
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing cyclotides to design and develop novel peptide GPCR ligands
Edin
Muratspahić
 ab,
Johannes
Koehbach
b,
Christian W.
Gruber
*a and
David J.
Craik
*b
ab,
Johannes
Koehbach
b,
Christian W.
Gruber
*a and
David J.
Craik
*b
aCenter for Physiology and Pharmacology, Institute of Pharmacology, Medical University of Vienna, Austria. E-mail: christian.w.gruber@meduniwien.ac.at
bInstitute for Molecular Bioscience, Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: d.craik@imb.uq.edu.au
First published on 22nd July 2020
Abstract
Cyclotides are plant-derived cyclic, disulfide-rich peptides with a unique cyclic cystine knot topology that confers them with remarkable structural stability and resistance to proteolytic degradation. Recently, cyclotides have emerged as promising scaffold molecules for designing peptide-based therapeutics. Here, we provide examples of how engineering cyclotides using molecular grafting may lead to the development of novel peptide ligands of G protein-coupled receptors (GPCRs), today's most exploited drug targets. Integrating bioactive epitopes into stable cyclotide scaffolds can lead to improved pharmacokinetics and oral activity as well as selectivity and high enzymatic stability. We also discuss and highlight the importance of engineered cyclotides as novel tools to study GPCR signaling.
 Edin Muratspahić | Edin Muratspahić received his MSc in Biological Chemistry at the University of Vienna (Austria). He performed his master thesis at the Institute of Cancer Research at the Medical University of Vienna (Austria) under the supervision of Hedwig Sutterlüty-Fall. In 2017, he started his PhD studies at the Institute of Pharmacology at the Medical University of Vienna under the guidance of Christian Gruber. His PhD studies involve discovery and development of novel nature-derived peptide GPCR ligands for treating therapeutic indications such as multiple sclerosis or pain. He is a Marietta Blau Fellow of the Austrian Federal Ministry of Education, Science and Research. |
 Johannes Koehbach | Johannes Koehbach obtained his PhD from the Medical University of Vienna (Austria) under the supervision of Christian Gruber and Michael Freissmuth, where he was studying the pharmacology of cyclotides on the oxytocin/vasopressin receptor family. He then moved to The University of Queensland (Australia) and was awarded a UQ Postdoctoral fellowship in the lab of Richard Clark. Since 2018, he is a postdoctoral Fellow with David Craik. His current research focus lies in the development and optimization of synthetic approaches to access complex peptide topologies such as cyclotides and other disulfide-rich or constrained structures. |
 Christian W. Gruber | Christian W. Gruber is research group leader and Associate Professor at the Medical University of Vienna (Austria). He studied Biochemistry at the University of Tübingen (Germany) and received a PhD in Molecular Biosciences from The University of Queensland (Australia). The research focus of his team is to study biological function, structure–activity relationship and pharmacological mechanism of nature-derived peptides isolated from plants and invertebrates (peptide hormones, neuropeptides and peptide toxins), and the development of novel peptide therapeutics. He is currently chief scientific advisor to Cyxone AB, a company which develops a cyclotide drug candidate for multiple sclerosis. Further information: http://www.meduniwien.ac.at/pharmakologie/ms/. |
 David J. Craik | David Craik is a Professor of Chemistry at IMB, UQ. He obtained his PhD in organic chemistry from La Trobe University in Australia and undertook postdoctoral studies at Florida State and Syracuse Universities, USA. He moved to UQ in 1995 to set up a biomolecular NMR laboratory and is currently an Australian Research Council (ARC) Laureate Fellow, Director of the ARC Centre of Excellence for Innovations in Peptide and Protein Science, and a Fellow of the Australian Academy of Science. His research focuses on applications of cyclic peptides, toxins and NMR in drug design. |
Introduction
Plants have historically been recognized as a rich source of therapeutic agents and they continue to profoundly inspire drug discovery. In fact, a myriad of today's drugs are plant-derived natural products or their derivatives.1 For instance, the sesquiterpene lactone artemisinin, isolated from Artemisia annua,2 and the indole alkaloids vinblastine and vincristine, derived from Catharanthus roseus,3 constitute mainstays of modern malaria and cancer treatment, respectively.In recent years, a considerable amount of literature has been accumulated, highlighting the potential of natural products as ligands of G protein-coupled receptors (GPCRs).4,5 These transmembrane receptors with more than 800 members encoded in the human genome are targeted by approximately 35% of US Food and Drug Administration-approved drugs, thus highlighting their druggability.6 Upon activation by a diverse array of ligands ranging from ions, and photons, small molecules and lipids to proteins and peptides, GPCRs interact with heterotrimeric G proteins composed of three subunits, Gα, Gβ and Gγ, to generate second messengers and initiate cellular signaling cascades.7 While the Gα subunit modulates adenylyl cyclase, phospholipase C-β, RHO-guanine nucleotide exchange factors or cGMP phosphodiesterase, the Gβγ complex is involved in the regulation of G protein-coupled inwardly rectifying potassium channels, voltage-gated Ca2+ channels, mitogen-activated protein kinases and many more downstream effector molecules.7 Binding of β-arrestins to activated GPCRs phosphorylated by GPCR kinases results in their uncoupling from heterotrimeric G proteins and targeting to clathrin-coated pits for endocytosis.7 Subsequently, GPCRs may encounter early endosomes, from where they either recycle to the cell membrane or undergo translocation for lysosomal degradation.8 Owing to their structural diversity and crucial role in a variety of physiological processes, GPCRs constitute the most attractive class of drug targets in modern pharmacology.
The vast majority of natural products acting as ligands of GPCRs are small molecules, but over the past few years nature-derived peptides have gained significant momentum in GPCR ligand discovery and development.9,10 Such peptide GPCR ligands have been mainly isolated from venomous animals, bacteria, fungi and plants and they provide a blueprint to produce novel peptidic GPCR ligands.9 Over 50 GPCR peptide drugs, most of which are chemically modified peptides, have been approved to date and more than 150 peptides have entered human clinical trials.10,11 Of approved drugs, five backbone-cyclic peptides targeting GPCRs have been approved as drugs.10 Bremelanotide is an example of a backbone-cyclic peptide selectively targeting the melanocortin 4 receptor (MC4R) whereas pasireotide, lanreotide, octreotide and vapreotide are backbone-cyclic peptides modulating the somatostatin GPCRs.10 In this article, we focus on a class of plant-derived cyclic, disulfide-rich peptides called cyclotides, and shed light on how they can be exploited to develop and engineer novel peptide GPCR ligands with improved pharmacological properties. We highlight the immense potential of plant-derived cyclotides as ligands of GPCRs.
Cyclotides
Structure, biosynthesis and function of native cyclotides
Cyclotides are the largest class of ribosomally synthesized and post-translationally modified plant-derived cyclic peptides.12 They are characterized by their unique cyclic cystine knot motif comprising a head-to-tail cyclic backbone and a cystine knot formed by six conserved cysteine (Cys) residues.13 This highly rigid structure provides them with exceptional stability against thermal, chemical or enzymatic degradation.14,15 Biosynthesis of cyclotides from gene-encoded precursors that possess a N-terminal signal peptide, N- and C-terminal flanking leader pro-peptides with single or multiple core peptide domains involves a cascade of events.16,17 Entering the secretory pathway via the endoplasmic reticulum enables proper folding of the cyclotide precursors and disulfide bond formation.18 Post-translational modifications of cyclotides including proteolytic cleavage and subsequent head-to-tail cyclization is thought to take place in the vacuole, leading to production of mature cyclotides.19 The key enzymes enabling cyclotide processing at the C-terminus and backbone cyclization, are asparaginyl endopeptidases that specifically target an Asn or Asp residue at the C-terminus, and have been elucidated over the last decade.20,21 More recently, Rehm and colleagues identified members of the papain-like cysteine proteases as cyclotide N-terminal processing enzymes which play a crucial role in the biosynthetic production of cyclotides.22So far, cyclotides have been discovered in five plant families including the Violaceae, Rubiaceae, Cucurbitaceae, Fabaceae and Solanaceae.23 They occur in all parts of the plant such as leaves, stems, flowers and roots, and a single plant species may express anything from just a few to over 150 individual cyclotides.24 Based on their sequences and structures, cyclotides can be classified into three major subfamilies: bracelet, Möbius and trypsin inhibitor subfamilies (Fig. 1).25 Compared to bracelet cyclotides, whose circular architecture mimics a polypeptide bracelet, Möbius cyclotides have a twist in their circular backbone due to a cis-prolyl peptide bond in loop 5 (Fig. 1).25 The trypsin inhibitor cyclotides are named for their potent inhibitory activity against the serine protease trypsin.26 They are associated with a lower degree of sequence homology to the bracelet and Möbius cyclotides and, in terms of sequences, are more similar to a family of linear peptides termed knottins.26,27 Accordingly, trypsin inhibitor cyclotides are alternatively referred to as cyclic knottins.27,28
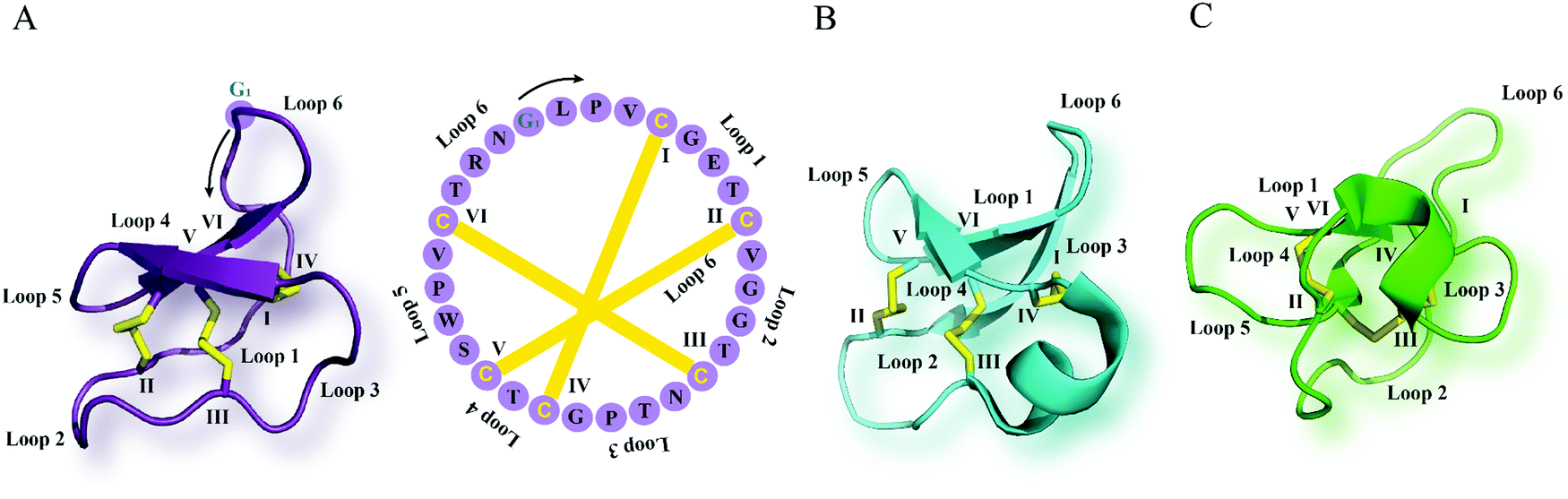 | ||
| Fig. 1 Cyclotide structure. NMR-derived three-dimensional structure and sequence of the (A) Möbius cyclotide kalata B1 (PDB: 1nb1), (B) bracelet cyclotide cycloviolacin O1 (PDB: 1nbj) and (C) trypsin inhibitor cyclotide MCoTI-II (PDB: 1ib9) are depicted. The cyclic cystine knot motif formed by three disulfide bonds is highlighted in yellow. The six cysteine residues are labeled I–VI, and the residues between adjacent cysteine residues represent loops 1–6. G1 indicates the starting point of the sequence while a black arrow points to the direction of the peptide chain starting from N- to C-terminus. MCoTI-II = Momordica cochinchinensis trypsin inhibitor-II; PDB = protein data bank. | ||
The discovery of cyclotides can be traced back to the early 1970s when kalata B1 (kB1), the prototypic member, was isolated from the herb Oldenlandia affinis, used in the Congo region of Africa as an uterotonic agent to induce contractions and accelerate childbirth.29,30 Since then, a plethora of biological functions of cyclotides have been reported.25,31,32 Studies demonstrated that kB1 exhibits insecticidal activity as it was capable of inhibiting the growth of an Australian cotton bollworm, Helicoverpa punctigera.33 Furthermore, other cyclotides have been shown to possess anthelmintic activity. For example kB6, isolated from O. affinis, was identified as the most potent inhibitor of gastrointestinal nematodes, Haemonchus contortus and Trichostrongylus colubriformis.34,35 These insecticidal and anthelminthic properties of cyclotides point towards their primary function as defense molecules in plants. Additionally, several cyclotides have been reported to have cytotoxic and anticancer properties. Bioactivity-guided fractionation of a Viola tricolor extract led to the isolation of three cyclotides, varv A, varv E and vitri A with anticancer properties, with vitri A showing the most pronounced cytotoxic effects.36 In a recent study, three novel cyclotides (poca A, poca B and CyO4) derived from Psychotria calceolaria were cytotoxic to cancer cells in a low μM range.37 Likewise, cyclotides from other plant families including the Rubiaceae and Fabaceae also demonstrated anticancer activities.38,39 In 1999, Tam and colleagues revealed salt-dependent antimicrobial activity of four cyclotides including kB1, circulin A and B and cyclopsychotride.40 All four cyclotides tested against nine strains of microbes showed activity against Gram-positive and negative bacteria, moderate activity against two fungi strains, and no activity against Candida albicans.40 Antimicrobial activity of cyclotides has been confirmed by other studies41–43 but it remains unclear whether this activity is biologically relevant as often the tested conditions did not include physiologically relevant media or bacteria relevant to plants.44 Recently, Slazak et al. examined the activity and mode of action of cycloviolacin cyclotides isolated from V. odorata and V. uliginosa against phytopathogens, thereby emphasizing their primary role in plant defense mechanisms.45
Molecular targets and mechanism of action
Having identified numerous biological activities of cyclotides, it was of vital importance to uncover their molecular targets. An early study characterizing the interaction of cyclotides with lipid membranes was reported by Kamimori and colleagues where kB1 and kB6 exhibited higher affinity towards dimyristoyl-L-α-phosphatidylethanolamine compared to other lipids consisting of phosphatidylcholine or phosphatidylglycerol.46 NMR spectroscopy was also exploited to determine the binding of kB1 to dodecylphosphocholine micelles.47 Moreover, using surface plasmon resonance, Henriques et al. observed a high affinity of kB1 to phosphatidylethanolamine (PE)-rich bilayers (Fig. 2) but weak binding to phosphatidylcholine bilayers, anionic bilayers and phosphatidylinositol-containing bilayers.48 Biophysical studies based on model and biological membranes provide evidence that kB1 targets cell membranes by a bimodal mode of action, whereby the bioactive face targets the PE headgroup, while the hydrophobic patch is responsible for the insertion into lipid bilayers.49–51 It is also worth mentioning that the synthetic enantiomer of kB1, D-kB1, shows slightly lower potency in hemolytic, insecticidal or anti-HIV assays than natural L-kB1.34,52 In a recent follow up study, Henriques and colleagues unveiled this chirality dependence by comparing peptide–lipid binding using mirror image model membranes.53 Synthesis of non-natural chiral phospholipids deciphered that the native L-kB1 is able to bind with higher affinity to phospholipids with the same chirality that is observed in eukaryotic membranes, thereby suggesting that the chiral environment of lipid bilayers is capable of modulating the function of membrane-active peptides (Fig. 2).53 In addition to Möbius cyclotides, the interaction of bracelet cyclotides with membranes has been also studied.54 In contrast to Möbius cyclotides kB1 and kB2, bracelet cyclotide cycloviolacin O2 was shown to be more active and selective towards anionic membranes containing dioleoylphosphocholine and dioleoylphosphate.54 Furthermore, interaction of this cyclotide with membrane lipids of E. coli resulted in a selective extraction of PE-containing lipids from the plasma membrane.54 | ||
| Fig. 2 Molecular targets and mechanism of action of cyclotides. L-Kalata B1 (purple, PDB: 1nb1) strongly interacts with the phosphatidylethanolamine-rich plasma membrane (straight arrow, left) whereas D-kalata B1's (blue, PDB: 2jue) interaction with the phosphatidylethanolamine-rich plasma membrane is significantly reduced (dashed arrow, left). By contrast, L-kalata B1 exhibits affinity towards a GPCR (straight arrow, right) but whether D-kalata B1 shows decreased receptor interaction remains to be determined (dashed arrow, right). Disulfide bonds are shown in yellow and phosphatidylethanolamine-rich plasma membrane and a GPCR are colored in red. GPCR = G protein-coupled receptor; PE = phosphatidylethanolamine; PDB = protein data bank. | ||
The unique interaction of cyclotides with PE confers them with the ability to enter into cells. Mutational studies on fluorescently labeled kB1 analogs revealed that in a MCF-7 breast cancer cell line the cytotoxic mutant [T20K]kB1 exhibits the ability to cross membranes more efficiently than the analog [V25K]kB1 with decreased cytotoxic activity.55 Moreover, cellular uptake of labeled [T20K]kB1 resulted in no cell internalization at 4 °C, whereas treatment with a macropinocytosis inhibitor allowed for penetration, suggesting that kB1 enters cells via endocytosis.55 In a follow up study, the underlying cell penetrating mechanism was elucidated using membrane-active or membrane-inactive kB1 variants labeled with a fluorophore via their Lys residue.56 [T20K]kB1 was shown to internalize into a HeLa cervical cancer cell line at a nontoxic peptide concentration compared to the inactive mutants [E7K]kB1, [T16K]kB1, and [V25K]kB1 which were impaired in their capability to penetrate the cells.56 Reduced cellular uptake was observed at 4 °C and thus was demonstrated to be due to changes in membrane fluidity and the inhibition of endocytosis which was partially dependent on both dynamin and actin. Overall, these studies suggest that kB1 enters cells either via direct translocation or endocytosis by binding to the membrane through the interaction with PE lipids.56
While cyclotides primarily interact with PE-rich membranes, GPCRs have been identified as another target (Fig. 2). In the area of cyclotide GPCR drug discovery an appropriate selection of controls used in the pharmacological assays ensures specific interaction of cyclotides with GPCRs, however, their interaction with membrane may represent a limitation for their use in GPCR drug development as this could lead to side effects. A seminal study on discovery of cyclotides as ligands of GPCRs was conducted by Koehbach and colleagues who unveiled the mechanisms underlying the oxytocic activity of cyclotides and their molecular targets.57 Bioactivity-guided fractionation and in-depth pharmacological characterization of an O. affinis extract identified kB7 as being able to trigger contractions of uterine smooth muscle cells by partially activating human oxytocin and vasopressin V1a receptors in the low μM concentration range (Table 1).57 Sequence analysis of kB7 and oxytocin revealed significant homology and thus enabled design of an oxytocin-like peptide with nM affinity and selectivity towards the oxytocin receptor. In a more recent study, Keov et al. showed for the first time that kB7 directly interacts with an insect GPCR, the inotocin receptor, of Tribolium castaneum and Lasius niger by partially agonizing these receptors with an EC50 of 7.2 and 2.2 μM, respectively (Table 1).58 The capability of cyclotides to modulate GPCR signaling has recently led to the isolation of several peptides from Carapichea ipecacuanha.59 One of these peptides, caripe 8, elicited antagonistic activity at the corticotropin releasing factor type I receptor, a class B GPCR (Table 1).59
| Cyclotide | Sequence | Target (MOA) | Affinity/potency (μM) | Ref. |
|---|---|---|---|---|
| GPCRs are named according to UniProt. More details on GPCR nomenclature are available in IUPHAR/BPS Guide to Pharmacology. n.d. refers to not determined ligand's mode of action (MOA). Grafted epitope sequences are highlighted in bold and italics.a Kalata B7 has been shown to interact with an insect GPCR, inotocin receptor, with an EC50 of 7.2 μM (Tribolium castaneum) and 2.2 μM (Lasius niger), respectively.58b B, X, and p denote the amino acid residues 2-naphthylalanine, citruline, and D-Pro, respectively.c B, X, and p represent the amino acid residues 2-naphthylalanine, citruline, and D-Pro, respectively, while Z indicates 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA).d X is L-2,3-diaminopropionic acid residue. | ||||
| Cyclopsychotride A | cyclo-CGESCVFIPCTVTALLGCSCKSKVCYKNSIP | NTR1 (antagonist) | IC50 3 | 127 |
| Kalata B7a | cyclo-CGETCTLGTCYTQGCTCSWPICKRNGLPV | OXTR, V1AR (partial agonist) | K i 50/EC50 12 | 57 |
| K i 12/EC50 4.8 | ||||
| Caripe 8 | cyclo-CGESCVFIPCITAAIGCSCKKKVCYRNGVIP | CRFR1 (antagonist) | n.d. | 59 |
| ckb-kal | cyclo-CGETCVGGTCNTPGCTCSWPVCKRPPGFSPL | BKRB1 (antagonist) | n.d. | 76 |
| [GHFRWG;23-28]kB1 | cyclo-CGETCVGGTCNTPGCTCSWPVCGHFRWGV | MC4R (agonist) | K i 0.029/EC50 0.580 | 89 |
| MCo-CVX-5Cb | cyclo-CPKILQRCRRDSDCPGACICRGNGYCGSYRXCRGpRRBCYXKV | CXCR4 (antagonist) | IC50 0.019 | 95 |
| MCo-CVX-6Dc | cyclo-CPZILQRCRRDSDCPGACICRGNGYCGSYRXCRGpRRBCYXKV | CXCR4 (antagonist) | IC50 0.00015 | 99 |
| MCo-AT1-7d | cyclo-CPKILQRCRRDSDCPGACICRGNGYCGSGEIYVRXGGV | MAS1R (agonist) | n.d. | 105 |
Cutting-edge structural biology techniques and advanced in silico methods developed over the past decade have broadened our knowledge of GPCR structure and function.6,60 With more than 20 GPCR structures in complex with peptides available, there are ample opportunities for rational, structure-based design of peptides.10,61 Moreover, these new technologies are being implemented to perform homology modelling and peptide–receptor interaction studies. For instance, Koehbach et al. utilized homology modelling to shed more light on the interaction of cyclotides with the oxytocin receptor.57 In line with the data of neurotensin (8–13) interacting with the extracellular surface of the neurotensin receptor,62 they revealed interactions of cyclotides with the extracellular loops of the oxytocin receptor and thus possible penetration into the ligand-binding pocket of peptide GPCRs despite their bulky size.57,63
Cyclotides as grafting scaffolds for GPCR ligand design and development
The development of peptide-based GPCR drugs, and peptide drugs in general, has been limited by the typically poor pharmacokinetic profiles of peptides (e.g. enzymatic instability, renal clearance) as well as low oral bioavailability and poor membrane permeability.9 Thus, novel strategies to tackle these challenges of peptides as therapeutics are vitally required. In this regard, the structural diversity, exceptional stability and multiple biological activities of cyclotides make them promising starting points for the discovery and development of novel GPCR ligands with unique pharmacological features. The concept of “molecular grafting” which delineates the insertion of a foreign epitope derived from a bioactive peptide or protein into the cyclotide scaffold,64 thereby endowing cyclotides with new biological activities, has created much interest in the field of cyclotide research (Fig. 3 and 4). Grafting epitopes onto cyclotides, in particular kB1 (Fig. 3) and Momordica cochinchinensis trypsin inhibitor-I (MCoTI-I) (Fig. 4), has been successfully applied to a wide range of molecular targets, including receptors and enzymes;65 however, herein we focus on its application and amenability in the context of designing and developing peptide GPCR ligands. The evidence based on the published knowledge of GPCR structures bound to endogenous peptides implies that α-, β-, or γ-turn motifs are the most common patterns of ligand recognition by GPCRs.66 Since these stable structural elements govern peptides interactions with GPCRs66 and as cyclotides have independently evolved these structural features in their framework,67 then it is likely cyclotides can not only act as defense molecules in plants but also serve mankind as a valuable source for developing peptide GPCR therapeutics. We now provide examples of how cyclotides, used as drug design scaffolds, can help to increase enzymatic stability, oral activity and selectivity of bioactive epitopes towards their target GPCRs. | ||
| Fig. 3 Examples of kB1 grafts. (A) A bioactive peptide epitope (orange) is inserted into loop 6 of kB1 (PDB: 1nb1) leading to stable peptide GPCR ligands with desired biological activity. Amino acid residues of native kB1 are shown in purple while grafted sequences are indicated in orange. Start and direction of the sequence is highlighted by a cysteine residue and a black arrow, respectively. Sequences and positions of bioactive epitopes grafted in loop 6 of kB1 discussed in this review are shown. Disulfide bonds or cysteine residues are highlighted in yellow and are labeled I–VI. Loops of cyclotides are labeled 1–6. (f) Represents the amino acid residue D-Phe. The starting point of the sequence has been taken from published studies. (B) Cartoon and surface overlay of kB1 and [GHFRWG; 23–28]kB1 (PDB: 2lur) as melanocortin pharmacophore highlighting the only key difference in the grafted loop 6. PDB = protein data bank; kB1 = kalata B1. | ||
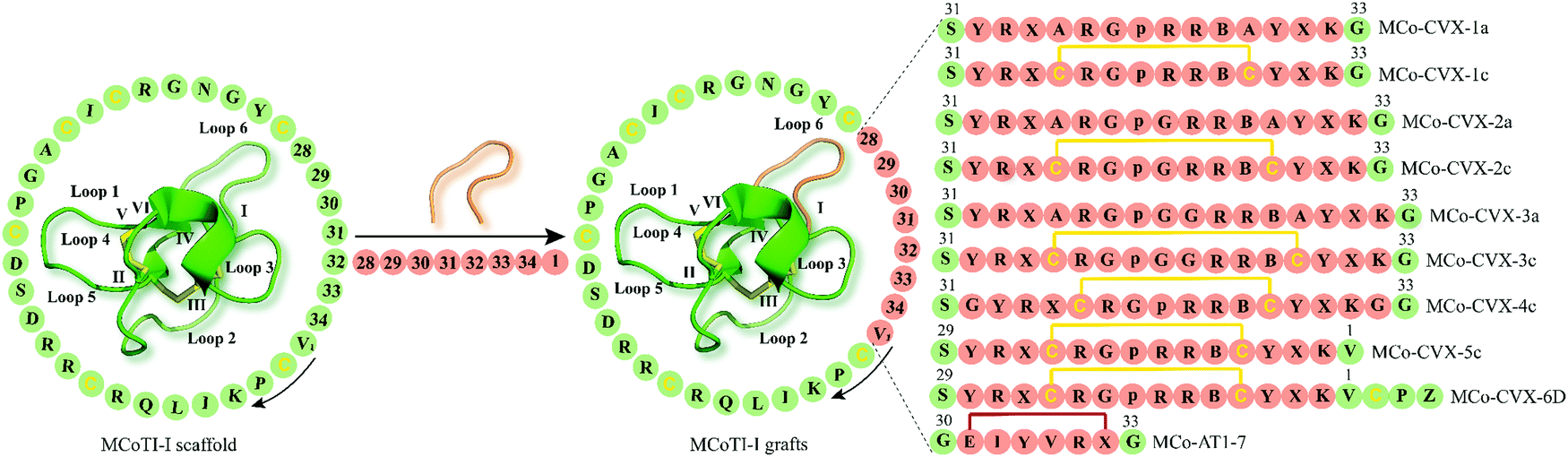 | ||
| Fig. 4 Examples of cyclotide GPCR ligands grafted onto loop 6 of MCoTI-I scaffold (MCoTI-II NMR structure was used as template, PDB: 1ib9). Amino acid residues of native MCoTI-I are shown in green while grafted sequences are highlighted in orange. Start and direction of the sequence is indicated by a valine residue and a black arrow, respectively. The starting point of the sequence has been taken from published studies. Disulfide bonds or cysteine residues are shown in yellow and are labeled I–VI. B and p indicate the amino acid residues 2-naphthylalanine and D-Pro, respectively, while Z represents 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA). X denotes citrulline residue except for MCo-AT1-7 where X is an L-2,3-diaminopropionic acid residue. The isopeptide bonds formed between the glycine residues and epitope are shown in red. MCoTI-I = Momordica cochinchinensis trypsin inhibitor-I; PDB = protein data bank. | ||
Targeting bradykinin receptors by a cyclotide scaffold to treat chronic pain
Chronic pain represents one of today's most challenging issues faced by the health-care sector, with more than 1.9 billion people worldwide suffering from this medical condition.68 Peptides derived from venomous animals such as cone-snails, snakes, spiders or scorpions have been shown to be a valuable source for the development of peptide therapeutics to treat chronic pain.69,70 Ziconotide, a synthetic version of ω-conotoxin MVIIA (ω-MVIIA) isolated from cone-snail Conus magus, is an example of a peptide drug approved for the treatment of chronic pain.71 However, ziconotide, – a blocker of N-type voltage-sensitive calcium-channels, Cav2.2 – develops centrally mediated side effects and thus needs to be intrathecally administered, restricting its use to only a limited number of patients with severe chronic pain.71 In this context, antagonists of bradykinin receptors, the prototypical GPCRs, have attracted increasing interest as therapeutic agents for the treatment of chronic and inflammatory pain.72,73 Bradykinin, kallidin and their des-Arg metabolites (des-Arg9-bradykinin and Lys-des-Arg10-bradykinin), collectively referred to as kinins, are short-lived peptide mediators with a half-life in blood of less than 30 seconds which are generated by virtue of the serine protease kallikrein that proteolytically cleaves the precursor protein kininogen.73 Kinins mediate their pharmacological effects by targeting bradykinin B1 and B2 GPCRs.73 Although the bradykinin B2 receptor is ubiquitously expressed in healthy tissues, the expression of bradykinin B1 receptor is predominantly associated with pathological tissue damage, suggesting that targeting bradykinin B1 receptors might be a promising approach to managing pain.74 Icatibant (HOE 140) is a potent and long-acting peptidomimetic bradykinin B2 receptor antagonist that has been approved to treat hereditary angioedema.75 However, as treatment with icatibant requires injection by the parenteral route, developing selective long-lived and orally active bradykinin B1 receptor peptide therapeutics represents an attractive strategy yet with significant challenges.Wong and colleagues have explored grafting onto cyclotides as a strategy to design and develop novel orally active and selective bradykinin B1 receptor peptide antagonists.76 Prompted by previous studies reporting des-Arg10-[Leu9]-kallidin (DALK) as a bradykinin B1 receptor antagonist,77 they opted to graft this epitope as well as des-Arg9-kinestatin (DAK) onto loop 6 of the kB1 framework which has been previously shown to be amenable to molecular grafting (Fig. 3A).78 Accordingly, loop 6 of kB1 (ckb) was substituted with either DALK (kal) or DAK (kin) resulting in two cyclic bradykinin antagonists: ckb-kal and ckb-kin (Fig. 3A).76 Additionally, a linear analog of ckb-kin, kb-kin, was designed to corroborate the necessity of the cyclic backbone to engineer stable, orally active bradykinin B1 receptor antagonists. Compared to the control peptide DALK with a half-life of less than five minutes, ckb-kal, ckb-kin and the linear variant kb-kin showed enhanced in vitro stability in human serum and lasted longer than six hours.76 These data provide evidence that the cystine knot architecture of kB1 may be effective in preventing enzymatic degradation of the designed peptide analogs. The toxicity of engineered cyclic and linear peptides against NIH3T3 cells was assessed, whereby the treatment with linear kb-kin led to about 10% cell death after 48 hours when using concentrations higher than 1 μM.76 Subsequent fluorescence-based functional assays were performed to elucidate the mode of action of the engineered peptides. All three peptides were capable of inhibiting intracellular calcium levels upon receptor activation with a bradykinin B1 receptor agonist des-Arg9-bradykinin (Table 1), but no antagonistic effect was observed following cell treatment with the bradykinin B2 receptor agonist bradykinin.76 The receptor subtype selectivity was further corroborated in a competition binding study: all three engineered bradykinin antagonists competitively displaced a fluorescein-des-Arg9-bradykinin agonist in HEK293 cells transiently expressing the bradykinin B1 receptor. The analgesic activity of the kB1-based peptides was then evaluated in an in vivo model of visceral pain induced by acetic acid thus leading to abdominal constriction (writhing) in mice.76 Both intraperitoneal (1 mg kg−1) and oral administration (10 mg kg−1) of grafted cyclotides demonstrated that ckb-kal acts as the most potent antagonist in the abdominal constriction assay followed by ckb-kin and then the linear control kb-kin.76 Intriguingly, intraperitoneal treatment with the DALK control resulted in analgesic activity similar to the cyclotide grafts, but no inhibition of writhing frequency was observed after oral application. Two-way analysis of variance on oral activity revealed no significant differences between intraperitoneal and oral administration for ckb-kal and ckb-kin, while there was a significant difference for the linear analogue kb-kin and the control peptide DALK. This study thus underpins the importance of the cyclic backbone of kB1 to retain and augment oral activity.
Exploiting the cyclotide framework to generate stable and selective melanocortin 4 receptor agonists
Obesity is a medical condition imposing a staggering burden on society and public health. Over the past few decades, obesity has reached pandemic proportions, with more than 100 million children and 600 million adults suffering from obesity (body mass index >30 kg m−2) with about 4 million deaths and 120 million disability-adjusted life-years as a consequence of excess body weight.79,80 Furthermore, obesity is a high-risk factor for comorbidities, including cardiovascular disease, diabetes mellitus, kidney dysfunction, cancer and musculoskeletal disorders.79 Liraglutide, a glucagon-like peptide 1 (GLP-1) analogue, has been recently approved to treat obesity,81 presumably acting by modulating appetite and body weight via activation of the GLP-1 receptor (GLP-1R) within glutamatergic neurons.82 In addition to targeting GLP-1R, the melanocortin system emerged as an alternative target to treat obesity83 and comprises five GPCRs, the melanocortin receptors (MCR 1–5). The endogenous peptide agonists include the adrenocorticotropic hormone and α, β-, and γ-melanocyte-stimulating hormones (MSH), which are all produced by proteolytic cleavage of the precursor protein proopiomelanocortin.83 Interestingly, the melanocortin system is unique in that it is the only GPCR system having two endogenous inverse agonists, i.e. agouti protein and agouti-related protein.84 Whereas MC1R, MC2R and MC5R are mainly expressed in melanocytes, the adrenal cortex and various peripheral tissues, respectively, high expression levels of MC3R and MC4R are detected in the central nervous system.85 MC4R is of particular interest as a target for the pharmacological treatment of obesity since activation of this GPCR leads to reduced food intake.86 In fact, setmelanotide is a potent MC4R peptide agonist that has completed phase II clinical trials to treat obesity and hyperphagia in patients with genetic defects in the leptin-melanocortin signaling pathways.87Given an unmet clinical need for MCR agonists, Eliasen et al. inserted a tetrapeptide sequence (His-Phe-Arg-Trp) of MSH and adrenocorticotropic hormones that is required to activate the MCRs88 into loop 6 of the kB1 scaffold (Fig. 3A).89 The tetrapeptide was flanked either by the remaining native amino acids in loop 6, i.e. T at the N-terminus and PV at the C-terminus or between G residues which were used instead of the native kB1 residues to prevent possible nonactive conformations of the tetrapeptide owing to direct replacements (Fig. 3A).89 In total, four cyclotide grafts were synthesized: [GHFRWG; 23–28]kB1, [GHRfWG; 23–28]kB1, [HFRW; 24–27]kB1 and [HfRW; 24–27]kB1 (Fig. 3A) and were subjected to an in-depth pharmacological characterization. All four peptides were tested in a radioligand binding assay and [GHFRWG; 23–28]kB1 was shown to displace a radiolabeled ligand from the MC4R in a concentration-dependent manner with a Ki of 29 nM (Table 1).89 Importantly, [GHFRWG; 23–28]kB1 exhibited 107- and 314-fold increased affinity towards MC4R over the MC1R and MC5R, respectively, while no binding to MC3R was detected at ligand concentrations up to 10 μM. To assess the ability of [GHFRWG; 23–28]kB1 to activate the MC4R, a functional cAMP assay was carried out. Compared to the endogenous ligand α-MSH with a potency of 3.7 nM, [GHFRWG; 23–28]kB1 was demonstrated to induce the activation of MC4R with an EC50 of 580 nM (Table 1).89 These data suggest that incorporation of the tetrapeptide into the kB1 scaffold results in a possible steric hindrance with the receptor and may account for the lower potency of the grafted cyclotide. Structural analysis by nuclear magnetic resonance spectroscopy revealed similar structural elements of [GHFRWG; 23–28]kB1 and kB1 indicating that [GHFRWG; 23–28]kB1 retained a native cyclotide fold (Fig. 3B). Additionally, surface-exposed hydrophobic residues of [GHFRWG; 23–28]kB1 such as His, Phe or Trp were surrounded by polar or charged residues while the Arg residue in loop 6 was shown to be highly solvent-exposed.89 Compared to α-MSH, [GHFRWG; 23–28]kB1 exhibited a significantly improved in vitro proteolytic stability with more than 80% of the peptide being intact after five hours. This study underscores the great potential of the kB1 scaffold to develop stable and selective agonists targeting MC4R for the treatment of therapeutic indications such as obesity.
Using cyclotide scaffolds to generate antagonists of CXC-motif-chemokine receptor 4
The CXC-motif-chemokine receptor 4 (CXCR4) was initially identified in 1996 as a co-receptor required for the entry of the human immunodeficiency virus type 1 (HIV-1) into host cells.90 Along with its endogenous chemokine CXCL12, CXCR4 participates in the control of cellular migration, organogenesis, hematopoiesis, and immune responses.91,92 Malfunctioning of CXCR4 is associated with a variety of medical conditions such as cancer, autoimmune diseases and atherosclerosis.91–93 Consequently, multiple synthetic CXCR4 ligands have been developed as antagonists of the CXCR4 (e.g. plerixafor/AMD3100 is an approved CX4R antagonist).94A decade ago, Aboye et al. reported the design and synthesis of a cyclotide-based antagonist of the HIV-1 viral replication capable of selectively targeting CXCR4.95 Using a MCoTI-I scaffold they grafted a circularly permutated version of CVX15 that was designed by fusing the N- and C-termini directly or via a flexible Glyn linker (n = 1, 2) and eliminating D-Pro8 and Pro9 residues, thereby resulting in the new N- and C-termini with Tyr10 and Lys7, respectively (Fig. 4).95 CVX15 is a cyclic peptide analog of the horseshoe crab peptide, polyphemusin, that has been previously shown to inhibit HIV by antagonizing CXCR4.96 Furthermore, in addition to substitution of Gln at position 6 by citrulline (CVX15 Gln6Cit), native Cys residues of CVX15 forming the disulfide bridge were replaced with Ala residues (Fig. 4). In a CXCR4-β-lactamase U2OS cell-based fluorescence assay, all cyclotide grafts concentration-dependently inhibited CXCR4 activated by endogenous CXCL12, yet with disparate potency ranging from 23.8 ± 0.3 μM (MCo-CVX-3a) to 19 ± 3 nM (MCo-CVX-5c) (Table 1).95 Compared to the most potent inhibitor MCo-CVX-5c, the peptide CVX15 Gln6Cit showed an EC50 value of 71 ± 13 nM. Furthermore, cyclotides MCo-CVX-1c and MCo-CVX-3c were approximately 10 times more potent than the cyclotide mutant versions substituted with an Ala residue instead of Cys (MCo-CVX-1a and MCo-CVX-3a), highlighting the importance of Cys residues for biological activity.95 Additionally, the length of the Gly linker was critical for the biological activity of the cyclotide grafts. For instance, the MCo-CVX-1c was the most potent antagonist with an EC50 of 0.10 ± 0.01 μM, which was designed by linking directly the N- and C-termini of the CVX15 peptide.95 Incorporation of additional residues on MCo-CVX-2c and MCo-CVX-3c generated moderately potent molecules with EC50 values of 2 and 3 μM, respectively, suggesting that Gly residues enhance flexibility of the peptide thus affecting the potency by presumably leading to a greater entropic penalty.95 Moreover, the position of the epitope in loop 6 was important for the biological activity of the grafted cyclotides. The most active cyclotide was MCo-CVX-5c (EC50 = 19 ± 3 nM) (Table 1), grafted between Gly residues 30 and 34, while MCo-CVX-1c (grafted at Asn32) and MCo-CVX-4c (grafted at Asn32 and with additional Gly linker) showed EC50 values of 102 ± 12 nM and 39 ± 1 nM, respectively. Also, MCo-CVX-5c inhibited not only CXC12-induced Erk phosphorylation and internalization of CXCR4 but also the entry and replication of CXCR4-tropic HIV-1 in human lymphocyte MT4 cells (EC50 = 2.0 ± 0.3 nM).95 Importantly, MCo-CVX-5c exhibited about three times higher potency than raltegravir, an approved drug to treat HIV infection.97 Determination of peptide in vitro stability in human serum revealed that MCo-CVX-5c is more stable than the native MCoTI-I, the grafted peptide CVX15 Gln6Cit or the linear, reduced, and alkylated version of MCo-CVX-5c. Biolayer interferometry analysis also indicated that the cyclotide MCo-CVX-5c is capable of binding to serum proteins with association and dissociation rate constants of (3.6 ± 0.7) × 103 M−1 s−1 and (1.4 ± 0.2) × 10−2 s−1, respectively.95 Serum binding has been reported to be a valuable strategy to prolong serum half-life of peptides.98
In a follow-up study, the grafted cyclotide MCo-CVX-5c was used as a template to synthesize MCo-CVX-6 in which a Lys residue in loop 6 was replaced by an Arg residue.99 MCo-CVX-6 was then conjugated to the metal-chelating agent 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraaceticacid (DOTA) via its ε-amine group of the Lys residue in loop 1, resulting in MCo-CVX-6D (Fig. 4). MCo-CVX-6D showed increased in vitro binding affinity for CXCR4 (Ki = 0.15 ± 0.01 nM) (Table 1) and was exploited in a radiolabeled cyclotide with 64Cu2+ ([64Cu]MCo-CVX-6D) suitable for positron emission tomography-computed tomography (PET-CT) imaging studies in mice carrying CXCR4 glioblastoma xenografts.99 Subsequent in vivo studies demonstrated significant uptake of radiolabeled MCo-CVX-6D in tumors expressing CXCR4, whereas the MCoTI-I control peptide labeled with [64Cu-DOTA] mainly accumulated in the kidneys.99 These data are in line with a report by Wang et al. who determined renal clearance of MCoTI-II as the major elimination route.100 These studies highlight the ability of MCoTI-I framework to accommodate sequence variations to develop stable and potent antagonists of the CXCR4.
Design of cyclotide-based peptides with angiotensin-like activity
The renin–angiotensin system orchestrates essential physiological processes in the human body including blood pressure and fluid balance.101 These physiological events are executed and maintained by a number of enzymes and endogenous peptides. Angiotensin I is produced by the protease renin which cleaves the precursor protein angiotensinogen, whereas angiotensin II is generated by angiotensin-converting enzyme which cleaves angiotensin I.101 Angiotensin-converting enzyme in turn is capable of cleaving angiotensin I and angiotensin II to form angiotensin 1–9 and angiotensin 1–7, respectively.101 Accumulating evidence underpins the implication of the renin-angiotensin system in carcinogenesis and its modulation by endogenous angiotensin (AT) 1–7.102 Studies have shown that the activation of the proto-oncogene MAS 1 receptor (MAS1R) induced by AT 1–7 significantly inhibits human lung cancer growth both in vitro and in vivo.103 Hence, targeting MAS1R provides an appealing strategy to develop therapies against lung cancer.Driven by these studies and owing to the poor pharmacodynamic and pharmacokinetic profiles of AT 1–7,104 Aboye and colleagues grafted an AT 1–7-like peptide into loop 6 of the MCoTI-I cyclotide, with Asp1 and Pro7 being replaced by diaminopropionic acid and Glu, respectively (Fig. 4).105 The AT 1–7 analogue was placed between Gly30 and Gly33 residues of MCoTI-I using the β-amino and γ-carboxylic groups, thereby generating two isopeptide bonds in the new MCo-AT 1–7 cyclotide (Fig. 4). This enabled the AT 1–7 analog to retain its positively and negatively charged groups at the N- and C-termini.105 The synthesized MCo-AT 1–7 showed striking in vitro stability in human serum with a half-life of 39 hours compared to its linearized, reduced and alkylated version, which degraded within 57 minutes.105 These results further stress the significance of the CCK motif for the enzymatic stability of cyclotides.
The ability of the grafted cyclotide to modulate the MAS1R signaling was assessed in a cell-based fluorescence assay in a CHO cell line stably expressing the receptor and measuring intracellular NO levels. The MCo-AT 1–7 cyclotide was shown to act as an agonist of the MAS1R by increasing intracellular NO in a concentration-dependent manner (Table 1).105 Co-incubation of MCo-ATI 1–7 with the MAS1R peptide antagonist A779 reinforced the specificity of the cyclotide graft as no increase in the intracellular NO concentration was observed. Overall, the findings of this study support the idea of using the MCoTI-I framework as a springboard for the development of peptide GPCR ligands with angiotensin-like activity.
Conclusions and perspective
Cyclotides have the potential to address some of the major challenges of peptide therapeutics including serum as well as gastrointestinal stability, oral activity and membrane permeability. In this review, we have elaborated on several examples in which grafting of bioactive epitopes onto cyclotide templates has proven to be a powerful strategy to facilitate the development of peptide GPCR ligands with enhanced pharmacological properties such as oral activity, receptor selectivity and/or resistance to proteolysis (Fig. 3, 4 and Table 1). Besides the potential of grafted cyclotides as therapeutics, we have referred to a study in which cyclotides have been shown to be a promising diagnostic tool by targeting GPCRs to enable the early detection of cancer and to monitor its progression.However, several limitations and challenges associated with molecular grafting need to be addressed in future research. Correct folding of grafted cyclotides is one of the key challenges which may explain why no engineered cyclotide targeting a GPCR of interest has yet entered human clinical trials. This being the case, a broader understanding of the processes involved in oxidative folding of cyclotides and how grafting influences folding would be valuable. Exploiting de novo design of peptides,106 recently discovered cyclization enzymes107,108 or hybrid methods relying on nature-inspired biosynthetic reactions109 are promising strategies to facilitate the folding of grafted cyclotides. In another relevant development, Yin et al. recently established a method based on the structure of anchor residues of the epitope and scaffold to guide form and function in grafted peptides.110 Another challenge is that the development of cyclotides bioengineered to unleash immune responses suggests that there is a possibility of inadvertent immunogenicity occurring for some cyclotide constructs.111,112 Compared to protein therapeutics,113 grafted cyclotides are less prone to be immunogenic due to their small size and resistance to proteolysis; however, careful assessment of preclinical immunogenicity studies prior to clinical evaluation is regarded as imperative.
Despite these challenges, grafted cyclotides represent powerful ligands and tools to illuminate GPCR pharmacology. For instance, it is known that native cyclotides do not cross the blood–brain barrier (BBB),100 however, this feature may be advantageous to target GPCRs specifically expressed in the peripheral nervous system. Difelikefalin is an excellent example of a peptide drug with poor penetration of the BBB, that was introduced into the clinic in 2019 to treat postoperative pain by selectively targeting κ-opioid receptors.10 Targeting κ-opioid receptors in the periphery has emerged as an appealing strategy to develop novel analgesics without centrally mediated side effects including sedation, dysphoria and hallucinations.114 Given the remarkable stability of cyclotides and their suitability for molecular grafting as well as their limited capability to penetrate the BBB, there is great potential to develop novel peripherally restricted cyclotide-based peptide GPCR therapeutics.
In recent years, cell penetrating peptides have gained impetus as a class of peptides capable of carrying and translocating large proteins or hydrophilic molecules across cell membranes.115 Intriguingly, cyclotides have been demonstrated to possess cell penetrating properties. For example, Ji et al. chemically engineered a cyclotide that antagonized an intracellular interaction of the tumor suppressor p53 and the oncoprotein Hdm2 both in vitro and in vivo.116 However, it has been known for a long time that cell penetrating peptides can remain trapped in endosomes during endocytosis, leading to their lysosomal degradation and limited ability to reach their target sites and to convey biological activity.115 Whether and how cyclotides escape endosomes remains to be determined, but the work of Ji et al. demonstrates that grafted cyclotides are active in the cytoplasm.116 Furthermore, Huang and colleagues have optimized the cell penetrating properties of cyclotides in order to foster their intracellular uptake.117 It is thus of interest to explore if the capacity of cyclotides to penetrate cell membranes can be leveraged in the future to engineer and develop cyclotide-based peptide ligands able to target GPCRs beyond barriers and reach areas such as the central nervous system (Fig. 5). Recent studies offer evidence that canonical GPCR signaling is not confined to the plasma membrane but may also occur in endosomes, suggesting that targeting endosomal GPCRs is a feasible strategy with therapeutic potential.118–121 This raises the further question of whether cyclotides equipped with a given bioactive epitope may modulate endosomal GPCR signaling (Fig. 5).
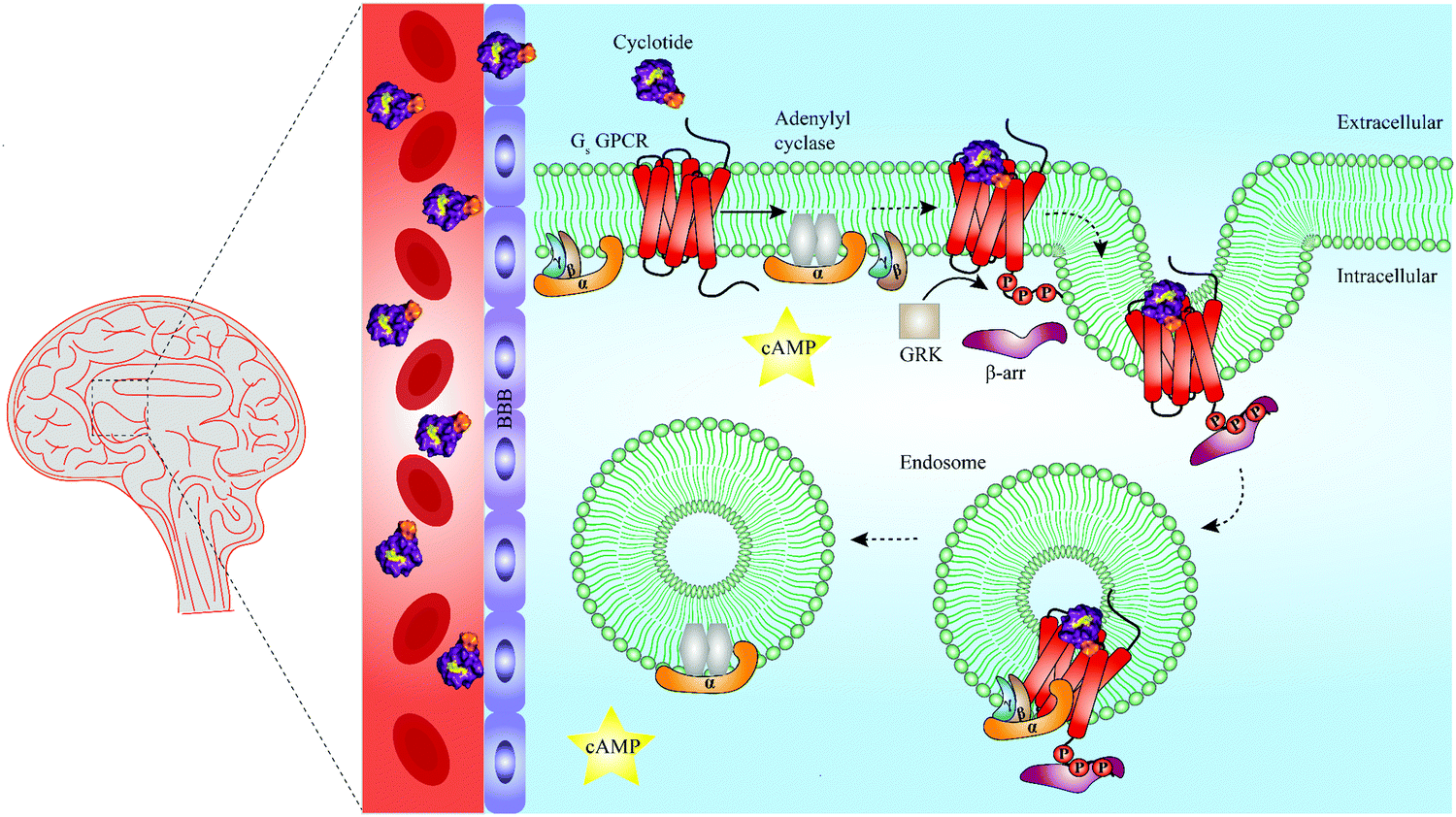 | ||
| Fig. 5 Summary of unresolved functions of engineered cyclotides as GPCR ligands. Native cyclotides have restricted capabilities to cross the blood–brain barrier (BBB), yet they are suitable to develop cyclotide grafts to target GPCRs in the central nervous system by penetrating the BBB. They may be harnessed as a tool to study biased signaling of GPCRs by either activating G protein-dependent or β-arrestin-dependent signaling pathways. Additionally, cyclotides may be able to initiate endosomal GPCR signaling of Gs protein-coupled receptors (Gs GPCR). For instance, following Gs GPCR activation by a grafted cyclotide, conformational change of a Gs GPCR leads to dissociation of the heterotrimeric G protein complex and production of cAMP (second messenger) via activation of adenylyl cyclase. While modulation of G protein-dependent pathways by cyclotides is well-studied (straight arrow), β-arrestin recruitment and thus β-arrestin-dependent pathways are poorly investigated (dashed arrows). Phosphorylation of a Gs GPCR by GRKs results in β-arrestin recruitment and initiation of clathrin-mediated endocytosis. According to a recent study published by Nguyen et al., once internalized, Gs GPCR and β-arrestin form a megaplex that activates Gs protein to enable a sustained endosomal Gs-mediated signaling.121 GRK = G protein-coupled receptor kinases, cAMP = cyclic adenosine monophosphate, Gs = stimulatory G protein, GPCR = G protein-coupled receptor. | ||
In the context of developing GPCR drugs devoid of centrally mediated deleterious side effects, the past five years have witnessed a renaissance in studies of the biased signaling of GPCRs. The ability of GPCR ligands to selectively activate one pathway over another122 has led to development of several peptide biased ligands preferably activating either G protein-dependent or β-arrestin-dependent signaling pathways linked to either beneficial or detrimental effects. For example, Zhang et al. identified a G protein-biased peptide agonist of the glucagon-like peptide-1 receptor by screening large combinatorial libraries suggesting that G protein-biased peptide agonists targeting GLP-1R are promising molecules to treat type II diabetes.123 On the other hand, β-arrestin biased peptide agonists targeting angiotensin II receptor type I have been proposed to have therapeutic potential for the treatment of cardiomyopathy.124 The ability of engineered cyclotides to engage in β-arrestin-biased signaling remains an unexplored area that warrants further investigation (Fig. 5). All cyclotide grafts discussed in this review have been chemically synthesized based on either Möbius or trypsin inhibitor cyclotide frameworks due to the fact that bracelet cyclotides are more difficult to fold in vitro,125 albeit they are more prevalent in nature than Möbius or trypsin inhibitor cyclotides.126 Hence, it remains an elusive question for now to know if grafting bioactive epitopes onto bracelet cyclotide scaffolds would lead to the development of cyclotide GPCR ligands with distinct pharmacological properties, e.g. improved potency or target selectivity.
Given the importance of GPCRs as drug targets,6 the unique diversity of naturally occurring cyclotides as well as advances in molecular grafting methodologies,110 we are confident that plant-derived cyclotides will provide new opportunities for GPCR drug development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research in the laboratory of C. W. G. is funded by the Austrian Science Fund (FWF) through project P32109. Work in the laboratory of D. J. C. on cyclotides is funded by grants from the Australian Research Council (DP150100443) and the National Health and Medical Research Council (GNT1084965 and GNT1164412). D. J. C. is an ARC Australian Laureate Fellow (FL150100146). E. M. is a Marietta Blau Fellow of Austrian Federal Ministry of Education, Science and Research (ICM-2019-13441).References
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs from 1981 to 2014, J. Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed
.
- Y. Tu, The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine, Nat. Med., 2011, 17, 1217 CrossRef CAS PubMed
- M. Gorman, N. Neuss, G. H. Svoboda, A. J. Barnes Jr and N. J. Cone, A note on the alkaloids of Vinca rosea Linn. (Catharanthus roseus G. Don.). II. Catharanthine, lochnericine, vindolinine, and vindoline, J. Am. Pharm. Assoc., 1959, 48, 256 CrossRef CAS PubMed
- A. Gupta, I. Gomes, E. N. Bobeck, A. K. Fakira, N. P. Massaro, I. Sharma, A. Cave, H. E. Hamm, J. Parello and L. A. Devi, Collybolide is a novel biased agonist of kappa-opioid receptors with potent antipruritic activity, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6041 CrossRef CAS PubMed
- Y. Kuroda, K. J. Nicacio, I. A. da Silva-Jr, P. R. Leger, S. Chang, J. R. Gubiani, V. M. Deflon, N. Nagashima, A. Rode and K. Blackford,
et al., Isolation, synthesis and bioactivity studies of phomactin terpenoids, Nat. Chem., 2018, 10, 938 CrossRef CAS PubMed
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schioth and D. E. Gloriam, Trends in GPCR drug discovery: new agents, targets and indications, Nat. Rev. Drug Discovery, 2017, 16, 829 CrossRef CAS PubMed
- D. Hilger, M. Masureel and B. K. Kobilka, Structure and dynamics of GPCR signaling complexes, Nat. Struct. Mol. Biol., 2018, 25, 4 CrossRef CAS PubMed
- K. Eichel and M. von Zastrow, Subcellular organization of GPCR signaling, Trends Pharmacol. Sci., 2018, 39, 200 CrossRef CAS PubMed
- E. Muratspahić, M. Freissmuth and C. W. Gruber, Nature-derived peptides: a growing niche for GPCR ligand discovery, Trends Pharmacol. Sci., 2019, 40, 309 CrossRef PubMed
- A. P. Davenport, C. C. G. Scully, C. de Graaf, A. J. H. Brown and J. J. Maguire, Advances in therapeutic peptides targeting G protein-coupled receptors, Nat. Rev. Drug Discovery, 2020, 19, 389–413 CrossRef CAS PubMed
- J. L. Lau and M. K. Dunn, Therapeutic peptides: Historical perspectives, current development trends, and future directions, Bioorg. Med. Chem., 2018, 26, 2700 CrossRef CAS PubMed
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis and J. Clardy,
et al., Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature, Nat. Prod. Rep., 2013, 30, 108 RSC
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif, J. Mol. Biol., 1999, 294, 1327 CrossRef CAS PubMed
- O. Saether, D. J. Craik, I. D. Campbell, K. Sletten, J. Juul and D. G. Norman, Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1, Biochemistry, 1995, 34, 4147 CrossRef CAS PubMed
- M. L. Colgrave and D. J. Craik, Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: the importance of the cyclic cystine knot, Biochemistry, 2004, 43, 5965 CrossRef CAS PubMed
- A. G. Poth, M. L. Colgrave, R. E. Lyons, N. L. Daly and D. J. Craik, Discovery of an unusual biosynthetic origin for circular proteins in legumes, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10127 CrossRef CAS PubMed
- J. L. Dutton, R. F. Renda, C. Waine, R. J. Clark, N. L. Daly, C. V. Jennings, M. A. Anderson and D. J. Craik, Conserved structural and sequence elements implicated in the processing of gene-encoded circular proteins, J. Biol. Chem., 2004, 279, 46858 CrossRef CAS PubMed
- C. W. Gruber, M. Cemazar, R. J. Clark, T. Horibe, R. F. Renda, M. A. Anderson and D. J. Craik, A novel plant protein-disulfide isomerase involved in the oxidative folding of cystine knot defense proteins, J. Biol. Chem., 2007, 282, 20435 CrossRef CAS PubMed
- B. F. Conlan, A. D. Gillon, B. L. Barbeta and M. A. Anderson, Subcellular targeting and biosynthesis of cyclotides in plant cells, Am. J. Bot., 2011, 98, 2018 CrossRef CAS PubMed
- I. Saska, A. D. Gillon, N. Hatsugai, R. G. Dietzgen, I. Hara-Nishimura, M. A. Anderson and D. J. Craik, An asparaginyl endopeptidase mediates in vivo protein backbone cyclization, J. Biol. Chem., 2007, 282, 29721 CrossRef CAS PubMed
- M. A. Jackson, E. K. Gilding, T. Shafee, K. S. Harris, Q. Kaas, S. Poon, K. Yap, H. Jia, R. Guarino and L. Y. Chan,
et al., Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases, Nat. Commun., 2018, 9, 2411 CrossRef CAS PubMed
- F. B. H. Rehm, M. A. Jackson, E. De Geyter, K. Yap, E. K. Gilding, T. Durek and D. J. Craik, Papain-like cysteine proteases prepare plant cyclic peptide precursors for cyclization, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7831 CrossRef CAS PubMed
- C. W. Gruber, A. G. Elliott, D. C. Ireland, P. G. Delprete, S. Dessein, U. Göransson, M. Trabi, C. K. Wang, A. B. Kinghorn and E. Robbrecht,
et al., Distribution and evolution of circular miniproteins in flowering plants, Plant Cell, 2008, 20, 2471 CrossRef CAS PubMed
- R. Hellinger, J. Koehbach, D. E. Soltis, E. J. Carpenter, G. K. Wong and C. W. Gruber, Peptidomics of circular cysteine-rich plant peptides: analysis of the diversity of cyclotides from Viola tricolor by transcriptome and proteome mining, J. Proteome Res., 2015, 14, 4851 CrossRef CAS PubMed
- S. J. de Veer, M. W. Kan and D. J. Craik, Cyclotides: from structure to function, Chem. Rev., 2019, 119, 12375 CrossRef CAS PubMed
- M. E. Felizmenio-Quimio, N. L. Daly and D. J. Craik, Circular proteins in plants: solution structure of a novel macrocyclic trypsin inhibitor from Momordica cochinchinensis, J. Biol. Chem., 2001, 276, 22875 CrossRef CAS PubMed
- A. Heitz, J. F. Hernandez, J. Gagnon, T. T. Hong, T. T. Pham, T. M. Nguyen, D. Le-Nguyen and L. Chiche, Solution structure of the squash trypsin inhibitor MCoTI-II. A new family for cyclic knottins, Biochemistry, 2001, 40, 7973 CrossRef CAS PubMed
- D. J. Craik, Circling the enemy: cyclic proteins in plant defence, Trends Plant Sci., 2009, 14, 328 CrossRef CAS PubMed
- L. Gran, On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus, Acta Pharmacol. Toxicol., 1973, 33, 400 CrossRef CAS PubMed
- L. Gran, Oxytocic principles of Oldenlandia affinis, Lloydia, 1973, 36, 174 CAS
- D. J. Craik, M. H. Lee, F. B. H. Rehm, B. Tombling, B. Doffek and H. Peacock, Ribosomally-synthesised cyclic peptides from plants as drug leads and pharmaceutical scaffolds, Bioorg. Med. Chem., 2018, 26, 2727 CrossRef CAS PubMed
- Y. H. Huang, Q. Du and D. J. Craik, Cyclotides: Disulfide-rich peptide toxins in plants, Toxicon, 2019, 172, 33 CrossRef CAS PubMed
- C. Jennings, J. West, C. Waine, D. Craik and M. Anderson, Biosynthesis and insecticidal properties of plant cyclotides: the cyclic knotted proteins from Oldenlandia affinis, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10614 CrossRef CAS PubMed
- M. L. Colgrave, A. C. Kotze, Y. H. Huang, J. O'Grady, S. M. Simonsen and D. J. Craik, Cyclotides: natural, circular plant peptides that possess significant activity against gastrointestinal nematode parasites of sheep, Biochemistry, 2008, 47, 5581 CrossRef CAS PubMed
- M. L. Colgrave, Y. H. Huang, D. J. Craik and A. C. Kotze, Cyclotide interactions with the nematode external surface, Antimicrob. Agents Chemother., 2010, 54, 2160 CrossRef CAS PubMed
- E. Svangard, U. Göransson, Z. Hocaoglu, J. Gullbo, R. Larsson, P. Claeson and L. Bohlin, Cytotoxic cyclotides from Viola tricolor, J. Nat. Prod., 2004, 67, 144 CrossRef PubMed
- M. E. F. Pinto, J. Z. G. Najas, L. G. Magalhaes, A. F. Bobey, J. N. Mendonca, N. P. Lopes, F. M. Leme, S. P. Teixeira, M. Trovo and A. D. Andricopulo,
et al., Inhibition of breast cancer cell migration by cyclotides isolated from Pombalia calceolaria, J. Nat. Prod., 2018, 81, 1203 CrossRef CAS PubMed
- S. L. Gerlach, R. Burman, L. Bohlin, D. Mondal and U. Göransson, Isolation, characterization, and bioactivity of cyclotides from the Micronesian plant Psychotria leptothyrsa, J. Nat. Prod., 2010, 73, 1207 CrossRef CAS PubMed
- Z. Sen, X. K. Zhan, J. Jing, Z. Yi and Z. Wanqi, Chemosensitizing activities of cyclotides from Clitoria ternatea in paclitaxel-resistant lung cancer cells, Oncol. Lett., 2013, 5, 641 CrossRef PubMed
- J. P. Tam, Y. A. Lu, J. L. Yang and K. W. Chiu, An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8913 CrossRef CAS PubMed
- M. Pranting, C. Loov, R. Burman, U. Göransson and D. I. Andersson, The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria, J. Antimicrob. Chemother., 2010, 65, 1964 CrossRef PubMed
- M. Zarrabi, R. Dalirfardouei, Z. Sepehrizade and R. K. Kermanshahi, Comparison of the antimicrobial effects of semipurified cyclotides from Iranian Viola odorata against some of plant and human pathogenic bacteria, J. Appl. Microbiol., 2013, 115, 367 CrossRef CAS PubMed
- S. Z. Malik, M. Linkevicius, U. Göransson and D. I. Andersson, Resistance to the cyclotide Cycloviolacin O2 in Salmonella enterica caused by different mutations that often confer cross-resistance or collateral sensitivity to other antimicrobial peptides, Antimicrob. Agents Chemother., 2017, 61, e00684 CrossRef CAS PubMed
- S. Troeira Henriques and D. J. Craik, Cyclotide structure and function: the role of membrane binding and permeation, Biochemistry, 2017, 56, 669 CrossRef CAS PubMed
- B. Slazak, M. Kapusta, A. A. Stromstedt, A. Slomka, M. Krychowiak, M. Shariatgorji, P. E. Andren, J. Bohdanowicz, E. Kuta and U. Göransson, How does the sweet Violet (Viola odorata L.) fight pathogens and pests - cyclotides as a comprehensive plant host defense system, Front. Plant Sci., 2018, 9, 1296 CrossRef PubMed
- H. Kamimori, K. Hall, D. J. Craik and M. I. Aguilar, Studies on the membrane interactions of the cyclotides kalata B1 and kalata B6 on model membrane systems by surface plasmon resonance, Anal. Biochem., 2005, 337, 149 CrossRef CAS PubMed
- Z. O. Shenkarev, K. D. Nadezhdin, V. A. Sobol, A. G. Sobol, L. Skjeldal and A. S. Arseniev, Conformation and mode of membrane interaction in cyclotides. Spatial structure of kalata B1 bound to a dodecylphosphocholine micelle, FEBS J., 2006, 273, 2658 CrossRef CAS PubMed
- S. T. Henriques, Y. H. Huang, K. J. Rosengren, H. G. Franquelim, F. A. Carvalho, A. Johnson, S. Sonza, G. Tachedjian, M. A. Castanho and N. L. Daly,
et al., Decoding the membrane activity of the cyclotide kalata B1: the importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-HIV activities, J. Biol. Chem., 2011, 286, 24231 CrossRef CAS PubMed
- Y. H. Huang, M. L. Colgrave, R. J. Clark, A. C. Kotze and D. J. Craik, Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity, J. Biol. Chem., 2010, 285, 10797 CrossRef CAS PubMed
- S. T. Henriques, Y. H. Huang, M. A. Castanho, L. A. Bagatolli, S. Sonza, G. Tachedjian, N. L. Daly and D. J. Craik, Phosphatidylethanolamine binding is a conserved feature of cyclotide-membrane interactions, J. Biol. Chem., 2012, 287, 33629 CrossRef CAS PubMed
- C. K. Wang, H. P. Wacklin and D. J. Craik, Cyclotides insert into lipid bilayers to form membrane pores and destabilize the membrane through hydrophobic and phosphoethanolamine-specific interactions, J. Biol. Chem., 2012, 287, 43884 CrossRef CAS PubMed
- L. Sando, S. T. Henriques, F. Foley, S. M. Simonsen, N. L. Daly, K. N. Hall, K. R. Gustafson, M. I. Aguilar and D. J. Craik, A Synthetic mirror image of kalata B1 reveals that cyclotide activity is independent of a protein receptor, ChemBioChem, 2011, 12, 2456 CrossRef CAS PubMed
- S. T. Henriques, H. Peacock, A. H. Benfield, C. K. Wang and D. J. Craik, Is the mirror image a true reflection? Intrinsic membrane chirality modulates peptide binding, J. Am. Chem. Soc., 2019, 141, 20460 CrossRef CAS PubMed
- R. Burman, A. A. Stromstedt, M. Malmsten and U. Göransson, Cyclotide-membrane interactions: defining factors of membrane binding, depletion and disruption, Biochim. Biophys. Acta, 2011, 1808, 2665 CrossRef CAS PubMed
- L. Cascales, S. T. Henriques, M. C. Kerr, Y. H. Huang, M. J. Sweet, N. L. Daly and D. J. Craik, Identification and characterization of a new family of cell-penetrating peptides: cyclic cell-penetrating peptides, J. Biol. Chem., 2011, 286, 36932 CrossRef CAS PubMed
- S. T. Henriques, Y. H. Huang, S. Chaousis, M. A. Sani, A. G. Poth, F. Separovic and D. J. Craik, The prototypic cyclotide kalata B1 has a unique mechanism of entering cells, Chem. Biol., 2015, 22, 1087 CrossRef CAS PubMed
- J. Koehbach, M. O'Brien, M. Muttenthaler, M. Miazzo, M. Akcan, A. G. Elliott, N. L. Daly, P. J. Harvey, S. Arrowsmith and S. Gunasekera,
et al., Oxytocic plant cyclotides as templates for peptide G protein-coupled receptor ligand design, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(52), 21183 CrossRef CAS PubMed
- P. Keov, Z. Liutkeviciute, R. Hellinger, R. J. Clark and C. W. Gruber, Discovery of peptide probes to modulate oxytocin-type receptors of insects, Sci. Rep., 2018, 8, 10020 CrossRef PubMed
- M. Fahradpour, P. Keov, C. Tognola, E. Perez-Santamarina, P. J. McCormick, A. Ghassempour and C. W. Gruber, Cyclotides isolated from an ipecac root extract antagonize the corticotropin releasing factor type 1 receptor, Front. Pharmacol., 2017, 8, 616 CrossRef PubMed
- I. Shimada, T. Ueda, Y. Kofuku, M. T. Eddy and K. Wuthrich, GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures, Nat. Rev. Drug Discovery, 2019, 18, 59 CrossRef CAS PubMed
- M. Congreve, C. de Graaf, N. A. Swain and C. G. Tate, Impact of GPCR structures on drug discovery, Cell, 2020, 181, 81 CrossRef CAS PubMed
- J. F. White, N. Noinaj, Y. Shibata, J. Love, B. Kloss, F. Xu, J. Gvozdenovic-Jeremic, P. Shah, J. Shiloach and C. G. Tate,
et al., Structure of the agonist-bound neurotensin receptor, Nature, 2012, 490, 508 CrossRef CAS PubMed
- J. Koehbach and C. W. Gruber, From ethnopharmacology to drug design, Commun. Integr. Biol., 2013, 6, e27583 CrossRef PubMed
- C. K. Wang and D. J. Craik, Designing macrocyclic disulfide-rich peptides for biotechnological applications, Nat. Chem. Biol., 2018, 14, 417 CrossRef CAS PubMed
- D. J. Craik and J. Du, Cyclotides as drug design scaffolds, Curr.
Opin. Chem. Biol., 2017, 38, 8 CrossRef CAS PubMed
- G. Ruiz-Gomez, J. D. Tyndall, B. Pfeiffer, G. Abbenante and D. P. Fairlie, Update 1 of: over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure, Chem. Rev., 2010, 110, PR1–41 CrossRef PubMed
- K. J. Rosengren, N. L. Daly, M. R. Plan, C. Waine and D. J. Craik, Twists, knots, and rings in proteins. Structural definition of the cyclotide framework, J. Biol. Chem., 2003, 278, 8606 CrossRef CAS PubMed
- S. E. E. Mills, K. P. Nicolson and B. H. Smith, Chronic pain: a review of its epidemiology and associated factors in population-based studies, Br. J. Anaesth., 2019, 123, e273 CrossRef PubMed
- M. W. Pennington, A. Czerwinski and R. S. Norton, Peptide therapeutics from venom: current status and potential, Bioorg. Med. Chem., 2018, 26, 2738 CrossRef CAS PubMed
- A. H. Jin, M. Muttenthaler, S. Dutertre, S. W. A. Himaya, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Conotoxins: chemistry and biology, Chem. Rev., 2019, 119, 11510 CrossRef CAS PubMed
- A. Schmidtko, J. Lotsch, R. Freynhagen and G. Geisslinger, Ziconotide for treatment of severe chronic pain, Lancet, 2010, 375, 1569 CrossRef CAS
- J. B. Calixto, R. Medeiros, E. S. Fernandes, J. Ferreira, D. A. Cabrini and M. M. Campos, Kinin B1 receptors: key G-protein-coupled receptors and their role in inflammatory and painful processes, Br. J. Pharmacol., 2004, 143, 803 CrossRef CAS PubMed
- H. Huang and M. R. Player, Bradykinin B1 receptor antagonists as potential therapeutic agents for pain, J. Med. Chem., 2010, 53, 5383 CrossRef CAS PubMed
- S. D. Kuduk and M. G. Bock, Bradykinin B1 receptor antagonists as novel analgesics: a retrospective of selected medicinal chemistry developments, Curr. Top. Med. Chem., 2008, 8, 1420 CrossRef CAS PubMed
- M. Cicardi, A. Banerji, F. Bracho, A. Malbran, B. Rosenkranz, M. Riedl, K. Bork, W. Lumry, W. Aberer and H. Bier,
et al., Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema, N. Engl. J. Med., 2010, 363, 532 CrossRef CAS PubMed
- C. T. Wong, D. K. Rowlands, C. H. Wong, T. W. Lo, G. K. Nguyen, H. Y. Li and J. P. Tam, Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment, Angew. Chem. Int. Ed. Engl., 2012, 51, 5620 CrossRef CAS PubMed
- D. Regoli, G. Drapeau, P. Rovero, S. Dion, P. D'Orleans-Juste and J. Barabe, The actions of kinin antagonists on B1 and B2 receptor systems, Eur. J. Pharmacol., 1986, 123, 61 CrossRef CAS PubMed
- S. Gunasekera, F. M. Foley, R. J. Clark, L. Sando, L. J. Fabri, D. J. Craik and N. L. Daly, Engineering stabilized vascular endothelial growth factor-A antagonists: synthesis, structural characterization, and bioactivity of grafted analogues of cyclotides, J. Med. Chem., 2008, 51, 7697 CrossRef CAS PubMed
- G. B. D. O. Collaborators, A. Afshin, M. H. Forouzanfar, M. B. Reitsma, P. Sur, K. Estep, A. Lee, L. Marczak, A. H. Mokdad and M. Moradi-Lakeh,
et al., Health effects of overweight and obesity in 195 countries over 25 years, N. Engl. J. Med., 2017, 377, 13 CrossRef PubMed
- B. A. Swinburn, V. I. Kraak, S. Allender, V. J. Atkins, P. I. Baker, J. R. Bogard, H. Brinsden, A. Calvillo, O. De Schutter and R. Devarajan,
et al., The Global syndemic of obesity, undernutrition, and climate change: the lancet commission report, Lancet, 2019, 393, 791 CrossRef
- X. Pi-Sunyer, A. Astrup, K. Fujioka, F. Greenway, A. Halpern, M. Krempf, D. C. Lau, C. W. le Roux, R. Violante Ortiz and C. B. Jensen,
et al., A randomized, controlled trial of 3.0 mg of liraglutide in weight management, N. Engl. J. Med., 2015, 373, 11 CrossRef PubMed
- J. M. Adams, H. Pei, D. A. Sandoval, R. J. Seeley, R. B. Chang, S. D. Liberles and D. P. Olson, Liraglutide modulates appetite and body weight through glucagon-Like peptide 1 receptor-expressing glutamatergic neurons, Diabetes, 2018, 67, 1538 CrossRef CAS PubMed
- J. W. Hill and L. D. Faulkner, The role of the melanocortin system in metabolic disease: new developments and advances, Neuroendocrinology, 2017, 104, 330 CrossRef CAS PubMed
- R. A. Adan and M. J. Kas, Inverse agonism gains weight, Trends Pharmacol. Sci., 2003, 24, 315 CrossRef CAS PubMed
- Y. Yang, Structure, function and regulation of the melanocortin receptors, Eur. J. Pharmacol., 2011, 660, 125 CrossRef CAS PubMed
- W. Fan, B. A. Boston, R. A. Kesterson, V. J. Hruby and R. D. Cone, Role of melanocortinergic neurons in feeding and the agouti obesity syndrome, Nature, 1997, 385, 165 CrossRef CAS PubMed
- P. Kuhnen, H. Krude and H. Biebermann, Melanocortin-4 receptor signalling: importance for weight regulation and obesity treatment, Trends Mol. Med., 2019, 25, 136 CrossRef PubMed
- V. J. Hruby, B. C. Wilkes, M. E. Hadley, F. Al-Obeidi, T. K. Sawyer, D. J. Staples, A. E. de Vaux, O. Dym, A. M. Castrucci and M. F. Hintz,
et al., alpha-Melanotropin: the minimal active sequence in the frog skin bioassay, J. Med. Chem., 1987, 30, 2126 CrossRef CAS PubMed
- R. Eliasen, N. L. Daly, B. S. Wulff, T. L. Andresen, K. W. Conde-Frieboes and D. J. Craik, Design, synthesis, structural and functional characterization of novel melanocortin agonists based on the cyclotide kalata B1, J. Biol. Chem., 2012, 287, 40493 CrossRef CAS PubMed
- Y. Feng, C. C. Broder, P. E. Kennedy and E. A. Berger, HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor, Science, 1996, 272, 872 CrossRef CAS PubMed
- H. J. Anders, P. Romagnani and A. Mantovani, Pathomechanisms: homeostatic chemokines in health, tissue regeneration, and progressive diseases, Trends Mol. Med., 2014, 20, 154 CrossRef CAS PubMed
- R. Janssens, S. Struyf and P. Proost, The unique structural and functional features of CXCL12, Cell. Mol. Immunol., 2018, 15, 299 CrossRef CAS PubMed
- F. Guo, Y. Wang, J. Liu, S. C. Mok, F. Xue and W. Zhang, CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks, Oncogene, 2016, 35, 816 CrossRef CAS PubMed
- W. Zhou, S. Guo, M. Liu, M. E. Burow and G. Wang, Targeting CXCL12/CXCR4 axis in tumor immunotherapy, Curr. Med. Chem., 2019, 26, 3026 CrossRef CAS PubMed
- T. L. Aboye, H. Ha, S. Majumder, F. Christ, Z. Debyser, A. Shekhtman, N. Neamati and J. A. Camarero, Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity, J. Med. Chem., 2012, 55, 10729 CrossRef CAS PubMed
- S. J. DeMarco, H. Henze, A. Lederer, K. Moehle, R. Mukherjee, B. Romagnoli, J. A. Robinson, F. Brianza, F. O. Gombert and S. Lociuro,
et al., Discovery of novel, highly potent and selective beta-hairpin mimetic CXCR4 inhibitors with excellent anti-HIV activity and pharmacokinetic profiles, Bioorg. Med. Chem., 2006, 14, 8396 CrossRef CAS PubMed
- R. T. Steigbigel, D. A. Cooper, P. N. Kumar, J. E. Eron, M. Schechter, M. Markowitz, M. R. Loutfy, J. L. Lennox, J. M. Gatell and J. K. Rockstroh,
et al., Raltegravir with optimized background therapy for resistant HIV-1 infection, N. Engl. J. Med., 2008, 359, 339 CrossRef PubMed
- S. C. Penchala, M. R. Miller, A. Pal, J. Dong, N. R. Madadi, J. Xie, H. Joo, J. Tsai, P. Batoon and V. Samoshin,
et al., A biomimetic approach for enhancing the in vivo half-life of peptides, Nat. Chem. Biol., 2015, 11, 793 CrossRef CAS PubMed
- W. G. Lesniak, T. Aboye, S. Chatterjee, J. A. Camarero and S. Nimmagadda, In vivo evaluation of an engineered cyclotide as specific CXCR4 imaging reagent, Chemistry, 2017, 23, 14469 CrossRef CAS PubMed
- C. K. Wang, S. Stalmans, B. De Spiegeleer and D. J. Craik, Biodistribution of the cyclotide MCoTI-II, a cyclic disulfide-rich peptide drug scaffold, J. Pept. Sci., 2016, 22, 305 CrossRef CAS PubMed
- M. Paz Ocaranza, J. A. Riquelme, L. Garcia, J. E. Jalil, M. Chiong, R. A. S. Santos and S. Lavandero, Counter-regulatory renin-angiotensin system in cardiovascular disease, Nat. Rev. Cardiol., 2020, 17, 116 CrossRef PubMed
- J. Menon, D. R. Soto-Pantoja, M. F. Callahan, J. M. Cline, C. M. Ferrario, E. A. Tallant and P. E. Gallagher, Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2, Cancer Res., 2007, 67, 2809 CrossRef CAS PubMed
- P. E. Gallagher and E. A. Tallant, Inhibition of human lung cancer cell growth by angiotensin-(1-7), Carcinogenesis, 2004, 25, 2045 CrossRef CAS PubMed
- D. Iusuf, R. H. Henning, W. H. van Gilst and A. J. Roks, Angiotensin-(1-7): pharmacological properties and pharmacotherapeutic perspectives, Eur. J. Pharmacol., 2008, 585, 303 CrossRef CAS PubMed
- T. Aboye, C. J. Meeks, S. Majumder, A. Shekhtman, K. Rodgers and J. A. Camarero, Design of a MCoTI-Based Cyclotide with Angiotensin (1-7)-Like Activity, Molecules, 2016, 21, 152 CrossRef PubMed
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. Pulavarti, Q. Kaas and A. Eletsky,
et al., Accurate de novo design of hyperstable constrained peptides, Nature, 2016, 538, 329 CrossRef CAS PubMed
- G. K. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis, Nat. Chem. Biol., 2014, 10, 732 CrossRef CAS PubMed
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden and D. J. Craik,
et al., Efficient backbone cyclization of linear peptides by a recombinant asparaginyl endopeptidase, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed
- K. Li, H. L. Condurso, G. Li, Y. Ding and S. D. Bruner, Structural basis for precursor protein-directed ribosomal peptide macrocyclization, Nat. Chem. Biol., 2016, 12, 973 CrossRef CAS PubMed
- H. Yin, D. J. Craik and C. K. Wang, Anchor residues guide form and function in grafted peptides, Angew. Chem., Int. Ed., 2019, 58, 7652 CrossRef CAS PubMed
- C. K. Wang, C. W. Gruber, M. Cemazar, C. Siatskas, P. Tagore, N. Payne, G. Sun, S. Wang, C. C. Bernard and D. J. Craik, Molecular grafting onto a stable framework yields novel cyclic peptides for the treatment of multiple sclerosis, ACS Chem. Biol., 2014, 9, 156 CrossRef CAS PubMed
- K. Thell, R. Hellinger, E. Sahin, P. Michenthaler, M. Gold-Binder, T. Haider, M. Kuttke, Z. Liutkeviciute, U. Göransson and C. Grundemann,
et al., Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3960 CrossRef CAS PubMed
- C. Yanover, N. Jain, G. Pierce, T. E. Howard and Z. E. Sauna, Pharmacogenetics and the immunogenicity of protein therapeutics, Nat. Biotechnol., 2011, 29, 870 CrossRef PubMed
- L. M. Snyder, M. C. Chiang, E. Loeza-Alcocer, Y. Omori, J. Hachisuka, T. D. Sheahan, J. R. Gale, P. C. Adelman, E. I. Sypek and S. A. Fulton,
et al., Kappa opioid receptor distribution and function in primary afferents, Neuron, 2018, 99, 1274 CrossRef CAS PubMed
- G. Guidotti, L. Brambilla and D. Rossi, Cell-Penetrating Peptides: from basic research to clinics, Trends Pharmacol. Sci., 2017, 38, 406 CrossRef CAS PubMed
- Y. Ji, S. Majumder, M. Millard, R. Borra, T. Bi, A. Y. Elnagar, N. Neamati, A. Shekhtman and J. A. Camarero, In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide, J. Am. Chem. Soc., 2013, 135, 11623 CrossRef CAS PubMed
- Y. H. Huang, S. Chaousis, O. Cheneval, D. J. Craik and S. T. Henriques, Optimization of the cyclotide framework to improve cell penetration properties, Front. Pharmacol., 2015, 6, 17 Search PubMed
- R. Irannejad, J. C. Tomshine, J. R. Tomshine, M. Chevalier, J. P. Mahoney, J. Steyaert, S. G. Rasmussen, R. K. Sunahara, H. El-Samad and B. Huang,
et al., Conformational biosensors reveal GPCR signalling from endosomes, Nature, 2013, 495, 534 CrossRef CAS PubMed
- A. R. B. Thomsen, B. Plouffe, T. J. Cahill 3rd, A. K. Shukla, J. T. Tarrasch, A. M. Dosey, A. W. Kahsai, R. T. Strachan, B. Pani and J. P. Mahoney,
et al., GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling, Cell, 2016, 166, 907 CrossRef CAS PubMed
- D. D. Jensen, T. Lieu, M. L. Halls, N. A. Veldhuis, W. L. Imlach, Q. N. Mai, D. P. Poole, T. Quach, L. Aurelio and J. Conner,
et al., Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief, Sci. Transl. Med., 2017, 9, eaal3447 CrossRef PubMed
- A. H. Nguyen, A. R. B. Thomsen, T. J. Cahill 3rd, R. Huang, L. Y. Huang, T. Marcink, O. B. Clarke, S. Heissel, A. Masoudi and D. Ben-Hail,
et al., Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex, Nat. Struct. Mol. Biol., 2019, 26, 1123 CrossRef CAS PubMed
- J. S. Smith, R. J. Lefkowitz and S. Rajagopal, Biased signalling: from simple switches to allosteric microprocessors, Nat. Rev. Drug Discovery, 2018, 17, 243 CrossRef CAS PubMed
- H. Zhang, E. Sturchler, J. Zhu, A. Nieto, P. A. Cistrone, J. Xie, L. He, K. Yea, T. Jones and R. Turn,
et al., Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects, Nat. Commun., 2015, 6, 8918 CrossRef CAS PubMed
- D. M. Ryba, J. Li, C. L. Cowan, B. Russell, B. M. Wolska and R. J. Solaro, Long-term biased beta-arrestin signaling improves cardiac structure and function in dilated cardiomyopathy, Circulation, 2017, 135, 1056 CrossRef CAS PubMed
- S. Gunasekera, N. L. Daly, R. J. Clark and D. J. Craik, Dissecting the oxidative folding of circular
cystine knot miniproteins, Antioxid. Redox Signaling, 2009, 11, 971 CrossRef CAS PubMed
- J. Weidmann and D. J. Craik, Discovery, structure, function, and applications of cyclotides: circular proteins from plants, J. Exp. Bot., 2016, 67, 4801 CrossRef CAS PubMed
- K. M. Witherup, M. J. Bogusky, P. S. Anderson, H. Ramjit, R. W. Ransom, T. Wood and M. Sardana, Cyclopsychotride A, a biologically active, 31-residue cyclic peptide isolated from Psychotria longipes, J. Nat. Prod., 1994, 57, 1619 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |