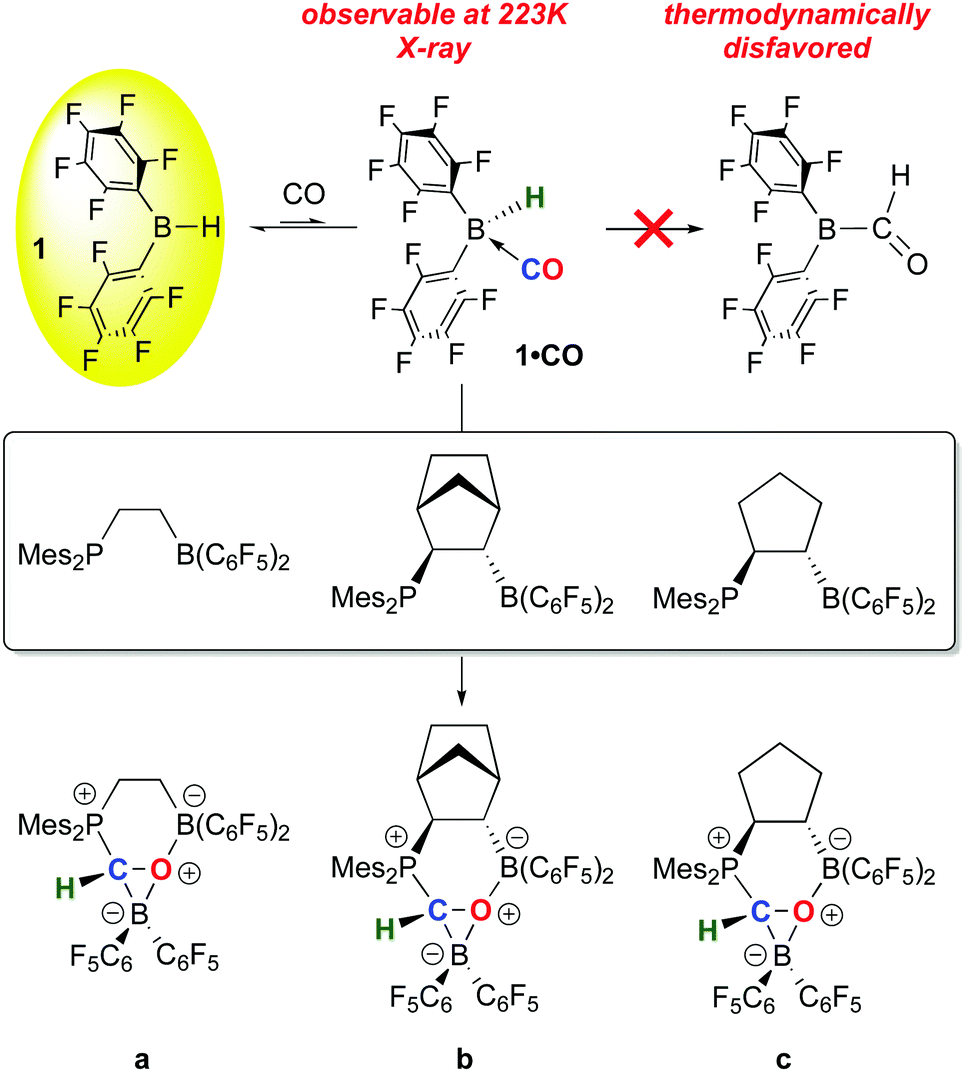 | ||
| Scheme 10 FLP activation of carbon monoxide coordinated to 1. | ||
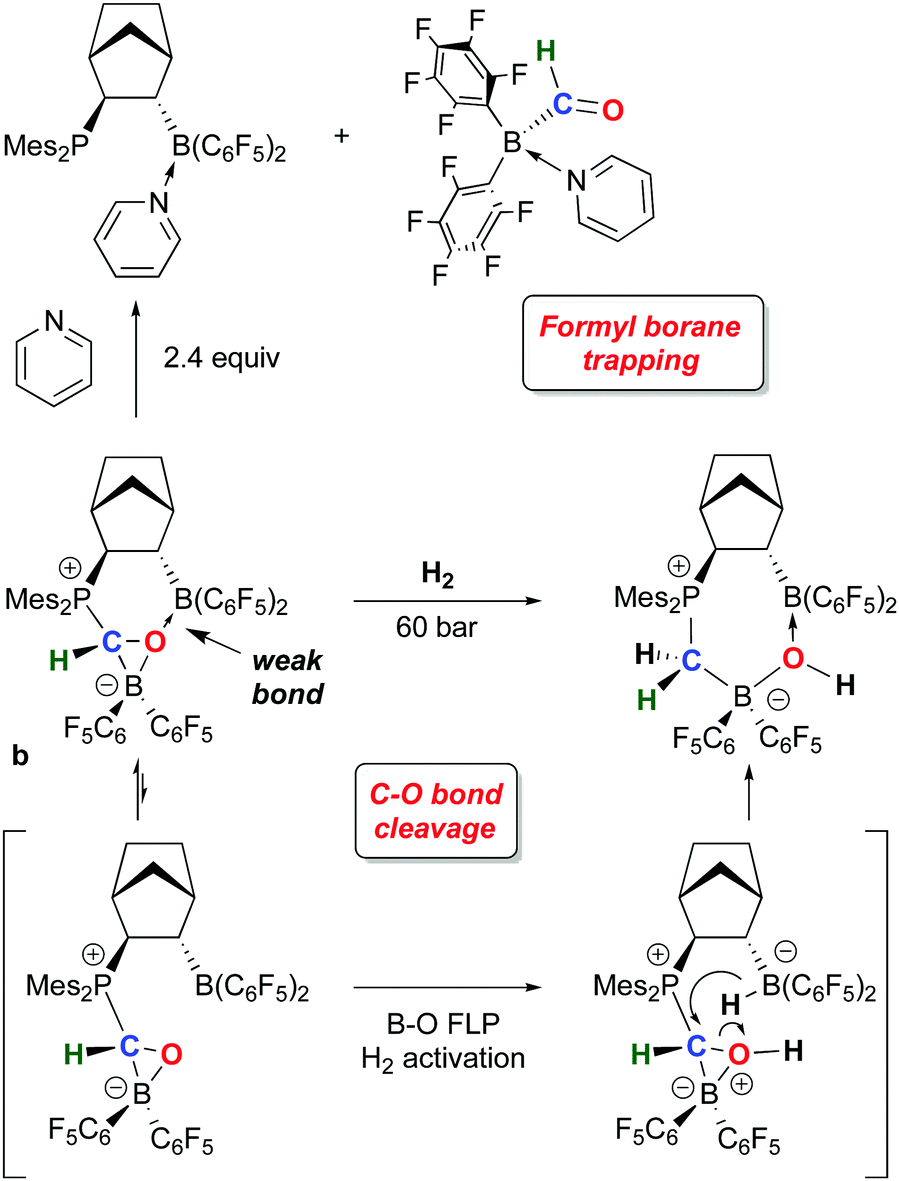 | ||
| Scheme 11 Formyl borane trapping and C–O bond cleavage reactions of FLP-stabilized formyl borane b. | ||
The bonding of CO to 1 is relatively weak but 1·CO is clearly reactive enough to undergo subsequent reactions and in this sense 1 itself engages in small molecule activation. For example, further studies by the Erker group have shown that the coordinated CO in 1·CO can also be hydrozirconated using the bulky Cp*2Zr(H)OMes reagent, producing formylhydridoborates that undergo reactions with a variety of small molecules (CO2, H2, PhNSO) and leading to fully reduced or highly functionalized CO.98 The ability to activate and functionalize carbon monoxide raises the prospect of metal free dinitrogen activation,99 and 1 has also played a role in this developing area. For example, it has been shown by Simonneau et al. that coordinated dinitrogen in Mo and W phosphine complexes can be functionalized by FLP-type addition of the H–B bond in 1 to the terminal N atom (Scheme 12).100 The B(C6F5)3 adducts of these dinitrogen complexes readily form via reaction of (Ph2PCH2CH2PPh2)2M(N2)2 with B(C6F5)3 and while the coordinated N2 assumes a diazenido-like structure, the lack of reactive groups on boron precludes functionalization of the activated N2 molecule. Use of 1, however, does allow for chemical functionalization of the N2; through dissociation of B(C6F5)3 and coordination of 1 to the terminal N atom, the borane hydride is abstracted by the free B(C6F5)3 to yield the products shown in which a B–N covalent bond has been forged. In a related study, Stephan and co-workers, showed that 1 undergoes adduct formation and 1,1-hydroboration of diphenyldiazomethane to give products related to probable intermediates in the FLP bonding of dinitrogen.101 While this remains a challenging problem for main group chemistry, these studies are suggesting new directions for metal free N2 reductive functionalizations.
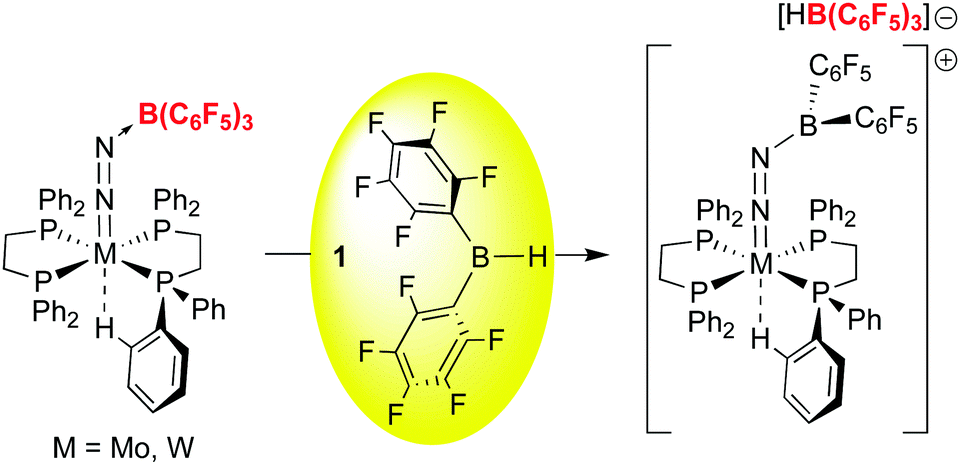 | ||
| Scheme 12 Functionalization of coordinated dinitrogen with 1. | ||
Another arena in which bis-pentafluorophenylborane has found recent application is in the activation of strong bonds, such as C–O and C–F. In the case of the latter, the Stephan group found that 1 is an effective stoichiometric reagent for reducing the C–F bonds in tertiary, secondary and primary alkyl fluorides,102 constituting a rare example of catalyst-free Csp3–F borylation. The ability of 1 to dissociate readily into monomeric (C6F5)2BH was key to its effective reactivity with these strong C–F bonds.
In contrast to these stoichiometric reactions, borane 1 has found more extensive application in the catalytic activation of C–O bonds, both by itself and as a catalyst for hydrosilation chemistry. In our original studies on B(C6F5)3 catalysed hydrosilation of carbonyl functions, we noted that with excess silane, exhaustive deoxygenation of substrates was possible.77 The scope of these reactions was subsequently more fully developed by Gevorgyan and Yamamoto for the reductive deoxygenation of simple alcohol103 and carbonyl104 substrates. Some years later, Gagne and co-workers applied this methodology to the metal free deoxygenation of carbohydrates and carbohydrate polymers derived from biomass, generating mixtures of hydrocarbons.105 These catalytic hydrosilations tended to be rather unselective, although by tuning the steric properties of the silane employed, some selectivity for specific C–O bonds in the carbohydrate substrates could be realized.106,107 In order to improve selectivity, Chang and co-workers postulated that, rather than tuning the reaction with the silane reagent employed, use of perfluoroaryl boranes with different substituents would modulate selectivity through tuning the Lewis acidity and steric properties of the hydridoborate anion that serves as the primary hydride delivery agent in these reductions upon activation of the silane.76,77,108 They turned to the readily prepared borinic acid (C6F5)2B–OH109,110 as a possible alternative, but found that it is rapidly converted to 1 in the presence of silanes (see also Scheme 1a above) and that 1 is the actual catalytic species in the selective C–O bond cleavage of a variety of sugars.19 This was among the first demonstrations that 1 is capable of frustrated Lewis pair activation of silanes by analogy to the manner we discovered in 1996 for B(C6F5)3.76 The higher selectivity of 1vs. B(C6F5)3 in these reactions was ascribed to both the lower steric bulk and higher hydricity of the [H2B(C6F5)2]− anion compared to its [HB(C6F5)3]− counterpart. The rates and conversions were also affected by the specific stereochemistry of the carbohydrate substrate, an effect that was ascribed to conformational steric effects in the key hydride delivery step from the [H2B(C6F5)2]− anion to the carbon atom of the C–O bond being cleaved. The reaction was further applied to the regioselective hydrosilative reduction of disaccharides.
The C–O cleavages described above illustrate how moderation of the boron Lewis acidity in B(C6F5)3 by substituting one of the pentafluorophenyl groups with a different group can influence both activity and selectivity of these catalytic silations. Bis-pentafluorophenyl borane 1 can serve in this role either by itself or as a versatile catalyst precursor. Further studies by the Chang group on the ring opening hydrosilation of epoxides offer another key recent example of the use of 1 in this way.22 In our full report concerning the synthesis and properties of 1,13 we noted that in ethereal solvents such as tetrahydrofuran, ring opening by formal addition of the B–H bond in 1 across the C–O bond occurred at temperatures above 80 °C to form the butyl borinic ester (C6F5)2BOnBu (Scheme 13a, top). We also noted in our 1997 review on perfluoroarylboranes4 that this reaction is much more facile for epoxides like styrene oxide, occurring rapidly at room temperature, presumably because of the extra driving force from the epoxide ring strain (Scheme 13a, bottom). The mechanism of this reaction was not studied in detail but presumably involves the THF and epoxide adducts of 1; such an adduct is observed spectroscopically for the THF base. These stoichiometric reactions were not pursued in detail by us, but the recent Chang report22 demonstrated that 1 is a highly effective catalyst precursor for ring opening of a variety of epoxide substrates and provides quite different selectivity in comparison to the same reactions mediated by B(C6F5)3. The mechanism of the reaction (Scheme 13b) involves the previously mentioned generation of 1 from (C6F5)2BOH and silane; in the presence of the epoxide substrate shown, 1 is rapidly converted to the cyclopentylborinic ester designated as the resting state in Scheme 13b. Separate experiments show that reaction of this species with silane in the absence of substrate to regenerate 1 is slow. However, in the presence of epoxide, the bis-pentafluorophenyl borinic ester is sufficiently Lewis acidic to engage in rapid FLP type activation of the silane Si–H bond to give the epoxide-stabilised silylium ion partnered with a strongly basic hydrido-alkoxyborate anion. This ion pair rapidly forms product to regenerate the borinic ester resting state. Because of the high hydricity of this hydrido borate (in comparison to [HB(C6F5)3]−),111 the cation does not have time to undergo rearrangements that occur in B(C6F5)3 mediated reactions. This results in different, complementary selectivity in the reactions with 1 as the catalyst precursor.
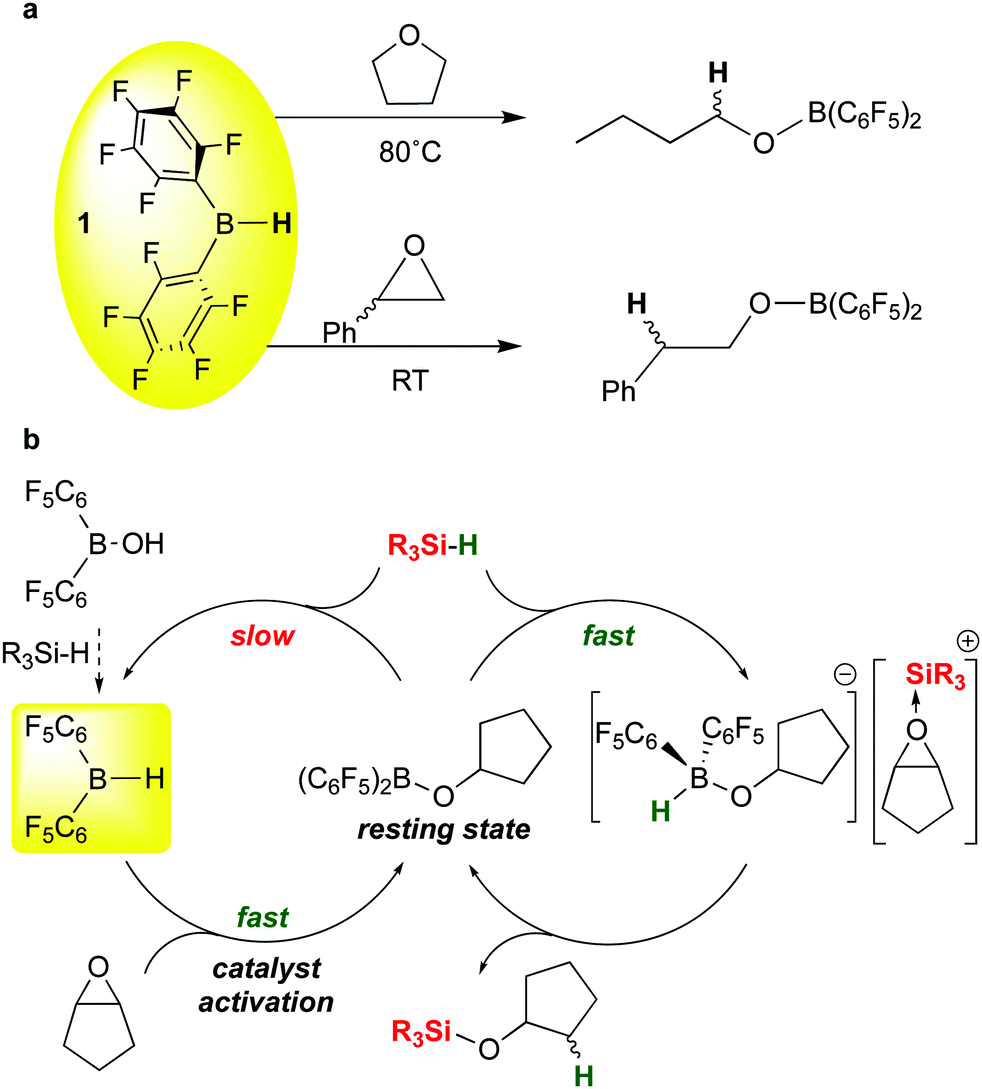 | ||
| Scheme 13 Stoichiometric and catalytic ring opening of epoxides with 1. | ||
Another recent example of how moderated Lewis acidity can direct reactivity stems from studies by the Gagne group on the selective deoxygenation of organic amides using borane/silane systems where the borane is generated from 1 and an olefin through hydroboration (Scheme 14). This work grew out of impressively selective late stage functionalizations using the borane catalysed FLP silation of highly functionalised complex natural products.107 Using an alkyl borane formed from 1 and 3-hexene as the catalyst, unprecedented chemoselectivity for hydrosilation of acetamide moieties was observed. The modularity of the RB(C6F5)2 catalysts available through variation of the olefin partnered with 1 allowed for the screening of a number of catalysts for this transformation, and led to the identification of (C6F5)2B(CH2)3Bpin as an effective catalyst for a number of amide substrates (Scheme 14a).112 While B(C6F5)3 can mediate this reaction,113,114 it generally is less selective and requires higher temperatures. This is likely due to the fact that organic amides form strong adducts with B(C6F5)3115 and the dissociation to free borane required for silane activation is disfavoured. By utilizing less Lewis acidic boranes, more free borane is present to activate silane, which is attacked by the most basic organic functional group in the molecule, accounting for the remarkable chemoselectivity for the amide functions in complex molecules (Scheme 14b).
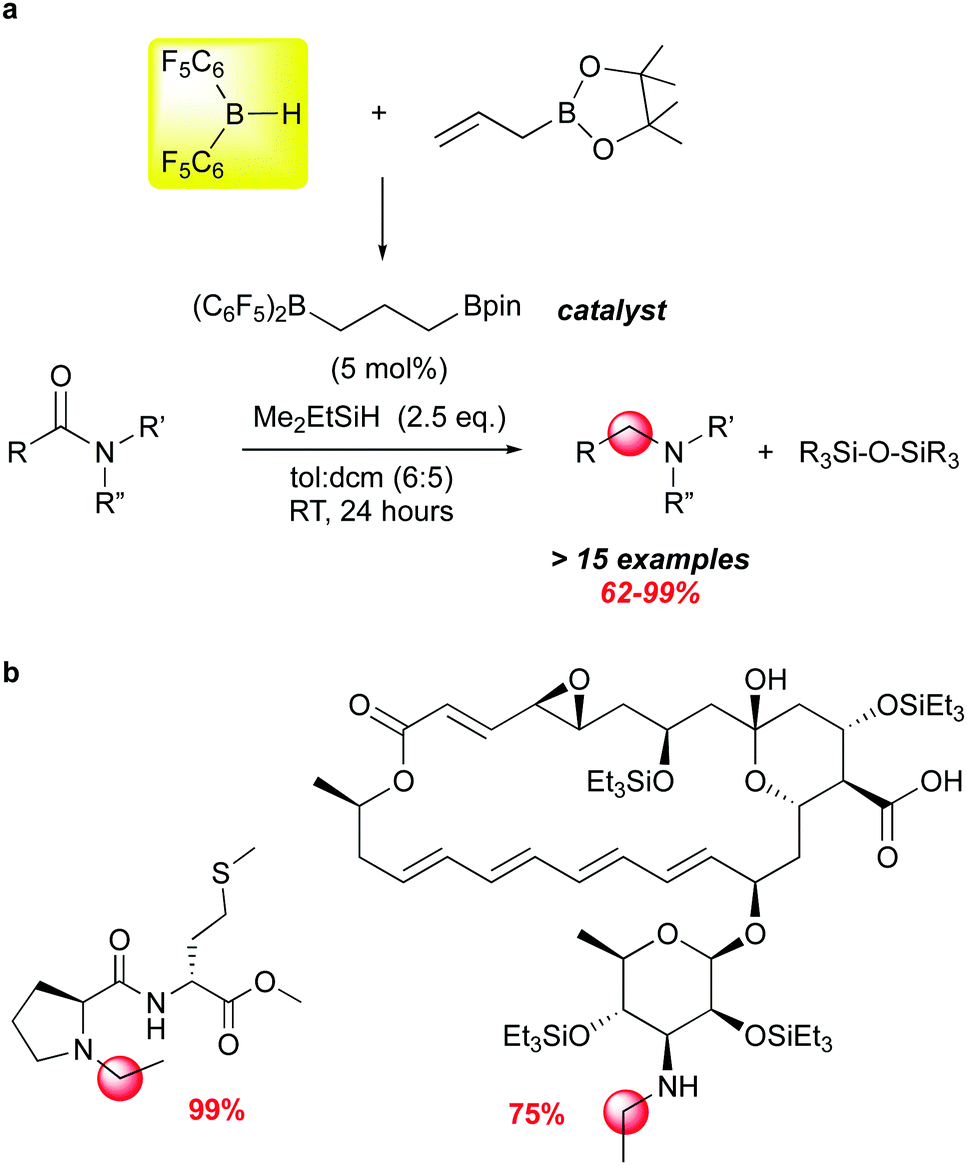 | ||
| Scheme 14 Selective hydrosilative reduction of organic amides using alkyl bis-pentafluorophenylboranes derived from 1 and an olefin via hydroboration. | ||
Incorporation of –B(C6F5)2 units into materials
The above sections have shown that 1 can serve as an effective reagent for generating catalysts, or behave as a catalyst in and of itself, for a variety of important and interesting transformations. Recently, it has also been used to incorporate –B(C6F5)2 units into the structures of organic materials. The incorporation of boron into π-conjugated organic molecules and polymers, particularly in the form of B–N units that are isoelectronic with C–C,116–119 has been a fruitful avenue of research in this area over the past 15 years. These compounds, in comparison to the all carbon analogs, tend to absorb at lower energies into the visible region of the spectrum and are generally easier to reduce. Indeed, incorporation of BN units along with perfluoroaryl substituents allows for use of some of these species as non-fullerene acceptors in certain applications.To this end, some researchers in this field have used 1 as a means of installing –B(C6F5)2 units into conjugated molecules and polymers. Pammer and co-workers have been particularly active in this regard and some of their molecules and materials are summarized in Scheme 15. The general strategy (Scheme 15a) involves preparation of a variety of molecular and polymeric precursors that incorporate ortho-styrenyl pyridine moieties that can be regioselectively hydroborated using 1 (and other boranes).120,121 The hydroboration is directed to the α styrenyl carbon by substituting the β carbon with one or two alkyl groups, and allows the borane centre produced to coordinate with the pyridyl nitrogen, thus installing the B–N unit. In some instances, the hydroboration with 1 required heating to overcome kinetic adduct formation with a pyridyl unit on the substrate. Using this methodology, more π extended B–N ladder compounds based on pyrimidine,122 pyrazine123 and quaterpyridine122 frameworks could be prepared as rac/meso mixtures that in some instances could be separated via crystallization. The compounds incorporating the –B(C6F5)2 groups using 1 exhibited low energy LUMOs (below −4.0 eV) in the range amenable for use as electron acceptor materials in organic solar cells. To this end, the Pammer group has prepared polymers of modest molecular weight incorporating this unit.124 While PCE performance of the devices prepared using these polymers was modest, this is a potentially novel class of non-fullerene acceptor material for use in organic solar cells that is under active investigation.125
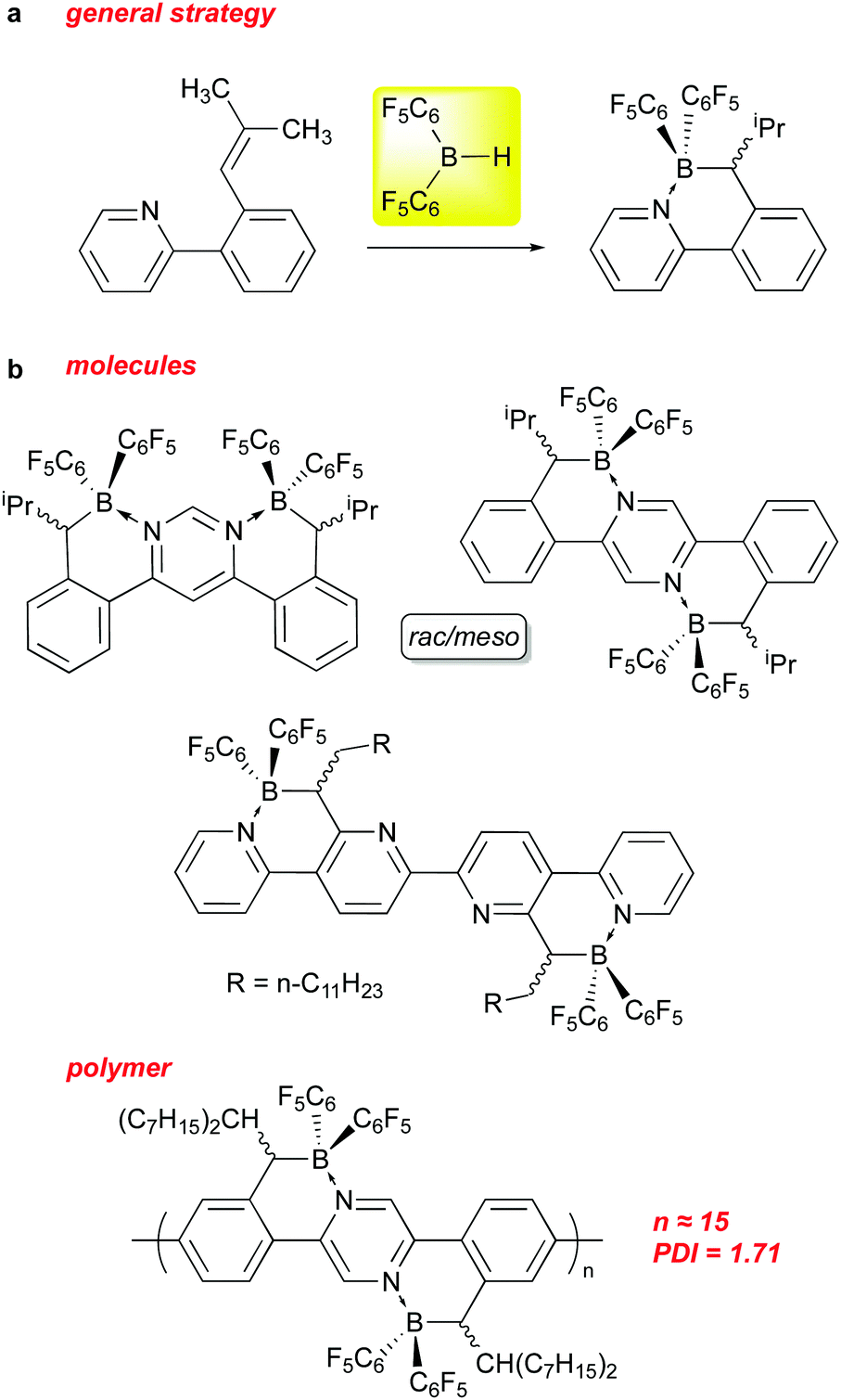 | ||
| Scheme 15 Use of 1 to install B–N units in molecular and polymeric electron acceptor materials. | ||
We end with a very recent report from Chang and co-workers that suggests that bis-pentafluorophenyl borane may play a growing role in the synthesis of B–N materials in the future. Applications of these molecules is to some degree being thwarted by a lack of general and efficient methods to the preparation of larger quantities.119 One widely used method is electrophilic borylation, but often this requires use of corrosive boron halides. Chang et al. have shown that 1 can be used to forge B–C bonds via electrophilic borylation and that the products can be catalytically converted to B–N heterocycles (Scheme 16).126 Here, they use their method of generating 1in situ from (C6F5)2B–OH in the presence of various 1,1-biphenyl dimethyl amino substrates. A survey of various amino groups showed that the dimethyl amino substituent was the most suitable, forming weaker adducts with 1 while allowing for enough of an interaction to direct borylation to the ortho site shown; this is accompanied by loss of H2. The presumed product ArB(C6F5)2 then undergoes H/C6F5 exchange with the excess PhSiH3 present to give the observed intramolecular amine–borane adducts which were all fully characterized, many also by X-ray crystallography.
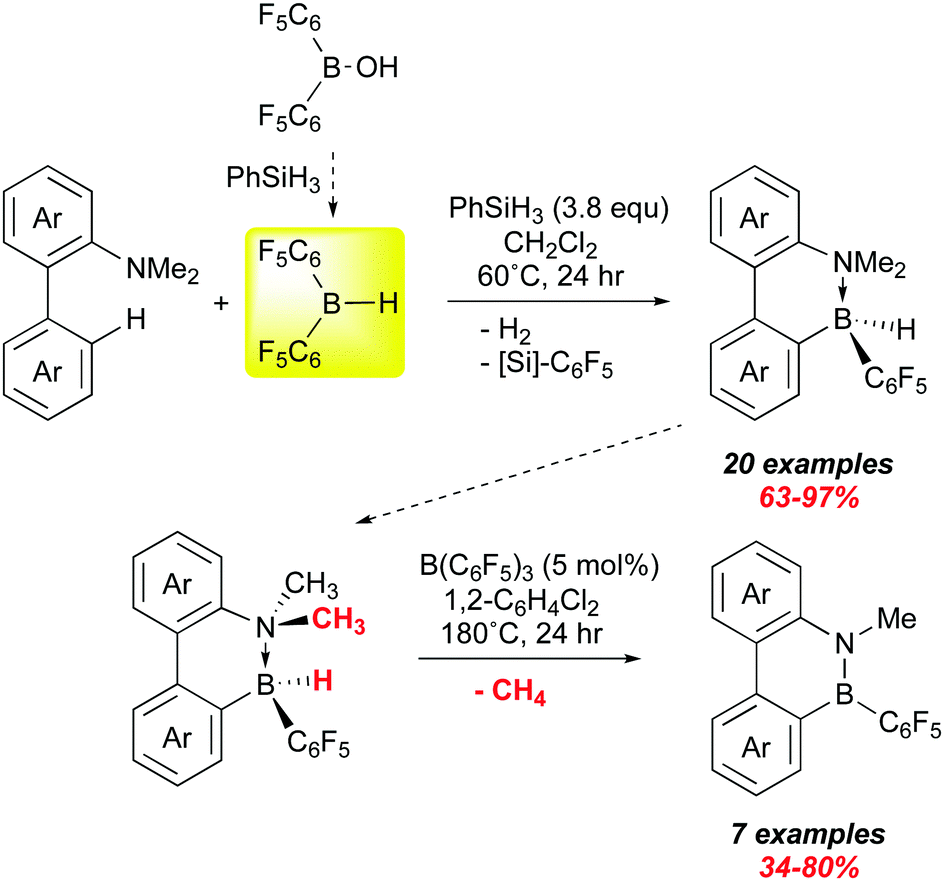 | ||
| Scheme 16 Electrophilic borylation of NMe2 substituted 1,1′-biaryls with 1. | ||
Loss of methane from these compounds to form the fully π conjugated B–N heterocyclic products was determined to be thermodynamically favourable by DFT, but prone to high kinetic barriers. The authors found, however, that catalytic amounts of B(C6F5)3 could mediate these tranformations, albeit under fairly harsh conditions. The borane abstracts hydride from the hydridoborane and the resulting ion pair releases methane irreversibly. Despite the high temperatures necessary, several amino boranes were converted to the planar B–N heterocycles in good to excellent yields, including more sophisticated, multifunctional substrates, leading to more extended B–N π systems. Indeed, it was possible to conduct both steps sequentially in one pot, providing a rapid and efficient path to relatively sophisticated materials on a gram scale. New routes to these compounds are key to their further exploitation as building blocks for acceptor materials in organic electronic applications.
Conclusions and future outlook
The emerging role of perfluoroaryl boranes in olefin polymerization catalysis in the late 1980's and early 1990's gave chemists ready access to the parent B(C6F5)3 compound and encouraged exploration of other boranes in this family. Since its first appearance 25 years ago, bis-pentafluorophenyl borane, 1, has emerged as one of the more useful members of this class of compounds because of its easy synthesis from B(C6F5)3 and its versatility for incorporating Lewis acidic –B(C6F5)2 units into molecules via hydroboration or electrophilic substitution processes. This article has highlighted some of its fundamental chemistry and many applications over the past two and a half decades. The two original papers10,13 introducing this reagent have been cited by ≈440 different publications and so the discussion we present is necessarily selective and non-comprehensive, but we hope conveys the diversity of areas in which it has been utilized. Much of the chemistry covered is from the past five years, attesting to the current interest in the chemistry and applications of 1, which promise to continue into the next decades.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from this work was provided by NSERC of Canada in the form of a Discovery Grant to W. E. P., who also thanks the Canada Research Chair secretariat for a Tier I CRC (2013–2020). E. A. P. thanks Alberta Innovates for scholarship support. W. E. P. would like to thank all the talented and dedicated students and postdoctoral fellows who worked on the chemistry from our labs described herein. As this article has a retrospective flavour, the senior author would also like to thank Prof. Dietmar Seyferth, who was the handling editor at Organometallics for our full paper describing the synthesis and properties of bis-pentafluorophenylborane, 1.13 The paper was reviewed quite favourably by two giants of organoborane chemistry and Prof. Seyferth took the initiative to ask the reviewers if he might reveal their identities to me; I still have the signed referee reports from Professors George Kabalka and Herbert C. Brown. This kindness and support to an early career researcher was typical of Prof. Seyferth and it meant a great deal to me at that stage of my career. W. E. P. would therefore like to dedicate this Feature Article to Prof. Deitmar Seyferth.Notes and references
- W. E. Piers, Adv. Organomet. Chem., 2005, vol. 52, pp. 1–76 Search PubMed.
- A. G. Massey, A. J. Park and F. G. A. Stone, Proc. Chem. Soc., 1963, 212 CAS.
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS.
- W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC.
- G. Erker, Dalton Trans., 2005, 1883–1890 RSC.
- R. L. Melen, Chem. Commun., 2014, 50, 1161–1174 RSC.
- M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- D. J. Parks, R. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809–811 CrossRef CAS.
- U. Blaschke, G. Erker, R. Frohlich and O. Meyer, Eur. J. Inorg. Chem., 1999, 2243–2247 CrossRef CAS.
- W. E. Piers, Chem. – Eur. J., 1998, 4, 13–18 CrossRef CAS.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef CAS.
- D. J. Parks and W. E. Piers, Tetrahedron, 1998, 54, 15469–15488 CrossRef CAS.
- A. Schnurr, K. Samigullin, J. M. Breunig, M. Bolte, H. W. Lerner and M. Wagner, Organometallics, 2011, 30, 2838–2843 CrossRef CAS.
- A. M. Fuller, D. L. Hughes, S. J. Lancaster and C. M. White, Organometallics, 2010, 29, 2194–2197 CrossRef CAS.
- S. Rubinsztajn, J. Chojnowski, M. Cypryk, U. Mizerska, W. Fortuniak and I. I. Bak-Sypien, J. Catal., 2019, 379, 90–99 CrossRef CAS.
- A. Ueno, J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Chem. – Eur. J., 2018, 24, 10044–10048 CrossRef CAS PubMed.
- J. Zhang, S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 13757–13761 CrossRef CAS PubMed.
- J. M. Breunig, F. Lehmann, M. Bolte, H. W. Lerner and M. Wagner, Organometallics, 2014, 33, 3163–3172 CrossRef CAS.
- E. A. Jacobs, R. Chandrasekar, D. A. Smith, C. M. White, M. Bochmann and S. J. Lancaster, J. Organomet. Chem., 2013, 730, 44–48 CrossRef CAS.
- J. Zhang, S. Park and S. Chang, Chem. Commun., 2018, 54, 7243–7246 RSC.
- P. E. Romero, PhD thesis, University of Calgary, 2006.
- M. Mendez and A. Cedillo, Comput. Theor. Chem., 2013, 1011, 44–56 CrossRef CAS.
- G. I. Nikonov, S. F. Vyboishchikov and O. G. Shirobokov, J. Am. Chem. Soc., 2012, 134, 5488–5491 CrossRef CAS PubMed.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- D. Winkelhaus, B. Neumann, H. G. Stammler and N. W. Mitzel, Dalton Trans., 2012, 41, 8609–8614 RSC.
- K. Samigullin, M. Bolte, H. W. Lerner and M. Wagner, Organometallics, 2014, 33, 3564–3569 CrossRef CAS.
- Z. P. Lu, Z. H. Cheng, Z. X. Chen, L. H. Weng, Z. H. Li and H. D. Wang, Angew. Chem., Int. Ed., 2011, 50, 12227–12231 CrossRef CAS PubMed.
- X. S. Tu, N. N. Zeng, R. Y. Li, Y. Q. Zhao, D. Z. Xie, Q. Peng and X. C. Wang, Angew. Chem., Int. Ed., 2018, 57, 15096–15100 CrossRef CAS PubMed.
- H. Y. Ye, Z. P. Lu, D. You, Z. X. Chen, Z. H. Li and H. D. Wang, Angew. Chem., Int. Ed., 2012, 51, 12047–12050 CrossRef CAS PubMed.
- K. K. Yan, B. M. Upton, J. Zhu, A. Ellern and A. D. Sadow, Organometallics, 2013, 32, 6834–6843 CrossRef CAS.
- K. D. Conroy, P. G. Hayes, W. E. Piers and M. Parvez, Organometallics, 2007, 26, 4464–4470 CrossRef CAS.
- P. A. Chase, W. E. Piers and M. Parvez, Organometallics, 2000, 19, 2040–2042 CrossRef CAS.
- R. E. V. Spence, D. J. Parks, W. E. Piers, M. A. Macdonald, M. J. Zaworotko and S. J. Rettig, Angew. Chem., Int. Ed. Engl., 1995, 34, 1230–1233 CrossRef.
- R. E. V. Spence, W. E. Piers, Y. M. Sun, M. Parvez, L. R. MacGillivray and M. J. Zaworotko, Organometallics, 1998, 17, 2459–2469 CrossRef CAS.
- Y. M. Sun, W. E. Piers and S. J. Rettig, Organometallics, 1996, 15, 4110–4112 CrossRef CAS.
- L. W. M. Lee, W. E. Piers, M. Parvez, S. J. Rettig and V. G. Young, Organometallics, 1999, 18, 3904–3912 CrossRef CAS.
- G. Ballmann, J. Martin, J. Langer, C. Färber and S. Harder, Z. Anorg. Allg. Chem., 2019 DOI:10.1002/zaac.201900179.
- J. Scott and D. J. Mindiola, Dalton Trans., 2009, 8463–8472 RSC.
- V. I. Minkin, R. M. Minyaev and R. Hoffmann, Usp. Khim., 2002, 71, 989–1014 Search PubMed.
- U. Radius, S. J. Silverio, R. Hoffmann and R. Gleiter, Organometallics, 1996, 15, 3737–3745 CrossRef CAS.
- R. R. Schrock and P. R. Sharp, J. Am. Chem. Soc., 1978, 100, 2389–2399 CrossRef CAS.
- K. S. Cook, W. E. Piers and S. J. Rettig, Organometallics, 1999, 18, 1575–1577 CrossRef CAS.
- K. S. Cook, W. E. Piers, T. K. Woo and R. McDonald, Organometallics, 2001, 20, 3927–3937 CrossRef CAS.
- K. S. Cook, W. E. Piers and R. McDonald, J. Am. Chem. Soc., 2002, 124, 5411–5418 CrossRef CAS PubMed.
- G. Parkin, Organometallics, 2006, 25, 4744–4747 CrossRef CAS.
- P. R. Sharp, S. J. Holmes, R. R. Schrock, M. R. Churchill and H. J. Wasserman, J. Am. Chem. Soc., 1981, 103, 965–966 CrossRef CAS.
- R. G. Carlson, M. A. Gile, J. A. Heppert, M. H. Mason, D. R. Powell, D. Vander Velde and J. M. Vilain, J. Am. Chem. Soc., 2002, 124, 1580–1581 CrossRef CAS PubMed.
- E. F. van der Eide, W. E. Piers, P. E. Romero, M. Parvez and R. McDonald, Organometallics, 2004, 23, 314–316 CrossRef CAS.
- P. E. Romero, W. E. Piers and R. McDonald, Angew. Chem., Int. Ed., 2004, 43, 6161–6165 CrossRef CAS PubMed.
- S. Mellino, L. C. Stevenson, E. Clot and P. Mountford, Organometallics, 2017, 36, 3329–3342 CrossRef CAS.
- T. G. Ostapowicz, C. Merkens, M. Holscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2013, 135, 2104–2107 CrossRef CAS PubMed.
- M. W. Drover, E. G. Bowes, J. A. Love and L. L. Schafer, Organometallics, 2017, 36, 331–341 CrossRef CAS.
- R. Spence and W. E. Piers, Organometallics, 1995, 14, 4617–4624 CrossRef CAS.
- G. Erker, Acc. Chem. Res., 2001, 34, 309–317 CrossRef CAS PubMed.
- W. E. Piers, Y. M. Sun and L. W. M. Lee, Top. Catal., 1999, 7, 133–143 CrossRef CAS.
- Y. M. Sun, R. Spence, W. E. Piers, M. Parvez and G. P. A. Yap, J. Am. Chem. Soc., 1997, 119, 5132–5143 CrossRef CAS.
- D. Huerlander, N. Kleigrewe, G. Kehr, G. Erker and R. Frohlich, Eur. J. Inorg. Chem., 2002, 2633–2642 CrossRef CAS.
- R. S. Rojas, B. C. Peoples, A. R. Cabrera, M. Valderrama, R. Frohich, G. Kehr, G. Erker, T. Wiegand and H. Eckert, Organometallics, 2011, 30, 6372–6382 CrossRef CAS.
- M. Hill, G. Kehr, R. Frohlich and G. Erker, Eur. J. Inorg. Chem., 2003, 3583–3589 CrossRef CAS.
- L. Chen, G. Kehr, R. Frohlich and G. Erker, Eur. J. Inorg. Chem., 2008, 73–83 CrossRef.
- M. Hill, G. Erker, G. Kehr, R. Frohlich and O. Kataeva, J. Am. Chem. Soc., 2004, 126, 11046–11057 CrossRef CAS PubMed.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Frohlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS PubMed.
- B.-H. Xu, G. Kehr, R. Fröhlich, B. Wibbeling, B. Schirmer, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7183–7186 CrossRef CAS PubMed.
- C. M. Momming, S. Fromel, G. Kehr, R. Frohlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 12280–12289 CrossRef PubMed.
- C. M. Momming, E. Otten, G. Kehr, R. Frohlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed.
- M. Sajid, A. Klose, B. Birkmann, L. Y. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Frohlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- A. Stute, L. Heletta, R. Fröhlich, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2012, 48, 11739–11741 RSC.
- M. Sajid, A. Lawzer, W. Dong, C. Rosorius, W. Sander, B. Schirmer, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2013, 135, 18567–18574 CrossRef CAS PubMed.
- A. J. P. Cardenas, B. J. Culotta, T. H. Warren, S. Grimme, A. Stute, R. Frohlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7567–7571 CrossRef CAS PubMed.
- A. J. P. Cardenas, Y. Hasegawa, G. Kehr, T. H. Warren and G. Erker, Coord. Chem. Rev., 2016, 306, 468–482 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- D. J. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131 RSC.
- D. J. Chen, Y. T. Wang and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475–9478 CrossRef CAS PubMed.
- J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed.
- D. J. Chen, V. Leich, F. F. Pan and J. Klankermayer, Chem. – Eur. J., 2012, 18, 5184–5187 CrossRef CAS PubMed.
- K. Y. Ye, X. W. Wang, C. G. Daniliuc, G. Kehr and G. Erker, Eur. J. Inorg. Chem., 2017, 368–371 CrossRef.
- W. Meng, X. Q. Feng and H. F. Du, Acc. Chem. Res., 2018, 51, 191–201 CrossRef CAS PubMed.
- S. L. Li, G. Li, W. Meng and H. F. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed.
- M. Fleige, J. Mobus, T. vom Stein, F. Glorius and D. W. Stephan, Chem. Commun., 2016, 52, 10830–10833 RSC.
- C. Jiang, O. Blacque and H. Berke, Organometallics, 2010, 29, 125–133 CrossRef CAS.
- X. Tao, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2017, 56, 1376–1380 CrossRef CAS PubMed.
- G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839–1850 RSC.
- X. Tao, C. G. Daniliuc, D. Dittrich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2018, 57, 13922–13926 CrossRef CAS PubMed.
- M. Sajid, G. Kehr, T. Wiegand, H. Eckert, C. Schwickert, R. Pöttgen, A. J. P. Cardenas, T. H. Warren, R. Fröhlich, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 8882–8895 CrossRef CAS PubMed.
- K.-Y. Ye, C. G. Daniliuc, S. Dong, G. Kehr and G. Erker, Organometallics, 2017, 36, 5003–5012 CrossRef CAS.
- A. Ueno, X. Tao, C. G. Daniliuc, G. Kehr and G. Erker, Organometallics, 2018, 37, 2665–2668 CrossRef CAS.
- P. Moquist, G. Q. Chen, C. Muck-Lichtenfeld, K. Bussmann, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Sci., 2015, 6, 816–825 RSC.
- Y. L. Liu, G. Kehr, C. G. Daniliuc and G. Erker, Chem. Sci., 2017, 8, 1097–1104 RSC.
- M. Sajid, L.-M. Elmer, C. Rosorius, C. G. Daniliuc, S. Grimme, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 2243–2246 CrossRef CAS PubMed.
- M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118–1121 CrossRef CAS PubMed.
- Z. B. Jian, G. Kehr, C. G. Daniliuc, B. Wibbeling, T. Wiegand, M. Siedow, H. Eckert, M. Bursch, S. Grimme and G. Erker, J. Am. Chem. Soc., 2017, 139, 6474–6483 CrossRef CAS PubMed.
- A. J. Ruddy, D. M. C. Ould, P. D. Newman and R. L. Melen, Dalton Trans., 2018, 47, 10377–10381 RSC.
- A. Simonneau, R. Turrel, L. Vendier and M. Etienne, Angew. Chem., Int. Ed., 2017, 56, 12268–12272 CrossRef CAS PubMed.
- C. N. Tang, Q. M. Liang, A. R. Jupp, T. C. Johnstone, R. C. Neu, D. T. Song, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 16588–16592 CrossRef CAS PubMed.
- K. L. Bamford, S. S. Chitnis, Z. W. Qu and D. W. Stephan, Chem. – Eur. J., 2018, 24, 16014–16018 CrossRef CAS PubMed.
- V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS PubMed.
- V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed.
- L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646–1649 CrossRef CAS PubMed.
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, ACS Catal., 2016, 6, 8399–8403 CrossRef CAS.
- T. A. Bender, P. R. Payne and M. R. Gagné, Nat. Chem., 2018, 10, 85–90 CrossRef CAS PubMed.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983 CrossRef CAS PubMed.
- R. D. Chambers and T. Chivers, J. Chem. Soc., 1965, 3933 RSC.
- T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Organometallics, 2003, 22, 1588–1590 CrossRef CAS.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- M. T. Peruzzi, Q. Q. Mei, S. J. Lee and M. R. Gagne, Chem. Commun., 2018, 54, 5855–5858 RSC.
- R. C. Chadwick, V. Kardelis, P. Lim and A. Adronov, J. Org. Chem., 2014, 79, 7728–7733 CrossRef CAS PubMed.
- E. Blondiaux and T. Cantat, Chem. Commun., 2014, 50, 9349–9352 RSC.
- D. J. Parks, W. E. Piers, M. Parvez, R. Atencio and M. J. Zaworotko, Organometallics, 1998, 17, 1369–1377 CrossRef CAS.
- M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. – Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed.
- M. M. Morgan and W. E. Piers, Dalton Trans., 2016, 45, 5920–5924 RSC.
- M. Grandl, T. Kaese, A. Krautsieder, Y. Sun and F. Pammer, Chem. – Eur. J., 2016, 22, 14373–14382 CrossRef CAS PubMed.
- F. Pammer, J. Schepper, J. Glockler, Y. Sun and A. Orthaber, Dalton Trans., 2019, 48, 10298–10312 RSC.
- M. Grandl, Y. Sun and F. Pammer, Org. Chem. Front., 2018, 5, 336–352 RSC.
- M. Grandl, B. Rudolf, Y. Sun, D. F. Bechtel, A. J. Pierik and F. Pammer, Organometallics, 2017, 36, 2527–2535 CrossRef CAS.
- M. Grandl, J. Schepper, S. Maity, A. Peukert, E. von Hauff and F. Pammer, Macromolecules, 2019, 52, 1013–1024 CrossRef.
- M. M. Morgan, M. Nazari, T. Pickl, J. M. Rautiainen, H. M. Tuononen, W. E. Piers, G. C. Welch and B. S. Gelfand, Chem. Commun., 2019, 55, 11095–11098 RSC.
- J. Zhang, H. Jung, D. Kim, S. Park and S. Chang, Angew. Chem., Int. Ed., 2019, 58, 7361–7365 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |