
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAquaporin-driven hydrogen peroxide transport: a case of molecular mimicry?†
Darren
Wragg
a,
Stefano
Leoni
*b and
Angela
Casini
 *a
*a
aDepartment of Chemistry, Technical University of Munich, Lichtenbergstr. 4, 85748 Garching, Germany. E-mail: angela.casini@tum.de
bSchool of Chemistry, Cardiff University, Park Place, CF103AT Cardiff, UK. E-mail: leonis@cf.ac.uk
First published on 25th November 2020
Abstract
Aquaporins (AQPs) are membrane proteins that have evolved to control cellular water uptake and efflux, and as such are amongst the most ancient biological “devices” in cellular organisms. Recently, using metadynamics, we have shown that water nanoconfinement within aquaporin channels results into bidirectional water movement along single file chains, extending previous investigations. Here, the elusive mechanisms of H2O2 facilitated transport by the human ‘peroxiporin’ AQP3 has been unravelled via a combination of atomistic simulations, showing that while hydrogen peroxide is able to mimic water during AQP3 permeation, this comes at a certain energy expense due to the required conformational changes within the channel. Furthermore, the intrinsic water dynamics allows for host H2O2 molecule solvation and transport in both directions, highlighting the fundamental role of water nanoconfinement for successful transduction and molecular selection. Overall, the bidirectional nature of the water flux under equilibrium conditions along with the mimicking behavior of hydrogen peroxide during a conductance event introduce a new chemical paradigm never reported so far in any theoretical paper involving any aquaporin isoform.
Introduction
The transport of H2O2 across cellular membranes by specific transmembrane proteins, named aquaporins (AQPs), has been considered one of the last milestones in the timeline of hydrogen peroxide discoveries in biochemistry.1 H2O2 can be dangerous or acts as a signaling molecule in various cellular processes, including cell migration and stem cell proliferation.2 In general, AQPs are responsible for water permeation across biomembranes,1 and are involved in a variety of important physiological processes. Therefore, they are also considered promising targets for therapeutic intervention.1,3,4 To date, the mechanism of hydrogen peroxide conductance is not fully understood. Water and H2O2 are highly related molecules, featuring similar dipole moment and hydrogen-bonding capability. Based on this similarity, it has been postulated that a permeating H2O2 molecule replacing a H2O molecule within the AQP channel should be energetically costless. Beitz and co-workers suggested that in principle all water-permeable AQPs can be H2O2 channels, and studying wild-type and mutated AQPs showed a positive correlation between the extent of water and H2O2 permeability in different isoforms.5 The few molecular dynamics (MD) studies reported so far investigated H2O2 permeation across AQP16,7 – an orthodox aquaporin which has not yet been fully validated experimentally as a hydrogen peroxide channel5,8 – or using plant AQPs isoforms,6 which are functionally and structurally very different from the mammalian isoforms.The functional unit of human AQPs is a tetramer with each monomer providing an independent channel consisting of three topological elements: an extracellular and a cytoplasmic vestibule connected by an extended narrow pore (Fig. 1A). Various structural and computational studies revealed water molecules passing through the AQPs channel in a single file.1,9,10 Two main constriction sites have been identified within the channel, which are responsible for substrate selectivity,1,9,11,12 namely the aromatic/arginine (ar/R) selectivity filter (SF), in proximity of the extracellular entrance, forming the narrowest part of the pore, and two conserved Asn-Pro-Ala (NPA) motifs in the middle of the channel, where the positive N-terminal ends of two half helices meet (Fig. 1A). Water that enters this region is re-oriented by the dipoles of the emanating half helices, such that hydrogen bonds between neighbouring water molecules in the chain are disrupted. Concerning other substrates, such as glycerol, the AQPs filtering can be to some extent explained by size selection due to steric hindrance, a venue that has been extensively explored in former approaches.1 This framework however, fails to justify how a membrane protein, evolved for water transport and osmotic pressure equilibration, can also selectively conduct ‘small’ signalling molecules as H2O2.
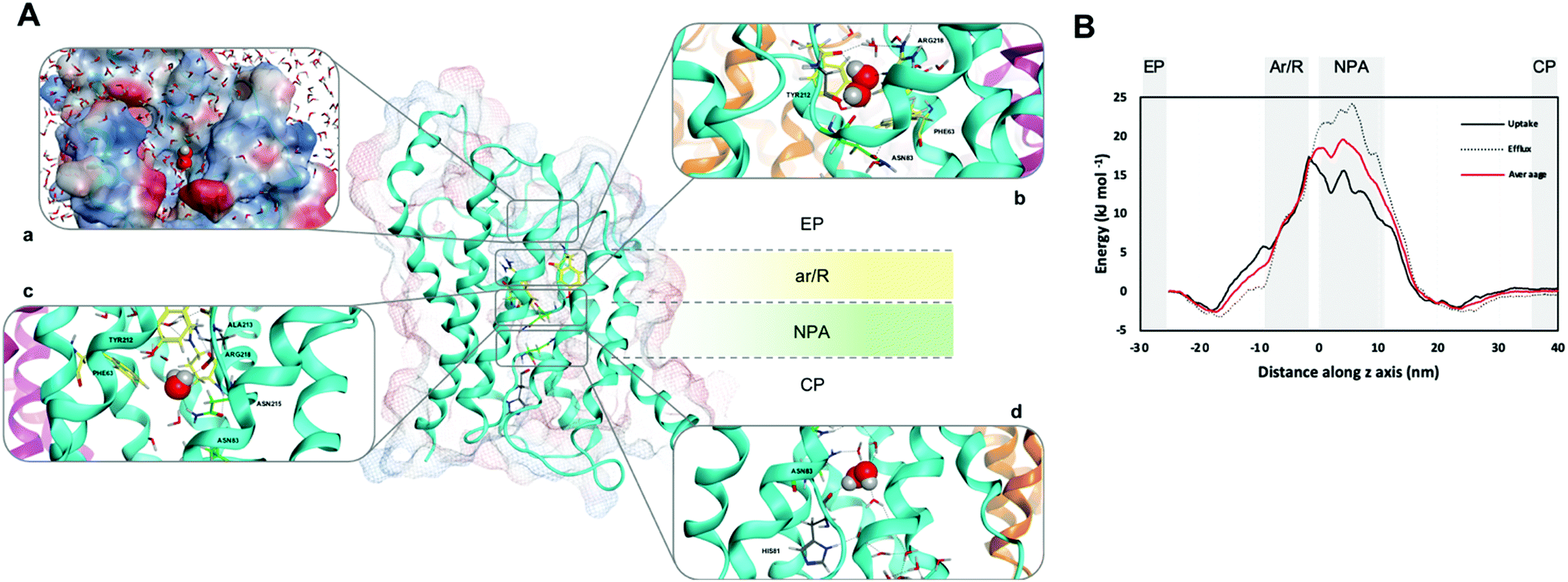 | ||
| Fig. 1 (A) hAQP3 monomer showing the ar/R (yellow) and NPA (green) selectivity filters. (a) Position of H2O2 as it enters the extracellular pocket of hAQP3 with lipophilic/hydrophilic molecular surface (hydrophilic = blue, lipophilic = red). (b) H2O2 orientation within the ar/R SF including H-bond network with residues Tyr212, Ala213 and Arg218. (c) H2O2 orientation during translocation between ar/R and NPA selectivity filters, interacting with residues Ala213 and Asn215 of the NPA motif. (d) H2O2 orientation when leaving the NPA selectivity filter, including H-bond network involving His81 and a bridging water molecule to Asn83. Figure generated using the MOE software.23 (B) Average free-energy surface of H2O2 uptake (solid line) and efflux (dashed line), calculated using metadynamics sampling. Averaged curve shown in red. | ||
To further expand our understanding of AQPs conductance, a combination of theoretical atomistic methods is applied here to attempt the elucidation of the mechanism of hydrogen peroxide permeation via human aquaglyceroporin-3 (AQP3).1 While this isoform is known to permeate both water and glycerol, it also facilitates uptake of H2O213,14 and has an important role in cancer development.3
Results
In details, a homology model of AQP3 was built following previously reported procedures.15,16 Thus, both biased and unbiased in silico techniques were used to study the permeation of H2O2 through the protein channel, namely metadynamics,17,18 to provide both free energy and mechanistic trajectories of conductance events, and transition path sampling (TPS),19 to achieve unbiased trajectory information.Metadynamics is an atomistic simulation technique enabling, within the same framework, acceleration of rare events along selected reaction coordinates, so-called collective variables (CV), and estimation of the free energy of complex molecular systems.17,18 This technique makes it possible to accelerate conformational transitions between metastable states, broadening the scope of MD simulations, and it has been already successfully applied to calculate the free-energy surface (FES) for the interactions of substrate/drugs with biomolecules, including DNA secondary structures.20 Recently, using metadynamics simulations, we investigated the mechanisms of glycerol permeation by the human AQP3 isoform.21 The obtained results showed that single-file water permeation through AQP3 is always bidirectional at equilibrium conditions, and that individual water molecules are able to ‘hop’ over each other (‘leap-frog’ mechanism), whilst traveling in opposite directions in certain key areas of the pore. Such mechanism for water transport may be essential to understand aquaporins ability to switch from uptake to efflux quickly under changing physiological conditions.21 Interestingly, it appeared that glycerol molecules exploit an existing water conduction mechanism in AQP3.
The herewith performed metadynamics calculations consisted of manifold 200 ns simulations to capture as many H2O2 permeation events as possible (see Experimental for details, ESI†). Overall, 12 uptake and 10 efflux events of H2O2 molecules were successfully observed from a total of 1.2 μs of combined simulation time. The averaged free-energy surfaces for both H2O2 uptake and efflux are shown in Fig. 1B and indicate the NPA region of the AQP3 channel as the one with the highest energy barrier during hydrogen peroxide conductance. This result suggests the NPA region as selectivity site for uncharged solutes, at variance with previously reported classical MD studies,22 whereby the ar/R SF was identified as the primary filter for other uncharged substrates. Specifically, the calculated absolute free-energy ΔG values for H2O2 match the trend previously observed for water and glycerol permeation, with H2O2 values fitting between those of the other two substrates21 (Table 1 and Fig. 1B).
Previous studies reported the experimentally calculated permeability values for water and glycerol permeation via different AQPs, including AQP3, in different cells.1,24–26 Moreover, a few studies also included hydrogen peroxide transport rates via AQP3.14,27 In general, permeability values for water are in the order of 10−3 cm s−1, while glycerol and H2O2 are in the range of 10−6 cm s−1. It should be noted that while experimental permeability values are usually assessed using an osmotic flux, our calculated free energy values are estimated at equilibrium conditions. Due to such crucial differences and various factors affecting substrate permeability, including lipid membrane composition that supports basal fluidity,28 it is impossible to draw a direct correlation between ΔG values and substrate permeabilities at this stage. Further metadynamics studies including a direct implementation of the osmotic flux through AQP3 in large scale simulations, within which substrate affinity can be measured, are warranted.
Analysis of the H-bond network for H2O2 permeation and residence times (Fig. 2A) shows that interactions are similar to those previously reported for water21 (Fig. S1, ESI†) throughout a permeation event, particularly in the NPA and cytoplasmic pocket (CP). From an in-depth analysis of the metadynamics’ data, the observed H2O2 entrance route into the AQP3 pore features less defined interactions with respect to glycerol permeation21 (see Movie M1, ESI†), which could be due to the H2O2 similarity in hydrogen bonding capability with water, facilitating the insertion of hydrogen peroxide molecules in the water file. As for glycerol permeation,21 during the passage of H2O2, we observe water molecules traversing the pore in either direction simultaneously, with little overall disruption (Movie M1, ESI†). In fact, H2O2 mimics water movement and allows the ‘leap-frog’ water mechanism to be preserved.
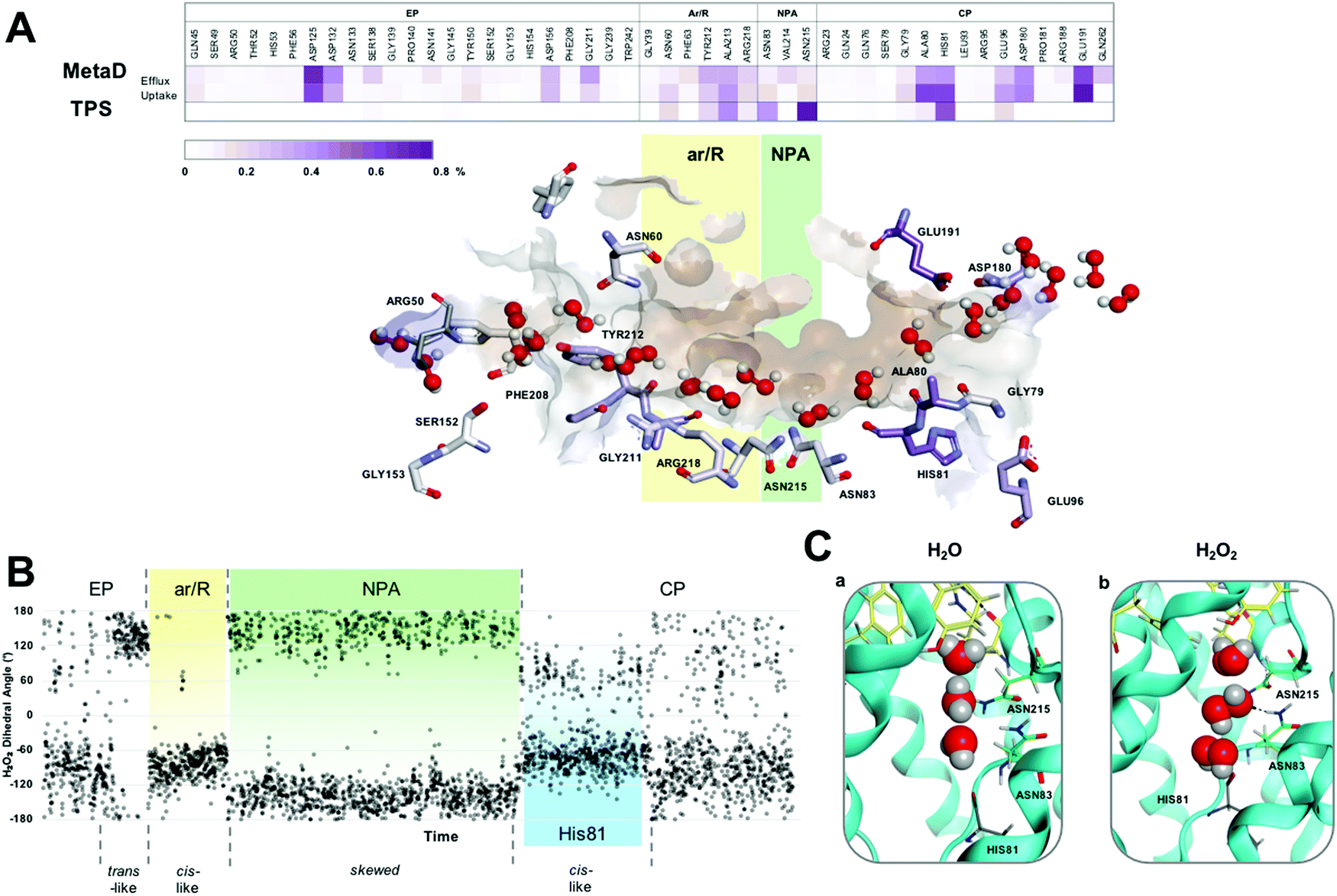 | ||
| Fig. 2 (A) H2O2 permeation routes and H-bond patterns and RT (%) from metadynamics and TPS calculations. Average H-bond RT (%) of H2O2 during uptake and efflux (on a scale of 0–0.8%) shown in a gradient purple colour. Multiple H2O2 molecules snapshots, taken from one representative simulation, are overlaid in one structure to create one single path. Amino acids that form crucial H-bonds are explicitly shown. Carbons are colour-mapped (white to purple) according to their corresponding RT (short to long). Pore colour representation based on hydrophobicity of the pore surface, blue = hydrophilic, brown = hydrophobic. (B) H2O2 dihedral angle change throughout a conductance event. (C) Water (a) and H2O2 (b) orientation when passing through the NPA motif (carbons coloured green) of hAQP3. Figure generated using MOE software.23 | ||
A detailed, representative H2O2 molecule pore permeation mechanism was obtained with the transition path sampling (TPS) method, enabling tackling of rare-events, which generates true dynamic reactive trajectories. Intermediate configurations from metadynamics provided a starting point, set between the ar/R and NPA selectivity filters, from which unbiased reactive trajectories were systematically shot off in both directions of time.
Analysis of reactive TPS trajectories enabled a greater insight into the interactions, at an atomic level, of the position and orientation of H2O2 during a conductance event. Single trajectory mechanistic steps compare favorably with those observed from the global metadynamics simulations, including similar H-bond interactions and residence times (Fig. 2A). Entering the AQP3 channel, H2O2 was found to interact mostly with water molecules within the extracellular pocket (EP, Fig. 2A), with little interplay with the protein residues until it approached the ar/R SF.
The preferred conformation of hydrogen peroxide is the nonplanar gauche or skew form;28 however, during the entire permeation event, H2O2 was observed to switch conformation whilst maintaining H-bond interactions with surrounding water molecules, as shown by the variation of the dihedral angle of the peroxide molecule (Fig. 2B and Fig. S2, Movie M2 in ESI†). Specifically, as H2O2 approaches the ar/R SF, it adopts a close to trans orientation to H-bond residues Tyr 212, Ala 213 and Arg218 (Fig. 2B and Fig. S3A, ESI†). Within the ar/R SF, H2O2 prefers a cis-like conformation (Fig. 1A(b), 2B and Fig. S3B, ESI†), which is maintained until approaching the NPA selectivity filter, with the oxygen atom facing downwards towards the NPA motif, mimicking water molecules (Fig. 1A(c), 2C and Fig. S3C, ESI†). Within the NPA region H2O2 reverts back to the preferred skew orientation (Fig. 2B, C and Fig. S3D, ESI†).
Finally, as H2O2 leaves the NPA region it again adopts a cis-like orientation with the oxygen atoms facing upwards, fully mimicking the water flipping mechanism (Fig. 1A(d), 2C and Fig. S3E, ESI†). In this region, H2O2 interacts with the backbone oxygen of the highly conserved His81 (Fig. 1A(d) and 2B). This interaction is observed globally in metadynamics and specifically in TPS calculations, and has been previously reported for the glycerol transport mechanism in hAQP321 and GlpF,29 respectively. TPS also confirms that water molecules coordinate to the backbone oxygen of the His81 residue located below the NPA selectivity filter (Movie M3 in ESI†). Whilst doing so, adjacent water molecules are able to hop over each other and re-join the water chain ahead of the coordinating molecule, or compete for binding to His81 (Video M3 in the ESI†). Interestingly, H2O2 can also bind to His81 and, mimicking water movement, allow adjacent waters to ‘hop’ over itself, therefore, maintaining the bidirectional water mechanism (Movie M2 in ESI†).
Overall, the extended residence time and multiple orientation and conformational changes observed for H2O2 passing through the NPA motif can explain the increase in free energy observed within this region (Fig. 1B). As H2O2 enters and exits the NPA, to fully mimic the water movement it must acquire a cis-type configuration, which has been shown to be the higher energy conformation for the peroxide molecule.30 These higher energy conformations, whilst essential for matching mechanistic rules of interaction with both the water chain and pore residues, are in line with the observed free energy increase. Therein, differences seen between calculated FES of individual trajectories, can therefore, be attributed to more or less strong fluctuations in the population of high-energy conformers. The constant switching of orientation to maintain optimal favorable interactions appears to be a justified energy expenditure for the conductance mechanism.
Conclusions
Recently, metadynamics showed us that water nanoconfinement within aquaporin channels results into bidirectional water movement along single file chains.21 This structure and intrinsic water dynamics allows for host molecule solvation and transport in both directions, highlighting the fundamental role of water nanoconfinement for successful transduction and molecular selection. Here, we shed light onto mechanistic steps of hydrogen peroxide facilitated transport by the human AQP3 using a combination of metadynamics and TPS methods, unveiling how mimicking water is key to efficiently negotiate internal energy barriers, while turning bare water transport into a signalling gateway. Moreover, our study further pinpoints the existence of a basic mechanism of water transport, over which guest/solute molecules are translocated. This allows for a direct comparison of peroxide with glycerol and for an atomistic elucidation of the mechanistic basis of selectivity, which is rooted into specific protein–guest molecule–water interactions. In summary, by harvesting details of water-mediated small molecule transport, we are laying the foundation for a molecular understanding of selective permeation in aquaporins, which is key for drug (inhibitor) design. We hope our work will stimulate the use of such advanced atomistic simulations to compute relevant quantities such as substrate/transporter binding free energies, and to generate very detailed transition path ensembles for in-depth understanding of elusive steps of solute transports across biomembranes, as well as for the design of bioinspired synthetic water channels.31Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
This work has been performed using resources provided by the “Cambridge Service for Data Driven Discovery” (CSD3, http://csd3.cam.ac.uk) system operated by the University of Cambridge Research Computing Service (http://www.hpc.cam.ac.uk) funded by EPSRC Tier-2 capital grant EP/P020259/1. We gratefully acknowledge the support of NVIDIA Corporation with the donation of a Quadro P5000 GPU used for this research. We thank ARCCA for access to computational facilities.References
- G. Soveral, S. Nielsen and A. Casini, Aquaporins in Health and Disease: New Molecular Targets for Drug Discovery, CRC Press, Taylor & Francis Group, 2017 Search PubMed.
- G. P. Bienert and F. Chaumont, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 1596–1604 CrossRef CAS.
- B. Aikman, A. De Almeida, S. M. Meier-Menches and A. Casini, Metallomics, 2018, 10, 696–712 RSC.
- C. Prata, S. Hrelia and D. Fiorentini, Int. J. Mol. Sci., 2019, 20, 1–15 Search PubMed.
- A. Almasalmeh, D. Krenc, B. Wu and E. Beitz, FEBS J., 2014, 281, 647–656 CrossRef CAS.
- R. M. Cordeiro, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 1786–1794 CrossRef CAS.
- M. Yusupov, J. Razzokov, R. M. Cordeiro and A. Bogaerts, Oxid. Med. Cell. Longevity, 2019, 2019, 2930504 Search PubMed.
- G. P. Bienert, A. L. B. Møller, K. A. Kristiansen, A. Schulz, I. M. Møller, J. K. Schjoerring and T. P. Jahn, J. Biol. Chem., 2007, 282, 1183–1192 CrossRef CAS.
- H. Sui, B. G. G. Han, J. K. J. K. K. Lee, P. Walian and B. K. B. K. K. Jap, Nature, 2001, 414, 872–878 CrossRef CAS.
- L. Janosi and M. Ceccarelli, PLoS One, 2013, 8, e59897 CrossRef CAS.
- K. Murata, K. Mitsuoka, T. Hiral, T. Walz, P. Agre, J. B. Heymann, A. Engel and Y. Fujiyoshi, Nature, 2000, 407, 599–605 CrossRef CAS.
- D. Fu, A. Libson, L. J. W. Miercke, C. Weitzman, P. Nollert, J. Krucinski and R. M. Stroud, Science, 2000, 290, 481–486 CrossRef CAS.
- E. W. Miller, B. C. Dickinson and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15681–15686 CrossRef CAS.
- J. R. Thiagarajah, J. Chang, J. A. Goettel, A. S. Verkman and W. I. Lencer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 568–573 CrossRef CAS.
- A. De Almeida, A. F. Mó, D. Wragg, M. Wenzel, P. Kavanagh, G. Barone, S. Leoni, G. Soveral and A. Casini, Chem. Commun., 2017, 53, 3830–3833 RSC.
- A. De Almeida, A. P. Martins, A. F. Mósca, H. J. Wijma, C. Prista, G. Soveral and A. Casini, Mol. BioSyst., 2016, 12, 1564–1573 RSC.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 2, 200–212 CrossRef.
- A. Barducci, M. Bonomi and M. Parrinello, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 826–843 CAS.
- P. G. Bolhuis and C. Dellago, in Advanced Computer Simulation Approaches for Soft Matter Sciences III, ed. K. Kremer and C. Holm, Springer, Berlin, Heidelberg, 2009, pp. 167–233 Search PubMed.
- D. Wragg, A. de Almeida, R. Bonsignore, F. E. Kühn, S. Leoni and A. Casini, Angew. Chem., 2018, 14524–14528 CrossRef CAS.
- D. Wragg, A. de Almeida, A. Casini and S. Leoni, Chem. – Eur. J., 2019, 25, 8713–8718 CAS.
- J. S. Hub and B. L. de Groot, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1198–1203 CrossRef CAS.
- C. C. Group, Inc. Montr. Quebec, Canada., 2018.
- I. V. Da Silva, M. Barroso, T. Moura, R. Castro and G. Soveral, Int. J. Mol. Sci., 2018, 19, 130–143 CrossRef.
- A. Serna, A. Galán-Cobo, C. Rodrigues, I. Sánchez-Gomar, J. J. Toledo-Aral, T. F. Moura, A. Casini, G. Soveral and M. Echevarría, J. Cell. Physiol., 2014, 229, 1787–1801 CrossRef CAS.
- L. Méndez-Giménez, S. Becerril, S. P. Camões, I. V. Da Silva, C. Rodrigues, R. Moncada, V. Valentí, V. Catalán, J. Gómez-Ambrosi, J. P. Miranda, G. Soveral, G. Frühbeck and A. Rodríguez, Int. J. Obes., 2017, 41, 1394–1402 CrossRef.
- C. Rodrigues, C. Pimpao, A. Coxixo, D. Lopes, P. A. Pedersen, F. Antunes and G. Soveral, Cancers, 2019, 11, 1–17 Search PubMed.
- G. P. Bienert, J. K. Schjoerring and T. P. Jahn, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 994–1003 CrossRef CAS.
- M. O. Jensen, S. Park, E. Tajkhorshid and K. Schulten, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6731–6736 CrossRef CAS.
- L. Song, M. Liu, W. Wu, Q. Zhang and Y. Mo, J. Chem. Theory Comput., 2005, 1, 394–402 CrossRef CAS.
- I. Kocsis, Z. Sun, Y. M. Legrand and M. Barboiu, npj Clean Water, 2018, 1, 1–11 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00160k |
| This journal is © The Royal Society of Chemistry 2020 |